

Summary
Brassica rapa comprises many important cultivated vegetables and oil crops. However, Chiifu v3.0, the current B. rapa reference genome, still contains hundreds of gaps. Here, we presented a near‐complete genome assembly of B. rapa Chiifu v4.0, which was 424.59 Mb with only two gaps, using Oxford Nanopore Technology (ONT) ultralong‐read sequencing and Hi‐C technologies. The new assembly contains 12 contigs, with a contig N50 of 38.26 Mb. Eight of the ten chromosomes were entirely reconstructed in a single contig from telomere to telomere. We found that the centromeres were mainly invaded by ALE and CRM long terminal repeats (LTRs). Moreover, there is a high divergence of centromere length and sequence among B. rapa genomes. We further found that centromeres are enriched for Copia invaded at 0.14 MYA on average, while pericentromeres are enriched for Gypsy LTRs invaded at 0.51 MYA on average. These results indicated the different invasion mechanisms of LTRs between the two structures. In addition, a novel repetitive sequence PCR630 was identified in the pericentromeres of B. rapa. Overall, the near‐complete genome assembly, B. rapa Chiifu v4.0, offers valuable tools for genomic and genetic studies of Brassica species and provides new insights into the evolution of centromeres.
Keywords: Brassica rapa, near‐complete genome assembly, LTRs, centromeres, evolution
Introduction
Decoding complete genome sequence information is vital for understanding genome structure and further facilitating the genetic improvement of critical agronomic traits. Recent advances in long‐read sequencing technologies, such as Pacific Biosciences (PacBio) and Oxford Nanopore Technology (ONT), have led to a paradigm shift in our ability to obtain chromosome sequences from telomere to telomere. Recently, the Telomere‐to‐Telomere (T2T) consortium proposed a complete sequence of a human genome using PacBio HiFi and ONT ultralong‐read sequencing (Nurk et al., 2022). In plants, telomere‐to‐telomere and gapless genomes were presented using PacBio HiFi or ONT reads for Arabidopsis thaliana (Hou et al., 2022; Naish et al., 2021; Wang et al., 2021), rice (Li et al., 2021b; Song et al., 2021; Zhang et al., 2022b), and watermelon (Deng et al., 2022).
The Brassica genus encompasses many vegetables and oil crops. Six Brassica species, comprising three diploid species, Brassica rapa (A genome), Brassica nigra (B genome), and Brassica oleracea (C genome), as well as the three amphidiploid species, Brassica juncea (A and B genomes), Brassica napus (A and C genomes), and Brassica carinata (B and C genomes), form the famous “triangle of U” (Nagaharu, 1935). Brassica species not only shared the whole genome duplication event at ~13–17 million years ago (MYA) with their close relative A. thaliana but also underwent the Brassica‐specific whole‐genome triplication event at ~5–9 MYA (Wang et al., 2011b). B. rapa formed many different subspecies with highly diverse morphotypes, such as heading Chinese cabbage, non‐heading pak choi, enlarged turnip tuber and oil seed crop yellow sarson (Cheng et al., 2016).
Using Chiifu‐401‐42 (Chinese cabbage), we achieved the first reference genome, Chiifu v1.5, among the Brassica species (Wang et al., 2011b). With the development of sequencing technologies, two updated versions of genome assemblies (Chiifu v2.5 and v3.0) and one updated version of genome annotation (Chiifu v3.5) were generated for B. rapa (Cai et al., 2017; Zhang et al., 2018, 2022c). Among these versions, Chiifu v1.5 and v2.5 were assembled with Illumina short reads (Cai et al., 2017; Wang et al., 2011b). Chiifu v3.0 was generated using a combination of PacBio, optical maps and chromosome conformation capture (Hi‐C) technologies (Zhang et al., 2018). Chiifu v3.5 is the updated annotation of Chiifu v3.0 using full‐length PacBio RNA sequencing technology (Zhang et al., 2022c). Recently, other B. rapa morphotypes have been sequenced using PacBio or ONT technologies, such as Chinese cabbage (Sun et al., 2022), pak choi (Li et al., 2020, 2021a,c; Xu et al., 2022), turnip (Park et al., 2021; Yang et al., 2022) and yellow sarson (Istace et al., 2021). In addition, a pan‐genome of B. rapa was released, including 16 genomes assembled using Illumina and PacBio reads (Cai et al., 2021). These B. rapa genome assemblies essentially promoted comparative genomics and genetic breeding studies of Brassica species. However, missing sequences and hundreds of gaps remain in these released genomes.
Centromeres are constricted regions of chromosomes responsible for attaching chromosomes to spindle microtubules during cell division (Comai et al., 2017). Centromeres and flanking pericentromeres comprise repetitive DNA sequences, such as long terminal repeats (LTRs) and satellite DNA (Talbert and Henikoff, 2022). In addition, centromeres contain many active genes (Nagaki et al., 2004). The completeness of centromeres in model plants Arabidopsis and rice has offered an intriguing understanding of their structure and function, including the enrichment of satellite DNA (Naish et al., 2021) and high methylation levels (Song et al., 2021). Recent studies have also shown that centromeres are highly variable in terms of organization and sequences, even among very closely related species, reflecting the rapid evolution of centromeres (Talbert and Henikoff, 2022). It was reported that ATHILA LTRs invade Arabidopsis centromeres CEN4 and CEN5 (Naish et al., 2021), and young ALE LTRs are predominantly amplified in the centromeres of B. nigra (Perumal et al., 2020). The centromere‐specific repeats (Cent‐SRs), including CentBr1, CentBr2, CRB and TR805 (Koo et al., 2011; Lim et al., 2005, 2007), were widely used to determine the centromere boundary in B. rapa (Li et al., 2021a,b,c; Sun et al., 2022; Yang et al., 2022; Zhang et al., 2018), B. nigra (CRB, Perumal et al., 2020), B. oleracea (Cai et al., 2020; Guo et al., 2021), B. juncea (Kang et al., 2021) and B. napus (Rousseau‐Gueutin et al., 2020). Based on bacterial artificial chromosome clones, Lim et al. (2007) characterized the pericentromere‐specific repeats (Peri‐SRs) of B. rapa, including PCRBr and TR238. Due to the poor assembly of the repetitive regions in the previously published Brassica reference genomes, a global view of the size, structure and evolution of centromeres and pericentromeres remains elusive.
In this study, we presented a near‐complete genome assembly of B. rapa using a combination of ONT ultralong‐read sequencing and Hi‐C technologies. Our new assembly, B. rapa Chiifu v4.0, achieves the highest continuity and completeness among the published B. rapa genomes, which provides the opportunity for global analysis of centromeres and pericentromeres. Centromeres were mainly invaded by ALE and CRM LTRs and showed high divergence among different B. rapa genomes. Moreover, the LTRs in pericentromeres are much older than those in centromeres of B. rapa.
Results
A near‐complete B. rapa genome assembly
To achieve a high‐quality B. rapa genome assembly, Chinese cabbage (Chiifu‐401‐42) was sequenced using ONT technologies. In total, 90.24 Gb (~180 × coverage) of ONT reads were generated from the Promethion platform. We assembled ONT long reads using NextDenovo (v2.5, https://github.com/Nextomics/NextDenovo) and polished the resulting contigs with corrected ONT reads and Illumina reads. We filled the gaps with corrected ONT long reads and generated 12 contigs with a contig N50 of 38.26 Mb (Table 1). After scaffolding using our previous Hi‐C data (Zhang et al., 2018), we anchored all contigs onto ten chromosomes. Our final genome assembly, termed B. rapa Chiifu v4.0, had 424.59 Mb sequences with only two gaps on chromosomes A05 and A08, while the other eight chromosomes were reconstructed in a single contig from telomere to telomere (Table 1).
Table 1.
