

Abstract
Coenzyme Q (CoQ) is a remarkably hydrophobic, redox-active lipid that empowers diverse cellular processes. Although most known for shuttling electrons between mitochondrial electron transport chain (ETC) complexes, the roles for CoQ are far more wide-reaching and ever-expanding. CoQ serves as a conduit for electrons from myriad pathways to enter the ETC, acts as a cofactor for biosynthetic and catabolic reactions, detoxifies damaging lipid species, and engages in cellular signaling and oxygen sensing. Many open questions remain regarding the biosynthesis, transport, and metabolism of CoQ, which hinders our ability to treat human CoQ deficiency. Here, we recount progress in filling these knowledge gaps, highlight unanswered questions, and underscore the need for novel tools to enable discoveries and improve the treatment of CoQ-related diseases.
Keywords: Coenzyme Q, Ubiquinone, Complex Q, Oxidative Phosphorylation, Lipids, Mitochondria
Coenzyme Q in Biochemistry and Beyond
Coenzyme Q (CoQ, ubiquinone; see Glossary) is an essential redox-active lipid with extraordinary biochemical properties. Decades of research have linked CoQ to numerous cellular processes, human diseases, and therapeutic regimens, and recent efforts are revealing surprising new roles. Despite being discovered 65 years ago in Madison, WI, USA [1] and Liverpool, UK [2], many open questions regarding the biosynthesis, noncanonical function, and transport of CoQ continue to limit our ability to treat human diseases of CoQ deficiency. We have previously provided an in-depth review of the biochemistry of CoQ synthesis [3]; here we will focus on an overview of the biosynthetic pathway, discuss the current state of knowledge on how CoQ is assimilated and distributed throughout the cell, and describe current efforts to overcome CoQ deficiency in disease, highlighting key findings in the past five years and underscoring aspects that remain unsolved.
Known Cellular Roles of Coenzyme Q
We begin by recounting established CoQ functions that highlight its diverse contributions to cellular homeostasis.
CoQ as a Mitochondrial Cofactor
CoQ performs its canonical role in the mitochondrial electron transport chain (ETC) by relaying electrons from oxidative phosphorylation (OxPhos) complexes I and II to complex III, helping to generate the proton motive force that drives ATP synthesis [3]. Although often overlooked, CoQ also accepts electrons as a cofactor for enzymes involved in pyrimidine biosynthesis, proline catabolism, glycolysis, fatty acid oxidation, and sulfide detoxification (Figure 1a), thereby providing diverse pathways for electrons to reach complex III [4]. In certain contexts, CoQ’s cofactor function is paramount; for example, certain human cancer cells rely on complex III more for CoQ regeneration to drive DHODH activity and the TCA cycle than for assisting NAD+ regeneration or promoting ATP production [5]. Relatedly, a hypothesis surrounding CoQ segmentation has emerged, proposing that mitochondrial CoQ may exist in two functionally distinct pools: one population of CoQ that associates with respiratory super-complexes would receive their electrons from NADH via complex I, while the remaining freely diffusing IMM CoQ would receive electrons from FAD-dependent enzymes [6]. This topic is still highly debated, yet it is tempting to speculate that maintaining distinct CoQ pools might have important functional consequences, including modulating mitochondrial respiratory capacity and redox signaling [7].
Figure 1. Cellular Roles of Coenzyme Q.
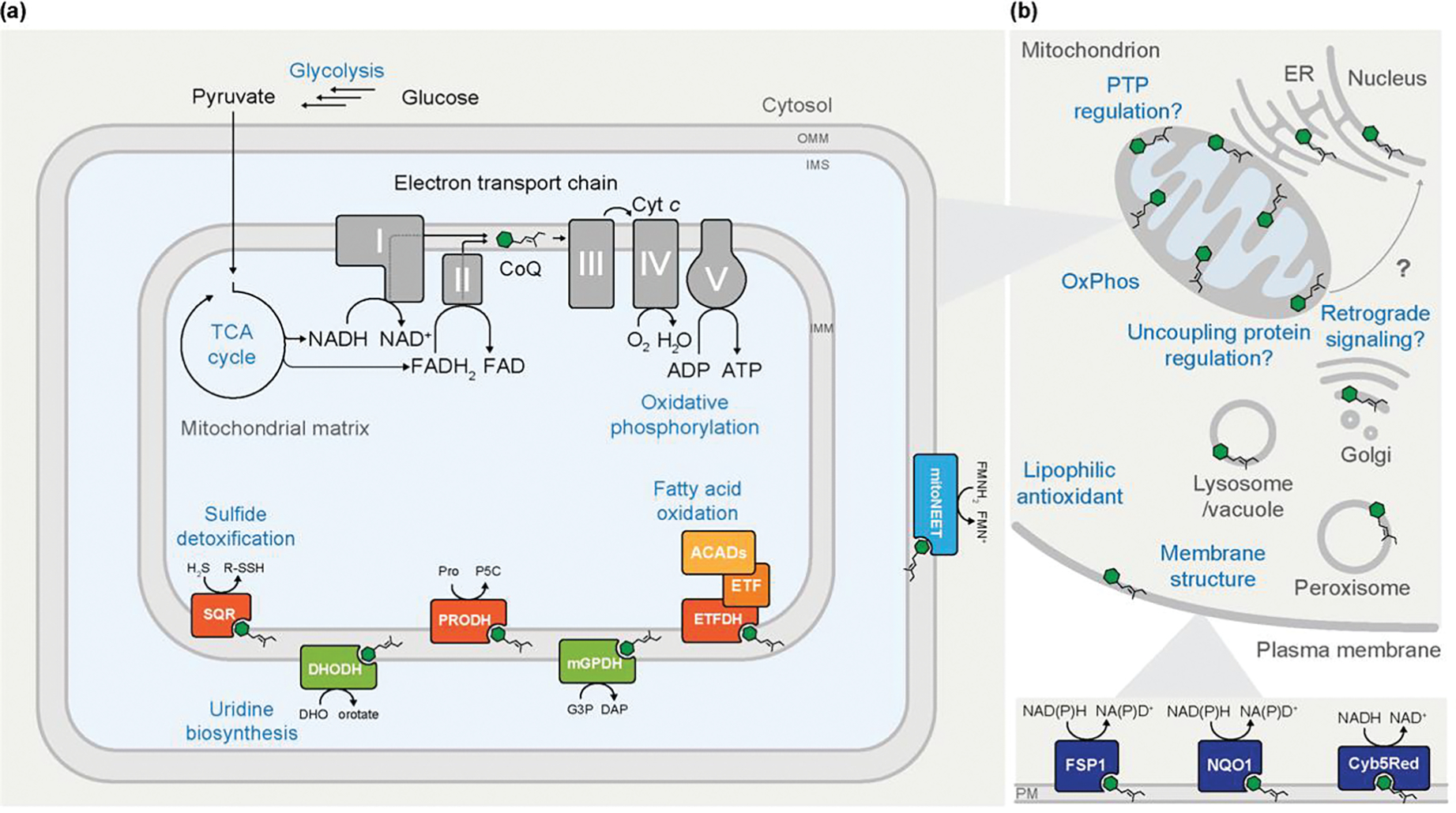
(A) Although coenzyme Q (CoQ) is typically regarded for its central role in the mitochondrial electron transport chain and oxidative phosphorylation (OxPhos), it additionally acts as a cofactor for multiple mitochondrial enzymes. As part of central energy metabolism, the tricarboxylic acid (TCA) cycle provides reducing equivalents that enable the reduction of CoQ at OxPhos complexes I and II. Reduction of CoQ by enzymes involved in uridine biosynthesis, fatty acid oxidation, and sulfide detoxification, among others, provides additional routes of electron entry to complex III of the ETC, which ultimately drives ATP synthesis. (B) An overview of the widespread cellular functions of CoQ. CoQ is present in nearly all cellular membranes. Some extramitochondrial roles of CoQ are depicted, including modulating membrane structure dynamics and regulating the uncoupling protein, though many of the roles are incompletely characterized. At the plasma membrane (PM), reduced CoQH2 acts as lipophilic antioxidant where it can be reduced by various oxidoreductases to combat lipid peroxidation and oxidative damage.
Combatting Ferroptosis
Extramitochondrial CoQ has been implicated in combatting ferroptosis, a form of nonapoptotic cell death caused by iron-dependent phospholipid peroxidation [8]. Oxidative stress from environmental and metabolic stimuli can catalyze the formation of toxic phospholipid hydroperoxides (PLOOHs) from polyunsaturated fatty acids (PUFAs), resulting in a loss of membrane integrity, oxidative damage to macromolecules, and eventually cell death [9]. In mammals, GPX4, a phospholipid hydroperoxide glutathione peroxidase, provides a major form of defense against ferroptosis by neutralizing PLOOHs [10]. Recent studies identified a GPX4-independent mechanism of ferroptosis protection via FSP1, an oxidoreductase that reduces plasma membrane (PM) CoQ to the hydroquinone form (CoQH2), which in turn suppresses PLOOHs [11,12].
Extramitochondrial Antioxidation
Before the discovery of FSP1, CoQ was long recognized as an important PM lipophilic antioxidant in both yeast and mammalian systems. The primary oxidoreductases responsible for maintaining the reduced PM CoQ pool are NAD(P)H:Quinone Oxidoreductase 1 (NQO1) and NADH-cytochrome b5 reductase (Cyb5Red) [13] (Figure 1b). NQO1 catalyzes the two-electron reduction of CoQ as part of the PM redox system, and can furthermore function as a superoxide reductase, generating NAD+ and modulating various signaling enzymes, including HIF1-α, p53, and sirtuins [14]. Cyb5Red, localized at both the PM and ER, is induced under stress conditions and catalyzes electron transfer from NADH to CoQ, thus contributing to the protective antioxidant pool of CoQ [15]. While this antioxidant function of Cyb5Red has been studied primarily at the PM [16], its presence in ER membranes suggests that it could likewise maintain the balance of CoQ/CoQH2 in this organelle. Very little is known concerning how the redox state of CoQ is dictated in organellar membranes beyond mitochondria and the PM, nor the functional consequences of maintaining a precise CoQ/CoQH2 ratio in these contexts. CoQ is also present in lysosomal membranes and can be reduced via a lysosomal ETC [17], although the proteins involved have yet to be identified. CoQ deficiency has been linked to aberrant lysosomal acidification [18], but how this relates to the lysosomal ETC is unknown.
