

Abstract
The uptake and metabolism of nutrients supports fundamental cellular process from bioenergetics to biomass production and cell fate regulation. Initial studies on metabolic networks in animal tissues revealed major differences in how tumors and normal tissue engaged central metabolic pathways. The discovery that oncogenes and tumor suppressors directly regulate cellular metabolic networks cemented the narrative that cancer metabolism is inherently different from metabolism in normal cells. Nevertheless, studies in cancer metabolism have revealed fundamental principles of mammalian cell metabolism, including the architecture and regulation of major metabolic networks and the intersection between metabolic pathways and cell fate control. As the field of cancer metabolism expands to include the stromal cells of tumors and even normal, differentiated tissue, it is increasingly clear that the principles of metabolism elucidated in cancer cells are relevant to a wide range of mammalian cells and that metabolic pathways can be rerouted to meet the demands of cells in particular lineages or states of development. The goal of this review is to provide an overview of how the field of cancer metabolism has provided a framework for revealing basic principles of cell metabolism and for dissecting the metabolic networks that allow cells to meet their specific demands. Understanding context-specific metabolic preferences and liabilities will enable continued development of approaches to specifically target cancer cells to help improve patient care.
Introduction
The field of cancer metabolism centers on the hypothesis that cellular metabolic networks support key aspects of cancer cell function. Many of the features that distinguish cancer cells from normal cells—aberrant proliferation, survival, migration, and cell fate control—are either directly controlled by cell metabolism or amenable to regulation by specific metabolites. Consequently, there is great interest in determining how cancer cells regulate metabolic networks and whether the metabolic demands of cancer cells can be exploited for therapeutic gain. Initial studies, based largely on in vitro models, led to the notion that cancer cells harbored a distinct metabolic state relative to normal cells. As the field has expanded into more diverse models and in vivo studies, it is increasingly clear that metabolism is not one size fits all. Just as normal cells from different tissues exhibit a wide array of metabolic profiles, cancer cells also exhibit enormous metabolic diversity and plasticity. As a result, few metabolic signatures reliably distinguish cancer cells from normal cells. Ultimately, the only unifying principle of cancer metabolism is the cancer patient themselves. Therefore, the goal of this review is to provide an overview of how studies in cancer metabolism have revealed fundamental principles of cell metabolism, to highlight the diversity of metabolic strategies employed by cancer cells, and to discuss the future of the field of cancer metabolism—namely, studying and targeting metabolism in patients to improve cancer care.
Main
Origins of cancer metabolism
The field of cancer metabolism began about 100 years ago when Otto Warburg discovered that tumor slices produced lactate from glucose even in the presence of ample oxygen1. This discovery was surprising because oxygen normally permits efficient glucose oxidation and thus suppresses fermentation—a paradigm known as the Pasteur effect (Figure 1). Here, Warburg showed that tumor tissue slices continued to produce lactate even when oxygen was abundant, a phenomenon of aerobic glycolysis now dubbed the Warburg effect. Warburg interpreted the inability of oxygen to fully repress lactate production as a sign that oxygen consumption—respiration—was fundamentally deficient in cancer cells. We now know that Warburg’s interpretation was not correct: respiration is rarely damaged in cancer cells2. Rather, aerobic glycolysis is essentially overflow metabolism—a product of cells taking up nutrients in excess of their demands3, 4. Nevertheless, the Warburg effect is the most famous feature of cancer metabolism because of its visibility and sheer ubiquity: almost every cultured cell line—not just transformed cells—produces lactate despite supraphysiological oxygen, so long as glucose is abundant.
Figure 1. Warburg and Pasteur effects.
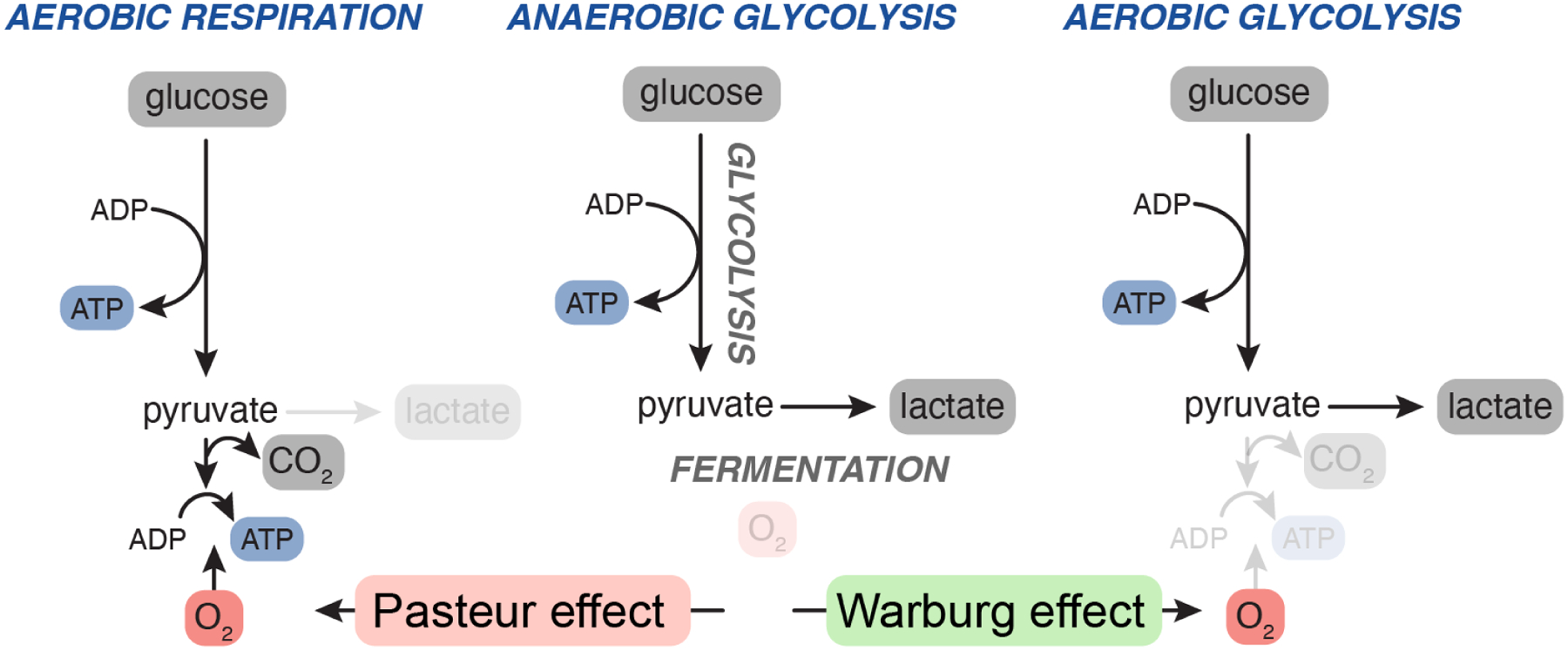
When oxygen is not present, the glycolytic breakdown of sugars (such as glucose) is sustained by converting the glycolytic end-product pyruvate to ethanol or lactate in a process known as fermentation (middle). Fermentation allows organisms to generate ATP from sugar without oxygen. The Pasteur effect describes the phenomenon in which oxygen suppresses fermentation by allowing organisms to oxidize pyruvate to carbon dioxide (left). The Warburg effect refers to the persistent fermentation (lactate production) of mammalian cells even when oxygen is abundant (right).
The perception that cancer metabolism is different from normal cell metabolism is due in part to the historical origin of both fields. Fundamental principles of cell metabolism were largely elucidated using differentiated animal tissues as ex vivo model systems. Throughout the middle of the 20th century, seminal work from Warburg, Cori and Cori, Krebs, Mitchell and others unraveled the core pathways of glucose metabolism, nitrogen handling, nutrient oxidation and ATP production5. This golden age of biochemistry was precipitated by technical advances such as Warburg’s pioneering of tissue slices and the Coris careful tissue mincing, which allowed the field to move beyond intact organisms and use tissues such as pigeon or frog muscle and rat liver to dissect metabolic networks ex vivo. Of the available model systems, only tumors provided an opportunity to study metabolism in actively diving cells. Accordingly, the earliest conception of cancer metabolism as ‘perverted’6 is based largely on the comparison to the metabolism of post-mitotic, differentiated tissues. It wasn’t until decades later that Eagle and colleagues uncovered principles required to propagate mammalian cells in vitro. Eagle’s work presaged major themes in cancer metabolism, identifying nutrients such as glutamine as being limiting for rapid proliferation7.
Despite the advance of cell culture, the field of cell metabolism lay relatively fallow until the end of the 20th century when metabolic enzymes were revealed as direct targets of growth factor signaling cascades and oncogenic transcription factors8. Early studies attempting to discover the origins for high glucose uptake in cancer cells led to the discovery that the common oncogenes ras and src induce expression of glucose transport proteins9. Not long after, the oncogenic transcription factor c-Myc, which is frequently amplified in human cancer, was shown to directly transactivate LDHA, the enzyme that converts glucose-derived pyruvate to lactate, thereby providing a direct link between oncogene expression and activation of aerobic glycoysis10. Importantly, this work also showed that LDHA was required for c-Myc to induce features of transformation, such as growth in three dimensions, demonstrating that metabolic regulation contributes to the aberrant features of cancer cell growth10. These studies directly linked oncogenic mutations to metabolic networks, thereby cementing Warburg’s hypothesis that metabolism is fundamentally altered in cancer cells, and ushered in an explosion of work charting the metabolic pathways that sustain cancer cell growth.
More recently, technological advances have broadened the field of cell metabolism beyond cancer cells in vitro. Advances in cell culture models have facilitated study of cell types beyond cancer, such as stem cells and immune cells, and improvements in in vivo isotope tracing now allow for quantitative mapping of metabolic networks in living animals11—a full circle return to the earliest days of cell metabolism in which living organisms were the only possible model systems. These studies have provided enormous insight into how metabolic networks are regulated and increasingly demonstrate that cancer metabolism is neither monolithic nor fundamentally different from metabolism of normal cells.
Cancer as a model system
Over the past several decades, cancer cells provided the dominant—and in many ways, ideal—model system for unraveling principles of cell metabolism. Cancer cells are readily amenable to cell culture, which permits intricate metabolic tracing and mechanistic dissection; there are numerous approaches to model cancer in vivo and thereby probe the role of metabolism in distinct stages of tumor progression, and the vast array of genetic mutations acquired by cancer cells provides an intrinsic experiment to link metabolic gene function to tumor phenotypes. As a result, most of our modern understanding of cell metabolism is rooted in cancer biology, further reinforcing the notion that cancer is synonymous with metabolic rewiring.
Metabolic determinants of cell growth
The essentially unlimited growth of cancer cells in culture provides the ideal system to determine which factors enable cell growth and proliferation. While the precise demands may vary depending on cell type or growth rate, studies of cancer cells have identified several conserved requirements for mammalian cell growth.
Nutrient uptake.
Cell proliferation requires net biomass increase, which in turn is fueled by nutrient uptake. While unicellular organisms take up nutrients in proportion to their environmental abundance, thereby tying growth to nutrient availability, most metazoan cells require additional signals to facilitate nutrient uptake. Consequently, proliferation is licensed by growth factors that allow cells to take up nutrients that support cell growth12 (Figure 2). Growth factor signaling through PI3K/AKT drives membrane localization of transporters required for uptake of critical nutrients such as glucose, amino acids, and iron13–17. AKT also directly phosphorylates hexokinase, thereby inducing the phosphorylation and capture of glucose for further glycolytic metabolism18. Growth factor signaling further activates glucose metabolism, and induces aerobic glycolysis, through myriad mechanisms including direct phosphorylation or allosteric regulation of glycolytic enzymes19–21. Mutations driving constitutive activation of PI3K/AKT are among the most common mutations in human cancer, and oncogenic transcription factors such as MYC and HIF can also activate nutrient acquisition by increasing expression of genes involved in the capture and metabolism of critical nutrients such as glucose and glutamine12, 22, 23. Accordingly, cancer cells are often hardwired for constitutive, cell-autonomous nutrient uptake.
Figure 2. Mechanisms of nutrient uptake.
