

Supplemental Digital Content is Available in the Text.
Experimental disruption of sleep induces heightened pain sensitivity, which is mediated by a loss of slow-wave, N3 sleep, and increases in cellular inflammation.
Keywords: Sleep loss; Sleep deprivation; Slow wave sleep; Inflammation; Monocyte, Interleukin-6; Tumor necrosis factor; Pain; Pain sensitivity; Hyperalgesia; Mediation
Abstract
Sleep loss heightens pain sensitivity, but the pathways underlying this association are not known. Given that experimental sleep disruption induces increases in cellular inflammation as well as selective loss of slow wave, N3 sleep, this study examined whether these mechanisms contribute to pain sensitivity following sleep loss in healthy adults. This assessor-blinded, cross-over sleep condition, single-site, randomized clinical trial enrolled 95 healthy adults (mean [SD] age, 27.8 [6.4]; female, 44 [53.7%]). The 2 sleep conditions were 2 nights of undisturbed sleep (US) and 2 nights of sleep disruption or forced awakening (FA, 8 pseudorandomly distributed awakenings and 200 minutes wake time during the 8-hour sleep opportunity), administered in a cross-over design after 2 weeks of washout and in a random order (FA-US; US-FA). Primary outcome was heat pain threshold (hPTH). Sleep architecture was assessed by polysomnography, and morning levels of cellular inflammation were evaluated by Toll-like receptor-4 stimulated monocyte intracellular proinflammatory cytokine production. As compared with US, FA was associated with decreases in the amount of slow wave or N3 sleep (P < 0.001), increases in Toll-like receptor-4 stimulated production of interleukin-6 and tumor necrosis factor-α (P = 0.03), and decreases in hPTH (P = 0.02). A comprehensive causal mediation analysis found that FA had an indirect effect on hPTH by decreases in N3 sleep and subsequent increases in inflammation (estimate=−0.15; 95% confidence interval, −0.30 to −0.03; P < 0.05) with the proportion mediated 34.9%. Differential loss of slow wave, N3 sleep, and increases in cellular inflammation are important drivers of pain sensitivity after sleep disruption.
Clinical Trials Registration: NCT01794689.
1. Introduction
Frequent difficulties with sleep occur in about one third of the population of the United States,45,49 with higher prevalence among patients who have comorbid pain symptoms.13,65 Observational data implicate sleep disturbance in the emergence, progression, and persistence of pain symptoms.1,13,42,46,68 One experimental model of sleep disturbance—sleep disruption by forced nocturnal awakenings—heightens pain sensitivity in humans.35,56,58,60 Understanding the mechanisms that underlie the association between sleep disruption and pain sensitivity is needed to develop targeted interventions to mitigate the risk of chronic pain.
Sleep disturbance is associated with elevated levels of inflammation,8,29 and experimental studies have found that even a modest amount of sleep loss activates inflammatory transcriptional signaling,26,30,31 which leads to aberrant increases in the production of proinflammatory cytokines by monocytes,27,30 the major contributor of inflammatory cytokines in peripheral blood. In turn, increases in inflammation are believed to heighten pain sensitivity; basic research has demonstrated that expression of proinflammatory cytokines amplify the pain message.52,63,69 However, it is not known whether inflammatory activation mediates the pathway between sleep disruption and heightened pain sensitivity in humans.
Experimental sleep disruption also induces alterations in sleep architecture including loss of slow wave or N3 sleep and loss of rapid eye movement (REM) sleep,60 which have separately been found to be associated with heightened pain sensitivity.38,50,54 Furthermore, loss of N3 sleep, but not REM sleep, is associated with increases in markers of inflammation.6,26 No study has used a multilevel approach to examine the separate and joint contributions of N3 sleep and inflammation on pain sensitivity.
Together, these findings highlight 3 mechanisms which are hypothesized to underlie the association between sleep disruption and pain sensitivity: differential change in amount of N3 sleep (ie, sleep mediation hypothesis), differential increase in inflammation (ie, inflammation mediation hypothesis), or a differential effect which is conditional on joint changes in N3 sleep and inflammation (ie, double mediation hypothesis). To disentangle these mechanisms, causal mediation analyses36,37 can be used to evaluate the extent to which the association between exposure to sleep disruption and heightened pain sensitivity can be decomposed into a direct effect of sleep disruption (ie, independent of sleep architecture, inflammation, and covariates), indirect effects through mediating mechanisms (ie, decreases in N3 sleep and increases in inflammation), and joint effects (ie, increases in inflammation related to decreases in N3 sleep). Pathways between sleep disruption, N3 sleep, inflammation, and pain sensitivity typically have been evaluated independently, yet they are not mutually exclusive and have different treatment implications; it is, therefore, important to disentangle them.
To address these aims, we used a comprehensive causal mediation model (Fig. 1) to clarify whether changes in the amount of N3 sleep and cellular inflammation mediate thermal pain sensitivity (ie, heat pain threshold, hPTH) in response to experimental sleep disruption. Sleep disruption was accomplished with the forced awakening (FA) procedure because it models sleep disturbance observed in patients with inflammation and pain. Moreover, FA has been found to produce a robust and reliable decrease in hPTH.60
Figure 1.

Hypothetical mediation model linking sleep disruption to pain sensitivity. The hypothetical mediation model shows the proposed pathway from sleep disruption or forced awakening to loss of N3 sleep, to activation and increase of cellular inflammation, and to heightened pain sensitivity.
2. Methods
2.1. Trial design and oversight
This study is a planned secondary analysis of a randomized controlled trial as previously reported.61 In brief, the primary study was a mixed-methods randomized controlled trial (NCT01794689) with a cross-over component for sleep condition (within-subject factor; uninterrupted sleep, US vs FA) to determine whether sleep condition altered morphine analgesia. The order administration of the sleep condition was randomized (ie, US-FA vs FA-US).
In this secondary analysis, we modeled whether cellular inflammation, as indexed by morning increases in the production of proinflammatory cytokines by monocytes after ligation of the Toll-like receptor (TLR)-4 with lipopolysaccharide, mediate hPTH in response to sleep loss. Finally, we tested whether decreases in N3 sleep lead to increases in cellular inflammation, which are then associated with increases in pain sensitivity (ie, decrease in hPTH).
The primary outcome, hPTH, was selected for the following reasons. In a prior report in a smaller cohort (n = 85), we found that FA leads to decreases in hPTH. Morning levels of cellular inflammation, obtained immediately after the sleep condition (8 am), were proximal to assessment of hPTH (10 am) (see protocol timeline, Fig. 1S, available at http://links.lww.com/PAIN/B740). Two other pain measures, pressure pain threshold (pPTH) and mechanical temporal summation (mTS), were obtained along with hPTH. FA did not alter pPTH, and this outcome was not included in the absence of a main effect of sleep disruption. FA had a sex-dependent effect on mTS,60 and an exploratory analysis was included for this outcome, although sex-dependent effects were not tested because the causal mediation model is not adequately powered to test effects moderated by sex. Our previous report also examined pain measures including cold pain tolerance, skin flare, and secondary hyperalgesia. However, each of these measures was obtained at more than 3 hours after assessment of morning levels of cellular inflammation (1130 am) (Fig. 1S, available at http://links.lww.com/PAIN/B740). During this morning interval, sleep loss induced activation of cellular inflammation returns to levels comparable with levels after uninterrupted sleep.27,30 Furthermore, during this interval, there was a heat capsaicin sensitization period; this intervening experimental procedure might confound the relationship between morning levels of cellular inflammation and distal measures of cold pain tolerance, skin flare, and secondary hyperalgesia.
