

Summary
High‐quality radish (Raphanus sativus) genome represents a valuable resource for agronomical trait improvements and understanding genome evolution among Brassicaceae species. However, existing radish genome assembly remains fragmentary, which greatly hampered functional genomics research and genome‐assisted breeding. Here, using a NAU‐LB radish inbred line, we generated a reference genome of 476.32 Mb with a scaffold N50 of 56.88 Mb by incorporating Illumina, PacBio and BioNano optical mapping techniques. Utilizing Hi‐C data, 448.12 Mb (94.08%) of the assembled sequences were anchored to nine radish chromosomes with 40 306 protein‐coding genes annotated. In total, 249.14 Mb (52.31%) comprised the repetitive sequences, among which long terminal repeats (LTRs, 30.31%) were the most abundant class. Beyond confirming the whole‐genome triplication (WGT) event in R. sativus lineage, we found several tandem arrayed genes were involved in stress response process, which may account for the distinctive phenotype of high disease resistance in R. sativus. By comparing against the existing Xin‐li‐mei radish genome, a total of 2 108 573 SNPs, 7740 large insertions, 7757 deletions and 84 inversions were identified. Interestingly, a 647‐bp insertion in the promoter of RsVRN1 gene can be directly bound by the DOF transcription repressor RsCDF3, resulting into its low promoter activity and late‐bolting phenotype of NAU‐LB cultivar. Importantly, introgression of this 647‐bp insertion allele, RsVRN1 In‐536, into early‐bolting genotype could contribute to delayed bolting time, indicating that it is a potential genetic resource for radish late‐bolting breeding. Together, this genome resource provides valuable information to facilitate comparative genomic analysis and accelerate genome‐guided breeding and improvement in radish.
Keywords: Raphanus sativus, Genome assembly, RsNBS‐LRRs, Structure variation, RsVRN1, Differential bolting time
Introduction
Radish (Raphanus sativus L., 2 n = 2x = 18), an annual or biennial dicot belonging to the Brassicaceae family, is an economically important root vegetable crop worldwide (Luo et al., 2020). During the long‐term artificial selection from the 13th century BC, a large number of local and commercial radish varieties varying in root size, shape, colour and flavour have been developed in the world (Mitsui et al., 2015). The fleshy taproot, as the most agronomically important edible organ, provided many beneficial nutrients composed of a wide range of carbohydrates, minerals, riboflavin, phytochemicals and dietary fibres (Shirasawa et al., 2020). However, despite the biological uniqueness and favourable nutritional value, the molecular genetic studies of radish are progressing much slower compared with its close relative Brassica crops (Cai et al., 2022; Liu et al., 2014; Lu et al., 2019; Song et al., 2021), which partially attributes to the limited information of genome assembly and annotation gains to date.
Construction of a high‐quality genome assembly is a fundamental step for dissecting genomic variations that contribute to explore the genetic and molecular basis of desirable traits in plants (Nunn et al., 2022). In the past decades, several genome assemblies of R. sativus and R. raphanistrum were released that consist of thousands of small arbitrarily ordered contigs and scaffolds (Kitashiba et al., 2014; Mitsui et al., 2015; Moghe et al., 2014). Using traditional genetic mapping, a proportion of draft radish genomes were assembled at the pseudo‐chromosome level with low coverage of the euchromatin regions (Jeong et al., 2016; Shirasawa et al., 2020; Zhang et al., 2015). Recently, leveraging PacBio single‐molecule real‐time (SMRT) sequencing and high‐throughput chromatin conformation capture (Hi‐C) mapping approach, Zhang et al. (2021) reported a new radish genome of Xin‐li‐mei with 446 MB mapped to nine chromosomes, providing valuable genetic resources for molecular mapping and gene cloning in radish. Fine‐scale physical mapping of centromeres is a fundamental step for high‐quality genome assembly in plants (Gong et al., 2012). CENTROMERE SPECIFIC HISTONE H3 (CENH3) is an epigenetic signature that specifies centromere location in plant kingdoms (Maheshwari et al., 2017; Naish et al., 2021). Although the ChIP‐seq has becoming a promising approach to assist assembly of CENH3‐associated centromeric chromatin for plant genomes (Hu et al., 2019; Schneider et al., 2016), no ChIP‐seq data have been deployed to assist the reference centromeres assembly in radish. To gain better understanding of genetic bases underlying the diversity of Raphanus, it is crucial to generate another high‐quality radish genome assembly of distinctive cultivar for identifying genetic variations likely contributed to the trait differences.
As a crucial developmental milestone of the plant life cycle, the timing of transition from vegetative to floral meristems is governed by different complex genetic pathways such as photoperiod, vernalization and gibberellic acid pathways (Chen et al., 2021; Cho et al., 2017; Shang et al., 2020). These pathways converged on key floral integrators (e.g. FT and SOC1) and induced the meristem‐identity genes (e.g. FRUITFULL, LFY and AP1) to initiate flower bud formation and anthesis (Lu et al., 2020; Srikanth and Schmid, 2011; Zhang et al., 2019). Vernalization is a long‐term cold‐mediated acquisition of flowering competence for the winter‐annual plants (Kang et al., 2021; Kyung et al., 2022). In Arabidopsis, the FLOWERING LOCUS C (FLC) acted as the central regulator of vernalization, and its stable repression and histone modifications could be achieved by the AtVRN1 gene during the winter (Cho et al., 2017; Levy et al., 2002). It has been extensively established that CDF (CYCLING DOF FACTOR) transcription factor (TF) is one of the most important participators in the transcriptional regulatory networks of bolting and flowering in plants (Renau‐Morata et al., 2020; Xu et al., 2021). Previous studies reported that a range of CDFs, including AtCDF1, AtCDF2, AtCDF3 and SlCDF3, were involved in photoperiodic regulation of flowering in Arabidopsis and tomato through a CO/FT‐dependent manner (Goralogia et al., 2017; Imaizumi et al., 2005; Xu et al., 2021). However, whether CDF TF genes mediate the regulatory network of vernalization pathways remains largely unclear in radish.
Genomic structural variations (SVs), including insertions, deletions, duplications and inversions, are an important resource of genetic variations determining important agronomic traits in crops (Alonge et al., 2020; Li et al., 2022; Wang et al., 2020). Identification of SVs among different varieties of Raphanus and investigating their evolutionary dynamics can shed light on the contribution of SVs to critical horticultural traits in radish domestication and breeding. In this study, a radish advanced inbred line ‘NAU‐LB’ was used to assemble an improved chromosome‐scale reference genome. Comparing with these radish varieties employed in previous radish pan‐genome study (Zhang et al., 2021), the ‘NAU‐LB’ genotype harbours an extremely late‐bolting phenotype that requires a long‐term vernalization process. Under normal environmental condition, it takes approximately 90–100 d from days after transplanting to bolting, making it as a desirable genotype to dissect the genomic architecture conditioning bolting time variation in radish. Despite comparative genomic and evolutionary analysis, genomic SVs between the NAU‐LB and Xin‐li‐mei genome were comprehensively identified. More importantly, we addressed a specific 647‐bp insertion in the RsVRN1 gene promoter that might be responsible for the late‐bolting phenotype. Our findings would provide valuable genome resources for genome evolution studies and pave the way for genomic selections of desirable target traits like late‐bolting in radish and other species in the Brassicaceae family.
Results
Assembly of a high‐quality radish genome
The k‐mer analysis indicated that the NAU‐LB genome was highly homozygous with heterozygosity of 0.14%, which was suitable for improving the quality of genome assembly (Table S1). A total of 54.01 Gb of reads (N50 length of 18.06 Kb) were generated with the PacBio RSII platform, amounting to 104.96× coverage of the estimated 514.57 Mb genome. Then, the consensus sequences were polished with 64.17 Gb (124.71 × coverage) of Illumina reads and 63.47 Gb (123.35 × coverage) of 10× Genomics data (Table S2). We initially assembled the genome into 1821 contigs (~ 483.68 Mb) and 872 scaffolds (~ 489.8 Mb) with a contig N50 and scaffold N50 of 1.76 and 2.86 Mb, respectively. Next, the long reads assembly was integrated with ~171.55 Gb BioNano optical map data (~182.45 × coverage, Table S3), creating a hybrid assembly consisting of 797 scaffolds (~468.61 Mb) with a scaffold N50 of 4.39 Mb (Table S4).
To construct the chromosome‐scale scaffolds, ~77.37 Gb of Hi‐C data (~150.36× coverage, Tables S2 and S5) was used to categorize these assemblies into super scaffolds. As expected, the Hi‐C interaction matrices displayed a distinct anti‐diagonal pattern for the intrachromosomal interactions (Figure 1a). The size of final genome assembly was 476.32 Mb (265 super‐scaffolds) with a scaffold N50 value reaching 56.88 Mb (Tables 1 and S6). The largest 9 super‐scaffolds representing pseudo‐chromosomes totalled 448.12 Mb (94.08%) of the radish genome with GC content of 34.24% (Tables 1 and S7), consistent with the chromosome order reported previously (Jeong et al., 2016). Collinearity analysis indicated good co‐linearity of chromosomes between this assembly and other previously radish chromosome‐level genomes (Figure S1), confirming the high accuracy of the contig orientation.
Figure 1.
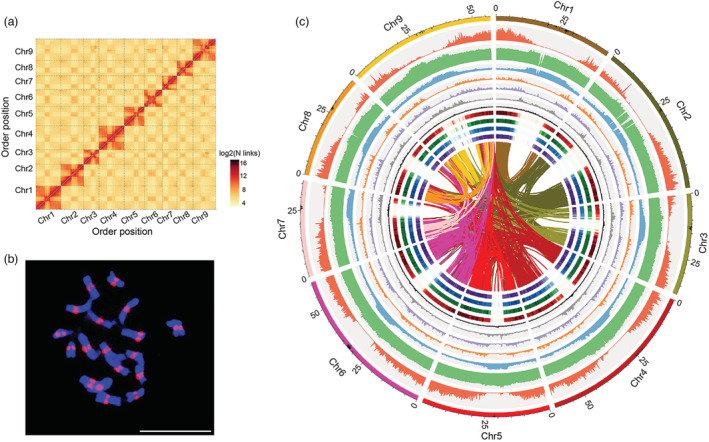
Overview of the radish genome assembly. (a) Genome‐wide Hi‐C map of the NAU‐LB genome. Post‐clustering heatmap shows density of Hi‐C interactions between contigs. (b) Fluorescence in situ hybridization (FISH) assay using NAU‐LB ChIP DNA. FISH signals (red) are visible in the centromeres on the chromosomes (blue). Scale bar: 10 μm. (c) Circos plot of the multidimensional topography for the NAU‐LB genome assembly. The outermost layer of coloured blocks was a circular representation of the 9 pseudochromosomes with each scale mark labelling of 5 Mb. Concentric circles from outermost to innermost show chromosome length with centromere region, gene density, GC content, TE density, LINE density, DNA density, Ty3‐gypsy LTR density, Ty1‐copia LTR density, root gene expression, leaf gene expression, stamen gene expression and pistil gene expression. The inner lines indicate syntenic blocks in the genome.
Table 1.