Additional Understudied Roles of CoQ
Beyond its ETC, cofactor, and antioxidant activities, CoQ has been linked to mammalian energy regulation pathways, though these connections are often incompletely understood. Brown adipose tissue (BAT) can engage in facultative thermogenesis, whereby uncoupling proteins (UCPs) act to dissipate the mitochondrial proton gradient to release heat [19]. Early studies characterized CoQ as a requisite activator of proton transport by UCP1 [20] and UCP2/UCP3 [21] in reconstituted liposomes, though these findings have been disputed [22]. More recently, studies in BAT have demonstrated that the scavenger receptor (SR) protein CD36 is required for CoQ uptake and normal BAT function, with Cd36−/− mice exhibiting decreased CoQ content, defective non-shivering thermogenesis, and cold intolerance [23], yet the molecular mechanisms at play are still elusive. An emerging theme surrounding CoQ homeostasis is that the ratio of oxidized CoQ to reduced CoQH2 can have important consequences in sensing and reflecting different metabolic states of the cell [24]. In mouse chemoreceptor cells, the CoQH2/CoQ ratio has been linked to acute oxygen sensing downstream of complex I-derived NADH and ROS [25]. Similarly, in states of hypoxia or impaired ETC in mice, high CoQH2/CoQ ratio can trigger reversal of complex II, resulting in the reduction of fumarate as an alternative electron acceptor [26]. Further work will be required to unravel if these may be mechanisms to link CoQ redox state to the regulation of nucleotide pools or downstream gene expression.
Coenzyme Q Biosynthesis and Regulation
CoQ is among the most hydrophobic molecules in nature, with a long polyisoprenoid lipid tail capped by a redox-active quinone head group (Figure 2a–b). These distinctive properties of CoQ present cells with significant challenges for its biosynthesis. Nonetheless, organisms across nearly all domains of life synthesize CoQ endogenously [27], with production in eukaryotes taking place primarily at the inner mitochondrial membrane (IMM) [28].
Figure 2. Coenzyme Q Structure and Eukaryotic Biosynthesis.
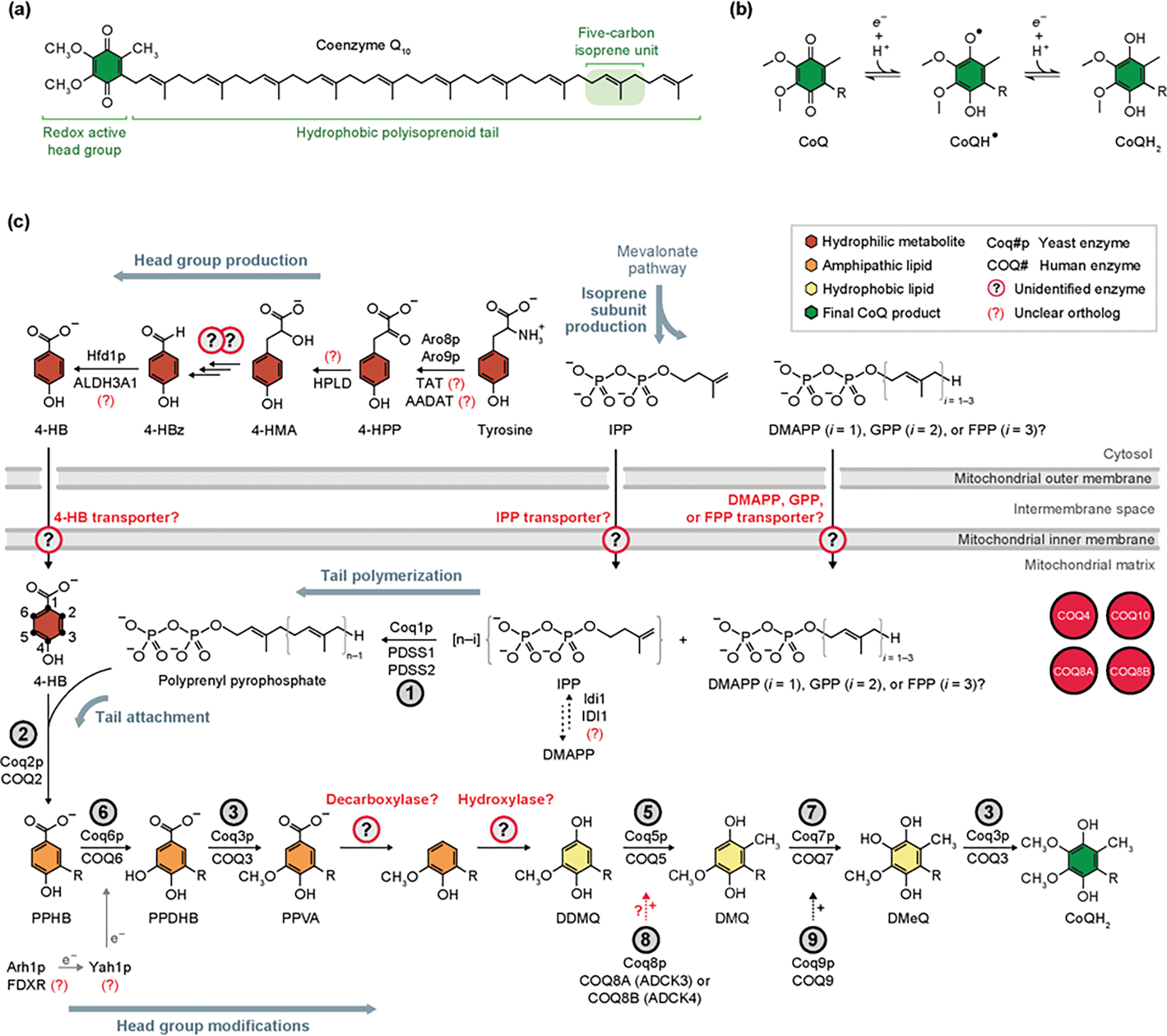
(A) The chemical structure of coenzyme Q10 (CoQ10). (B) Single electron transfer redox reactions of the CoQ head group, whereby CoQ can cycle through the oxidized (CoQ), radical (CoQH•), and fully reduced (CoQH2) forms. This redox activity allows CoQ function as a cofactor for numerous enzymes, relay electrons in the ETC, and act as an antioxidant. ‘R’ indicates the polyisoprenoid tail. (C) The primary eukaryotic pathway, conserved from S. cerevisiae to humans, of CoQ biosynthesis is depicted. CoQ production begins with the head group precursor 4-HB, derived from tyrosine, and tail subunit IPP, derived from the mevalonate pathway, which are both transported into the mitochondrial matrix by unknown means. Following tail polymerization and head group attachment, CoQ intermediates are processed through a series of head group modifications to yield mature CoQ. Biosynthetic enzymes that catalyze each reaction are denoted by the circled number above each arrow. Unidentified enzymes and transporters indicated by circled question marks. Auxiliary proteins with some unclear involvement in CoQ biosynthesis are depicted in red circles. ‘+’ symbol near dashed arrows designates a hypothesized supportive role for the indicated reaction. Abbreviations: 4-HBz, 4-hydroxybenzaldehyde; 4-HMA, 4-hydroxymandelate; 4-HPP, 4-hydroxyphenylpyruvate; AADAT, mitochondrial alpha-aminoadipate aminotransferase; ALDH3A1, aldehyde dehydrogenase 3A1; DDMQ, demethoxy-demethyl-coenzyme Q; DMQ, demethoxy-coenzyme Q; DMeQ, demethyl-coenzyme Q; DMAPP, dimethylallyl pyrophosphate; FDX2 (FDX1L), mitochondrial ferredoxin 2 (ferredoxin 1-like); FDXR, adrenodoxin reductase; FPP, farnesyl pyrophosphate; GPP, geranyl pyrophosphate; HPLD, hydroxyphenylpyruvate dioxygenase-like protein; IDI1, isopentenyl-diphosphate delta isomerase 1; IPP, isopentenyl pyrophosphate; PDSS1, prenyl (decaprenyl) diphosphate synthase subunit 1; PPDHB, polyprenyl-dihydroxybenzoate; PPHB, polyprenyl-hydroxybenzoate; PPVA, polyprenyl-vanillic acid; TAT, tyrosine aminotransferase.
Recap of Biosynthetic Pathway and Updates
CoQ biosynthesis occurs in four stages: (i) head group production, (ii) isoprenoid tail production, (iii) attachment of the head group precursor to the tail, and (iv) head group modifications (Figure 2c). The universally conserved head group precursor, 4-hydroxybenzoate (4-HB), is derived from tyrosine in mammals, however yeast and bacteria can also produce it de novo via the shikimate pathway [3]. Though many of the steps in the tyrosine-to-4-HB pathway remain undefined, Hfd1p catalyzes the dehydrogenation of 4-hydroxybenzaldehyde (4-HBz) to 4-HB [29–31] in yeast. Efforts to elucidate missing enzymes in this pathway have encountered significant enzymatic redundancy (Box 1), with up to five aminotransferases capable of catalyzing the tyrosine transamination to 4-hydroxyphenylpyruvate (4-HPP) in vivo [31]. Recent work involving heavy O2 tracing in human cells established 4-hydroxymandelate (4-HMA), produced from 4-HPP by hydroxyphenylpyruvate dioxygenase-like protein (HPLD), as an intermediate in this pathway [32], consistent with intermediates observed in yeast [31]. Work on the mandelate pathway in yeast suggests that there are multiple enzymes and additional steps required to convert 4-HPP to 4-HMA [33].
Box 1. Why is CoQ biosynthesis still not fully characterized?
The study of CoQ biology has been an active area of research for over 65 years, yet fundamental questions regarding its biosynthesis, transport, and functions remain unanswered. Why have such facets of CoQ biochemistry evaded discovery for decades, despite incredible advances in methodology? The challenges for the CoQ field are a microcosm of a grander challenge in the post-genomic age: to define functions for all proteins. Some CoQ-related proteins may have evaded detection due to redundancy, whereby one or more proteins can compensate for their loss. The issue of redundancy is illustrated in the Tyr-to-4-HB pathway in yeast, where multiple enzymes can transaminate tyrosine to 4-HPP under the tested conditions [31]. On the converse, one protein can perform multiple functions and participate in an array of cellular processes, making it difficult to delineate its function in CoQ biosynthesis. Gene essentiality poses an additional hurdle in genetic screening efforts because genes essential for survival will drop out in traditional negative-selection screening approaches. Alternatively, it is possible that select genes are not essential for CoQ biosynthesis under the standard laboratory conditions. These challenges are further compounded by the difficulties in purifying membrane-associated CoQ proteins and reconstituting their individual enzyme activity in vitro.