The protocol was approved by the Johns Hopkins University School of Medicine Institutional Review Board and the University of California Los Angeles School of Medicine Institutional Review Board in compliance with the Declaration of Helsinki and registered with clinicaltrials.gov (NCT01794689). All participants completed informed consent before participation. All data were deidentified. Trial data monitoring and steering committees of the Johns Hopkins University Clinical Translational Sciences Institute oversaw the study, which was undertaken according to the intention-to-treat principle. This study adhered to the Consolidated Standards of Reporting Trials (CONSORT) reporting guidelines.66
2.2. Participants
All participants were recruited from the community using social media platforms, print advertisements, and community fliers as previously described.60,61 Enrolled participants met the following eligibility criteria: adults aged 18 to 48 years, Research Diagnostic Criteria for Normal Sleepers,12 stable sleep phase between 21:00 and 10:00 hours, no evidence of sleep impairment (ie, Pittsburgh Sleep Quality Index, PSQI < 5), and self-reported total sleep time (TST) between 6.5 and 8.5 hours per night with a sleep efficiency > 85%. We excluded participants with a body mass index > 35 kg/m2, history of chronic pain, acute pain, significant medical or psychiatric morbidity within the past 6 months, lifetime history of substance abuse or dependence, or positive toxicology screen. After polysomnography (PSG), we also excluded those with apnea–hypopnea index >10/h or periodic limb movements with arousal > 15/h. Full inclusion and exclusion criteria are previously reported.60,61
2.3. Trial procedures
After completing eligibility evaluation including PSG screening with full respiratory and electromyography procedures to diagnose any sleep disordered breathing and periodic limb movement disorder as previously described,60,61 participants were randomized to 2 groups, defined by order of administration of US or FA (ie, US-FA vs FA-US). Participants underwent 2 consecutive nights of US (or FA), followed by a 2-week washout interval in their home environment, and then completed 2 consecutive nights of the opposing sleep condition FA (or US). The morning after the second night of each sleep condition, US-FA or FA-US, participants underwent blood sampling for assessment of cellular inflammation as indexed by TLR-4 stimulated monocytic production of proinflammatory cytokines. We have previously found that experimental sleep loss induces an activation of this cellular marker of inflammation,27,30 whereas there are only mixed effects of sleep loss on circulating levels of interleukin (IL)-6 possibly due to the delayed effects of acute sleep deprivation on the production and then release of proinflammatory cytokines in to the circulation4,26,29; monocyte expression of intracellular proinflammatory cytokines is upstream of systemic levels of proinflammatory cytokines. Although other proinflammatory (ie, IL-1β) as well as anti-inflammatory cytokines (ie, IL-10) have been found to alter pain sensitivity,14 evidence linking sleep disturbance to these measures is limited or null.26,29
After blood sampling, participants then completed QST to determine hPTH as well as pPTH and mTS. After exposure to heat and capsaicin sensitization, additional QST was completed including cold pain tolerance, skin flare, and secondary hyperalgesia. The timeline of the study protocol is shown in the Supplement (Fig. 1S, available at http://links.lww.com/PAIN/B740). The full study protocol has been previously reported.60
Sleep condition was either FA or US. The FA condition was administered on each of 2 consecutive nights and involved an 8 hours sleep opportunity period, which was divided into 8, 1-hour interval after lights out. One of the intervals was randomly determined to be a 60-minute awakening, during which no sleep was permitted, and each of the remaining 7 60-minute intervals were subdivided into tertiles (three 20-minute blocks). A forced 20-minute awakening was randomly scheduled to occur in either the first, second, or third tertile of each hour. During FA, nursing staff kept participants awake by having them sit up in bed to reduce the chance of microsleep. During FA, maximum TST was 280 minutes. During US, participants were given an 8-hour period of sleep opportunity or undisturbed sleep with a maximum TST of 480 minutes.
2.4. Polysomnography assessment
During each of the 2 nights of US and the 2 nights of FA, sleep was monitored by PSG (Embla N7000, Natus Medical Incorporated) in accordance with standardized acquisition and sleep scoring procedures,39 using Embla REMLogic Software. In brief, certified PSG technicians used standard procedures to obtain the recording montage, using normal placement of electrocardiography, electrooculography, and electromyography following the American Academy of Sleep Medicine guidelines. All records were scored according to the American Academy of Sleep Medicine guidelines, by a registered PSG technician and reviewed by a board-certified sleep medicine physician. Sleep continuity and minutes and percentage of sleep architecture measures were summed over the 2 nights within each US and FA.
2.5. Assessment of cellular inflammation or Toll-like receptor-4 stimulated monocytic cytokine production
Approximately 30 minutes after participants awoke after the second night of the sleep condition (US vs FA; ie, 730 am), a nurse inserted a single port lumen peripheral catheter in an upper extremity vein and obtained a blood sample after 30 minutes of rest (ie, 8 am). Within 2 hours of blood sampling, heparin-treated blood (1 mL) was mixed with 100 pg/mL lipopolysaccharide (Sigma, St. Louis, MO) and 10 μg/mL brefeldin A (Sigma, St. Louis, MO) and incubated for 4 hours at 37°C and fixed. A parallel sample was incubated without addition of lipopolysaccharide to quantify unstimulated levels of cytokine production, representative of monocytic activation in vivo.
Monocytic production of IL-6 and tumor necrosis factor (TNF)-α was then assessed by flow cytometry using peridinin chlorophyll protein-labeled CD14 monoclonal antibody, allophycocyanin-labeled anti-TNF-α monoclonal antibody, and phycoerythrin-labeled IL-6 antibody, as previously described.30,32 Red blood cells were lysed in fluorescence-activated cell sorting lysing solution (BD Biosciences, San Jose, CA); remaining cells were permeabilized in fluorescence-activated cell sorting permeabilizing buffer (BD Biosciences), and fluorescence-conjugated antibodies were added for 30 minutes at room temperature in the dark. Cells were then washed and resuspended in 1% paraformaldehyde for assay on a Coulter Elite flow cytometer using the Coulter Elite software. Cells were gated on total viable leukocytes, based on a SSC vs FSC plot, and then a SSC vs CD14+ plot was used to define the monocyte population. About 5000 CD14+ events were counted by 3-color flow cytometry (FACScan, BD Biosciences). Using quadrant statistics on Cell Quest Pro Software (BD Biosciences),32 the percentage of cytokine expression was determined for unstimulated and stimulated monocyte cells as defined by 3 unique cellular subsets, including monocytes expressing IL-6, monocytes cells expressing TNF-α, and monocytes simultaneously coexpressing IL-6 and TNF-α. In addition, a composite of cellular inflammation was generated, which included each of the 3 cellular subsets or monocytes expressing IL-6 or TNF-α. For the mediation analyses, the composite of the 3 subsets was used.
2.6. Quantitative sensory testing
Quantitative sensory testing of hPTH occurred on the morning after the second night of sleep manipulation (ie, US vs FA) and about 2 hours after blood sampling. As previously described,60 hPTH was assessed by a computer-driven, Peltier element–based stimulator (Medoc TSA II, neurosensory Analyzer, Ramat Yishai, Israel) with a 9 cm2 advanced thermal stimulation thermode, with assessment on the ventral forearm using an ascending method of limits paradigm (ie, from a 32°C baseline, the temperature was steadily increased at 0.5°C/second until the participant reported the first sensation of pain). Two trials were conducted separated by greater than 3 minutes; the thermode affixed by a Velcro strap was repositioned to an adjacent site after each trial to avoid sensitization. Two temperatures from each trial were averaged to index hPTH.