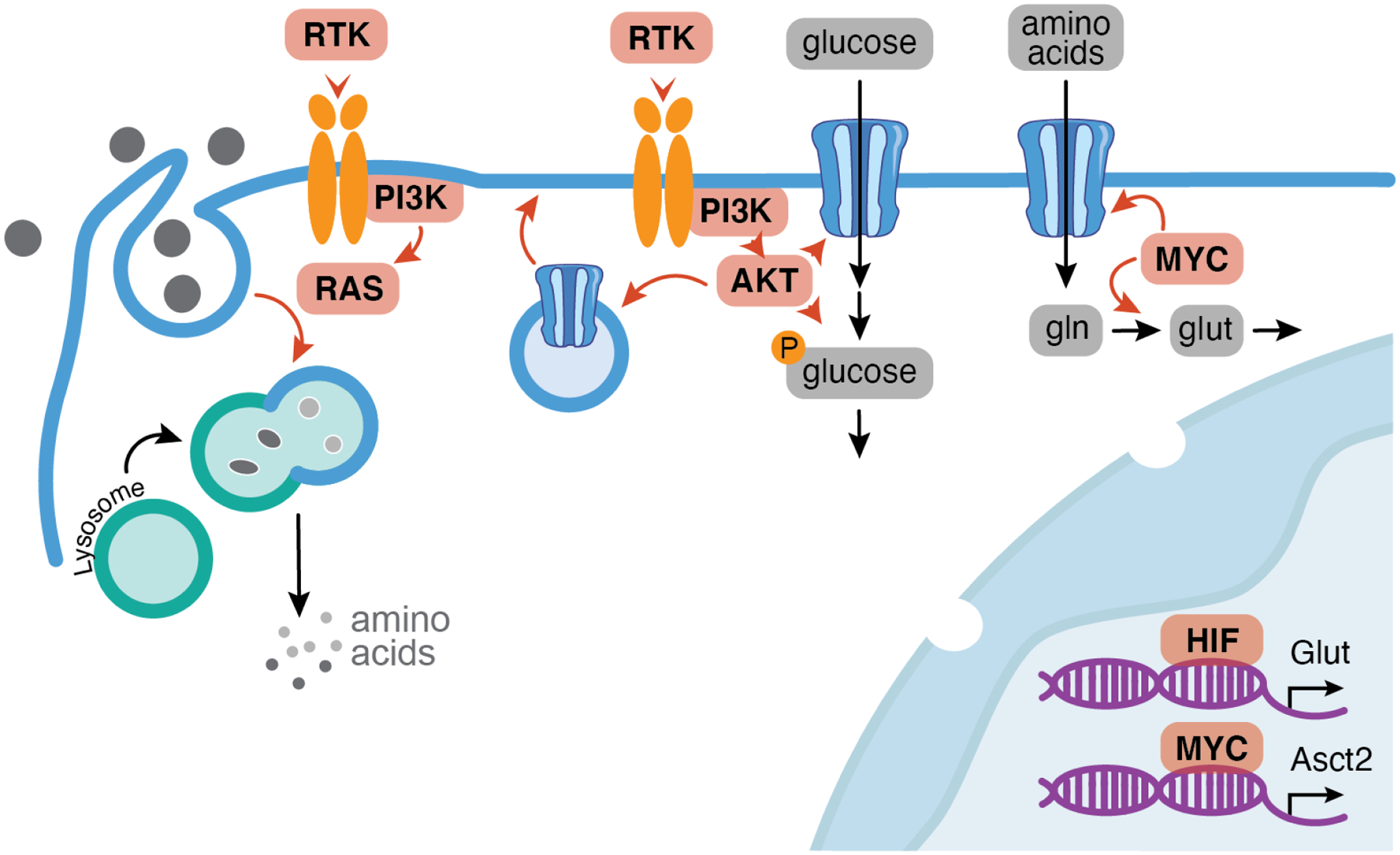
Nutrient uptake is largely initiated by receptor tyrosine kinases (RTK) which activate signaling cascades that promote the membrane localization of nutrient transporters and activity of enzymes that metabolize—and therefore help capture—nutrients. Signaling pathways downstream of PI3K, most notably RAS, can also direct bulk uptake of macromolecules via macropinocytosis. Nutrient uptake is also facilitated by transcriptional induction of membrane transporters via oncogenic transcription factors hypoxia inducible factor (HIF) and MYC. Genes frequently mutated or activated in cancer are colored in pink.
Increased nutrient uptake is so fundamental to cancer biology that imaging uptake of a radioactive glucose analog is a mainstay of cancer imaging24. In cultured cells, glutamine is the second most consumed nutrient after glucose, and radioactive glutamine analogs are emerging as a useful alternative to image tumors in tissues with constitutively high glucose uptake25. Growth factor signaling can also direct non-canonical forms of nutrient uptake when conventional soluble nutrients are limiting. For example, both PI3K and RAS signaling can activate macropinocytosis, a non-selective form of bulk endocytosis that enables engulfment and delivery of exogenous macromolecules to the lysosome for digestion26, 27. This alternative nutrient acquisition strategy enables cancer cell survival in nutrient deprived microenvironments and also supports growth of normal cells, including rapidly proliferating T cells and developing embryos27–30. Indeed, elevated nutrient uptake extends far beyond cancer cells: even within a tumor, the (likely non-proliferating) immune cells can take up more glucose than the cancer cells31. These results add nuance to the general paradigm that cancer cells exhibit high nutrient uptake, demonstrating roles for oncogenic mutations, lineage, and the local microenvironment in shaping overall nutrient consumption profiles.
Macromolecule biosynthesis.
Once taken into cells, nutrients are funneled into metabolic networks that produce the energy, reducing equivalents and molecular building blocks required for cell growth. To grow and divide, cells must duplicate membranes (lipids), DNA and RNA (nucleotides) and proteins (amino acids) (Figure 3). While cells can directly take up some of the components for these macromolecules, such as lipids, amino acids and even salvaged nucleotides, proliferating cells inevitably must synthesize de novo the critical components for cell growth. Unsurprisingly, transcriptional and signaling networks controlling such biosynthetic pathways are frequently deregulated in cancer (Figure 3). For example, Myc directly activates genes involved in purine synthesis32; NRF2—a transcription factor activated in many human tumors by genetic loss of negative feedback regulation—induces genes involved in serine biosynthesis and the pentose phosphate pathway, both of which supply metabolic components required for nucleotide synthesis33; and SREBP, a transcriptional regulator of lipid synthesis, is activated by mTORC1 downstream of PI3K/AKT34. The PI3K/AKT/mTORC1 axis broadly regulates many components of anabolic metabolism, including direct stimulation of protein synthesis, post-translational activation of enzymes that generate key metabolic building blocks, and transcriptional repression of catabolic processes that antagonize growth35. In some cases, tumors acquire genetic alterations in metabolic enzymes themselves: PHGDH, which encodes the enzyme that initiates serine synthesis, is frequently amplified in human tumors and PHGDH expression supports tumor growth especially in tissues that have limiting levels of environmental serine36–38.
Figure 3. Biosynthetic networks.
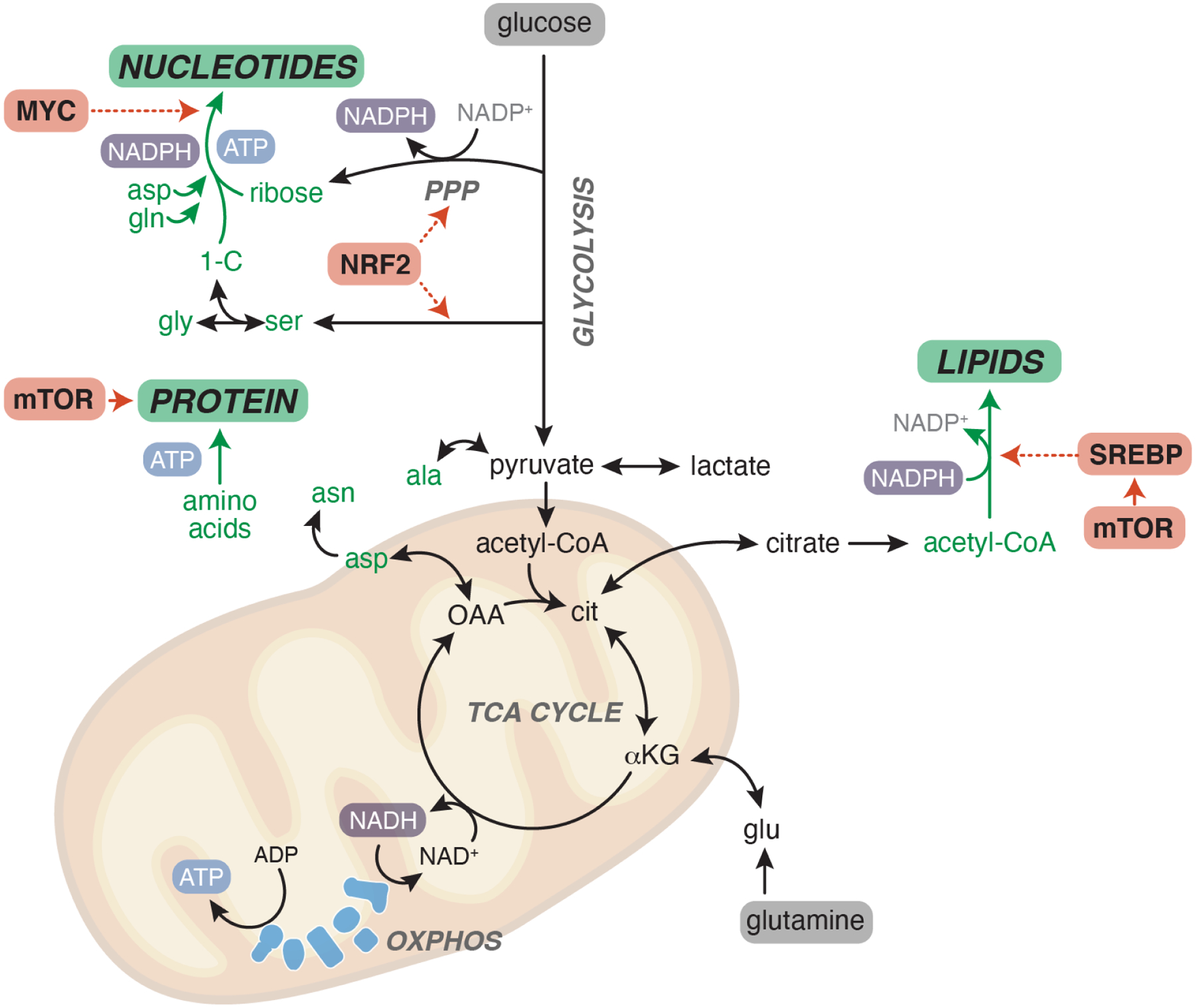
Once inside the cell, nutrients—most notably glucose and glutamine—are funneled into metabolic networks that provide the building blocks necessary to produce the major macromolecules for growth: lipids, nucleotides and protein. Critical metabolic building blocks are shown in green. Major routes for ATP and reducing equivalent (NADPH) production are highlighted. Metabolic regulators commonly activated in cancer are shown in pink. Dashed lines represent transcriptional control of metabolic pathways by indicated transcription factors. PPP, pentose phosphate pathway. OXPHOS, oxidative phosphorylation.
Beyond the building blocks themselves, anabolic metabolism requires reducing equivalents and energy to help glue metabolites together into macromolecules. As nutrients are oxidized, their high-energy electrons (reducing equivalents) are deposited onto electron carriers including NAD+, NADP+ and FAD. Reduced electron carriers, most notably NADPH, can donate electrons to reduce intracellular metabolites as part of key biosynthetic (e.g. fatty acid synthesis, nucleic acid biosynthesis) or homeostatic pathways (e.g. regeneration of antioxidants such as glutathione and thioredoxin). Electron carriers (especially NADH and FADH2) also funnel electrons into the electron transport chain (ETC) in the inner mitochondrial membrane. Electron passage through the ETC generates an electrochemical gradient that is harnessed to drive ATP synthesis in a process known as oxidative phosphorylation (OXPHOS) (FIG). ATP can also be made through glycolysis, albeit with greatly reduced efficiency.
ATP fuels most macromolecular biosynthesis, including nucleotide biosynthesis, DNA replication and protein translation. This has led to a common perception that ATP may be limiting for cell proliferation. In fact, the opposite may be true: ATP allosterically inhibits several metabolic enzymes including the glycolytic enzyme phosphofructokinase 1, providing a mechanism for cells to prevent excess nutrient catabolism when energy is plentiful. The notion that cells harbor significant excess capacity to generate ATP is strengthened by numerous observations that OXPHOS is not required for cancer cell proliferation in vitro or for tumor growth in vivo39–41. Whether the notable plasticity in ATP sourcing characteristic of many cancer cells extends to normal cells, and whether this plasticity is enabled by high nutrient uptake, remains to be determined, although early evidence in regulatory T cells suggests that OXPHOS independence may not be unique to cancer cells42.
Although many of the principles governing biomass generation have been elucidated in cancer cells, these principles also extend to normal cells, which likewise depend on continuous protein, RNA and lipid synthesis to maintain homeostasis, regardless of whether or not there is a net gain in biomass. Consistently, few differences emerged when comparing metabolite fluxes in transformed cells and normal cells induced to proliferate at an equivalent rate43. Even absent cell proliferation, cellular macromolecules will turn over at a certain rate and some cells will require additional macromolecule or ATP production depending on the specialized demands of the cell. For example, exocrine cells may have high requirement for continuous protein synthesis while contractile or excitatory cells may have high ATP demand. Even DNA synthesis, which is largely restricted to actively dividing cells, is not unique to cancer cells as many normal cells (immune cells, epithelial cells) also undergo periods of rapid proliferation. Thus, whether cancer cells—especially those growing in vivo—have higher demand for biomass generation compared to normal cells remains unclear, especially as not all cancer cells within a tumor are actively dividing. Combining quantitative isotope tracing to macromolecules with comprehensive profiling of tissue health following administration of metabolic inhibitors will help to reveal the extent to which different cell types or organs rely on specific metabolic networks to support biomass production.
Electron carrier regeneration.
Increasing evidence indicates that regeneration of oxidized electron carriers, rather than ATP production, constrains cell growth. Maintaining flux through any oxidative pathway requires continuous supply of electron acceptors, although some enzymes will be more sensitive to changes in electron acceptor availability than others (Figure 4A)44. In the cytosol, both glycolysis and serine synthesis require NAD+; accordingly, conditions that impair cytosolic NAD+ regeneration restrain serine synthesis and induce serine auxotrophy in mammalian cells (Figure 4B)45, 46. Within the mitochondria, the tricarboxylic acid (TCA) cycle serves as the terminal hub of nutrient oxidation, reducing electron carriers that are re-oxidized by depositing electrons into the ETC. Regenerating oxidized electron carriers is emerging as one of the most important functions of the ETC in mammalian cells: ETC inhibition prevents electron carrier regeneration and stalls the TCA cycle, and this stalling is reversed by providing cells orthogonal methods to regenerate mitochondrial NAD+ (ref47). The TCA cycle is a source of several key intermediates for biosynthetic pathways, including oxaloacetate for aspartate and nucleotide biosynthesis, citrate for lipid synthesis, and succinyl-CoA for heme biosynthesis (Figure 3). As oxaloacetate is the most oxidized TCA cycle intermediate, it is perhaps not surprising that aspartate is the biggest vulnerability of cells experiencing ETC disruption; consequently, providing cells exogenous aspartate can be sufficient to restore growth in ETC-inhibited conditions48, 49. Consistently, heterologous genetic strategies to enable mitochondrial NAD+ regeneration are likewise sufficient to restore tumor growth despite ETC disruption41. Collectively, these data demonstrate that sustained electron flow through cellular metabolic networks is required to unleash the full potential of cell growth.