How, then, can we address such obstacles that have hindered our understanding of this critical cofactor for so long? Such issues necessitate the development of new tools and probes: to inhibit and activate the biosynthetic pathway, to map the movement of CoQ throughout the cell, and to track which proteins interact with CoQ. As many proteins involved in CoQ biosynthesis are understudied, many are lacking sufficient reagents, such as antibodies and cell lines that would empower their study. Addressing these issues will also require rethinking and updating traditional strategies to overcome prior limitations, such as by performing positive-selection, CRISPRi/a, or phenotypic genetic screening to home in on genes related to CoQ biosynthesis. Existing approaches could also be revisited in contexts that more closely resemble physiological conditions, for example performing genetic screens in human plasma-like medium [106]. Finally, integrative ‘systems biochemistry’ approaches [64,107] present a potential avenue to guide new hypothesis-driven studies to probe the molecular function of uncharacterized proteins in CoQ biosynthesis and associate new proteins with this process. We hope that in combination, these advancements in tools, strategies, and experimental frameworks will provide the keys to filling the open gaps in knowledge in CoQ biosynthesis.
The isoprenoid tail, composed of isoprene repeats, is derived from isopentenyl pyrophosphate (IPP) or dimethylallyl pyrophosphate (DMAPP) subunits from the mevalonate pathway [3]. It is still unknown how the 4-HB and isoprene precursors are transported from the cytosol into the mitochondrial matrix. Upon precursor entry into the matrix, Coq1p/PDSS1/PDSS2 polymerizes isoprenes to form the full-length tail, whose length varies by species. Coq2p/COQ2 then attaches the tail to 4-HB to yield polyprenyl-hydroxybenzoate (PPHB) [3].
The final stage of CoQ biosynthesis involves a series of modifications of the PPHB headgroup, including decarboxylation, hydroxylation, and methylation reactions carried out by Coq3-Coq9p/COQ3-COQ9 [3] (Figure 2C). The proposed order of these reactions is based on the accumulation of CoQ intermediates in mutant yeast [34] and bacterial strains [35]. However, given the accumulation of certain intermediates in both human and yeast COQ6 mutant cells, there is some ambiguity concerning the order of reactions, namely, whether the decarboxylation and hydroxylation steps are executed before or after the Coq6-mediated hydroxylation reaction [36,37]. One outstanding question is what eukaryotic enzyme(s) catalyze the decarboxylation and hydroxylation steps, theoretically converting polyprenyl-vanillic acid (PPVA) to demethoxy-demethyl-coenzyme Q (DDMQ) (Box 1). Another active area of research seeks to elucidate the roles of auxiliary proteins in CoQ biosynthesis, including Coq4p/COQ4, Coq8p/COQ8A/COQ8B, Coq9p/COQ9, and Coq10/COQ10A/COQ10B. COQ8 is a member of the highly conserved UbiB family of kinase-like proteins with strong links to prenyl-lipid biosynthesis [38]. Coq8p has been hypothesized to be a protein kinase, and coq8Δ yeast exhibit alterations in phosphorylation of Coq proteins [39]. However, mammalian COQ8A lacks protein kinase activity in vitro, and instead displays ATPase activity that can be modulated by lipids and small molecules [40]. How Coq8 functions as either a kinase or ATPase to support CoQ biosynthesis remains vague (Box 2). Although the lipid-binding protein Coq10p is not essential for CoQ biosynthesis, it is necessary for efficient CoQ synthesis and its utilization in OxPhos [41]. Further efforts are required to elucidate how Coq10p and other auxiliary proteins supports CoQ biosynthesis in yeast and mammalian systems.
Box 2. The UbiB Protein Family.
The UbiB family is an enigmatic group of kinase-like proteins strongly tied to the biosynthesis of CoQ and other prenyl lipids [38,108–110]. This protein family is conserved throughout all domains of life (Figure Ia), yet little functional characterization exists for its members. All five human homologs (COQ8A/B, ADCK1/2/5) have been associated with human diseases [80] (Figure Ib), underscoring the need to understand their precise molecular function. Although originally predicted to be protein kinases, the crystal structure of human COQ8A revealed conserved structural features that sterically occlude the active site [101] (Figure Ic). COQ8A exhibited low levels of autophosphorylation, but lacked any kinase activity in trans. Instead, COQ8A may leverage its ATPase activity to enable proper CoQ biosynthesis through an unknown mechanism [40]. Additional studies have demonstrated that Coq8p binds isoprenoid lipids that resemble CoQ intermediates, and that its ATPase activity is activated by cardiolipin-containing membranes [101,102], which further suggest an unorthodox kinase-like activity in maintaining CoQ biosynthesis. Overexpression of Coq8p in different Coq yeast deletions strains can stabilize Coq protein subcomplexes and enable the accumulation of late-stage CoQ intermediates [111]. Taken together, these data suggest a model in which Coq8p leverages its ATPase activity to support the formation of complex Q, potentially coupled with extraction of lipid intermediates from the IMM (Figure Id). Additional yeast UbiB family members, Cqd1/2p, have been implicated in CoQ distribution [78], pointing towards broader roles of the UbiB family proteins in CoQ homeostasis (Figure Ie). Although their precise mechanism of action remails elusive, the recent development of probes to target specific UbiB family proteins promises to help unravel their roles in CoQ biosynthesis, distribution, and more.
CoQ Biosynthetic Complex
The enzymes responsible for CoQ head-group modification form a complex at the matrix face of the IMM [42–45]. This complex, termed ‘complex Q’ or the ‘CoQ-synthome,’ is a fascinating example of a ‘metabolon’ [46]—a dynamic enzyme complex carrying out the sequential steps of a metabolic pathway by cluster channeling [47]. Posited roles of metabolons include enhancing flux due to enzyme proximity and substrate channeling, and sequestering toxic intermediates. Indeed, early CoQ intermediates in yeast have demonstrated toxicity and their accumulation impairs trafficking of exogenously supplied CoQ [48]. CoQ exists almost entirely in the hydrophobic portion of the bilayer and its head-group structure is carefully designed to enable its precise redox processes. The CoQ metabolon could serve to access the early precursor PPHB, orchestrate sequential modifications, and prevent premature release of intermediates back into the bilayer.
Studies have identified Coq3-9p/COQ3-9 as core components of complex Q [43,45], with additional evidence to implicate the uncharacterized yeast proteins Coq11p [44] and Coq21p [49]. However, the complete composition and stoichiometry of the enzymes and auxiliary proteins in complex Q are not known. COQ8A and COQ9 also bind lipid intermediates [40,50], supporting the notion that complex Q is composed of proteinacious and non-proteinacious components. Studies in yeast have demonstrated that CoQ proteins exhibit interdependency for their stability [42], and have led to the hypothesis that CoQ or and/or its biosynthetic intermediates may also be required for complex Q formation and stability.
Due to the difficulties in purifying and reconstituting this complex in vitro, combined with the lack of detailed structural information on individual CoQ proteins, complex Q has evaded isolation and structural characterization. It is unclear whether complex Q is one uniform complex, or a varying collection of smaller sub-complexes. Recently, a cryo-electron microscopy structure of an octameric complex of human COQ7 and COQ9 was solved, providing structural insight into a complex of CoQ proteins at a lipid bilayer [51]. COQ9 is an isoprenoid lipid-binding protein and its disruption results in accumulation of the COQ7 substrate, DMQ, thus establishing the hypothesis that COQ9 may act as a lipid-presenting protein [50,52]. Consistently, this complex structure, along with molecular dynamics simulations, support a model in which a COQ7:COQ9 tetramer could bind and remodel membranes to extract CoQ lipid intermediates and help present them to other complex Q proteins for further modification [51]. Despite these advances, much remains obscure regarding the architecture and assembly of complex Q (Figure 3a and see Outstanding questions).
Figure 3. Models Framing Knowledge Gaps in Complex Q Organization and CoQ Distribution.