Statistics for radish genome assembly and annotation of NAU‐LB
| Assembly and annotation feature | Statistics |
|---|---|
| Assembly features | |
| Assembled genome size (Mb) | 476.32 |
| Number of scaffolds | 265 |
| Scaffold N50 (Mb) | 56.88 |
| Scaffold N90 (Mb) | 36.29 |
| Longest scaffold (Mb) | 62.08 |
| Anchored to chromosome (Mb) | 448.12 |
| Scaffold GC content (%) | 34.24 |
| Anchored % of assembly | 94.08 |
| Total repetitive sequences (%) | 52.31 |
| Gene models | |
| Number of protein‐coding genes | 40 306 |
| Mean coding sequence length (bp) | 1114.86 |
| Mean number of exons per gene | 4.98 |
Notable improvement of centromere regions and gene annotation
To evaluate the quality of this genome assembly, we firstly mapped 99.68% of Illumina short reads to the assembly with 98.82% overall coverage (Table S8). CEGMA (Core Eukaryotic Genes Mapping Approach) and BUSCO (Benchmarking Universal Single Copy Orthologs) analysis indicated that 241 (97.18%) and 1376 (95.50%) conserved core eukaryotic genes were captured in the whole‐genome assembly (Table S9 and S10). Moreover, 99.49% of Expressed sequence tag (EST) sequences assembled from transcriptome sequencing were mapped to the genome assembly, from which 88.85% of ESTs were considered as complete (more than 90% of the transcript sequences aligned to one scaffold) (Table S11). For the centromeres region assembly, a total of 39.27 million reads generated from centromere‐specific Histone 3 (CenH3) ChIP‐seq library were mapped to the unique sites in NAU‐LB genome. Fluorescence in situ hybridization (FISH) assay showed that several high‐intensity and distinct signals were observed in the centromeric region for each radish chromosome (Figure 1b), confirming highly specific enrichment of the CenH3 ChIP DNA sequences. With the exception of Chr.1, the CenH3 ChIP‐seq data were mapped to a sharp interval on eight chromosomes (Figure 1c), which might be attributed to the fragmented nature of Chr.1 centromeric regions. Moreover, long terminal repeat (LTR) annotation indicated an LTR assembly index (LAI) score of assembly was 16.07 (Figure S2), reaching to the criterion of reference quality. Together, the extensive coverage of core plant genes and centromere sequence continuity indicated the high completeness and accuracy of our genome assembly, ensuring the reliability of subsequent comparative genomic analysis.
Combing ab initio, homology‐based and RNA sequence‐aided prediction methods, a total of 40 306 protein‐coding genes were annotated with an average gene length of 2196.62 bp, coding sequence length of 1114.86 bp and exon number of 4.98 (Tables 1 and S12). Notably, 39 290 (97.48%) of the protein‐coding genes had a match in at least one databases, including 97.41%, 80.64%, 76.62% and 75.28% of genes matching GenBank NR, InterPro, SwissProt and Pfam entries, respectively (Table S13).
Dynamic evolution of LTR retrotransposons
In plants, the polyploidization driven by whole‐genome duplication (WGD) events and proliferation of transposable elements (TEs) are major sources for genome size expansion (Van de Peer et al., 2009). In the current study, 249.14 Mb (52.31%) of the assembled genome comprised of repetitive sequences, which is larger than that reported for two B. rapa assemblies: NHCC001 (213.04 Mb) and Chiifu v3.0 (133.95 Mb). Among these repetitive elements, the LTR retrotransposons, including Ty1_Copia (22.01%) and Ty3_Gypsy (21.93%), were the most abundant (combined: 30.31%), followed by the DNA TEs (4.87%) and LINEs (3.47%) (Table S14). These TEs exhibited an inverse correlation with gene density (Figure 1c). In particular, 9276 intact LTR‐RTs with an average length of 7141.43 bp were identified, much more than the number classified in the A. thaliana and B. rapa genomes (Figure 2a). In comparison with A. thaliana and B. rapa, the frequency distribution of intact radish LTR‐RTs insertion time had a burst in less than 1 million years ago (MYA) (Figure 2b and Figure S3). These insertions took place in radish specifically, as the timing of the insertion event was much less than the estimated divergence time of A. thaliana and B. rapa. In addition, 2970 radish genes contained TE insertions (Figure 2c), with 1138 intact LTR‐RTs (38.32%) reside within 2 Kb upstream of the gene body (Figure 2d), followed by 1120 and 301 inserted into the 3′‐terminus region (37.71%) and encoding region (10.13%), respectively. The proximity to genes indicated their potential roles in regulating gene expression in radish.
Figure 2.
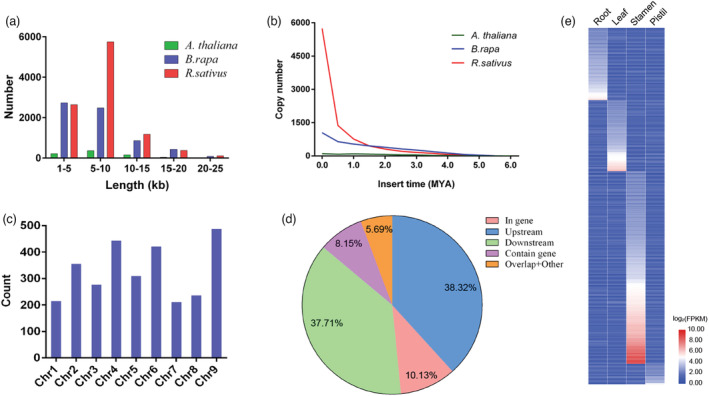
Evolution of LTR retrotransposons in the NAU‐LB genome. (a) Length distribution of the intact LTR‐RTs in radish, Arabidopsis thaliana and Brassica rapa. (b) Distribution of insertion times of LTR retrotransposons in radish, A. thaliana and B. rapa. (c) Count of LTR‐RTs in each chromosome. (d) The proportion of the LTR‐RTs located in different regions of genes. (e) Tissue expression patterns of the LTR‐inserted genes in radish root, leaf, stamen and pistil.
Comparative genomic and genome evolutionary analysis
Using 1576 shared single‐copy orthologous gene families, phylogenetic analysis showed that the R. sativus diverged from B. rapa and B. oleracea approximately 7.1–10.4 MYA (Figure 3a). Gene family evolution analysis showed that 1078 and 4445 gene families were significantly expanded and contracted in radish, respectively (P < 0.01) (Figure 3b). For the enriched GO terms, the expanded genes were significantly involved in translation and inorganic cation transmembrane transport in biological process category (P < 0.05, FDR <0.05; Table S15). Using the single‐copy orthologous gene families (Figure 3c), analyses of transversions at four‐fold degenerate sites (4DTv) and Ks values indicated that, except for the recent WGT event (4DTv distance ≈ 0.15, Ks peak value ≈ 0.4) shared among the Brassicaceae species (Hu et al., 2011), no additional specific WGD event was occurred in the evolutionary process of R. sativus (Figures 4a and S4). In addition, 14 175 gene families were shared by R. sativus, A. thaliana, B. rapa and B. oleracea, whereas 661 gene families containing 1319 genes were specific to R. sativus (Figure 4b),
Figure 3.
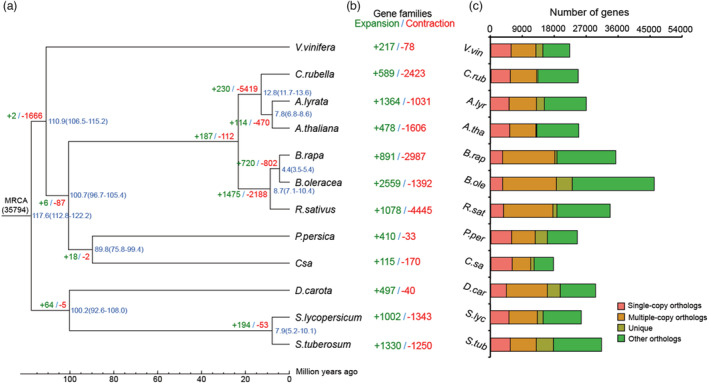
Phylogenetic tree and gene family identification in radish and some other related species. (a) Inferred phylogenetic tree constructed from single‐copy orthologue gene families in NAU‐LB and other eleven additional plant species. Divergence timings (million years ago, MYA) were indicated at each node, representing 95% credibility intervals of the estimated dates. (b) Expansions and contractions of gene families. The number of expanded and contracted gene families was marked with plus and minus ahead the digitals, respectively. (c) Clusters of orthologous and paralogous gene families in NAU‐LB and other eleven plant species.
Figure 4.
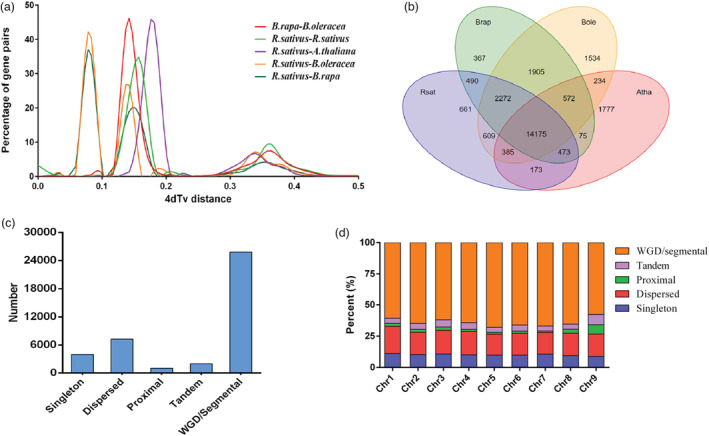
Comparative genomic and genome evolutionary analysis of the radish genome. (a) 4dTv distance distribution of duplicated gene pairs in syntenic blocks within the genomes of Raphanus sativus, Arabidopsis thaliana and Brassica rapa and B. oleracea. (b) Venn diagram of shared orthologous gene families among R. sativus, A. thaliana, B. rapa and B. oleracea. (c) Classification of gene duplicates origin in the NAU‐LB genome. The origins of gene duplicates were classified into five types: whole genome/segmental duplication (collinear genes in collinear blocks), tandem duplication (consecutive repeat), proximal duplication (two duplicated genes are distributed adjacent to each other on chromosomes, with no more than 10 genes spaced but not adjacent), dispersed duplication (duplication type other than WGD/segmental, tandem and proximal) and singleton (no duplication). (d) Number of gene duplicates on each chromosome of the NAU‐LB genome.
Classification of gene duplicates origin indicated that WGD/segmental duplication is the predominant duplicate gene origin type (64.02%, 25 802), followed by dispersed duplication (18.00%, 7255), tandem duplication (5.03%, 2029) and proximal duplication (2.63%, 1061) (Figure 4c). Interestingly, the distribution of duplicate gene origin type varied among nine radish chromosomes. The Chr. 9 had the highest proportion of proximal and tandem duplication, while the Chr. 5 had the highest proportion of WGD/segmental duplication (67.96%, 3864) (Figure 4d). Functional enrichment analysis revealed that the genes from different duplication origins have biological function preference. The gene duplicates originated from tandem duplication were enriched in the GO terms related to biotic and/or abiotic stress response, such as response to biotic stimulus and defence response to fungus (Table S16). Considering the tandemly arrayed genes might tend to be volatile after gene duplication (Miao et al., 2021), the retained tandem genes may play vital functional roles in regulating important biological process of radish plants.
Identification and evolution of radish NBS‐LRR genes
In plants, most of the cloned disease resistance genes are encode nucleotide‐binding‐site‐leucine‐rich‐repeat (NBS‐LRR) proteins, which play a pivotal role in host resistance to disease in horticultural plants (Bayer et al., 2019; Golicz et al., 2016). Based on searching against the protein sequences in our NAU‐LB assembled genome with NBS domain (Pfam: PF00931), a total of 110 NBS‐LRR resistance genes (RsNBS001–RsNBS110) were identified in this study (Figure S5), which were clustered and dispersed into nine radish chromosomes (Figure S6) with Chr. 6 contained the largest number of genes (22, 20%). Phylogenetic analysis showed that 45 and 65 RsNBS genes were classified into CNL (CNL‐A ~ CNL‐D) and TNL (TNL‐A ~ TNL‐H) subfamilies (Table S17), respectively. Transcriptome analysis showed that a proportion of RsNBS genes exhibited tissue‐specific gene expression in radish. For instance, the RsNBS045, RsNBS085 and RsNBS097 genes were highly expressed in leaves, while the RsNBS011, RsNBS063 and RsNBS096 genes exhibited high expression profile in root (Figure S7). Interestingly, MCScanX analysis showed that 36 (32.7%) RsNBS genes were classified into 16 tandem duplication events, which were unevenly distributed on eight radish chromosomes except for Chr. 3 (Figure S6). Moreover, 77 pairs of whole genome duplication (WGD)/segmental events consisted of 55 RsNBS genes (e.g. RsNBS001, RsNBS011, RsNBS065 and RsNBS079) were identified in all chromosomes (Figure S8), indicating the important role of tandem duplication and WGD/segmental duplication in driving NBS gene expansion in radish.
