

Abstract
Forcing budding yeast to chromatinize their DNA with human histones manifests an abrupt fitness cost. We previously proposed chromosomal aneuploidy and missense mutations as two potential modes of adaptation to histone humanization. Here, we show that aneuploidy in histone‐humanized yeasts is specific to a subset of chromosomes that are defined by their centromeric evolutionary origins but that these aneuploidies are not adaptive. Instead, we find that a set of missense mutations in outer kinetochore proteins drives adaptation to human histones. Furthermore, we characterize the molecular mechanism underlying adaptation in two mutants of the outer kinetochore DASH/Dam1 complex, which reduce aneuploidy by suppression of chromosome instability. Molecular modeling and biochemical experiments show that these two mutants likely disrupt a conserved oligomerization interface thereby weakening microtubule attachments. We propose a model through which weakened microtubule attachments promote increased kinetochore‐microtubule turnover and thus suppress chromosome instability. In sum, our data show how a set of point mutations evolved in histone‐humanized yeasts to counterbalance human histone‐induced chromosomal instability through weakening microtubule interactions, eventually promoting a return to euploidy.
Keywords: aneuploidy, centromere dysfunction, histones, kinetochore, Saccharomyces cerevisiae
Subject Categories: Cell Cycle; Chromatin, Transcription & Genomics; DNA Replication, Recombination & Repair
Increased kinetochore‐microtubule turnover suppresses chromosome segregation defects caused by budding yeast chromatinization with human histones.
Introduction
Evolution is punctuated by moments of turmoil followed by rapid adaptation and stasis (Gould & Eldredge, 1993). Genomic studies have revealed a mode of “creation by crisis,” with mechanisms ranging from whole genome duplication in yeasts to chromothripsis and telomere crisis in cancer cells (Counter et al, 1992; Wolfe & Shields, 1997; Baca et al, 2013). These episodic bursts of innovation typically play out at the level of large‐scale DNA mutation (Heasley et al, 2021). Cross‐species genome hybridization via molecular engineering presents a dramatic new example of lab‐directed genetic crisis (Kachroo et al, 2015; Laurent et al, 2020; Boonekamp et al, 2022). Because histone proteins are intimately associated with DNA, the complete exchange of native histone genes with non‐native histone genes poses a substantial genetic barrier. We have previously shown that the budding yeast, Saccharomyces cerevisiae, can subsist with human histone comprised chromatin (Truong & Boeke, 2017). However, a large bottleneck limits how readily this occurs, with just ~ 1 in 109 cells surviving the initial histone swap (Haase et al, 2019). The histone‐humanized cells are initially extremely unfit, with a generation time ranging from 8 to 12 h, but they quickly adapt and fitness improves. Adaptation is associated with the acquisition of distinct bypass mutations and accumulation of aneuploid chromosomes, but how these processes contribute to improved fitness is unclear. However, the high levels of chromosome instability suggest critical defects in machinery responsible for chromosome segregation in histone humanized strains.
Accurate segregation of chromosomes relies on ensuring proper centromere–kinetochore–microtubule connections. After replication, sister chromosomes must be captured by the spindle microtubules, bioriented, and segregated equally into daughter cells (Nicklas, 1997). The establishment of kinetochore biorientation is the critical step in ensuring faithful chromosome segregation. Failure to establish correct orientation causes chromosome missegregation and aneuploidy, which leads to decreased cellular fitness in lab yeasts (Torres et al, 2007, 2010; Hose et al, 2020) and underlies many human maladies (Oromendia & Amon, 2014; Antonarakis, 2017).
Centromeres serve as the coupling point of chromosomes to the spindle microtubules—an interaction, which is bridged by the megadalton kinetochore complex (Biggins, 2013). The centromeric variant histone H3 (Cse4 in S. cerevisiae, CENP‐A in humans) plays a central role in the process of chromosome segregation by defining the region of centromeric DNA in the majority of species (Steiner & Henikoff, 2015). In S. cerevisiae, centromeres are defined as a single Cse4‐containing nucleosome that wraps a specific sequence of ~ 125 base pairs (bp) of DNA. Coupling to a single microtubule is achieved through the association of a Cse4‐containing nucleosome with the inner kinetochore protein complexes CCAN and Cbf3 (Cottarel et al, 1989; Winey et al, 1995; Furuyama & Biggins, 2007; Biggins, 2013). From here adaptor complexes, MINDMIS12 and CNN1CENP‐T, bridge the gap to link to the outer kinetochore complexes Ndc80c and DASH/Dam1c that interface with microtubules (Jenni et al, 2017).
Kinetochore‐microtubule attachments made by Ndc80c and DASH/Dam1c are highly regulated to ensure incorrect attachments are not over‐stabilized (Tien et al, 2010). Directed destabilization of incorrect attachments allows for attachments to be released and corrected. This regulation is achieved through kinetochore‐microtubule turnover driven by the kinase activity of Aurora B kinase (Tanaka et al, 2002; Cimini et al, 2006; Pinsky et al, 2006; Carmena et al, 2012). Aurora B (Ipl1) forms the chromosomal passenger complex (CPC), alongside INCENP (Sli15), Borealin (Bir1), and Survivin (Nbl1), whose recruitment to centromeric chromatin stimulates correction of incorrect microtubule attachments (Kawashima et al, 2010; Yamagishi et al, 2010; Carmena et al, 2012). As these mechanisms of CPC recruitment involve the direct interaction with nucleosomes (Abad et al, 2019), it is, thus, plausible human histones may disrupt this pathway and lead to chromosome instability in histone humanized yeasts.
Whole genome sequencing of histone‐humanized yeasts hinted toward mutation of outer kinetochore genes and aneuploidy as two potential paths of adaptation (Truong & Boeke, 2017). Here we set out to answer the relative contributions of mutation and aneuploidy to adaptation to human histones. We find that aneuploidy is altogether non‐adaptive and that aneuploidy accumulation is biased to a non‐random subset of chromosomes based on their centromeric evolutionary origins. Instead, using genetic, molecular modeling, and biochemistry techniques we show that a set of DASH/Dam1c mutants are adaptive to yeasts with human histones. Together our data support a mechanism whereby DASH/Dam1c mutants disrupt their oligomerization, which weakens kinetochore‐microtubule attachments thereby suppressing chromosome instability and reducing the incidence of aneuploidy.
Results
DASH/Dam1c mutants are dominant suppressors of human histones
Histone‐humanized yeasts are generated by using a low background dual‐plasmid histone shuffle assay followed by counterselection of the yeast histone genes, which are linked to the URA3 marker, with 5‐FOA (Figs 1A and EV1A; Truong & Boeke, 2017; Haase et al, 2019). After approximately 2–4 weeks of growth, we observed candidate humanized colonies as indicated by the appearance of small colonies, whereas larger colonies retained yeast histone genes (Fig EV1B). We validated eight new lineages (yHs9‐16) as bona fide humanized clones by the loss of the yeast histone genes as determined by PCR analysis (Fig EV1B) and whole genome sequencing (Fig EV1C). To see how these lineages adapt to human histones we continually passaged them in rich medium and observed a dramatic fitness improvement within a brief period of 30 generations (corresponding to five passages lasting a total of approximately 25 days; Figs 1A and EV1D). For example, in the histone‐humanized lineage yHs13, the mean growth rate significantly improved over 17‐fold between the ancestral and the generation 30 descendant (Fig 1B). We found that lineage yHs13 initially evolved a mutation in the DAM1 gene—a component of the outer kinetochore complex DASH/Dam1c (Fig 1C). Additionally, we identified 60 unique nucleotide variants from the ancestral and evolved lineages (Table EV1; Appendix Table S1), more than doubling the total number of candidate suppressors of histone humanization from previously reported (Truong & Boeke, 2017). On average, each strain had three mutations (Fig EV1E) and we observed only two genes, URA2 and GEX2, with more than one mutation (Truong & Boeke, 2017 and Table EV1). This set of genes was biased toward processes related to the cell cycle, chromosome segregation, rRNA processes, and chromatin remodeling (Appendix Fig S1). Across all of our histone‐humanized lineages we isolated six independent lineages with mutations in the outer kinetochore protein complexes DASH/Dam1c, Ndc80c, and Spc105c, suggesting that alteration to microtubule attachments may be a potent route for adaptation (Fig 1C).
Figure 1. DASH/Dam1c mutants are dominant genetic suppressors of histone humanization.
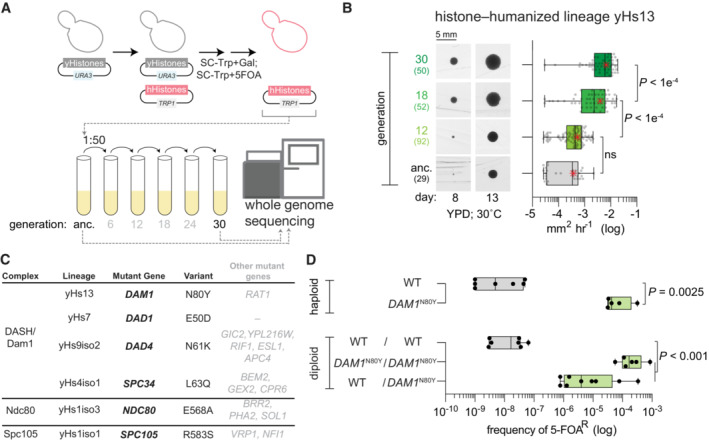
- Dual‐plasmid histone shuffle strategy used to generate histone‐humanized yeasts (see Materials and Methods for details). Humanized isolates were passaged in a rich medium for at least 5 cycles, at which point the ancestral isolate and evolved were sequenced.
- Growth assay of ancestral and evolved histone‐humanized lineage yHs13. Cells from the ancestral populations and evolved populations (indicated by generations in rich medium) were restruck onto a rich medium agar plate and colonies were imaged for up to 3 weeks to observe the change in colony size (left images). The average growth rate in mm2 h−1 was calculated by taking the change in colony size between time points divided by the time interval (right graph). Numbers in parentheses indicate the number of colonies analyzed, the central band represents the median, the box extends from the 25th to 75th percentiles, the whiskers represent minimum to maximum, and the asterisks represent the mean. The significance of mean differences in growth rates was determined with an ordinary one‐way ANOVA multiple comparisons with Turkey correction of multiple hypothesis tests. Scale bar 5 mm.
- Table of histone‐humanized lineages that evolved a mutation in an outer kinetochore complex. The variant column shows the observed nonsynonymous alteration of the bolded gene in the mutant gene column. Other genes that were mutated in each lineage are shown in non‐bolded font, details of these mutations can be found in Table EV1 and Appendix Table S1.
- Sufficiency validation for the DAM1 N80Y mutation demonstrates the DAM1 N80Y mutation significantly increases the rate of humanization over the wild type (P value = 0.0093). Green‐filled bars indicate successful isolation and confirmation of humanized yeasts. Each point represents a single biological replicate, the central band represents the median, the box extends from the 25th to 75th percentiles and the whiskers represent the minimum and maximum. The significance of the mean difference in 5–FOAR frequency was determined with the Mann–Whitney test.
Figure EV1. Isolation and verification of histone‐humanized yeasts.
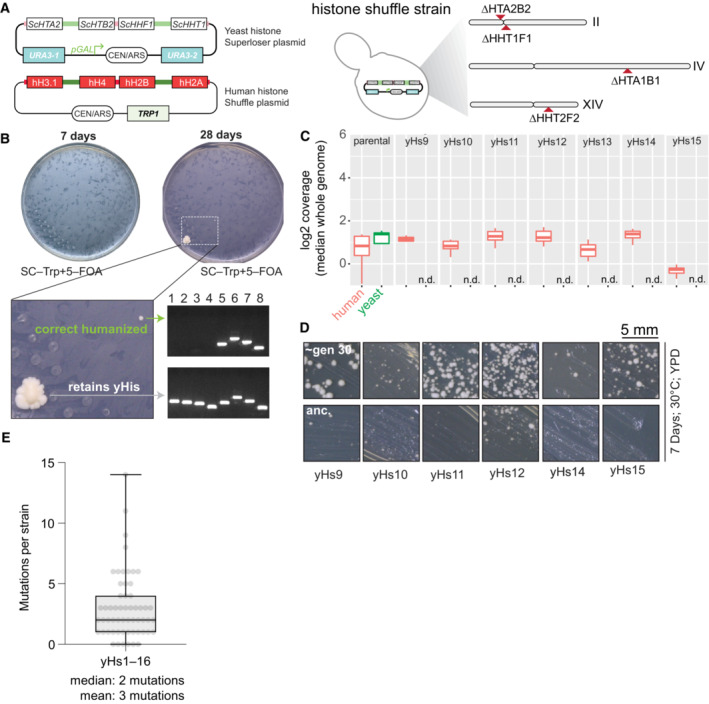
- Dual‐plasmid histone shuffle assay overview. First, a strain with all core histone gene clusters deleted is maintained with a single set of yeast histone genes on the counter‐selectable URA3 plasmid (Haase et al, 2019). To shuffle out the yeast histones for human histones, a second plasmid encoding the four core human histones and with a TRP1 selectable marker is transformed into the shuffle strain. The shuffle strains in then grown on media containing 5–FOA to force the cells to grow with exclusively human histones.
- Example humanization experiment results. Plates are shown at two time points to illustrate the severe growth defects upon the initial humanization event. Two colonies were isolated, one retaining all yeast histones (either through inactivation of URA3 or some plasmid recombination event) and a bona fide histone‐humanized clone as verified by PCR genotyping (Haase et al, 2019). Lanes 1–4, PCR genotyping of yeast H2A, H2B, H4, and H3, respectively; lanes 5–8 PCR genotyping of human H2A, H2B, H3, and H4, respectively.
- Whole genome sequencing coverage plots of the two histone plasmids (human histones, red; yeast histones, green) are shown for the parental shuffle strain prior to humanization and for histone‐humanized clones (yHs9–yHs15). Box and whisker plot of the sliding window coverage for each plasmid from the indicated strain (n = 1), the central band represents the median coverage of all windows, the box extends from the 25th to 75th percentile, and the whiskers represent the 95% confidence interval.
- YPD plate growth assays for histone‐humanized clones from the original unevolved glycerol stock and the evolved descendants after ~ 30 generations of growth. The scale bar represents 5 mm.
- Box and whisker plots of number of mutations observed per histone‐humanized lineages. Each dot represents the number of mutations observed in a histone‐humanized isolate (biological replicate, n = 57), the central band represents the median, the box extends from the 25th to 75th percentile, and the whiskers represent the minimum and maximum.
