

Abstract
Preeclampsia (PE) is a relatively common but severe pregnancy disorder (with very limited effective treatments) characterized by hypertension (HTN) and usually proteinuria (PRO) or other organ damage. Genome-wide association studies (GWAS) of PE, HTN, and PRO have mostly identified risk loci single nucleotide polymorphisms (SNPs) located in noncoding genomic regions, likely impacting the regulation of distal gene expression. The latest GWAS associated (P < 1 × 10−6) SNPs to PE (n = 25), HTN (n = 1926), and PRO (n = 170). Our algorithmic analysis (CoDeS3D) used chromatin connection data (Hi-C) derived from 70 cell lines followed by analysis of two expression quantitative trail loci (eQTL) cohorts: GTEx (838 donors, 54 tissues, totaling 15 253 samples) and DICE (91 donors, 13 blood tissue types). We identified spatially constrained eQTLs which implicate gene targets in PE (n = 16), HTN (n = 3561), and PRO (n = 335). By overlapping these target genes and their molecular pathways (protein–protein interaction networks), we identified shared functional impacts between PE and HTN, which are significantly enriched for regulatory interactions which target genes intolerant to loss-of-function mutations. While the disease-associated SNP loci mostly do not overlap, the regulatory signals (target genes and pathways) overlap, informing on PE risk mechanisms. This demonstrates a model in which genetic predisposition to HTN and PRO lays a molecular groundwork toward risk for PE pathogenesis. This overlap at the gene regulatory network level identifies possible shared therapeutic targets for future study.
Keywords: genetics, gene regulation, epigenetics, preeclampsia, hypertension, proteinuria
Our model suggests that the better studied genetic predisposition to hypertension shares a molecular basis with preeclampsia risk, suggesting shared pathways for treatment.
Graphical Abstract
Graphical Abstract.

Introduction
Preeclampsia (PE) is a severe disease that affects 2–8% of all pregnancies, impacting pregnant women who were normotensive prior to pregnancy [1]. The onset of PE typically occurs after 20 weeks of gestation, and PE can be categorized into subtypes according to the age of onset: early (<34 weeks) and late (≥34 weeks) [2]. Early-onset PE is considered a fetal disorder (placental dysfunction). In contrast, late-onset PE is considered a maternal disorder (underlying maternal constitutional disorder with a normal placenta) [2]. Thus, it is hypothesized that fetal and maternal risk factors contribute asymmetrically to early and late PE, respectively. Consistent with this, maternal genetic variant rs4769612 (near FLT1) has a stronger association with cases of late-onset PE.
Clinically, PE is characterized by high blood pressure (hypertension; HTN) with a systolic blood pressure (SBP) ≥140 mmHg and/or diastolic blood pressure (DBP) ≥90 mmHg in addition to signs of organ damage/dysfunction, which are commonly observed through proteinuria (PRO; proteins excreted through the urine) [3]. Left untreated, PE can lead into the development of eclampsia, which is characterized by severe seizures and an increased maternal and fetal morbidity and mortality. Preeclampsia increases the risk that the fetus will be born preterm or exhibit intrauterine growth restriction/fetal growth restriction [4]. Despite the seriousness of PE, the only widely accepted treatment is induction of labor [5].
Preeclampsia is a complex disease, with numerous genetic and environmental factors [6]. Preeclampsia heritability has been estimated to be 54.4% maternal/42.5% fetal in Central Asians and 38.1% maternal/21.3% fetal in Europeans [7]. The genetic variants that are associated with PE can impact on the pathophysiology in two key ways: (1) aberrant fetal-derived cytotrophoblast invasion in the uterine wall in early pregnancy can lead to failed remodeling of the maternal spiral arteries perfusing the placenta; and (2) hypoxia and/or oxidative stress to the placenta causes the release of syncytiotrophoblast-derived factors (e.g., receptor for vascular endothelial growth factor, VEGFR-1 (otherwise referred to as Flt-1), endothelin, and proinflammatory cytokines [8]) into the maternal circulation leading to an exaggerated inflammatory response [9]. The release of proinflammatory cytokines contributes to the systemic inflammation associated with pregnancy, whilst Flt-1 and endothelin cause endothelial dysfunction (the likely cause of maternal HTN) [8]. Endothelial dysfunction is not unique to PE; it is also a principal pathogenic mechanism in coronary artery disease and stroke (which share HTN as a risk factor) [10]. This association has led to the study of shared genetics factors between HTN and PE [11, 12]. Similarly, sFlt-1 has also been linked with proteinuria [13]. As such, there is likely to be an interplay of genetic factors and/or pathways that predispose to PE, HTN, and PRO.
Preeclampsia involves a complex interplay between the maternal, paternal, and fetal genomes. The paternal contribution is illustrated by the finding that fathers whose mothers had PE, when carrying them, have an increased chance of the pregnancies they father being preeclamptic [14]. Genetic variants attributed to maternal and fetal contributions to risk of developing PE have been identified by GWAS [7, 15–17]. These variants include a fetal copy-number deletion near PSG11 and the INHBB locus [15, 17]. Five maternally derived variants associated with PE (rs259983, rs1421085, rs4769613, rs12050029, rs149427560) also predispose individuals for HTN, consistent with the existence of a shared genetic risk between PE and HTN [7, 16]. As GWAS of PE are far smaller than similar studies in HTN (sample sizes multitudes larger than PE) [18], it remains likely that this genetic overlap will increase as the genetics of PE are studied further.
Beyond sample size limitations, there is a need to characterize the functional roles and effects of PE-associated variants. PE-associated SNPs have typically been associated with the closest genes in the linear sequence (or the closest phenotypically relevant gene) [7, 15–17, 19]. However, in these regions, SNPs that associate with transcription levels of genes that are distant to the SNP locus [19] potentially drive more of the heritable component of disease [20]. For example, genes associated with trans-acting eQTLs are enriched for loss-of-function intolerance [21]. Thus, a better way to annotate non-coding variants to impacted genes, protein levels, and pathways is to investigate their enrichment in control elements and assign functions via methods which attribute these variants to gene regulation [22–24]. One approach to achieve this is to integrate information on the three-dimensional arrangement of DNA into the functional analyses [25]. By leveraging our knowledge of these 3D connections in relationship to loci tagged by variants associated with PE, we can better understand their role within hubs of regulation with spatially proximal genes, irrespective of their linear distance from the locus in question.