Figure 4. Electron carrier regeneration.
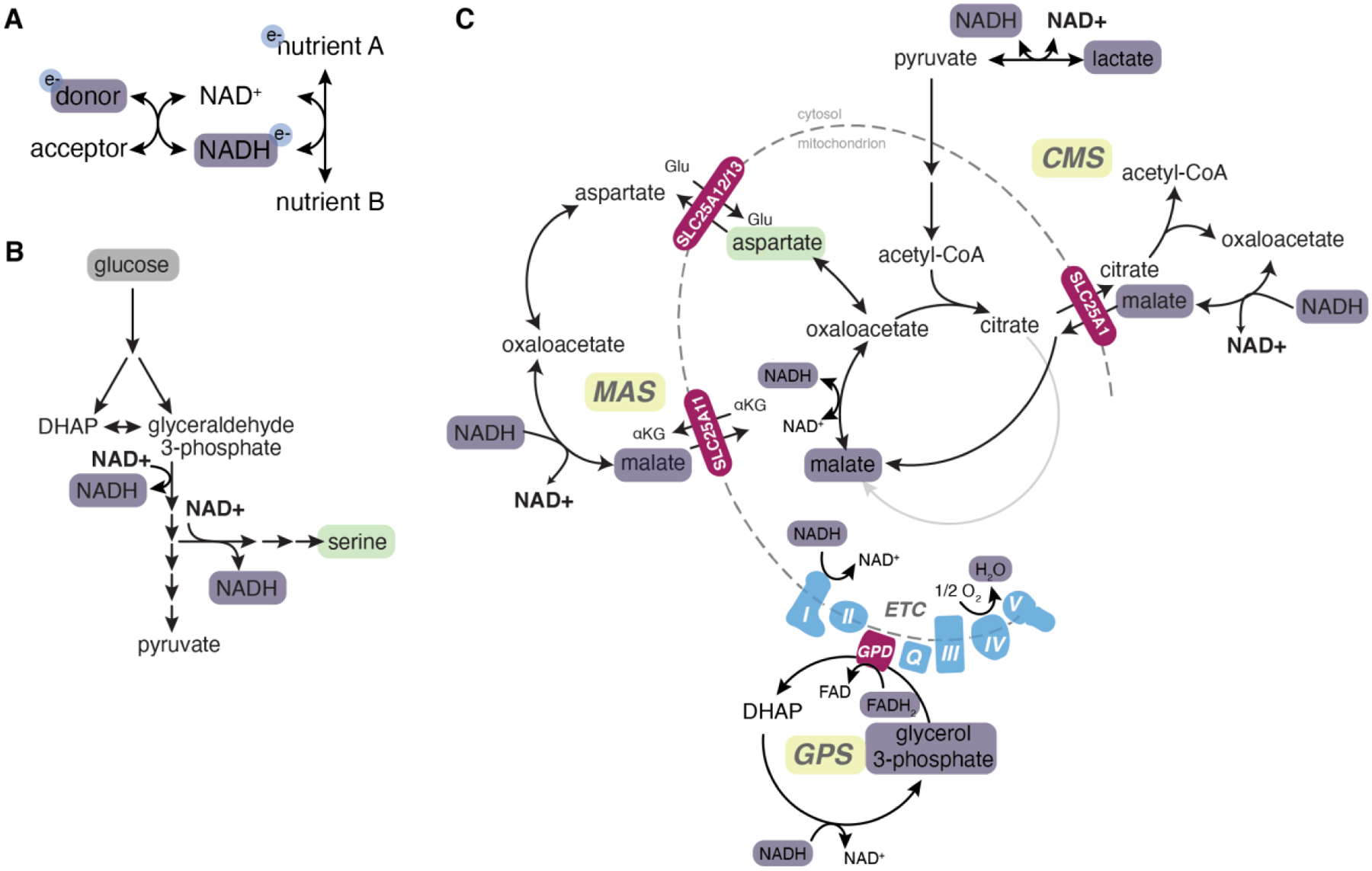
All metabolic pathways require a continuous supply of electron donors and acceptors. A, Basic principles of redox reactions. For a more reduced metabolite (A) to be converted to a more oxidized metabolite (B), an electron carrier (commonly NAD+) is required to accept reducing equivalents from A. Continued production of B depends on electron carrier regeneration, which can be achieved by the reduced electron carrier (here, NADH) donating electrons to another electron acceptor (for example, complex I of the electron transport chain). B, Major cytosolic sources of NADH are glycolysis (which produces 2 NADH for each molecule of glucose) and de novo serine synthesis. Continued flux through these pathways requires regeneration of oxidized NAD+. C, Reduction of pyruvate to lactate via LDH is a major source of cytosolic NAD+ regeneration, although if pyruvate is produced via glycolysis this reaction is redox neutral. Electrons can be transferred to the cytosol via electron shuttles. The glycerol 3-phosphate (GPS) shuttle couples reduction of dihydroxyacetone phosphate (DHAP) to glycerol 3-phosphate with oxidation of glycerol 3-phosphate back to DHAP thereby transferring reducing equivalents into the mitochondrial electron transport chain (ETC). Both the citrate-malate shuttle (CMS) and malate-aspartate shuttles (MAS) transfer reducing equivalents into mitochondria via reduction of cytosolic oxaloacetate to malate, which is imported into the mitochondria to regenerate oxaloacetate and NADH. The ETC is a major source of electron carrier regeneration and electrons are ultimately used to reduce oxygen to form water. Purple boxes, reduced electron carriers. Green boxes, oxidized metabolites that become limiting in reductive conditions.
Where do electrons end up? In most cases: oxygen. Electrons deposited into the ETC ultimately reduce oxygen to form water. Even reducing equivalents generated in the cytosol can enter the mitochondrial ETC through electron shuttles such as the glycerol 3-phosphate, malate-aspartate, and citrate-malate shuttles (Figure 4C). When oxygen is limiting, cells can employ alternative strategies to cope with excess electrons, such as increasing proline synthesis to divert electrons away from the ETC, enabling continued TCA cycle flux50. In rare cases where electrons cannot move forward through the ETC, they can reverse and reduce alternative acceptors such as fumarate51, 52.
The second major electron acceptor is pyruvate. In the cytosol, lactate dehydrogenase (LDH) reduces pyruvate to form lactate, thereby regenerating NAD+ (Figure 4C). When oxygen is limiting, electron shuttles cannot function and LDH becomes the dominant mechanism for cytosolic NAD+ regeneration; accordingly, cells excrete pyruvate as lactate during hypoxia—a phenomenon known as anaerobic glycolysis. Electron shuttles can saturate even in abundant oxygen—for example, when mitochondrial ADP is not present at sufficient levels to drive OXPHOS and flux through the ETC—and in this scenario cytosolic NADH instead reduces pyruvate, producing lactate3, 4. Therefore, the Warburg effect is likely a necessary outcome of demand for NAD+ regeneration in proliferating cells.
Metabolic control of cell identity
Metabolites do more than support cell growth. Increasingly, metabolites are recognized to play critical roles regulating gene expression programs that control cell fate. While gene expression programs are ultimately dictated by transcription factors, chemical modifications on DNA and histones can shape transcriptional outcomes by modulating chromatin organization and recruitment of effector proteins such as those involved in chromatin remodeling or transcriptional regulation. The deposition and removal of chromatin modifications is directly linked to cell metabolism: select intracellular metabolites provide the substrates for the chemical modifications, and others are required for their enzymatic removal (Figure 5). All methylation requires the universal methyl donor S-adenosylmethionine, which is derived from methionine, and methionine starvation routinely decreases several histone methylation marks in cultured cells in vitro and in vivo53. Similarly, acetylation requires acetyl-CoA, and starving cells of nucleocytosolic acetyl-CoA by limiting glucose availability or inhibiting the major enzymatic source of nucleocytosolic acetyl-CoA (ACL) likewise decreases acetylation on several histone residues54. Acetylation is usually removed by simple hydrolysis, although in some cases sirtuins remove acetylation in an NAD+-dependent fashion. Methylation removal is more complicated: certain mono- and di-methylation can be removed by lysine-specific demethylase 1 (LSD1), which uses FAD as a cofactor that is re-oxidized by molecular oxygen. More generally, methylation is controlled by a family of alpha-ketoglutarate (αKG)-dependent dioxygenases which use αKG and molecular oxygen as obligate co-substrates to facilitate demethylation; accordingly, depriving cells of glutamine, the dominant source of αKG, induces hypermethylation at select histone lysine residues55. Collectively, these initial studies in cultured mammalian cells demonstrated proof of concept: metabolic interventions can affect the landscape of chromatin modifications and, ultimately, gene expression programs.
Figure 5. Intersection between metabolism and chromatin.
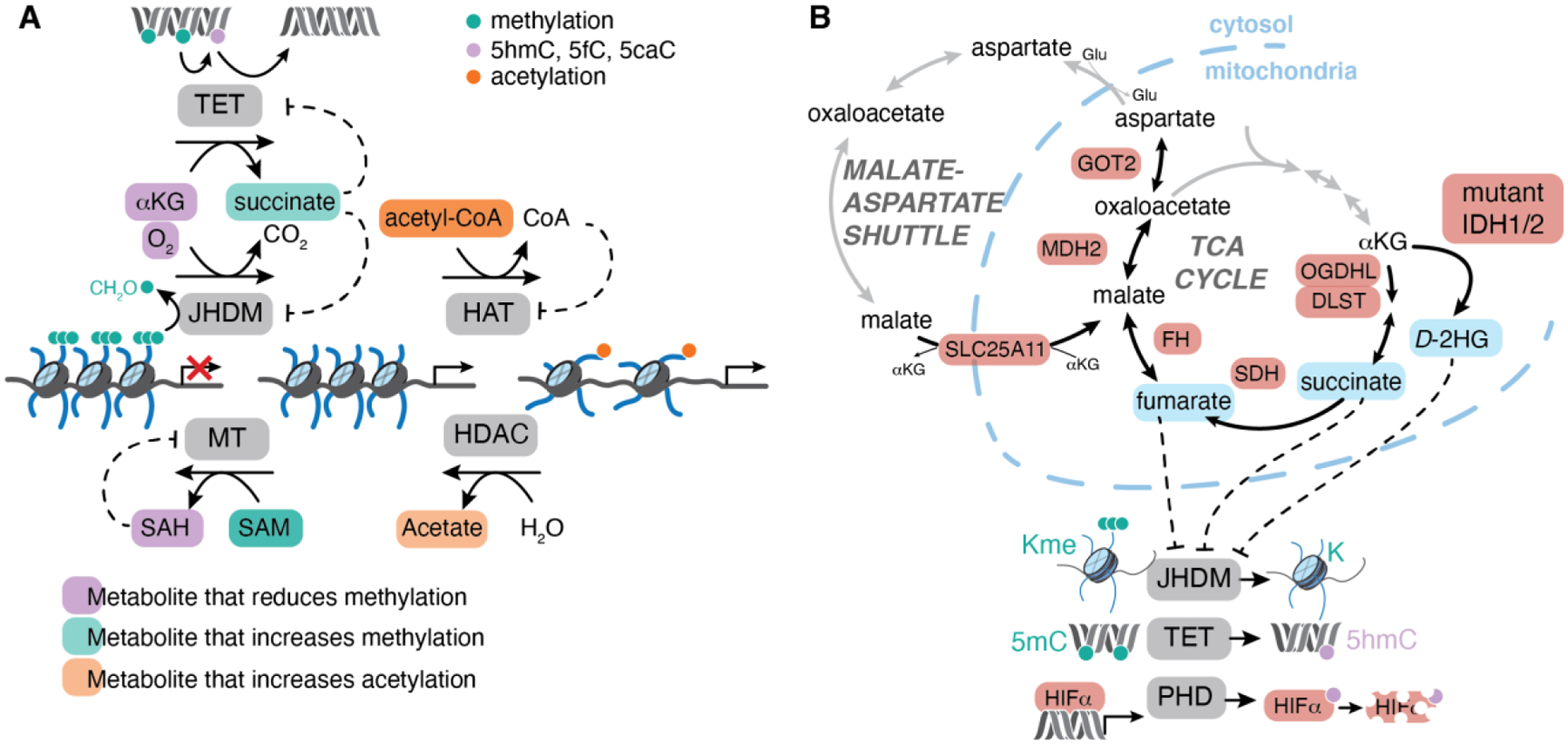
A, Overview of metabolites that are substrates and inhibitors of major chromatin-modifying enzymes. DNA and histone methylation is deposited by methyltransferases (MT) that use S-adenosylmethionine (SAM) as a methyl donor, producing S-adenosylhomocysteine (SAH) as a product. Histone acetyltransferases (HAT) use acetyl-CoA or other related acyl-CoAs as substrates to modify histone lysine residues. Most histone deacetylases (HDAC) use water to catalyze simple hydrolysis of acetylated lysine, yielding acetate. Some mono- and dimethylated histone lysine residues can be removed by lysine-specific demethylase 1 (LSD1), which uses FAD as a cofactor that is reoxidized by molecular oxygen. Most lysine and arginine methylation is removed by jumonji-domain containing histone demethylases (JHDM) that use alpha-ketoglutarate (αKG) and molecular oxygen as co-substrates to hydroxylate methylated residues, yielding succinate and carbon dioxide as products. The unstable hydroxyl-methyl intermediate is lost as formaldehyde. DNA methylation is removed by the ten-eleven translocation (TET) family of enzymes that hydroxylate DNA methylcytosine to 5-hydroxymethylcytosine (5hmC). 5hmC can lead to passive loss of DNA methylation during cell division or can be iteratively oxidized to 5-formylcytosine (5fC) or 5-carboxycytosine (5caC) which can be actively removed and replaced with unmodified cytosine. Metabolites whose abundance is linked to changes in chromatin modifications are highlighted in orange (promoting acetylation), teal (promoting methylation) or purple (reducing methylation). B, Mutations in genes encoding components of the TCA cycle or malate-aspartate shuttle (highlighted in red) are found in human cancer patients. Somatic mutations in isocitrate dehydrogenase (IDH1/2) enable production of D-2HG from αKG. Germline mutations in components of succinate dehydrogenase complex (SDH) or fumarate hydratase (FH) are associated with tumor predisposition syndromes marked by loss of heterozygosity and accumulation of succinate or fumarate, respectively. All 3 oncometabolites, highlighted in orange, can inhibit αKG-dependent dioxygenases including the JHDM, TET or prolyl hydroxylase (PHD) proteins and are associated with hypermetylation of histones (Kme) or DNA (5mC) and normoxic stabilization of hypoxia inducible factor alpha subunits (HIFα).