2.7. Statistical analysis
Intervention effects on outcomes of polysomnographic sleep, cellular inflammation, and pain outcomes were tested on an intention-to-treat basis using a mixed model approach. Data from all randomly assigned participants were included. The distribution of residuals for each model was inspected for normality, and the distribution of dependent variables was found to show normality. For each of the models, the fixed effect was group (order of sleep condition: FA-US vs US-FA) and the repeated effect was sleep condition (FA and US). A 2-way interaction was also examined and not found to be present for any of the outcomes. All models were controlled for the following covariates: sex, age, BMI, race (dichotomized Black vs others), and ethnicity (Hispanic/Latino vs non-Hispanic/Latino), given evidence that these covariates have previously been shown to be related to pain outcome after exposure to FA.60,61 We used type III fixed effects (F and t) and set the statistical significance at P < 0.05.
To test mediation, the methods of Hayes et al. and Hayes PROCESS macro (version 4.1) was used.16,18,19,53 The hypothesized double mediation model was tested (ie, Model 6),17 in which the effect of FA on hPTH was mediated by the loss of N3 sleep and increase in cellular inflammation as temporally ordered and shown in the hypothesized pathway (Fig. 1). Mediational models also adjusted for the above set of covariates. Exploratory analyses examined whether inclusion of N2 and REM sleep in the model altered the pathway linking N3 sleep, cellular inflammation, and hPTH.
3. Results
3.1. Participants
From June 2013 to January 2018, 1802 persons were screened, 255 were enrolled and underwent medical eligibility assessment, 205 advanced to baseline QST eligibility assessment, and 102 advanced to a night of polysomnography screening. After these assessments, 1 additional participant was excluded and another declined. Hence, 51 were randomly assigned to FA-US and 49 to US-FA. Before allocation to the first experimental session, 3 participants discontinued and 2 were excluded due to ECG abnormalities. Hence, 95 participants were randomized and allocated, with 46 to FA-US and 49 to US-FA (Fig. 2).
Figure 2.

Participant flowchart. Details regarding screening, eligibility assessment, experimental manipulation of sleep including forced awakening (FA) vs uninterrupted sleep (US), and completion of testing including polysomnography (PSG), quantitative sensory testing (QST), and cellular inflammation are given in the Methods.
During the protocol including 2 sessions of QST and PSG, 1 participant in FA-US was excluded and 4 discontinued and 3 were excluded in US-FA; hence, QST and PSG were completed with 45 in FA-US and 42 in US-FA. Blood sampling for the assessment of inflammation was completed with 40 in FA-US and 42 in US-FA. Analyses and causal mediational models included 40 FA-US and 42 US-FA. Order of sleep condition groups (ie, FA-US vs US-FA) showed balanced baseline characteristics (Table 1). There were no adverse events.
Table 1.
Demographic and clinical characteristics at baseline.*
| Characteristic | Overall (N = 82) | FA-US (N = 40) | US-FA (N = 42) | Difference |
|---|---|---|---|---|
| Age, y | 27.8 (6.4) | 28.0 (7.0) | 27.6 (5.8) | t(80)= 0.29, 0.77 |
| Female sex, no. (%) | 44 (53.7) | 24 (60.0) | 20 (47.6) | X2(1, 82)= 1.26, 0.26 |
| Race group, no. (%)† | X2(4, 81)= 3.96, 0.41 | |||
| Asian | 11 (13.6) | 4 (10.3) | 7 (16.7) | |
| Black | 28 (34.6) | 14 (35.9) | 14 (33.3) | |
| White | 37 (45.7) | 20 (51.3) | 17 (40.5) | |
| Multiracial | 3 (3.7) | 0 (0.0) | 3 (7.1) | |
| Others | 2 (2.5) | 1 (2.6) | 1 (2.4) | |
| Ethnicity, no (%)† | X2(1, 82)= 1.01, 0.38 | |||
| Non-Hispanic | 69 (84.1) | 32 (80.0) | 37 (88.1) | |
| Hispanic | 13 (15.9) | 8 (20.0) | 5 (11.9) | |
| Marital status, no. (%) | X2(4, 82)= 1.72, 0.79 | |||
| Single | 68 (82.9) | 32 (80.0) | 36 (85.7) | |
| Married | 5 (6.1) | 2 (5.0) | 3 (7.1) | |
| Divorced | 2 (2.4) | 1 (2.5) | 1 (2.4) | |
| Separated | 3 (3.7) | 2 (5.0) | 1 (2.4) | |
| Living w/partner | 4 (4.9) | 3 (7.4) | 1 (2.4) | |
| Education, no. (%) | X2(6, 81)= 3.33, 0.77 | |||
| Doctoral degree | 6 (7.4) | 2 (5.0) | 4 (9.8) | |
| Master's degree | 10 (12.3) | 6 (15.0) | 4 (9.8) | |
| College graduate | 39 (48.1) | 19 (47.5) | 20 (48.8) | |
| Some college | 19 (23.5) | 9 (22.5) | 10 (24.4) | |
| Some college (current student) | 1 (1.2) | 1 (2.5) | 0 (0.0) | |
| Tech. School grad | 1 (1.2) | 1 (2.5) | 0 (0.0) | |
| High school graduate | 5 (6.2) | 2 (5.0) | 3 (7.3) | |
| Employment, no.(%) | X2(6, 81)= 3.70, 0.72 | |||
| Student (full time) | 6 (7.4) | 3 (7.5) | 3 (7.3) | |
| Student (full or part time) | 25 (30.9) | 11 (27.5) | 14 (34.1) | |
| Student (part time) | 1 (1.2) | 0 (0.0) | 1 (2.4) | |
| Unemployed | 10 (12.3) | 6 (15.0) | 4 (9.8) | |
| Working for pay (full time) | 19 (23.5) | 11 (27.5) | 8 (19.5) | |
| Working for pay (part time) | 19 (23.5) | 8 (20.0) | 11 (26.8) | |
| Homemaker | 1 (1.2) | 1 (2.5) | 0 (0.0) | |
| Body mass index‡ | 25.7 (3.8) | 25.5 (3.9) | 25.9 (3.6) | t(80)= −0.52, 0.61 |
| Pittsburgh Sleep Quality Index§ | 1.9 (1.35) | 2.0 (1.4) | 1.7 (1.3) | t(80)= 0.96, 0.35 |
Values are means +/−(SD), unless otherwise specified with 95% confidence intervals.
Race and ethnicity were reported by the participants.
The body mass index is the weight in kilograms divided by the square of the height in meters.
Pittsburgh Sleep Quality Index measures severity of sleep disturbance; a score of <5 indicates no clinically significant sleep disturbance.
3.2. Sleep continuity and sleep architecture
Measures of sleep continuity and sleep architecture obtained by PSG for the 2 groups (FA-US, US-FA) and for the 2 sleep conditions (FA vs US) are shown in Table 2. There were no group effects indicating that the order of exposure to FA did not alter measures of sleep continuity or sleep architecture.
Table 2.