Here, we have used Contextualizing Developmental SNPs in three Dimensions (CoDeS3D) to investigate the spatially constrained eQTLs and their target genes that are associated with PE, HTN, and PRO to better understand the genetic overlap and putative biological mechanisms associated with these complex conditions. Our results identified gene candidates not previously associated with PE by published GWAS, or candidate gene studies. We then used STRING, LOEUF scores, g:Profiler, and literature queries to show that these genes converge on biological pathways and protein–protein interaction networks (PPINs) that involve PE-, HTN-, and PRO-associated eQTL targeted genes [26–29].
Methods
Retrieval of PE-, HTN-, and PRO-associated SNPs
Unique SNPs associated (P ≤ 1 × 10−6) with either PE, HTN, or PRO were identified from published literature (March 2021, Search terms: “Preeclampsia GWAS,” “Hypertension GWAS,” “Proteinuria GWAS,” “Preeclampsia associated SNPs,” “Hypertension associated SNPs,” “Proteinuria associated SNPs”) and the NHGRI-EBI GWAS Catalogue (https://www.ebi.ac.uk/gwas/; downloaded March 2021; specific SNP sources found in Supplementary Table S1) [30].
SNPs were filtered to only include those with a P-value of ≤1 × 10−6. SNPs were cleaned prior to use to ensure that they were associated with the phenotype in question. For PE, reported traits for SNP associations included fetal factor PE, maternal factor PE, early-onset PE, and late-onset PE. For HTN, reported traits for all SNPs were HTN, pulse pressure measurement, DBP, SBP, early-onset HTN, treatment resistant HTN, primary HTN, and mean arterial pressure. For PRO, reported traits included PRO, albuminuria, moderate albuminuria, urinary albumin to creatine ratio, and urinary albumin excretion. For HTN, some SNPs were associated with factors considered to be out of scope for an overlap with PE, and these were therefore excluded from further analysis. These included SNPs associated with pulmonary arterial HTN, bevacizumab induced HTN, idiopathic intracranial HTN, and blood pressure response to thiazide diuretics, candesartan, calcium-channel blockers, and hydrochlorothiazide. For both HTN and PRO, studies that only reported associations in males were removed as, by definition, PE only occurs in females.
Expression QTL and targeted gene identification and analysis
The SNP sets from each of the three conditions (PE, HTN, or PRO) were independently analyzed for the existence of spatially constrained eQTL. For this analysis, we used CoDeS3D to identify the regulatory impacts of genetic variants by assessing them for spatially constrained gene regulation [29]. CoDeS3D first identifies spatial associations between sections of DNA using information from Hi-C contact data across 70 different cell lines and tissues (Supplementary Table S2) [31]. These spatial contacts were then interrogated for an association with gene expression (eQTL) from the Genotype-Tissue Expression database (GTEx), which encompasses 49 human tissues (GTEx V8) [32]. These eQTL target gene associations were divided into three classes according to the separation distance: (1) SNP and gene <1 Mb apart from one another (cis interactions); (2) SNP and gene >1 Mb apart (trans-intrachromosomal); and (3) SNP and gene on different chromosomes (trans-interchromosomal). Significance was corrected to control for false positives (Benjamini–Hochberg false discovery rate; FDR < 0.05). All SNP IDs and chromosomal positions used in this manuscript are labeled according to GRCh38.
LOEUF score and expression variation between interaction types
LOEUF scores were obtained for all spatially constrained eQTL target genes (gnomAD database; https://gnomad.broadinstitute.org, v2.1.1 [28]). A Kruskal–Wallis H test (KW) was performed to identify significant differences (P ≤ 0.05) in underlying LOEUF score distribution for each interaction type (cis, trans-intrachromosomal, trans-interchromosomal). Where significant KW results were identified, we subsequently performed a Dunn post-hoc test to determine which pairwise differences were significant (Benjamini–Hochberg FDR < 0.05).
Mapping protein–protein interactions between PE eQTL targeted genes and directly associated HTN and PRO eQTL targeted genes
Functional protein-level connections between the spatially constrained eQTL target genes were identified using STRING (Search Tool for the Retrieval of Interacting Genes/Proteins) (https://string-db.org, version 11.0) [26]. Default settings were used, with the exception of the minimum required interaction score, which was set to 0.700 (high confidence), disconnected nodes were hidden, and active interaction sources were altered to include only text mining, experiments, databases, and co-expression. The analysis included all target genes. However, for simplification purposes, the final output for the HTN and PRO target genes was filtered to include only target genes that directly interacted with at least one PE target.
Pathway enrichment for HTN/PRO proteins directly associated with PE proteins
Biological pathways that are impacted both by the individual genes and pathways that represent a convergence of affected genes were identified using g:Profiler (FDR < 0.05; g:GOSt – Functional profiling; https://biit.cs.ut.ee/gprofiler/gost, version e104_eg51_p15_3922dba) [27] and STRING protein–protein interaction enrichment data. Only biological pathways annotated within KEGG, Reactome, and WikiPathways were considered.
Expression QTL targeted gene expression in arteries, blood, and immune cell subtypes
We compared RNA expression and eQTL detection of the eQTL targeted genes within arteries (aorta, coronary, and tibial) and whole blood, both identified in GTEx, whilst also identifying blood-specific spatially constrained cis-eQTLs identified within 13 different immune cell types using the Database of Immune Cell EQTLs/Expression/Epigenomics (DICE; FDR P-value ≤0.05; https://dice-database.org, latest build: January 7, 2020) [33]. All trans-interactions were recorded as “not detected” in DICE. Genes with TPM ≥ 0.5 are commonly considered to be expressed in a specific tissue [34].
Data and code availability
All datasets generated for this study are included in the Supplementary Tables and Files available in figshare with the identifier (https://doi.org/10.17608/k6.auckland.17082647.v1).
This study makes use of data generated by the GTEx Project [32] and DICE [33].
The CoDeS3D pipeline is available at https://github.com/Genome3d/codes3d-v1 [29].
Results
PE shares disease-associated SNPs, spatially constrained eQTLs and eQTL targeted genes with HTN, but not with PRO
The major diagnostic criteria for PE include elevated blood pressure (HTN) and organ damage detected through protein in the urine (PRO). Therefore, we wanted to study the potential genetic convergence of these three conditions at the SNP, gene, and protein level. SNPs associated (P ≤ 1 × 10−6) with PE (n = 25), HTN (n = 1926), and PRO (n = 170; Supplementary Table S1) were identified. Six SNPs are associated with both PE and HTN (Figure 1A; Supplementary Table S1), and an additional six SNPs were associated with both HTN and PRO (Figure 1A; Supplementary Table S1). None of the identified SNPs were shared between PE and PRO.