The hypothesis that chromatin responds to metabolic regulation has gained great attention, but several important biochemical and conceptual questions remain to be addressed. First, are physiological shifts in metabolite availability sufficient to alter the chromatin landscape? In other words—absent extreme nutrient deprivation—are co-substrates ever limiting for activity of chromatin-modifying enzymes? The answer to this question depends on the affinity of the enzyme for the metabolite substrate (KM) and the local concentration of the metabolite that is available to the enzyme in question. In many cases, the average cellular concentration of metabolite substrates is lower than (or in range of) the enzyme KM for that metabolite, supporting the notion that metabolite availability may indeed restrict activity of these enzymes56. Even when average cellular metabolite concentration exceeds typical enzyme KM values (as, for example, in the case of αKG whose intracellular concentration often vastly exceeds the KM of αKG-dependent dioxygenases), many additional biochemical considerations combine to determine the likelihood that a metabolite limits enzyme activity57. Notably, measures of average cellular metabolite concentration obscure important—and potentially large—intracellular variations in metabolite levels. The inherent compartmentalization of metabolic networks ensures that many metabolites are unevenly distributed across cells, and emerging approaches to profile organelle-specific metabolite concentrations are underscoring the differential abundance of metabolites across subcellular compartments58, 59. Even within the nucleus, hyper-local production and consumption of specific metabolites will alter the amount of substrate available to a given enzyme. Furthermore, each of these chromatin-regulatory enzymes is inhibited by its product (S-adenosylhomocysteine, CoA, or succinate, respectively), which compete with substrate for the enzyme active site (Figure 5). Accordingly, concentration of both substrates and products must be considered when determining whether a metabolite will be able to control enzyme activity.
Second, how can metabolic fluctuations induce specific cellular outcomes? The same chemical modification on different histone residues can convey vastly different information and yet all enzymes regulating a given modification require the same metabolic co-substrates. For example, histone 3 lysine 4 trimethylation (H3K4me3) is associated with gene activation, while H3K27me3 and H3K9me3 are associated with gene repression; how, then, can a metabolite specifically affect one or the other modification? Importantly, not all enzymes in each family have the same affinity for their metabolic substrates. For example, in the case of αKG-dependent dioxygenases, lysine demethylase 6A (KDM6A) which removes H3K27me3 has the lowest affinity for molecular oxygen and therefore is most limited of all dioxygenases when cells experience hypoxia60. Enzymes in the same family can exhibit affinities for substrates and/or inhibitors that are separated by an order of magnitude, raising the possibility that there is a hierarchy of responses depending on the severity of the metabolic alteration and that it is the relative balance of substrates and inhibitors—rather than the absolute concentration of each—that controls enzyme activity. Additionally, individual modifications can be regulated by multiple enzymes that may not be equally expressed in all cellular contexts; in this scenario, cells that express enzymes with lower affinity for specific metabolites may be more responsive to perturbations of that metabolite than cells who express high-affinity homologs. Similarly, within a cell, modifications guarded by low-affinity enzymes will be more likely to respond to metabolite shifts than modifications guarded by high-affinity enzymes, and this pattern of responsiveness may vary depending on the relative expression of relevant chromatin-modifying enzymes.
Third, for chromatin regulation to exert coherent effects on gene expression profiles, modifications need to be controlled at the right genetic locus: if metabolites induce global shifts in a certain modification, how is regional specificity achieved? One possibility is that regional specificity is determined by the pre-existing chromatin landscape in a cell. For example, compact heterochromatin may be relatively devoid of chromatin-modifying enzymes and thus resistant to metabolic perturbations over short timescales; in contrast, accessible or dynamic regions of the genome may have greater local concentrations of chromatin modifying-enzymes and thus be more amenable to metabolic control. Alternatively, transcription factors may themselves respond to metabolic perturbations, thereby providing an important layer of specificity. Transcription factors, like histones, can be acetylated or methylated and these modifications could conceivably alter overall DNA binding activity or cofactor recruitment, thereby modulating gene expression programs. A striking example of the role for transcription factors in directing local chromatin regulation was found in glioblastoma cell lines, where increasing acetyl-CoA availability induced H3K27 acetylation most notably at genomic loci bound by nuclear factor of activated T cells (NFAT) 161. Mechanistically, acetyl-CoA induced intracellular signaling cascades that control NFAT1 nuclear localization, and the combined effect of nuclear NFAT1 alongside plentiful acetyl-CoA availability fueled local deposition of H3K27ac at NFAT1 target genes61. In this manner, fluctuations in metabolite levels may control upstream pathways that direct transcription factor localization and/or activity, while remaining permissive for continued activation of chromatin-modifying enzymes.
Some of the best evidence linking metabolites to cell fate control comes from studies of human cancer, where pathological accumulation of specific TCA-cycle related metabolites (succinate, fumarate, and 2-hydroxyglutarate) is associated with development of specific tumor types. Notably, all of these “oncometabolites”—defined as metabolites with a clear link to tumor development through plausibly defined mechanisms8—serve as competitive inhibitors of αKG-dependent dioxygenases, suggesting that these enzymes are particularly susceptible to metabolic control (Figure 5). Unraveling which dioxygenases respond to metabolic perturbations, and how dioxygenase inhibition supports tumor growth, is revealing fundamental principles of metabolic control of cell fate that extend well beyond cancer cells.
Hereditary cancer syndromes.
Heterozygous germline mutations in genes encoding components of the TCA cycle enzymes succinate dehydrogenase (SDH) and fumarate hydratase (FH) are associated with familial cancer syndromes marked by predisposition to a subset of tumors such as phaeochromocytomas, paragangliomas, and renal cell carcinoma among others62. In all cases, tumors are marked by loss of heterozygosity of the remaining wild-type allele, leading to complete loss of enzyme function and large accumulation of succinate or fumarate, respectively. While the precise mechanisms linking succinate and fumarate accumulation to tumorigenesis remain unknown, and are likely context-specific, metabolite accumulation is clearly associated with inhibition of several αKG-dependent dioxygenases including the jumonji-domain containing family of histone demethylases, the TET family of methylcytosine dioxygenases that iteratively oxidize DNA methylcytosine, and the prolyl hydroxylases that target the labile α subunit of hypoxia inducible factor (HIF) for degradation (Figure 5).
The discovery that both succinate and fumarate stabilize HIFα represented the first evidence that αKG-dependent dioxygenases are sensitive to TCA cycle metabolite levels in mammalian cells63, 64. While HIFα stabilization induces metabolic rewiring that may be advantageous to tumors, HIFα is unlikely to be the sole effector of oncometabolites in human tumors; indeed, renal cyst formation driven by FH mutation does not require HIF65. Both SDH- and FH-deficient tumors are broadly associated with chromatin hypermethylation that favors silencing of genes involved in normal differentiation62. Additionally, SDH and FH inhibition can trigger the epithelial-mesenchymal transition, whereby epithelial cells take on features of more migratory mesenchymal cells, by repressing the miR-200 locus that is normally activated by TET proteins66. Oncometabolite-driven inhibition of KDM4A/B also hampers DNA damage repair, which may further contribute to tumor progression67, 68. Fumarate may exert effects beyond dioxygenase inhibition: as an electrophile, it can directly modify reactive cysteines, including those within KEAP1 that are required to destabilize NRF2, thus activating antioxidant gene expression programs65, 69.
More recently, germline mutations in related genes, including genes encoding components of the OGDH complex and the malate-aspartate shuttle, have likewise been identified in patients with familial cancer syndromes (Figure 5)62. Several of these tumors also display chromatin hypermethylation and/or HIFα stabilization, analogously to SDH/FH-deficient tumors. Intriguingly, these components all revolve around the TCA cycle generally, and αKG metabolism specifically. Whether this convergence is because these mutations are relatively well-tolerated in certain contexts compared to other metabolic disruptions, or because of a central role for αKG-dependent dioxygenases or other αKG-regulated processes in antagonizing tumorigenesis, remains to be determined. Taken together, familial cancer syndromes provide robust evidence that metabolic networks control cancer susceptibility and represent critical model systems for elucidating principles linking metabolites to cell fate control in diverse lineages.
Somatic mutations in metabolic enzymes.
The third oncometabolite, 2-hydroxyglutarate (2HG), is produced in tumors harboring specific somatic mutations in genes encoding isocitrate dehydrogenase 1/2 (IDH1/2). These active-site mutations induce neomorphic enzymatic activity, enabling IDH1/2 to reduce αKG, forming D-2HG (Figure 5). Like succinate and fumarate, D-2HG accumulates at high (millimolar) levels in tumors and is associated with inhibition of a panel of αKG-dependent dioxygenases. IDH1/2 mutations are found at high frequency in acute myeloid leukemia (AML), chondrosarcoma, cholangiocarcinoma, glioma, and other tumor types at somewhat lower frequencies70. Chromatin hypermethylation and impaired differentiation is a common feature of IDH-mutant tumors of all lineages, although the chromatin modifications and genetic loci affected vary depending on lineage. Consistently, the dioxygenases implicated as targets of D-2HG vary between tumor types. For example, TET enzymes are implicated in AML, where TET and IDH1/2 mutations rarely co-occur, while enzymes controlling histone methylation are implicated in other tumor types71, 72. Several drugs targeting mutant IDH1/2 isoforms are already in the clinic and have shown efficacy especially in the context of AML, where they induce cancer cell differentiation. In some cases, tumor relapse is associated with acquisition of mutations that restore D-2HG production, indicating that—at least in AML—continued production of D-2HG facilitates malignant self-renewal70, 73.
D-2HG exerts effects beyond chromatin regulation. D-2HG production can be associated with additional metabolic alterations including transaminase inhibition and decrease in intracellular NAD+, each of which can be exploited pharmacologically, although whether these changes contribute to tumorigenesis remain unclear70, 74. More broadly, D-2HG is produced at such high levels that it can leave cancer cells and affect surrounding cells within a tumor, where it is associated with reduced T-cell infiltration and effector function75–78. These studies provide important reminders that metabolic changes rarely occur in a vacuum and that linking specific metabolic alterations to tumor outcomes requires a precise combination of biochemical and genetic approaches to identify relevant targets of individual metabolites.
Endogenous metabolic networks.
Even without mutations in metabolic enzymes, tumors can benefit from the links between metabolism and the chromatin landscape. In some cases, this link is directly regulated by oncogenes and tumor suppressors. For example, AKT directly phosphorylates and activates ACL, leading to enhanced nucleocytosolic acetyl-CoA and elevated histone acetylation driven by PI3K/AKT signaling in multiple tumor types79. ACL-dependent histone acetylation in turn supports the fate changes that occur early in pancreatic adenocarcinoma (PDAC) development: in pancreatic acinar cells, oncogenic Kras induces histone acetylation and acinar-ductal metaplasia, and inhibiting ACL or the reading of histone acetylation (via bromodomains) blocks this fate transition80. Similarly, the tumor suppressor p53 (TP53) controls αKG levels in PDAC, and both p53 and αKG are sufficient to induce PDAC cells to differentiate to a premalignant state81. Enforcing succinate accumulation prevents p53 from inducing differentiation and allows tumors to grow despite robust p53 accumulation, suggesting that activation of one or more αKG-dependent dioxygenases is required for effective tumor suppression by p53 in PDAC81.
In addition to oncogenic mutations, local environmental cues can alter metabolites that control cancer cell fate. During hypoxia, the combined effect of low pH and reductive stress (high NADH/NAD+) favors promiscuous reduction of αKG to L-2HG by LDH and malate dehydrogenase (MDH)82–84. Even though hypoxic L-2HG accumulation is lower than that of D-2HG in IDH-mutant cancers, L-2HG can inhibit αKG-dependent dioxygenases more potently than D-2HG82, and L-2HG accumulation is necessary and sufficient for hypoxia-induced hypermethylation at select histone residues82. Suggestively, many tissue stem cells reside within hypoxic niches, but whether L-2HG contributes to the effects of hypoxia on homeostatic or malignant self-renewal remains to be determined85, 86. Other stimuli besides oxygen availability regulate L-2HG levels: L-2HG also accumulates following downregulation of L2HGDH, the enzyme that re-oxidizes L-2HG to αKG, or ETC disruption87. Thus, L-2HG may signal reductive stress in contexts beyond cancer.