Effects of experimental sleep conditions, forced awakening vs uninterrupted sleep, on measures of sleep continuity, and sleep architecture by 2 groups.*
| FA-US (N = 46) | US-FA (N = 49) | Group df = (1, 69) | Condition df = (1, 65) | |||
|---|---|---|---|---|---|---|
| FA | US | US | FA | |||
| Sleep continuity | ||||||
| Sleep onset latency | 25.9 ± 23.8 | 17.1 ± 16.9 | 14.7 ± 12.8 | 22.3 ± 16.2 | F = 1.58, P = 0.22 | F = 7.43, P = 0.008 |
| Total sleep time | 248.2 ± 24.9 | 433.5 ± 36.7 | 441.7 ± 34.3 | 248.3 ± 29.9 | F = 1.97, P = 0.17 | F = 1684, P < 0.001 |
| Sleep efficiency, % | 52.2 ± 5.2 | 91.4 ± 6.4 | 92.3 ± 7.7 | 52.3 ± 6.4 | F = 1.47, P = 0.23 | F = 1945, P < 0.001 |
| Wake after sleep onset | 205.5 ± 33.8 | 24.7 ± 19.2 | 23.6 ± 31.9 | 208.3 ± 28.1 | F = 0.02, P = 0.90 | F = 1914, P < 0.001 |
| Sleep architecture | ||||||
| N1 | 20.5 ± 8.8 | 17.4 ± 10.0 | 18.3 ± 14.2 | 20.8 ± 9.9 | F = 0.02, P = 0.90 | F = 5.09, P = 0.027 |
| N2 | 116.2 ± 17.1 | 216.1 ± 34.7 | 214.1 ± 43.3 | 119.0 ± 22.8 | F = 0.01, P = 0.93 | F = 497.8, P<0.001 |
| N3 | 67.2 ± 21.7 | 106.7 ± 35.7 | 108.0 ± 41.0 | 65.1 ± 21.0 | F = 0.39, P = 0.54 | F = 139.6, P < 0.001 |
| REM | 44.3 ± 16.3 | 93.3 ± 24.7 | 101.3 ± 24.1 | 43.3 ± 17.4 | F = 1.73, P = 0.20 | F = 451.8, P < 0.001 |
| N1% | 8.4 ± 3.3 | 4.3 ± 2.6 | 4.1 ± 3.5 | 8.3 ± 4.2 | F = 0.04, P = 0.84 | F = 97.8, P < 0.001 |
| N2% | 47.4 ± 7.5 | 50.5 ± 7.7 | 48.6 ± 9.9 | 48.2 ± 8.4 | F = 0.08, P = 0.78 | F = 3.16, P = 0.08 |
| N3% | 26.4 ± 7.3 | 23.8 ± 7.1 | 23.6.4 ± 8.5 | 26.7 ± 7.7 | F = 0.00, P = 0.97 | F = 10.08, P = 0.002 |
| REM% | 17.8 ± 6.0 | 21.4 ± 5.0 | 23.6 ± 4.9 | 16.8 ± 6.2 | F = 0.25, P = 0.63 | F = 60.3, P < 0.001 |
Sleep measures are shown in mean minutes or percentage (+/− SD).
In models adjusting for age, race, ethnicity, and body mass index, FA was associated with significant reduction of TST and sleep efficiency, and an increase in wake after sleep onset, as compared with US. (Table 2) Moreover, FA altered measures of sleep architecture. In adjusted models, FA was associated with an increase in the amounts of N1 sleep and a decrease in the amounts of N2, N3, and REM sleep, as compared with US.(Table 2) By contrast, FA was associated with increases in the percentage of N1 and N3 sleep and decreases in the percentage of REM sleep(Table 2).
3.3. Unstimulated and stimulated monocyte expression of inflammatory cytokines
Measures of cellular inflammation as indexed by unstimulated and stimulated monocyte expression of IL-6, TNF-α, coexpression of IL-6 and TNF-α, and the composite of these cellular subsets were evaluated for the 2 order groups and for the 2 sleep conditions. Unstimulated monocyte expression of IL-6 and TNF-α represents the level of in vivo activation of monocytes, whereas stimulated monocyte expression of IL-6 and TNF-α indicates the ability of monocytes to respond to an inflammatory challenge; both measures have been found to be altered by experimental sleep loss.30,32
There were no group effects on unstimulated and stimulated monocyte expression of proinflammatory cytokines for each of the cellular subsets or the composite (all P's > 0.05; Fig. 2S, available at http://links.lww.com/PAIN/B740).
In adjusted models, FA was associated with an increase in the stimulated monocyte expression of IL-6 (F = 2.8, df = 1, 69.9, P = 0.09), an increase in stimulated expression of TNF-α (F = 5.2 df = 1, 74.9, P = 0.03), an increase in stimulated coexpression of IL-6 and TNF-α (F = 3.9 df = 1, 74.5, P = 0.05), and an increase in the composite of these 3 cellular subsets. (F = 4.9 df = 1, 71.4, P = 0.03; Fig. 3). There were no condition effects on unstimulated monocyte expression of proinflammatory cytokines for each of the cellular subsets or the composite (all P's > 0.05; Fig. 3S, available at http://links.lww.com/PAIN/B740).
Figure 3.
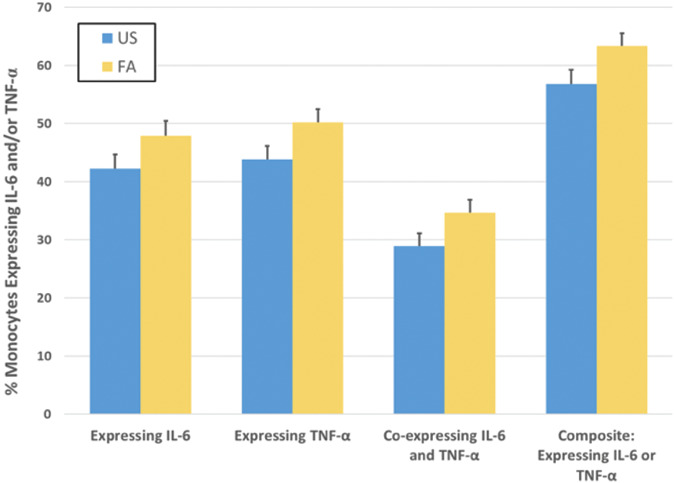
Cellular inflammation as indexed by stimulated monocyte expression of proinflammatory cytokines in response to forced awakening (FA), as compared with uninterrupted sleep (US). Adults underwent 2 nights of forced awakening (FA) or 2 nights of uninterrupted sleep (US) with the assessment of stimulated monocyte expression of proinflammatory cytokines in the morning after the second night of the assigned sleep condition. As compared with US, FA was associated with an increase in the stimulated monocyte expression of IL-6 (F = 2.8, df = 1, 69.9, P = 0.09), an increase in stimulated expression TNF-α (F = 5.2, df = 1, 74.9, P = 0.03), an increase in stimulated coexpression of IL-6 and TNF-α (F = 3.9, df = 1, 74.5, P = 0.05), and an increase in the composite of these 3 cellular subsets. (F = 4.9, df = 1, 71.4, P = 0.03). Statistical analyses adjusted for sex, age, BMI, Black race, and Hispanic or Latino ethnicity. Values shown are marginalized means and standard error bars.
3.4. Pain outcomes
There were no group effects for hPTH, indicating that the order of administration of FA vs US did not alter the pain outcome (P > 0.05; Fig. 4S, available at http://links.lww.com/PAIN/B740). In adjusted models, FA was associated with a significant decrease in hPTH (F = 5.52, df = 1, 65, P = 0.02; Fig. 4).