Figure 1.
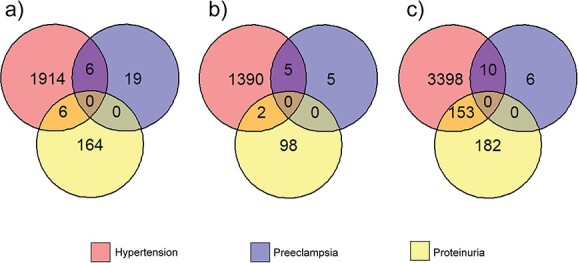
Overlaps between PE, HTN, and PRO at the (A) disease-associated SNP, (B) eQTL, and (C) gene level. Numbers indicate exact counts.
Spatially constrained eQTLs were identified by screening the disease-associated SNPs through CoDeS3D (FDR q < 0.05; Figure 1B; Supplementary Tables S3–S5) [29]. Of the 25 PE-associated SNPs, 15 and 4 were identified in cis- and trans eQTL-gene pairs, respectively (Table 1). Similarly, of the 1926 HTN-associated SNPs, 4402 and 1799 were identified in cis- and trans eQTL-gene pairs, respectively. Likewise, 326 cis eQTL-gene pairs and 125 trans eQTL-gene pairs were identified for the 170 PRO-associated SNPs. The number of eQTL-gene pairs can be greater than the number of disease-associated SNPs as eQTLs can have multiple gene targets in the same, or different tissues (Table 1; Supplementary Table S5). Five of the six SNPs that were shared between PE and HTN were identified as eQTLs (Figure 1A and B). Similarly, two of the six SNPs that were shared between HTN and PRO were identified as eQTLs.
Table 1.
Summary of eQTLs associated with PE, HTN, and PRO.
| PE | HTN | PRO | |
|---|---|---|---|
| No. of disease-associated SNPsa | 25 | 1926 | 170 |
| No. of eQTL-gene pairsb | 41 | 28 632 | 2412 |
| No. of unique eQTL-gene pairsc | 19 | 6201 | 451 |
| No. of unique cis eQTL-gene pairsd | 15 | 4402 | 326 |
| No. of unique trans eQTL-gene pairse | 4 | 1799 | 125 |
| No. of eQTL targeted genesf | 16 | 3561 | 335 |
aNumber of SNPs associated with PE, HTN, or PRO from the literature. PE = preeclampsia; HTN = hypertension; PRO = proteinuria.
beQTL-gene pairs were defined as an SNP that is an eQTL and its linked gene occuring in a specific tissue; therefore, the same eQTL-gene pair in more than one tissue were recorded as separate eQTL-gene pairs (FDR < 0.05).
cUnique is defined as eQTL-gene pairs that occur in a minimum of one GTEx tissue (if present in more than one, the eQTL-gene pair is only counted once).
dUnique eQTLs that act in cis; between an SNP and gene that are <1 Mb apart.
eUnique trans eQTL-gene pairs where the eQTL and gene are >1 Mb apart, this includes both trans-intrachromosomal and trans-interchromosomal.
feQTL targeted genes are the genes where the expression levels are at least partially associated with the base identity of the SNP.
PE-associated eQTLs targeted 16 genes, HTN-associated eQTLs targeted 3561 genes, and PRO-associated eQTLs targeted 335 genes (Figure 1C). Of the 16 PE target genes, 10 were also targeted by HTN loci (Figure 1C). Four of these 10 shared eQTL targeted genes were not due to shared SNPs, or SNPs in linkage disequilibrium, between the two conditions (Supplementary Table S8). While HTN and PRO shared 153 target genes, PE and PRO shared 0 gene.
Mean LOEUF scores and eQTL targeted gene expression levels vary with the type of spatially constrained eQTL interaction
We used measures of mutational constraint to assign LOEUF scores (gnomAD database v2.1.1) to the spatially constrained eQTL target genes. We then tested for differences in LOEUF scores for the three interaction types (cis, trans-intrachromosomal, and trans-interchromosomal; Figure 2A–C, respectively; Supplementary Table S10). Mean LOEUF scores were observed to decrease as the distance between the eQTL and the target gene increased (from cis to trans-intrachromosomal to trans-interchromosomal; Figure 2). This is consistent with the trans-eQTLs being enriched for associations with genes that are intolerant to loss-of-function mutations. However, the PE results are inconclusive due to the limited number of eQTLs that were identified (Figure 2A).
Figure 2.

LOEUF score and expression distributions differed for genes targeted by trans-acting and cis-acting eQTLs associated with HTN and PRO. LOEUF scores were plotted for genes according to the interaction type (cis, trans-intrachromosomal, or trans-interchromosomal) for (A) preeclampsia (PE), (B) hypertension (HTN), and (C) proteinuria (PRO). The red line denotes the threshold between tolerant and intolerant genes (LOEUF 0.35, as defined by gnomAD [28]). The bars in the box plots denote the quartiles and median. Expression levels of the eQTL targeted genes in 49 tissues (Supplementary Tables S3–S5) were plotted for (D) PE, (E) HTN, and (F) PRO according to eQTL interaction type (e.g. cis, trans-intrachromosomal, or trans-interchromosomal). •, the mean LOEUF score (A–C) or expression (D–F). *, P-value ≤ 0.05.
The mean expression levels of the eQTL target genes expression varied depending on interaction type, with significant differences for HTN (Figure 2E) and PRO (Figure 2F). Again, PE numbers prevented us testing for a significant difference (Figure 2D). Collectively, these results suggest that expression of the HTN- and PRO-associated eQTL targeted genes varies significantly depending on the associating eQTL interaction type.
Expression QTL targeted gene products interact within PPINs for PE-HTN and PE-PRO
We tested for phenotypic overlap with PE (PE-HTN and PE-PRO) arising through the interaction of gene products within PPINs. Convergence was observed, as clusters of interacting proteins encoded by PE-associated eQTL target genes directly interacted with proteins encoded by both HTN- and PRO-associated eQTL target genes (Figure 3A and B, respectively). For both the PE-HTN and PE-PRO PPINs, the top 10 enriched pathways (P < 0.05) include intracellular signaling by second messengers, diseases of signal transduction by growth factor receptors and second messengers, and dopaminergic synapse (Supplementary Table S9). The large cluster within the PE-HTN PPIN contains all five PE genes that are also present in the PE-PRO PPIN (GDNF, GSK3B, SLC2A3, FTO, and ALDH2; blue and purple nodes, Figure 3A and B). Nine of the 16 proteins within the PE-PRO PPIN were also identified within the PE-HTN PPIN. The four shared eQTL target genes between HTN and PRO (orange nodes, Figure 3) are targeted by different eQTLs (i.e., different SNPs targeting the same gene). Notably, the PPIN (i.e., biological pathway) is the first biological level at which we see an interaction between PE and PRO, following our observations that no SNPs, eQTLs, or targeted genes are shared between these two conditions.