Summary of Brassica rapa genome assemblies
| Item | Chinese cabbage | Chinese cabbage | Chinese cabbage | Pak choi | Pak choi | Pak choi | Turnip | Yellow sarson |
|---|---|---|---|---|---|---|---|---|
| Chiifu v4.0 (This study) | Chiifu v3.0 (Zhang et al., 2018) | assembly “A03” (Sun et al., 2022) | PC‐fu (Xu et al., 2022) | NHCC001 (Li et al., 2020) | ZYCX (Li et al., 2021c) | ECD04 (Yang et al., 2022) | Z1 v2 (Istace et al., 2021) | |
| Estimated genome size (Mb) | 455 | 455 | 455 | 478 | 478 | 478 | 518 | 529 |
| Assembly size (Mb) | 424.59 | 353.14 | 403.20 | 411.40 | 405.33 | 370.42 | 350.34 | 443.95 |
| Contig number | 12 | 1498 | 1222 | 2288 | 602 | 1985 | 1275 | 299 |
| Contig N50 (kb) | 38 257 | 1446 | 4290 | 4700 | 2830 | 2820 | 1520 | 10 256 |
| Gap‐free chromosome number | 8 | None | None | None | None | None | None | None |
| Gaps number | 2 | 407 | 1160 | 986 | 291 | 993 | 1203 | 85 |
| Gene models | 47 531 | 46 250 | 47 779 | 52 511 | 48 158 | 45 363 | 48 094 | 56 073 |
| GC Content (%) | 37.59 | 36.83 | 36.83 | 37.68 | 37.13 | 37.12 | 36.78 | 37.20 |
| TE proportion (%) | 53.78 | 45.84 | 50.99 | 63.30 | 46.15 | 39.80 | 42.71 | 52.97 |
| Completeness (% BUSCO) | 99.40 | 97.70 | 98.60 | 99.20 | 99.07 | 98.10 | 97.50 | 96.30 |
| LTR assembly index score | 15.05 | 9.69 | 7.61 | 3.99 | 11.19 | 8.11 | 12.19 | 5.64 |
The accuracy and completeness of Chiifu v4.0 were validated using multiple methods. First, the Hi‐C heatmap shows high consistency across all chromosomes, demonstrating the correct ordering and orientation of contigs in the new assembly (Figure S1). Second, the new assembly has high collinearity with Chiifu v3.0 (Figure 1). Third, genome accuracy was demonstrated by the high mapping rates of two raw sequences on the new assembly, including 99.73% (1 657 704/1 662 217) of ONT reads and 100% (1083/1083) of BAC sequences mapped on the new assembly. The sequence error after correction was 0.46%, estimated by Qualimap (v.2.2.1; Okonechnikov et al., 2016). Finally, for gene content assessment, our assembly captured 99.40% of the BUSCO 1614 reference gene set (Simao et al., 2015; Table 1). In addition, misassembled regions (371 kb) of Chiifu v3.0 were corrected in this new assembly, which were further validated by the Hi‐C heatmap and ONT reads (Figure S2).
Figure 1.
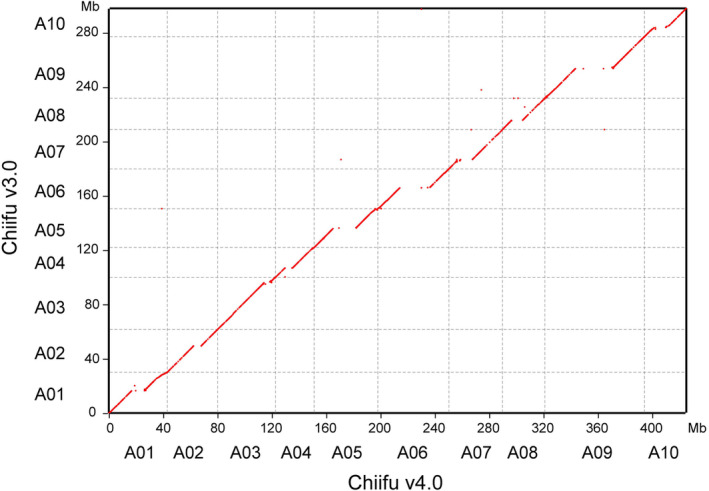
Chromosome collinearity between Brassica rapa Chiifu v4.0 and v3.0. The collinear regions between Chiifu v4.0 and v3.0 are shown in red dots. Chromosomes of Chiifu v4.0 are shown on the x‐axis, and chromosomes of Chiifu v3.0 are shown on the y‐axis.
We employed MAKER‐P (Campbell et al., 2014) to annotate the new assembly with the same evidence used to annotate Chiifu v3.0 (Zhang et al., 2018). To maintain consistency across different Chiifu versions, 47 233 protein‐coding genes (99.97%) were lifted from Chiifu v3.5 (47 249; Zhang et al., 2022c). Combining 298 gene models annotated with MAKER‐P in the newly assembled regions, the final annotation of the new assembly contained 47 531 gene models. We used EDTA (Ou et al., 2019) to annotate the repetitive sequences in the new assembly. In total, 393 202 transposable elements (TEs) were identified in the new assembly, accounting for ~53.78% (228.35 Mb/424.59 Mb) of Chiifu v4.0, approximately 10% greater than that of Chiifu v3.0 (Table 1).
Compared with Chiifu v3.0, we added ~71.45 Mb of novel sequences in the near‐complete assembly, almost all of which (98.64%, 70.48 Mb/71.45 Mb) were in the centromeres and pericentromeres (Figure S3). Moreover, all two gaps of Chiifu v4.0 were in the centromeric regions (Figure S4). We further compared Chiifu v4.0 with the other B. rapa genome assemblies based on long‐read sequencing, including a Chinese cabbage (assembly “A03”; Sun et al., 2022), three pak choi (PC‐fu, Xu et al., 2022; NHCC001, Li et al., 2020; ZYCX, Li et al., 2021c), a turnip (ECD04, Yang et al., 2022) and a yellow sarson (Z1 v2, Istace et al., 2021). Among them, Chiifu v4.0 had not only the longest contig N50 (38.26 Mb) and the fewest gaps (2) but also the highest values of the BUSCO (99.40%) and LTR assembly index (LAI) score (15.05) (Figures 2 and S5; Table 1). These results suggested that Chiifu v4.0 achieved the highest continuity and completeness among B. rapa genome assemblies.
Figure 2.
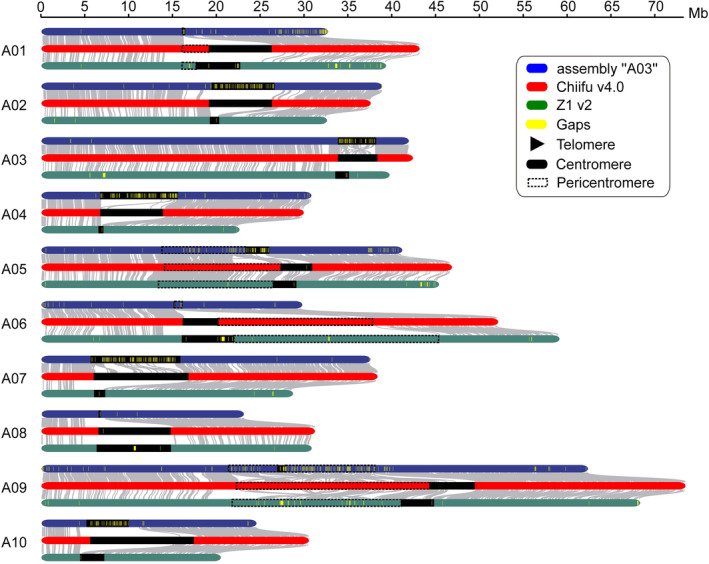
Chromosome collinearity between Brassica rapa Chiifu v4.0, assembly “A03” and Z1 v2. Grey lines link the collinear regions, and yellow blocks show the gaps. Black blocks indicate centromeres, and dashed blocks indicate pericentromeres.