Genomic SVs between NAU‐LB and Xin‐li‐mei genome
Through collinear blocks alignment, the genetic variants consisted of SNPs, small InDels (<50 bp in size) and SVs (large insertion, deletion and inversions, ≥50 bp in size) were identified between the NAU‐LB and Xin‐li‐mei genome. Using NAU‐LB genome as the reference, a total of 2 108 573 SNPs were identified with an average density of 4.71 SNPs per Kb (Table S18). In all, 365 143 (17.32%) of SNPs were located in the exonic region, among which 132 894 (6.30%) were nonsynonymous SNPs (Table S19). Moreover, 5496 (1.05%) of the identified 521 225 small InDels caused changes of start/stop codons, splicing sites or frameshifts (Table S20), which likely resulted in the divergent gene functions in the two genetic backgrounds. In total, 15 581 SVs ranging from 51 bp to 14.4 Mb were identified (Figure 5a, b), including 7740 insertions, 7757 deletions and 84 inversions (Table S21 and S22), which together covered 32.00% (~143.41 Mb) of the total genome. Interestingly, approximately 31.35% of the SV sequences were LTR retrotransposons (Figure 5c), suggesting that SVs occurred frequently in genome regions occupied by LTR retrotransposons. In addition, 47 large inversions longer than 100 Kb were located on Chr. 2, 4, 5, 6, 7 (Table S22), which might contribute to relatively lower collinearity of these chromosomes between the two radish genomes.
Figure 5.
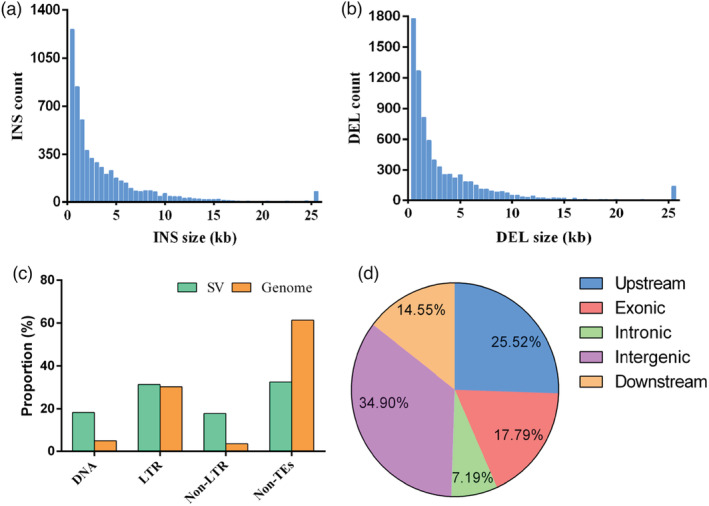
Features of structural variants between the NAU‐LB and Xin‐li‐mei genome. (a) Distribution of insertion size. (b) Distribution of deletion size. (c) Contents of different categories of transposable elements in the SV regions and NAU‐LB genome. (d) The proportion of SVs located in different regions of genes.
As shown in Figure 5d, 17.79% (2761 out of 15 517) and 25.52% (3960 out of 15 517) of the annotated SVs were overlapped with the coding sequences (CDS) and promoter regions (defined as 2 Kb upstream of gene body), notably lower than the proportion in the intergenic regions (34.90%, 5415 out of 15 517), which was consistent with the previous reports of peach and rice genomes (Fuentes et al., 2019; Guan et al., 2021). Only a small proportion of SVs were retained in the CDS regions might partially attribute to the fact that SVs occurring in CDS might lead into loss‐of‐function effect and faced strong purifying selection during plant genome evolution (Guan et al., 2021).
A 647‐bp insertion in promoter region leads into late‐bolting phenotype in NAU‐LB
As an important breeding objective in modern breeding of Brassicaceae crops, the trait of bolting and flowering was continually selected during radish domestication (Jeong et al., 2016). Using NAU‐LB assembly as the reference genome, one 39‐bp insertion was found in the intron of FUL gene of Xin‐li‐mei genome, while one 16‐bp deletion and one 18‐bp deletion were identified in the intron of LFY and FT gene, respectively (Table S23). One 9‐bp deletion and two insertions (8 and 9 bp) were detected in the intron and promoter of FLC3 gene, respectively. For the VRN2 gene, one 14‐bp deletion was detected in the promoter region, while one 15‐bp insertion and two deletions (6 bp and 42 bp) were detected in the intron region. In addition, three and five nonsynonymous SNVs were identified in the exon of LFY and VRN2 gene, respectively. Considering these genes were major determines in the photoperiod or vernalization pathways, it's reasonably to hypothesize that these genomic variants might contribute to differences of bolting and flowering time during radish evolution and domestication.
In the Brassicaceae family, several winter‐annual plant ecotypes can only start transition to flowering state after fulfilling the vernalization requirement. By comparing the coding region of RsVRN1 gene from NAU‐LB genome and two other radish genomes (Xin‐li‐mei and WK10039), only eight synonymous SNVs were found in the ‘WK10039’ genome (Figure S9), resulting in no frameshift mutation or amino acid changes of the RsVRN1 gene (Figure S10). To validate the reliability of genome sequences, the coding regions of RsVRN1 gene were cloned and sequenced from the late‐bolting genotype ‘NAU‐LB’ and early‐bolting genotype ‘Xin‐li‐mei’ using PCR approach. The RsVRN1 gene contains an open reading frame (ORF) of 1032 bp that encodes 343 amino acids (Figure S10), and no nucleotide sequence difference was observed in the coding regions between the two radish genotypes. Interestingly, a 647‐bp insertion was identified in the promoter region of RsVRN1 gene in ‘NAU‐LB’ genotype (Figures 6a, b and S11), which was further verified by PCR amplification using the primer pairs listed in Table S24. The expression profile of the RsVRN1 gene was increased during the prolonged vernalization time in ‘NAU‐LB’ genotype (P < 0.01; Figure 6c). To investigate whether the promoter sequence differences affect promoter activity of RsVRN1 gene, the corresponding promoters without or with the 647‐bp insertion (pRsVRN1 Del‐536/S1, pRsVRN1 In‐536/S2) were fused to luciferase gene, respectively (Figure 6d). Interestingly, the pRsVRN1 In‐536‐LUC exhibited significantly low LUC activity than the pRsVRN1 Del‐536‐LUC in N. benthamiana leaves (P < 0.001; Figure 6e), indicating that the 647‐bp insertion reduces promoter activity of the RsVRN1 gene in NAU‐LB.
Figure 6.
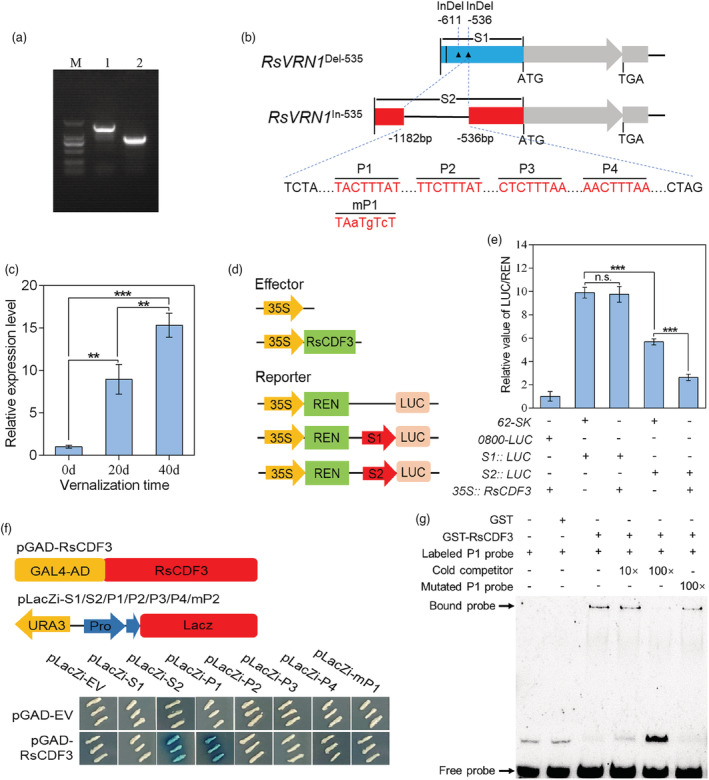
RsCDF3 can directly binds to the promoter of RsVRN1 In‐536 allele. (a) PCR‐based cloning of the RsVRN1 promoter from ‘NAU‐LB’ and ‘Xin‐li‐mei’ genotype, respectively. M: DL2000 marker. 1: NAU‐LB; 2: Xin‐li‐mei. (b) Schematic diagram of the RsVRN1 Del‐536 and RsVRN1 In‐536 promoter fragments used for construction of the transient expression vector. The vertical line and triangle represent one SNP and two InDels in the RsVRN1 promoter of Xin‐li‐mei compared with NAU‐LB, respectively. Four potential DOF binding elements (P1, P2, P3 and P4) within the 647‐bp insertion are indicated in red. The mutated nucleotides are presented in lowercase in mP1 fragment sequences. (c) The expression profile of the RsVRN1 gene during prolonged vernalization period in ‘NAU‐LB’ genotype. (d) Schematic diagrams of the LUC, S1 and S2 reporter constructs used for transient expression assay. (e) Transient expression assays of different promoter fragments from two RsVRN1 genotypes. (f) Yeast one‐hybrid assays showing that RsCDF3 binds to P1 fragment within the 647‐bp insertion. The prey and bait vectors used for the assays are indicated at the top. (g) Analysis of RsCDF3 binding to the P1 fragment of the 647‐bp insertion in an EMSA system. Biotin‐labelled probes were incubated with GST or GST‐tagged RsVRN1. 10× and 100× unlabelled competitor fragments were added to evaluate binding specificity. Values are mean ± SD from three independent biological replicates. Asterisks indicate statistically significant differences using two‐sided Student's t test (**P < 0.01; ***P < 0.001).
RsCDF3 directly binds to the RsVRN1 In ‐536 allele and inhibits bolting time
By aligning to the PLACE database, some key cis‐acting elements targeted by several transcription factors were found in the 647‐bp insertion (Table S25). To test if the promoter variations of RsVRN1 gene associate with differential binding and regulation by several specific TFs, a yeast one‐hybrid (Y1H) library was screened using the 647‐bp insertion promoter fragment as the bait. Interestingly, the RsCDF3, belonging to D subfamily of the DOF TFs, was identified in the screens. The Y1H assays with the S1, S2 promoter fragments and potential DOF binding sites (P1, P2, P3, P4) of RsVRN1 indicated direct binding of RsCDF3 to the P1 site in the 647‐bp insertion (Figure 6f). Moreover, a dual‐luciferase reporter assay showed that RsCDF3 significantly repressed the transactivation of the RsVRN1 In‐536 promoter variant compared with the RsVRN1 Del‐536 promoter variants (Figure 6e). Furthermore, the EMSA assay indicated that RsCDF3‐dependent mobility shifts were detected with biotin‐labelled P1 probes and competed by an unlabelled cold competitor probe in a dose‐dependent manner, but not by a mutated P1 probe (Figure 6g), further supporting that RsCDF3 specifically binds to the P1 fragment of the 647‐bp insertion. Together, these results indicated that RsCDF3 can specifically bind the DOF binding elements (5′‐TACTTTAT‐3′) in the 647‐bp insertion of RsVRN1 In‐536 promoter and suppress its transcription activity.