Figure 3.

Protein–protein interaction networks occurring between eQTL targeted genes. Protein–protein interaction network for (A) all PE-associated eQTL targeted genes and HTN-associated eQTL targeted genes; and (B) all PE-associated eQTL targeted genes and PRO-associated eQTL targeted genes that directly interact with at least one PE-associated eQTL targeted gene. Expression QTL-targeted genes are labeled blue (PE only), purple (PE and HTN), and orange (shared HTN and PRO). Proteins annotated with a stripe are connected to the PPIN in (A) but are not (B), despite being present in the dataset. Grey nodes are HTN only or PRO only eQTL targeted genes, in A and B, respectively.
Expression and eQTL detection in artery subtypes, whole blood, and immune cells
PE is a disease of the placental arteries [35]. Therefore, altered expression of PE-, HTN-, or PRO-associated eQTL targeted genes within the blood, arteries, or immune cells may be biologically relevant. From the PE-HTN PPIN (Figure 3), three clusters were present, one large cluster containing 116 proteins (cluster PH1), cluster PH2 containing four proteins (IBA57, NFS1, NFU1, and HSCB), and cluster PH3 containing OBSCN and RHOT2. Of the 116 proteins in cluster PH1, 109 were observed to have their RNA expressed (transcript levels ≥0.5 transcripts per million [TPM]) in at least one of the artery types (tibial, aorta, and coronary) from the GTEx database (v8) (Supplementary Table S7), and 32 were targeted by a spatial eQTL detected in at least one artery subtype. In whole blood, 88 of the 116 proteins in cluster PH1 showed RNA expression (GTEx), whilst 9 were targeted by a spatial eQTL in whole blood (Supplementary Table S7). Of note, FGF5, in cluster PH1, showed no expression or eQTLs in any artery subtype or whole blood but is targeted by an eQTL in naïve CD4+ T cells (Treg) in the DICE database (Supplementary Table S6). All four proteins in cluster PH2 were expressed in all three artery subtypes and whole blood in the GTEx database. However, only IBA57 was targeted by a spatial eQTL, in whole blood. This IBA57 eQTL was also detected in CD4+ T cell (Th17) in the DICE database (Supplementary Table S6). From cluster PH3, both proteins were also expressed in all artery types and whole blood in the GTEx database, but only RHOT2 was targeted by a spatial eQTL, in the tibial artery.
For the PE-PRO PPIN, four small clusters were observed: cluster PP1 (GDNF, SLC2A3, RAP1A, STBD1, FRS2, and VAMP8), cluster PP2 (GSK3B, PPP2R5C, AKT2, PSME4, and PSMD3), cluster PP3 (ALDH2, GATM, and GLYCTK), and cluster PP4 (FTO and GNPDA2). For cluster PP1, all proteins except for GDNF were expressed in all three artery types and whole blood in the GTEx database. Yet, spatial eQTLs were only detected for STBD1 and GDNF in the tibial artery (Supplementary Table S7). For cluster PP2, all proteins are expressed in the three artery types and whole blood, but there were no spatial eQTLs targeting these genes in any artery type nor whole blood. All three genes in cluster PP3 were observed to be expressed in all three artery subtypes and whole blood in the GTEx database. Spatial eQTLs targeting all three proteins were present in at least one of the artery types, and a spatial eQTL targeting GATM and GLYCTK in whole blood. For cluster PP4, both genes were expressed in all artery types and whole blood. However, we only identified a spatial eQTL for GNPDA2 in the tibial artery.
Discussion
The 3D conformation of the genome impacts on transcription regulation through the spatial associations that occur between regulatory elements and genes located across the genome [25]. The integration of spatial information into the assignment of putative functions to the genetic variants associated with PE-, HTN-, and PRO identified a significant number of cis- and trans-acting eQTLs, with clear overlaps between PE and HTN at both the gene level and within signaling pathways. Collectively, these findings provide insights into the pathways and mechanisms that may be involved in the pathogenesis of PE and its relationships to HTN and PRO, potential targets for therapeutic intervention.
PE and PRO converge at the pathway level but not the genetic level
Preeclampsia is a hypertensive disorder with a co-occurrence of PRO in most PE cases [3, 14]. The identification of overlap at the genetic level for PE and HTN (but not PRO) suggests that the GWAS-identified PE variants have more in common with HTN than PRO. The observed overlap between PE and HTN has been identified before, where genetic factors associated with HTN have also been associated with PE [7]. The lack of overlap at the genetic level for PE and PRO was overcome through a convergence upon shared biological pathways in the PPIN. However, the lack of any clear connection between PE and PRO at the genetic level provides support that the mechanism of PRO in PE is more as a result of the maternal HTN induced by the placenta, rather than more established kidney disease as is observed in nonpregnant populations. Thus, this provides additional support for the American Society of Obstetrics and Gynecology’s recommendation for PRO to not be a diagnostic criterion for PE [3, 36].
Key pathways elucidated by blood- and artery-specific contributions to gene regulation in PE
The placenta is widely thought to have a significant and central role in the pathogenesis of PE. Therefore, it is important to detect whether expression changes occurred in placental tissue as a result of the disease-associated SNPs identified [37]. However, placental (trophoblast) expression and eQTLs are not present in GTEx. Instead, we have used eQTL data for the three artery subtypes and whole blood from GTEx and verified those findings in eQTLs from the DICE database. The consideration of these tissues as significant tissues is due to the hypothetical involvement and dysfunction of these various arterial tissues in PE [38, 39].