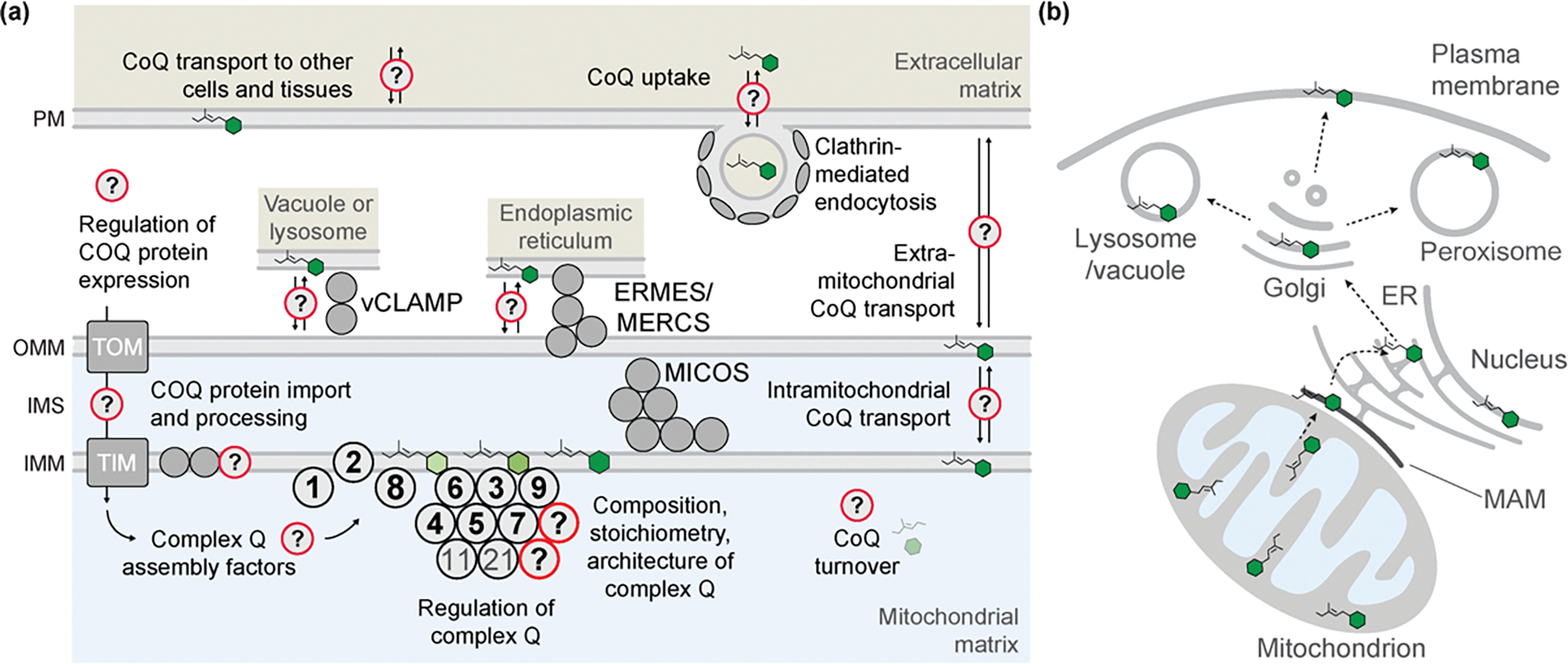
(A) A depiction of outstanding questions regarding complex Q composition and assembly, along with possible mechanisms contributing to CoQ transport and cellular distribution. Although there is much evidence to support the existence and identify core components of complex Q, its full composition, stoichiometry, and architecture are undefined, and how its assembly is regulated is unknown. All COQ genes are nuclearly encoded, but many questions exist regarding factors that regulate their gene expression and how they are processed and imported into mitochondria via TIM and TOM complexes. As complex Q has been observed in proximity to MICOS and ERMES, it is hypothesized that these complexes could facilitate import of CoQ precursors to mitochondria or export of mature CoQ to other organelles. Studies involving administration of exogenous CoQ has suggested that clathrin-mediated endocytosis machinery may be involved in CoQ assimilation. (B) Depiction of a putative transport pathway of CoQ throughout the cell. Following synthesis at the IMM, CoQ passes through mitochondrial-associated membranes (MAM) to the ER. There, CoQ encounters the endomembrane system and is packaged in the Golgi for widespread mobilization to different organelles and cellular membranes. Abbreviations: ERMES, ER-mitochondria encounter structure; MERCS, mitochondria-ER contact sites; MICOS, mitochondrial contact site and cristae organization system; TIM, translocase of the inner mitochondrial membrane; TOM, translocase of the outer mitochondrial membrane; vCLAMP, vacuolar and mitochondrial patch.
Outstanding Questions.
How are the precursors to CoQ biosynthesis transported into mitochondria?
What enzyme carries out the decarboxylation and hydroxylation of PPVA to DDMQ? Is this transformation achieved by a single reaction mechanism or two distinct steps?
What are the molecular functions of uncharacterized mitochondrial proteins related to CoQ biosynthesis?
How is CoQ biosynthesis coordinated with mitochondrial biogenesis and OxPhos?
What are the transcriptional, post-transcriptional, and post-translational mechanisms that regulate CoQ biosynthesis?
How is CoQ degraded, and how is this turnover coordinated with biosynthesis?
What is the complete composition of proteins, lipids, and metabolites of complex Q, and what is their stoichiometry?
How is complex Q formed and maintained? Are there dedicated assembly factors?
Is complex Q a uniform, stable complex or a dynamic assembly of sub-complexes?
What is the function of complex Q? Does it enable substrate channeling or sequester potentially toxic intermediates?
What is the functional difference between complex Q and CoQ domains?
What role does the spatial positioning of complex Q at ER-mitochondrial contact sites serve?
Can CoQ be synthesized in other organelles, such as the Golgi, and if so, what enzymes catalyze this process?
How is exogenous CoQ assimilated by the cell?
How is CoQ transported from mitochondria and distributed throughout the cell?
What role do mitochondrial sites, such as ERMES and MICOS, as well as the endomembrane system play in CoQ distribution?
Are there additional functions of CoQ in other organellar membranes?
Can CoQ intermediates or CoQ analogs substitute for non-ETC functions of CoQ?
Can CoQ inhibition be a beneficial therapeutic strategy in some contexts?
Why do different tissues have different CoQ/CoQH2 ratio? Could the ratio control the direction of enzymatic reactions in which CoQ participates as a cofactor?
Regulation of CoQ Biosynthesis
As an essential metabolic pathway, CoQ production is regulated at the post-transcriptional, post-translational, and epigenetic levels, which have been reviewed in detail elsewhere [53]. Early studies placed CoQ biosynthetic genes under the control of the metabolic transcriptional regulators PPARα [54] and NF-κB [55] in mouse and human cell culture, respectively. These factors may link regulation of CoQ biosynthesis with other branches of lipid metabolism, or drive biosynthesis as a response to oxidative stress, but further work is required to fully illustrate the contexts in which these mechanisms predominate. Studies in zebrafish embryos have identified CoQ biosynthesis genes as direct targets of the essential erythropoietic transcription factor TIFγ, with tifγ mutant embryos displaying decreased levels of CoQ [56]. The mammalian RNA-binding protein CLUH binds mRNA transcripts of many mitochondrial proteins, including COQ3 and COQ6, and has been implicated in the control of mitochondrial biogenesis [57]. More directly, CoQ abundance has been linked to the yeast RNA-binding protein Puf3p, which exerts post-transcriptional control by binding to 3’UTRs of mRNAs encoding proteins involved in mitochondrial biogenesis [58] and COQ5 [59].
Post-translation modifications (PTMs), including phosphorylation and acylation, have been observed for numerous COQ proteins [60], although these lack direct evidence of functional relevance. Additionally, post-translational processing of COQ proteins is emerging as a regulatory mechanism for proper stability and function. The yeast mitoprotease Oct1p directly processes Coq5p into its mature form [61]. Deletion of another mitochondrial protease, PARL, has been tied to decreased CoQ levels and defective complex III activity in mice [62]. On the converse, deletion of a yeast phosphatidylethanolamine N-methyltransferase gene, cho2, significantly increased cellular CoQ levels, potentially stemming from elevated mitochondrial methylation capacity due to increased SAM levels [63]. Finally, loss of the uncharacterized protein human PYURF was connected to a defect in both CoQ biosynthesis and complex I [64]. PYURF physically interacts with COQ5 and NDUFAF5, both SAM-dependent methyltransferases [64], nominating PYURF as a chaperone for concerted support of complex I and CoQ pathways. It is important to note that each example above describes a protein found either in yeast or mammals, but not yet in both. Further work is required to identify the functional orthologs that drive these processes.
CoQ Uptake, Distribution, and Transport
Remarkably, CoQ is found in nearly all cellular membranes [65] (Figure 1b). Despite its ubiquitous presence, key gaps in knowledge remain regarding the proteins and pathways that are responsible for distributing CoQ throughout the cell.
CoQ Domains
A recent study in yeast to uncover the in vivo spatial organization of CoQ biosynthetic proteins revealed that CoQ head-group modifying enzymes colocalize to multiple discrete foci, termed ‘domains’ [66]. The formation of CoQ domains is dependent on the presence of essential COQ proteins, and they exhibit the ability to assemble and disassemble in response to lipid substrate flux [66]. CoQ domains are positioned adjacent to ER-mitochondria contact sites, and defects in such contact sites disrupt CoQ domain formation and lead to the accumulation of CoQ biosynthetic intermediates [66]. CoQ domains have been found in proximity to the ER-mitochondria encounter structure (ERMES) complex, and consistently, deletion of ERMES subunits results in destabilization of domains [67]. These findings suggest that the proximity of CoQ domains to ER-mitochondrial contact sites may stem from the need to coordinate metabolic pathways via the exchange of substrates and products, or the distribution of mature CoQ throughout the cell via the endomembrane system (Figure 3a).
Implication of Endomembrane System and Mitochondrial Contact Sites in CoQ Distribution
CoQ is extremely hydrophobic and embedded within lipid bilayers, although there is no clear consensus on its exact orientation within the membrane [68]. As such, it is highly unlikely that it is able to traverse the aqueous cellular milieu without dedicated protein machinery. CoQ species with six or more isoprene units are unable to freely exchange between synthetic liposomes, unlike the more hydrophilic artificial CoQ analogs with two or four isoprene units [48]. Despite this observation, the pathway required for CoQ transport has yet to be characterized. In a study to track cellular CoQ distribution, treatment of human cells with the headgroup precursor [14C]4-HB resulted in accumulation of labeled CoQ first at mitochondria, subsequently in mitochondrial associated membranes, ER, and lastly at the PM [69]. Treatment with Brefeldin A, an inhibitor of ER-Golgi transport, disrupted incorporation of newly synthesized CoQ in the PM [69]. This work suggests a model in which CoQ is synthesized at the IMM, transported to the ER, then distributed throughout the cell via the endomembrane system, although specific proteins involved in bidirectional CoQ trafficking remain elusive (Figure 3b). As mitochondrial contact sites within and between organelles have been implicated in phospholipid trafficking throughout the cell [70,71], it is possible that they too play a role in CoQ distribution.
Studies on the Uptake of Exogenous CoQ
Considering the need for improved CoQ delivery strategies for the treatment of mitochondrial dysfunction arising from CoQ deficiencies, it is critical to understand the way in which exogenous CoQ is assimilated by the cell. In a coq3Δ yeast strain dependent on exogenous CoQ for respiratory growth, deletion of four factors in the endocytic pathway (Erg2, Pep12, Tlg2, and Vps45) abrogated rescue by CoQ [72]. More recently, targeted screening in a coq2Δ background revealed additional factors required for uptake of exogenous CoQ, including the previously identified genes and factors involved in clathrin-mediated endocytosis [48]. Many of these studies have been performed in yeast due to the well characterized pathways and ease of genetic manipulation, but studies in mammalian systems will be required to determine if these observations are conserved. In a human clinical trial involving long-term CoQ supplementation, polymorphisms in ABCB1, CD36, CYP7A1, and NPC1L1 were found to be associated with higher levels of serum CoQ and beneficial effects of CoQ supplementation [73,74]. Given the established roles of CD36 and ABCB1 in fatty acid transport [75,76], and NPC1L1 in cholesterol uptake [77], these genes are encouraging targets for further mechanistic investigations into CoQ assimilation. Future work will be required to appreciate the role of these targets, and others, in facilitating CoQ uptake.