The DASH/Dam1c mutations arose in lineages with additional mutations (Fig 1C). We, therefore tested the sufficiency of these mutations in suppressing the fitness defect associated with histone humanization. First, we used CRISPR‐Cas9 mediated mutagenesis to scarlessly introduce each of the nonsynonymous mutations into an isogenic haploid histone shuffle strain and then derived both heterozygous and homozygous diploid histone shuffle strains by mating (Fig EV2A and B; Appendix Table S1). The majority of mutants had no growth defects in the wild‐type (WT) background, except for a general growth defect of the dad4 N61K mutant and cold sensitivity at 4°C for the DAM1 N80Y mutant (Fig EV2C, where dominant mutations appear capitalized). The sufficiency of each mutation was tested using our histone plasmid shuffle assay (Figs 1A and EV1A). We defined sufficiency as the ability of a mutant to robustly generate histone‐humanized colonies that, upon counter‐selection of the yeast histone plasmid with 5‐FOA, are resistant to 5‐FOA (5‐FOAR) and devoid of yeast histones. For example, in a haploid strain, the DAM1 N80Y mutant increased the frequency of 5‐FOAR over ~ 4,000 times that of the WT (Fig 1D). Likewise, the homozygous diploid DAM1 N80Y mutant generated 5‐FOAR colonies at ~ 10,000 times the diploid WT frequency. Further, we found that DAM1 N80Y is dominant to WT, as the heterozygous diploid strain produced 5‐FOAR colonies at a frequency ~ 3,000 times higher than the diploid WT (Fig 1D). We obtained similar results for all tested mutants except for dad4 N61K, which only weakly humanized in the heterozygous diploid background (Fig EV2D–E; all calculated humanization frequencies can be found in Table EV2). Importantly, these mutants lead to faster growth. We observed that humanized colonies appeared within 14 days upon counterselection (Fig EV2D), in contrast to the WT background, where colonies can take over 21 days to first appear (Truong & Boeke, 2017). These data show that the DASH mutants are dominant and significantly increased the frequency of generating histone humanized yeasts. We next sought to disentangle the contributions of aneuploidy and the DASH mutants to adaptation to human histones.
Figure EV2. CRISPR‐Cas9 mediated mutation of outer kinetochore genes and suppression validation.
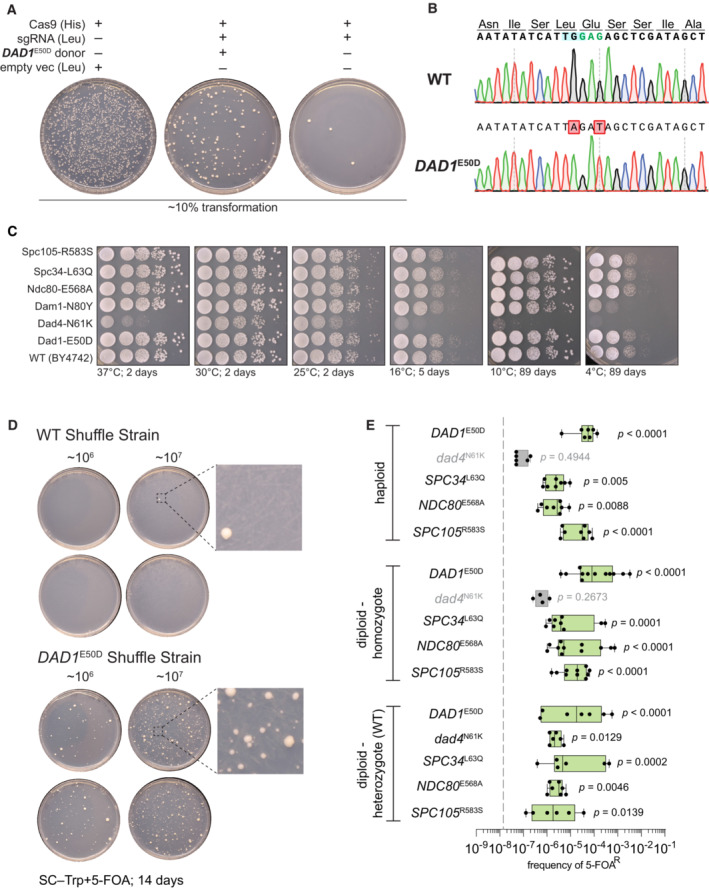
- Example CRISPR/Cas9 editing transformation to scarlessly introduce each point missense mutation to an isogenic histone shuffle strain. Note in the absence of dsDNA donor the sgRNA targeting DAD1 results in a severe killing phenotype and upon co‐transformation with a proper dsDNA, the killing phenotype is rescued.
- Example Sanger tracks for the edited DAD1 E50D mutation and wild‐type sequence. The targeting PAM is highlighted in blue, the edited codon in green, and the modified base pairs in red.
- Growth assay on rich medium (YPD) for the indicated histone shuffle strains with yeast histones. Spots are 10‐fold serial dilutions from starting OD600 of 1.0.
- Example suppressor mutation sufficiency histone‐humanization experiment is shown for the DAD1 E50D mutation. Two concentrations of cells were plated (106 and 107 ml−1).
- Histone humanizations for all tested suppressor mutations are shown. The average rates for wild‐type strains are plotted as a gray‐dashed line. Significance was determined with a Kruskal–Wallis test of the mean frequency of 5‐FOAR for each mutant versus the mean frequency of 5‐FOAR of wild type, with multiple‐comparison corrections with the false discovery rate method. Green boxes represent suppressors who significantly increased the rate of humanization above wild‐type level. Each dot represents a biological replicate of the histone humanization assay (n ≥ 4), the central band represents the median, the box extends from the 25th to 75th percentile, and the whiskers represent the minimum and maximum.
Ancient paralogous centromere origins best explain the aneuploid frequency
A prominent feature in all ancestral humanized lineages is aneuploidy, which we observed as gains of chromosomes (Fig 2A; Appendix Fig S2 and Table S2; Truong & Boeke, 2017). The majority of humanized lineages maintained their aneuploidies as their fitness improved (Fig EV1D; Appendix Fig S2). We observed that the frequency of aneuploid in histone humanized yeast showed a weak negative correlation with chromosome size (Fig EV3A; Pearson's r = 0.51; P = 0.037). Intriguingly these aneuploids occurred nonrandomly, with eight chromosomes (I, III, XVI, II, XII, XI, IX, and V) displaying chromosomal gains most frequently (Fig 2A and B). Given that S. cerevisiae is a product of an allopolyploidization event occurring ~ 93 million years ago we decided to investigate the difference in aneuploidy between the eight centromere/chromosome paralog pairs. (Wolfe & Shields, 1997; Marcet‐Houben & Gabaldón, 2015; Shen et al, 2018). Each of the 16 chromosomes can be assigned to one of eight centromere paralog pairs (Fig 2B and D), which are inferred from synteny analysis (Gordon et al, 2011). Surprisingly, we find that aneuploid frequency in our histone‐humanized yeasts is split between centromere paralog pairs, with one member of each pair being frequently aneuploid (Fig 2A and B). We defined two groups, with one group consisting of the paralogs that display frequent aneuploidy in histone humanized yeasts (group A) and the other group composed of the paralogs that display a low frequency of aneuploidy in histone humanized yeasts (group B). One paralog pair, CEN15—CEN13, both presented zero aneuploidies in our data set, thus we excluded the pair from subsequent analysis (Fig 2B).
Figure 2. Ancient paralogous centromere pairs explain aneuploid frequency.
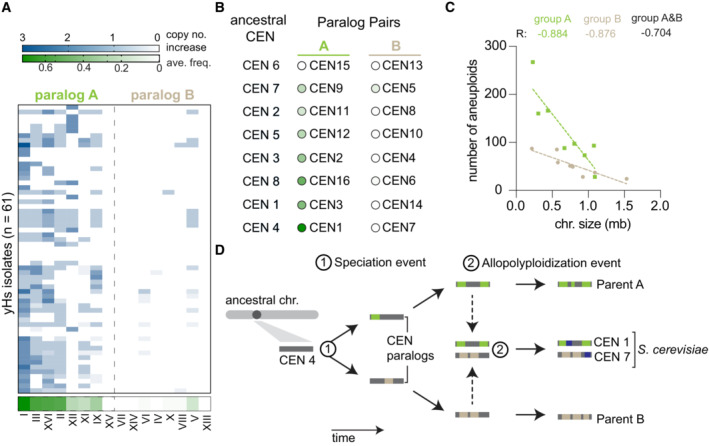
- Aneuploid frequency in histone‐humanized yeasts. Chromosomes displayed are divided into two groupings based on paralogs (i.e., Chr I is the paralog of Chr VII). The average frequency of aneuploidy in histone humanized yeast for each chromosome is shown at the bottom of the green‐colored heat map. Each row represents the chromosomal copy number for a single humanized lineage either from this study and Truong and Boeke (2017).
- Difference in aneuploid frequency between paralog pairs, colored circles next to chromosomes indicate the average aneuploid frequency in histone humanized yeast as defined in panel (A). Paired t‐test between the mean aneuploid frequency of paralog group A and B P = 0.004. Ancestral CENs and paralog pairs are taken from Gordon et al, 2011.
- Number of observed aneuploidies for each chromosome as a function of chromosome size. The Pearson correlation coefficient (R) is shown for the three models (only group A chromosomes, green; only group B chromosomes, beige; all chromosomes, black). The data are best explained by two models that consider chromosomes from group A and B separately, extra sum‐of‐squares F test P = 0.0203.
- Model of paralogous chromosome/centromere evolution. An initial speciation event (1) led to the creation of paralogous centromeres. Following speciation, each lineage accumulated specific modifications of its chromosomes and centromeres (indicated by the accumulation of colored bars on the ancestral gray bar). After millions of years of evolution (Marcet‐Houben & Gabaldón, 2015) these two lineages hybridized, producing the ancestor of S. cerevisiae (2). This new species would have 16 centromeres, with 8 arising from each parental lineage, each carrying with them the lineage‐specific modifications. We hypothesize that given the bias in aneuploid frequency between these paralog pairs in modern S. cerevisiae, the ancient origins of centromeres in S. cerevisiae may still retain functional consequences today.
Figure EV3. Chromosome size alone does not explain the frequency of aneuploidy in yeasts.
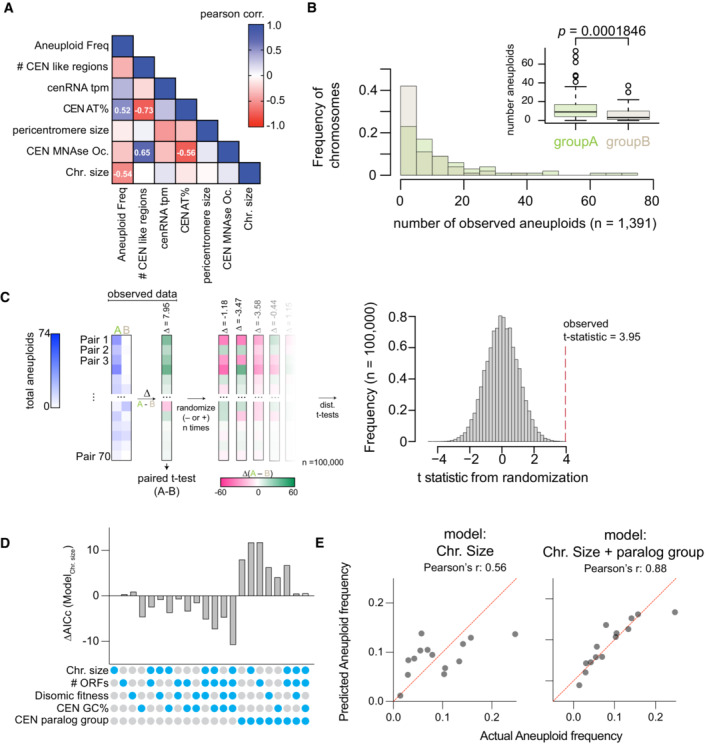
- Histogram of the number of aneuploidies per chromosome from this study and nine additional studies (see text). Inset shows a boxplot of the same data with paired t‐test of the mean difference in aneuploid frequency between groups A and B. Each dot represents the observed number of aneuploids from our study and nine additional studies (n = 80 for each group), the central band represents the median, the box extends from the 25th to 75th percentile, and the whiskers represent the 95% confidence interval.
- Randomizations of the mean difference in aneuploidy occurrence between centromere paralog pairs. Left panel, schematic of the analysis. Briefly, we examined the mean difference in aneuploidy frequency between centromere paralog pairs (Group A—Group B, e.g., CEN1—CEN7) in our study and nine additional studies (Table EV3; Kao et al, 2010; McCulley & Petes, 2010; Selmecki et al, 2015; Gallone et al, 2016; Zhu et al, 2016; Jaffe et al, 2017; Duan et al, 2018; Peter et al, 2018; Sharp et al, 2018). Next, from these 70 paired aneuploid values (note, we excluded the comparison of pair CEN15‐CEN13), we calculated the difference in aneuploidy occurrence between each group A and group B pairs. We then performed a paired t‐test on the mean difference in aneuploid occurrence between group A and group B pairs (t = 3.95, P = 0.0001846). To evaluate how “extreme” the observed t‐statistic is, we performed randomized allocations of the observed differences in aneuploid occurrence. Succinctly put, we randomly assigned the sign (− or +) to the differences between group A and B paralog pairs and then calculated t‐statistics from 100,000 randomized such allocations. The randomizations yielded a distribution of t‐statistics of the mean difference in aneuploid occurrence between pairs assuming the null hypothesis is true (i.e., there is no difference in aneuploid frequency between the two groups). Right panel: histogram of the t‐statistics from 100,000 randomizations of the difference in aneuploid occurrence. The observed t‐statistic is shown with a red dashed line (P < 0.00004).
- Comparison of linear regression models of aneuploid frequency explained by the indicated factors. Regression models were compared using the Akaike information criterion (AIC) method.
- Example predictions from linear regression models based on chromosome size with or without the centromere paralog information.
To test whether the difference in aneuploid frequency between groups A and B chromosomes is a trend observed in other yeast strains (non‐histone‐humanized), we analyzed the frequency of chromosomal aneuploidy across a diverse set of 1,767 yeast strains (Table EV3; Kao et al, 2010; McCulley & Petes, 2010; Selmecki et al, 2015; Gallone et al, 2016; Zhu et al, 2016; Jaffe et al, 2017; Duan et al, 2018; Peter et al, 2018; Sharp et al, 2018). We observed a total of 1,549 occurrences of aneuploidy across all chromosomes in these strains (chromosome gains) and that the frequency of aneuploidy is significantly greater for chromosomes of centromere paralog group A than of group B (Fig EV3B; paired t‐test mean difference in aneuploid frequencies P = 0.0001846). Furthermore, random pairings of the data demonstrated that the increased incidence of aneuploidy for chromosomes in group A is significantly greater than chance alone would predict (Fig EV3C; P < 0.00004).
To examine the factors underlying chromosomal aneuploidy further, we investigated linear regression models of aneuploid frequency across all 1,767 yeast strains. We considered factors such as chromosome size, number of open reading frames (ORFs) per chromosome, relative fitness of strains with specific disomic chromosomes (Beach et al, 2017), centromere GC percentage, and centromere paralog groups. In all cases, linear regression models which incorporated the centromere paralog group factor performed well, with models further incorporating chromosome size or number of ORFs in addition to the centromere paralog group factor performing the best (Fig EV3D). Lastly, the number of aneuploids per chromosome across all 1,767 examined strains showed a significant negative correlation with chromosome size when we considered the two distinct groupings (Fig 2C), suggesting that group B chromosomes have unexpectedly lower frequency of aneuploidy than chromosome size alone would predict. These analyses show that chromosome‐specific aneuploidy is biased between centromere paralog pairs and is influenced by both chromosome size and gene‐specific differences between the tolerance of chromosomes. In sum, these data suggest that yeast have a non‐random aneuploidy landscape shaped by the evolutionary history of their chromosomes and centromeres (Fig 2D), which may be one factor in explaining the observed frequency of aneuploidy in our data and the meta‐analysis of 1,767 yeast strains.