We observed a number of proteins whose genes had altered regulation due to disease-associated eQTLs in whole blood, one of the three artery subtypes, and/or immune cell subtypes. We refer to these proteins as “key” proteins due to their potential to be points of pathway disruption. From the PE-HTN PPIN, there were 41 key proteins in cluster PH1, 2 from cluster PH2 (IBA57 and NFU1), and 1 from cluster PH3 (RHOT2). From a literature search, it was found that seven proteins in PH1 (CALML4, PIK3CB, NCAM1, GNAO1, NFATC3, FGF5, and GAB2) are altered in PE patients or are involved in certain PE related mechanisms (e.g., endothelial dysfunction). Decreased RNA expression of CALML4 and PIK3CB has been observed in tissue from the maternal–fetal interface of PE patients and from the decidua for early-onset PE patients, respectively [40, 41]. Similarly, NCAM1 has an increased mRNA and protein expression in placentas from PE patients, and its suppression within human umbilical vein endothelial cells led to increased invasiveness and migration of these cells and suppression of an MAPK signaling pathway [42]. GNAO1 is significantly associated with early-onset PE [43]. NFATC3 has been previously observed to be involved in secretion of sFlt-1 as well as expression of proinflammatory cytokines from cytotrophoblasts [44]. FGF5 has been previously detected as being targeted by PE- and HTN-associated variants located at the FGF5 locus [7]. Lastly, GAB2 is upregulated in the placenta in response to systemic hypoxia and might also have a role in angiogenesis, both of which may contribute to the PE pathophysiology [45, 46]. Thus, PE-associated SNPs altering regulatory mechanisms that target these key proteins could result in a ripple effect of small alterations adding up to PE risk.
From the PE-PRO PPIN, there were seven key proteins identified: three (GDNF, STBD1, and VAMP8) in PP1, three (ALDH2, GATM, and GLYCTK) in cluster PP3, and one (GNPDA2) in cluster PP4. Altered regulation of GATM and STBD1 has previously been observed to be associated with the PE phenotype, with one study showing increased GATM mRNA levels within PE placentae, and a different study reporting that STBD1 expression was associated with PE placentae [47, 48]. Importantly for our PE-PRO analysis, GATM is specifically involved in a creatine synthesis step that occurs specifically within the kidneys. The same kidney damage in PE that causes PRO may damage the ability of the kidneys to produce sufficient creatine, and therefore, the increased placental expression levels of GATM could be a consequence of kidney damage having occurred [47, 49].
Signaling pathways are the predominant pathways enriched for in the PPINs
The majority of pathways enriched for in both the PE-HTN and PE-PRO PPINs were signaling pathways. Second messengers can impact PE development, such as an increase in sensitivity to Ca2+ in arteries, which is influenced by numerous second messengers activating pathways such as MAPK [38]. Additionally, two intracellular kinases (signaling pathways), PKB/Akt and MAPK/ERK, are thought to hold a strategic role in the pathogenesis of PE [50]. This is consistent with our analysis identifying the biological pathways, “PIP3 activates Akt signaling,” “MAPK family signaling cascade,” “MAPK1/MAPK3 signaling,” and “RAF/MAP kinase cascade” being enriched for in the PE-PRO PPIN. In addition to this, the previously discussed key gene/protein, NCAM1, was observed to be overexpressed in PE placentas, with suppression of the gene reported to suppress a particular MAPK signaling pathway, p38MAPK [42]. Similarly, dysregulation of the Wnt/β-catenin signaling pathway has recently been identified to be a potential contributor to PE pathogenesis, which is consistent with the identification of enrichment for this pathway from the PE-HTN PPIN [51]. Enrichment of “neurotrophin signaling pathways” within our PE-PRO PPIN is supported by findings that brain-derived neurotrophic factor (a neurotrophin) has a role in placental development, angiogenesis, and the inflammatory response, as well as decreased expression in PE patients [52].
The potential for so many pathways to be implicated in PE contributes to the complexity of understanding the disease process, as it highlights that there may be a range of different routes through which PE could develop. This suggests the possibility that PE may be able to be divided into subtypes depending on the disease variants present in the individuals, as these will cause specific genes to have altered regulation and therefore specific pathways that may be more likely to be disrupted. This idea of genetic subtypes within a disease has already been demonstrated through cancer genetic studies that have been able to identify subtypes of patients according to the genetic variants present, allowing tailored treatments to be created depending on the subtype, and improvements in survival rates occurring as a result of this [53, 54].
Proposed model for genetic influences on PE
We hypothesized a model for the genetic influences in PE pathogenesis, which involves tissue-specific gene targets as well as maternal- and fetal-derived genetic factors (Figure 4).
Figure 4.

Proposed model of genetic contributions to PE development. The proteins altered in PE (blue), HTN (red), and PRO (yellow) or shared between conditions (purple or orange) contribute to the onset of PE in arterial and blood tissues. The differential patterns of genetic influence highlight the diversity of processes that may be impacted within the various tissues impacted in PE pathogenesis. Other tissues = suprapubic not sun exposed skin, lower leg sun exposed skin, esophagus mucosa, cultured fibroblast cells, skeletal muscle, EBV transformed lymphocyte cells, and kidney cortex. Figure created on draw.io, as deployed at https://app.diagrams.net
We propose that maternal risk for PE arises through two different mechanisms: (i) genes/proteins regulated by both HTN and PRO; and (ii) only genes impacting HTN influence PE, with organ damage occurring as a secondary effect. For both of these mechanisms, key genes/proteins are disrupted in tissues important to both the maternal and fetal sides of the placental interface, resulting in PE in the mother. Mechanism (i) is supported by the sharing of four proteins between HTN and PRO at the protein and pathway level (STBD1, RAP1A, FRS2, and PPP2R5C). GSK3B, FTO, and SLC2A3 are found in both PPINs and have functions that can have impact on PE pathogenesis. GSK3B is activated in response to hypoxia, an environment observed in PE patients due to inadequate placental implantation. Additionally, GSK3B, when activated, is involved in degrading a protein called GCM1, known to have a role in placental development [55]. FTO could impact on PE through the same pathway as BMI, an independent risk factor for PE [56, 57]. SLC2A3 is a glucose transporter found within the placenta, where glucose uptake aids placental development [58, 59]. Mechanism (ii) is supported by our finding that HTN and PE form much larger networks in the PPIN, with five proteins found in the PE-HTN PPIN only (ITPR2, FGF5, INTS9, IBA57, and OBSCN). ITPR2 may contribute to PE development through its influence on vascular tone and blood pressure through Ca2+ release, a pathway shared by HTN and PE [60, 61]. The model also indicates the presence of key genes modified by fetal genetic risk factors, with potential roles for ITPR2 and GSK3B previously discussed as maternal factors above.
It should be noted that the exact contribution each of these genes may have to the pathogenesis of PE is unknown. However, each key gene is thought to contribute fractionally to its development, and it is a combinatorial effect of a number of them that results in PE pathology.