Local variation in nutrients beyond oxygen are also emerging as important regulators of cancer cell fate. In the core of some solid tumors, limited glutamine availability compromises αKG-dependent demethylation and favors de-differentiation that is reversed by exogenous glutamine supplementation88. αKG dependent dioxygenases are also dependent upon ascorbate (vitamin C), a cofactor that supports activity presumably by facilitating regeneration of the ferrous iron required for catalysis, and ascorbate supplementation slows leukemic progression in a partially TET-dependent manner89, 90. Many local factors can combine to determine intracellular metabolite levels. For example, de novo serine synthesis produces αKG as an obligate byproduct, and dietary serine restriction induces αKG-dependent histone demethylation and cancer cell differentiation in models of squamous cell carcinoma45. Blocking de novo serine synthesis favored expansion of stem-like cancer cells, even on a serine-replete diet, suggesting that, in this lineage, spontaneous activation of de novo serine synthesis can regulate cancer cell fate45.
Collectively, the above studies support a model in which environmental or genetic conditions that favor αKG-dependent dioxygenase activity are permissive for tumor suppressive differentiation, and this metabolic control of differentiation can be coopted in cancer cells. While many of the links between metabolism and chromatin were forged in cancer cells, the underlying biochemical principles are relevant to all mammalian cell types. Therefore, an important area of future work is determining the degree to which endogenous metabolic networks modulate normal development, and whether developing organisms can insulate cell fate transitions from metabolic control.
Diversity of metabolic strategies
While studies in cancer cells have outlined the principles of cell metabolism—how metabolites support cell growth and identity—recent advances have underscored that metabolism is rarely “one-size-fits-all”. Rather, cells can adopt different metabolic strategies depending on their lineage, developmental stage, or environmental context. Advances in in vivo isotope tracing have allowed the field of cancer metabolism to move beyond studies of metabolism in cultured cells and directly monitor metabolic networks in vivo. These studies have helped to overturn many of the initial conceptions of cancer metabolism and even helped to revise major assumptions about metabolism in normal tissues. Together, these studies emphasize that metabolic pathways are not static but rather context specific: cells can use different nutrient sources and rewire intracellular metabolic pathways to help them surmount challenges or meet their specific demands
Environmental context
Tracing metabolic substrate use in human tumors led to two major surprises in apparent contradiction to the Warburg effect: first, many human tumors may oxidize glucose even more than surrounding normal tissue; and second, some human tumors use lactate as a metabolic fuel91, 92. In these studies, the observation that metabolites downstream of pyruvate—including lactate and TCA cycle-related metabolites—were more labeled from infused glucose than were upstream glycolytic intermediates led to the model that lactate derived from circulating glucose represented a major carbon source for several tumors and tissues. Consistently, xenografts formed from cancer cells harboring genetic deletion of the plasma membrane monocarboxylate transporter that facilitates lactate uptake showed reduced incorporation of lactate-derived carbons into downstream metabolic networks92. A similar trend for lactate labeling exceeding that of upstream glycolytic intermediates was also seen in tumors from patients with triple negative breast cancer, although in this case tumor lactate was also more labeled than circulating lactate, implying high local lactate production that is poorly mixed with surrounding cells93. Concurrently, studies in normal tissues revealed that circulating lactate is a major source of TCA cycle intermediates for many organs, thereby providing a route for cells to maintain homeostatic TCA cycle flux without requirement for growth-factor directed uptake of nutrients such as glucose and glutamine94.
The discovery that tumors and other tissues use lactate as a fuel turned the Warburg effect from a perceived universal feature of cancer to a context-specific phenomenon and provided a dramatic example of the impact of environmental context on cellular metabolic networks. Clearly, different tumors exhibit different propensity for glucose oxidation. While human lung tumors that oxidize glucose more than surrounding normal tissue91, glucose oxidation is suppressed in clear cell renal cell carcinoma, a tumor driven by VHL deficiency that enforces pseudohypoxic HIFα stabilization and transcriptional activation of metabolic genes that favor aerobic glycolysis over glucose oxidation (Ldha, Pdk1)95. Even within a single tumor, there is regional heterogeneity in glucose oxidation that is largely correlated to perfusion91. Traditional tracing studies that average metabolite profiles across a sample will struggle to account for such heterogeneity; advances in spatial mass spectrometry may help to resolve gradients in metabolic activity across tissues96.
Nutrient availability is likely to vary considerably both within and between tumors, as even entire organs have notable differences in nutrient consumption and release profiles97, 98. Accordingly, metabolic preferences may be dependent on anatomic location and tumor size, perfusion, and cell type composition. Recent studies are increasingly corroborating the notion that metabolic dependencies are context specific. For example, glutamate-oxaloacetate transaminase 2 (GOT2), a central component of the malate-aspartate shuttle that facilitates aspartate synthesis, is required for many cancer cells in vitro but largely dispensable in vivo; in part, this is because cells in vivo have altered demands for electron shuttles and alternative protein sources for aspartate scavenging99, 100. Simply changing nutrient availability is likewise sufficient to reorganize metabolic networks and shift metabolic dependencies in cultured cells101–103. These results do not negate the importance and insight of metabolic studies performed in cultured cells. Indeed, CRISPR screens demonstrate that PDAC cells growing in culture and as tumors share many metabolic dependencies104, 105. Both the similarities and the differences are informative: similarities may reflect universal metabolic requirements or intrinsic demands of a particular cell type; differences may reveal metabolic limitations in specific conditions or locations. In the case of PDAC, the observation that heme synthesis is limiting specifically in vivo led to the discovery that cultured cells can share heme precursors and that heme degradation is increased in vivo104, 105. Thus, while cell culture provides an excellent model system for understanding the regulation and architecture of metabolic networks, comparing metabolic preferences and dependencies across different growth conditions is revealing the myriad metabolic strategies that support cell function.
Cell intrinsic determinants
While many cancer cells exhibit a convergent metabolic phenotype, vestiges of normal tissue metabolism remain. Studies comparing gene expression programs across human tumors showed that most tumors exhibit metabolic gene signatures that are similar to their corresponding normal tissue106, 107. Indeed, the tissue of origin plays a major role determining cancer cell metabolism. For example, lung adenocarcinomas depend on branched chain amino acids more than pancreatic adenocarcinomas, even when both tumors are driven by identical oncogenic lesions108. These tissue-specific preferences can persist even as tumors progress and metastasize: the metabolic profiles of metastatic tumors are similar to their primary tumor counterparts, and metastatic cancer cells retain metabolic phenotypes and preference for growth in their primary site even after propagation in the metastatic site109. It is tempting to speculate that the metabolic preferences of cancer cells endowed by their tissue of origin restricts growth in different tissues and thereby contributes to the notable organ tropism of many human tumors.
Lineage is clearly not the only determinant of cancer metabolism. The transforming oncogene also plays a role, as shown by comparing metabolic profiles of liver and lung tumors harboring c-Myc or c-Met oncogenes110. Nevertheless, isotope tracing in over 80 human non-small cell (NSCLC) lung cancer cells showed that oncogenotype alone cannot predict metabolic profiles, indicating that other factors—epigenetic, transcriptional, or proteomic—also shape metabolic networks111. Likely, developmental stage contributes to metabolic differences, as metabolic rewiring frequently accompanies differentiation112. Suggestively, in the breast, normal basal, luminal progenitor and mature luminal populations subtypes harbor different preferences for glycolysis and OXPHOS that persist in established tumors113. Thus, the metabolic phenotype of the cancer cell-of-origin may be retained throughout tumor progression.
Despite cell-intrinsic metabolic preferences, cancer cells can rewire metabolic pathways in response to changing metabolic demands. Quantitative flux modeling suggests that multiple tumor types reduce ATP production compared to normal tissue; in the pancreas, this may be because PDAC tumors have lower protein synthesis rates, which would in turn decrease ATP demand114. Whether this metabolic switch occurs after transformation, or is an intrinsic feature of cells susceptible to transformation, remains to be determined. Even within the cancer cells of a tumor, cells can adopt different metabolic configurations according to their cell state. For example, quiescent cells rely on glutamate dehydrogenase (GDH) as a major route of glutamine anaplerosis; in contrast, transamination reactions drive most anaplerosis in proliferating cells115. Frequently, the stem-like cells that reside within a tumor show increased reliance on OXPHOS, which may be targeted therapeutically116. These results raise the possibility that phenotypic heterogeneity in cancer cells may be accompanied by targetable metabolic heterogeneity.
The best example of metabolic plasticity in cancer is the metabolic transitions that accompany various stages of metastasis. As cancer cells extravasate into the vasculature, travel to distant organs, and ultimately recolonize a new site, they are subject to changing environments that each impose distinct metabolic stresses. In particular, the oxidative stress imposed by circulation appears to be a major bottleneck to efficient metastasis. Reversible activation of NADPH-generating pathways enables successful metastasis, which is further heightened by providing exogenous antioxidants117. Compared to cells with low metastatic potential, efficiently metastasizing cells take up more lactate, which decreases intracellular pH and activates the pentose phosphate pathway, thereby increasing NADPH production118. Importantly, the role of oxidative stress in metastasis may be highly context specific: in models of pancreatic cancer, reactive oxygen species promote metastatic spread but also impairs tumor growth at secondary sites119.
Metastatic colonization imposes another set of significant metabolic challenges that vary depending on the site of colonization. The same tumor metastasizing to different organs will give rise to secondary tumors with distinct, organ-specific transcriptional profiles, potentially reflecting selection of cells with capacity to reseed a given organ or organ-induced metabolic rewiring following colonization120, 121. In some cases, local metabolite availability can support metastatic seeding. When breast cancer cells migrate to the lung, they take advantage local pyruvate availability to produce αKG via pyruvate transamination; in turn, αKG-dependent prolyl hydroxylases induce collagen crosslinking and extracellular matrix maturation that favors metastatic outgrowth122. Further discussion on the role of antioxidants and metabolic reprogramming during metastasis can be found in recent reviews123.
Expanding metabolic networks
Our increasing appreciation of metabolic diversity in cancer cells has helped to annotate and even extend our knowledge of intracellular metabolism, revealing additional nutrient sources new pathway configurations. Just as protein has emerged as an important substrate to support cancer cells when amino acids are limiting, unconventional substrates can also support sugar-dependent metabolic networks when glucose is limiting. For example, cancer cells can salvage ribose required for nucleotide biosynthesis and redox homeostasis from extracellular uridine or even RNA, and hyaluronic acid—a major component of the extracellular matrix—can provide glucosamine and glucuronic acid sugars via hexosamine salvage pathways124–127. Reduced nitrogen—critical for biosynthesis of amino acids and nucleotides—can also be obtained by recycling ammonia that is otherwise a metabolic waste product ultimately discarded in the form of urea. Investigating oncogenotype-specific metabolic dependencies in NSCLC revealed that Kras/Lkb1-mutant NSCLC are particularly dependent upon carbamoyl phosphate synthase-1 (CPS1), an initiating enzyme in the urea cycle traditionally considered to be restricted to hepatocytes. CPS1 condenses ammonia and bicarbonate to form carbamoyl phosphate that is required for de novo pyrimidine synthesis, thus allowing Kras/Lkb1-mutant NSCLC cells to capture ammonia for nucleotide synthesis128. Ammonia can also be incorporated into central carbon metabolism by reverse glutamate dehydrogenase flux in breast cancer cells129, thereby circumventing potentially toxic ammonia accumulation while salvaging valuable reduced nitrogen to support cell proliferation.
More broadly, many steps of metabolic pathways—including glycolysis and the TCA cycle—are close to thermodynamic equilibrium and thus readily reversible according to metabolite supply and demand130, 131. During hypoxia or ETC disruption, when traditional oxidative TCA cycle metabolism is constrained, cells can shift to reductive glutamine metabolism in which glutamine-derived αKG is carboxylated in an NADPH-dependent manner via reverse IDH activity to sustain citrate production required for cytosolic acetyl-CoA and de novo lipid synthesis132, 133. Such reductive carboxylation allows cells to generate citrate from glutamine-derived carbons without the concomitant generation of NADH, thus providing an efficient adaptation for cells under conditions of impaired NAD+ regeneration. Cells can also circumvent several NADH-generating steps of the TCA cycle by engaging the citrate-malate shuttle, rather than the conventional TCA cycle, as an alternative cycle of continuous citrate regeneration (Figure 4C)134. Isotope tracing studies in cancer cells and immune cells demonstrate variable preference for each route of citrate regeneration, and the choice between canonical TCA cycle and citrate-malate shuttle can shift as stem cells undergo commitment and differentiation134, 135. These and similar studies collectively help erode traditional views of metabolism embedded in textbooks and reveal the diverse metabolic configurations that support cell function in various environmental contexts and cell state configurations.
Additional roles of metabolic enzymes
A handful of metabolic enzymes have functions beyond those of their traditional catalytic activity. Such ‘moonlighting’ allows metabolic enzymes to participate in cellular processes beyond metabolism, potentially expanding the sphere of influence of metabolic networks to other cellular functions. There are two classic examples of moonlighting activity: cytochrome c and aconitase 1. Cytochrome c normally resides on the inner mitochondrial membrane where it carries electrons between complex III and IV of the electron transport chain. Upon initiation of apoptotic cascades, cytochrome c is released to the cytosol where it binds protease-activating factor 1 (Apaf1), forming the apoptosome. The critical role of this moonlighting function—rather than electron transport function—in execution of cell death programs was demonstrated by mutant forms of cytochrome c that retain electron transfer ability while disrupting Apaf1 interactions136. Analogously, under homeostatic conditions, cytosolic aconitase (ACO1) catalyzes the interconversion of citrate and aconitate. Upon iron starvation, ACO1 gains the ability to bind mRNAs with specific recognition structures known as iron-responsive elements (IREs); in this capacity, ACO1 is often referred to as IRE binding protein (IRE-BP) or iron-responsive protein (IRP) 1. IRE binding allows IRP1 to alter stability of mRNAs involved in iron uptake and storage, and the switch between aconitase and IRE binding capacity is fully reversible depending on assembly of the [4Fe4S] cluster required for aconitase activity137. Here, the functional switch from aconitase to IRE binding allows for a clearly observable distinction between conventional aconitase function and IRE binding activity.