To investigate the role of RsCDF3 in bolting and flowering, we generated transgenic T3 Arabidopsis plants (Figure 7a) constitutively expressing RsCDF3 driven by the CaMV 35 S promoter. Under long‐day condition, both total rosette leaf number and days from germination to flowering indicated that the RsCDF3‐overexpressed plants exhibited dramatically delayed floral initiation compared with that in WT plants (Figure 7b, c), providing clear evidence supporting a negative role for RsCDF3 in regulating floral initiation. We then evaluate the transcriptional activity of RsCDF3 in vitro and in vivo. In yeast, the transactivation activity of VP16 was significantly repressed when RsCDF3 was fused to the VP16 activator (Figure 7d). In N. benthamiana leaves, RsCDF3 significantly repressed expression of the LUC reporter and VP16 activator in comparison to the effect of pBD and pBD‐VP16 vector, respectively (P < 0.001; Figure 7e–g). These results indicated that RsCDF3 had transcriptional repression activity in both yeast and plant cells. Considering that VRN1 gene is critical to accelerate flowering post‐vernalization (Sharma et al., 2020), it is reasonable to conclude that RsCDF3‐mediated repression of RsVRN1 due to the 647‐bp insertion is likely responsible for late‐bolting phenotype of the ‘NAU‐LB’ genotype.
Figure 7.
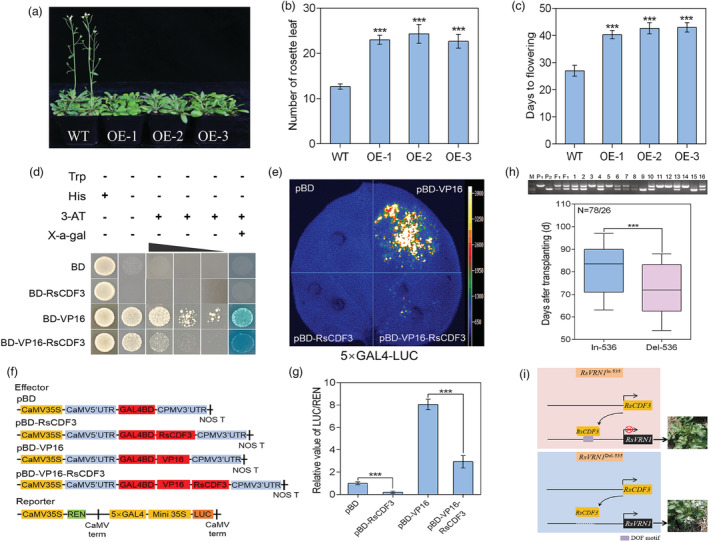
The overexpression, transcriptional repressor activity of RsCDF3 as well as introgression of the RsVRN1 In‐536 allele in radish. (a) Representative images of wild‐type (WT) and RsCDF3‐OE plants under long‐day (LD) condition. (b, c) Rosette leaf number (b) and days to flowering (c) of WT and RsCDF3‐OE plants under LD condition. (d) Transcriptional repression assay of RsCDF3 in yeast. The transformants were streaked on SD/‐Trp, SD/‐Trp‐His, SD/‐Trp‐His+3‐AT and SD/‐Trp‐His+3‐AT+X‐gal plates. (e) Transcriptional repression assay of RsCDF3 in N. benthamiana leaves. pBD and pBD‐VP16 were used as a negative and positive control, respectively. (f) Schematic diagrams of the effector and reporter constructs used for the transcriptional activity analysis. 5 × GAL4, five GAL4 binding domains; LUC, firefly luciferase; REN, Renilla luciferase. (g) The relative value of LUC/REN measured in the transcriptional activity analysis. (h) Genotyping of proRsVRN1 (up) and boxplot of bolting time (bottom) in the F2 population generated by crossing ‘NAU‐LB’ carrying the RsVRN1 In‐536 allele and ‘NAU‐YH’ carrying the RsVRN1 Del‐536 allele. (i) A proposed model for RsVRN1 In‐536/RsVRN1 Del‐536 allele‐mediated bolting time difference in radish. Values are mean ± SD from three biological replicates. Asterisks indicate statistically significant differences using two‐sided Student's t test (***P < 0.001).
Introgression of the RsVRN1 In ‐536 allele could inhibit radish bolting time
To investigate whether the insertion allele of RsVRN1 In‐536 can make a contribution to late‐bolting, a F2 population consisted of 104 individuals was generated by crossing ‘NAU‐LB’ carrying the RsVRN1 In‐536 allele and an early‐bolting radish genotype ‘NAU‐YH’ carrying the RsVRN1 Del‐536 allele. Evaluation of bolting time confirmed that the F2‐RsVRN1 In‐536 plants from F2 lines exhibited late bolting phenotype compared with their F2‐RsVRN1 Del‐536 siblings (P < 0.001; Figures 7h and S12), indicating that the RsVRN1 In‐536 allele can contribute to late bolting in radish breeding. Therefore, the RsVRN1 In‐536 allele would facilitate the development of genetic marker for early/late‐bolting selection at an early stage in radish.
Discussion
Comparison of NAU‐LB genome to other previous radish genomes
High‐quality chromosome‐level genome assembly is a vital prerequisite for molecular breeding and decoding the molecular basis of economically important traits in plants (Guo et al., 2020; Xia et al., 2020; Zhang et al., 2022). In recent years, the Hi‐C sequencing technology, relying on the linkage information across a range of length scales spanning tens of megabases, had becoming an efficient approach to assemble chromosome‐scale scaffolds for many large eukaryotic genomes (Cai et al., 2021; Ghurye et al., 2019). Radish is an important root vegetable crop of the Brassicaceae family. Although several radish genomes have been assigned to the pseudo‐chromosome level (Jeong et al., 2016; Shirasawa et al., 2020; Zhang et al., 2015, 2021), only one chromosome‐level genome of Xin‐li‐mei was constructed using the Hi‐C mapping (Zhang et al., 2021), which greatly hampered the discovery of functional genomic variations and dissection of the genetic determinants of several complex traits in radish.
In this study, we generated a new highly accurate radish genome of 476.32 Mb with a scaffold N50 value reaching 56.88 Mb (Table S6 and S7), which was slightly longer than the corresponding values (459.83 Mb with scaffold N50 of 49.37 Mb) of the Xin‐li‐mei radish genome (Table S26). Moreover, leveraging CENH3 localizes exclusively to functional centromeres (Maheshwari et al., 2017; Naish et al., 2021), we firstly assembled the repetitive‐DNA‐rich centromeric regions of radish genome by combining the CenH3 ChIP‐seq and long‐read sequencing approach, revealing insights into centromere architecture and chromatin organization for Raphanus species. Interestingly, despite good co‐linearity between NAU‐LB and Xin‐li‐mei genome (Figure S1), the genetic variants (SNPs, small InDels and SVs) conditioning differential bolting and flowering time were addressed in radish. In addition, the dynamic evolution of LTR retrotransposons and duplicate gene classification were well characterized. With the rapid advance of sequencing technologies, it is urgent to characterize the genetic variations among a number of radish species, sub‐species and varieties and dissect their impact on genetic control of vital horticultural traits in Raphanus (Zhang et al., 2021). Taken together, the availability of this complete radish genome assembly provides a valuable genome resource for further genetic breeding and evolutionary and comparative studies in genus Raphanus.
A specific RsVRN1 In ‐536 allele is responsible for late‐bolting phenotype in radish
Increasing the number of studies indicated that SVs can cause major phenotypic and morphological variance by affecting a few critical gene dosage, function and regulation in crop species (Renner et al., 2021; Wang et al., 2017; Yang et al., 2019). In this study, the assembled radish genotype ‘NAU‐LB’ exhibited an extremely late‐bolting phenotype compared with ‘Xin‐li‐mei’ genotype. In Arabidopsis, the AtVRN1 gene was critical for stable repression and histone modifications of the FLC gene (Sharma et al., 2020). Although the biological functions of VRN1 and its downstream signalling networks associated with plant bolting and flowering had been addressed (Kyung et al., 2022; Sharma et al., 2020), the vital molecular switches regulating VRN1 gene expression under vernalization pathway remain largely unclear. In the current study, we found that a 647‐bp indel in the RsVRN1 promoter was responsible for variation of RsVRN1 promoter activity between early‐ and late‐bolting radish genotypes (Figure 6e). Interestingly, this 647‐bp insertion harbouring specific DOF binding elements (5′‐TACTTTAT‐3′) was directly bound by RsCDF3 to delay bolting time (Figures 6f, g and 7h). Overexpression of RsCDF3 significantly inhibits bolting time in Arabidopsis (Figure 7a–c). Several CDFs (e.g. AtCDF1, AtCDF2 and AtCDF3) were participated in the flowering‐time control via modulating the expression of CO and/or FT genes in Arabidopsis (Corrales et al., 2017; Fornara et al., 2009; Goralogia et al., 2017). To our knowledge, this study firstly revealed an RsCDF3‐RsVRN1 module, which involved in CDF‐mediated bolting and flowering process via a CO/FT‐independent manner in plants.
Notably, the replacement of RsVRN1 Del‐536 allele in early‐bolting radish varieties with RsVRN1 In‐536 allele significantly represses bolting time under normal growth condition (Figure 7h), indicating that introgression of this RsVRN1 In‐536 alleles could provide an effective and flexible strategy for the development of elite late‐bolting radish cultivars. A working model for RsVRN1 In‐536 allele‐mediated bolting time difference was proposed in radish (Figure 7i). In detail, RsCDF3 directly binds to the DOF binding elements within the 647‐bp insertion of the RsVRN1In‐536 promoter, leading to significantly decreased activity of pRsVRN1 and late‐bolting phenotype. In contrast, RsCDF3 does not bind to the RsVRN1 Del‐536 promoter, leading to relatively high activity of pRsVRN1 and early‐bolting phenotype. Further functional characterization of the adaptive advantages of RsVRN1 In‐536 and RsVRN1 Del‐536 would fine‐tune the gene regulatory network of vernalization pathway in radish breeding programs.
In conclusion, a new high‐quality radish genome was generated with 448.12 Mb (94.08%) assembled into nine radish chromosomes. The assembly of centromeric regions, dynamic evolution of LTR retrotransposons and duplicate gene classification were characterized. Among the identified SNPs, small INDELs and SVs between NAU‐LB and Xin‐li‐mei radish genome, a 647‐bp insertion in the promoter region of RsVRN1 gene resulted in its low promoter activity, which was partially conferring the late‐bolting phenotype of the ‘NAU‐LB’ genotype. These results would not only facilitate comprehensive identification of genetic variants associated with key horticultural traits, but also provided rich genomic resources to establish efficient gene‐targeted strategies to improve desirable traits in radish.
Experimental procedures
Plant materials and genome sequencing
Germinated seeds of an extremely late‐bolting genotype ‘NAU‐LB’ were planted in a growth chamber with a photoperiod cycle of 14 h/25 °C light and 10 h/18 °C dark (Xu et al., 2020). Genomic DNA was extracted from young fresh leaf tissues at the four true‐leaf stage using the DNAsecure Plant Kit (Tiangen Biotech, Beijing, China). A SMRTbell DNA library with 20 kb insertion was constructed using the SMRTbell template prep kit and sequenced on the PacBio Sequel platform (Pacific Biosciences, CA). For BioNano optical mapping, the genomic DNA was labelled with Nb.BssSI and subjected to optical scanning on the BioNano Genomics Saphyr System (Renner et al., 2021). For Hi‐C sequencing, the isolated DNA from fresh young leaves was fixed, cross‐linked and biotinylated following previous report (Miao et al., 2021). The libraries of Illumina, 10 × Genomics and Hi‐C sequencing were sequenced on an Illumina HiSeq×Ten platform (Illumina, San Diego, CA).