Figure 4.

Heat pain threshold in response to forced awakening (FA), as compared with uninterrupted sleep (US). Adults underwent 2 nights of forced awakening (FA) or 2 nights of uninterrupted sleep (US) with the assessment of heat pain threshold (hPTH) in the morning after the second night of the assigned sleep condition. Statistical analyses adjusted for sex, age, BMI, Black race, and Hispanic or Latino ethnicity. Difference is statistically significant (F = 5.52, df = 1, 65, P = 0.02). Values shown are marginalized means and standard error bars.
3.5. Forced awakening and heat pain threshold: sleep and inflammatory mediational pathways
Table 3 presents the results of the mediation analyses, simultaneously modeling the experimental condition (ie, US vs FA), amounts N3 sleep, and the composite of cellular inflammation (ie, stimulated monocytic production of proinflammatory cytokines) on hPTH, adjusting for the above set of covariates. As shown, there is a direct effect of cellular inflammation on hPTH. In addition, consistent with the hypothesized model (Fig. 1), the effect of experimental sleep loss on hPTH was mediated by a loss of N3 sleep and increases in cellular inflammation (unstandardized coefficient, −0.15, 95% CI, −0.30 to −0.03; Fig. 5) The proportion mediated by N3 sleep was 25.0%, and the proportion mediated by cellular inflammation was 19.0%; in the double mediation model, the proportion mediated was 34.9%. By contrast, when percentage of N3 sleep was modeled, the pathway, FA > percentage N3 sleep > cellular inflammation > hPTH, was not significant (0.03, 95% CI, −0.001 to 0.08; Fig. 5S, available at http://links.lww.com/PAIN/B740), indicating that only loss N3 amount, but not changes in the percentage of N3 sleep, mediated the effect of FA on pain sensitivity.
Table 3.
Direct and indirect mediation effects of sleep condition, N3 sleep, and cellular inflammation on heat pain threshold.
| Variable | Direct effects | Indirect effects | ||||
|---|---|---|---|---|---|---|
| FA-US | N3 sleep | Inflammation | FA-US/N3 sleep | FA-US/inflammation | FA-US/N3 sleep/inflammation | |
| Heat pain threshold | −0.43 (−1.24 to 0.38) | 0.00 (−0.01 to 0.01) | −0.02* (−0.04 to −0.01) | -0.24 (-0.64 to 0.15) | −0.02 (−0.45 to 0.39) | −0.15* (−0.30 to −0.03) |
Values are unstandardized coefficients and 95% confidence intervals. Covariates include sex, age, BMI, Black race, and Hispanic or Latino ethnicity. *P < .05.
Figure 5.

Mediational model linking sleep disruption to pain sensitivity. The mediational model links experimentally induced sleep disruption or forced awakening (FA) to heightened pain sensitivity as measured by heat pain threshold (hPTH) through indirect paths of loss of N3 sleep and increases in cellular inflammation. The model shows a significant direct effect of FA to decrease in N3 sleep, decrease of N3 sleep to increase in cellular inflammation, and increase in cellular inflammation to decrease in hPTH, with an overall significant mediational path. Values are unstandardized coefficients. Covariates include sex, age, BMI, Black race, and Hispanic or Latino ethnicity. *P < 0.05; **P < 0.01.
Although neither loss of N2 nor REM sleep have been found to be associated with increases in markers of inflammation,6,26 exploratory analyses were performed to evaluate whether the mediational pathway (ie, FA > N3 sleep > cellular inflammation > hPTH) was altered with inclusion of N2 sleep or REM sleep. When N2 sleep was included in the model, the pathway, FA > N3 sleep > cellular inflammation > hPTH, remained significant (unstandardized coefficient, −0.11, 95% CI, −0.28 to −0.00: Fig. 6S, available at http://links.lww.com/PAIN/B740); the pathway FA > N2 sleep > cellular inflammation > hPTH was not significant (unstandardized coefficient, 0.16, 95% CI, −0.28 to 0.49; Fig. 6S, available at http://links.lww.com/PAIN/B740). In addition, when REM sleep was included in the model, the pathway, FA > N3 sleep > cellular inflammation > hPTH, remained significant (unstandardized coefficient, −0.14, 95% CI, −0.29 to −0.02); the pathway, FA > REM sleep > cellular inflammation > hPTH, was not significant (unstandardized coefficient, −0.15, 95% CI, −0.4 to 0.06; Fig. 7S, available at http://links.lww.com/PAIN/B740). Finally, we explored whether the effect of FA on hPTH was mediated by loss of total sleep time. In the mediational model, the pathway FA > total sleep time > cellular inflammation > hPTH was not significant (unstandardized coefficient, −0.29, 95% CI, −1.00 to 0.17; Fig. 8S, available at http://links.lww.com/PAIN/B740).
Secondary exploratory analyses examined mTS because we have previously found that FA has effects, albeit sex dependent, on this pain outcome. In the mediational model, the pathway FA > N3 sleep > cellular inflammation > mechanical temporal summation was not significant (unstandardized coefficient, −0.01, 95% CI, −0.04 to 0.05; Fig. 9S, available at http://links.lww.com/PAIN/B740).
4. Discussion
The current study is novel in comprehensively evaluating pathways, including loss of N3 sleep and activation of cellular inflammation, which are hypothesized to contribute to increases in pain sensitivity after sleep disruption. In healthy adults, experimental disruption of sleep due to the administration of FA induced a significant decrease in heat pain threshold, as compared with responses after US. Experimental manipulation of sleep with FA also led to disturbance in sleep continuity and changes in sleep architecture, including loss of N3 sleep. Moreover, in the morning after FA, there was a robust activation of cellular inflammation, as indicated by increases in TLR-4 stimulated monocytic production of IL-6 and TNF-α. Notably, increases in cellular inflammation had a direct effect in predicting decreases in heat pain threshold (ie, increases in sensitivity to heat pain). Finally, in a comprehensive mediation model, the effect of FA on hPTH was doubly mediated by a loss of N3 sleep and subsequent increases in cellular inflammation.