Chromosome aneuploidies are not adaptive to human histones
The nonrandom aneuploidy landscape driven by paralog type, predicts aneuploidy is non‐adaptive in histone humanized yeasts, we, therefore, explicitly tested this prediction. For this experiment, we used the histone humanized strain yHs5, which has the scc4 D65Y mutation and has eight persistently aneuploid chromosomes (Chromosomes I, II, III, V, IX, XI, XII, XVI; Truong & Boeke, 2017). We hypothesized that if aneuploidy were adaptive, then a strain with preexisting aneuploidy would generate histone humanized yeast at a higher rate than isogenic a euploid strain. To generate the isogenic strains we “captured” yHs5 clones in various ploidy states by “re‐yeastification” of its histones. This was achieved by transforming yHs5 with a plasmid encoding yeast histones (Fig EV4A). We assessed 12 “re‐yeastified” transformants for aneuploidy using flow cytometry and observed that eight transformants had heavy aneuploid loads and four transformants were either euploid or near‐euploid (Fig EV4B). Our prediction, if aneuploidy were adaptive, was that when we humanized these strains again the ‘re‐yeastified’ strains with preexisting aneuploids would humanize at a higher frequency than the euploid counterparts. On the contrary, we observed that the level of preexisting aneuploidy did not affect the humanization rate at all, as both isogenic aneuploid and euploid shuffle strains humanized at the same rate (Fig EV4C). From this experiment, we conclude that aneuploidy does not provide a selective advantage for histone‐humanization. These data are consistent with the idea that the missense scc4 D65Y mutation is the driving force for adaptation to histone humanization.
Figure EV4. Aneuploidy is non‐adaptive for histone‐humanization.
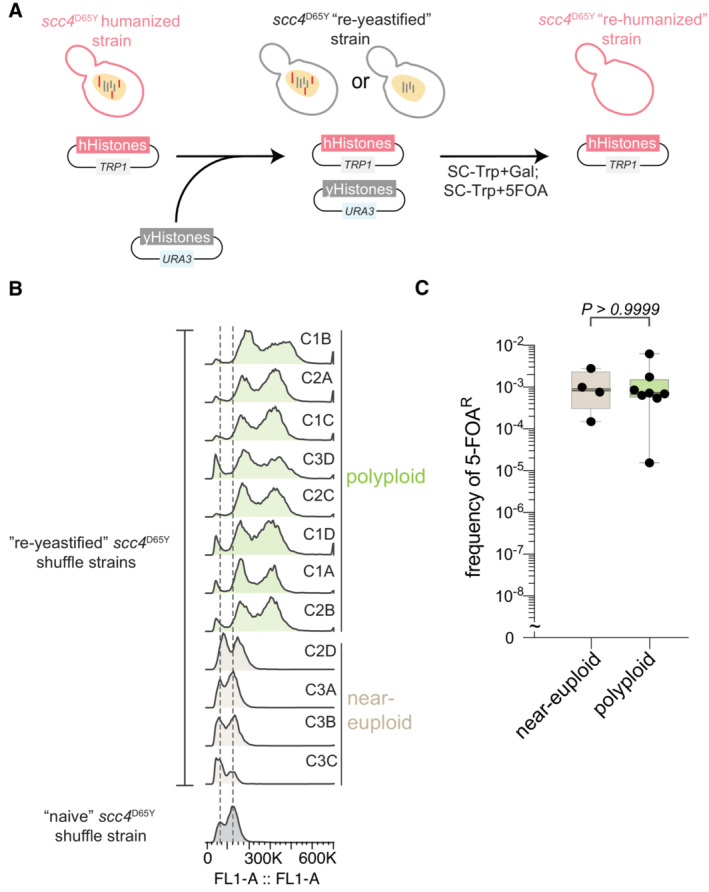
- Schematic of the re‐yeastification process. First an already histone‐humanized strain is transformed with the plasmid encoding all four yeast core histones, then is immediately re‐humanized by counterselection of the same.
- Ploidy analysis using flow cytometry of the re‐yeastified scc4 D65Y strains and the parental strain that has never had human histones.
- Frequency of 5–FOAR‐resistant colonies following histone humanization of the re‐yeastified strains. Kruskal–Wallis test with Dunn's multiple corrections; test against the parent scc4 D65Y strain show that both euploid and polyploidy strains humanize at significantly higher rates, P = 0.009 and P = 0.0034, respectively. Each dot represents a biological replicate of the histone humanization assay (n ≥ 4), the central band represents the median, the box extends from the 25th to 75th percentile, and the whiskers represent the minimum to maximum.
Histone‐humanized yeasts display chromosome instability caused by centromere dysfunction
The above results suggest that human histones induce chromosome instability. Our previous work showed that centromeric DNA is sensitized to micrococcal nuclease (MNase) digestion in the histone‐humanized yeasts (Truong & Boeke, 2017). This is consistent with lower occupancy of the centromeric nucleosome. Normally, the 16 centromere‐kinetochore attachments are clustered throughout the entirety of the cell cycle and transcriptional activity at centromeres, which is tightly regulated, is important for establishing these attachments (Ling & Yuen, 2019; Hedouin et al, 2022). We reasoned that human histones may expose centromeric DNA and create more transcriptionally open chromatin. We first, reanalyzed MNase digested chromatin sequencing data to infer nucleosome occupancy. We observed that centromeric nucleosome occupancy is severely depleted in histone‐humanized yeasts (Fig 3A). This was true of both humanized lineages tested (yHs7 and yHs5, with the DAD1 E50D and scc4 D65Y mutations, respectively), with no significant differences between the two humanized lineages.
Figure 3. Histone humanization cause centromere dysfunction in yeast.
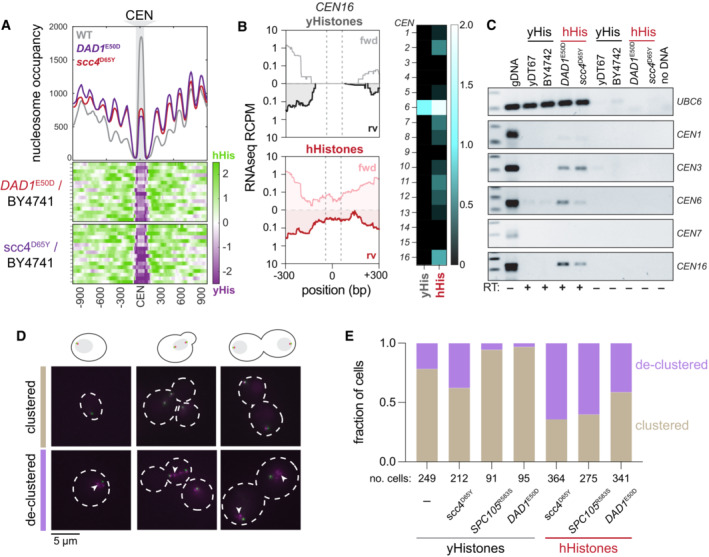
- Centromeric nucleosome occupancy. The relative nucleosome occupancy plots are given for each strain in the top plot (gray, wild type; red, DAD1 E50D humanized; purple, scc4 D65Y humanized). Values represent the chromosome mean occupancy 900 bp up and downstream of all 16 centromeres (each centromere occupancy values being an average of three biological replicates). Bottom plots, log2 ratio nucleosome occupancy enriched in humanized (green) and depleted in humanized (purple), each row represents 1 of the 16 centromeric regions.
- Total RNA sequencing of cenRNAs in yeasts. Left; read counts per million tracks for CEN16 in wild type (black, top) and DAD1 E50D humanized (red, bottom) yeasts. Right; heatmap of cenRNAs transcripts per million counts across all chromosomes in wildtype and DAD1 E50D humanized strains. Centromere VI is marked with an asterisk to indicate that the plasmid encoding the histone plasmid also encodes a CEN6 sequence, which reads likely emanant from (See Appendix Fig S3).
- RT‐PCRs of the indicated RNAs. Lane 1 is genomic DNA, lane 2 is wildtype shuffle strain (yDT67), lane 3 is BY4742, lane 4 is DAD1 E50D humanized, lane 5 is scc4 D65Y humanized, lanes 6–9 are the same as 2–5, but without reverse transcriptase added, lane 10 is no DNA.
- Example images from strains with fluorescent tags on the microtubule organizing center (Spc110‐GFP) and the kinetochore (Nuf2‐RFP). Images were taken from cultures in mid‐log phase of growth.
- Bar plots of the fraction of cells with clustered or declustered Nuf2‐RFP foci. Brown shading indicates a fraction of cells with correct clustering, and the purple shading indicates a fraction of cells with declustered Nuf2‐RFP foci. The number of cells analyzed for each genotype is shown below.
We next assessed the levels of CEN transcription genome‐wide by total‐RNA sequencing on WT (those with yeast histones) and histone humanized yeasts. Since CEN RNAs are rare in WT yeast, this assay is expected to fail to detect any meaningful amount of CEN RNAs (Hedouin et al, 2022). In our data, we were able to identify robust CEN RNA transcription from the humanized lineages tested, while failing to detect such transcription in WT cells (Fig 3B and C). CEN6 RNA was the most robustly transcribed centromere (Fig 3B; right). This result can be attributed to reads that emanate from the plasmid‐borne centromere sequence—plasmids encoding the histone genes also encode a minimal CEN6 sequence—and not to chromosomal CEN6. Indeed we observed fewer reads mapping to chromosomal CEN6 in WT cells versus humanized cells (Appendix Fig S3A). These results were confirmed by RT‐PCR on a subset of yeast CEN RNAs (Fig 3C). Importantly we used a polyT18 oligo for the RT reaction as CEN RNAs are polyadenylated (Ling & Yuen, 2019). The two humanized lineages tested showed robust CEN RNA expression for chromosomes III, VI, and XVI, whereas the two WT controls showed little to no expression (Fig 3C). Given that the RT‐PCR primers used are not complementary to the plasmid‐borne CEN6 sequence, we observed little amplification in the WT strains. We confirmed these results for CEN6 RNA by quantitative RT‐PCR. We observed, on average, a ~ 5‐fold increase in CEN6 RNA in histone humanized yeasts versus WT (Appendix Fig S3B). In sum, these results suggest that elevated CEN RNA transcription is a general defect caused by human histones. Altogether our results indicate that the structure of the histone‐humanized centromeric chromatin is under a persistent state of dysfunction, as indicated by the increased MNase sensitivity and transcription of the centromeres.
Given the strong functional relationship between centromere and kinetochore, we wondered if their coupling could also be compromised in the histone humanized yeasts. We next investigated kinetochore clustering by imaging log phase cells with an RFP‐tagged kinetochore protein Nuf2 in WT and humanized backgrounds (Fig 3D). We first compared kinetochore clustering in four different genetic backgrounds (WT, DAD1 E50D, SPC105 R583S, and scc4 D65Y) with yeast histones. We observed moderate levels of Nuf2‐RFP foci declustering in both WT (note, WT here is the histone shuffle strain) and scc4 D65Y strains. In comparison, we observed improved Nuf2‐RFP clustering in both the DAD1 E50D and SPC105 R583S strains (Fig 3E).
We next generated histone‐humanized strains with Nuf2‐RFP for the three mutants (DAD1 E50D, SPC105 R583S, and scc4 D65Y) and performed the same imaging experiment. In our three histone‐humanized strains, we observed substantial Nuf2‐RFP declustering, with over 70% of cells in the scc4 D65Y background exhibiting declustered Nuf2‐RFP foci (Fig 3E). These data are consistent with centromere dysfunction driving higher rates of chromosome instability. Intriguingly, the DAD1 E50D background showed only 30% of cells with declustered Nuf2‐RFP foci, whereas SPC105 R583S background showed over 60% with declustered Nuf2‐RFP foci. Despite the marked improvement to kinetochore clustering in the humanized DAD1 E50D mutant, it showed little difference with the scc4 D65Y mutant in terms of CEN RNA transcription or CEN MNase sensitivity (Fig 3A–C and E). This suggests that DAD1 E50D mutant, and perhaps other DASH mutants, rescues chromosome instability independently of directly rescuing the human histone‐induced centromere dysfunction.
DASH/Dam1c mutants suppress chromosome instability and lead to euploidy
We were curious if the DASH mutants suppress chromosome instability, as suggested by the kinetochore clustering data. We, therefore, sought to understand the dynamics of aneuploidy from the ancestral to the evolved histone humanized strains in lineages that evolved DASH mutations. Strikingly, the lineage yHs13, which evolved the DAM1 N80Y mutation, progressed from presenting chromosome XII aneuploidy to complete euploidy in the evolved strain (Fig 4A). Across all lineages sequenced, we identified a total of four lineages (yHs4, yHs7, yHs9, and yHs13) with mutant DASH/Dam1c genes (Appendix Table S1 and Truong & Boeke, 2017), and on average these evolved lineages had significantly fewer aneuploid chromosomes compared to all other evolved lineages (Fig 4B). Furthermore, these four lineages saw a decrease, on average, of 2–3 fewer aneuploid chromosomes when comparing the evolved to the ancestral isolates (Fig 4C). These results suggest that the DASH mutants suppress chromosome instability and have an “aneuploidy‐reducing” phenotype.
Figure 4. DASH/Dam1c mutations are sufficient for aneuploid reduction.
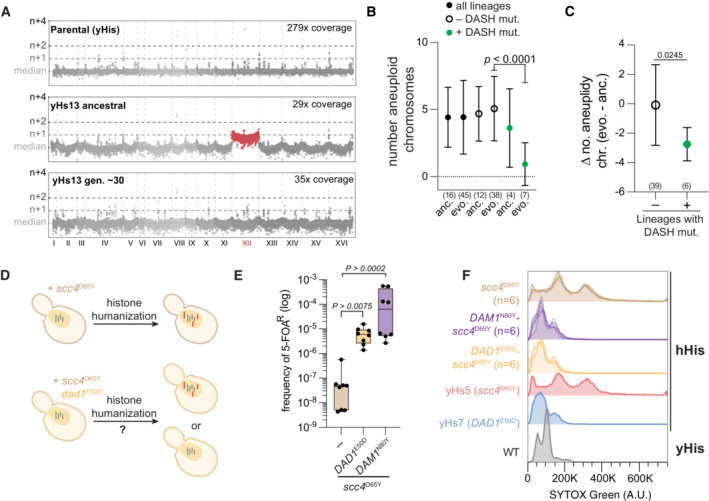
- Genome‐wide chromosome coverage maps for the parental (yeast histones) and the ancestral and evolved yHs13 strains (note the loss of aneuploid chromosome XII in the evolved isolate). Note that yHs13 is the lineage that evolved the mutation DAM1 N80Y. Chromosome coverage copy was normalized as the log2 ratio of the average coverage of a 1 Kb window by the genome median coverage.
- Evolution of aneuploid chromosomes in histone‐humanized strains. The number of aneuploid chromosomes is given for each of the indicated grouping of humanized strains. The sample size is given below each (number of histone‐humanized lineages), Dunnett's tests of the mean differences between the three ancestral‐evolved pairs are provided. Error bars report the standard deviation.
- Change in the average number of aneuploid chromosomes in humanized lineages with or without DASH/Dam1c mutations. The sample size is given below each, unpaired t‐test of the mean difference is provided. Error bars report the standard deviation.