There is growing appreciation that the dysregulated or heterogeneous expression of metabolic enzymes in cancer cells may do more than just control flux through their respective pathways. For example, the gluconeogenic enzyme fructose-1,6-bisphosphatase 1 (FBP1) is strongly downregulated in clear cell renal cell carcinoma (ccRCC)138. Perhaps unsurprisingly given its role in gluconeogenesis, restoring FBP1 activity in ccRCC cells antagonizes forward glycolytic flux and slows ccRCC growth. More surprisingly, catalytic dead versions of FBP1 also restrained glucose metabolism and slowed tumor growth, indicating that functions of FBP1 beyond its traditional enzymatic role contribute to its tumor suppressive ability. Indeed, in normal kidney tubules, FBP1 can localize to the nucleus where it binds and inhibits HIF, blocking transcription of hypoxia responsive genes138. As many metabolic enzymes are highly abundant, there is ample potential for enzymes to interact with, and potentially regulate, proteins in diverse functional pathways. For example, PHGDH can bind phosphofructokinase (PFK) and potentiate its glycolytic activity; accordingly, breast cancer cells with low PHGDH expression have reduced PFK flux139. PHGDH-low cells divert glycolytic intermediates upstream of PFK to the sialic acid pathway which in turn promotes integrin signaling and cancer metastasis, providing a mechanistic explanation for the increased metastatic propensity of breast cancer cells with low PHGDH expression139.
Distinguishing between the contributions of enzymatic activity and moonlighting activity to cancer phenotypes requires generation of catalytic-dead mutants and/or mutants that specifically impair moonlighting function. Notably, overexpressing both wild-type and catalytic-dead PHGDH restrained breast cancer cell metastasis, supporting the notion that PHGDH harbors non-enzymatic moonlighting activities139. Similarly, excluding FBP1 from the nucleus prevented its ability to slow tumor growth, underscoring the importance of this nuclear moonlighting role of FBP1138. In contrast, while many metabolic enzymes have been reported to harbor unconventional catalytic activities (for example, metabolite kinases using proteins as alternative substrates), the importance of these additional functions is difficult to parse absent genetic mutations that enable separation of function140. Taken together, while it is tempting to speculate that these moonlighting functions allow for conjoined regulation of distinct cellular pathways, future work should address the degree to which these additional activities are generalizable, required for specific outcomes, and/or or under selective pressure.
What distinguishes cancer metabolism from cell metabolism?
As discussed above, metabolic networks take many forms in both cancer cells and normal cells, and pathways can be dynamically regulated according to environmental context or cell state. Thus, absent specific metabolic mutations, there is considerable overlap between the metabolic states available to cancer cells and normal cells. What, then, truly distinguishes cancer metabolism from normal cell metabolism? The major feature that reliably distinguishes the study of cancer metabolism is the cancer patient themselves. Accordingly, putting cancer cell metabolism in context of the host is essential for understanding cancer evolution and identifying therapeutic opportunities.
Intersection with host physiology
Host physiology has clear impact on tumor progression: age, diet, exercise, and even gut microbiota can affect tumor incidence and progression116, 141, 142. Both systemic and local factors likely play important roles in tumor development and can do so by directly acting on cancer cells or indirectly by acting on immune or other stromal cells that can control cancer development. Recently, the interaction between cancer and immune cells has gained great attention as a potential mechanism by which host physiology may affect tumor biology. For example, high fat diet dampens anti-tumor immune responses in mice as a result of metabolic competition within the tumor microenvironment: when exposed to a high fat diet, cancer cells increase fatty acid uptake and metabolism, paradoxically depriving cytotoxic T-cells of critical nutrients. Accordingly, blocking cancer cell fatty acid oxidation restores anti-tumor immunity in mice fed a high-fat diet143. Dietary composition can also directly affect cancer cells, conceivably by providing a fitness advantage to cancer cells in one cell state over another or by inducing signaling or transcriptional pathways that control cancer cell state. For example, high fat diets promote emergence of colorectal cancer at least in part by increasing self-renewal capacity of intestinal progenitors144, 145. Likewise, dietary fructose is readily taken up by the intestinal epithelium where it signals to control glycolysis and promote intestinal hyperplasia and tumorigenesis146, 147. Following excessive fructose consumption, gut microbiota metabolize fructose to acetate148, which is reported to be an important fuel for several tumor types149. Taken together, it is increasingly clear that complex interactions underlie the relationship between host physiology and tumor progression and this topic is discussed in depth in recent reviews116, 141, 142.
Cancer can also affect the host. The most obvious example is cachexia, a process in which advanced tumors induce host tissue wasting. Cachexia induces devastating loss of quality of life and often accelerates patient mortality. Consequently, there is great interest in identifying strategies to delay or reverse cachexia; nevertheless, why tumors induce cachexia remains essentially unknown150. Central hypotheses include altered circulating metabolites due to tumor growth or changes in circulating factors (either derived from the tumor, immune system, or other impacted organ) that ultimately induce wasting of skeletal muscle and other organs. Likely, all of these factors may come into play, and may also be tumor—or even patient—specific. Continued dissection of how tumors arising in specific organs signal throughout the host will reveal novel insight into how mammals maintain organismal homeostasis and how this is disrupted in cancer patients.
Targeting tumor metabolism
The ultimate goal of studying cancer metabolism is to improve cancer care. Metabolic networks that sustain cancer cell proliferation or malignancy are attractive targets, but several challenges have hampered development of inhibitors that trigger effective and durable responses. One central challenge is general toxicity: as both cancer cells and normal cells rely on many of the same metabolic networks, it can be difficult to identify a therapeutic window that enables specific targeting of cancer cells. Metabolic plasticity poses an additional, formidable challenge. Cells harbor multiple redundant mechanisms to sustain key metabolic fluxes and can often, with time, circumvent toxicity induced by pathway inhibition by rerouting metabolic networks to cope with the interruption or transitioning to an alternate cell state less reliant on the targeted pathway. Even if plasticity is constrained—not all metabolic networks are equally accessible to all cell types—cancer cells, like normal cells, exist in heterogeneous cell states that are associated with different metabolic signatures151. Cancer cell heterogeneity and phenotypic plasticity heighten the difficulty of identifying metabolic pathways that are both uniformly required for the diverse population of cancer cells in a tumor and significantly different from the vast array of normal cells in the body.
Despite these challenges, there are several promising strategies for targeting cancer metabolism (Figure 6): 1) inhibiting a cancer-specific mutant enzyme whose activity is required to sustain tumor growth; 2) blocking a metabolic pathway that has become essential because of therapeutic interventions or genetic mutations accrued by cancer cells, and 3) targeting critical or redundant metabolic pathways to hobble an essential output. Blocking high flux, essential metabolic pathways has proven to be highly effective in certain contexts and is the basis for the oldest chemotherapies: anti-metabolites, which interfere with nucleotide synthesis or use and are still in clinical use today. Many ongoing efforts focus on other high-flux pathways that are critical to cancer cells, including OXPHOS and glutamine metabolism. Given that these pathways are features of normal cells, identifying a therapeutic window with acceptable toxicity can be challenging152. Nevertheless, antimetabolites are clearly effective components of therapy in some tumor types indicating that targeting metabolism is a viable strategy for some patients153, 154.
Figure 6. Strategies for targeting metabolism.
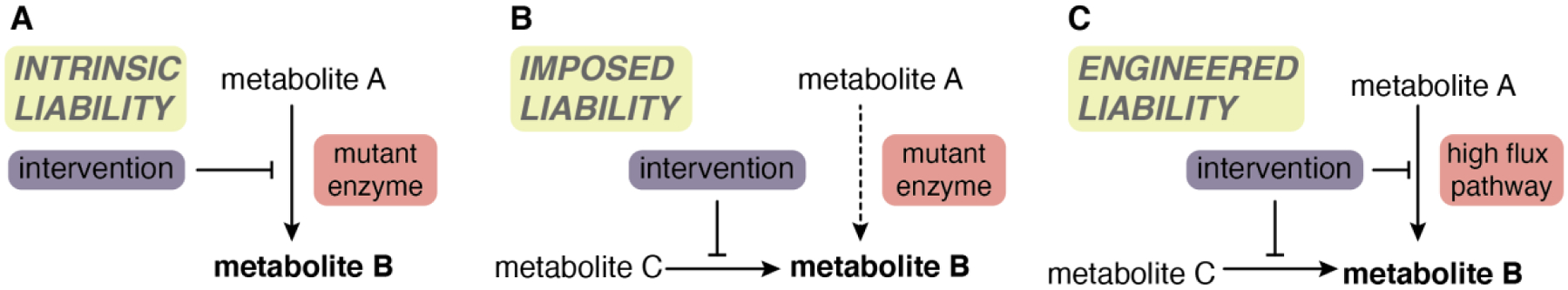
A, Metabolic interventions hold particular promise in tumors in which oncogenic mutations induce reliance on a specific metabolic pathway. For example, IDH1/2-mutant tumors use mutant IDH1/2 to produce D-2HG from αKG. Blocking this cancer-specific pathway has shown therapeutic efficacy in some patients. B, Some tumors have metabolic liabilities that arise as a result of their oncogenic mutations. For example, the truncated TCA cycle in SDH- and FH-deficient tumors imposes reliance on alternative metabolic pathways that may therefor represent a selective liability in these cancer cells relative to normal cells. C, Targeting a major metabolic pathway (e.x. glutamine usage or OXPHOS) may hold promise but efficacy may be constrained by alternative mechanisms that allow cells to circumvent such inhibition. In this scenario, dual targeting of compensatory pathways could represent an effective strategy, although toxicity to normal cells is likely to be a major risk.
Some cell types may harbor inherent liabilities: acute lymphoblastic leukemia cells have high asparagine demand but low capacity to synthesize asparagine de novo; accordingly, asparaginase treatment is a component of effective therapy in these tumors68, 155. In other cases, it may be possible to target liabilities induced by specific mutations. For example, SDH- and FH-deficient tumors are dependent upon metabolic networks that allow them to cope with TCA cycle truncation; targeting these alternative strategies or exploiting their oncometabolite-induced deficiencies in homologous recombination DNA repair may represent a therapeutic opportunity156. Perhaps the biggest success in cancer metabolism to date is therapeutic targeting of mutant IDH. Here, small molecules that specifically target the cancer-specific IDH mutations reduce 2HG production and have shown efficacy in certain tumor types, although relapse does occur70. Given the diversity and plasticity of metabolic networks that support cancer growth, it is unlikely any single metabolic intervention will elicit durable outcomes. More likely, metabolic interventions in combination with orthogonal approaches will provide an avenue to treat specific tumors, likely by crippling cells that are spared by conventional agents.
More recently, dietary manipulation of host metabolism has emerged as a novel strategy to improve anti-tumor therapies141. Dietary methionine restriction limits influx into the folate cycle that supports nucleotide synthesis and increases response to chemotherapy or radiation in mouse models of colorectal cancer and sarcoma157. Similarly, ketogenic diets increase efficacy of PI3K inhibitors in multiple mouse models of cancer by limiting the transient hyperglycemia and insulin signaling rebound that otherwise follows PI3K inhibition158. The above interventions, excepting therapies that target cancer-specific mutations, will target all cells within the tumor—not just cancer cells. As immune and cancer cells often share similar metabolic demands, such interventions may also affect anti-tumor immunity154. For example, T cell effector function is limited by methionine availability, and increasing circulating methionine improved anti-tumor immune responses in mouse models of colon cancer159. Therefore, effectively targeting cancer metabolism requires a thorough understanding of the local metabolic interactions that support tumor growth as well as consideration of long-term effects on normal cell populations.
Challenges and opportunities
Studies in cancer cells have shown how metabolites support multiple steps of oncogenesis, from facilitating aberrant cell fate regulation, fueling growth and proliferation, and enabling survival in diverse microenvironments. Although these metabolic outputs are rarely transforming in themselves, they can support cancer growth and progression and so are frequently nominated as therapeutic targets. However, the degree to which metabolism represents a targetable node for cancer therapy relies on the extent to which cancer cells harbor metabolic preferences that are significantly different from normal cells critical for organismal health and homeostasis. Therefore, a central challenge for the field moving forward is to apply the same level of rigor and interest as has been applied to cancer cells to unravel the metabolic networks that support the emergence and maintenance of normal tissues. While funders may be hesitant to see the value of such studies, they are necessary to understand how metabolism is remodeled during diseases and a prerequisite for designing safe, effective therapies that spare normal tissue function. Advances in functional genomics, such as the capacity to perform CRISPR-mediated screening in tissues in adult mice160, will facilitate comparison of how cancer cells and normal cells respond to acute perturbation of metabolic genes. Isotope tracing studies in animals and human patients are beginning to illuminate metabolic networks employed by tissues and tumors in vivo, although these studies can be hampered by the complexities of host metabolism and the potential for highly variable perfusion and cell composition across different samples. Here, spatial imaging approaches and/or tracing to metabolic endpoints that remain stable even after cell isolation161 will provide opportunities to dissect metabolic pathways of specific regions or cell types. A continued challenge will be to capture rare or transient metabolic states that may nevertheless be critical for cell function or tissue homeostasis. Given that to date most of our understanding of how metabolism supports cell proliferation is based on studies in cancer cells, such experiments querying the metabolic strategies employed by non-transformed cells are required to identify metabolic signatures that truly distinguish cancer cells from normal cells.