Studies on the Inter-Organellar Distribution of CoQ
A recent forward genetics screen in yeast leveraged the role of extramitochondrial CoQ in combating oxidative stress to identify genes involved in cellular CoQ distribution. This screen, combined with biochemical fractionation, lipid profiling, and yeast genetics, linked two IMM proteins, Cqd1p and Cqd2p, to CoQ distribution, and revealed that they work in a reciprocal fashion: deletion of Cqd1p results in low CoQ mitochondrial levels and higher PM CoQ levels, whereas loss of Cqd2p shows the opposite effects [78]. Cqd1p and Cqd2p are members of the UbiB family of proteins, and their conserved ATPase residues are required for their CoQ distribution functions [78]. This could suggest a model in which the ATPase activity of Cqd1p/Cqd2p is coupled to extraction of CoQ from the IMM to facilitate its transport. Notably, CoQ transport out of mitochondria still occurred in the absence of Cdq1/2p and was furthermore unaffected by deletion of genes involved in ERMES, MICOS, vCLAMP, or lipid homeostasis, which supports the possible redundancy or overlapping functions of these pathways in CoQ distribution [78]. This finding is consistent with previous studies that have demonstrated genetic interactions of Cdq2 with ERMES complex proteins [76] and genes involved in mitochondrial lipid metabolism [79], further underscoring the potential functional link between contact site proteins and those involved in CoQ transport.
CoQ Therapeutics and Tools
Stemming from its critical cellular roles, CoQ has wide-ranging impacts on human health and disease. Genetic defects in CoQ biosynthesis result in varied diseases, including nephropathies, ataxias, and myopathies [80]. Furthermore, secondary CoQ deficiency is observed in a broad array of mitochondrial disorders [81]. Conversely, genetic disruption of CoQ biosynthesis in C. elegans [82] and mice [83] has been shown to increase lifespan, though the mechanism is unknown. Due to poor bioavailability of CoQ [84] (Figure 4a), oral supplementation of exogenous CoQ is largely ineffective [85], posing a significant barrier to the treatment of CoQ deficiencies in the clinic (Figure 4b–c). To move towards more effective therapeutics, there is an urgent need to develop novel tools to improve cellular uptake, bypass dysfunctional steps, or manipulate the pathway. While various formulations and CoQ analogs have been reviewed extensively elsewhere [86,87], here we will focus on a handful of encouraging strategies to achieve these goals.
Figure 4. Challenges in Uptake of Exogenous CoQ and Strategies to Overcome Them to Treat CoQ Deficiency.
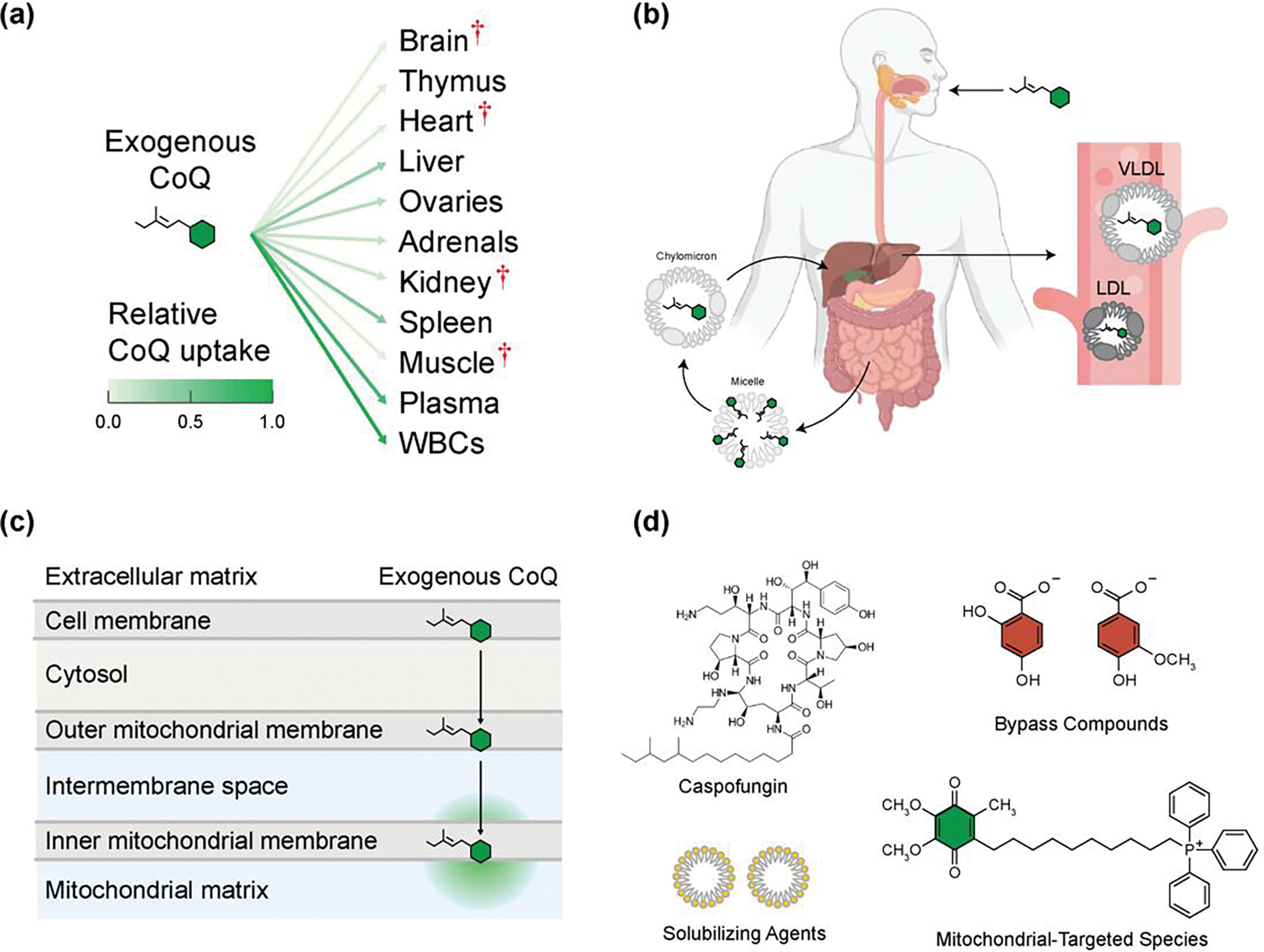
(A) Exogenous CoQ is taken up poorly by most rat tissues after intraperitoneal injection. Red cross denotes organs most commonly affected in CoQ deficiency. Data from [Bentinger et al.] [112]. (B) Following oral intake of exogenous CoQ, it is absorbed in the small intestine and packaged into chylomicrons, then taken up by the liver and repackaged into low-density lipoproteins (LDL) and very low-density lipoproteins (VLDL), which can then enter blood circulation. Created with BioRender.com. (C) Once exogenous CoQ is in the bloodstream, it then must be transported across multiple lipid membranes and aqueous compartments to reach the mitochondrial inner membrane. (D) Depicted are examples of strategies to increase the solubility and uptake of exogenous CoQ by packaging with solubilizing agents like micelles, in novel formulations such as with caspofungin, or increasing organellar localization with the attachment of mitochondrial-targeting species like TPP. Alternative, CoQ deficiencies could by treated with bypass compounds that supersede the need for enzymes in the biosynthetic pathway that may be mutant or defective.
Novel Delivery Strategies
Various strategies have been explored to overcome the biophysical barriers associated with CoQ delivery, including mitochondrial targeting moieties (e.g., triphenylphosphonium (TPP)), encapsulation in liposomes, nanoparticles, micelles, and nanoemulsions [86] (Figure 4d). The most widely studied TPP compound, MitoQ [88], is a potent antioxidant with various clinical applications, but it is unable to rescue CoQ’s ETC function, likely because the bulky TPP group prevents MitoQ’s membrane incorporation [87]. Alternative tactics link TPP to the surface of CoQ-encapsulated nanoparticles [89], but the ability of such agents to restore respiration has yet to be examined. An emerging strategy is to encapsulate CoQ in mitochondrial-targeted liposomes, such as the MITO-Porter system, that are designed to fuse directly with the mitochondrial membrane [90,91]. Despite progress, these delivery modules remain highly lipophilic and subject to the biophysical barriers of delivery. A recent approach surmounts this problem via micellization of CoQ with the fungicide caspofungin (CF). This significantly improves the water solubility and cellular uptake of CoQ [92] resulting in increased tissue levels of CoQ and improved overall survival of a CoQ-deficient mouse model; however, overcoming the need for many rounds of IV administration may present a challenge to therapeutic adoption [92] (Figure 4d). An alternative strategy to overcome the poor bioavailability of CoQ is to utilize water soluble CoQ head group analogs that can bypass specific enzymatic defects [93–95] (Figure 4d). Bypass compounds represent an appealing route because they are soluble, their incorporation into the native biosynthetic pathway results in proper cellular distribution of downstream CoQ, and they have demonstrated efficacy in restoring CoQ levels in human cell culture and extending lifespan in mouse models [37,95–97].
Small Molecule Modulators of CoQ Biosynthesis
A complementary approach to supplementation is the development of molecular tools that can modulate the CoQ pathway and help elucidate the molecular functions of poorly characterized CoQ-related proteins. In efforts to study the role of CoQ limitation and supplementation in aging-related disorders, various inhibitors of CoQ biosynthesis have been developed, including 4-nitrobenzoate (COQ2 inhibitor) [98], clioquinol (COQ7 inhibitor) [99], and zoledronic acid (farnesyl diphosphate synthase inhibitor) [100]. Recently, Coq8p and COQ8A have been prominent targets of chemical modulation. Though its ATPase activity is required for CoQ production [40,101], the precise molecular role of these COQ8 homologs remains obscure. A covalent inhibitor targeting an engineered Coq8p mutant has provided a probe to manipulate the CoQ biosynthetic pathway in yeast for mechanistic studies, though it is limited by lack of efficacy against the WT protein [102]. More recently, a selective COQ8A inhibitor has been developed and is able to robustly inhibit de novo CoQ production in a mammalian cellular context [103]. Although CoQ is a critical cofactor for energy generation and other metabolic processes, there are some potential benefits to inhibiting its biosynthesis. Studies in C. elegans [82] and mice [83] have demonstrated that CoQ limitation can increase lifespan; CoQ pathway inhibitors could assist in unraveling this surprising phenomenon. Additionally, there has been a surge in efforts to manipulate ferroptosis in pathological settings, specifically various cancers [104]. Limiting the amount of CoQ available for FSP1 reduction might therefore be an effective anticancer strategy. In contrast, the CoQ headgroup-like phenolic compound 2-propylphenol has been characterized as a direct activator of the ATPase activity of COQ8A [105]. Taken together, these novel probes expand the chemical toolkit for modulating the CoQ pathway in cells and provide a foundation for elucidating the role of auxiliary proteins in CoQ biosynthesis, with the potential to provide insights for future therapeutic development.