- Aneuploidy accumulation assay. Histone humanization in the scc4 D65Y genetic background results in persistent aneuploidy (red chromosomes), does the presence of a second mutation, DAD1 E50D or DAM1 N80Y, stop the accumulation of aneuploidies to maintain euploidy (gray chromosomes)?
- DASH mutants increase histone humanization in the scc4 D65Y background. Humanization rates of the scc4 D65Y mutant with and without DAD1 E50D or DAM1 N80Y. The significance of the mean difference in 5‐FOAR frequency was determined with the Mann–Whitney test. Each dot represents one biological replicate (n = 8), the central band represents the median, the box extends from the 25th to 75th percentiles, and the whiskers represent the minimum and maximum.
- Ploidy analysis by flow cytometry in the scc4 D65Y humanized mutants from panel (D). Flow cytometry data from humanized lineages yHs5 and yHs7 are shown for aneuploid and euploid controls, respectively.
We next performed a laboratory evolution experiment to test the sufficiency of the DASH mutants apparent “aneuploidy‐reducing” phenotype (Appendix Fig S4A). To this end, we took an already histone‐humanized lineage (yHs16), which harbored seven aneuploid chromosomes in the ancestral strain (Appendix Figs S4A and S5A), and transformed in a plasmid containing either DAD1 WT or the mutant DAD1 E50D sequence. Three transformants each were selected and continually passaged in media to select for the plasmid over the course of 4 months, totaling ~ 90 generations. The ancestral strains, and evolved populations at ~ 30 and ~ 90 generations were frozen and whole genome sequencing was performed to determine chromosomal copy number. The clones transformed with the DAD1 WT plasmid did not show a significant change in the copy number of the ancestral aneuploid chromosomes, highlighting the persistent nature of aneuploidy in humanized strains (Appendix Figs S4A–C and S5B). In contrast, 2 of the 3 clones transformed with mutant DAD1 E50D showed nearly complete loss of all aneuploid chromosomes by 90 generations. Notably, one clone achieved euploidy almost immediately after acquiring DAD1 E50D, while the other more slowly reduced aneuploidies as the experiment progressed (Appendix Figs S4A–C and S5C). Interestingly, clone one, which immediately returned to euploidy, eventually lost the DAD1 E50D mutant plasmid at generation 90 (Appendix Fig S4D). Further, the loss of DAD1 E50D plasmid coincided with the appearance of two new mutations including Sli15 D331Y, a component of the chromosomal passenger complex (CPC) that regulates kinetochore‐microtubule attachments, and Smc5 H984N, subunit of the Smc5‐Smc6 complex involved in chromosome separation (Appendix Fig S4E and F). The return‐to‐euploidy in both clones was confirmed by computing the log2 ratio median chromosome coverage, which returned to a log2 genome median of ~ 0 (Appendix Fig S4C). This experiment was conducted in the presence of the native genomic DAD1 gene, where the mutant gene—encoded on an episomal plasmid—is in the context of additional mutations (Appendix Table S1, see yHs16). Therefore, we cannot exclude the hypothesis of reduced phenotypic penetrance of DAD1 E50D in this experiment. These results suggest that at least some mutants of the DASH complex are sufficient for aneuploidy reduction.
We next asked if these DASH mutants could prevent aneuploid chromosomes from accumulating (Fig 4D). To assess this, we compared the humanization frequency of the scc4 D65Y mutant shuffle strain in the presence or absence of DASH mutants (DAD1 E50D and DAM1 N80Y). While the rate of humanization for scc4 D65Y mutant alone was low (~ 1–10 per million cells), in the presence of either the DAD1 E50D or DAM1 N80Y mutants the humanization rate increased 100–1,000‐fold (Fig 4E). In the shuffle strain with only the mutation scc4 D65Y, we observed concomitant increases of the ploidy levels upon humanization (Fig 4F). In stark contrast, when the combination of mutations of either scc4 D65Y with DAD1 E50D or DAM1 N80Y were humanized, neither clone showed evidence of aneuploidy, as measured by DNA content by flow cytometry (Fig 4F). Therefore, we conclude that both DAD1 E50D and DAM1 N80Y mutants are sufficient to suppress chromosome instability and cure aneuploidy in the histone‐humanized yeasts.
DASH/Dam1c mutants suppress ipl1‐2 driven chromosome instability
The Aurora B kinase, IPL1 in yeast, is the master regulator of kinetochore‐microtubule attachments, the disruption of which leads to defects in chromosome segregation (Biggins et al, 1999; Pinsky et al, 2006, p. 1). We, therefore, examined the interaction with the temperature‐sensitive ipl1‐2 allele and our DASH/Dam1 mutants. The mutant ipl1‐2 is known to increase ploidy by chromosome missegregation, causes severe growth defects at non‐permissive temperatures and sensitivity to benomyl (Chan & Botstein, 1993). We grew strains at permissible (24°C) and non‐permissible temperatures (32°C) on YPD and also at the non‐permissible temperature with the addition of the microtubule depolymerizing agent, benomyl. The combination of the non‐permissive temperature and benomyl led to lethality in the ipl1‐2 background (Fig 5A; Appendix Fig S6). We next constructed double mutants of ipl1‐2 in combination with the outer kinetochore mutations and tested for the rescue of the ipl1‐2 phenotype (note: these experiments were done in a genetic background with yeast histones encoded at the normal chromosomal loci). We observed that the tested DASH mutants rescued the temperature and benomyl sensitivities (Fig 5A; Appendix Fig S6A). We note ipl1‐2 temperature sensitivity was only slightly rescued by SPC105 R583S and NDC80 E568A mutants. These results are consistent with the Nuf2 clustering data that the DASH mutants strongly suppress chromosome instability, whereas the other outer kinetochore complex mutants do not.
Figure 5. DASH/Dam1c mutants rescue Aurora B kinase and SAC mutants.
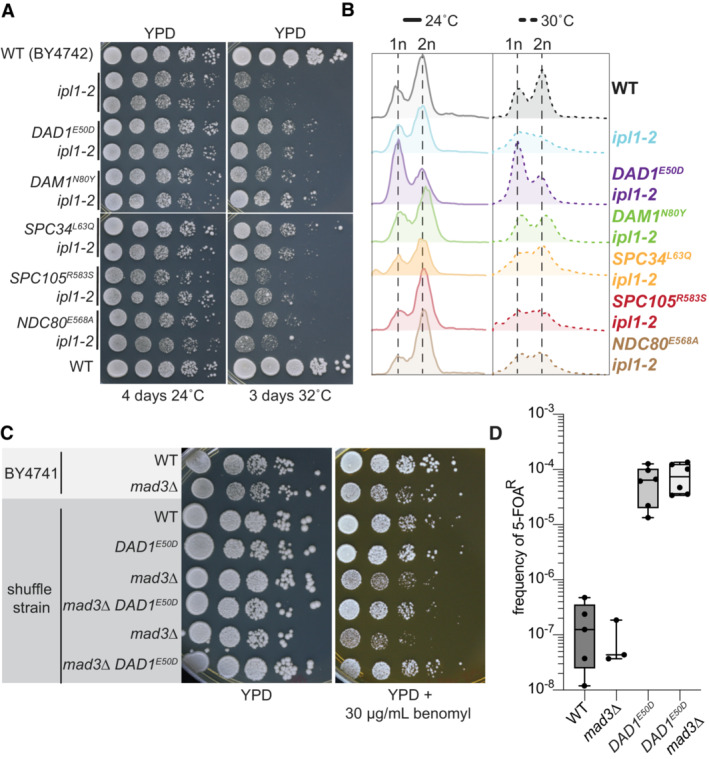
- Growth assays of wild type, ipl1‐2 mutants, and ipl1‐2 kinetochore double mutants at permissive (24°C) and non‐permissive temperature (32°C). Ten‐fold serial dilutions of ~ 1.0 OD600 yeast cultures were spotted onto YPD solid media and grown at the indicated temperatures for 4 and 3 days, respectively.
- Flow cytometry analysis of wild type, ipl1‐2 mutants, and ipl1‐2 kinetochore double mutants at the permissive (24°C) and non‐permissive temperature (30°C). Yeast cultures were grown to logarithmic phase at 24°C and divided and grown at 24 and 30°C for 6 h before cells were collected and processed for DNA content analysis using Sytox Green stain.
- Growth assays of wild type, mad3∆ mutants, and mad3∆ DAD1 E50D double mutants on YPD with and without 30 μg/ml benomyl. Ten‐fold serial dilutions of ~ 1.0 OD600 yeast cultures were spotted onto the indicated solid media and grown at 30°C for 3 days.
- Humanization of mad3∆ mutant and double mad3∆ DAD1 E50D mutant. Each dot represents a biological replicate (n ≥ 3), the central band represents the median, the box extends from the 25th to 75th percentile, and the whiskers represent the minimum and maximum.
Chromosome missegregation in the ipl1‐2 strain can be assayed by flow cytometry (Chan & Botstein, 1993). When grown at the non‐permissible temperature the ipl1‐2 mutant lost the distinctive 1n and 2n ploidy peaks of the cycling population (Fig 5B). In agreement with the spot assays, flow cytometry of cells grown at the permissive or non‐permissive temperatures showed that the DASH/Dam1c mutants were able to fully rescue the defective cycling peaks of the ipl1‐2 mutant (Fig 5B). However, the NDC80 E568A and SPC105 R583S mutants failed to rescue the cycling peaks. Importantly, in the absence of ipl1‐2 the DASH/Dam1c mutants and both NDC80 E568A and SPC105 R583S showed normal 1n and 2n peaks at either 24 or 30°C (Appendix Fig S6B). We conclude that the DASH/Dam1c mutants not only stabilize ploidy levels outside of the genetic background of histone humanized yeast but also effectively suppress aneuploidy in ipl1‐2 cells.
We next investigated whether the DASH/Dam1c mutants could suppress the phenotypic defect of a spindle assembly checkpoint (SAC) mutant, which delays the onset of sister chromosome separation to allow for corrections of unattached or incorrect kinetochore‐microtubule connections (Musacchio & Salmon, 2007). We found that the DAD1 E50D mutation is able to suppress the benomyl sensitivity of the SAC mutant, mad3∆ (Fig 5C). The humanization of a DAD1 E50D mad3∆ strain showed that SAC activity was dispensable for robust humanization (Fig 5D). This suggests that the activity of the SAC is not required for histone humanization in the DAD1 E50D background and that the DAD1 E50D mutant suppresses chromosome instability independently of this regulatory pathway.
Molecular basis for suppression of histone‐humanization by DAD1 E50D and DAM1 N80Y
To gain insights into the molecular mechanism of the suppression of chromosome instability, we mapped our mutants onto the cryo‐EM structure of Chaetomium thermophilum's DASH/Dam1 complex (Jenni & Harrison, 2018). The mutant residues of Dad1 and Dam1 are separated by less than ~ 4 Å (Fig 6A), we thus hypothesized that their interaction may underlie a shared molecular mechanism through which they suppress chromosome instability. First, we took a molecular modeling approach to gain insight into the structural changes imparted by the DAD1 E50D and DAM1 N80Y mutations. In the cryo‐EM structure, the residue Glu50 of Dad1 is stabilized by interactions with the neighboring residue Arg47 (Fig 6A), which is not conserved in S. cerevisiae (Fig 6E). We next constructed a homology model of S. cerevisiae's DASH/Dam1c complex to understand how residue Glu50 is arranged in the absence of residue Arg47. In our model, the residue Glu50 of Dad1 interacts exclusively with the residue Asn80 of Dam1 (Fig 6A), potentially via a weak hydrogen bond. Given that either mutation, DAD1 E50D or DAM1 N80Y, has the potential to disrupt this interaction, we modeled both mutations onto the homology structure of S. cerevisiae's DASH complex. In either case, the mutations resulted in a loss of the WT Glu50‐Asn80 interaction (Fig 6A). Secondly, we experimentally mutated the Asn80 residue of Dam1 to every other amino acid, with the idea being that any mutation at position Asn80 of Dam1 will disrupt the interaction with Glu50 of Dad1. Remarkably, any mutation that we introduced to position 80 of Dam1 significantly improved the rate of humanization over that of WT Dam1, although the original DAM1 N80Y mutation was superior (Fig 6B; Appendix Fig S7). These data support our model that the loss of the Glu50‐Asn80 interaction is a key feature for suppression.
Figure 6. Molecular basis for suppression of histone humanization by DAD1 E50D and DAM1 N80Y .
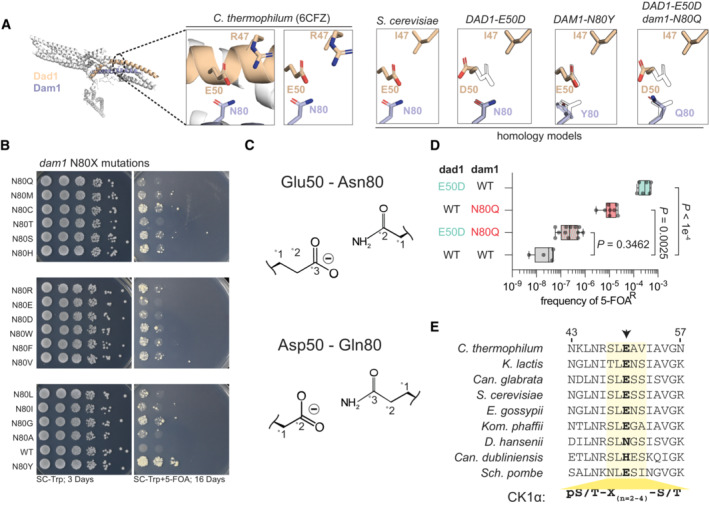
- Homology models of S. cerevisiae DASH/Dam1c complex. The decametric DASH/Dam1c Cyro‐EM structure of C. thermophilum is shown, with Dad1 and Dam1 highlighted. The corresponding homology model is shown to the right, with various mutant models shown (with outlined WT residues for reference). Note the relative positioning of Dad1 residue 50 and Dam1 residue 80.
- Histone‐humanization assay for various dam1 N80X mutants. 5‐FOA is used to counter‐select the yeast histone plasmid, forcing growth with the human histone plasmid. Yeast was serially diluted from a starting culture of 1.0 OD600.
- Cartoon interactions for complementary pairings of Dad1 residue 50 and Dam1 residue 80.
- The dual mutant Dad1‐Asp50—Dam1‐Gln80 DASH/Dam1c fails to humanize, while the single mutants readily humanize. The significance of the mean difference in 5–FOAR frequency was determined with the Mann–Whitney test. Each dot represents a biological replicate (n ≥ 4), the central band represents the median, the box extends from the 25th to 75th percentile, and the whiskers represent the minimum and maximum.
- Protein alignment of Dad1 orthologs, highlighting a conserved casein 1 kinase consensus phosphorylation site (yellow‐shaded region). Arrow indicates the mutant residue 50 of Dad1.