Genome assembly and Hi‐C scaffolding
Raw contigs were de novo assembled and corrected with PacBio long reads using FALCON (ver. 0.3.0) and Arrow (ver. 2.1.0), respectively. Then, Pilon (https://github.com/nanoporetech/ont‐assembly‐polish) was used to polish PacBio‐corrected contigs with the Illumina short reads. The 10 × Genomics linked‐reads were mapped to the consensus assembly using Burrows‐Wheeler Aligner (Li and Durbin, 2009). FragScaff was used to extend contigs into initial scaffolds (Renner et al., 2021). Raw BioNano data were assembled into a consensus physical map using the IrysView package (BioNano Genomics, San Diego), and the hybrid scaffold assembly was constructed using the IrysSolve software (BioNano Genomics, San Diego). Gap filling was performed using FGAP (ver. 1.8.1) with PacBio subreads (Piro et al., 2014).
After removing low‐quality and adapter sequences, clean Hi‐C reads were aligned to the assembly using BWA (ver. 0.7.17) with default parameters (Li and Durbin, 2009). The deduplicated list of Hi‐C reads was generated using Juicer pipeline (ver. 1.5.7) (Durand et al., 2016). Draft genome scaffolds were clustered with valid interaction read pairs using the 3D de novo assembly (3D‐dna) pipeline (Dudchenko et al., 2017). The heatmap for Hi‐C interaction was generated by the 3D‐DNA visualize module and Juicebox (ver. 1.9.0) (Durand et al., 2016).
Chromosomal immunofluorescence, FISH and ChIP‐seq
Chromosomal immunofluorescence and FISH experiments were carried out following a previous report (Li et al., 2018). In brief, the amplified DNAs were labelled with nick‐translation using biotin‐16‐dUTP or digoxigenin‐11‐dUTP (Roche Diagnostics).
On the basis of hybridization, the signals of biotin‐labelled and digoxigenin‐labelled probes were identified with Alexa Fluor™ 488 streptavidin (Thermo Fisher Scientific, Waltham, MA) and rhodamine‐conjugated anti‐digoxigenin (Roche Diagnostics), respectively. The chromosomes were counterstained with 4′6‐Diamidino‐2‐phenylindole (DAPI) in a vectashield antifade solution.
ChIP and ChIP‐seq were conducted according to the previous studies by Nagaki et al. (2003) and Huang et al. (2021). The nuclei isolated from young radish leaves were digested with 0.5 U micrococcal nuclease (MNase; Sigma‐Aldrich) and used for immunoprecipitation with antibodies. The untreated chromatin was employed as input control. The library was constructed using the ChIP and input control DNA samples following the construction protocol from NEBNext Ultra™ DNA Library Prep Kit (New England BioLabs, Ipswich, MA) and sequenced on the Illumina HiSeq platform (Illumina, San Diego, CA).
Genome evaluation and RNA‐seq analysis
To assess genome assembly quality, we mapped the Illumina paired‐end reads to the assembly using BWA (ver. 0.7.17). Genome assembly completeness was evaluated using the Bench marking Universal Single‐copy Orthologs (BUSCO) (ver. 4.0.6) and Core Eukaryotic Genes Mapping Approach (CEGMA) analysis (ver. 2.5) (Parra et al., 2007; Simao et al., 2015). Construction of four cDNA libraries from leaf, root, pistil and stamen tissues was performed according to previous report (Xu et al., 2020). Clean RNA‐seq reads were aligned to the genome assembly using Bowtie2 (Langmead and Salzberg, 2012). Gene expression level was quantified as FPKM (fragments per kilobase of transcript per million mapped reads) value using Cufflinks (ver. 2.1.1) (Trapnell et al., 2012). Genes with a twofold change and adjusted P < 0.05 were considered as differentially expressed.
Repeat annotation
Transposable elements (TEs) were identified at both the DNA and protein levels using a combination of de novo and sequence similarity‐based strategies. A de novo repeat database was constructed using the RepeatModeler software (http://www.repeatmasker.org/RepeatModeler/). RepeatMasker (ver. 4.0.7) (http://www.repeatmasker.org) was used to screen TEs from the Repbase database (ver. 19.06) (http://www.girinst.org/repbase), MIPS Repeat Element Database (ver. 9.3) and the de novo repeat library. Assembly sequences were searched against the repetitive element protein database using the WU‐BLASTX package (Tarailo‐Graovac and Chen, 2009). Tandem repeats were annotated using Tandem Repeats Finder (TRF, ver. 4.09). The candidate long terminal repeat retrotransposons (LTR‐RTs) were identified using LTR_FINDER (ver. 1.0.7) (Xu and Wang, 2007). Intact LTR‐RTs were classified into Copia (PF07727) and Gypsy (PF000078) superfamilies using HMMER (http://hmmer.org) with E‐value of 1 e‐5. The insertion timing of intact LTR‐RTs was estimated using LTR_retriever (Ou et al., 2018a; Ou and Jiang, 2018b).
Gene prediction and genome annotation
For de novo gene prediction, the Genscan (hollywood.mit.edu/GENSCAN.html), Augustus (Nachtweide and Stanke, 2019), GeneID (Blanco et al., 2018), GlimmerHMM (Majoros et al., 2004) and SNAP (Korf, 2004) were used to scan the genome. For the similarity‐based approach, the assembled scaffolds were searched against nonredundant protein sequences using GeMoMa (Keilwagen et al., 2016). For RNA‐seq‐based prediction, the transcriptome sequences from four tissues were aligned to the genomes using TopHat (ver. 2.1.1) (Trapnell et al., 2012). Nonredundant gene set was generated by integrating three gene models using EVidenceModeler (EVM) (Haas et al., 2008). Gene functional annotation was achieved by performing a BLASTP search against sequences from the NCBI‐nr (https://www.ncbi.nlm.nih.gov/), Swiss‐Prot (http://www.uniprot.org/), GO (http://www.geneontology.org/) and KEGG (http://www.genome.jp/kegg/) and InterProScan (www.ebi.ac.uk/interpro/) databases with an E‐value threshold of 1 e‐5.
Phylogenetic, synteny analysis and divergence time estimation
To investigate the genome evolutionary history of R. sativus, the orthologous genes between radish and 11 other plant species including four Brassicaceae species (Arabidopsis thaliana, Arabidopsis lyrate, Brassica rapa and B. oleracea) and seven eudicots clade (Capsella rubella, Cucumis sativus, Prunus persica, Vitis vinifera, Solanum tuberosum, S. lycopersicum and Daucus carota) were identified using OrthoMCL (ver. 2.0.9) (Li et al., 2003) with default parameters. The protein sequences of 1527 single‐copy orthologous groups were aligned using MUSCLE (ver. 3.8.31) (Edgar, 2004). For each orthologous group, a maximum likelihood phylogenetic tree was constructed using RaxML (ver. 8.2.12) (Stamatakis, 2014) with 400 bootstrap replicates.
Paralogous and orthologous genes were identified using all‐against‐all BLASTP with an E‐value threshold of 1 e‐5. Based on identifying syntenic blocks using MCScanX (Wang et al., 2012), the 4DTv value of orthologous/paralogous gene pair was calculated to determine WGD events (Maere et al., 2005). The synonymous substitution (Ks) values of syntenic gene pair were calculated using the yn00 program in PAML package (Yang, 2007). Species divergence time was estimated using MCMCTree in PAML package. Speciation event dates for monocot‐eudicot and A. thaliana–B. rapa split time were used to calibrate the divergence time. Based on the phylogenetic tree topology, significant expansion and contraction of radish gene families were identified using CAFE (ver. 4.2.1) (De Bie et al., 2006).
Identification of NBS‐LRR resistance gene
A set of NBS‐LRR proteins was identified from the NAU‐LB genome using the Hidden Markov Model (HMM) of the NBS family (NB‐ACR; Pfam: PF00931). The conserved domains of RsNBS proteins were identified using Pfam 33.0 (http://pfam.xfam.org/) and SMART (http://smart.embl‐heidelberg.de/). Chromosomal location and gene structure of RsNBS genes were analysed using the MapInspect Software 9 and GSDS (http://gsds.cbi.pku.edu.cn/), respectively. MEME (http://meme‐suite.org/) was used to isolate candidate motifs from the NBS‐LRRs. The NBS‐LRRs were subdivided into CNL and TNL groups following the previous report (Tirnaz et al., 2020). The predicted proteins were trimmed at ~10 aa N terminal to the first Gly before the P‐loop motif and ~ 30 aa beyond the MHDV motif. Based on multiple sequence alignments, a phylogenetic tree of NBS‐LRR proteins was constructed using RaxML (Stamatakis, 2014).
Structural variants identification
The Nucmer program (Marcais et al., 2018) was used to align the Xin‐li‐mei genome to the NAU‐LB assembly with the parameters ‘‐‐mum ‐g 1000 ‐c 90 ‐l 40’, and the alignment block filter was performed using delta‐filter with one‐to‐one alignment mode (‐1). SNPs and InDels were extracted from one‐to‐one block using show‐snp with parameter settings ‘‐ClrH’ of the MUMmer4 toolkit. Functional effects of SNPs and InDels were annotated using the SnpEff package (Cingolani et al., 2012). Translocation and inversion events (≥1 Kb) were detected based on the nonallelic similarity blocks from the resulting alignments using MUMmer4 following the previous reports (Liu et al., 2020). The functional annotations of SVs were carried out using the ANNOVAR package (Wang et al., 2010).
cDNA library screening and yeast one‐hybrid (Y1H) assay
The 647‐bp insertion of the RsVRN1 promoter was cloned and inserted into the pHIS2 vector for yeast library screening. In brief, 5 μg of the bait plasmid pHIS2‐proRsVRN1 and 10 μg of the cDNA library plasmid were co‐transformed into the yeast strain Y187 using the Matchmarker™ Library Construction & Screening Kits (Clontech). The yeast cells were coated with synthetic define (SD) medium lacking His, Leu and Ade with an appropriate 3‐amino‐1,2,4‐triazole (3‐AT) concentration. After incubating in a 28 °C oven for 3–5 days, single colonies were picked for PCR detection. Only single‐band PCR products were sequenced. Sequencing fragments were aligned with the Arabidopsis Information Resource (TAIR) database and NAU‐LB radish genome.
For Y1H assay, the 647‐bp insertion of the RsVRN1 promoter was cloned into the pLacZi vector, while the full‐length ORF of RsCDF3 was inserted into the pJG vector. To detect positive clones, different combinations of plasmids were co‐transformed into the yeast strain EGY48. Yeast cells were grown on SD/‐Trp‐Ura selection plates for 3 days. Positive interactions were identified on SD medium containing X‐a‐gal.
Generation of Arabidopsis plants overexpressing RsCDF3
The ORF of RsCDF3 was PCR amplified and inserted into the pCAMBIA1300‐GFP vector to produce the pCAMBIA1300‐RsCDF3‐GFP recombinant expression vectors.
Agrobacterium tumefaciens strain GV3101 carrying the construct 35 S::RsCDF3 was transformed into Arabidopsis Col‐0 plants using the floral dip method. The T1 seedlings were raised on Murashige and Skoog medium with 30 mg/L hygromycin. Based on validating the positive transformants, three T3 transgenic Arabidopsis lines were further employed for the phenotypic analyses.