Limitations of the GTEx tissue data and future directions
The predominate limitation of using GTEx data is that the tissue donor characteristics are not a reflection of the characteristics of individuals in which PE occurs. GTEx samples were isolated from a cohort of male (67.1%) and female (32.9%) tissue donors, none of whom were pregnant (GTEx database, V8 Donor info). Numerous studies have demonstrated that age has an impact on gene expression, suggesting that data sourced from individuals who are beyond child-bearing age are not ideal when looking at a condition that only occurs in females of reproductive age [62]. Therefore, the extrapolation of these results to PE pathogenesis requires further experimental and longitudinal sampling during pregnancy. It would be advantageous to replicate these gene regulation studies in placental tissue subsets (e.g., trophoblasts) across varying gestational ages (e.g., first versus third trimester), as this would allow identification of eQTLs that may be only occurring in the placenta at specific developmental timepoints. This would help identify pathways specific to certain PE subtypes (e.g., early vs late PE), built on the basis that these subtypes may have different development pathways.
Another limitation of this work is that PE has shown much clinical heterogenicity (e.g., fetal factor PE, maternal factor PE, early-onset PE, and late-onset PE), but this work does not distinguish the genetic effects from early-onset from those found in late-onset PE. This results in the model not distinguishing which gene/pathway is more associated with which subtype of PE.
Notwithstanding these limitations, we were able to demonstrate tissue-specificity of our disease-associated eQTLs and were able to highlight potential differences in contributions from both the maternal and fetal genomes.
Conclusion
In conclusion, we were able to identify a significant number of eQTLs associated with PE, HTN, and PRO and the biological pathways they affect. The identification of overlap between HTN and PE at the genetic and protein levels distinguishes HTN from PRO in terms of their relationships with PE.
Authors’ contributions
G.B. contributed to conceptualization, performed analyses, interpreted the data, and co-wrote the manuscript revision. W.S. and J.O.S. supervised G.B., conceptualized, and co-wrote the manuscript. All authors contributed to the article and approved the submitted version.
Conflict of interest
The authors have declared that no conflict of interest exists.
Supplementary Material
Footnotes
† Grant Support: J.O.S. was funded by donations from the Dines Family Trust. W.S. was supported by a postdoctoral fellowship from the Auckland Medical Research Foundation (grant ID 1320002) and a Royal Society of New Zealand Marsden Grant (20-UOA-002). This work contains data from the Genotype-Tissue Expression (GTEx) Project, which was supported by the Common Fund of the Office of the Director of the National Institutes of Health, and by NCI, NHGRI, NHLBI, NIDA, NIMH, and NINDS.
Contributor Information
Genevieve Boom, Liggins Institute, The University of Auckland, Auckland, New Zealand.
Justin M O’Sullivan, Liggins Institute, The University of Auckland, Auckland, New Zealand; The Maurice Wilkins Centre, The University of Auckland, Auckland, New Zealand; Australian Parkinson’s Mission, Garvan Institute of Medical Research, Sydney, NSW, Australia; MRC Lifecourse Epidemiology Unit, University of Southampton, Southampton, UK; Singapore Institute for Clinical Sciences, Agency for Science, Technology and Research (A*STAR), Singapore.
William Schierding, Liggins Institute, The University of Auckland, Auckland, New Zealand; The Maurice Wilkins Centre, The University of Auckland, Auckland, New Zealand.
References
- 1. Jeyabalan A. Epidemiology of preeclampsia: impact of obesity. Nutr Rev 2013; 71:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Raymond D, Peterson E. A critical review of early-onset and late-onset preeclampsia. Obstet Gynecol Surv 2011; 66:497–506. [DOI] [PubMed] [Google Scholar]
- 3. Dong X, Gou W, Li C, Wu M, Han Z, Li X, Chen Q. Proteinuria in preeclampsia: not essential to diagnosis but related to disease severity and fetal outcomes. Pregnancy Hypertens 2017; 8:60–64. [DOI] [PubMed] [Google Scholar]
- 4. Fox R, Kitt J, Leeson P, Aye CYL, Lewandowski AJ. Preeclampsia: risk factors, diagnosis, management, and the cardiovascular impact on the offspring. J Clin Med 2019; 8:1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. English FA, Kenny LC, McCarthy FP. Risk factors and effective management of preeclampsia. Integr Blood Press Control 2015; 8:7–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Redman C. Pre-eclampsia: a complex and variable disease. Pregnancy Hypertens An Int J Women’s Cardiovasc Heal 2014; 4:241–242. [DOI] [PubMed] [Google Scholar]
- 7. Steinthorsdottir V, McGinnis R, Williams NO, Stefansdottir L, Thorleifsson G, Shooter S, Fadista J, Sigurdsson JK, Auro KM, Berezina G, Borges MC, Bumpstead S, et al. Genetic predisposition to hypertension is associated with preeclampsia in European and central Asian women. Nat Commun 2020; 11:5976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Han C, Han L, Huang P, Chen Y, Wang Y, Xue F. Syncytiotrophoblast-derived extracellular vesicles in pathophysiology of preeclampsia. Front Physiol 2019; 10:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Roberts JM, Hubel CA. The two stage model of preeclampsia: variations on the theme. NIH Public Access 2010; 30:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rodie VA, Freeman DJ, Sattar N, Greer IA. Pre-eclampsia and cardiovascular disease: metabolic syndrome of pregnancy? Atherosclerosis 2004; 175:189–202. [DOI] [PubMed] [Google Scholar]