Our results provide insight into the mechanisms that are believed to mediate increases in pain sensitivity after sleep disruption. Although there is evidence that sleep disruption, including FA, can induce decreases in N3 sleep,21,59,60 with additional separate studies showing that partial night sleep deprivation can activate cellular markers of inflammation,30,32 this is the first study that has used a multilevel approach to examine these mechanisms and their role in mediating the link between sleep disruption and pain sensitivity. These findings suggest that interventions that mitigate loss of N3 sleep or attenuate increases in cellular inflammation among persons who are experiencing sleep disturbance have the potential to reduce the risk of heightened pain sensitivity, even in the context of 2 nights of exposure to sleep disruption. The significance of these experimental observations is further strengthened by evidence that the present sleep disruption paradigm models a ubiquitous pattern of sleep loss experienced by persons with either chronic pain, insomnia, or chronic stress due to demanding work schedules or interpersonal difficulties.11,41,64
Preclinical research has demonstrated that proinflammatory cytokines such as IL-6 and TNF-α have receptor-mediated sensitizing effects on afferent nociceptive pathways in spinal dorsal horn and dorsal root ganglia.2,57 Furthermore, these proinflammatory cytokines also mediate muscle and joint hyperalgesia10,44 and contribute to increases in pain sensitivity possibly by sensitizing nociceptors in peripheral nerve terminals.44,48 Translation of this basic research to clinical models has been limited, but experimental endotoxemia, which induces systemic inflammation, heightens pain sensitivity across multiple testing modalities in humans.3,9,20,33 Furthermore, results from large population-based studies indicate that increases in levels of systemic inflammation may be associated with decreased cold pain tolerance.23,55 In addition, several cross-sectional, observational studies have shown that chronic pain populations, including those with fibromyalgia, knee osteoarthritis, complex regional pain syndrome, or postamputation pain, exhibit elevated peripheral levels of proinflammatory mediators, compared with controls.7,22,40,51,67 In addition to the effects on pain sensitivity, increases in cellular inflammation are linked to a number of chronic inflammatory disorders, with additional effects on infectious disease, given that IL-6 is a pleiotropic cytokine that influences both antigen-specific immune responses and inflammatory reactions.34
Our findings are consistent with a prior study demonstrating that prolonged sleep restriction elevates serum IL-6, which in turn was positively associated with subjective ratings of spontaneous daily pain.15 This study is novel by showing that experimental sleep disruption activates upstream sources of inflammation and that such activation of monocytic, cellular inflammation mediates increases in pain sensitivity (ie, hPTH) in humans. Moreover, this effect of FA is doubly mediated by selective loss of N3 sleep. Although FA also led to changes in REM sleep, inclusion of this measure in the comprehensive mediational model showed that REM sleep did not mediate the effects of FA on hPTH and did REM sleep did not alter the mediational role of N3 on hPTH. Furthermore, loss of N3 amounts, but not changes in the percentage of N3, contributed to increases in pain sensitivity, suggesting that the amount of N3 is a determinant of increases in cellular inflammation and subsequent heightened pain sensitivity. Given that the total sleep time did not mediate changes in cellular inflammation and hPTH in response to FA, and that the findings seem to be specific for loss of N3 minutes, further research is needed to evaluate whether this mediational model extends to other models of experimental sleep restriction in which total sleep time is reduced with relative preservation of N3 sleep. Although the effector mechanisms underlying the pathway between loss of N3 sleep and increases in cellular inflammation are not known, experimental sleep disruption as well as naturally occurring sleep disturbance are known to augment sympathetic nervous system activity.24–26 In turn, sympathetic activation leads to increased release of norepinephrine, binding to β-adrenergic receptors on monocytes,47 increases in activity of the transcriptional factor, nuclear factor (NF)κB,5 upregulation of inflammatory transcriptional profiles, and expression of inflammatory mediators.28,43 Given that N3 sleep is associated with a downregulation of sympathetic output as compared with other sleep stages,25,62 loss of amount of N3 in response to FA may lead to further increases in sympathetic output, which together contribute to increases in TLR-4 stimulated monocytic production of inflammatory cytokines.
In interpreting these results, causal interpretations should be avoided and consideration of a number of limitations is warranted. First, there are multiple assumptions used in causal mediation models including the absence of unmeasured confounders for the relationships between sleep disruption and pain sensitivity outcomes, sleep disruption to mediators (ie, N3 sleep and inflammation), and mediators to pain sensitivity outcomes. In addition, although selection of the covariates was guided by prior research on the effects of sleep disruption on pain outcomes, residual confounding by unmeasured risk factors is possible. Second, the sample is limited to healthy adults, and it is not known whether these results generalize to older adults, persons with chronic sleep disturbance, or those with underlying comorbidities and elevated levels of inflammation. Third, FA was delivered over 2 nights with assessment of acute effects in the morning after the sleep manipulation. Whether these effects are perpetuated by chronic sleep disturbance or across multiple nights of sleep fragmentation or whether increases in inflammation and hyperalgesia persist beyond a night of recovery sleep require further study. Fourth, acute sleep disturbance leads to sex-dependent sensitization across a number of QST outcomes60 but not hPTH; however, the sample size was not adequate to test sex-stratified mediational pathways for these other pain outcomes.
5. Conclusions
Overall, the results presented above demonstrate that the differential loss of N3 sleep and increases in cellular inflammation may be important drivers of pain sensitivity in response to sleep disruption. Identifying those who are likely to show a loss of N3 sleep such as older adults or those who have underlying inflammatory disorders or exaggerated inflammatory responses to challenge has implications for priority monitoring of heightened pain sensitivity and risk of progression to states of chronic pain after exposure to sleep disruption. Future work should endeavor to understand the relative importance of these sleep and inflammatory mediators within the context of other contributors to pain sensitivity and how these pathways together operate across the life course to influence pain outcomes.
Conflict of interest statement
The authors have no conflicts of interest to declare.
Appendix A. Supplemental digital content
Supplemental digital content associated with this article can be found online at http://links.lww.com/PAIN/B740.
Acknowledgments
This project was funded by National Institutes of Health/National Institute on Drug Abuse (NIH/NIDA) R01DA0329922 (to M.T.S. and M.R.I.) and by the Norman Cousins Center for Psychoneuroimmunology.
Footnotes
Sponsorships or competing interests that may be relevant to content are disclosed at the end of this article.
Supplemental digital content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal's Web site (www.painjournalonline.com).
Contributor Information
Richard Olmstead, Email: richard.olmstead@ucla.edu.
Martin F. Bjurstrom, Email: martin.bjurstrom@med.lu.se.
Patrick H. Finan, Email: pfinan1@jhu.edu.
Michael T. Smith, Email: msmith62@jhmi.edu.