Transcriptional activity assays
The coding sequence of RsCDF3 was inserted into pGBKT7 and BD‐VP16 vector to evaluate transcriptional activity of RsCDF3 in yeast. The fusion plasmid constructs were then transformed into yeast strain AH109. The pGBKT7 and BD‐VP16 were used as negative and positive controls, respectively. Transfected yeast cells were grown on SD/‐Trp‐His selection plates for 3 d at 30 °C. The β‐galactosidase activity was examined by X‐gal staining. To further validate transcription activity of RsCDF3 in N. benthamiana, the coding sequence of RsCDF3 was inserted into pBD and pBD‐VP16 effector to generate effector plasmid. After introducing into Agrobacterium GV3101, the effector was co‐infiltrated into N. benthamiana leaves with a double‐reporter vector. The pBD and pBD‐VP16 were used as negative and positive controls, respectively. The primers used in transactivation assays are listed in Table S24.
Dual luciferase reporter assay
The promoter region of RsVRN1 gene was amplified from the NAU‐LB and Xin‐li‐mei genomic DNA, respectively. Both sets of fragments were ligated into pGreenII‐0800‐LUC vector to generate the ProRsVRN1‐NAU‐LB‐LUC and ProRsVRN1‐Xin‐li‐mei‐LUC construct, respectively. The constructed vector and an empty vector (control) were transformed into A. tumefaciens strain GV3101, which were further infiltrated into N. benthamiana leaves. Luciferase signalling was tested using a living fluorescence imager (Lb985, Berthold, Germany). Transient expression was determined as the ratio of firefly luciferase (LUC) to Renilla luciferase (REN) activities (Fan et al., 2020) using Dual Luciferase Reporter Assay Kit (Vazyme, Nanjing, China). Three biological replicates were prepared for each sample. The primers for vector construction are shown in Table S24.
Electrophoretic mobility shift assay (EMSA)
For EMSA assay, the CDS of RsCDF3 was cloned into the pGEX4T‐1 vector and expressed in E. coli strain Rosetta (DE3, Promega). The recombinant protein was induced in transformed E. coli using 0.6 mM isopropylthio‐b‐galactoside at OD600 = 0.6, and the cells were incubated at 28 °C for 12 h. The GST fusion protein was purified with GST‐Sefinose resin (Promega) following the manufacturer's instructions. EMSA reactions were performed using the Light Shift Chemiluminescent EMSA Kit (Thermo Fisher Scientific) according to the manufacturer's instructions. The biotin‐labelled, unlabelled and mutated DNA probes are listed in Table S24.
Conflicts of interest
The authors declare that they have no conflicts of interest.
Author contributions
L.X. and L.L. conceived and designed the project. J.D., W.Z., M.T. and L.W. conducted the data analyses. K.W. and K.X. performed the ChIP‐seq experiment. W.Z., X.Z. and Y.M. carried out phenotype investigation and gene structure variation analysis. Q.H., X.Z. and K.W. performed the dual luciferase reporter assay. L.X. wrote the manuscript. Y.C., Y.W. and L.L. reviewed and revised the manuscript. All authors read and approved the final manuscript.
Supporting information
Figure S1. Genome collinearity of NAU‐LB with other previous radish genomes. (a), (b), (c) and (d) represents the mummer plot of NAU‐LB chromosomes with WK10039, RSAskr_r1.0, Xin‐li‐mei and XYB36‐2, respectively. The chromosomes of NAU‐LB were numbered according to the WK10039, which is different with that of Xin‐li‐mei and XYB36‐2 in the Chr1 and Chr6.
Figure S2. LTR assembly index (LAI) evaluation of each chromosome for the NAU‐LB genome assembly.
Figure S3. Estimated times of insertion for the Copia and Gypsy LTR‐RTs in radish. MYA: million years ago.
Figure S4. The ks distance distribution of duplicated gene pairs in syntenic blocks within the genomes of Raphanus sativus, Arabidopsis thaliana, Brassica rapa and B. oleracea.
Figure S5. Phylogenetic relationship of NBS‐LRRs between radish and Arabidopsis.
Figure S6. The location of 110 RsNBS‐LRRs in nine radish chromosomes. The red line indicates the RsNBS‐LRRs that evolved tandem duplication events.
Figure S7. Heat map for expression profiling of 110 RsNBS‐LRRs gene in root, leaf, stamen and pistil of radish.
Figure S8. The RsNBS‐LRRs genes evolved whole genome duplication (WGD)/segmental events in radish.
Figure S9. The coding sequence alignment of RsVRN1 gene from NAU‐LB, Xin‐li‐mei and WK10039 genome.
Figure S10. The amino acid alignment of RsVRN1 gene from NAU‐LB, Xin‐li‐mei and WK10039 genome.
Figure S11. The promoter alignment of RsVRN1 gene from NAU‐LB and Xin‐li‐mei.
Figure S12. Genotyping of proRsVRN1 in the F2 population generated by crossing ‘NAU‐LB’ carrying the RsVRN1 In‐536 allele and ‘NAU‐YH’ carrying the RsVRN1 Del‐536 allele.
Table S1. Survey statistics of radish genome using the k‐mer analysis.
Table S2. Summary of the Illumina, PacBio, 10 × Genomics, BioNano and Hi‐C data for the radish genome assembly.
Table S3. Summary of BioNano molecule quality and assembly data.
Table S4. Statistics of the radish genome assembly using the BioNano data.
Table S5. Summary of the Hi‐C reads mapping data.
Table S6. Statistics of the radish genome assembly using the Hi‐C data.
Table S7. Statistics of nine pseudochromosomes constructed from the Hi‐C interaction matrices.
Table S8. Assessment of genome consistency based on the Illumina reads.
Table S9. CEGMA analysis of the radish genome completeness.
Table S10. BUSCO analysis of the radish genome completeness.
Table S11. Assessment of radish genome assembly by the EST assembled transcripts.
Table S12. Statistics of the protein‐coding genes from the NAU‐LB genome.
Table S13. Summary of functional annotation for the predicted genes.
Table S14. Summary of repetitive elements from the NAU‐LB genome assembly.
Table S15. The significantly enriched GO terms of the expanded genes identified from the NAU‐LB genome.
Table S16. The significantly enriched GO terms of the tandem duplicated genes identified from the NAU‐LB genome.
Table S17. The statistics of the identified NBS‐LRRs from the NAU‐LB genome.
Table S18. The category of SNPs identified from the NAU‐LB and Xin‐li‐mei genome.
Table S19. The statistics of nonsynonymous SNV identified from the NAU‐LB and Xin‐li‐mei genome.
Table S20. The statistics of small InDels causing changes of start/stop codons, splicing sites or frameshifts identified from the NAU‐LB and Xin‐li‐mei genome.
Table S21. The statistics of large indels (≥50 bp) identified from the NAU‐LB and Xin‐li‐mei genome.
Table S22. The statistics of large inversions identified from the NAU‐LB and Xin‐li‐mei genome.
Table S23. Detailed information of SNPs and indels identified from the bolting and flowering‐related genes between the NAU‐LB and Xin‐li‐mei genome.
Table S24. The primer pairs used in this study.
Table S25. Detailed information of putative cis‐acting elements identified from 647‐bp insertion in the promoter region of the RsVRN1 gene.
Table S26. Comparison of the NAU‐LB genome assembly and four previously reported radish genomes.
Acknowledgements
We would thank Prof. Shinhan Shiu from Michigan State University for revising the manuscript comprehensively. This work was supported by grants from the National Natural Science Foundation of China (32172579; 32272710), National Key Technology R&D Program of China (2018YFD1000800), Jiangsu Seed Industry Revitalization Project [JBGS(2021)071], the Jiangsu Agricultural S&T Innovation Fund [CX(21)2020; CX(22)3169], the earmarked fund for Jiangsu Agricultural Industry Technology System [JATS(2022)463], and the Priority Academic Program Development of Jiangsu Higher Education Institutions.
Data availability statement
Genome assembly and gene annotations of the NAU‐LB had been deposited in the Genome Warehouse (GWH) database of the National Genomics Data Center (https://bigd.big.ac.cn) under the BioProject number PRJCA011486. The original ChIP‐seq and RNA‐seq data had been deposited in the Genome Sequence Archive (GSA) database of the National Genomics Data Center under accession number CRA007987 and CRA007992, respectively.
References
- Alonge, M. , Wang, X. , Benoit, M. , Soyk, S. , Pereira, L. , Zhang, L. , Suresh, H. et al. (2020) Major impacts of widespread structural variation on gene expression and crop improvement in tomato. Cell, 182, 145–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayer, P.E. , Golicz, A.A. , Tirnaz, S. , Chan, C.K.K. , Edwards, D. and Batle, J. (2019) Variation in abundance of predicted resistance genes in the Brassica oleracea pangenome. Plant Biotechnol. J. 17, 789–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco, E. , Parra, G. and Guigó, R. (2018) Using gene id to identify genes. Curr. Protoc. Bioinform. 64, e56. [DOI] [PubMed] [Google Scholar]
- Cai, X. , Chang, L. , Zhang, T. , Chen, H. , Zhang, L. , Lin, R. , Liang, J. et al. (2021) Impacts of allopolyploidization and structural variation on intraspecific diversification in Brassica rapa . Genome Biol. 22, 166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai, X. , Lin, R.M. , Liang, J.L. , King, G.J. , Wu, J. and Wang, X.W. (2022) Transposable element insertion: a hidden major source of domesticated phenotypic variation in Brassica rapa . Plant Biotechnol. J. 20, 1298–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, Y.L. , Zhang, L.P. , Zhang, H.Y. , Chen, L.G. and Yu, D.Q. (2021) ERF1 delays flowering through direct inhibition of FLOWERING LOCUS T expression in Arabidopsis . J. Integr. Plant Biol. 63, 1712–1723. [DOI] [PubMed] [Google Scholar]
- Cho, L.H. , Yoon, J. and An, G. (2017) The control of flowering time by environmental factors. Plant J. 90, 708–719. [DOI] [PubMed] [Google Scholar]
- Cingolani, P. , Platts, A. , Wang, L.L. , Coon, M. , Nguyen, T. , Wang, L. , Land, S.J. et al. (2012) A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso‐2; iso‐3. Fly, 6, 80–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corrales, A.R. , Carrillo, L. , Lasierra, P. , Nebauer, S.G. , Dominguez‐Figueroa, J. , Renau‐Morata, B. , Pollmann, S. et al. (2017) Multifaceted role of cycling DOF factor 3 (CDF3) in the regulation of flowering time and abiotic stress responses in Arabidopsis . Plant Cell Environ. 40, 748–764. [DOI] [PubMed] [Google Scholar]
- De Bie, T. , Cristianini, N. , Demuth, J.P. and Hahn, M.W. (2006) CAFE: a computational tool for the study of gene family evolution. Bioinformatics, 22, 1269–1271. [DOI] [PubMed] [Google Scholar]
- Dudchenko, O. , Batra, S.S. , Omer, A.D. , Nyquist, S.K. , Hoeger, M. , Durand, N.C. , Shamim, M.S. et al. (2017) De novo assembly of the Aedes aegypti genome using Hi‐C yields chromosome‐length scaffolds. Science, 356, 92–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand, N.C. , Robinson, J.T. , Shamin, M.S. , Machol, I. , Mesirov, J.P. , Lander, E.S. and Aiden, E.L. (2016) Juicer provides a one‐click system for analyzing loop‐resolution Hi‐C experiments. Cell Syst. 3, 95–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar, R.C. (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan, L.X. , Wang, Y. , Xu, L. , Tang, M.J. , Zhang, X.L. , Ying, J.L. et al. (2020) A genome‐wide association study uncovers a critical role of the RsPAP2 gene in red‐skinned Raphanus sativus L. Hortic. Res. 7, 164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fornara, F. , Panigrahi, K.C. , Gissot, L. , Sauerbrunn, N. , Ruhl, M. , Jarillo, J.A. and Coupland, G. (2009) Arabidopsis DOF transcription factors act redundantly to reduce CONSTANS expression and are essential for a photoperiodic flowering response. Dev. Cell, 17, 75–86. [DOI] [PubMed] [Google Scholar]