We reasoned that we could restore the Glu50‐Asn80 hydrogen bond interaction via an alternative amino acid pairing, noting that it may be restored by the similar duo of amino acids, Dad1Asp50‐Dam1Gln80 (Fig 6C). In agreement, our molecular modeling suggested that the Asp50‐Gln80 pairing forms an interaction similar to the WT Glu50‐Asn80 interaction (Fig 6A), therefore, the yeast containing both mutations should fail to humanize or humanize at a lower rate than either single mutation alone. As before, we found that the single Dad1‐Asp50 or Dam1‐Gln80 mutations humanized at rates ~ 10,000‐ and ~ 370‐fold, respectively, over WT (Fig 6D). As predicted, the double Dad1‐Asp50 Dam1‐Gln80 mutant only weakly humanized at a rate of only ~ 8.5‐fold over WT, significantly worse than either mutation alone, showing that restoration of this hydrogen bond is sufficient to lower the ability to undergo histone humanization (Fig 6D). However, loss of this hydrogen bond is not the only mechanism at play as it does not account for the difference in humanization rates between the DAM1 N80Y and dam1 N80Q variants. We, therefore, tested if the glutamic acid at residue 50 of Dad1 was essential for humanization by the DAM1 N80Y mutation. To this end, we generated a dad1 E50A and DAM1 N80Y shuffle strains and performed the histone humanization assay. As expected the single dad1 E50A mutation resulted in an elevated rate of histone humanization, as predicted due to disruption of the hydrogen bond (Appendix Fig S7B). Furthermore, we observed that humanization in the double dad1 E50A DAM1 N80Y mutant strain was significantly reduced in comparison to the DAM1 N80Y alone (Appendix Fig S7B). These data suggest that suppression of histone humanization by DAM1 N80Y functions through additional means that are dependent on the glutamic acid at residue 50 of Dad1. Taken together, these data point to disruption of the hydrogen bond between Dad1‐Glu50 and Dam1‐Asn80 being one critical aspect for aneuploidy suppression by the DAD1 E50D and DAM1 N80Y mutations.
DAD1 E50D disrupts DASH complex oligomerization and weakens microtubule attachments
What is the mechanism of suppression for the DAD1 E50D mutation downstream of the loss of the hydrogen bond? We note that the Glu50 residue of Dad1 falls into the middle of an evolutionarily conserved region that lies precisely at a critical interface of DASH/Dam1c oligomerization (Figs 6E and 7A; Jenni & Harrison, 2018). This suggested that these mutations may alter the oligomerization of the DASH complex. To this end, we purified recombinant WT and mutant DAD1 E50D DASH complexes to characterize the effect of the E50D mutation in vitro. The DASH complex makes visually impressive rings around microtubules in vitro (Miranda et al, 2005) and we wondered if the DAD1 E50D mutant might perturb this feature. We, therefore, reconstituted both WT and mutant DAD1 E50D DASH complexes on microtubules and imaged them using negative stain electron microscopy. We observed that both the WT and mutant DAD1 E50D DASH complexes formed complete rings around microtubules, but noted that the DAD1 E50D rings appeared less distinct (Figs 7B and EV5A). Analysis of single rings confirmed that mutant DAD1 E50D DASH complex formed rings that are less well defined as indicated by a decrease in the average intensity of pixels near the edges of the microtubule lattice (Fig 7B and C).
Figure 7. DAD1 E50D disrupts DASH complex oligomerization and weakens microtubule attachments.
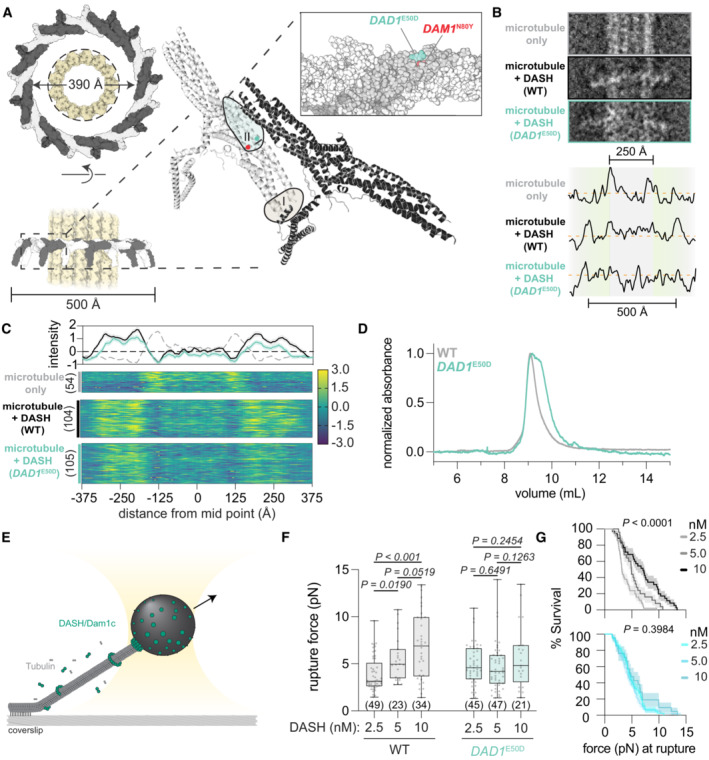
- Structure of the 17‐member DASH/Dam1c ring of C. thermophilum around a microtubule lattice is shown (6CFZ and 5SYF, respectively). The ring, with a 390 Å interior circumference, encircles a single microtubule with a width of 250 Å (dashed line). Individual protomers oligomerize through interactions of two conserved interfaces, with the DAD1 E50 and DAM1 N80 residues highlighted in the zoomed‐in region of interface II.
- Analysis of DASH complex rings around microtubules. Top, example images are shown for the three cases measured (bare microtubules, WT DASH, and DAD1 E50D DASH). The bare microtubule lattice was measured from the same micrographs with a DASH complex present, where sections that showed no DASH complex were measured. Below the z‐score normalized pixel intensities for the example EM micrographs. Note the increased intensity outside the range of the diameter of the microtubule lattice for samples with the DASH complex.
- Heatmaps show the z‐score normalized pixel intensities along a straight edge bisecting the width of the microtubule or microtubule + DASH complex. The number of particles analyzed is shown in parentheses. Above are the average profiles of pixel intensity with 95% CI. The dashed gray line is the average profile of pixel intensity of bare microtubules.
- Size exclusion chromatography of the indicated DASH complexes.
- Schematic of the optical trap experiment with wildtype and mutant DAD1 E50D DASH complex on microtubules.
- Rupture force assay from experiments at three concentrations of DASH complex incubated with the beads. Briefly, 3.5 pM of beads were incubated with the indicated concentrations of DASH complex (representing a ratio of DASH complex to beads of 714:1, 1,428:1, and 2,860:1). These DASH‐decorated beads were then washed before suspending in a reaction mix containing free tubulin and 5 nM free DASH complex. Two‐way ANOVA tests of the mean difference in rupture force are shown with multiple test corrections (false discovery rate). Each dot represents an individual rupture force measurement (n ≥ 21; the exact number shown below each box between parentheses), the central band represents the median, the box extends from the 25th to 75th percentile, and the whiskers represent the minimum and maximum.
- Survival curves for each DASH complex are shown at the indicated concentrations. Log‐rank tests were performed to test whether the survival curves were significantly different from one another. Error bars represent the standard error and are displayed as the shaded area.
Figure EV5. DAD1 E50D disrupts DASH/Dam1c oligomerization and reduces its dimerization.
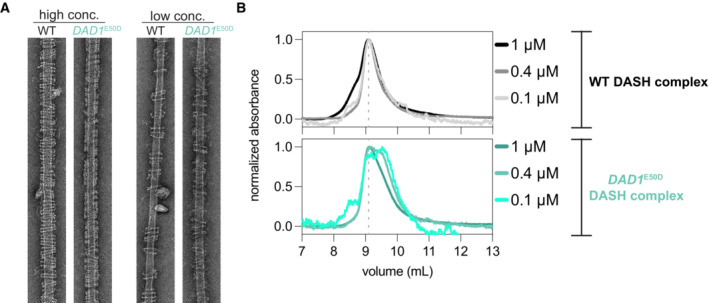
- Example EM images of DASH rings taken at low and high concentrations. High concentration > 25 nM complex and low concentration 11–16 nM complex. Microtubules are at a concentration of 20 nM tubulin dimer.
- Size exclusion chromatography of WT (upper graph) and mutant DAD1 E50D (lower graph) DASH complexes. Three dilutions of the DASH complex are shown. SEC analysis at lower concentrations of the mutant complex, but not for the WT complex, reveal the emergence of a second peak due to a species with a lower apparent molecular weight.
While the DAD1 E50D mutant still formed rings, the above single‐ring analysis suggested oligomerization may be subtly compromised. We hypothesized that the DAD1 E50D mutant might affect the dimerization of the complex in the absence of microtubules. To test this, we performed size exclusion chromatography for both the WT and mutant DAD1 E50D complexes. Previous studies have demonstrated that the WT DASH complex forms a dimer in solution (Umbreit et al, 2014). Indeed, we observed a single major peak eluting at ~ 9 ml, corresponding to the WT DASH complex dimer (Figs 7D and EV5B). However, for the DAD1 E50D mutant complex, we observed a broadening of this peak with a second major peak appearing at ~ 9.5 ml, which more clearly appeared at lower concentrations of complex (Figs 7D and EV5B). From these data, we conclude that the DAD1 E50D mutation weakens the interaction between DASH/Dam1c protomers, which we observe as a decrease in the dimer fraction and the appearance of a second species of lower apparent molecular weight.
We predicted that the decrease in dimerization ability of the DAD1 E50D DASH complex would weaken microtubule attachments. We tested the strength of DASH microtubule attachments using a rupture force assay (Fig 7E). We observed for the WT DASH complex that the load‐bearing strength increased when higher concentrations of the complex were added to the beads—in agreement with oligomerization driving stronger attachments (Fig 7F and G; Umbreit et al, 2014). However, for the mutant DAD1 E50D DASH complex we observed no scaling in the strength of attachment, as the mean rupture force did not significantly differ at the tested concentrations (Fig 7F and G). Put together our biochemical and rupture force assays suggest that the DAD1 E50D mutant weakens the interaction interface between individual protomers, which leads to reduced dimerization and ultimately reduced oligomerization and ring formation to drive weakened microtubule attachments.
Trade‐off between mitotic chromosome segregation versus successful meiosis
We have shown that the subtle DAD1 E50D mutation improves the fidelity of mitotic chromosome segregation in certain conditions. Why then is the glutamic acid at residue 50 of Dad1 nearly invariant across species (Fig 6E), if DAD1 E50D appears more fit in mitosis? To this end, we considered meiosis as a potential explanation for why this residue is so well conserved—hypothesizing that the DAD1 E50D mutation would be detrimental to meiosis. We thus made DAD1 E50D heterozygous and homozygous diploid mutants in the sporulation‐competent SK1 background and compared sporulation efficiency to WT SK1 diploid (Fig 8A). We observed that the DAD1 E50D mutation reduced sporulation efficiency in either a heterozygous or homozygous state, however, sporulation efficiency was severely impaired in the heterozygous mutant diploid (Fig 8A). Additionally, both heterozygous and homozygous mutants produced a greater fraction of abnormal and immature asci after 5 days in sporulation medium (Appendix Table S6). Next, we dissected asci which contained four fully‐formed spores to test for viability. We observed a significant decrease in spore viability specifically in the DAD1 E50D heterozygous mutant (Fig 8B). In contrast, spore viability was not significantly reduced in the homozygous mutants (Fig 8B). Intriguingly, in tetrads from heterozygous diploids which produced only two viable spores the frequency of the DAD1 E50D mutation was significantly enriched over the expected 1:1 ratio to DAD1 WT (Fig 8C; Appendix Fig S8). These data support the idea that WT DAD1 and DAD1 E50D are incompatible, with their interaction leading to an imbalance in the strength of microtubule attachments, perhaps explaining the poor viability of spores. Furthermore, the reduced sporulation efficiency in both DAD1 E50D hetero‐ and homozygous strains supports the idea that the weakened microtubule interaction of DAD1 E50D is not optimal for meiosis.
Figure 8. DAD1 E50D decreases the efficiency of sporulation and spore viability.
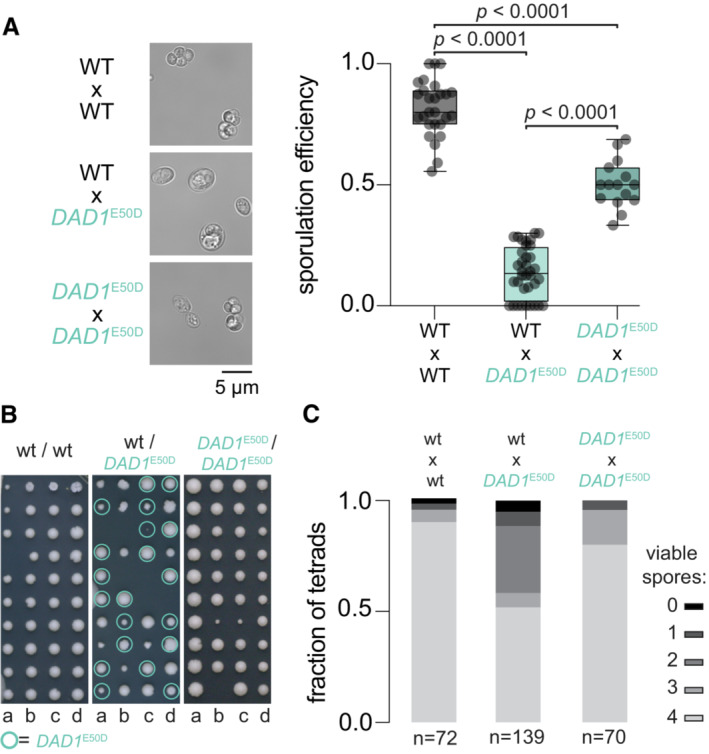
- Sporulation efficiency after 5 days in sporulation medium at 25°C. Example micrographs are shown to the left for both wildtype and DAD1 E50D SK1 strains. Right, quantification of average sporulation efficiency from all micrographs. Two‐tailed unpaired t‐test of the mean difference in sporulation efficiency. Each dot represents the fraction of cells sporulated in a single field of view (technical replicates; n ≥ 15), the central band represents the median, the box extends from the 25th to 75th percentile, and the whiskers represent the minimum and maximum.
- Spore viability assay. Individual tetrads were micromanipulated on an agar surface and spores were dissected and arrayed. Genotype was determined by PCR genotyping of the DAD1 locus. Quantification from all tetrad dissections performed, total are shown below. The one‐tailed probability of observing 51% spore viability in DAD1 E50D SK1 over a total of 139 tetrads is P < 0.000001, assuming spore viability of ~ 90% from wild‐type SK1.
- Genotyping of spores isolated from tetrads with either four viable spores or two viable spores. A fraction of spores with the indicated allele (gray DAD1 WT and pink DAD1 E50D) is displayed. The one‐tailed binomial test is shown for the probability of observing the frequency of the DAD1 E50D allele given equal segregation (DAD1 WT, P = 0.46; DAD1 E50D, P = 0.0011). Numbers below each bar indicates the total number of spores genotyped.
Discussion
Here we mechanistically determine how the budding yeast S. cerevisiae adapts to life with human histones. Following the initial humanization event the genome is shaped by chromosome instability and as a result a high burden of aneuploidy. Selective advantage for any mutation which enhances life with human chromatin is strong, with fitness improving rapidly, but gradually plateauing. We isolate and characterize a class of mutations that arose following the initial humanization event. These mutants increase the fidelity of chromosome segregation in response to centromere dysfunction and ultimately lead to ploidy stabilization and subsequently aneuploidy reduction.