References
- [1].Afolalu EF, Ramlee F, Tang NKY. Effects of sleep changes on pain-related health outcomes in the general population: a systematic review of longitudinal studies with exploratory meta-analysis. Sleep Med Rev 2018;39:82–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Basbaum AI, Bautista DM, Scherrer G, Julius D. Cellular and molecular mechanisms of pain. Cell 2009;139:267–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Benson S, Kattoor J, Wegner A, Hammes F, Reidick D, Grigoleit JS, Engler H, Oberbeck R, Schedlowski M, Elsenbruch S. Acute experimental endotoxemia induces visceral hypersensitivity and altered pain evaluation in healthy humans. PAIN 2012;153:794–9. [DOI] [PubMed] [Google Scholar]
- [4].Besedovsky L, Lange T, Haack M. The sleep-immune crosstalk in health and disease. Physiol Rev 2019;99:1325–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Bierhaus A, Wolf J, Andrassy M, Rohleder N, Humpert PM, Petrov D, Ferstl R, von Eynatten M, Wendt T, Rudofsky G, Joswig M, Morcos M, Schwaninger M, McEwen B, Kirschbaum C, Nawroth PP. A mechanism converting psychosocial stress into mononuclear cell activation. Proc Natl Acad Sci U S A 2003;100:1920–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Bjurstrom MF, Olmstead R, Irwin MR. Reciprocal relationship between sleep macrostructure and evening and morning cellular inflammation in rheumatoid arthritis. Psychosom Med 2016;79:24–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Chamessian A, Van de Ven T, Buchheit T, Hsia HL, McDuffie M, Gamazon ER, Walsh C, Bruehl S, Buckenmaier C, III, Shaw A. Differential expression of systemic inflammatory mediators in amputees with chronic residual limb pain. PAIN 2017;158:68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Cho HJ, Seeman TE, Kiefe CI, Lauderdale DS, Irwin MR. Sleep disturbance and longitudinal risk of inflammation: moderating influences of social integration and social isolation in the Coronary Artery Risk Development in Young Adults (CARDIA) study. Brain Behav Immun 2015;46:319–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].de Goeij M, van Eijk LT, Vanelderen P, Wilder-Smith OH, Vissers KC, van der Hoeven JG, Kox M, Scheffer GJ, Pickkers P. Systemic inflammation decreases pain threshold in humans in vivo. PLoS One 2013;8:e84159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Dina OA, Green PG, Levine JD. Role of interleukin-6 in chronic muscle hyperalgesic priming. Neuroscience 2008;152:521–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Drewes AM, Svendsen L, Taagholt SJ, Bjerregard K, Nielsen KD, Hansen B. Sleep in rheumatoid arthritis: a comparison with healthy subjects and studies of sleep/wake interactions. Br J Rheumatol 1998;37:71–81. [DOI] [PubMed] [Google Scholar]
- [12].Edinger JD, Bonnet MH, Bootzin RR, Doghramji K, Dorsey CM, Espie CA, Jamieson AO, McCall WV, Morin CM, Stepanski EJ, American Academy of Sleep Medicine Work G. Derivation of research diagnostic criteria for insomnia: report of an American Academy of sleep medicine work group. Sleep 2004;27:1567–96. [DOI] [PubMed] [Google Scholar]
- [13].Finan PH, Goodin BR, Smith MT. The association of sleep and pain: an update and a path forward. J Pain 2013;14:1539–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Grace PM, Hutchinson MR, Maier SF, Watkins LR. Pathological pain and the neuroimmune interface. Nat Rev Immunol 2014;14:217–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Haack M, Sanchez E, Mullington JM. Elevated inflammatory markers in response to prolonged sleep restriction are associated with increased pain experience in healthy volunteers. Sleep 2007;30:1145–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Hayes AF. An index and test of linear moderated mediation. Multivariate Behav Res 2015;50:1–22. [DOI] [PubMed] [Google Scholar]
- [17].Hayes AF. Introduction to mediation, moderation, and conditional process analysis: A regression-based approach. Guilford Press, 2022. [Google Scholar]
- [18].Hayes AF, Preacher KJ. Quantifying and testing indirect effects in simple mediation models when the constituent paths are nonlinear. Multivariate Behav Res 2010;45:627–60. [DOI] [PubMed] [Google Scholar]
- [19].Hofmann SG, Curtiss JE, Hayes SC. Beyond linear mediation: toward a dynamic network approach to study treatment processes. Clin Psychol Rev 2020;76:101824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Hutchinson MR, Buijs M, Tuke J, Kwok YH, Gentgall M, Williams D, Rolan P. Low-dose endotoxin potentiates capsaicin-induced pain in man: evidence for a pain neuroimmune connection. Brain Behav Immun 2013;30:3–11. [DOI] [PubMed] [Google Scholar]
- [21].Iacovides S, George K, Kamerman P, Baker FC. Sleep fragmentation hypersensitizes healthy young women to deep and superficial experimental pain. J Pain 2017;18:844–54. [DOI] [PubMed] [Google Scholar]
- [22].Imamura M, Ezquerro F, Marcon Alfieri F, Vilas Boas L, Tozetto-Mendoza TR, Chen J, Ozcakar L, Arendt-Nielsen L, Rizzo Battistella L. Serum levels of proinflammatory cytokines in painful knee osteoarthritis and sensitization. Int J Inflam 2015;2015:329792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Iordanova Schistad E, Kong XY, Furberg AS, Backryd E, Grimnes G, Emaus N, Rosseland LA, Gordh T, Stubhaug A, Engdahl B, Halvorsen B, Nielsen CS. A population-based study of inflammatory mechanisms and pain sensitivity. PAIN 2020;161:338–50. [DOI] [PubMed] [Google Scholar]
- [24].Irwin M, Clark C, Kennedy B, Gillin J, Ziegler M. Nocturnal catecholamines and immune function in insomniacs, depressed patients, and control subjects. Brain Behav Immun 2003;17:365–72. [DOI] [PubMed] [Google Scholar]
- [25].Irwin M, Thompson J, Miller C, Gillin J, Ziegler M. Effects of sleep and sleep deprivation on catecholamine and interleukin-2 levels in humans: clinical implications. J Clin Endocrin Metab 1999;84:1979–85. [DOI] [PubMed] [Google Scholar]
- [26].Irwin MR. Sleep and inflammation: partners in sickness and in health. Nat Rev Immunol 2019;19:702–15. [DOI] [PubMed] [Google Scholar]
- [27].Irwin MR, Carrillo C, Olmstead R. Sleep loss activates cellular markers of inflammation: sex differences. Brain Behav Immun 2010;24:54–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Irwin MR, Cole SW. Reciprocal regulation of the neural and innate immune systems. Nat Rev Immunol 2011;11:625–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Irwin MR, Olmstead R, Carroll JE. Sleep disturbance, sleep duration, and inflammation: a systematic review and meta-analysis of cohort studies and experimental sleep deprivation. Biol Psychiatry 2016;80:40–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Irwin MR, Wang M, Campomayor CO, Collado-Hidalgo A, Cole S. Sleep deprivation and activation of morning levels of cellular and genomic markers of inflammation. Arch Intern Med 2006;166:1756–62. [DOI] [PubMed] [Google Scholar]
- [31].Irwin MR, Wang M, Ribeiro D, Cho HJ, Olmstead R, Breen EC, Martinez-Maza O, Cole S. Sleep loss activates cellular inflammatory signaling. Biol Psychiatry 2008;64:538–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Irwin MR, Witarama T, Caudill M, Olmstead R, Breen EC. Sleep loss activates cellular inflammation and signal transducer and activator of transcription (STAT) family proteins in humans. Brain Behav Immun 2015;47:86–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Karshikoff B, Lekander M, Soop A, Lindstedt F, Ingvar M, Kosek E, Olgart Hoglund C, Axelsson J. Modality and sex differences in pain sensitivity during human endotoxemia. Brain Behav Immun 2015;46:35–43. [DOI] [PubMed] [Google Scholar]