Invasion of B. rapa centromeres by ALE and CRM LTRs
Through ONT ultralong‐read sequencing, eight of the ten chromosomes were assembled as gap‐free, and the remaining two centromeres were significantly improved in Chiifu v4.0 (Table S1). It provides an unprecedented opportunity to study the landscape of centromeres in B. rapa. To identify the location and sequence of centromeres in our new assembly, we used the enrichment of Cent‐SRs, including CentBr1, CentBr2, CRB and TR805 (Koo et al., 2011; Lim et al., 2005, 2007), which was directly associated with BrCENH3 proteins in B. rapa (Wang et al., 2011a). Of the ten centromeres, two centromeres on chromosomes A03 and A05 were enriched for CenBr2, while the other eight centromeres were enriched for CenBr1 (Figure S6). Although the chromosome name of previous fluorescence in situ hybridization (FISH) experiments did not correspond well with that of Chiifu v4.0, eight centromeres enriching for CenBr1 and two for CenBr2 were consistent with the previous FISH experiments (Lim et al., 2007). Sequence analysis revealed that 94.23% (65.01 Mb/68.99 Mb) of the centromeric region was occupied by LTRs (Figure 3a; Table S2). Among these 555 centromeric genes of Chiifu v4.0, 17.66% (98/555) of them were transcribed, much lower than the gene transcription ratio of the whole genome (45.57%, 21 659/47 531).
Figure 3.
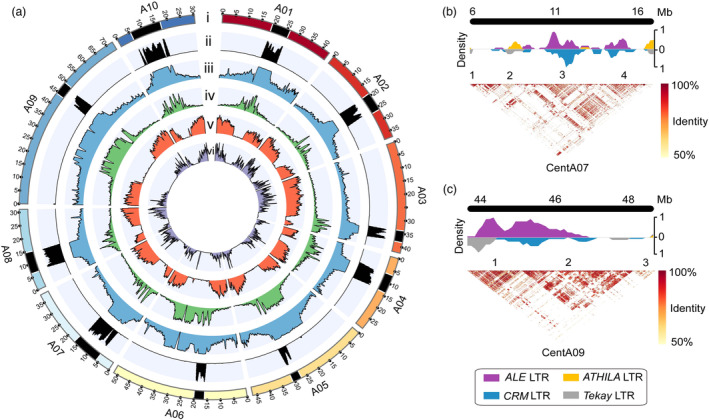
Characterization of the centromeres in Brassica rapa Chiifu v4.0. (a) Circos plot of Chiifu v4.0. i: Ten chromosomes of Chiifu v4.0. Centromeres are shown as black blocks. ii: Distribution of centromere‐specific repeats along chromosomes. iii: TE density across the chromosomes. iv: LTRs density across the chromosomes. v: Gene density of the chromosomes. vi: Expression level of genes along the chromosomes. (b,c) LTR distribution and heatmap of sequence identity in centromeres CentA07 (b) and CentA09 (c). Heatmap shows pairwise sequence identity between all non‐overlapping 10 kb regions of centromere. Numbers 1–4 indicate the sequence regions in centromeres. All data were calculated by a 500 kb sliding window and 100 kb step size.
To better understand the long‐range organization of centromeres, we generated a heatmap showing the pairwise sequence identity along the centromeres. The results showed that centromeres were disrupted into different regions in the centromeric sequences in Chiifu v4.0 (Figures 3b,c and S7). In A. thaliana, the centromeres CEN4 and CEN5 are invaded by ATHILA LTRs (Naish et al., 2021). In Chiifu v4.0, we identified 3256 full‐length long terminal repeat retrotransposons (FL‐LTR‐RTs) for the whole genome and grouped them into 12 families based on repeat domain protein homology (Figure S8; Table S3). We detected 974 FL‐LTR‐RTs in centromeres (Table S3), and 34.49% (336/974) contained Cent‐SRs. Notably, among the 12 FL‐LTR‐RTs families, 539 ALE (Copia) and 281 CRM (Gypsy) LTRs were specifically increased in copy number within these invaded regions in the centromeres (Figures 3b,c and S7). These results indicated that the centromeres were mainly invaded by ALE and CRM LTRs, further shaping the centromere structures in B. rapa.
Diversity of centromeres among B. rapa genomes
In our newly assembly Chiifu v4.0, eight complete centromeres with no gap were assembled, namely CentA01 (7.06 Mb), CentA02 (7.10 Mb), CentA03 (4.44 Mb), CentA04 (7.09 Mb), CentA06 (4.00 Mb), CentA07 (10.75 Mb), CentA09 (5.10 Mb) and CentA10 (11.75 Mb). For comparison with other B. rapa genome assemblies, the same criteria of Chiifu v4.0 were used to define the centromere boundaries of other genome assemblies (Table S1). The results showed that all centromeres have gaps in other B. rapa genome assemblies, except for the centromere of chromosome A08 in NHCC001 (Figures 2 and S5; Table S1). Furthermore, we found that Chiifu v4.0, assembly “A03”, PC‐fu, ECD04 and Z1 v2 had significantly more assembled Cent‐SRs than NHCC001 and ZYCX. However, PC‐fu had many Cent‐SRs on the end of chromosomes, indicating the misassembled Cent‐SRs in its genome (Figure S6; Table S4). Thus, Chiifu v4.0, assembly “A03”, ECD04 and Z1 v2 were used for our subsequent analysis.
We found that the centromere length varied significantly among different B. rapa genomes. To avoid making an incorrect conclusion due to the incomplete assembly, we only compared the eight gap‐free centromeres of Chiifu v4.0 with the orthologous centromeres of the other three assemblies (assembly “A03”, ECD04 and Z1 v2). After removing gaps, CentA04 of assembly “A03” (8.76 Mb) and CentA06 of Z1 v2 (5.56 Mb) were still longer than the orthologous centromeres of Chiifu v4.0 (7.09 Mb of CentA04; 4.00 Mb of CentA06; Table S1).
Chromosomal collinearity analysis showed that the orthologous centromeres had little or no sequence collinearity among the B. rapa assemblies. For example, unlike the chromosomal arms, the centromeres CentA04 between Chiifu v4.0 and assembly “A03” had almost no sequence collinearity (Figure 4a). Little sequence collinearity was also observed when we compared the other seven complete centromeres of Chiifu v4.0 with the orthologous centromeres of assembly “A03”, ECD04 and Z1 v2 (Figure S9). Together, these results indicated that the centromeres are highly variable among different B. rapa genomes.
Figure 4.
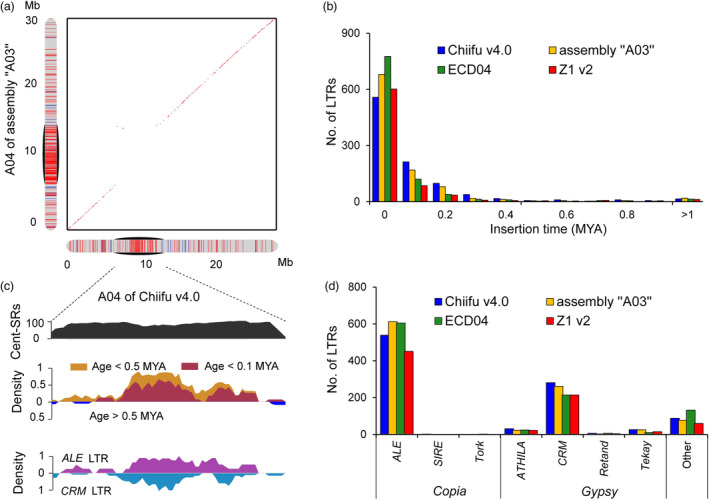
Comparison of centromeres among Brassica rapa genome assemblies. (a) Collinearity of chromosome A04 of Chiifu v4.0 and assembly “A03”. Black ovals indicate centromeres. (b) Insertion time of FL‐LTR‐RTs in centromeres among different assemblies. (c) Distribution of features on the centromere CentA04 in Chiifu v4.0, including Cent‐SRs, LTR age and family. The density was calculated using a 500 kb sliding window and 100 kb step size. (d) Copy number of FL‐LTR‐RT families in centromere regions of B. rapa genome assemblies.