At the same time, cultured cancer cells continue to provide an outstanding model system to delineate metabolic networks in mammalian cells, and much remains to be discovered regarding how cells wire metabolic pathways in response to diverse stimuli. For example, CRISPR screens are helping to de-orphanize a raft of transporters and proteins involved in central metabolism, and screens performed in different cells or conditions are illuminating the various metabolic configurations that support cell function. Isotope tracing in cultured cells avoids many of the limitations inherent in in vivo experiments and allows for precise mechanistic dissection of the intracellular networks that support cell growth. Similarly, rapid organelle purification has facilitated discovery of metabolic alterations that would not be detectable when averaging signal across all cellular compartments162. The rapid turnover of many metabolites relative to the timescales of purification, however quick, limits the metabolites that can be reliably assessed with such approaches; nevertheless mass spectrometry methods employing specific isotopic standards to correct for post-harvest metabolism163, development of metabolite sensors and the expanding toolkit of heterologous enzymes that target specific enzymatic capacity to distinct subcellular compartments are collectively driving unprecedented insight into the metabolic networks that occur throughout living cells. Moving forward, the development and refinement of complex cell culture models of both normal cells and tumor cells—namely, organoids or tumoroids—will reveal how cell metabolism is affected in more complex ecosystems and demonstrate how metabolism shapes relationships between different cell types. A deeper understanding of how metabolic networks support cell function will provide opportunities to better tailor treatments to patients and, hopefully, improve therapeutic outcomes.
Acknowledgements.
I am grateful to Navdeep Chandel for discussion and Julia Brunner for help with figure preparation. I apologize that due to space limitations I could not cite many important studies in the field. L.W.S.F. is a New York Stem Cell Foundation - Robertson Investigator. This work was supported by the NYSCF, the Pershing Square Sohn Prize for Cancer Research and the NIH/NCI (R37 CA252305) as well as the Memorial Sloan Kettering Cancer Center Support Grant P30CA008748.
Footnotes
Conflict of interest. L.W.S.F. holds a patent and is an author on patent applications related to cellular metabolism and cell fate control.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Warburg O On the origin of cancer cells. Science 123, 309–314 (1956). [DOI] [PubMed] [Google Scholar]
- 2.DeBerardinis RJ & Chandel NS We need to talk about the Warburg effect. Nat Metab 2, 127–129 (2020). [DOI] [PubMed] [Google Scholar]
- 3.Luengo A et al. Increased demand for NAD(+) relative to ATP drives aerobic glycolysis. Molecular cell 81, 691–707 e696 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang Y et al. Saturation of the mitochondrial NADH shuttles drives aerobic glycolysis in proliferating cells. Molecular cell (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.DeBerardinis RJ & Thompson CB Cellular metabolism and disease: what do metabolic outliers teach us? Cell 148, 1132–1144 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cori CF MAMMALIAN CARBOHYDRATE METABOLISM. Physiological Reviews 11, 143–275 (1931). [Google Scholar]
- 7.Eagle H Nutrition needs of mammalian cells in tissue culture. Science 122, 501–514 (1955). [DOI] [PubMed] [Google Scholar]
- 8.DeBerardinis RJ & Chandel NS Fundamentals of cancer metabolism. Sci Adv 2, e1600200 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Flier JS, Mueckler MM, Usher P & Lodish HF Elevated levels of glucose transport and transporter messenger RNA are induced by ras or src oncogenes. Science 235, 1492–1495 (1987). [DOI] [PubMed] [Google Scholar]
- 10.Shim H et al. c-Myc transactivation of LDH-A: implications for tumor metabolism and growth. Proceedings of the National Academy of Sciences of the United States of America 94, 6658–6663 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bartman CR, TeSlaa T & Rabinowitz JD Quantitative flux analysis in mammals. Nat Metab 3, 896–908 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.DeBerardinis RJ, Lum JJ, Hatzivassiliou G & Thompson CB The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell metabolism 7, 11–20 (2008). [DOI] [PubMed] [Google Scholar]
- 13.Rathmell JC, Vander Heiden MG, Harris MH, Frauwirth KA & Thompson CB In the absence of extrinsic signals, nutrient utilization by lymphocytes is insufficient to maintain either cell size or viability. Molecular cell 6, 683–692 (2000). [DOI] [PubMed] [Google Scholar]
- 14.Edinger AL & Thompson CB Akt maintains cell size and survival by increasing mTOR-dependent nutrient uptake. Mol Biol Cell 13, 2276–2288 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Plas DR, Talapatra S, Edinger AL, Rathmell JC & Thompson CB Akt and Bcl-xL promote growth factor-independent survival through distinct effects on mitochondrial physiology. The Journal of biological chemistry 276, 12041–12048 (2001). [DOI] [PubMed] [Google Scholar]
- 16.Barata JT et al. Activation of PI3K is indispensable for interleukin 7-mediated viability, proliferation, glucose use, and growth of T cell acute lymphoblastic leukemia cells. The Journal of experimental medicine 200, 659–669 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Avissar NE, Sax HC & Toia L In human entrocytes, GLN transport and ASCT2 surface expression induced by short-term EGF are MAPK, PI3K, and Rho-dependent. Dig Dis Sci 53, 2113–2125 (2008). [DOI] [PubMed] [Google Scholar]
- 18.Gottlob K et al. Inhibition of early apoptotic events by Akt/PKB is dependent on the first committed step of glycolysis and mitochondrial hexokinase. Genes & development 15, 1406–1418 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Christofk HR, Vander Heiden MG, Wu N, Asara JM & Cantley LC Pyruvate kinase M2 is a phosphotyrosine-binding protein. Nature 452, 181–186 (2008). [DOI] [PubMed] [Google Scholar]
- 20.Sale EM, White MF & Kahn CR Phosphorylation of glycolytic and gluconeogenic enzymes by the insulin receptor kinase. J Cell Biochem 33, 15–26 (1987). [DOI] [PubMed] [Google Scholar]
- 21.Reiss N, Kanety H & Schlessinger J Five enzymes of the glycolytic pathway serve as substrates for purified epidermal-growth-factor-receptor kinase. The Biochemical journal 239, 691–697 (1986). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wise DR et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proceedings of the National Academy of Sciences of the United States of America 105, 18782–18787 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ebert BL, Firth JD & Ratcliffe PJ Hypoxia and mitochondrial inhibitors regulate expression of glucose transporter-1 via distinct Cis-acting sequences. The Journal of biological chemistry 270, 29083–29089 (1995). [DOI] [PubMed] [Google Scholar]
- 24.Vander Heiden MG, Cantley LC & Thompson CB Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324, 1029–1033 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Venneti S et al. Glutamine-based PET imaging facilitates enhanced metabolic evaluation of gliomas in vivo. Sci Transl Med 7, 274ra217 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Palm W, Araki J, King B, DeMatteo RG & Thompson CB Critical role for PI3-kinase in regulating the use of proteins as an amino acid source. Proceedings of the National Academy of Sciences of the United States of America 114, E8628–E8636 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Commisso C et al. Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature 497, 633–637 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Palm W et al. The Utilization of Extracellular Proteins as Nutrients Is Suppressed by mTORC1. Cell 162, 259–270 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Charpentier JC et al. Macropinocytosis drives T cell growth by sustaining the activation of mTORC1. Nature communications 11, 180 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Todorova PK et al. Amino acid intake strategies define pluripotent cell states. bioRxiv, 2022.2011.2016.516803 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Reinfeld BI et al. Cell-programmed nutrient partitioning in the tumour microenvironment. Nature 593, 282–288 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Muhar M et al. SLAM-seq defines direct gene-regulatory functions of the BRD4-MYC axis. Science 360, 800–805 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Harris IS & DeNicola GM The Complex Interplay between Antioxidants and ROS in Cancer. Trends Cell Biol 30, 440–451 (2020). [DOI] [PubMed] [Google Scholar]
- 34.Duvel K et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Molecular cell 39, 171–183 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Valvezan AJ & Manning BD Molecular logic of mTORC1 signalling as a metabolic rheostat. Nat Metab 1, 321–333 (2019). [DOI] [PubMed] [Google Scholar]
- 36.Possemato R et al. Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature 476, 346–350 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Locasale JW et al. Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nature genetics 43, 869–874 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sullivan MR et al. Increased Serine Synthesis Provides an Advantage for Tumors Arising in Tissues Where Serine Levels Are Limiting. Cell metabolism 29, 1410–1421 e1414 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sullivan LB et al. Supporting Aspartate Biosynthesis Is an Essential Function of Respiration in Proliferating Cells. Cell 162, 552–563 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Birsoy K et al. An Essential Role of the Mitochondrial Electron Transport Chain in Cell Proliferation Is to Enable Aspartate Synthesis. Cell 162, 540–551 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Martinez-Reyes I et al. Mitochondrial ubiquinol oxidation is necessary for tumour growth. Nature 585, 288–292 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Weinberg SE et al. Mitochondrial complex III is essential for suppressive function of regulatory T cells. Nature 565, 495–499 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Keibler MA et al. Differential substrate use in EGF- and oncogenic KRAS-stimulated human mammary epithelial cells. The FEBS journal 288, 5629–5649 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Williamson DH, Lund P & Krebs HA The redox state of free nicotinamide-adenine dinucleotide in the cytoplasm and mitochondria of rat liver. The Biochemical journal 103, 514–527 (1967). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Baksh SC et al. Extracellular serine controls epidermal stem cell fate and tumour initiation. Nat Cell Biol 22, 779–790 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Diehl FF, Lewis CA, Fiske BP & Vander Heiden MG Cellular redox state constrains serine synthesis and nucleotide production to impact cell proliferation. Nat Metab 1, 861–867 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pachnis P et al. In vivo isotope tracing reveals a requirement for the electron transport chain in glucose and glutamine metabolism by tumors. Sci Adv 8, eabn9550 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sullivan LB et al. Aspartate is an endogenous metabolic limitation for tumour growth. Nat Cell Biol 20, 782–788 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Garcia-Bermudez J et al. Aspartate is a limiting metabolite for cancer cell proliferation under hypoxia and in tumours. Nat Cell Biol 20, 775–781 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Westbrook RL et al. Proline synthesis through PYCR1 is required to support cancer cell proliferation and survival in oxygen-limiting conditions. Cell Rep 38, 110320 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Spinelli JB et al. Fumarate is a terminal electron acceptor in the mammalian electron transport chain. Science 374, 1227–1237 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chouchani ET et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 515, 431–435 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mentch SJ et al. Histone Methylation Dynamics and Gene Regulation Occur through the Sensing of One-Carbon Metabolism. Cell metabolism 22, 861–873 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wellen KE et al. ATP-citrate lyase links cellular metabolism to histone acetylation. Science 324, 1076–1080 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Carey BW, Finley LW, Cross JR, Allis CD & Thompson CB Intracellular alpha-ketoglutarate maintains the pluripotency of embryonic stem cells. Nature 518, 413–416 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Reid MA, Dai Z & Locasale JW The impact of cellular metabolism on chromatin dynamics and epigenetics. Nat Cell Biol 19, 1298–1306 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Baksh SC & Finley LWS Metabolic Coordination of Cell Fate by α-Ketoglutarate-Dependent Dioxygenases. Trends Cell Biol 31, 24–36 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chen WW, Freinkman E, Wang T, Birsoy K & Sabatini DM Absolute Quantification of Matrix Metabolites Reveals the Dynamics of Mitochondrial Metabolism. Cell 166, 1324–1337 e1311 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Trefely S et al. Quantitative subcellular acyl-CoA analysis reveals distinct nuclear metabolism and isoleucine-dependent histone propionylation. Molecular cell 82, 447–462 e446 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chakraborty AA et al. Histone demethylase KDM6A directly senses oxygen to control chromatin and cell fate. Science 363, 1217–1222 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lee JV et al. Acetyl-CoA promotes glioblastoma cell adhesion and migration through Ca(2+)-NFAT signaling. Genes & development 32, 497–511 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sciacovelli M, Schmidt C, Maher ER & Frezza C Metabolic Drivers in Hereditary Cancer Syndromes. Annual Review of Cancer Biology 4, 77–97 (2020). [Google Scholar]
- 63.Selak MA et al. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer cell 7, 77–85 (2005). [DOI] [PubMed] [Google Scholar]
- 64.Isaacs JS et al. HIF overexpression correlates with biallelic loss of fumarate hydratase in renal cancer: novel role of fumarate in regulation of HIF stability. Cancer cell 8, 143–153 (2005). [DOI] [PubMed] [Google Scholar]
- 65.Adam J et al. Renal cyst formation in Fh1-deficient mice is independent of the Hif/Phd pathway: roles for fumarate in KEAP1 succination and Nrf2 signaling. Cancer cell 20, 524–537 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sciacovelli M et al. Fumarate is an epigenetic modifier that elicits epithelial-to-mesenchymal transition. Nature 537, 544–547 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sulkowski PL et al. 2-Hydroxyglutarate produced by neomorphic IDH mutations suppresses homologous recombination and induces PARP inhibitor sensitivity. Sci Transl Med 9 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sulkowski PL et al. Krebs-cycle-deficient hereditary cancer syndromes are defined by defects in homologous-recombination DNA repair. Nature genetics 50, 1086–1092 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ooi A et al. An antioxidant response phenotype shared between hereditary and sporadic type 2 papillary renal cell carcinoma. Cancer cell 20, 511–523 (2011). [DOI] [PubMed] [Google Scholar]
- 70.Pirozzi CJ & Yan H The implications of IDH mutations for cancer development and therapy. Nat Rev Clin Oncol 18, 645–661 (2021). [DOI] [PubMed] [Google Scholar]
