

Abstract
The failure to repress transcription of repetitive genomic elements can lead to catastrophic genome instability and is associated with various human diseases. As such, multiple parallel mechanisms cooperate to ensure repression and heterochromatinization of these elements, especially during germline development and early embryogenesis. A vital question in the field is how specificity in establishing heterochromatin at repetitive elements is achieved. Apart from trans‐acting protein factors, recent evidence points to a role of different RNA species in targeting repressive histone marks and DNA methylation to these sites in mammals. Here, we review recent discoveries on this topic and predominantly focus on the role of RNA methylation, piRNAs, and other localized satellite RNAs.
Keywords: chromatin, development, repeats, RNA, transposons
Subject Categories: Chromatin, Transcription & Genomics; RNA Biology
This chapter of the 2023 RNA review series by Zylicz and colleagues covers the role of RNA in regulating heterochromatic silencing of repetitive elements in mammals.

Introduction
Genomes are complex ecosystems that have been shaped by the two major forces of natural selection and genetic drift. They are inhabited by diverse communities of selfish genetic elements as well as inert, opportunistic sequences. Opposite selective forces create a balance between parasitic expansion and host homeostasis, which eventually benefits both sides (Rebollo et al, 2012; Chuong et al, 2017; Platt et al, 2018). In line with this functional relevance, repetitive elements are not randomly distributed across the genome, but rather reside in specific chromatin regions, which allow their tight control (Bourque et al, 2018). The developing germline is considered the major battlefield in the war against these elements. Animals, such as mammals, in which the germ cells are specified from the soma, face an additional threat, as reprogramming of the epigenome provides windows of opportunity for their expansion (Ozata et al, 2019). Studies over the last decades have shown that most of these elements rely on transcription in one way or another to execute their function (Bourque et al, 2018; Smurova & De Wulf, 2018; Talbert & Henikoff, 2018). How cells harness this dependency to conspire their strategies against them is going to be the focus of this review.
Seminal studies in many model organisms such as fission yeast, nematodes, and flies, have established the eminent role of RNA molecules in this process (Holoch & Moazed, 2015; Martienssen & Moazed, 2015; Bhattacharjee et al, 2019; Weiser & Kim, 2019; Duempelmann et al, 2020; Padeken et al, 2022). Recent evidence from mammalian systems further supports the notion of context‐specific RNA‐mediated recruitment of chromatin modifying complexes as a general mechanism across species. Since the canonical mechanisms of silencing repetitive elements by heterochromatin have been extensively reviewed in the past, here we will only briefly discuss it and instead focus on the emerging role of RNA molecules in heterochromatic silencing in mammalian cells (Goodier & Kazazian, 2008; Rowe & Trono, 2011; Leung & Lorincz, 2012; Goodier, 2016; Groh & Schotta, 2017; Kwon & Chung, 2020; Padeken et al, 2022). In this review, we examine specifically how low‐level transcription of repetitive elements and the resulting RNA transcript contribute to heterochromatin formation in mouse and human cells. First, in the following chapter and accompanying boxes, we will introduce the different types of repetitive elements present in mammalian genomes and briefly outline the general schemes and patterns used to silence repeats across different species.
Diversity of repetitive elements and general mechanisms for their silencing
The sequence complexity of the mammalian genomes is diverse, and a relatively large portion of it comprises repetitive sequences. These sequences can be categorized based on their degree of repetitiveness, their organization, or their functional role. They can be found in both coding and non‐coding regions and can be arranged in tandem or dispersed across the genome (López‐Flores & Garrido‐Ramos, 2012; Garrido‐Ramos, 2017). Those that are arranged in tandem are mainly satellite sequences (Box 1), whereas transposable elements (TE, Box 2) comprise the dispersed portion of the repetitive sequences. According to the Repeat Masker database (Smit et al, n.d.), together both repeat groups account for ~52 and ~45% of the human and the mouse genome respectively. Satellite sequences and TEs are traditionally linked to constitutive heterochromatin (Box 3); however, their location within the genome might deviate, with regards to the evolutionary history of each repeat type (López‐Flores & Garrido‐Ramos, 2012; Sultana et al, 2017).
Box 1. Satellite sequences.