Rapidly amplified LTRs drove the evolution of centromeres
Sequence analysis revealed that TEs contributed 92.90%–97.17% of centromere sequences in different B. rapa genomes (Table S2). We further annotated the FL‐LTR‐RTs in assembly “A03”, ECD04 and Z1 v2. A total of 1001, 993 and 767 FL‐LTR‐RTs were identified in the centromeric regions of assembly “A03”, ECD04 and Z1 v2, which was similar to the quantity of FL‐LTR‐RTs in the centromeres of Chiifu v4.0 (974) (Figure 4b; Table S3). Analysing the insertion time of FL‐LTR‐RTs in centromeres showed that 78.83%–86.04% of FL‐LTR‐RTs were amplified ≤0.5 MYA and 38.57%–57.78% were amplified ≤0.1 MYA in Chiifu v4.0, assembly “A03”, ECD04 and Z1 v2. In comparison, 5.64%–7.86% were amplified >1 MYA in the centromeres of Chiifu v4.0, assembly “A03”, ECD04 and Z1 v2 (Figure 4b). Furthermore, we detected 539, 612, 605 and 451 ALE LTRs and 281, 261, 214 and 214 CRM LTRs in the centromeres of Chiifu v4.0, assembly “A03”, ECD04 and Z1 v2, respectively (Figure 4d). These findings suggested that LTRs are shared but exhibit different ages and copy numbers in the centromeres of B. rapa.
According to a recent study (Perumal et al., 2020), we defined FL‐LTR‐RTs with age ≤0.5 MYA as young LTRs and age >0.5 MYA as old LTRs. The age distribution analysis of FL‐LTR‐RTs showed that the centromere regions were enriched for young LTRs in B. rapa (Figures 5a and S10). Further comparison of the insertion time of LTRs in different chromosomal regions in Chiifu v4.0 showed that FL‐LTR‐RTs in centromeres were significantly younger (0.14 MYA on average) than those of the whole genome (0.32 MYA on average; Figure 5b). We found that LTRs in the center part of centromeres were much younger than other portions of centromeres in Chiifu v4.0 (Figures 5c and S11). Furthermore, we identified 83 nested insertion events of FL‐LTR‐RTs in Chiifu v4.0 (Table S5), which were much fewer than that of B. nigra (262 events, Perumal et al., 2020). Together, our results suggested that the LTRs were rapidly amplified in centromeres, which could drive the evolution of centromeres in B. rapa.
Figure 5.
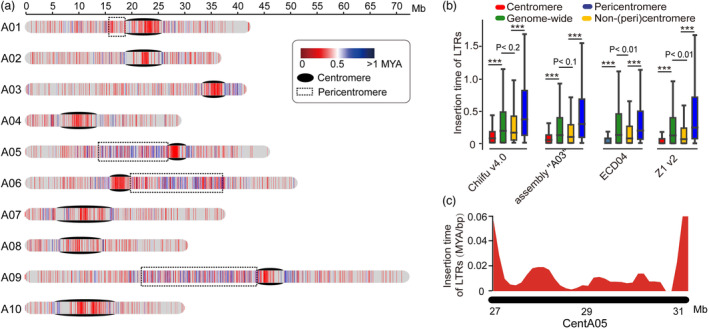
Young LTRs in the centromeres of Brassica rapa. (a) Age distribution of FL‐LTR‐RTs in Chiifu v4.0. Black ovals indicate centromeres, and dashed rectangles indicate pericentromeres. (b) Comparison of the insertion time of FL‐LTR‐RTs for the centromeres, pericentromeres, non‐(peri)centromeres and whole genome in different B. rapa genome assemblies. The P values were calculated using a two‐sided t‐test. ‘***’ indicates P < 0.001. (c) The density of insertion time of FL‐LTR‐RTs in centromere CentA05 of Chiifu v4.0. The density of insertion time was calculated using a 500 kb sliding window and 100 kb step size.
Old LTRs were enriched in B. rapa pericentromeres
Due to being highly repetitive, pericentromeres have rarely been studied in Brassica genomes. To determine the pericentromeres in B. rapa, the Peri‐SRs, including PCRBr and TR238 (Lim et al., 2007), were used to delimit the boundaries and sizes of pericentromeres in Chiifu v4.0, assembly “A03”, ECD04 and Z1 v2. Peri‐SRs were detected on chromosomes A01, A05, A06 and A09 in B. rapa genomes (Figures S6 and S12; Table S6). Our findings of four chromosomes with Peri‐SRs were consistent with the results of previous FISH experiments (Lim et al., 2007). Sequence analysis indicated that 90.27% (12 608/13 966) of rRNA sequences were located in the pericentromeric regions in Chiifu v4.0 (Figure S13). Among these 773 FL‐LTR‐RTs in pericentromeres, 37.00% (286) were CRM, and 34.41% (266) were Tekay LTRs, indicating that pericentromeres were invaded by CRM and Tekay LTRs (Figure S14). For these 263 pericentromeric genes, 19.39% (51/263) were found to be transcribed, which was much lower than that of the whole genome (45.57%) but slightly higher than that of centromeres (17.66%).
After comparing the insertion time of LTRs between pericentromeres and other chromosomal regions in Chiifu v4.0, we found that the insertion time of FL‐LTR‐RTs in pericentromeres was significantly older (0.51 MYA on average) than those of the whole genome (0.32 MYA on average) and further much older than those in centromeres (0.14 MYA on average) (Figure 5b). Similar patterns were found in assembly “A03”, ECD04 and Z1 v2 (Figure 5b). Comparing the LTRs between pericentromeres and centromeres revealed that pericentromeres were enriched for Gypsy LTRs while centromeres were enriched for more Copia than Gypsy LTRs (Figures 6c–e and S15). Furthermore, the insertion time of Gypsy LTRs (0.29–0.38 MYA on average) was prominent older than that of Copia LTRs (0.14–0.22 MYA on average; Figure 6f), which was probably why the LTRs in pericentromeres were older than those in the centromeres of B. rapa.
Figure 6.
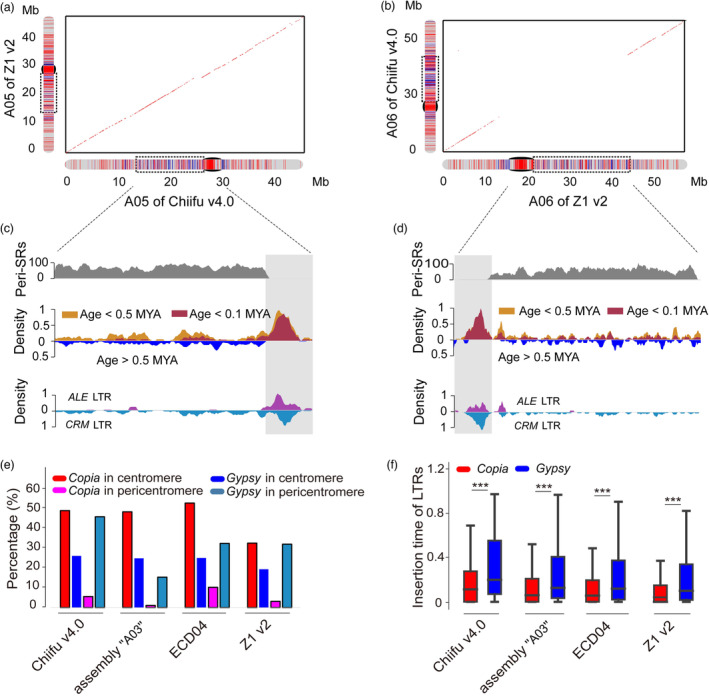
Old LTRs in the pericentromeres of Brassica rapa. (a) Collinearity of chromosome A05 of Chiifu v4.0 and Z1 v2. (b) Collinearity of chromosome A06 of Z1 v2 and Chiifu v4.0. Black rectangles indicate the centromeres, and dashed rectangles indicate the pericentromeres. (c, d) Distribution of various features in the pericentromere regions of chromosome A05 of Chiifu v4.0 (c) and A06 of Z1 v2 (d), including Peri‐SRs, LTR age and family. Grey blocks indicate the centromere regions. The density was calculated using a 500 kb sliding window and 100 kb step size. (e) Percentage of Copia and Gypsy LTRs in centromeres and pericentromeres. (f) Insertion time of the Copia and Gypsy LTRs. The P values were calculated using a two‐sided t‐test. ‘***’ indicates P < 0.001.