Concluding remarks
The remarkable biochemical properties of coenzyme Q are pivotal for cellular bioenergetics, biosynthesis, and homeostasis, and recent studies suggest further functions for this ubiquitous cofactor are likely to emerge. Given its key roles in mitochondria and beyond, it is imperative that we fully elucidate the biochemistry of CoQ synthesis and transport — processes that have remained partially or largely unclear for 65 years. Progress in these areas has been hampered by numerous challenges, including hydrophobicity barriers and enzyme redundancy, motivating a drive for new tools and experimental approaches. By bringing to light the varied, often overlooked cellular roles of CoQ, and by highlighting primary gaps in knowledge, we hope that this review will help focus new efforts in CoQ biology and therapeutic development.
Figure I. UbiB Protein Structure and Function.
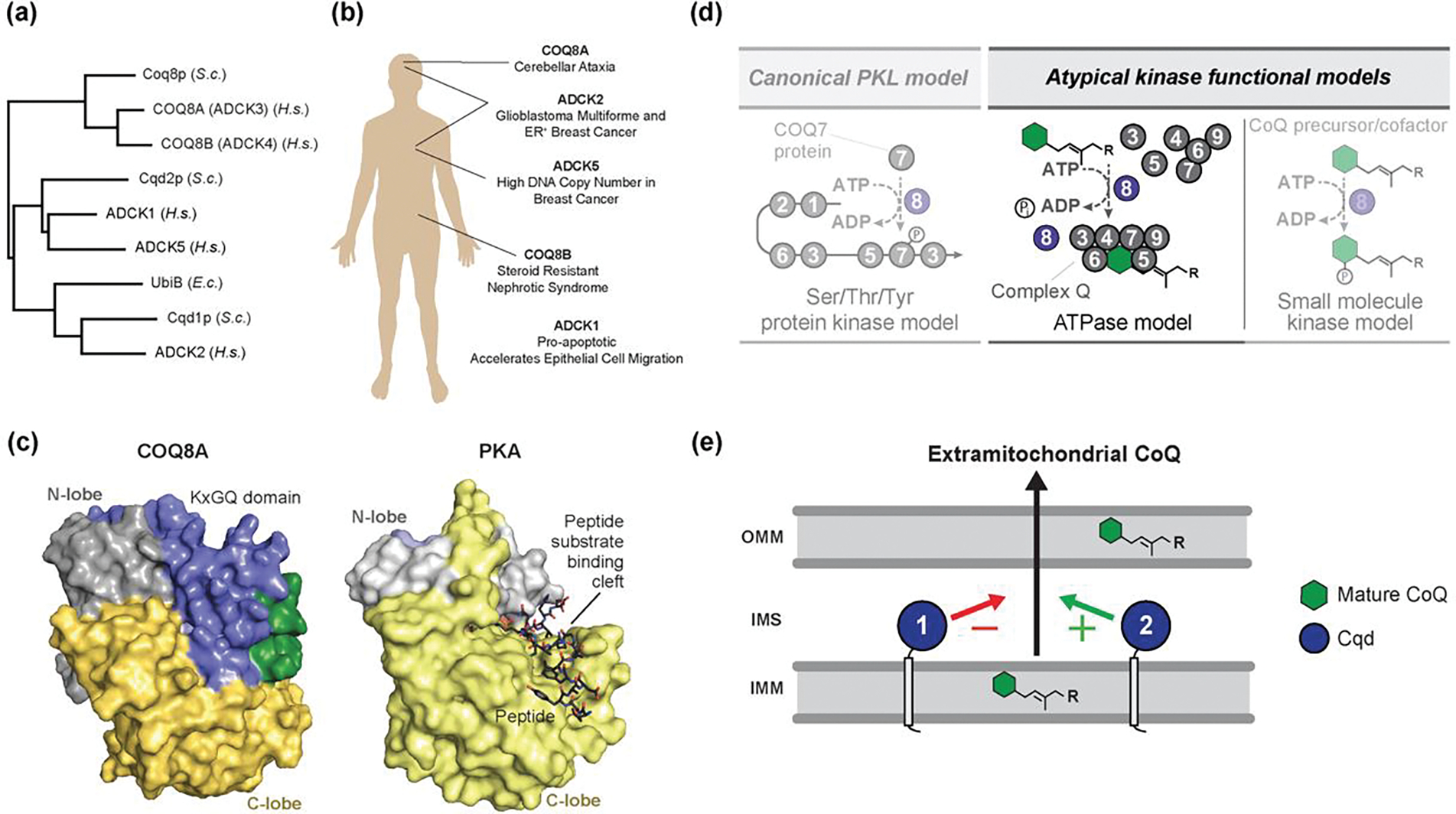
(A) UbiB proteins are conserved across all domains of life, from humans to bacteria. (B) Mutations in human UbiB family proteins have been linked to numerous diseases. (C) Surface representations of COQ8ANΔ254 (PDB=4PED) and Protein Kinase A (PDB=1ATP) showing occlusion of the canonical peptide binding site by the highly conserved and UbiB-specific KxGQ domain (adapted from Stefely et al. [101]. (D) Potential models for the role of COQ8 in supporting CoQ biosynthesis. COQ8 could act as a canonical protein kinase to phosphorylate an enzyme in the biosynthetic pathway and modulate its activity, but this is not supported by current data. Alternatively, COQ8 could leverage its ATPase activity to extract hydrophobic substrates from the membrane and seed the formation of complex Q. Finally, COQ8 may act as a small-molecule kinase to support CoQ production, but no such substrate has been identified (adapted from Stefely et al. [40]. (E) Model of UbiB protein function in CoQ cellular distribution. Localized at the IMM, yeast Cqd1p and Cqd2p are proposed to have opposing roles in the extramitochondrial transport of mature CoQ, though the mechanism has yet to be defined. Abbreviations: S.c., Saccharomyces cerevisiae; H.s., Homo sapiens; E.c., Escherichia coli.
Highlights.
Coenzyme Q (CoQ) plays major roles in cell metabolism beyond transferring electrons in the ETC, including acting as an antioxidant and enzyme cofactor. Its ubiquitous presence in cellular membranes suggests that more roles for CoQ are yet to be discovered.
Recent studies have identified a role for plasma membrane CoQ in combating ferroptosis. These findings have reinvigorated efforts to understand how CoQ is transported throughout the cell.
CoQ is synthesized in a metabolon-like complex that is proximal to mitochondrial contact sites, which may be important for facilitating CoQ transport out of mitochondria.
The CoQ biosynthetic pathway remains incompletely characterized, with multiple enzymatic and transport steps lacking associated proteins. Discovery efforts have met challenges including enzyme redundancy, essentiality, and hydrophobic barriers associated with synthesizing CoQ.
Acknowledgements
We thank current and former members of the Pagliarini Lab, especially fellow ‘Q Branch’ colleagues Mateusz Manicki and Jon Stefely, for helpful discussion. This work was supported by the NIH award R35 GM131795 (D.J.P.) and funds from the BJC Investigator Program (D.J.P.).
Glossary
- Cluster channeling
a feature of multienzyme metabolons in which sequential steps of a metabolic pathway can be carried out by many copies of a given enzyme rather than depending on the enzyme in closest physical proximity.
- Complex Q
Complex Q, also known as the CoQ-synthome, is the biosynthetic complex that performs he final stage of CoQ biosynthesis.
- CoQ6
Coenzyme Q (CoQ), also known as ubiquinone (UQ), is the redox-active lipid product of the biosynthetic pathway discussed here. The subscripted number specifies the number of isoprene units in the lipid tail (e.g. CoQ6 indicates 6 isoprene units). The redox state of the lipid can also be specified: the fully reduced form (the hydroquinone) as ‘CoQH2’ (ubiquinol, UQH2), the singly dehydrogenated radical form (the semiquinone) as ‘CoQH•’ (ubisemiquinone, UQH•), and the oxidized form (the quinone) as ‘CoQ’ (ubiquinone, UQ).
- COQ6
COQ6 is an example of a human enzyme require for CoQ biosynthesis.
- Coq6p
Coq6p is an example of a yeast enzyme required for CoQ biosynthesis.
- DHODH
dihydroorotate dehydrogenase, an enzyme that mediates the CoQ-dependent oxidation of dihydroorotate to orotate.
- Ferroptosis
A type of programmed cell death characterized by iron-dependent phospholipid peroxidation.