- 11. Roten LT, Fenstad MH, Forsmo S, Johnson MP, Moses EK, Austgulen R, Skorpen F. A low COMT activity haplotype is associated with recurrent preeclampsia in a Norwegian population cohort (HUNT2). Mol Hum Reprod 2011; 17:439–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Johansson Å, Curran JE, Johnson MP, Freed KA, Fenstad MH, Bjørge L, Eide IP, Carless MA, Rainwater DL, Goring HHH, Austgulen R, Moses EK, et al. Identification of ACOX2 as a shared genetic risk factor for preeclampsia and cardiovascular disease. Eur J Hum Genet 2011; 19:796–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. McElwain CJ, Tuboly E, McCarthy FP, McCarthy CM. Mechanisms of endothelial dysfunction in pre-eclampsia and gestational diabetes mellitus: windows into future cardiometabolic health? Front Endocrinol (Lausanne) 2020; 11:1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Williams PJ, Broughton Pipkin F. The genetics of pre-eclampsia and other hypertensive disorders of pregnancy. Best Pract Res Clin Obstet Gynaecol 2011; 25:405–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhao L, Triche EW, Walsh KM, Bracken MB, Saftlas AF, Hoh J, Dewan AT. Genome-wide association study identifies a maternal copy-number deletion in PSG11 enriched among preeclampsia patients. BMC Pregnancy Childbirth 2012; 12:61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. McGinnis R, Steinthorsdottir V, Williams NO, Thorleifsson G, Shooter S, Hjartardottir S, Bumpstead S, Stefansdottir L, Hildyard L, Sigurdsson JK, Kemp JP, Silva GB, et al. Variants in the fetal genome near FLT1 are associated with risk of preeclampsia. Nat Genet 2017; 49:1255–1260. [DOI] [PubMed] [Google Scholar]
- 17. Johnson MP, Brennecke SP, East CE, Göring HHH, Kent JW, Dyer TD, Said JM, Roten LT, Iversen AC, Abraham LJ, Heinonen S, Kajantie E, et al. Genome-wide association scan identifies a risk locus for preeclampsia on 2q14, near the inhibin, beta B gene. PLoS One 2012; 7:e33666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ehret GB, Ferreira T, Chasman DI, Jackson AU, Schmidt EM, Johnson T, Thorleifsson G, Luan J, Donnelly LA, Kanoni S, Petersen A-K, Pihur V, et al. The genetics of blood pressure regulation and its target organs from association studies in 342,415 individuals. Nat Genet 2016; 48:1171–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Brodie A, Azaria JR, Ofran Y. How far from the SNP may the causative genes be? Nucleic Acids Res 2016; 44:6046–6054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Liu X, Li YI, Pritchard JK. Trans effects on gene expression can drive Omnigenic inheritance. Cell 2019; 177:1022–1034.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Schierding W, Horsfield JA, O’Sullivan JM. Low tolerance for transcriptional variation at cohesin genes is accompanied by functional links to disease-relevant pathways. J Med Genet 2020; 0:jmedgenet-2020-107095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fadason T, Farrow S, Gokuladhas S, Golovina E, Nyaga D, O’Sullivan JM, Schierding W. Assigning function to SNPs: considerations when interpreting genetic variation. Semin Cell Dev Biol 2021; 121:135–142. 10.1016/j.semcdb.2021.08.008. [DOI] [PubMed] [Google Scholar]
- 23. Cano-gamez E, Trynka G. From GWAS to function : using functional genomics to identify the mechanisms underlying complex diseases. Front Genet 2020; 11:1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nica AC, Dermitzakis ET. Expression quantitative trait loci: present and future. Philos Trans Biol Sci 2013; 368:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zheng H, Xie W. The role of 3D genome organization in development and cell differentiation. Nat Rev Mol Cell Biol 2019; 20:535–550. [DOI] [PubMed] [Google Scholar]
- 26. Szklarczyk D, Gable AL, Nastou KC, Lyon D, Kirsch R, Pyysalo S, Doncheva NT, Legeay M, Fang T, Bork P, Jensen LJ, von Mering C. The STRING database in 2021: customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res 2021; 49:D605–D612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Raudvere U, Kolberg L, Kuzmin I, Arak T, Adler P, Peterson H, Vilo J. G:profiler: a web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res 2019; 47:W191–W198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, Collins RL, Laricchia KM, Ganna A, Birnbaum DP, Gauthier LD, Brand H, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020; 581:434–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fadason T, Schierding W, Lumley T, O’Sullivan JM. Chromatin interactions and expression quantitative trait loci reveal genetic drivers of multimorbidities. Nat Commun 2018; 9:5198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Buniello A, MacArthur JAL, Cerezo M, Harris LW, Hayhurst J, Malangone C, McMahon A, Morales J, Mountjoy E, Sollis E, Suveges D, Vrousgou O, et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res 2019; 47:D1005–D1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. van Berkum NL, Lieberman-Aiden E, Williams L, Imakaev M, Gnirke A, Mirny LA, Dekker J, Lander ES. Hi-C: a method to study the three-dimensional architecture of genomes. J Vis Exp 2010; 1869. 10.3791/1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. GTEx Consortium . The GTEx consortium atlas of genetic regulatory effects across human tissues. Science (80-) 2020; 369:1318–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Schmiedel BJ, Singh D, Madrigal A, Valdovino-Gonzalez AG, White BM, Zapardiel-Gonzalo J, Ha B, Altay G, Greenbaum JA, McVicker G, Seumois G, Rao A, et al. Impact of genetic polymorphisms on human immune cell gene expression. Cell 2018; 175:1701–1715.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. EMBL-EBI . Expression atlas - frequently asked questions. EMBL - EBI 2021; 1:1. [Google Scholar]
- 35. Khalil G. Preeclampsia: pathophysiology and the maternal-fetal risk. J Hypertens Manag 2017; 3:1–5. [Google Scholar]
- 36. Brown MA, Magee LA, Kenny LC, Karumanchi SA, McCarthy F, Saito S, Hall DR, Warren CE, Adoyi G, Ishaku S, International Society for the Study of Hypertension in Pregnancy (ISSHP) . The hypertensive disorders of pregnancy: ISSHP classification, diagnosis & management recommendations for international practice. Pregnancy Hypertens 2018; 13:291–310. [DOI] [PubMed] [Google Scholar]
- 37. Tannetta D, Sargent I. Placental disease and the maternal syndrome of preeclampsia: missing links? Curr Hypertens Rep 2013; 15:590–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Goulopoulou S. Maternal vascular physiology in preeclampsia. Hypertension 2017; 70:1066–1073. [DOI] [PubMed] [Google Scholar]