Named after the distinct characteristic “satellite” bands that they form when applied on density‐gradient of cesium chloride, satellite sequences consist of a variable number of tandem repeats (VNTRs). Conventionally, depending on the length of the repeat unit, VNTRs can be subdivided into microsatellites (< 10 nucleotides ‐ also known as Simple Sequence Repeats or Short Tandem Repeats), mini‐satellites (≥ 10 nt) or satellites (≥ 150 nt; Gemayel et al, 2010). The former two groups, share many common characteristics, such as that they can be found in protein‐coding, non‐coding, and regulatory regions, they exhibit higher mutation rates than the average in the genome, and they are highly polymorphic, that is, exhibiting variations in the number of repeat units among individuals in a population (Fan & Chu, 2007). It has been estimated that around 10–20% of eukaryotic genes and promoters contain unstable repeat series. Specific gene families are significantly enriched in such elements, which suggests that they might be subjected to higher evolution rates (Gemayel et al, 2010; López‐Flores & Garrido‐Ramos, 2012). Additionally, microsatellite instability has been linked to many genetic diseases such as Huntington disease, spinobulbar muscular atrophy (SBMA‐Kennedy disease), spinocerebellar ataxias, fragile X syndrome, Friedreich ataxia, myotonic dystrophy type 1 and 2 and others (Madireddy & Gerhardt, 2017; Orr & Zoghbi, 2007).
Satellite DNA is a major component of constitutive heterochromatin, mainly located in the centromeric, pericentric and subtelomeric regions of chromosomes. Typically, they consist of 150–180 or 300–360 bp, corresponding to mono‐ and di‐nucleosomes, and account for 2.6 and 0.2% of the human and mouse genome respectively (Garrido‐Ramos, 2017). Their monomers might include micro‐ and/or mini‐satellites and they are usually organized in long arrays of hundreds, or thousands, of not strictly identical copies. The primate‐specific α‐satellite elements are a remarkable example of centromeric repeats with functional relevance. While their monomers consist of 171‐bp, they are usually found as dimers. Most monomers contain a binding site for the Centromere Protein B (CENP‐B), a protein that enhances recruitment of the centromere‐specific H3 histone CENP‐A and facilitates kinetochore formation and function (Fachinetti et al, 2015; Otake et al, 2020). Satellite DNA sequences are also predicted to form secondary structures such as non‐beta form DNA, G‐quadruplexes, and dimeric i‐motif structures, which can potentially act as platforms of protein recruitment or exclusion. Such structures are thought to be involved in chromatid cohesion post replication and telomere maintenance (Thakur et al, 2021). Furthermore, different types of satellite sequences carry distinct heterochromatin components which are associated to specific epigenetic features such as replication timing, nuclear localization and chromatid cohesion (Guenatri et al, 2004; Otake et al, 2020). In mice for instance, centromeric minor satellite chromatin is organized with CENP‐A/B/C molecules while the pericentric major satellite repeats are not. Such distinct chromatin features allow major satellite sequences from different chromosomes to cluster together during interphase, and also prolong resolution of sister chromatids at these sites until metaphase (Guenatri et al, 2004). Thus, satellite sequences can play an important role in maintaining genome stability throughout the cell cycle.
Box 2. Transposable elements.
Transposable elements (TEs), or transposons, are interspersed repeat sequences which, unlike satellites, either have or had the ability to change their location around the genome. In principle, TEs carry the regulatory and the coding sequences to catalyze their self‐transposition. However, a large number of them rely on the functional machinery encoded by other copies of TEs, distinguishing them into autonomous and non‐autonomous transposons. Their ability to move and replicate affects the genome by generating plasticity and heterogeneity (Goodier & Kazazian, 2008; Bourque et al, 2018). They comprise about 48.49 and 41.73% of the human and mouse genome respectively, and can be classified into two major classes (retrotransposons and DNA transposons), based on the presence or absence of an RNA intermediate involved in their mechanism of transposition. According to the classification system proposed by Wicker et al (2007, 2009a, 2009b), TEs can be further subdivided into orders, based on the insertion mechanism they deploy, a distinction which best reflects major differences in their structural organization.