Footnotes
Declaration of interests
None are declared by the authors
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Crane FL et al. (1957) Isolation of a quinone from beef heart mitochondria. Biochim Biophys Acta 25, 220–221 [DOI] [PubMed] [Google Scholar]
- 2.Morton RA (1958) Ubiquinone. Nature 182, 1764–1767 [DOI] [PubMed] [Google Scholar]
- 3.Stefely JA and Pagliarini DJ (2017) Biochemistry of Mitochondrial Coenzyme Q Biosynthesis. Trends Biochem Sci 42, 824–843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baschiera E et al. (2021) The multiple roles of coenzyme Q in cellular homeostasis and their relevance for the pathogenesis of coenzyme Q deficiency. Free Radical Bio Med 166, 277–286 [DOI] [PubMed] [Google Scholar]
- 5.Martínez-Reyes I et al. (2020) Mitochondrial ubiquinol oxidation is necessary for tumour growth. Nature 585, 288–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lapuente-Brun E et al. (2013) Supercomplex Assembly Determines Electron Flux in the Mitochondrial Electron Transport Chain. Science 340, 1567–1570 [DOI] [PubMed] [Google Scholar]
- 7.Hernansanz-Agustín P and Enríquez JA (2021) Functional segmentation of CoQ and cyt c pools by respiratory complex superassembly. Free Radical Bio Med 167, 232–242 [DOI] [PubMed] [Google Scholar]
- 8.Dixon SJ et al. (2012) Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 149, 1060–1072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jiang X et al. (2021) Ferroptosis: mechanisms, biology and role in disease. Nat Rev Mol Cell Bio 22, 266–282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang WS et al. (2014) Regulation of Ferroptotic Cancer Cell Death by GPX4. Cell 156, 317–331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bersuker K et al. (2019) The CoQ oxidoreductase FSP1 acts in parallel to GPX4 to inhibit ferroptosis. Nature 575, 688–692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Doll S et al. (2019) FSP1 is a glutathione-independent ferroptosis suppressor. Nature 575, 693–698 [DOI] [PubMed] [Google Scholar]
- 13.Navas P et al. (2007) The importance of plasma membrane coenzyme Q in aging and stress responses. Mitochondrion 7 Suppl, S34–40 [DOI] [PubMed] [Google Scholar]
- 14.Ross D and Siegel D (2017) Functions of NQO1 in Cellular Protection and CoQ10 Metabolism and its Potential Role as a Redox Sensitive Molecular Switch. Front Physiol 8, 595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bello RI et al. (2003) Hydrogen Peroxide- and Cell-Density-Regulated Expression of NADH-Cytochrome b5 Reductase in HeLa Cells. J Bioenerg Biomembr 35, 169–179 [DOI] [PubMed] [Google Scholar]
- 16.Villalba JM et al. (1997) Role of cytochrome b5 reductase on the antioxidant function of coenzyme Q in the plasma membrane. Mol Aspects Med 18, 7–13 [DOI] [PubMed] [Google Scholar]
- 17.Gille L and Nohl H (2000) The Existence of a Lysosomal Redox Chain and the Role of Ubiquinone. Arch Biochem Biophys 375, 347–354 [DOI] [PubMed] [Google Scholar]
- 18.Heaton RA et al. (2020) The Effect of Cellular Coenzyme Q10 Deficiency on Lysosomal Acidification. J Clin Medicine 9, 1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Argyropoulos G and Harper M-E (2002) Invited Review: Uncoupling proteins and thermoregulation. J Appl Physiol 92, 2187–2198 [DOI] [PubMed] [Google Scholar]
- 20.Echtay KS et al. (2000) Coenzyme Q is an obligatory cofactor for uncoupling protein function. Nature 408, 609–613 [DOI] [PubMed] [Google Scholar]
- 21.Echtay KS et al. (2001) Uncoupling proteins 2 and 3 are highly active H+ transporters and highly nucleotide sensitive when activated by coenzyme Q (ubiquinone). Proc National Acad Sci 98, 1416–1421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Esteves TC et al. (2004) Ubiquinone is not required for proton conductance by uncoupling protein 1 in yeast mitochondria. Biochem J 379, 309–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Anderson CM et al. (2015) Dependence of Brown Adipose Tissue Function on CD36-Mediated Coenzyme Q Uptake. Cell Reports 10, 505–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Murphy MP and Chouchani ET (2022) Why succinate? Physiological regulation by a mitochondrial coenzyme Q sentinel. Nat Chem Biol 18, 461–469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Arias-Mayenco I et al. (2018) Acute O2 Sensing: Role of Coenzyme QH2/Q Ratio and Mitochondrial ROS Compartmentalization. Cell Metab 28, 145–158.e4 [DOI] [PubMed] [Google Scholar]
- 26.Spinelli JB et al. (2021) Fumarate is a terminal electron acceptor in the mammalian electron transport chain. Science 374, 1227–1237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lester RL and Crane FL (1959) The Natural Occurrence of Coenzyme Q and Related Compounds. J Biol Chem 234, 2169–2175 [PubMed] [Google Scholar]
- 28.Momose K and Rudney H (1972) 3-Polyprenyl-4-hydroxybenzoate Synthesis in the Inner Membrane of Mitochondria from p-Hydroxybenzoate and Isopentenyl pyrophosphate: a demonstration of isoprenoid synthesis in rat liver mitochondria. J Biol Chem 247, 3930–3940 [PubMed] [Google Scholar]
- 29.Payet L-A et al. (2016) Mechanistic Details of Early Steps in Coenzyme Q Biosynthesis Pathway in Yeast. Cell Chem Biol 23, 1241–1250 [DOI] [PubMed] [Google Scholar]
- 30.Stefely JA et al. (2016) Mitochondrial protein functions elucidated by multi-omic mass spectrometry profiling. Nat Biotechnol 34, 1191–1197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Robinson KP et al. (2021) Defining intermediates and redundancies in coenzyme Q precursor biosynthesis. J Biological Chem 296, 100643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Banh RS et al. (2021) The polar oxy-metabolome reveals the 4-hydroxymandelate CoQ10 synthesis pathway. Nature 597, 420–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Valera MJ et al. (2020) The Mandelate Pathway, an Alternative to the Phenylalanine Ammonia Lyase Pathway for the Synthesis of Benzenoids in Ascomycete Yeasts. Appl Environ Microb 86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fernández-del-Río L and Clarke CF (2021) Coenzyme Q Biosynthesis: An Update on the Origins of the Benzenoid Ring and Discovery of New Ring Precursors. Metabolites 11, 385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Abby SS et al. (2020) Advances in bacterial pathways for the biosynthesis of ubiquinone. Biochim Biophys Acta Bioenerg 1861, 148259. [DOI] [PubMed] [Google Scholar]
- 36.Ozeir M et al. (2011) Coenzyme Q Biosynthesis: Coq6 Is Required for the C5-Hydroxylation Reaction and Substrate Analogs Rescue Coq6 Deficiency. Chemistry & Biology 18, 1134–1142 [DOI] [PubMed] [Google Scholar]
- 37.Lopez MJA et al. (2019) Vanillic Acid Restores Coenzyme Q Biosynthesis and ATP Production in Human Cells Lacking COQ6. Oxid Med Cell Longev 2019, 3904905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Poon WW et al. (2000) Identification of Escherichia coli ubiB, a Gene Required for the First Monooxygenase Step in Ubiquinone Biosynthesis. J Bacteriol 182, 5139–5146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xie LX et al. (2011) Expression of the human atypical kinase ADCK3 rescues coenzyme Q biosynthesis and phosphorylation of Coq polypeptides in yeast coq8 mutants. Biochimica Et Biophysica Acta Bba - Mol Cell Biology Lipids 1811, 348–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stefely JA et al. (2016) Cerebellar Ataxia and Coenzyme Q Deficiency through Loss of Unorthodox Kinase Activity. Mol Cell 63, 608–620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tsui HS et al. (2019) Human COQ10A and COQ10B are distinct lipid-binding START domain proteins required for coenzyme Q function[S]. J Lipid Res 60, 1293–1310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hsieh EJ et al. (2007) Saccharomyces cerevisiae Coq9 polypeptide is a subunit of the mitochondrial coenzyme Q biosynthetic complex. Arch Biochem Biophys 463, 19–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.He CH et al. (2014) Coenzyme Q supplementation or over-expression of the yeast Coq8 putative kinase stabilizes multi-subunit Coq polypeptide complexes in yeast coq null mutants. Biochim Biophys Acta 1841, 630–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Allan CM et al. (2015) Identification of Coq11, a new coenzyme Q biosynthetic protein in the CoQ-synthome in Saccharomyces cerevisiae. J Biol Chem 290, 7517–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Floyd BJ et al. (2016) Mitochondrial Protein Interaction Mapping Identifies Regulators of Respiratory Chain Function. Mol Cell 63, 621–632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Srere P (1985) The metabolon. Trends Biochem Sci 10, 109–110 [Google Scholar]
- 47.Pareek V et al. (2021) Metabolic channeling: predictions, deductions, and evidence. Mol Cell 81, 3775–3785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fernández-del-Río L et al. (2020) Genes and lipids that impact uptake and assimilation of exogenous coenzyme Q in Saccharomyces cerevisiae. Free Radical Bio Med 154, 105–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Morgenstern M et al. (2017) Definition of a High-Confidence Mitochondrial Proteome at Quantitative Scale. Cell Reports 19, 2836–2852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lohman DC et al. (2019) An Isoprene Lipid-Binding Protein Promotes Eukaryotic Coenzyme Q Biosynthesis. Mol Cell 73, 763–774 e10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Manicki M et al. (2022) Structure and functionality of a multimeric human COQ7:COQ9 complex. Mol Cell 82, 1–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lohman DC et al. (2014) Mitochondrial COQ9 is a lipid-binding protein that associates with COQ7 to enable coenzyme Q biosynthesis. Proc Natl Acad Sci U S A 111, E4697–705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Villalba JM and Navas P (2021) Regulation of coenzyme Q biosynthesis pathway in eukaryotes. Free Radical Bio Med 165, 312–323 [DOI] [PubMed] [Google Scholar]
- 54.Turunen M et al. (2000) Influence of peroxisome proliferator-activated receptor α on ubiquinone biosynthesis. J Mol Biol 297, 607–614 [DOI] [PubMed] [Google Scholar]