We identified a novel pericentromeric repeat based on the FL‐LTR‐RT sequences in B. rapa. We used self‐self‐blast of the 773 pericentromere‐enriched FL‐LTR‐RTs and filtered out the repeats that occurred fewer than 50 times. Then we blasted the remaining sequences to the pericentromere sequences and kept the repeats which hit on the pericentromeres over 1000 times. After filtering, we identified a novel repeat, termed PCR630, which is 630 bp in length with 1550 hits on pericentromeres. There was no sequence similarity when it was compared with either PCRBr (1022 hits on pericentromeres) or TR238 (87 407 hits on pericentromeres). We further found that PCR630 had a similar pericentromere‐specific distribution pattern as PCRBr and TR238 in B. rapa (Figure S16).
Discussion
The B. rapa Chiifu genome has been the most widely used reference genome in the Brassica research community since it was published in 2011 (citations >2000 times based on Google Scholar of November 2022; Wang et al., 2011b; Cai et al., 2017; Zhang et al., 2018). However, Chiifu 3.0, the current reference genome of B. rapa, still has 407 gaps and a relatively short contig N50 of 1.45 Mb (Zhang et al., 2018). In this study, we presented a near‐complete genome assembly, B. rapa Chiifu v4.0, using ONT sequencing and Hi‐C technologies. The contig N50 of Chiifu v4.0 (38.26 Mb) is the largest among the published Brassica genomes up to November 2022. Long‐read sequencing provides more comprehensive genome coverage. In Chiifu v4.0, eight telomere‐to‐telomere chromosomes were well assembled, and only two gaps were located on the centromeres of the other two chromosomes (Table 1). We identified 18 telomeres on nine chromosomes by screening the telomere‐specific repeat (BrSTR, Koo et al., 2011; Figure S17). Compared with Chiifu v3.0, the new assembly added ~71.45 Mb novel sequences, ~10% more repeats, and 298 novel genes, almost all located in the centromeres and pericentromeres. The near‐complete reference genome, B. rapa Chiifu v4.0, representing the highest completeness, reliability and quality, will drive the future discovery of genome structure and functional genes in Brassica.
Centromeres show rapid divergence among B. rapa genomes. Centromeres comprise highly repetitive elements, which are structures essential for maintaining chromosome integrity during cell division and ensuring the fidelity of their inheritance of chromosome complements (Comai et al., 2017). Although functions of centromeres are highly conserved in chromosome segregation among eukaryotes, centromeres evolve with high variability and show little or no collinearity in sequence and DNA composition even among closely related species (Gao et al., 2015; Talbert and Henikoff, 2022). In this study, the size and sequence of centromeres on orthologous chromosomes varied among different B. rapa genomes (Figures 4a and S6), indicating that centromeres are highly variable not only among very closely related species but also between distinct accessions within the same species. These results are likely related to the low recombination ratio in centromeric regions (Zhang et al., 2018), which may prevent the exchange of centromeric sequences and result in high variability in centromeres.
Rapidly amplified LTRs drive the evolution of centromeres. In B. rapa, centromeres were mainly invaded by ALE and CRM LTRs in B. rapa (Figures 3 and S7), suggesting that these two LTR families might play an essential role in the rapid evolution of centromeres (Figure 4d). It was noted that Arabidopsis centromeres had been invaded by ATHILA LTRs (Naish et al., 2021), and ALE LTRs had significantly increased in the centromeres of B. nigra (Perumal et al., 2020). In this study, we found that LTRs were shared in the centromeres in different B. rapa genomes, but the abundances and ages of LTRs were vastly divergent, suggesting that the rapidly changed LTRs could drive the evolution of centromeres in B. rapa. In the future, more studies are required to fully establish the role of the recently amplified LTRs in centromeres in B. rapa.
The old LTRs were enriched in the pericentromeres. Previous studies have shown that LTRs in centromeres are younger than those of other chromosomal regions in B. nigra (Perumal et al., 2020) and cotton (Yang et al., 2021). The present study also showed younger LTRs in centromeres of B. rapa (Figure 5a). Interestingly, LTRs in pericentromeres were much older (0.37 MYA on average) than those in centromeres in B. rapa (Figure 5b). In humans, the mutation rate of centromeric sequences is accelerated more than 2.2‐fold compared with other portions of the chromosome (Altemose et al., 2022; Logsdon et al., 2021). It could imply that the rapid amplification of young LTRs in the centromeres could force the old LTRs out, leading to the residence of relatively old LTRs in the flanking pericentromeres in B. rapa.
Together, our near‐complete genome assembly, B. rapa Chiifu v4.0, provides a critical genome resource for the Brassica research community and reveals the rapid evolution of centromeres in B. rapa. Such resources will provide a solid foundation for elucidating the genome structure and functions of Brassica species.
Materials and methods
Genome sequencing and de novo assembly
Brassica rapa L. ssp. pekinensis inbred line (Chiifu‐401‐42) was used for whole‐genome sequencing in this study. In total, 500 mg of frozen leaf tissues were used to generate high‐quality genomic DNA. For the Chiifu genome, the R9.4.1 (SQK‐LSK110) genomic library was prepared following the nanopore protocol (https://community.nanoporetech.com/protocols). The libraries were then sequenced on a Promethion platform, and MinKnow with Guppy (v5.0.16) was used for base calling with default parameters. A total of 90.24 Gb of ONT long reads with ~180 × coverage was generated, including ~64× coverage of ultralong reads (>50 kb). The ONT read N50 was 40 kb, and the most extended read was 487 325 bp.
Subsequently, the raw ONT data were filtered for quality at Q10, and the resulting reads were de novo assembled using NextDenovo (v2.5, https://github.com/Nextomics/NextDenovo) with parameters: “read_cutoff = 5k” and “seed_cutoff = 75 000”. The raw ONT reads were error‐corrected using Canu (v1.5; Koren et al., 2017) with default parameters. The resulting contigs were polished using three iterations of Racon (v1.4.3; Vaser et al., 2017) with correct ONT reads and two iterations of Pilon (v1.22; Walker et al., 2014) with Illumina reads obtained from BRAD (http://brassicadb.org; Chen et al., 2022).
Contigs scaffolding and gap filling
The B. rapa Hi‐C data from our previous study were used to correct and scaffold polished contigs (Zhang et al., 2018). We first aligned these Hi‐C reads to raw contigs using bowtie2 (v2.3.3; Langmead and Salzberg, 2012). Contact maps for all contigs produced by HiC‐Pro (v3.1.0; Servant et al., 2015) were drawn using the ggplot2 package (http://ggplot2.org/). We then checked the interaction signals for each contig with the others and split them when they had a strong signal with distant sequences. Finally, the corrected contigs were used as input for scaffolding by 3D‐DNA (v180922; Dudchenko et al., 2017) with default parameters. Scaffolds were manually checked and refined with Juicebox (v1.11.08; Durand et al., 2016). The gaps in these scaffolds were closed by TGS_GapCloser (v1.1.1; Xu et al., 2020) with corrected ONT long reads. We finally filled the gaps with the corrected ONT, BAC (PRJEA28961) and corrected PacBio reads (Zhang et al., 2018) of B. rapa Chiifu‐401‐42 using a python script (https://github.com/zhangleiworld/gapfill_by_reads).
Genome annotation
Genes were lifted from Chiifu v3.5 with Liftoff (v1.5.1; Shumate and Salzberg, 2021). MAKER‐P (v3; Campbell et al., 2014) was used to annotate the newly assembled regions in Chiifu v4.0. TEs were identified using EDTA (v1.9.6; Ou et al., 2019). FL‐LTR‐RTs were identified using LTR_retriever (v2.9.0; Ou and Jiang, 2018b) as described previously (Perumal et al., 2020) and further classified by TEsorter (v1.1.1; Zhang et al., 2022a). The insertion time of the FL‐LTR‐RTs was calculated as previously described (Liu et al., 2014). The FL‐LTR‐RTs were manually analysed to identify nested TE insertion following a previous study (Perumal et al., 2020). The LTR assembly index score was calculated by LAI (vbeta3.2; Ou et al., 2018a).