- Fuentes, R.R. , Chebotarov, D. , Duitama, J. , Smith, S. , De la Hoz, J.F. , Mohiyuddin, M. , Wing, R.A. et al. (2019) Structural variants in 3000 rice genomes. Genome Res. 29, 870–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghurye, J. , Az, R. , Walenz, B.P. , Schmitt, A. , Selvaraj, S. , Pop, M. , Phillippy, A.M. et al. (2019) Integrating Hi‐C links with assembly graphs for chromosome‐scale assembly. PLoS Comput. Biol. 15, e1007273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golicz, A.A. , Bayer, P.E. , Barker, G.C. , Edger, P.P. , Kim, H. , Martinez, P.A. , Chan, C.K.K. et al. (2016) The pangenome of an agronomically important crop plant Brassica oleracea . Nat. Commun. 7, 13390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong, Z. , Wu, Y. , Koblizkova, A. , Torres, G.A. , Wang, K. , Iovene, M. , Neumann, P. et al. (2012) Repeatless and repeat‐based centromeres in potato: implications for centromere evolution. Plant Cell, 24, 3559–3574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goralogia, G.S. , Liu, T.K. , Zhao, L. , Panipinto, P.M. , Groover, E.D. , Bains, Y.S. and Imaizumi, T. (2017) CYCLING DOF FACTOR 1 represses transcription through the TOPLESS co‐repressor to control photoperiodic flowering in Arabidopsis . Plant J. 92, 244–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan, J.T. , Xu, Y.G. , Yu, Y. , Fu, J. , Ren, F. , Guo, J.Y. , Zhao, J.B. et al. (2021) Genome structure variation analyses of peach reveal population dynamics and a 1.67 Mb causal inversion for fruit shape. Genome Biol. 22, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, J. , Cao, K. , Deng, C. , Li, Y. , Zhu, G. , Fang, W. , Chen, C. et al. (2020) An integrated peach genome structural variation map uncovers genes associated with fruit traits. Genome Biol. 21, 258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas, B.J. , Salzbery, S.L. , Zhu, W. , Pertea, M. , Allen, J.E. , Orvis, J. , White, O. et al. (2008) Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biol. 9, R7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu, T.T. , Pattyn, P. , Bakker, E.G. , Cao, J. , Chen, J.F. , Clark, R.M. , Fahlgren, N. et al. (2011) The Arabidopsis lyrata genome sequence and the basis of rapid genome size change. Nat. Genet. 43, 476–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu, Y. , Chen, J.D. , Fang, L. , Zhang, Z.Y. , Ma, W. , Niu, Y.C. , Ju, L.Z. et al. (2019) Gossypium barbadense and Gossypium hirsutum genomes provide insights into the origin and evolution of allotetraploid cotton. Nat. Genet. 51, 739–748. [DOI] [PubMed] [Google Scholar]
- Huang, Y. , Ding, W.J. , Zhang, M.Q. , Han, J.L. , Jing, Y.F. , Yao, W. , Hasterok, R. et al. (2021) The formation and evolution of centromeric satellite repeats in Saccharum species. Plant J. 106, 616–629. [DOI] [PubMed] [Google Scholar]
- Imaizumi, T. , Schultz, T.F. , Harmon, F.G. , Ho, L.A. and Kay, S.A. (2005) FKF1 F‐box protein mediates cyclic degradation of a repressor of CONSTANS in Arabidopsis. Science, 309, 293–297. [DOI] [PubMed] [Google Scholar]
- Jeong, Y.M. , Kim, N. , Ahn, B.O. , Oh, M. , Chun, W.H. , Chun, H. , Jeong, S. et al. (2016) Elucidating the triplicated ancestral genome structure of radish based on chromosome‐level comparison with the Brassica genomes. Theor. Appl. Genet. 129, 1357–1372. [DOI] [PubMed] [Google Scholar]
- Kang, L. , Qian, L.W. , Zheng, M. , Chen, L. , Chen, H. , Yang, L. , You, L. et al. (2021) Genomic insights into the origin, domestication and diversification of Brassica juncea . Nat. Genet. 53, 1392–1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keilwagen, J. , Wenk, M. , Erickson, J.L. , Schattat, M.H. , Grau, J. and Hurtung, F. (2016) Using intron position conservation for homology‐based gene prediction. Nucleic Acids Res. 44, e89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitashiba, H. , Li, F. , Hirakawa, H. , Kawanaba, T. , Zou, Z. , Hasegawa, Y. , Tonosaki, K. et al. (2014) Draft sequences of the radish (Raphanus sativus L.) genome. DNA Res. 21, 481–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korf, I. (2004) Gene finding in novel genomes. BMC Bioinform. 5, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyung, J. , Jeon, M. , Jeong, G. , Shin, Y. , Seo, E. and Yu, J. (2022) The two clock proteins CCA1 and LHY activate VIN3 transcription during vernalization through the vernalization‐responsive cis‐element. Plant Cell, 34, 1020–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead, B. and Salzberg, S.L. (2012) Fast gapped‐read alignment with Bowtie 2. Nat. Methods, 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy, Y.Y. , Mesnage, S. , Mylne, J.S. , Gendall, A.R. and Dean, C. (2002) Multiple roles of Arabidopsis VRN1 in vernalization and flowering time control. Science, 297, 243–246. [DOI] [PubMed] [Google Scholar]
- Li, H. and Durbin, R. (2009) Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinformatics, 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, L. , Stoeckert, C.J. and Roos, D.S. (2003) OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Y. , Zuo, S. , Zhang, Z. , Li, Z. , Han, J. , Chu, Z. , Hasterok, R. et al. (2018) Centromeric DNA characterization in the model grass Brachypodium distachyon provides insights on the evolution of the genus. Plant J. 93, 1088–1101. [DOI] [PubMed] [Google Scholar]
- Li, H.B. , Wang, S.H. , Chai, S. , Yang, Z. , Zhang, Q. , Xin, H. , Xu, Y. et al. (2022) Graph‐based pan‐genome reveals structural and sequence variations related to agronomic traits and domestication in cucumber. Nat. Commun. 13, 682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, S. , Liu, Y. , Yang, X. , Tong, C. , Edwards, D. , Parkin, I.A. et al. (2014) The Brassica oleracea genome reveals the asymmetrical evolution of polyploid genomes. Nat. Commun. 5, 3930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, Y.C. , Du, H.L. , Li, P.C. , Shen, Y. , Peng, H. , Liu, S. , Zhou, G. et al. (2020) Pan‐Genome of Wild and Cultivated Soybeans. Cell, 182, 1–15. [DOI] [PubMed] [Google Scholar]
- Lu, K. , Wei, L. , Li, X. , Wang, Y. , Wu, J. , Liu, M. , Zhang, C. et al. (2019) Whole‐genome resequencing reveals Brassica napus origin and genetic loci involved in its improvement. Nat. Commun. 10, 1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, J. , Sun, J.J. , Jiang, A.Q. , Bai, M.J. , Fan, C.G. , Liu, J.Y. , Ning, G.G. et al. (2020) Alternate expression of CONSTANS‐LIKE 4 in short days and CONSTANS in long days facilitates day‐neutral response in Rosa chinensis . J. Exp. Bot. 71, 4057–4068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo, X.B. , Xu, L. , Wang, Y. , Dong, J.H. , Chen, Y.L. , Tang, M.J. , Fan, L.X. et al. (2020) An ultra‐high density genetic map provides insights into genome synteny, recombination landscape and taproot skin color in radish (Raphanus sativus L.). Plant Biotech. J. 18, 274–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maere, S. , De Bodt, S. , Raes, J. , Casneuf, T. , Van Montagu, M. , Kuiper, M. and Van de Peer, Y. (2005) Modeling gene and genome duplications in eukaryotes. Proc. Natl. Acad. Sci. U. S. A. 102, 5454–5459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maheshwari, S. , Ishii, T. , Brown, C.T. , Houben, A. and Comai, L. (2017) Centromere location in Arabidopsis is unaltered by extreme divergence in CENH3 protein sequence. Genome Res. 27, 471–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majoros, W.H. , Pertea, M. and Salzberg, S.L. (2004) TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene‐finders. Bioinformatics, 20, 2878–2879. [DOI] [PubMed] [Google Scholar]
- Marcais, G. , Delcher, A.L. , Phillippy, A.M. , Coston, R. , Salzberg, S.L. and Zimin, A. (2018) MUMmer4: A fast and versatile genome alignment system. PLoS Comput. Biol. 14, e1005944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao, J.H. , Feng, Q. , Li, Y. , Zhao, Q. , Zhou, C. , Lu, H. , Fan, D. et al. (2021) Chromosome‐scale assembly and analysis of biomass crop Miscanthus lutarioriparius genome. Nat. Commun. 12, 2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsui, Y. , Shimomura, M. , Komatsu, K. , Namiki, N. , Shibata‐Hatta, M. , Imai, M. , Katayose, Y. et al. (2015) The radish genome and comprehensive gene expression profile of tuberous root formation and development. Sci. Rep. 5, 10835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moghe, G.D. , Hufnagel, D.E. , Tang, H. , Xiao, Y. , Dworkin, L. , Town, C.D. , Conner, J.K. et al. (2014) Consequences of whole‐genome triplication as revealed by comparative genomic analyses of the wild radish Raphanus raphanistrum and three other Brassicaceae species. Plant Cell, 26, 1925–1937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nachtweide, S. and Stanke, M. (2019) Multi‐genome annotation with AUGUSTUS. Methods Mol. Biol. 1962, 139–160. [DOI] [PubMed] [Google Scholar]
- Nagaki, K. , Talbert, P.B. , Zhong, C.X. , Dawe, R.K. , Henikoff, S. and Jiang, J. (2003) Chromatin immunoprecipitation reveals that the 180‐bp satellite repeat is the key functional DNA element of Arabidopsis thaliana centromeres. Genetics, 163, 1221–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naish, M. , Alonge, M. , Wlodzimierz, P. , Tock, A.J. , Abramson, B.W. , Schmücker, A. , Mandáková, T. et al. (2021) The genetic and epigenetic landscape of the Arabidopsis centromeres. Science, 374, eabi7489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunn, A. , Rodríguez‐Arévalo, I. , Tandukar, Z. , Frels, K. , Contreras‐Garrido, A. , Carbonell‐Bejerano, P. , Zhang, P.P. et al. (2022) Chromosome‐level Thlaspi arvense genome provides new tools for translational research and for a newly domesticated cash cover crop of the cooler climates. Plant Biotech. J. 20, 944–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou, S. and Jiang, N. (2018b) LTR_retriever: a highly accurate and sensitive program for identification of long terminal repeat retrotransposons. Plant Physiol. 176, 1410–1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou, S. , Chen, J. and Jiang, N. (2018a) Assessing genome assembly quality using the LTR Assembly Index (LAI). Nucleic Acids Res. 46, e126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parra, G. , Bradnam, K. and Korf, I. (2007) CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics, 23, 1061–1067. [DOI] [PubMed] [Google Scholar]