- 39. Tarca AL, Romero R, Erez O, Gudicha DW, Than NG, Benshalom-Tirosh N, Pacora P, Hsu CD, Chaiworapongsa T, Hassan SS, Gomez-Lopez N. Maternal whole blood mRNA signatures identify women at risk of early preeclampsia: a longitudinal study. J Matern neonatal Med Off J Eur Assoc Perinat Med Fed Asia Ocean Perinat Soc Int Soc Perinat Obstet 2021; 34:3463–3474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lian IA, Langaas M, Moses E, Johansson Å. Differential gene expression at the maternal-fetal interface in preeclampsia is influenced by gestational age. PLoS One 2013; 8:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tong J, Niu Y, Chen ZJ, Zhang C. Comparison of the transcriptional profile in the decidua of early-onset and late-onset pre-eclampsia. J Obstet Gynaecol Res 2020; 46:1055–1066. [DOI] [PubMed] [Google Scholar]
- 42. Zhang XL, Xu FX, Han XY. siRNA-mediated NCAM1 gene silencing suppresses oxidative stress in pre-eclampsia by inhibiting the p38MAPK signaling pathway. J Cell Biochem 2019; 120:18608–18617. [DOI] [PubMed] [Google Scholar]
- 43. Benny P, Yamasato K, Yunits B, Zhu X, Ching T, Garmire LX, Berry MJ, Towner D. Maternal cardiovascular-related single nucleotide polymorphisms, genes, and pathways associated with early-onset preeclampsia. PLoS One 2019; 14:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ye L, Gratton A, Hannan NJ, Cannon P, Deo M, Palmer KR, Tong S, Kaitu'u-Lino T'J, Brownfoot FC. Nuclear factor of activated T-cells (NFAT) regulates soluble fms-like tyrosine kinase-1 secretion (sFlt-1) from human placenta. Placenta 2016; 48:110–118. [DOI] [PubMed] [Google Scholar]
- 45. Arroyo J, Torry RJ, Torry DS. Deferential regulation of placenta growth factor (PlGF)-mediated signal transduction in human primary term trophoblast and endothelial cells. Placenta 2004; 25:379–386. [DOI] [PubMed] [Google Scholar]
- 46. Trollmann R, Rehrauer H, Schneider C, Krischke G, Huemmler N, Keller S, Rascher W, Gassmann M. Late-gestational systemic hypoxia leads to a similar early gene response in mouse placenta and developing brain. Am J Physiol Regul Integr Comp Physiol 2010; 299:1489–1499. [DOI] [PubMed] [Google Scholar]
- 47. Ellery SJ, Murthi P, Gatta PAD, May AK, Davies-Tuck ML, Kowalski GM, Callahan DL, Bruce CR, Wallace EM, Walker DW, Dickinson H, Snow RJ. The effects of early-onset pre-eclampsia on placental creatine metabolism in the third trimester. Int J Mol Sci 2020; 21:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Brew O, Sullivan MHF, Woodman A. Comparison of normal and pre-eclamptic placental gene expression: a systematic review with meta-analysis. PLoS One 2016; 11:1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Joncquel-Chevalier Curt M, Voicu PM, Fontaine M, Dessein AF, Porchet N, Mention-Mulliez K, Dobbelaere D, Soto-Ares G, Cheillan D, Vamecq J. Creatine biosynthesis and transport in health and disease. Biochimie 2015; 119:146–165. [DOI] [PubMed] [Google Scholar]
- 50. D'Oria R, Laviola L, Giorgino F, Unfer V, Bettocchi S, Scioscia M. PKB/Akt and MAPK/ERK phosphorylation is highly induced by inositols: novel potential insights in endothelial dysfunction in preeclampsia. Pregnancy Hypertens 2017; 10:107–112. [DOI] [PubMed] [Google Scholar]
- 51. Zhang Z, Wang X, Zhang L, Shi Y, Wang J, Yan H. Wnt/β-catenin signaling pathway in trophoblasts and abnormal activation in preeclampsia (review). Mol Med Rep 2017; 16:1007–1013. [DOI] [PubMed] [Google Scholar]
- 52. Perucci LO, Vieira ÉLM, Teixeira AL, Gomes KB, Dusse LM, Sousa LP. Decreased plasma concentrations of brain-derived neurotrophic factor in preeclampsia. Clin Chim Acta 2017; 464:142–147. [DOI] [PubMed] [Google Scholar]
- 53. Papaemmanuil E, Gerstung M, Bullinger L, Gaidzik VI, Paschka P, Roberts ND, Potter NE, Heuser M, Thol F, Bolli N, Gundem G, Van Loo P, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med 2016; 374:2209–2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Stone RM, Mandrekar SJ, Sanford BL, Laumann K, Geyer S, Bloomfield CD, Thiede C, Prior TW, Döhner K, Marcucci G, Lo-Coco F, Klisovic RB, et al. Midostaurin plus chemotherapy for acute myeloid leukemia with a FLT3 mutation. N Engl J Med 2017; 377:454–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Chiang MH, Liang FY, Chen CP, Chang CW, Cheong ML, Wang LJ, Liang CY, Lin FY, Chou CC, Chen H. Mechanism of hypoxia-induced GCM1 degradation: implications for the pathogenesis of preeclampsia. J Biol Chem 2009; 284:17411–17419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Loos RJF, Bouchard C. FTO: the first gene contributing to common forms of human obesity. Obes Rev 2008; 9:246–250. [DOI] [PubMed] [Google Scholar]
- 57. Lopez-Jaramillo P, Barajas J, Rueda-Quijano SM, Lopez-Lopez C, Felix C. Obesity and preeclampsia: common pathophysiological mechanisms. Front Physiol 2018; 9:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Hu M, Li J, Baker PN, Tong C. Revisiting preeclampsia: a metabolic disorder of the placenta. FEBS J 2021; 289:336–354. 10.1111/febs.15745. [DOI] [PubMed] [Google Scholar]
- 59. Novakovic B, Gordon L, Robinson WP, Desoye G, Saffery R. Glucose as a fetal nutrient: dynamic regulation of several glucose transporter genes by DNA methylation in the human placenta across gestation. J Nutr Biochem 2013; 24:282–288. [DOI] [PubMed] [Google Scholar]
- 60. Wilker E, Mittleman MA, Litonjua AA, Poon A, Baccarelli A, Suh H, Wright RO, Sparrow D, Vokonas P, Schwartz J. Postural changes in blood pressure associated with interactions between candidate genes for chronic respiratory diseases and exposure to particulate matter. Environ Health Perspect 2009; 117:935–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Lin Q, Zhao L, Jing R, Trexler C, Wang H, Li Y, Tang H, Huang F, Zhang F, Fang X, Liu J, Jia N, et al. Inositol 1,4,5-trisphosphate receptors in endothelial cells play an essential role in vasodilation and blood pressure regulation. J Am Heart Assoc 2019; 8:e011704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Viñuela A, Brown AA, Buil A, Tsai PC, Davies MN, Bell JT, Dermitzakis ET, Spector TD, Small KS. Age-dependent changes in mean and variance of gene expression across tissues in a twin cohort. Hum Mol Genet 2018; 27:732–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All datasets generated for this study are included in the Supplementary Tables and Files available in figshare with the identifier (https://doi.org/10.17608/k6.auckland.17082647.v1).
This study makes use of data generated by the GTEx Project [32] and DICE [33].
The CoDeS3D pipeline is available at https://github.com/Genome3d/codes3d-v1 [29].