We observed an elevated frequency of chromosomal aneuploidy for a non‐random set of chromosomes (Fig 2A). By examining the frequency of aneuploidy across 1,767 diverse strains we show that neither chromosome size (number of ORFs) or differences in the tolerance of specific disomic chromosomes explain the data best. Only when the model integrates centromere paralog pairs information does the model predict aneuploid frequency well. From these analyses, we propose that chromosome‐specific aneuploid frequency in S. cerevisiae may be driven in part by differences in ancient paralogous centromeres in combination with other factors such as gene‐specific tolerances and chromosome size. Our hypothesis suggests that functional differences between paralogous centromeres have been maintained since the ancient allopolyploidization event (Fig 2D), which informs a non‐random aneuploid landscape in yeast. Further study will be needed to investigate budding yeast centromere function in the light of centromere paralog evolution.
Successful chromosomal segregation hinges on correct kinetochore–microtubule attachments. As we show, centromere function is disrupted in histone‐humanized yeasts—regardless of the specific genetic background examined—yet, the DASH/Dam1c mutants display euploidy. We speculate that in the histone‐humanized cells the kinetochore–centromere attachments might be deficient, which could explain a few observations regarding the centromeres of histone‐humanized yeasts. First, the centromeres are extremely sensitive to MNase digestion, and second, they exhibit increased transcription. These observations suggest that the formation and maintenance of centromeric chromatin are perturbed by human histones—perhaps driven by species‐specific incompatibilities between human histones and the yeast centromeric histone H3, Cse4. Furthermore, the high rates of chromosome instability suggest that human histone‐induced centromere dysfunction may also disrupt the recruitment of additional factors to centromeres, such as Cohesin, Shugoshin, or the CPC (Cho & Harrison, 2012, p. 10; Verzijlbergen et al, 2014; Hinshaw et al, 2017).
How then do the DASH/Dam1c mutants suppress this centromere dysfunction? Our data suggest that the reduced occupancy of centromeric nucleosomes in histone humanized yeasts is concomitant with the reduction of Aurora B recruitment to centromeres and thus mitotic error correction. We propose that the DASH/Dam1c mutants suppress the loss of Aurora B activity through decreased microtubule binding which promotes increased kinetochore‐microtubule turnover. In this model, increased turnover of kinetochore‐microtubule attachments allows for reduced errors in segregation (Cimini et al, 2006; Zaytsev & Grishchuk, 2015). Increased cycles of attachment‐detachment caused by weakened microtubule‐kinetochore coupling may allow cells an opportunity to achieve bipolar attachment. Effectively, this can be thought of as a “blunt” corrective measure, that is potent when the initial rate of biorientation is low or when no mechanism of correction is functioning (i.e., lack of Aurora B activity). Thus, seemingly paradoxically, a compromised inner centromere is effectively rescued by weakening the strength of the outer kinetochore. Further study will reveal deeper insights into how histone‐humanized yeast adapt to human histones and the downstream mechanisms of the DAD1 E50D and DAM1 N80Y mutants. Of particular interest would be to assess whether kinetochores from histone‐humanized yeasts can sense tension. We speculate that the intrinsic catch bond‐like behavior of yeast kinetochores may remain functional in histone‐humanized yeasts, which when coupled alongside DAD1 E50D induced kinetochore‐microtubule turnover, may allow for stabilization of proper kinetochore‐microtubule attachments (Akiyoshi et al, 2010).
The phenotypic effects of the subtle DAD1 E50D mutation, from glutamic acid to aspartic acid, are dramatic. It underlies a potent adaptive route to life with human histones—which we propose is due to the suppression of centromere dysfunction. Furthermore, DAD1 E50D mutation is not only limited to the peculiarity of the histone‐humanized genetic system, as it suppresses chromosome instability and aneuploidy accumulation in genetic backgrounds with yeast histones (Fig 5). Our data collectively supports the notion that DAD1 E50D mutation enhances the fidelity of chromosome segregation during mitosis, especially during periods of chromosome instability (histone humanization or loss of Aurora B). However, the DAD1 E50D mutation is detrimental to the success of meiosis, especially in a heterozygous state. The meiotic defects we observed are likely a result of impairment to chromosome segregation during meiotic division, such as meiosis I. Interestingly, the DAD1 E50D mutation lies within a conserved predicted phosphorylation site of the casein kinase I Hrr25 (Fig 6E), which is a component of the meiosis I‐specific kinetochore crosslinking monopolin complex (Petronczki et al, 2006; Monje‐Casas et al, 2007; Corbett et al, 2010; Sarangapani et al, 2014). Whether or not Dad1 is a target of Hrr25 or if the DAD1 E50D mutation interferes with Hrr25 meiotic function is not known. However, the observed meiotic defects of the DAD1 E50D mutant suggest that this interface may be important for meiosis. It will be of interest to investigate if DASH/Dam1c oligomerization is regulated during the transition from meiosis I to meiosis II kinetochores.
During the preparation of our manuscript, Clarke et al (2023) reported strikingly similar conclusions from a completely orthogonal genetic screen for elevated rates of aneuploidy. Notably, they developed a screen for suppressors of deficiency in BIR1, encoding a component of the CPC, deletion of which leads to increased chromosome segregation defects and elevated aneuploidy. Remarkably, they isolated a distinct set of DASH complex mutants targeting the same oligomerization interface. Furthermore, they described a second pathway of bir1∆‐induced chromosome instability suppression through mutation of the CPC subunit Sli15 (G334S), which increases its microtubule binding independently of its centromere recruitment. We observed in our loss of aneuploidy experiment that the DAD1 E50D mutant was lost and supplanted by the Sli15 (D331Y) mutant, perhaps due to direct recruitment of the CPC to microtubules alleviating the benefit of the DAD1 E50D mutation. Our results complement and extend their findings to reveal the molecular and biochemical details demonstrating that weakened kinetochore–microtubule attachments provide a mechanistic path for the suppression of dysfunctional centromeres. In particular, our in vitro data directly illustrates that the DAD1 E50D mutation drives weakened microtubule attachments via impairment to DASH/Dam1c oligomerization.
Intriguingly, we exclusively isolated mutations in the outer kinetochore (DASH/Dam1c, Spc105c, and Ndc80c) and never in the inner kinetochore. This may be attributable to our genetic screen for suppressor mutants being undersaturated. We have to date only isolated mutants of the same gene twice (URA2 and GEX2), both of which are relatively large genes, and these mutations are seemingly inconsequential (synonymous and noncoding). Perhaps with more independent isolates of histone humanized yeasts, we would uncover mutants of the inner kinetochore. However, given that our evolution experiment is rather short (~ 30 generations) we do not expect to isolate mutants in the human histones or Cse4 (all encoded by small genes). Any mutation in the core histone genes is likely not to be fully penetrant since they are present episomally at 2–4 copies per cell. Finally, we have previously shown that multiple mutations to both human H3 and H2A are required to partially restore fitness to histone humanized yeasts (Truong & Boeke, 2017). Thus it is unlikely that any single missense mutation in either human histones or Cse4 would rescue incompatibility between human histones and yeast Cse4 at centromeric nucleosomes. Finally, it is important to consider what the outcome of the incompatibility at centromeric nucleosomes is. Our data suggests that dysfunction in the regulation of kinetochore‐microtubule attachments is the key phenotypic defect that arises from centromeric nucleosome incompatibility. Consequently, repeated isolations of outer kinetochore mutants and a lack of inner kinetochore mutants may be expected, as the former class of mutants is more likely to modify kinetochore‐microtubule attachments.
In sum, our data reveal a mechanism of adaptation to chromatin comprised of human histones in budding yeasts. We note that this is only one of many paths through which yeast may adapt to human histones, as we identified multiple cellular processes which are enriched in our list of candidate suppressor mutations. (Appendix Fig S1; Table EV1). Lastly, we highlight the unique evolutionary forces at play in histone humanized yeasts which not only illuminate the centrality of nucleosomes in eukaryotic life but afford a powerful system in which to probe biological functions.
Materials and Methods
Strains, plasmids and oligos
All strains used in this work are listed in Appendix Table S3 and are available upon request. To construct the haploid and diploid shuffle strains with human histone suppressor mutations we used CRISPR‐Cas9 to scarlessly introduce the mutation of interest. First, we designed sgRNAs targeting our genes of interest alongside repair templates to introduce the desired mutations (Fig EV2A). To clone the guide RNAs we designed a single oligo that consisted of 20 bp upstream the nearest NGG to our codon of interest, flanked by homology sequences for Gibson assembly into our expression plasmid (5′–TGAAAGATAAATGATC–20 bp–GTTTTAGAGCTAGAAA). For the Gibson assemblies, 1 μg of plasmid DNA was digested with NotI and CIP overnight and then purified with the Zymo DNA Clean and Concentrator kit (Zymo Research cat. D4030). Next, to the Gibson mix, we added 20 ng of digested plasmid with 1 μl of the sgRNA oligo (50 μM), mixed and incubated at 50°C for 1 h. The Gibson reaction was then diluted 1:10 and the dilution was transformed into chemically competent E. coli, the transformation was plated to the appropriate selection and clones were verified by sanger sequencing. We used a double‐stranded DNA (dsDNA) repair template to introduce the mutation and eliminate the PAM site. dsDNA was constructed from two oligos that are annealed and 5′ overhangs filled in using Klenow polymerase according to the manufacture's specifications (NEB cat. M0210L). All yeast transformations were carried out using the lithium‐acetate method. For each CRISPR edit, the strain of interest was first pre‐transformed with a Cas9 plasmid, and cells harboring Cas9 were transformed with the guide RNA plasmid and the dsDNA repair template. Edited cells were then selected by double selection of the Cas9 plasmid and the sgRNA plasmid. Strains were validated by sanger sequencing.
All plasmids are listed in Appendix Table S4. All oligos are listed in Appendix Table S5.
Humanization assay and evolution experiments
We humanized yeast's core histone genes as previously reported (Truong & Boeke, 2017; Haase et al, 2019). Briefly, a shuffle strain—which has the four histone gene clusters chromosomally deleted and has a single set of yeast histone genes on a URA3 plasmid—is transformed with a TRP1 plasmid encoding a single set of human histone genes. Importantly, the yeasts and human histone genes are expressed using orthogonal promoters and terminators. Next, each transformant is inoculated into 5 ml of SC‐Trp medium containing 2% galactose and 2% raffinose as the carbon source, this is to inactivate the conditional centromere on the yeast‐histone URA3 plasmid. Once the culture has reached saturation, a range of volumes (typically 1 μl for a robust humanizing strain or up to 1 ml for a weak humanizing strain) are plated to SC‐Trp agar plates containing 5‐FOA (1 mg ml−1), to counter‐select the URA3 marker (Haase et al, 2019). Plates are sealed in a Tupperware box with a damp paper towel and growth is monitored for up to 4 weeks, but longer in some cases. After a final count of colonies, which grew on the SC‐Trp + 5‐FOA plates, a humanization rate is calculated—the total number of colonies is divided by the estimated number of cells plated.
Colonies that appeared after 7 days of growth on plates with 5‐FOA were PCR genotyped to confirm the loss of the yeast histone genes (Haase et al, 2019). Confirmed clones were patched to rich media agar plates (YPD) and stored in glycerol at −80°C. A portion of each patch was then cultured in SC‐Trp and grown to saturation (generation 0). Cultures were then diluted by inoculating 5 ml of growth medium with 50 μl of saturated culture, this passaging was repeated for five cycles. At each time point a portion of the culture was stored in glycerol at −80°C.
Whole genome sequencing and variant analysis
Genomic DNA was extracted from stationary yeast cultures using a double phenol‐chloroform extraction method. Briefly, yeast pellets, from ~ 1.5–5 ml of culture, were disrupted by bead beating in a solution consisting of 225 μl 1× TES (TE buffer +0.5% SDS) plus 200 μl of 25:24:1 phenol:chloroform:isoamyl alcohol (Thermo Fisher cat. 15593031) in tubes with a pre‐aliquoted amount of 0.5 mm diameter yttria‐stabilized zirconium oxide beads (MP Bio cat. 116960050‐CF). A second step of phenol‐chloroform extraction was done for each sample and DNA was precipitated in 70% ethanol. Extracted genomic DNA (gDNA) was then resuspended in a solution containing 30 mg/ml RNAse A (Thermo Fisher cat. EN0531) to remove any RNA. Next, approximately 50 ng of gDNA was used as input to the NEB Ultra II FS Library Prep Kit for Illumina (NEB cat. E7805L). Libraries were then sequenced using either paired end 2 × 36 bp or 2 × 72 bp read chemistry. Single nucleotide variants were called as previously described (Truong & Boeke, 2017).
Ploidy estimate from whole genome sequencing data
Chromosomal ploidy was estimated from the sequencing coverage data. First, the median coverage of 1 kb windows across the genome was calculated. From these windows, we divided each by the median coverage of the euploid genome (those chromosomes for which we never observed aneuploidy in humanized strains: VII, XIV, XIII, and XV) and took the log2 ratio. The mean of these log2 ratio windows for each chromosome was used to determine ploidy counts. In some cases, especially for lower sequencing coverage samples, we observed a smiley pattern of coverage (Gallone et al, 2016), in such situations we had to manually annotate ploidy levels.
Growth assays
Because the humanized strains displayed rapid improvement in growth rates and variability across the population, we avoided measurements of growth in liquid culture to ensure that a “rare” fast grower would not take over the population. We, therefore, measured growth rate as the rate of change in the area of colonies grown on a YPD agar surface. To ensure accurate growth rate measurements of each generation, no preceding step of culturing was performed prior to measurement. To this end, we struck out each generation from −80°C glycerol stocks to YPD agar plates. Scans were acquired using the ScanMaker 9800XL Plus (Microtek International) for each generation at two time points to calculate the rate of change in mm2 h−1. Colony sizes were measured in the image analysis program Fiji (Schindelin et al, 2012), by manual analysis.
Ploidy assessment by flow cytometry analysis
Cultures were grown to mid‐log phase and 1–2 × 107 cells were collected by centrifugation and resuspended in 1.5 ml of water. Next, to fix and permeabilize the cells 3.5 ml of 100% ethanol was added slowly to each and left overnight at −20°C. Then, cells were pelleted and washed three times with water. To remove contaminating RNA, suspensions were centrifuged and resuspended in an RNAse A solution (15 mM NaCL, 10 mM Tris pH 8.0, 0.1 mg/ml RNAse A) and incubated at 37°C overnight. Cells were then pelleted and resuspended in 50 mM Tris. 0.5 ml of processed cells were then mixed with 0.5 ml of SYTOX Green stain (2 μM SYTOX Green [Thermo Fisher cat. S7020] in 50 mM Tris pH 7.5) and incubated for 1 h at 4°C in the dark. Cells were finally pelleted and resuspended in 1 ml of Tris pH 7.5 and sonicated. Flow cytometry analysis was performed on the BD Accuri C6 flow cytometer and the data was analyzed in the program FlowJo (v10.0.7).