- Piro, V.C. , Faoro, H. , Weiss, V. , Steffens, M.B. , Pedrosa, F.O. , Souza, E.M. and Raittz, R.T. (2014) FGAP: an automated gap closing tool. BMC. Res. Notes 7, 371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renau‐Morata, B. , Carrillo, L. , Dominguez‐Figueroa, J. , Vicente‐Carbajosa, J. , Molina, R.V. , Nebauer, S.G. and Medina, J. (2020) CDF transcription factors: plant regulators to deal with extreme environmental conditions. J. Exp. Bot. 71, 3803–3815. [DOI] [PubMed] [Google Scholar]
- Renner, S.S. , Wu, S. , Pérez‐Escobar, O.A. , Silber, M.V. , Fei, Z. and Chomicki, G. (2021) A chromosome‐level genome of a Kordofan melon illuminates the origin of domesticated watermelons. Proc. Natl. Acad. Sci. U. S. A. 118, e2101486118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider, K.L. , Xie, Z. , Wolfgruber, T.K. and Presting, G.G. (2016) Inbreeding drives maize centromere evolution. Proc. Natl. Acad. Sci. U. S. A. 113, 987–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang, J. , Tian, J. , Cheng, H. , Yan, Q. , Li, L. , Jamal, A. , Xu, Z. et al. (2020) The chromosome‐level wintersweet (Chimonanthus praecox) genome provides insights into floral scent biosynthesis and flowering in winter. Genome Biol. 21, 200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma, N. , Geuten, K. , Giri, B.S. and Varm, A. (2020) The molecular mechanism of vernalization in Arabidopsis and cereals: role of Flowering Locus C and its homologs. Physiol. Plant. 170, 373–383. [DOI] [PubMed] [Google Scholar]
- Shirasawa, K. , Hirakawa, H. , Fukino, N. , Kitashiba, H. and Isobe, S. (2020) Genome sequence and analysis of a Japanese radish (Raphanus sativus) cultivar named ‘Sakurajima Daikon’ possessing giant root. DNA Res. 27, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simao, F.A. , Waterhouse, R.M. , Ioannidis, P. , Kriventseva, E.V. and Zdobnov, E.M. (2015) BUSCO: assessing genome assembly and annotation completeness with single‐copy orthologs. Bioinformatics, 31, 3210–3212. [DOI] [PubMed] [Google Scholar]
- Song, X.M. , Wei, Y.P. , Xiao, D. , Gong, K. , Sun, P.C. , Ren, Y.M. , Yuan, J.Q. et al. (2021) Brassica carinata genome characterization clarifies U's triangle model of evolution and polyploidy in Brassica . Plant Physiol. 186, 388–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srikanth, A. and Schmid, M. (2011) Regulation of flowering time: all roads lead to Rome. Cellular Mol. Life Sci. 68, 2013–2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis, A. (2014) RAxML version 8: a tool for phylogenetic analysis and post‐analysis of large phylogenies. Bioinformatics, 30, 1312–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarailo‐Graovac, M. and Chen, N. (2009) Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinformatics, 25, 4.10.1–4.10.14. [DOI] [PubMed] [Google Scholar]
- Tirnaz, S. , Bayer, P.E. , Inturrisi, F. , Zhang, F. , Yang, H. , Aria Dolatabadian, A. , Neik, T.X. et al. (2020) Resistance gene analogs in the Brassicaceae: identification, characterization, distribution, and evolution. Plant Physiol. 184, 909–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell, C. , Roberts, A. , Goff, L. , Pertea, G. , Kim, D. , Kelley, D.R. , Pimentel, H. et al. (2012) Differential gene and transcript expression analysis of RNA‐seq experiments with TopHat and Cufflinks. Nat. Protoc. 7, 562–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van de Peer, Y. , Maere, S. and Meyer, A. (2009) The evolutionary significance of ancient genome duplications. Nat. Rev. Genet. 10, 725–732. [DOI] [PubMed] [Google Scholar]
- Wang, K. , Li, M. and Hakonarson, H. (2010) ANNOVAR: functional annotation of genetic variants from high‐throughput sequencing data. Nucl. Acids Res. 38, e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Y. , Tang, H. , Debarry, J.D. , Tan, X. , Li, J. , Wang, X. , Lee, T. et al. (2012) MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40, e49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, X. , Xu, Y. , Zhang, S. , Cao, L. , Huang, Y. , Cheng, J. , Wu, G. et al. (2017) Genomic analyses of primitive, wild and cultivated citrus provide insights into asexual reproduction. Nat. Genet. 49, 765–772. [DOI] [PubMed] [Google Scholar]
- Wang, X. , Gao, L. , Jiao, C. , Stravoravdis, S. , Hosmani, P.S. , Saha, S. , Zhang, J. et al. (2020) Genome of Solanum pimpinellifolium provides Insights into structural variants during tomato breeding. Nat. Commun. 11, 5817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia, E. , Tong, W. , Hou, Y. , An, Y. , Chen, L. , Wu, Q. , Liu, Y. et al. (2020) The reference genome of tea plant and resequencing of 81 diverse accessions provide insights into its genome evolution and adaptation. Mol. Plant, 13, 1013–1026. [DOI] [PubMed] [Google Scholar]
- Xu, Z. and Wang, H. (2007) LTR_FINDER: an efficient tool for the prediction of full‐length LTR retrotransposons. Nucleic Acids Res. 35, W265–W268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, L. , Zhang, F. , Tang, M.J. , Wang, Y. , Dong, J.H. , Ying, J.L. , Chen, Y.L. et al. (2020) Melatonin confers cadmium tolerance by modulating critical heavy metal chelators and transporters in radish plants. J. Pineal Res. 69, e12659. [DOI] [PubMed] [Google Scholar]
- Xu, D. , Li, X. , Wu, X. , Meng, L. , Zou, Z. , Bao, E. , Bian, Z. et al. (2021) Tomato SlCDF3 delays flowering time by regulating different FT‐like genes under long‐day and short‐day conditions. Front. Plant Sci. 12, 650068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Z. (2007) PAML4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591. [DOI] [PubMed] [Google Scholar]
- Yang, N. , Liu, J. , Gao, Q. , Gui, S. , Chen, L. , Yang, L. , Huang, J. et al. (2019) Genome assembly of a tropical maize inbred line provides insights into structural variation and crop improvement. Nat. Genet. 51, 1052–1059. [DOI] [PubMed] [Google Scholar]
- Zhang, X. , Yue, Z. , Mei, S. , Qiu, Y. , Yang, X. , Chen, X. , Cheng, F. et al. (2015) A de novo genome of a Chinese radish cultivar. Hortic. Plant J. 1, 155–164. [Google Scholar]
- Zhang, T. , Qiao, Q. , Novikova, Y.P. , Wang, Q. , Yue, J. , Guan, Y. , Ming, S. et al. (2019) Genome of Crucihimalaya himalaica, a close relative of Arabidopsis, shows ecological adaptation to high altitude. Proc. Natl. Acad. Sci. U. S. A. 116, 7137–7146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, X. , Liu, T. , Wang, J. , Wang, P. , Qiu, Y. , Zhao, W. , Pang, S. et al. (2021) Pan‐genome of Raphanus highlights genetic variation and introgression among domesticated, wild, and weedy radishes. Mol. Plant, 14, 1–24. [DOI] [PubMed] [Google Scholar]
- Zhang, X.N. , Lin, S.N. , Peng, D. , Wu, Q.S. , Liao, X.Z. , Xiang, K.L. et al. (2022) Integrated multi‐omic data and analyses reveal the pathways underlying key ornamental traits incarnation flowers. Plant Biotechnol. J. 20, 1182–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Genome collinearity of NAU‐LB with other previous radish genomes. (a), (b), (c) and (d) represents the mummer plot of NAU‐LB chromosomes with WK10039, RSAskr_r1.0, Xin‐li‐mei and XYB36‐2, respectively. The chromosomes of NAU‐LB were numbered according to the WK10039, which is different with that of Xin‐li‐mei and XYB36‐2 in the Chr1 and Chr6.
Figure S2. LTR assembly index (LAI) evaluation of each chromosome for the NAU‐LB genome assembly.
Figure S3. Estimated times of insertion for the Copia and Gypsy LTR‐RTs in radish. MYA: million years ago.
Figure S4. The ks distance distribution of duplicated gene pairs in syntenic blocks within the genomes of Raphanus sativus, Arabidopsis thaliana, Brassica rapa and B. oleracea.
Figure S5. Phylogenetic relationship of NBS‐LRRs between radish and Arabidopsis.
Figure S6. The location of 110 RsNBS‐LRRs in nine radish chromosomes. The red line indicates the RsNBS‐LRRs that evolved tandem duplication events.
Figure S7. Heat map for expression profiling of 110 RsNBS‐LRRs gene in root, leaf, stamen and pistil of radish.
Figure S8. The RsNBS‐LRRs genes evolved whole genome duplication (WGD)/segmental events in radish.
Figure S9. The coding sequence alignment of RsVRN1 gene from NAU‐LB, Xin‐li‐mei and WK10039 genome.
Figure S10. The amino acid alignment of RsVRN1 gene from NAU‐LB, Xin‐li‐mei and WK10039 genome.
Figure S11. The promoter alignment of RsVRN1 gene from NAU‐LB and Xin‐li‐mei.
Figure S12. Genotyping of proRsVRN1 in the F2 population generated by crossing ‘NAU‐LB’ carrying the RsVRN1 In‐536 allele and ‘NAU‐YH’ carrying the RsVRN1 Del‐536 allele.
Table S1. Survey statistics of radish genome using the k‐mer analysis.
Table S2. Summary of the Illumina, PacBio, 10 × Genomics, BioNano and Hi‐C data for the radish genome assembly.
Table S3. Summary of BioNano molecule quality and assembly data.
Table S4. Statistics of the radish genome assembly using the BioNano data.
Table S5. Summary of the Hi‐C reads mapping data.
Table S6. Statistics of the radish genome assembly using the Hi‐C data.
Table S7. Statistics of nine pseudochromosomes constructed from the Hi‐C interaction matrices.
Table S8. Assessment of genome consistency based on the Illumina reads.
Table S9. CEGMA analysis of the radish genome completeness.
Table S10. BUSCO analysis of the radish genome completeness.
Table S11. Assessment of radish genome assembly by the EST assembled transcripts.
Table S12. Statistics of the protein‐coding genes from the NAU‐LB genome.
Table S13. Summary of functional annotation for the predicted genes.
Table S14. Summary of repetitive elements from the NAU‐LB genome assembly.
Table S15. The significantly enriched GO terms of the expanded genes identified from the NAU‐LB genome.
Table S16. The significantly enriched GO terms of the tandem duplicated genes identified from the NAU‐LB genome.
Table S17. The statistics of the identified NBS‐LRRs from the NAU‐LB genome.
Table S18. The category of SNPs identified from the NAU‐LB and Xin‐li‐mei genome.
Table S19. The statistics of nonsynonymous SNV identified from the NAU‐LB and Xin‐li‐mei genome.
Table S20. The statistics of small InDels causing changes of start/stop codons, splicing sites or frameshifts identified from the NAU‐LB and Xin‐li‐mei genome.
Table S21. The statistics of large indels (≥50 bp) identified from the NAU‐LB and Xin‐li‐mei genome.
Table S22. The statistics of large inversions identified from the NAU‐LB and Xin‐li‐mei genome.
Table S23. Detailed information of SNPs and indels identified from the bolting and flowering‐related genes between the NAU‐LB and Xin‐li‐mei genome.
Table S24. The primer pairs used in this study.
Table S25. Detailed information of putative cis‐acting elements identified from 647‐bp insertion in the promoter region of the RsVRN1 gene.
Table S26. Comparison of the NAU‐LB genome assembly and four previously reported radish genomes.
Data Availability Statement
Genome assembly and gene annotations of the NAU‐LB had been deposited in the Genome Warehouse (GWH) database of the National Genomics Data Center (https://bigd.big.ac.cn) under the BioProject number PRJCA011486. The original ChIP‐seq and RNA‐seq data had been deposited in the Genome Sequence Archive (GSA) database of the National Genomics Data Center under accession number CRA007987 and CRA007992, respectively.