- 55.Brea-Calvo G et al. (2009) Cell Survival from Chemotherapy Depends on NF-κB Transcriptional Up-Regulation of Coenzyme Q Biosynthesis. Plos One 4, e5301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rossmann MP et al. (2021) Cell-specific transcriptional control of mitochondrial metabolism by TIF1γ drives erythropoiesis. Science 372, 716–721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gao J et al. (2014) CLUH regulates mitochondrial biogenesis by binding mRNAs of nuclear-encoded mitochondrial proteins. J Cell Biol 207, 213–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lee C-D and Tu BP (2015) Glucose-Regulated Phosphorylation of the PUF Protein Puf3 Regulates the Translational Fate of Its Bound mRNAs and Association with RNA Granules. Cell Reports 11, 1638–1650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lapointe CP et al. (2018) Multi-omics Reveal Specific Targets of the RNA-Binding Protein Puf3p and Its Orchestration of Mitochondrial Biogenesis. Cell Syst 6, 125–135 e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hornbeck PV et al. (2015) PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res 43, D512–D520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Veling MT et al. (2017) Multi-omic Mitoprotease Profiling Defines a Role for Oct1p in Coenzyme Q Production. Mol Cell 68, 970–977 e11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Spinazzi M et al. (2019) PARL deficiency in mouse causes Complex III defects, coenzyme Q depletion, and Leigh-like syndrome. Proc National Acad Sci 116, 277–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ayer A et al. (2021) Genetic screening reveals phospholipid metabolism as a key regulator of the biosynthesis of the redox-active lipid coenzyme Q. Redox Biol 46, 102127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rensvold JW et al. (2022) Defining mitochondrial protein functions through deep multiomic profiling. Nature 606, 382–388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kalén A et al. (1987) Ubiquinone biosynthesis by the microsomal fraction from rat liver. Biochimica Et Biophysica Acta Bba - Gen Subj 926, 70–78 [DOI] [PubMed] [Google Scholar]
- 66.Subramanian K et al. (2019) Coenzyme Q biosynthetic proteins assemble in a substrate-dependent manner into domains at ER-mitochondria contacts. J Cell Biol DOI: 10.1083/jcb.201808044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Eisenberg-Bord M et al. (2019) The Endoplasmic Reticulum-Mitochondria Encounter Structure Complex Coordinates Coenzyme Q Biosynthesis. Contact (Thousand Oaks) 2, 2515256418825409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Braasch-Turi MM et al. (2022) Chemistry of Lipoquinones: Properties, Synthesis, and Membrane Location of Ubiquinones, Plastoquinones, and Menaquinones. Int J Mol Sci 23, 12856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Fernandez-Ayala DJ et al. (2005) Coenzyme Q distribution in HL-60 human cells depends on the endomembrane system. Biochim Biophys Acta 1713, 129–37 [DOI] [PubMed] [Google Scholar]
- 70.Rampelt H et al. (2017) Role of the mitochondrial contact site and cristae organizing system in membrane architecture and dynamics. Biochimica Et Biophysica Acta Bba - Mol Cell Res 1864, 737–746 [DOI] [PubMed] [Google Scholar]
- 71.Dimmer KS and Rapaport D (2017) Mitochondrial contact sites as platforms for phospholipid exchange. Biochimica Et Biophysica Acta Bba - Mol Cell Biology Lipids 1862, 69–80 [DOI] [PubMed] [Google Scholar]
- 72.Padilla-Lopez S et al. (2009) Genetic evidence for the requirement of the endocytic pathway in the uptake of coenzyme Q6 in Saccharomyces cerevisiae. Biochim Biophys Acta 1788, 1238–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Takahashi M et al. (2021) CYP7A1, NPC1L1, ABCB1, and CD36 Polymorphisms Are Associated with Increased Serum Coenzyme Q10 after Long-Term Supplementation in Women. Antioxidants 10, 431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Takahashi M et al. (2022) CYP7A1, NPC1L1, ABCB1, and CD36 Polymorphisms Associated with Coenzyme Q10 Availability Affect the Subjective Quality of Life Score (SF-36) after Long-Term CoQ10 Supplementation in Women. Nutrients 14, 2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hao J-W et al. (2020) CD36 facilitates fatty acid uptake by dynamic palmitoylation-regulated endocytosis. Nat Commun 11, 4765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Helvoort A van et al. (1996) MDR1 P-Glycoprotein Is a Lipid Translocase of Broad Specificity, While MDR3 P-Glycoprotein Specifically Translocates Phosphatidylcholine. Cell 87, 507–517 [DOI] [PubMed] [Google Scholar]
- 77.Altmann SW et al. (2004) Niemann-Pick C1 Like 1 Protein Is Critical for Intestinal Cholesterol Absorption. Science 303, 1201–1204 [DOI] [PubMed] [Google Scholar]
- 78.Kemmerer ZA et al. (2021) UbiB proteins regulate cellular CoQ distribution in Saccharomyces cerevisiae. Nat Commun 12, 4769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Odendall F et al. (2019) The mitochondrial intermembrane space–facing proteins Mcp2 and Tgl2 are involved in yeast lipid metabolism. Mol Biol Cell 30, 2681–2694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Alcázar-Fabra M et al. (2018) Clinical syndromes associated with Coenzyme Q10 deficiency. Essays Biochem 62, 377–398 [DOI] [PubMed] [Google Scholar]
- 81.Navas P (2021) Secondary CoQ10 deficiency, bioenergetics unbalance in disease and aging. Biofactors 47, 551–569 [DOI] [PubMed] [Google Scholar]
- 82.Rodríguez-Aguilera JC et al. (2005) The role of ubiquinone in Caenorhabditis elegans longevity. Ageing Res Rev 4, 41–53 [DOI] [PubMed] [Google Scholar]
- 83.Takahashi K et al. (2014) Extended lifespan, reduced body size and leg skeletal muscle mass, and decreased mitochondrial function in clk-1 transgenic mice. Exp Gerontol 58, 146–153 [DOI] [PubMed] [Google Scholar]
- 84.Mantle D and Dybring A (2020) Bioavailability of Coenzyme Q10: An Overview of the Absorption Process and Subsequent Metabolism. Antioxidants 9, 386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wang Y and Hekimi S (2022) The efficacy of coenzyme Q10 treatment in alleviating the symptoms of primary coenzyme Q10 deficiency: A systematic review. J Cell Mol Med DOI: 10.1111/jcmm.17488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Pastor-Maldonado CJ et al. (2020) Coenzyme Q10: Novel Formulations and Medical Trends. Int J Mol Sci 21, 8432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Suárez-Rivero JM et al. (2021) Coenzyme Q10 Analogues: Benefits and Challenges for Therapeutics. Antioxidants 10, 236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kelso GF et al. (2001) Selective Targeting of a Redox-active Ubiquinone to Mitochondria within Cells: Antioxidant and Antiapoptotic Properties. J Biol Chem 276, 4588–4596 [DOI] [PubMed] [Google Scholar]
- 89.Ozbay HS et al. (2022) Mitochondria-targeted CoQ10 loaded PLGA-b-PEG-TPP nanoparticles: Their effects on mitochondrial functions of COQ8B−/− HK-2 cells. Eur J Pharm Biopharm 173, 22–33 [DOI] [PubMed] [Google Scholar]
- 90.Yamada Y et al. (2008) MITO-Porter: A liposome-based carrier system for delivery of macromolecules into mitochondria via membrane fusion. Biochimica Et Biophysica Acta Bba - Biomembr 1778, 423–432 [DOI] [PubMed] [Google Scholar]
- 91.Yamada Y et al. (2017) Packaging of the Coenzyme Q10 into a Liposome for Mitochondrial Delivery and the Intracellular Observation in Patient Derived Mitochondrial Disease Cells. Biological Pharm Bulletin 40, 2183–2190 [DOI] [PubMed] [Google Scholar]
- 92.Wang Y and Hekimi S (2020) Micellization of coenzyme Q by the fungicide caspofungin allows for safe intravenous administration to reach extreme supraphysiological concentrations. Redox Biol 36, 101680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Pierrel F (2017) Impact of Chemical Analogs of 4-Hydroxybenzoic Acid on Coenzyme Q Biosynthesis: From Inhibition to Bypass of Coenzyme Q Deficiency. Front Physiol 8, 436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Pesini A (2022) Mechanisms and Therapeutic Effects of Benzoquinone Ring Analogs in Primary CoQ Deficiencies. Antioxidants 11, 665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wang Y et al. (2015) Mitochondrial function and lifespan of mice with controlled ubiquinone biosynthesis. Nat Commun 6, 6393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Freyer C et al. (2015) Rescue of primary ubiquinone deficiency due to a novel COQ7 defect using 2,4–dihydroxybensoic acid. Journal of Medical Genetics 52, 779–783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wang Y et al. (2017) Pathogenicity of two COQ7 mutations and responses to 2,4-dihydroxybenzoate bypass treatment. J Cell Mol Med 21, 2329–2343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Forsman U et al. (2010) 4-Nitrobenzoate inhibits coenzyme Q biosynthesis in mammalian cell cultures. Nat Chem Biol 6, 515–517 [DOI] [PubMed] [Google Scholar]
- 99.Wang Y et al. (2009) The Anti-neurodegeneration Drug Clioquinol Inhibits the Aging-associated Protein CLK-1. J Biol Chem 284, 314–323 [DOI] [PubMed] [Google Scholar]
- 100.Fernández-del-Río L et al. (2021) Regulation of hepatic coenzyme Q biosynthesis by dietary omega-3 polyunsaturated fatty acids. Redox Biol 46, 102061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Stefely JA et al. (2015) Mitochondrial ADCK3 employs an atypical protein kinase-like fold to enable coenzyme Q biosynthesis. Mol Cell 57, 83–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Reidenbach AG et al. (2018) Conserved Lipid and Small-Molecule Modulation of COQ8 Reveals Regulation of the Ancient Kinase-like UbiB Family. Cell Chem Biol 25, 154–165 e11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Murray NH et al. (2022) Small-molecule inhibition of the archetypal UbiB protein COQ8. Nat Chem Biol DOI: 10.1038/s41589-022-01168-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Koppula P et al. (2022) A targetable CoQ-FSP1 axis drives ferroptosis- and radiation-resistance in KEAP1 inactive lung cancers. Nat Commun 13, 2206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Murray NH et al. (2022) 2-Propylphenol Allosterically Modulates COQ8A to Enhance ATPase Activity. Acs Chem Biol DOI: 10.1021/acschembio.2c00434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Rossiter NJ et al. (2021) CRISPR screens in physiologic medium reveal conditionally essential genes in human cells. Cell Metab 33, 1248–1263.e9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Sung AY et al. (2020) Systems Biochemistry Approaches to Defining Mitochondrial Protein Function. Cell Metabolism 31, 669–678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Do TQ (2001) A defect in coenzyme Q biosynthesis is responsible for the respiratory deficiency in Saccharomyces cerevisiae abc1 mutants. J Biol Chem 276, 18161–8 [DOI] [PubMed] [Google Scholar]
- 109.Lundquist PK et al. (2012) ABC1K atypical kinases in plants: filling the organellar kinase void. Trends Plant Sci 17, 546–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Pralon T et al. (2020) Mutation of the Atypical Kinase ABC1K3 Partially Rescues the Proton Gradient Regulation 6 Phenotype in Arabidopsis thaliana. Front Plant Sci 11, 337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Xie LX et al. (2012) Overexpression of the Coq8 kinase in Saccharomyces cerevisiae coq null mutants allows for accumulation of diagnostic intermediates of the coenzyme Q6 biosynthetic pathway. J Biol Chem 287, 23571–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Bentinger M et al. (2003) Distribution and breakdown of labeled coenzyme Q10 in rat. Free Radical Biology and Medicine 34, 563–575 [DOI] [PubMed] [Google Scholar]