RNA extraction, total RNA sequencing, and analysis
Yeast strains were grown at 30°C to mid‐log phase (between OD600 0.6 and 0.8) in biological triplicate. Cultures were immediately placed on ice, pelleted, and flash frozen in liquid nitrogen then stored at −80°C. Total RNA was then extracted by resuspending each cell pellet in RNA extraction buffer (50 mM Tris–HCl pH 8.0 and 100 mM NaCl) and transferred to a 2 ml screw‐capped tube containing 0.5 mm diameter yttria‐stabilized zirconium oxide beads and samples were mechanically disrupted at 4°C for 15 cycles of 5 s shaking and 30 s rest. DNA and RNA were then purified by phenol‐chloroform extraction, by mixing at a 1:0.95 ratio of lysate to a 25:24:1 mixture of phenol:chloroform:isoamyl alcohol. To precipitate the nucleic acid the aqueous fraction was then mixed with 99.5% ethanol to a final solution of 70% ethanol. Purified DNA and RNA were then treated with DNAse I (Agilent cat. 600031) to remove contaminating DNA from each sample. RNA quality was finally assessed on an agarose gel.
Purified RNA was then used for preparing total‐RNA stranded RNAseq libraries with the QIAseq Stranded Total RNA Lib Kit (Qiagen cat. 180745) according to the manufacturer's specification. Ribosomal rRNA was depleted using the QIAseq FastSelect –rRNA Yeast Kit (Qiagen cat. 334217). Libraries were sequenced as paired‐end 75 base pair reads on an Illumina NextSeq 500. Reads were first processed to remove Illumina barcodes and quality trimming with Trimmomatic (v0.39) and quality assessed with FastQC (v0.11.4). Processed reads were then aligned to the S288C reference genome (release R64‐2‐1) using the Kallisto pseudoalignment (v0.46.0) for CEN RNA transcript counts and STAR aligner (v 2.5.2) for CEN RNA genome tracks.
MNase sequencing analysis
MNase digested raw sequencing data was downloaded from our previous study and reads were processed as before (Truong & Boeke, 2017). Aligned reads were then used as input into the DANPOS program (Chen et al, 2013) to estimate nucleosome occupancy and positioning across the genome. Finally, profile tracks for the 16 centromeres were made using the profile function, and results were plotted using MATLAB. Orientation of all centromeres when plotted was kept to run from CDEI to CDEIII.
CEN RNA RT‐PCR and quantitative RT‐PCR analysis
Total RNA was extracted as before from log phase cultures. cDNA was generated using the SuperScript™ IV Reverse Transcriptase (Thermo Fisher cat. 18090010) according to the manufacturers specifications using a polydT20 oligo to enrich for polyadenylated transcripts including CEN RNAs. Reversed transcribed RNA was then used for PCR amplification using CEN RNA‐specific oligos and GoTaq® Master Mix. Amplification was performed as follows; 20 cycles of 98°C—15 s, 55°C—15 s, and 72°C—15 s. DNA was analyzed on 1% agarose TTE gel, and an image was acquired on a BioRad GelDoc.
For qRT‐PCR analysis of CEN6 RNA, we reverse transcribed ~ 500 ng of RNA from both WT and histone humanized yeast using a polydT20 oligo. cDNA was then used as a template for amplification with transcript‐specific primers for UBC6, CLN2, and CEN6 RNAs (Appendix Table S5). qRT‐PCR reactions were carried out using the LightCycler® 480 SYBR Green I Master Mix (Roche Cat no. 04707516001) according to the manufacturers specification. The relative levels of RNAs were normalized to the expression level of UBC6.
RFP‐Nuf2 strain construction and imaging
To image kinetochores and the spindle pole body we scarlessly tagged Nuf2 and Spc110 with RFP and GFP, respectively. Briefly, each histone shuffle strain, pre‐transformed with a plasmid encoding Cas9, was transformed with a guide RNA expressing plasmid targeting the 5′ end of the gene and a circular dDNA repair template that encoded the appropriate fluorescent protein. The repair templates encoded a fluorescent protein gene (ymScarlet or mNeonGreen) and homology sequences targeting 500 bp up and downstream from the stop codon. Correct repair of the double‐strand break results in C‐terminal fusion genes and strains were validated by imaging and PCR genotyping. Imaging was done in asynchronous populations grown to the mid‐log phase (OD600 0.6–0.8). Histone humanized strains were generated as above.
DASH complex homology modeling
Homology models were constructed in the SWISS‐MODEL suite (Waterhouse et al, 2018). To obtain models for the various Dad1 and Dam1 mutants we chose to model only the arm II of the complex (consisting of the proteins Dad11–94, Dam162–115, Duo159–121, Dad36–94, and Spc345–38) in a 1‐1‐1‐1‐1 hetero‐state, using the known structure 6cfz.1 as the template. For each mutant model considered, we did the same with the only difference being that the query peptide of interest was modified to contain the mutant residue.
Dam1 residue 80 CRISPR/cas9 editing
To generate mutations at residue 80 on Dam1 (N80X), we used CRISPR/cas9 genome editing as above. Briefly, a guide RNA targeting Dam1 was used to introduce a double‐strand break, which we repaired with dsDNA donor templates encoding both the mutation of interest and a PAM inactivating silent mutation. We were able to isolate 16 out of 18 possible mutations (excluding the wild‐type asparagine and the already cloned tyrosine variant). For two mutations, N80P and N80K, we only ever isolated clones that inactivated the PAM sequence but retained the wild‐type residue 80 sequence—we did not pursue these further.
DASH complex purification and tension measurement
Saccharomyces cerevisiae DASH complex was expressed in Escherichia coli using a single polycistronic vector, as previously described (Flores et al, 2022). The Spc34p component of the DASH/Dam1 complex contained either a C‐terminal FLAG‐tag or a His6‐tag. BL21 Rosetta 2 DE3 cells were transformed with either wild‐type DASH complex (pJT44—FLAG, or PC4—6X His) or with single mutations described in the manuscript. Cells were grown to OD600 of 0.6, where the addition of 2 mM IPTG induced protein expression. Cells were induced for 16 h at 18°C while shaking at 240 rpm. Cells were collected by centrifugation, resuspended in 50 mM sodium phosphate buffer (pH 6.9) containing 500 mM NaCl, 1 mM PMSF, and 1 protease tablet (Roche) and lysed with a French press. Lysates were cleared by centrifugation (20 min, 25,000 g, 4°C). Mutant and wild‐type DASH complexes were first purified via affinity chromatography using either FLAG resin (GE Healthcare Bioscience) or Nickel resin (Bio‐Rad). For FLAG purification, the lysate was applied onto FLAG resin under gravity flow and eluted with 50 mM sodium phosphate buffer (pH 6.9) containing 500 mM NaCl and 100 μg/ml FLAG peptide. For His‐tagged DASH complexes, samples were eluted in 50 mM sodium phosphate buffer containing 500 mM NaCl and 500 mM imidazole. Both His and FLAG‐tagged DASH constructs were further purified via size exclusion chromatography.
Recombinant His6‐tag DASH complex was linked to 0.56 μm‐diameter streptavidin‐coated polystyrene microbeads (Spherotech) using biotinylated His5‐antibody (Qiagen), as previously described (Asbury et al, 2006; Franck et al, 2007; Powers et al, 2009). For rupture force assays, a final concentration of 2.5, 5, or 10 nM DASH complex was incubated with 3.5 pM beads for 1 h. Beads were washed twice with AB solution (1X BRB80 [80 mM potassium PIPES buffer, pH 6.8, containing 1 mM MgCl2, and 1 mM EGTA], 2 mg/ml BSA, and 1 mM DTT) to remove unbound His6‐tagged DASH complex. Glass slides and functionalized coverslips were used to construct flow channels. Channels were functionalized by adding 5 mg/ml biotinylated BSA (Vector Labs) and incubated for 15 min inside a humidity chamber, washed with 1X BRB80, and then incubated with 0.3 mg/ml avidin DN (Vector Labs) for 5 min. Following, channels were further washed with 1X BRB80, and biotinylated microtubule seeds stabilized with GMPCPP were added and allowed to incubate for 5 min inside the humidity chamber. Growth buffer (1X BRB80, 8 mg/ml BSA, 1 mM GTP, and 1 mg/ml κ‐casein) was added and allowed to incubate in the chamber for 5 min. DASH‐coated beads were added to a reaction mix (1X BRB80, 8 mg/ml BSA, 1 mM GTP, 40 mM glucose, and 1 mM DTT) and sonicated for 10 s. Following sonication, an oxygen scavenging system (250 μg/ml glucose oxidase, 30 μg/ml catalase, and 4.5 mg/ml glucose) was added. Lastly, ~ 2–2.5 mg/ml of purified bovine brain tubulin and 5 nM FLAG‐tagged DASH complex were added to the reaction mixture. The reaction mixture was subsequently added to the channel, which was sealed with nail polish.
Data collection was performed as previously described (Flores et al, 2022). Briefly, data were collected for a total of 1 h after the addition of tubulin into the reaction mixture. To determine whether a DASH complex‐coated bead was able to bind to microtubules in the absence of force, it was placed onto a microtubule, and then the trap was shuttered. Microtubule‐attached beads were initially pulled at 1 pN of force. After, a gradual increasing force was applied (0.25 pN/s) until the bead ruptured from the microtubule tip or the maximum trapping force was reached (~ 20 pN under the conditions described here). Rupture forces were analyzed using Igor Pro (WaveMetrics).
Electron microscopy
EM was performed as described below, based on the methods outlined in Kim et al, 2017. Bovine brain tubulin was clarified at a concentration of 75 μM at 4°C in BRB80 (80 mM potassium PIPES buffer [pH 6.8], 1 mM EGTA, 1 mM MgCl2) at 90,000 rpm in a Beckman TLA‐100 rotor for 10 min. From the supernatant, 26.5 μl of clarified tubulin was removed and 1.5 μl 100 mM MgCl2, 1.5 μl DMSO plus 0.5 μl GTP was added. The resulting solution was incubated at 37°C for 30 min to allow polymerization of tubulin. A wide bore pipette was used to add 140 μl of BRB80 containing 10 μM taxol (BRB80‐T) to the resulting microtubules. The reaction was mixed by gentle pipetting and 100 μl was spun at 58,000 rpm at 37°C in a Beckman TLA‐100 rotor for 10 min to pellet the microtubules. The supernatant was removed and microtubules were resuspended in 100 μl BRB80‐T using a wide bore pipette at room temperature. MT concentration was calculated via BCA assay of the resuspended MT pellet.
Samples for EM were prepared in a final volume of 50 μl BRB80‐T containing 20 nM microtubules plus between 5 and 35 nM DASH complex (WT or E50D). Once mixed, MTs plus DASH complex were incubated at room temperature for 30 min. Carbon‐coated copper grids were negatively discharged in a glow discharge device. A 3 μl volume of sample was applied onto a discharged grid for 30 s before the excess liquid was blotted away with Whatman 1 filter papers. The grid was then washed with BRB80‐T and blotted three times prior to application and blotting of 30 μl of 2% uranyl formate three times. The stained grid was then air‐dried prior to imaging.
The grids were imaged on a Tecnai T12 transmission electron microscope (FEI, Hillsboro, OR) operating at 120 kV using a Tungsten filament electron source. Images were recorded on a bottom‐mounted Ultrascan 4000 (Gatan, Pleasanton, CA) camera at 42,000× nominal magnification. The Fiji implementation of ImageJ was used to export images in PNG format for publication.
Image analysis of oligomerized rings was carried out in Fiji using the plot profile function to obtain the intensity profiles along the distance of individual DASH complex rings (with the midpoint set at the center of the microtubule lattice). Intensity data were then normalized by subtraction of the average intensity divided by the standard deviation of intensity. The normalized average intensity profile of the unbound microtubules was then subtracted from each to obtain the profiles corresponding to the DASH complex rings.
DASH complex size exclusion chromatography
Size‐exclusion chromatography was carried out using a Superdex 200 Increase 10/300 GL column (Cytiva) equilibrated in DASH complex purification buffer (500 mM NaCl, 50 mM sodium phosphate buffer [pH 6.9]). The column was calibrated using a gel filtration markers kit for protein molecular weights 29,000–700,000 Da (Sigma‐Aldrich, product number MWGF1000).
Sporulation and ascus dissections
Diploid yeasts were grown for 2 days on solid GNA agar plates prior to sporulation induction. Yeast was then resuspended to 1 × 107 cells in 5 ml of sporulation medium (1% potassium acetate, 0.05% zinc acetate, with supplemented amino acids) and rotated for 5 days and 25°C. After 5 days, cultures were either immediately processed or stored at 4°C for up to 2 weeks. Asci were digested in 25 μl of 0.5 mg/ml zymolase‐20T in 1 M sorbitol for 6–7 min at 37°C, at which point 200 μl of 1 M sorbitol was gently added and digested asci placed on ice. Spores were then micromanipulated and arrayed onto YPD agar plates.
Author contributions
Max A B Haase: Conceptualization; data curation; formal analysis; validation; investigation; visualization; methodology; writing—original draft; project administration; writing—review and editing. Guðjón Ólafsson: Formal analysis; investigation; visualization; methodology; writing—review and editing. Rachel L Flores: Formal analysis; investigation; visualization; methodology; writing—review and editing. Emmanuel Boakye‐Ansah: Formal analysis; investigation; methodology. Alex Zelter: Formal analysis; investigation; methodology; writing—review and editing. Miles Sasha Dickinson: Formal analysis; writing—review and editing. Luciana Lazar‐Stefanita: Conceptualization; writing—review and editing. David M Truong: Conceptualization; writing—review and editing. Charles, L Asbury: Supervision; funding acquisition; writing—review and editing. Trisha N Davis: Supervision; funding acquisition; writing—review and editing. Jef D Boeke: Conceptualization; funding acquisition; writing—review and editing.
Disclosure and competing interests statement
Jef Boeke is a Founder and Director of CDI Labs, Inc., a Founder of and consultant to Neochromosome, Inc, a Founder, SAB member of and consultant to ReOpen Diagnostics, LLC and serves or served on the Scientific Advisory Board of the following: Sangamo, Inc., Logomix, Inc., Modern Meadow, Inc., Rome Therapeutics, Inc., Sample6, Inc., Tessera Therapeutics, Inc. and the Wyss Institute.
Supporting information
Appendix
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
PDF+
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Source Data for Figure 8
Acknowledgements
We thank Imani Sinclair and the NYU Reagent Preparation core for their support with growth media used in this study; Joel Quispe for help with EM and his maintenance of the UW Beckman CryoEM Center; Hannah Ashe, Gwen Ellis, Raven Luther, and Matthew Maurano for providing Illumina sequencing service. This work was supported by the National Science Foundation (URoL‐1921641) to J.D.B., NIGMS R35130293 to T.N.D. and NIGMS R35134842 to C.L.A.
The EMBO Journal (2023) 42: e112600
See also: Z Storchová (April 2023)
Data availability
All strains and plasmids are available by request to the corresponding author. Raw sequencing reads are deposited and available at the Sequence Read Archive (SRA—Bioproject accession number: PRJNA884379, http://www.ncbi.nlm.nih.gov/sra?term=PRJNA884379 and Accession numbers: SRR21712309, http://www.ncbi.nlm.nih.gov/sra?term=SRR21712309—SRR21712362, http://www.ncbi.nlm.nih.gov/sra?term=SRR21712362).