Class I elements, best known as retrotransposons, are the major contributors of the repetitive fraction in mammals and they can be categorized into five orders, that is, long terminal repeat (LTR) retrotransposons, the long and short interspersed nuclear elements (LINEs & SINEs), the Penelope‐like and the Dictyostelium intermediate repeat sequence‐like (DIR‐like) elements. Their transposition relies on an RNA intermediate which is reversed transcribed by a TE‐encoded reverse transcriptase. This mechanism is usually described as a “copy & paste” due to the fact that each transposition event produces a new copy, integrated into a new location. On the other hand, Class II elements, also known as DNA transposons, are found in all eukaryotes. Such TEs account for 3.7 and 1.4% of the human and the mouse genome respectively, and their mechanism of transposition can be categorized as “cut & paste” (Kapitonov & Jurka, 2008). In humans, Class II elements, that is, DNA transposons have been inert for the past 50 million years with the last evidence of activity before the divergence between humans and new world monkeys (Lander et al, 2001).
Despite their large numbers, the vast majority of TEs progressively erode until they become no longer capable of transposition. For instance, it has been estimated that more than 99.9% of the ~500,000 copies of human LINE1 elements are fixed and immobile, with about 100 of them, known as “hot LINE1s”, remaining active (Sassaman et al, 1997; Brouha et al, 2003). Still, transposable elements are an important source of mutations and polymorphisms in mammals. In laboratory mice, TE insertions are responsible for ~10% of all de novo mutations (Maksakova et al, 2006), while for humans it is estimated that this rate is ~1/21 births for 7SL‐Alu (Xing et al, 2009) and 1/95–1/270 births for LINE1 elements (Ewing & Kazazian, 2010). More than 120 of such insertions have been associated with human diseases such as hemophilia A & B, cystic fibrosis, Duchene muscular dystrophy, beta‐thalassemia, hypercholesterolemia, Apert syndrome, neurofibromatosis, leukemias and cancers (Cordaux & Batzer, 2009; Hancks & Kazazian, 2016). Thus, controlling TE expression is of great importance for genome stability and health.
Box 3. Heterochromatin in mammals.
Chromatin organization differs in space and time, both in terms of its epigenetic marks and the level of compaction. These differences influence accessibility to genetic information, thereby allowing for timely and selective expression of genes. Nevertheless, chromatin organization serves additional roles in the nucleus, such as facilitating attachment to nuclear lamina, chromosome segregation, prevention of telomere erosion, and silencing of repetitive elements (Allshire & Madhani, 2018; Janssen et al, 2018). These rather “housekeeping” functions are extremely important in maintaining genome stability and are particularly linked to condensed chromatin, termed as heterochromatin.
Differentiating it from euchromatin based on cytological examination, heterochromatin was initially described as the portion of chromatin that consistently fails to decondense following mitosis. Research over the past decades has shown that this definition better reflects a constitutive, rather than the cell‐type specific and developmentally regulated heterochromatin, now referred to as facultative heterochromatin. Different “flavors” of heterochromatin, as a concept, have been proposed and discussed in the past (Allshire & Madhani, 2018; Janssen et al, 2018; Thakur et al, 2021; Żylicz & Heard, 2020). Typically, heterochromatin forms at the centromeres, the telomeres, as well as their adjacent regions (pericentric/pericentromeric and subtelomeric respectively). The main features of constitutive heterochromatin in mammalian genomes are the enrichment for certain marks, for example, 5‐methyl‐cytocine (5mC) on DNA, histone H3 di‐ and tri‐ methylation on Lysine 9 (H3K9me2/3) as well as histone H4 Lysine 20 trimethylation (H4K20me3); the absence of histone acetylation; the presence of the heterochromatin protein 1 homologs (HP1α/β/γ); the distinct subnuclear localization in the periphery or in chromocenters; and the presence of repetitive sequences (Janssen et al, 2018). Most of these characteristics share a high degree of interdependence and constitute multiple lines of protection that secure the functionality of heterochromatin.
Efficient heterochromatinization depends primarily on histone modifications. For instance, H3K9me2/3 marks are essential for both establishment and maintenance of heterochromatin. Numerous enzymes involved in this process, including the G9A:GLP complex, which is responsible for deposition of H3K9 mono‐ and di‐methylation, and the SETDB1/2 and SUV39H1/2 complexes, which catalyze di‐ and tri‐ methylation of H3K9 residues (Janssen et al, 2018; Padeken et al, 2022). These Lysine methylation marks, in turn, can be recognized by proteins containing specific domains such as the chromodomain, the Tudor domain and others (Yap & Zhou, 2010, 2011). Such proteins can either be enzymes themselves or act as platforms for recruitment of additional factors. A remarkable example is the HP1 protein which can recognize H3K9me3 via its chromodomain, bind RNA molecules, and recruit additional HP1 units, as well as the SETDB1, SUV39H1/2 and SUV420H1/2 enzymes (Muchardt et al, 2002; Schultz et al, 2002; Yamamoto & Sonoda, 2003; Schotta et al, 2004; Bosch‐Presegué et al, 2017). These unique characteristics of HP1 place it at the center of heterochromatin formation, maintenance, and spreading.