Identification of synteny between Chiifu v4.0 and other assemblies
Chiifu v4.0 was aligned to other B. rapa assemblies using Mummer (v4.0.0beta2; Marçais et al., 2018) with parameter settings “‐‐mum ‐c 5 000 ‐l 2 000”. Then, we used the “delta‐filter‐1” parameter with the one‐to‐one alignment block option to filter the alignment results. Further, “show‐coords” were used to show the synteny's coordinate between Chiifu v4.0 and other B. rapa assemblies.
Identification of centromeres, pericentromeres, telomeres and rRNA
We used LASTZ (v1.04.00; http://www.bx.psu.edu/~rsharris/lastz/) to align the Cent‐SRs (CentBr1, CentBr2, CRB and TR805) and Peri‐SRs (PCRBr and TR238) to the reference genome (Koo et al., 2011; Lim et al., 2005, 2007). The signals of the Cent‐SRs and Peri‐SRs were used as evidence supporting the localization of the centromeres and pericentromeres in Chiifu v4.0. To identify the telomeres in B. rapa, the telomere‐specific sequence BrSTR (Koo et al., 2011) was aligned with Chiifu v4.0. The rRNA sequences were predicted by Infernal (v1.1.2; Nawrocki and Eddy, 2013) using the Rfam database. The same methods were used to identify the centromeres, pericentromeres, telomeres and rRNA sequences in the other B. rapa genome assemblies.
Identification of the novel pericentromeric repeat
The novel pericentromeric repeat was identified using a self‐self‐blast of the FL‐LTR‐RT sequences in pericentromeres of Chiifu v4.0. BLASTN was run using an e‐value cutoff of 1e‐5. Any repeat that occurred over 50 times and hit on the pericentromeres more than 1000 times was defined as the novel pericentromeric repeat. The sequence of novel pericentromeric repeat was listed in Table S7.
Conflicts of interest
No conflict of interest was declared.
Author contributions
X.W. designed the project; L.Z., J.W. and J.L. prepared materials and performed the experiments; L.Z., Z.Z., H.C. and X.W. performed the data analysis; L.Z. and X.W. wrote the manuscript; J.W. and J.L. revised the manuscript. All authors read and approved the final manuscript.
Supporting information
Figure S1 Whole‐genome Hi‐C heatmap of Brassica rapa Chiifu v4.0 at 100 kb resolution.
Figure S2 Comparison of the heatmap and gaps of the conflict regions between Brassica rapa Chiifu v4.0 and v3.0.
Figure S3 The position of the additional sequences in Brassica rapa Chiifu v4.0 relative to Chiifu v3.0.
Figure S4 The gap positions of Brassica rapa Chiifu v4.0.
Figure S5 Chromosome collinearity between Brassica rapa Chiifu v4.0 and the other assemblies.
Figure S6 The distribution of Cent‐SRs and Peri‐SRs in Brassica rapa genome assemblies.
Figure S7 Heatmap shows pairwise sequence identity between all non‐overlapping 10 kb regions of centromeres in Brassica rapa Chiifu v4.0.
Figure S8 Annotation of FL‐LTR‐RTs in different Brassica rapa genome assemblies.
Figure S9 Centromere collinearity between Brassica rapa Chiifu v4.0 and other assemblies.
Figure S10 The age distribution of FL‐LTR‐RTs in Brassica rapa genome assemblies.
Figure S11 The density of insertion time of FL‐LTR‐RTs in centromeres of Brassica rapa Chiifu v4.0.
Figure S12 Characterization of the pericentromeres in Brassica rapa Chiifu v4.0.
Figure S13 The distribution of rRNA sequences in Brassica rapa genome assemblies.
Figure S14 Heatmap shows pairwise sequence identity between all non‐overlapping 10 kb regions of pericentromeres in Brassica rapa Chiifu v4.0.
Figure S15 The family distribution of FL‐LTR‐RTs in Brassica rapa genome assemblies.
Figure S16 The distribution of PCR630 in Brassica rapa genome assemblies.
Figure S17 The distribution of telomere‐specific repeat in Brassica rapa genome assemblies.
Table S1 The location of centromeres in Brassica rapa genome assemblies.
Table S2 Summary of TEs and LTRs in the centromeres of Brassica rapa genome assemblies.
Table S3 Summary of FL‐LTR‐RTs in Brassica rapa genome assemblies.
Table S4 Summary of Cent‐SRs and Peri‐SRs in Brassica rapa genome assemblies.
Table S5 Summary of nested LTRs events in Brassica rapa genome assemblies.
Table S6 The location of pericentromeres in Brassica rapa genome assemblies.
Table S7 The sequence of PCR630 in Brassica rapa.
Acknowledgements
This work was supported by the National Key Research and Development Program of China (2022YFF1003003), the Agricultural Science and Technology Innovation Program (ASTIP), the Central Public‐interest Scientific Institution Basal Research Fund (Y2022PT23) and the China Postdoctoral Science Foundation (2019M650918).
Contributor Information
Jian Wu, Email: wujian@caas.cn.
Xiaowu Wang, Email: wangxiaowu@caas.cn.
Data availability statement
The raw ONT reads are freely available through the Genome Sequence Archive under accession number CRA008441 (https://ngdc.cncb.ac.cn/gsa/). The genome sequences are freely available through the BRAD website (http://39.100.233.196:82/download_genome/Brassica_Genome_data/Brara_Chiifu_V4.0/). All other data generated or analysed during this study are included in this published article and its supplementary information files.
References
- Altemose, N. , Logsdon, G.A. , Bzikadze, A.V. , Sidhwani, P. , Langley, S.A. , Caldas, G.V. , Hoyt, S.J. et al. (2022) Complete genomic and epigenetic maps of human centromeres. Science, 376, eabl4178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai, C.C. , Wang, X.B. , Liu, B. , Wu, J. , Liang, J.L. , Cui, Y.A. , Cheng, F. et al. (2017) Brassica rapa genome 2.0: a reference upgrade through sequence re‐assembly and gene re‐annotation. Mol. Plant, 10, 649–651. [DOI] [PubMed] [Google Scholar]
- Cai, X. , Wu, J. , Liang, J. , Lin, R. , Zhang, K. , Cheng, F. and Wang, X. (2020) Improved Brassica oleracea JZS assembly reveals significant changing of LTR‐RT dynamics in different morphotypes. Theor. Appl. Genet. 133, 3187–3199. [DOI] [PubMed] [Google Scholar]
- Cai, X. , Chang, L. , Zhang, T. , Chen, H. , Zhang, L. , Lin, R. , Liang, J. et al. (2021) Impacts of allopolyploidization and structural variation on intraspecific diversification in Brassica rapa. Genome Biol. 22, 1–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell, M.S. , Law, M.Y. , Holt, C. , Stein, J.C. , Moghe, G.D. , Hufnagel, D.E. , Lei, J.K. et al. (2014) MAKER‐P: a tool kit for the rapid creation, management, and quality control of plant genome annotations. Plant Physiol. 164, 513–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, H.X. , Wang, T.P. , He, X.N. , Cai, X. , Lin, R.M. , Liang, J.L. , Wu, J. et al. (2022) BRAD V3.0: an upgraded Brassicaceae database. Nucleic Acids Res. 50, D1432–D1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng, F. , Sun, R.F. , Hou, X.L. , Zheng, H.K. , Zhang, F.L. , Zhang, Y.Y. , Liu, B. et al. (2016) Subgenome parallel selection is associated with morphotype diversification and convergent crop domestication in Brassica rapa and Brassica oleracea . Nat. Genet. 48, 1218–1224. [DOI] [PubMed] [Google Scholar]
- Comai, L. , Maheshwari, S. and Marimuthu, M.P.A. (2017) Plant centromeres. Curr. Opin. Plant Biol. 36, 158–167. [DOI] [PubMed] [Google Scholar]
- Deng, Y. , Liu, S. , Zhang, Y. , Tan, J. , Li, X. , Chu, X. , Xu, B. et al. (2022) A telomere‐to‐telomere gap‐free reference genome of watermelon and its mutation library provide important resources for gene discovery and breeding. Mol. Plant, 15, 1268–1284. [DOI] [PubMed] [Google Scholar]