- 71.Figueroa ME et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer cell 18, 553–567 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lu C et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 483, 474–478 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Intlekofer AM et al. Acquired resistance to IDH inhibition through trans or cis dimer-interface mutations. Nature 559, 125–129 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.McBrayer SK et al. Transaminase Inhibition by 2-Hydroxyglutarate Impairs Glutamate Biosynthesis and Redox Homeostasis in Glioma. Cell 175, 101–116 e125 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kohanbash G et al. Isocitrate dehydrogenase mutations suppress STAT1 and CD8+ T cell accumulation in gliomas. The Journal of clinical investigation 127, 1425–1437 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Amankulor NM et al. Mutant IDH1 regulates the tumor-associated immune system in gliomas. Genes & development 31, 774–786 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bunse L et al. Suppression of antitumor T cell immunity by the oncometabolite (R)-2-hydroxyglutarate. Nature medicine 24, 1192–1203 (2018). [DOI] [PubMed] [Google Scholar]
- 78.Notarangelo G et al. Oncometabolite d-2HG alters T cell metabolism to impair CD8(+) T cell function. Science 377, 1519–1529 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lee JV et al. Akt-dependent metabolic reprogramming regulates tumor cell histone acetylation. Cell metabolism 20, 306–319 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Carrer A et al. Acetyl-CoA Metabolism Supports Multistep Pancreatic Tumorigenesis. Cancer discovery 9, 416–435 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Morris JP et al. α-Ketoglutarate links p53 to cell fate during tumour suppression. Nature 573, 595–599 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Intlekofer AM et al. Hypoxia Induces Production of L-2-Hydroxyglutarate. Cell metabolism 22, 304–311 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Oldham WM, Clish CB, Yang Y & Loscalzo J Hypoxia-Mediated Increases in L-2-hydroxyglutarate Coordinate the Metabolic Response to Reductive Stress. Cell metabolism 22, 291–303 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Intlekofer AM et al. L-2-Hydroxyglutarate production arises from noncanonical enzyme function at acidic pH. Nature chemical biology 13, 494–500 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Li Z et al. Hypoxia-inducible factors regulate tumorigenic capacity of glioma stem cells. Cancer cell 15, 501–513 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ito K & Suda T Metabolic requirements for the maintenance of self-renewing stem cells. Nature reviews. Molecular cell biology 15, 243–256 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Shim EH et al. L-2-Hydroxyglutarate: an epigenetic modifier and putative oncometabolite in renal cancer. Cancer discovery 4, 1290–1298 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Pan M et al. Regional glutamine deficiency in tumours promotes dedifferentiation through inhibition of histone demethylation. Nat Cell Biol 18, 1090–1101 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Cimmino L et al. Restoration of TET2 Function Blocks Aberrant Self-Renewal and Leukemia Progression. Cell 170, 1079–1095 e1020 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Agathocleous M et al. Ascorbate regulates haematopoietic stem cell function and leukaemogenesis. Nature 549, 476–481 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hensley CT et al. Metabolic Heterogeneity in Human Lung Tumors. Cell 164, 681–694 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Faubert B et al. Lactate Metabolism in Human Lung Tumors. Cell 171, 358–371 e359 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ghergurovich JM et al. Local production of lactate, ribose phosphate, and amino acids within human triple-negative breast cancer. Med (N Y) 2, 736–754 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hui S et al. Glucose feeds the TCA cycle via circulating lactate. Nature 551, 115–118 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Courtney KD et al. Isotope Tracing of Human Clear Cell Renal Cell Carcinomas Demonstrates Suppressed Glucose Oxidation In Vivo. Cell metabolism (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wang L et al. Spatially resolved isotope tracing reveals tissue metabolic activity. Nat Methods 19, 223–230 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Sullivan MR et al. Quantification of microenvironmental metabolites in murine cancers reveals determinants of tumor nutrient availability. Elife 8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Jang C et al. Metabolite Exchange between Mammalian Organs Quantified in Pigs. Cell metabolism 30, 594–606 e593 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kerk SA et al. Metabolic requirement for GOT2 in pancreatic cancer depends on environmental context. Elife 11 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Garcia-Bermudez J et al. Adaptive stimulation of macropinocytosis overcomes aspartate limitation in cancer cells under hypoxia. Nat Metab 4, 724–738 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Rossiter NJ et al. CRISPR screens in physiologic medium reveal conditionally essential genes in human cells. Cell metabolism 33, 1248–1263 e1249 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Cantor JR et al. Physiologic Medium Rewires Cellular Metabolism and Reveals Uric Acid as an Endogenous Inhibitor of UMP Synthase. Cell 169, 258–272 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Voorde JV et al. Improving the metabolic fidelity of cancer models with a physiological cell culture medium. Science Advances 5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Biancur DE et al. Functional Genomics Identifies Metabolic Vulnerabilities in Pancreatic Cancer. Cell metabolism 33, 199–210 e198 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zhu XG et al. Functional Genomics In Vivo Reveal Metabolic Dependencies of Pancreatic Cancer Cells. Cell metabolism 33, 211–221 e216 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Hu J et al. Heterogeneity of tumor-induced gene expression changes in the human metabolic network. Nat Biotechnol 31, 522–529 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Gaude E & Frezza C Tissue-specific and convergent metabolic transformation of cancer correlates with metastatic potential and patient survival. Nature communications 7, 13041 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Mayers JR et al. Tissue of origin dictates branched-chain amino acid metabolism in mutant Kras-driven cancers. Science 353, 1161–1165 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Sivanand S et al. Cancer tissue of origin constrains the growth and metabolism of metastases. bioRxiv, 2022.2008.2017.504141 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Yuneva MO et al. The metabolic profile of tumors depends on both the responsible genetic lesion and tissue type. Cell metabolism 15, 157–170 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Chen PH et al. Metabolic Diversity in Human Non-Small Cell Lung Cancer Cells. Molecular cell 76, 838–851 e835 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Intlekofer AM & Finley LWS Metabolic signatures of cancer cells and stem cells. Nature Metabolism 1, 177–188 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Mahendralingam MJ et al. Mammary epithelial cells have lineage-rooted metabolic identities. Nature Metabolism 3, 665–681 (2021). [DOI] [PubMed] [Google Scholar]
- 114.Bartman CR et al. Slow TCA flux implies low ATP production in tumors. bioRxiv, 2021.2010.2004.463108 (2021). [Google Scholar]
- 115.Coloff JL et al. Differential Glutamate Metabolism in Proliferating and Quiescent Mammary Epithelial Cells. Cell metabolism 23, 867–880 (2016). [DOI] [PubMed] [Google Scholar]
- 116.Brunner JS & Finley LWS Metabolic determinants of tumour initiation. Nature Reviews Endocrinology (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Piskounova E et al. Oxidative stress inhibits distant metastasis by human melanoma cells. Nature 527, 186–191 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Tasdogan A et al. Metabolic heterogeneity confers differences in melanoma metastatic potential. Nature 577, 115–120 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Cheung EC et al. Dynamic ROS Control by TIGAR Regulates the Initiation and Progression of Pancreatic Cancer. Cancer cell 37, 168–182 e164 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Basnet H et al. Flura-seq identifies organ-specific metabolic adaptations during early metastatic colonization. Elife 8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Fischer GM et al. Molecular Profiling Reveals Unique Immune and Metabolic Features of Melanoma Brain Metastases. Cancer discovery 9, 628–645 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Elia I et al. Breast cancer cells rely on environmental pyruvate to shape the metastatic niche. Nature 568, 117–121 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Faubert B, Solmonson A & DeBerardinis RJ Metabolic reprogramming and cancer progression. Science 368 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Campbell S et al. Glutamine deprivation triggers NAGK-dependent hexosamine salvage. Elife 10 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Kim PK et al. Hyaluronic acid fuels pancreatic cancer cell growth. Elife 10 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Ward MH et al. Nutrient profiling reveals extracellular uridine as a fuel for pancreatic cancer through uridine phosphorylase 1. bioRxiv, 2021.2006.2007.447448 (2021). [Google Scholar]
- 127.Jourdain AA et al. Salvage of Ribose from Uridine or RNA Supports Glycolysis when Glucose is Limiting. bioRxiv, 2021.2006.2010.447789 (2021). [Google Scholar]
- 128.Kim J et al. CPS1 maintains pyrimidine pools and DNA synthesis in KRAS/LKB1-mutant lung cancer cells. Nature 546, 168–172 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Spinelli JB et al. Metabolic recycling of ammonia via glutamate dehydrogenase supports breast cancer biomass. Science 358, 941–946 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Park JO et al. Near-equilibrium glycolysis supports metabolic homeostasis and energy yield. Nature chemical biology 15, 1001–1008 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Park JO et al. Metabolite concentrations, fluxes and free energies imply efficient enzyme usage. Nature chemical biology 12, 482–489 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Mullen AR et al. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature 481, 385–388 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Metallo CM et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 481, 380–384 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Arnold PK et al. A non-canonical tricarboxylic acid cycle underlies cellular identity. Nature 603, 477–481 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Assmann N et al. Srebp-controlled glucose metabolism is essential for NK cell functional responses. Nat Immunol 18, 1197–1206 (2017). [DOI] [PubMed] [Google Scholar]
- 136.Hao Z et al. Specific ablation of the apoptotic functions of cytochrome C reveals a differential requirement for cytochrome C and Apaf-1 in apoptosis. Cell 121, 579–591 (2005). [DOI] [PubMed] [Google Scholar]
- 137.Haile DJ et al. Reciprocal control of RNA-binding and aconitase activity in the regulation of the iron-responsive element binding protein: role of the iron-sulfur cluster. Proceedings of the National Academy of Sciences of the United States of America 89, 7536–7540 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Li B et al. Fructose-1,6-bisphosphatase opposes renal carcinoma progression. Nature 513, 251–255 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Rossi M et al. PHGDH heterogeneity potentiates cancer cell dissemination and metastasis. Nature 605, 747–753 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Pan C, Li B & Simon MC Moonlighting functions of metabolic enzymes and metabolites in cancer. Molecular cell 81, 3760–3774 (2021). [DOI] [PubMed] [Google Scholar]
- 141.Taylor SR, Falcone JN, Cantley LC & Goncalves MD Developing dietary interventions as therapy for cancer. Nature reviews. Cancer 22, 452–466 (2022). [DOI] [PubMed] [Google Scholar]
- 142.Fane M & Weeraratna AT How the ageing microenvironment influences tumour progression. Nature Reviews Cancer 20, 89–106 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Ringel AE et al. Obesity Shapes Metabolism in the Tumor Microenvironment to Suppress Anti-Tumor Immunity. Cell 183, 1848–1866 e1826 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Beyaz S et al. High-fat diet enhances stemness and tumorigenicity of intestinal progenitors. Nature 531, 53–58 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Mana MD et al. High-fat diet-activated fatty acid oxidation mediates intestinal stemness and tumorigenicity. Cell Rep 35, 109212 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Goncalves MD et al. High-fructose corn syrup enhances intestinal tumor growth in mice. Science 363, 1345–1349 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Taylor SR et al. Dietary fructose improves intestinal cell survival and nutrient absorption. Nature 597, 263–267 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Zhao S et al. Dietary fructose feeds hepatic lipogenesis via microbiota-derived acetate. Nature 579, 586–591 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Comerford SA et al. Acetate dependence of tumors. Cell 159, 1591–1602 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Biswas AK & Acharyya S Cancer-Associated Cachexia: A Systemic Consequence of Cancer Progression. Annual Review of Cancer Biology 4, 391–411 (2020). [Google Scholar]
- 151.Oren Y et al. Cycling cancer persister cells arise from lineages with distinct programs. Nature 596, 576–582 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Yap TA et al. Complex I inhibitor of oxidative phosphorylation in advanced solid tumors and acute myeloid leukemia: phase I trials. Nature medicine (2023). [DOI] [PubMed] [Google Scholar]
- 153.Luengo A, Gui DY & Vander Heiden MG Targeting Metabolism for Cancer Therapy. Cell Chem Biol 24, 1161–1180 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Stine ZE, Schug ZT, Salvino JM & Dang CV Targeting cancer metabolism in the era of precision oncology. Nature reviews. Drug discovery 21, 141–162 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Wilde BR et al. FH variant pathogenicity promotes purine salvage pathway dependence in kidney cancer. bioRxiv, 2022.2008.2015.504023 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Clavell LA et al. Four-agent induction and intensive asparaginase therapy for treatment of childhood acute lymphoblastic leukemia. The New England journal of medicine 315, 657–663 (1986). [DOI] [PubMed] [Google Scholar]
- 157.Gao X et al. Dietary methionine influences therapy in mouse cancer models and alters human metabolism. Nature 572, 397–401 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Hopkins BD et al. Suppression of insulin feedback enhances the efficacy of PI3K inhibitors. Nature 560, 499–503 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Bian Y et al. Cancer SLC43A2 alters T cell methionine metabolism and histone methylation. Nature 585, 277–282 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Keys HR & Knouse KA Genome-scale CRISPR screening in a single mouse liver. Cell Genom 2 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Lau AN et al. Dissecting cell-type-specific metabolism in pancreatic ductal adenocarcinoma. Elife 9 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Wang Y et al. SLC25A39 is necessary for mitochondrial glutathione import in mammalian cells. Nature 599, 136–140 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Trefely S et al. Subcellular metabolic pathway kinetics are revealed by correcting for artifactual post harvest metabolism. Mol Metab 30, 61–71 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]