References
- Abad MA, Ruppert JG, Buzuk L, Wear M, Zou J, Webb KM, Kelly DA, Voigt P, Rappsilber J, Earnshaw WC et al (2019) Borealin–nucleosome interaction secures chromosome association of the chromosomal passenger complex. J Cell Biol 218: 3912–3925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akiyoshi B, Sarangapani KK, Powers AF, Nelson CR, Reichow SL, Arellano‐Santoyo H, Gonen T, Ranish JA, Asbury CL, Biggins S (2010) Tension directly stabilizes reconstituted kinetochore‐microtubule attachments. Nature 468: 576–579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonarakis SE (2017) Down syndrome and the complexity of genome dosage imbalance. Nat Rev Genet 18: 147–163 [DOI] [PubMed] [Google Scholar]
- Asbury CL, Gestaut DR, Powers AF, Franck AD, Davis TN (2006) The Dam1 kinetochore complex harnesses microtubule dynamics to produce force and movement. Proc Natl Acad Sci USA 103: 9873–9878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baca SC, Prandi D, Lawrence MS, Mosquera JM, Romanel A, Drier Y, Park K, Kitabayashi N, MacDonald TY, Ghandi M et al (2013) Punctuated evolution of prostate cancer genomes. Cell 153: 666–677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beach RR, Ricci‐Tam C, Brennan CM, Moomau CA, hsin Hsu P, Hua B, Silberman RE, Springer M, Amon A (2017) Aneuploidy causes non‐genetic individuality. Cell 169: 229–242.e21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biggins S (2013) The composition, functions, and regulation of the budding yeast kinetochore. Genetics 194: 817–846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biggins S, Severin FF, Bhalla N, Sassoon I, Hyman AA, Murray AW (1999) The conserved protein kinase Ipl1 regulates microtubule binding to kinetochores in budding yeast. Genes Dev 13: 532–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boonekamp FJ, Knibbe E, Vieira‐Lara MA, Wijsman M, Luttik MAH, van Eunen K, Ridder MD, Bron R, Almonacid Suarez AM, van Rijn P et al (2022) Full humanization of the glycolytic pathway in Saccharomyces cerevisiae . Cell Rep 39: 111010 [DOI] [PubMed] [Google Scholar]
- Carmena M, Wheelock M, Funabiki H, Earnshaw WC (2012) The chromosomal passenger complex (CPC): from easy rider to the godfather of mitosis. Nat Rev Mol Cell Biol 13: 789–803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan CS, Botstein D (1993) Isolation and characterization of chromosome‐gain and increase‐in‐ploidy mutants in yeast. Genetics 135: 677–691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K, Xi Y, Pan X, Li Z, Kaestner K, Tyler J, Dent S, He X, Li W (2013) DANPOS: dynamic analysis of nucleosome position and occupancy by sequencing. Genome Res 23: 341–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho U‐S, Harrison SC (2012) Ndc10 is a platform for inner kinetochore assembly in budding yeast. Nat Struct Mol Biol 19: 48–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cimini D, Wan X, Hirel CB, Salmon ED (2006) Aurora kinase promotes turnover of kinetochore microtubules to reduce chromosome segregation errors. Curr Biol 16: 1711–1718 [DOI] [PubMed] [Google Scholar]
- Clarke MN, Marsoner T, MAY A, Ravichandran MC, Campbell CS (2023) Adaptation to high rates of chromosomal instability and aneuploidy through multiple pathways in budding yeast. EMBO J 42: e111500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbett KD, Yip CK, Ee L‐S, Walz T, Amon A, Harrison SC (2010) The Monopolin complex crosslinks kinetochore components to regulate chromosome‐microtubule attachments. Cell 142: 556–567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cottarel G, Shero JH, Hieter P, Hegemann JH (1989) A 125‐base‐pair CEN6 DNA fragment is sufficient for complete meiotic and mitotic centromere functions in Saccharomyces cerevisiae. Mol Cell Biol 9: 3342–3349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Counter CM, Avilion AA, LeFeuvre CE, Stewart NG, Greider CW, Harley CB, Bacchetti S (1992) Telomere shortening associated with chromosome instability is arrested in immortal cells which express telomerase activity. EMBO J 11: 1921–1929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan S‐F, Han P‐J, Wang Q‐M, Liu W‐Q, Shi J‐Y, Li K, Zhang X‐L, Bai F‐Y (2018) The origin and adaptive evolution of domesticated populations of yeast from Far East Asia. Nat Commun 9: 2690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores RL, Peterson ZE, Zelter A, Riffle M, Asbury CL, Davis TN (2022) Three interacting regions of the Ndc80 and Dam1 complexes support microtubule tip‐coupling under load. J Cell Biol 221: e202107016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franck AD, Powers AF, Gestaut DR, Gonen T, Davis TN, Asbury CL (2007) Tension applied through the Dam1 complex promotes microtubule elongation providing a direct mechanism for length control in mitosis. Nat Cell Biol 9: 832–837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuyama S, Biggins S (2007) Centromere identity is specified by a single centromeric nucleosome in budding yeast. Proc Natl Acad Sci USA 104: 14706–14711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallone B, Steensels J, Prahl T, Soriaga L, Saels V, Herrera‐Malaver B, Merlevede A, Roncoroni M, Voordeckers K, Miraglia L et al (2016) Domestication and divergence of Saccharomyces cerevisiae Beer yeasts. Cell 166: 1397–1410.e16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon JL, Byrne KP, Wolfe KH (2011) Mechanisms of chromosome number evolution in yeast. PLoS Genet 7: e1002190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould SJ, Eldredge N (1993) Punctuated equilibrium comes of age. Nature 366: 223–227 [DOI] [PubMed] [Google Scholar]
- Haase MAB, Truong DM, Boeke JD (2019) Superloser: a plasmid shuffling vector for Saccharomyces cerevisiae with exceedingly low background. G3 (Bethesda) 9: 2699–2707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heasley LR, Sampaio NMV, Argueso JL (2021) Systemic and rapid restructuring of the genome: a new perspective on punctuated equilibrium. Curr Genet 67: 57–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedouin S, Logsdon GA, Underwood JG, Biggins S (2022) A transcriptional roadblock protects yeast centromeres. Nucleic Acids Res 50: 7801–7815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinshaw SM, Makrantoni V, Harrison SC, Marston AL (2017) The kinetochore receptor for the Cohesin loading complex. Cell 171: 72–84.e13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hose J, Escalante LE, Clowers KJ, Dutcher HA, Robinson D, Bouriakov V, Coon JJ, Shishkova E, Gasch AP (2020) The genetic basis of aneuploidy tolerance in wild yeast. Elife 9: e52063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaffe M, Sherlock G, Levy SF (2017) iSeq: a new double‐barcode method for detecting dynamic genetic interactions in yeast. G3 (Bethesda) 7: 143–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenni S, Harrison SC (2018) Structure of the DASH/Dam1 complex shows its role at the yeast kinetochore‐microtubule interface. Science 360: 552–558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenni S, Dimitrova YN, Valverde R, Hinshaw SM, Harrison SC (2017) Molecular structures of yeast kinetochore subcomplexes and their roles in chromosome segregation. Cold Spring Harb Symp Quant Biol 82: 83–89 [DOI] [PubMed] [Google Scholar]
- Kachroo AH, Laurent JM, Yellman CM, Meyer AG, Wilke CO, Marcotte EM (2015) Systematic humanization of yeast genes reveals conserved functions and genetic modularity. Science 348: 921–925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao KC, Schwartz K, Sherlock G (2010) A genome‐wide analysis reveals No nuclear Dobzhansky‐muller pairs of determinants of speciation between S. cerevisiae and S. paradoxus, but suggests more complex incompatibilities. PLoS Genet 6: e1001038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawashima SA, Yamagishi Y, Honda T, Ishiguro K, Watanabe Y (2010) Phosphorylation of H2A by Bub1 prevents chromosomal instability through localizing Shugoshin. Science 327: 172–177 [DOI] [PubMed] [Google Scholar]
- Kim JO, Zelter A, Umbreit NT, Bollozos A, Riffle M, Johnson R, MacCoss MJ, Asbury CL, Davis TN (2017) The Ndc80 complex bridges two Dam1 complex rings. Elife 6: e21069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurent JM, Garge RK, Teufel AI, Wilke CO, Kachroo AH, Marcotte EM (2020) Humanization of yeast genes with multiple human orthologs reveals functional divergence between paralogs. PLoS Biol 18: e3000627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefrançois P, Auerbach RK, Yellman CM, Roeder GS, Snyder M (2013) Centromere‐like regions in the budding yeast genome. PLoS Genet 9: e1003209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling YH, Yuen KWY (2019) Point centromere activity requires an optimal level of centromeric noncoding RNA. Proc Natl Acad Sci USA 116: 6270–6279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcet‐Houben M, Gabaldón T (2015) Beyond the whole‐genome duplication: phylogenetic evidence for an ancient interspecies hybridization in the Baker's yeast lineage. PLoS Biol 13: e1002220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCulley JL, Petes TD (2010) Chromosome rearrangements and aneuploidy in yeast strains lacking both Tel1p and Mec1p reflect deficiencies in two different mechanisms. Proc Natl Acad Sci USA 107: 11465–11470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranda JL, Wulf PD, Sorger PK, Harrison SC (2005) The yeast DASH complex forms closed rings on microtubules. Nat Struct Mol Biol 12: 138–143 [DOI] [PubMed] [Google Scholar]
- Monje‐Casas F, Prabhu VR, Lee BH, Boselli M, Amon A (2007) Kinetochore orientation during meiosis is controlled by Aurora B and the Monopolin complex. Cell 128: 477–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musacchio A, Salmon ED (2007) The spindle‐assembly checkpoint in space and time. Nat Rev Mol Cell Biol 8: 379–393 [DOI] [PubMed] [Google Scholar]
- Nicklas RB (1997) How cells get the right chromosomes. Science 275: 632–637 [DOI] [PubMed] [Google Scholar]
- Oromendia AB, Amon A (2014) Aneuploidy: implications for protein homeostasis and disease. Dis Model Mech 7: 15–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paldi F, Alver B, Robertson D, Schalbetter SA, Kerr A, Kelly DA, Baxter J, Neale MJ, Marston AL (2020) Convergent genes shape budding yeast pericentromeres. Nature 582: 119–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peter J, De Chiara M, Friedrich A, Yue J‐X, Pflieger D, Bergström A, Sigwalt A, Barre B, Freel K, Llored A et al (2018) Genome evolution across 1,011 Saccharomyces cerevisiae isolates. Nature 556: 339–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petronczki M, Matos J, Mori S, Gregan J, Bogdanova A, Schwickart M, Mechtler K, Shirahige K, Zachariae W, Nasmyth K (2006) Monopolar attachment of sister kinetochores at meiosis I requires casein kinase 1. Cell 126: 1049–1064 [DOI] [PubMed] [Google Scholar]
- Pinsky BA, Kung C, Shokat KM, Biggins S (2006) The Ipl1‐Aurora protein kinase activates the spindle checkpoint by creating unattached kinetochores. Nat Cell Biol 8: 78–83 [DOI] [PubMed] [Google Scholar]
- Powers AF, Franck AD, Gestaut DR, Cooper J, Gracyzk B, Wei RR, Wordeman L, Davis TN, Asbury CL (2009) The Ndc80 kinetochore complex forms load‐bearing attachments to dynamic microtubule tips via biased diffusion. Cell 136: 865–875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarangapani KK, Duro E, Deng Y, Alves Fde L, Ye Q, Opoku KN, Ceto S, Rappsilber J, Corbett KD, Biggins S et al (2014) Sister kinetochores are mechanically fused during meiosis I in yeast. Science 346: 248–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda‐Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B et al (2012) Fiji: an open‐source platform for biological‐image analysis. Nat Methods 9: 676–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selmecki AM, Maruvka YE, Richmond PA, Guillet M, Shoresh N, Sorenson AL, De S, Kishony R, Michor F, Dowell R et al (2015) Polyploidy can drive rapid adaptation in yeast. Nature 519: 349–352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp NP, Sandell L, James CG, Otto SP (2018) The genome‐wide rate and spectrum of spontaneous mutations differ between haploid and diploid yeast. Proc Natl Acad Sci USA 115: E5046–E5055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen X‐X, Opulente DA, Kominek J, Zhou X, Steenwyk JL, Buh KV, Haase MAB, Wisecaver JH, Wang M, Doering DT et al (2018) Tempo and mode of genome evolution in the budding yeast subphylum. Cell 175: 1533–1545.e20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner FA, Henikoff S (2015) Diversity in the organization of centromeric chromatin. Curr Opin Genet Dev 31: 28–35 [DOI] [PubMed] [Google Scholar]
- Tanaka TU, Rachidi N, Janke C, Pereira G, Galova M, Schiebel E, Stark MJR, Nasmyth K (2002) Evidence that the Ipl1‐Sli15 (Aurora kinase‐INCENP) complex promotes chromosome Bi‐orientation by altering kinetochore‐spindle pole connections. Cell 108: 317–329 [DOI] [PubMed] [Google Scholar]
- Tien JF, Umbreit NT, Gestaut DR, Franck AD, Cooper J, Wordeman L, Gonen T, Asbury CL, Davis TN (2010) Cooperation of the Dam1 and Ndc80 kinetochore complexes enhances microtubule coupling and is regulated by aurora B. J Cell Biol 189: 713–723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres EM, Sokolsky T, Tucker CM, Chan LY, Boselli M, Dunham MJ, Amon A (2007) Effects of aneuploidy on cellular physiology and cell division in haploid yeast. Science 317: 916–924 [DOI] [PubMed] [Google Scholar]
- Torres EM, Dephoure N, Panneerselvam A, Tucker CM, Whittaker CA, Gygi SP, Dunham MJ, Amon A (2010) Identification of aneuploidy‐tolerating mutations. Cell 143: 71–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truong DM, Boeke JD (2017) Resetting the yeast epigenome with human nucleosomes. Cell 171: 1508–1519.e13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umbreit NT, Miller MP, Tien JF, Cattin Ortolá J, Gui L, Lee KK, Biggins S, Asbury CL, Davis TN (2014) Kinetochores require oligomerization of Dam1 complex to maintain microtubule attachments against tension and promote biorientation. Nat Commun 5: 4951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verzijlbergen KF, Nerusheva OO, Kelly D, Kerr A, Clift D, de Lima AF, Rappsilber J, Marston AL (2014) Shugoshin biases chromosomes for biorientation through condensin recruitment to the pericentromere. Elife 3: e01374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R, Heer FT, de Beer TAP, Rempfer C, Bordoli L et al (2018) SWISS‐MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res 46: W296–W303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winey M, Mamay CL, O'Toole ET, Mastronarde DN, Giddings TH, McDonald KL, McIntosh JR (1995) Three‐dimensional ultrastructural analysis of the Saccharomyces cerevisiae mitotic spindle. J Cell Biol 129: 1601–1615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe KH, Shields DC (1997) Molecular evidence for an ancient duplication of the entire yeast genome. Nature 387: 708–713 [DOI] [PubMed] [Google Scholar]
- Yamagishi Y, Honda T, Tanno Y, Watanabe Y (2010) Two histone Marks establish the inner centromere and chromosome Bi‐orientation. Science 330: 239–243 [DOI] [PubMed] [Google Scholar]
- Zaytsev AV, Grishchuk EL (2015) Basic mechanism for biorientation of mitotic chromosomes is provided by the kinetochore geometry and indiscriminate turnover of kinetochore microtubules. Mol Biol Cell 26: 3985–3998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu YO, Sherlock G, Petrov DA (2016) Whole genome analysis of 132 clinical Saccharomyces cerevisiae strains reveals extensive ploidy variation. G3 (Bethesda) 6: 2421–2434 [DOI] [PMC free article] [PubMed] [Google Scholar]