- Dudchenko, O. , Batra, S.S. , Omer, A.D. , Nyquist, S.K. , Hoeger, M. , Durand, N.C. , Shamim, M.S. et al. (2017) De novo assembly of the Aedes aegypti genome using Hi‐C yields chromosome‐length scaffolds. Science, 356, 92–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand, N.C. , Robinson, J.T. , Shamim, M.S. , Machol, I. , Mesirov, J.P. , Lander, E.S. and Aiden, E.L. (2016) Juicebox provides a visualization system for Hi‐C contact maps with unlimited zoom. Cell Syst. 3, 99–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao, D.Y. , Jiang, N. , Wing, R.A. , Jiang, J.M. and Jackson, S.A. (2015) Transposons play an important role in the evolution and diversification of centromeres among closely related species. Front. Plant Sci. 6, 216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, N. , Wang, S. , Gao, L. , Liu, Y. , Wang, X. , Lai, E. , Duan, M. et al. (2021) Genome sequencing sheds light on the contribution of structural variants to Brassica oleracea diversification. BMC Biol. 19, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou, X. , Wang, D. , Cheng, Z. , Wang, Y. and Jiao, Y. (2022) A near‐complete assembly of an Arabidopsis thaliana genome. Mol. Plant, 15, 1247–1250. [DOI] [PubMed] [Google Scholar]
- Istace, B. , Belser, C. , Falentin, C. , Labadie, K. , Boideau, F. , Deniot, G. , Maillet, L. et al. (2021) Sequencing and chromosome‐scale assembly of plant genomes, Brassica rapa as a use case. Biology, 10, 732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang, L. , Qian, L. , Zheng, M. , Chen, L. , Chen, H. , Yang, L. , You, L. et al. (2021) Genomic insights into the origin, domestication and diversification of Brassica juncea . Nat. Genet. 53, 1392–1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo, D.H. , Hong, C.P. , Batley, J. , Chung, Y.S. , Edwards, D. , Bang, J.W. , Hur, Y. et al. (2011) Rapid divergence of repetitive DNAs in Brassica relatives. Genomics, 97, 173–185. [DOI] [PubMed] [Google Scholar]
- Koren, S. , Walenz, B.P. , Berlin, K. , Miller, J.R. , Bergman, N.H. and Phillippy, A.M. (2017) Canu: scalable and accurate long‐read assembly via adaptive k‐mer weighting and repeat separation. Genome Res. 27, 722–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead, B. and Salzberg, S.L. (2012) Fast gapped‐read alignment with Bowtie 2. Nat. Methods, 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Y. , Liu, G.F. , Ma, L.M. , Liu, T.K. , Zhang, C.W. , Xiao, D. , Zheng, H.K. et al. (2020) A chromosome‐level reference genome of non‐heading Chinese cabbage [Brassica campestris (syn. Brassica rapa) ssp. chinensis]. Hortic. Res. 7, 212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, B. , Zhang, X.L. , Liu, Z.Q. , Wang, L.L. , Song, L.P. , Liang, X.M. , Dou, S.W. et al. (2021a) Genetic and molecular characterization of a self‐compatible Brassica rapa Line possessing a new class II S haplotype. Plan. Theory, 10, 2815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, K. , Jiang, W.K. , Hui, Y.Y. , Kong, M.J. , Feng, L.Y. , Gao, L.Z. , Li, P.F. et al. (2021b) Gapless indica rice genome reveals synergistic contributions of active transposable elements and segmental duplications to rice genome evolution. Mol. Plant, 14, 1745–1756. [DOI] [PubMed] [Google Scholar]
- Li, P.R. , Su, T.B. , Zhao, X.Y. , Wang, W.H. , Zhang, D.S. , Yu, Y.J. , Bayer, P.E. et al. (2021c) Assembly of the non‐heading pak choi genome and comparison with the genomes of heading Chinese cabbage and the oilseed yellow sarson. Plant Biotechnol. J. 19, 966–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim, K.B. , de Jong, H. , Yang, T.J. , Park, J.Y. , Kwon, S.J. , Kim, J.S. , Lim, M.H. et al. (2005) Characterization of rDNAs and tandem repeats in the heterochromatin of Brassica rapa . Mol. Cells, 19, 436–444. [PubMed] [Google Scholar]
- Lim, K.B. , Yang, T.J. , Hwang, Y.J. , Kim, J.S. , Park, J.Y. , Kwon, S.J. , Kim, J.A. et al. (2007) Characterization of the centromere and peri‐centromere retrotransposons in Brassica rapa and their distribution in related Brassica species. Plant J. 49, 173–183. [DOI] [PubMed] [Google Scholar]
- Liu, S.Y. , Liu, Y.M. , Yang, X.H. , Tong, C.B. , Edwards, D. , Parkin, I.A.P. , Zhao, M.X. et al. (2014) The Brassica oleracea genome reveals the asymmetrical evolution of polyploid genomes. Nat. Commun. 5, 3930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logsdon, G.A. , Vollger, M.R. , Hsieh, P. , Mao, Y. , Liskovykh, M.A. , Koren, S. , Nurk, S. et al. (2021) The structure, function and evolution of a complete human chromosome 8. Nature, 593, 101–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marçais, G. , Delcher, A.L. , Phillippy, A.M. , Coston, R. , Salzberg, S.L. and Zimin, A. (2018) MUMmer4: A fast and versatile genome alignment system. PLoS Comput. Biol. 14, e1005944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagaharu, U. (1935) Genome analysis in Brassica with special reference to the experimental formation of B. napus and peculiar mode of fertilization. Jpn. J. Bot. 7, 389–452. [Google Scholar]
- Nagaki, K. , Cheng, Z.K. , Ouyang, S. , Talbert, P.B. , Kim, M. , Jones, K.M. , Henikoff, S. et al. (2004) Sequencing of a rice centromere uncovers active genes. Nat. Genet. 36, 138–145. [DOI] [PubMed] [Google Scholar]
- Naish, M. , Alonge, M. , Wlodzimierz, P. , Tock, A.J. , Abramson, B.W. , Schmucker, A. , Mandakova, T. et al. (2021) The genetic and epigenetic landscape of the Arabidopsis centromeres. Science, 374, eabi7489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nawrocki, E.P. and Eddy, S.R. (2013) Infernal 1.1: 100‐fold faster RNA homology searches. Bioinformatics, 29, 2933–2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurk, S. , Koren, S. , Rhie, A. , Rautiainen, M. , Bzikadze, A.V. , Mikheenko, A. , Vollger, M.R. et al. (2022) The complete sequence of a human genome. Science, 376, 44–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okonechnikov, K. , Conesa, A. and García‐Alcalde, F. (2016) Qualimap 2: advanced multi‐sample quality control for high‐throughput sequencing data. Bioinformatics, 32, 292–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou, S. and Jiang, N. (2018b) LTR_retriever: a highly accurate and sensitive program for identification of long terminal repeat retrotransposons. Plant Physiol. 176, 1410–1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou, S. , Chen, J. and Jiang, N. (2018a) Assessing genome assembly quality using the LTR Assembly Index (LAI). Nucleic Acids Res. 46, e126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou, S. , Su, W. , Liao, Y. , Chougule, K. , Agda, J.R.A. , Hellinga, A.J. , Lugo, C.S.B. et al. (2019) Benchmarking transposable element annotation methods for creation of a streamlined, comprehensive pipeline. Genome Biol. 20, 275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park, S.G. , Noh, E. , Choi, S. , Choi, B. , Shin, I.G. , Yoo, S.I. , Lee, D.J. et al. (2021) Draft genome assembly and transcriptome dataset for European turnip (Brassica rapa L. ssp. rapifera), ECD4 carrying clubroot resistance. Front. Genet. 12, 1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perumal, S. , Koh, C.S. , Jin, L.L. , Buchwaldt, M. , Higgins, E.E. , Zheng, C.F. , Sankoff, D. et al. (2020) A high‐contiguity Brassica nigra genome localizes active centromeres and defines the ancestral Brassica genome. Nat. Plants, 6, 929–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousseau‐Gueutin, M. , Belser, C. , Da Silva, C. , Richard, G. , Istace, B. , Cruaud, C. , Istace, B. et al. (2020) Long‐read assembly of the Brassica napus reference genome Darmor‐bzh. GigaScience, 9, giaa137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Servant, N. , Varoquaux, N. , Lajoie, B.R. , Viara, E. , Chen, C.J. , Vert, J.P. , Heard, E. et al. (2015) HiC‐Pro: an optimized and flexible pipeline for Hi‐C data processing. Genome Biol. 16, 259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shumate, A. and Salzberg, S.L. (2021) Liftoff: accurate mapping of gene annotations. Bioinformatics, 37, 1639–1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simao, F.A. , Waterhouse, R.M. , Ioannidis, P. , Kriventseva, E.V. and Zdobnov, E.M. (2015) BUSCO: assessing genome assembly and annotation completeness with single‐copy orthologs. Bioinformatics, 31, 3210–3212. [DOI] [PubMed] [Google Scholar]