- [34].Kishimoto T. Interleukin-6: discovery of a pleiotropic cytokine. Arthritis Res Ther 2006;8(suppl 2):S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Krause AJ, Prather AA, Wager TD, Lindquist MA, Walker MP. The pain of sleep loss: a brain characterization in humans. J Neurosci 2019;39:2291–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Lange T, Rasmussen M, Thygesen LC. Assessing natural direct and indirect effects through multiple pathways. Am J Epidemiol 2014;179:513–18. [DOI] [PubMed] [Google Scholar]
- [37].Lange T, Vansteelandt S, Bekaert M. A simple unified approach for estimating natural direct and indirect effects. Am J Epidemiol 2012;176:190–5. [DOI] [PubMed] [Google Scholar]
- [38].Lautenbacher S, Kundermann B, Krieg JC. Sleep deprivation and pain perception. Sleep Med Rev 2006;10:357–69. [DOI] [PubMed] [Google Scholar]
- [39].Littner M, Hirshkowitz M, Kramer M, Kapen S, Anderson WM, Bailey D, Berry RB, Davila D, Johnson S, Kushida C, Loube DI, Wise M, Woodson BT, American Academy of Sleep M, Standards of Practice C. Practice parameters for using polysomnography to evaluate insomnia: an update. Sleep 2003;26:754–60. [DOI] [PubMed] [Google Scholar]
- [40].Lundborg C, Hahn-Zoric M, Biber B, Hansson E. Glial cell line-derived neurotrophic factor is increased in cerebrospinal fluid but decreased in blood during long-term pain. J Neuroimmunol 2010;220:108–13. [DOI] [PubMed] [Google Scholar]
- [41].Mahowald MW, Mahowald ML, Bundlie SR, Ytterberg SR. Sleep fragmentation in rheumatoid arthritis. Arthritis Rheum 1989;32:974–83. [DOI] [PubMed] [Google Scholar]
- [42].McBeth J, Lacey RJ, Wilkie R. Predictors of new-onset widespread pain in older adults: results from a population-based prospective cohort study in the UK. Arthritis Rheumatol 2014;66:757–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Miller GE, Chen E, Sze J, Marin T, Arevalo JM, Doll R, Ma R, Cole SW. A functional genomic fingerprint of chronic stress in humans: blunted glucocorticoid and increased NF-kappaB signaling. Biol Psychiatry 2008;64:266–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Milligan ED, Watkins LR. Pathological and protective roles of glia in chronic pain. Nat Rev Neurosci 2009;10:23–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Morin CM, LeBlanc M, Daley M, Gregoire JP, Merette C. Epidemiology of insomnia: prevalence, self-help treatments, consultations, and determinants of help-seeking behaviors. Sleep Med 2006;7:123–30. [DOI] [PubMed] [Google Scholar]
- [46].Mundal I, Grawe RW, Bjorngaard JH, Linaker OM, Fors EA. Psychosocial factors and risk of chronic widespread pain: an 11-year follow-up study-the HUNT study. PAIN 2014;155:1555–61. [DOI] [PubMed] [Google Scholar]
- [47].Murray DR, Irwin M, Rearden CA, Ziegler M, Motulsky H, Maisel AS. Sympathetic and immune interactions during dynamic exercise: mediation via Beta2-adrenergic dependent mechanism. Circulation 1992;83:203–13. [DOI] [PubMed] [Google Scholar]
- [48].Obreja O, Schmelz M, Poole S, Kress M. Interleukin-6 in combination with its soluble IL-6 receptor sensitises rat skin nociceptors to heat, in vivo. PAIN 2002;96:57–62. [DOI] [PubMed] [Google Scholar]
- [49].Ohayon MM. Epidemiology of insomnia: what we know and what we still need to learn. Sleep Med Rev 2002;6:97–111. [DOI] [PubMed] [Google Scholar]
- [50].Onen SH, Alloui A, Gross A, Eschallier A, Dubray C. The effects of total sleep deprivation, selective sleep interruption and sleep recovery on pain tolerance thresholds in healthy subjects. J Sleep Res 2001;10:35–42. [DOI] [PubMed] [Google Scholar]
- [51].Parkitny L, McAuley JH, Di Pietro F, Stanton TR, O'Connell NE, Marinus J, van Hilten JJ, Moseley GL. Inflammation in complex regional pain syndrome: a systematic review and meta-analysis. Neurology 2013;80:106–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Pinho-Ribeiro FA, Verri WA, Jr., Chiu IM. Nociceptor sensory neuron-immune interactions in pain and inflammation. Trends Immunol 2017;38:5–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Preacher KJ, Hayes AF. SPSS and SAS procedures for estimating indirect effects in simple mediation models. Behav Res Methods Instrum Comput 2004;36:717–31. [DOI] [PubMed] [Google Scholar]
- [54].Roehrs T, Hyde M, Blaisdell B, Greenwald M, Roth T. Sleep loss and REM sleep loss are hyperalgesic. Sleep 2006;29:145–51. [DOI] [PubMed] [Google Scholar]
- [55].Schistad EI, Stubhaug A, Furberg AS, Engdahl BL, Nielsen CS. C-reactive protein and cold-pressor tolerance in the general population: the Tromso Study. Pain 2017;158:1280–8. [DOI] [PubMed] [Google Scholar]
- [56].Schrimpf M, Liegl G, Boeckle M, Leitner A, Geisler P, Pieh C. The effect of sleep deprivation on pain perception in healthy subjects: a meta-analysis. Sleep Med 2015;16:1313–20. [DOI] [PubMed] [Google Scholar]
- [57].Segond von Banchet G, Boettger MK, Fischer N, Gajda M, Brauer R, Schaible HG. Experimental arthritis causes tumor necrosis factor-alpha-dependent infiltration of macrophages into rat dorsal root ganglia which correlates with pain-related behavior. PAIN 2009;145:151–9. [DOI] [PubMed] [Google Scholar]
- [58].Simpson NS, Scott-Sutherland J, Gautam S, Sethna N, Haack M. Chronic exposure to insufficient sleep alters processes of pain habituation and sensitization. PAIN 2018;159:33–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Smith MT, Edwards RR, McCann UD, Haythornthwaite JA. The effects of sleep deprivation on pain inhibition and spontaneous pain in women. Sleep 2007;30:494–505. [DOI] [PubMed] [Google Scholar]
- [60].Smith MT, Jr., Remeniuk B, Finan PH, Speed TJ, Tompkins DA, Robinson M, Gonzalez K, Bjurstrom MF, Irwin MR. Sex differences in measures of central sensitization and pain sensitivity to experimental sleep disruption: implications for sex differences in chronic pain. Sleep 2019;42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Smith MT, Mun CJ, Remeniuk B, Finan PH, Campbell CM, Buenaver LF, Robinson M, Fulton B, Tompkins DA, Tremblay JM, Strain EC, Irwin MR. Experimental sleep disruption attenuates morphine analgesia: findings from a randomized trial and implications for the opioid abuse epidemic. Sci Rep 2020;10:20121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Somers VK, Dyken ME, Mark AL, Abboud FM. Sympathetic-nerve activity during sleep in normal subjects. N Engl J Med 1993;328:303–7. [DOI] [PubMed] [Google Scholar]
- [63].Sommer C, Leinders M, Uceyler N. Inflammation in the pathophysiology of neuropathic pain. PAIN 2018;159:595–602. [DOI] [PubMed] [Google Scholar]
- [64].Tang NK. Insomnia Co-occurring with chronic pain: clinical features, interaction, assessments and possible interventions. Rev Pain 2008;2:2–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Taylor DJ, Mallory LJ, Lichstein KL, Durrence HH, Riedel BW, Bush AJ. Comorbidity of chronic insomnia with medical problems. Sleep 2007;30:213–18. [DOI] [PubMed] [Google Scholar]
- [66].Turner L, Shamseer L, Altman DG, Weeks L, Peters J, Kober T, Dias S, Schulz KF, Plint AC, Moher D. Consolidated standards of reporting trials (CONSORT) and the completeness of reporting of randomised controlled trials (RCTs) published in medical journals. Cochrane Database Syst Rev 2012;11:MR000030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Uceyler N, Hauser W, Sommer C. Systematic review with meta-analysis: cytokines in fibromyalgia syndrome. BMC Musculoskelet Disord 2011;12:245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Varallo G, Giusti EM, Manna C, Castelnuovo G, Pizza F, Franceschini C, Plazzi G. Sleep disturbances and sleep disorders as risk factors for chronic postsurgical pain: a systematic review and meta-analysis. Sleep Med Rev 2022;63:101630. [DOI] [PubMed] [Google Scholar]
- [69].Watkins LR, Maier SF, Goehler LE. Immune activation: the role of pro-inflammatory cytokines in inflammation, illness responses and pathological pain states. PAIN 1995;63:289–302. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental digital content associated with this article can be found online at http://links.lww.com/PAIN/B740.