- Song, J.M. , Xie, W.Z. , Wang, S. , Guo, Y.X. , Koo, D.H. , Kudrna, D. , Gong, C.B. et al. (2021) Two gap‐free reference genomes and a global view of the centromere architecture in rice. Mol. Plant, 14, 1757–1767. [DOI] [PubMed] [Google Scholar]
- Sun, X.X. , Li, X. , Lu, Y. , Wang, S. , Zhang, X.M. , Zhang, K. , Su, X.J. et al. (2022) Construction of a high‐density mutant population of Chinese cabbage facilitates the genetic dissection of agronomic traits. Mol. Plant, 15, 913–924. [DOI] [PubMed] [Google Scholar]
- Talbert, P.B. and Henikoff, S. (2022) The genetics and epigenetics of satellite centromeres. Genome Res. 32, 608–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaser, R. , Sovic, I. , Nagarajan, N. and Sikic, M. (2017) Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. 27, 737–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker, B.J. , Abeel, T. , Shea, T. , Priest, M. , Abouelliel, A. , Sakthikumar, S. , Cuomo, C.A. et al. (2014) Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One, 9, e112963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, G. , He, Q. , Liu, F. , Cheng, Z. , Talbert, P.B. and Jin, W. (2011a) Characterization of CENH3 proteins and centromere‐associated DNA sequences in diploid and allotetraploid Brassica species. Chromosoma, 120, 353–365. [DOI] [PubMed] [Google Scholar]
- Wang, X. , Wang, H. , Wang, J. , Sun, R. , Wu, J. , Liu, S. , Bai, Y. et al. (2011b) The genome of the mesopolyploid crop species Brassica rapa . Nat. Genet. 43, 1035–1039. [DOI] [PubMed] [Google Scholar]
- Wang, B. , Yang, X. , Jia, Y. , Xu, Y. , Jia, P. , Dang, N. , Wang, S. et al. (2021) High‐quality Arabidopsis thaliana genome assembly with Nanopore and HiFi long reads. Genomics, Proteomics Bioinf. 20, 4–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, M.Y. , Guo, L.D. , Gu, S.Q. , Wang, O. , Zhang, R. , Peters, B.A. , Fan, G.Y. et al. (2020) TGS‐GapCloser: a fast and accurate gap closer for large genomes with low coverage of error‐prone long reads. Gigascience, 9, giaa094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, H. , Wang, C. , Shao, G. , Wu, S. , Liu, P. , Cao, P. , Jiang, P. et al. (2022) The reference genome and full‐length transcriptome of pak choi provide insights into the cuticle formation and heat adaption. Hortic. Res. 9, uhac123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Z. , Ge, X. , Li, W. , Jin, Y. , Liu, L. , Hu, W. , Liu, F. et al. (2021) Cotton D genome assemblies built with long‐read data unveil mechanisms of centromere evolution and stress tolerance divergence. BMC Biol. 19, 115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Z.Q. , Jiang, Y.F. , Gong, J.F. , Li, Q. , Dun, B.C. , Liu, D.X. , Yin, F.F. et al. (2022) R gene triplication confers European fodder turnip with improved clubroot resistance. Plant Biotechnol. J. 20, 1502–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, L. , Cai, X. , Wu, J. , Liu, M. , Grob, S. , Cheng, F. , Liang, J.L. et al. (2018) Improved Brassica rapa reference genome by single‐molecule sequencing and chromosome conformation capture technologies. Hortic. Res. 5, 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, R.G. , Li, G.Y. , Wang, X.L. , Dainat, J. , Wang, Z.X. , Ou, S. and Ma, Y. (2022a) TEsorter: an accurate and fast method to classify LTR‐retrotransposons in plant genomes. Hortic Res. 9, uhac017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Y. , Fu, J. , Wang, K. , Han, X. , Yan, T. , Su, Y. , Li, Y. et al. (2022b) The telomere‐to‐telomere gap‐free genome of four rice parents reveals SV and PAV patterns in hybrid rice breeding. Plant Biotechnol. J. 20, 1642–1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Z.C. , Guo, J. , Cai, X. , Li, Y.F. , Xi, X. , Lin, R.M. , Liang, J.L. et al. (2022c) Improved reference genome annotation of Brassica rapa by Pacific Biosciences RNA sequencing. Front. Plant Sci. 13, 841618. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Whole‐genome Hi‐C heatmap of Brassica rapa Chiifu v4.0 at 100 kb resolution.
Figure S2 Comparison of the heatmap and gaps of the conflict regions between Brassica rapa Chiifu v4.0 and v3.0.
Figure S3 The position of the additional sequences in Brassica rapa Chiifu v4.0 relative to Chiifu v3.0.
Figure S4 The gap positions of Brassica rapa Chiifu v4.0.
Figure S5 Chromosome collinearity between Brassica rapa Chiifu v4.0 and the other assemblies.
Figure S6 The distribution of Cent‐SRs and Peri‐SRs in Brassica rapa genome assemblies.
Figure S7 Heatmap shows pairwise sequence identity between all non‐overlapping 10 kb regions of centromeres in Brassica rapa Chiifu v4.0.
Figure S8 Annotation of FL‐LTR‐RTs in different Brassica rapa genome assemblies.
Figure S9 Centromere collinearity between Brassica rapa Chiifu v4.0 and other assemblies.
Figure S10 The age distribution of FL‐LTR‐RTs in Brassica rapa genome assemblies.
Figure S11 The density of insertion time of FL‐LTR‐RTs in centromeres of Brassica rapa Chiifu v4.0.
Figure S12 Characterization of the pericentromeres in Brassica rapa Chiifu v4.0.
Figure S13 The distribution of rRNA sequences in Brassica rapa genome assemblies.
Figure S14 Heatmap shows pairwise sequence identity between all non‐overlapping 10 kb regions of pericentromeres in Brassica rapa Chiifu v4.0.
Figure S15 The family distribution of FL‐LTR‐RTs in Brassica rapa genome assemblies.
Figure S16 The distribution of PCR630 in Brassica rapa genome assemblies.
Figure S17 The distribution of telomere‐specific repeat in Brassica rapa genome assemblies.
Table S1 The location of centromeres in Brassica rapa genome assemblies.
Table S2 Summary of TEs and LTRs in the centromeres of Brassica rapa genome assemblies.
Table S3 Summary of FL‐LTR‐RTs in Brassica rapa genome assemblies.
Table S4 Summary of Cent‐SRs and Peri‐SRs in Brassica rapa genome assemblies.
Table S5 Summary of nested LTRs events in Brassica rapa genome assemblies.
Table S6 The location of pericentromeres in Brassica rapa genome assemblies.
Table S7 The sequence of PCR630 in Brassica rapa.
Data Availability Statement
The raw ONT reads are freely available through the Genome Sequence Archive under accession number CRA008441 (https://ngdc.cncb.ac.cn/gsa/). The genome sequences are freely available through the BRAD website (http://39.100.233.196:82/download_genome/Brassica_Genome_data/Brara_Chiifu_V4.0/). All other data generated or analysed during this study are included in this published article and its supplementary information files.