In addition to histone modification, DNA methylation is also the major type of chromatin modification and it has been reported that the enzymes required for H3K9 methylation, such as G9a, SETDB1 & SUV39H1/2, can influence de novo DNA methylation, which is highly correlated with gene silencing (Lehnertz et al, 2003; Dong et al, 2008; Leung et al, 2011; Liu et al, 2014; Du et al, 2015). In almost all somatic cells of mice and human, ~70% of every cytosine in the context of CpG dinucleotides is found to be methylated (Li & Zhang, 2014). During germline development this DNA methylation is erased nearly completely, only to be re‐established in the gametes in a sex‐specific manner. Following fertilization, these levels will once again become severely reduced before establishing somatic patterns during implantation (Smith & Meissner, 2013; Greenberg & Bourc'his, 2019; Zeng & Chen, 2019). The enzymes responsible for deposition of the mark and maintenance through multiple cell cycles are DNMT3A/B:DNMT3L complexes and DNMT1 respectively. Genetic ablation of DNMT1 in somatic cells or the mouse embryo is lethal due to increased genomic instability highlighting its role in TE repression (Li et al, 1992; Jackson‐Grusby et al, 2001; Chen et al, 2007). Additionally, both the mark itself and the DNMT proteins can directly or indirectly recruit chromatin modifying complexes, such as histone deacetylases (HDACs) and the heterodimeric histone methyltransferase complex G9a:GLP (Jones et al, 1998; Nan et al, 1998; Fuks et al, 2001; Deplus et al, 2002; Estève et al, 2006). Together, this chromatin machinery allows for stable repression of heterochromatic regions particularly keeping repetitive loci in check.
Molecular mechanisms controlling the expression of repeats always require sophisticated pathways that can discern these elements from their own genes. One such pathway relates to intronless transcription, a particular feature of repeat RNAs which rely on reverse transcription. Importantly, this feature is sufficient to initiate a specific innate immunity response, a cellular mechanism also deployed against retroviruses (Seczynska et al, 2022). Other mechanisms for repeat recognition have also evolved and relate to the DNA sequence of repeats themselves. Both processes allow for the deployment of distinct strategies to old better adapted elements, and young, less mutated, and likely more deleterious ones. The suppression of elements via DNA‐binding proteins, such as transcription factors (TF), is frequently used to target evolutionary old families with higher mutational burden, even if they are not actively transcribed. However, a recent study argues for the importance of TF‐based suppression for young families as well (Wolf et al, 2020). While mechanisms resulting in repeat silencing have diverged among species these basic RNA‐ and DNA‐dependent strategies of repeat targeting are used throughout the animal kingdom.
Three major RNA‐based strategies are deployed against repetitive elements across species, generally referred to as transcriptional, co‐transcriptional, and post‐transcriptional gene silencing (TGS, CTGS, and PTGS). This review will focus on the TGS, where the major mode of action involves retention of the nascent unspliced RNA repeat transcripts on chromatin. These transcripts in turn act as platforms for co‐operative assembly of complexes that facilitate heterochromatin establishment and spreading over repetitive elements. Once the heterochromatin environment is established, chromatin‐binding proteins further stabilize the retention of repeat RNAs on chromatin, hence sustaining the cycle of transcription‐dependent chromatin silencing. As these nascent transcripts can in principle be deleterious for cells, it is of no surprise that this type of TGS is often found tightly interconnected with CTGS and PTGS, which are in essence sophisticated nuclear or cytoplasmic RNA decay systems respectively. Exciting findings revealed additional layers of regulation at the level of transcription elongation, splicing control, and translation; however, such mechanisms fall out of the scope of this review (Guang et al, 2010; Wilson & Doudna, 2013; Wang et al, 2015; Attig et al, 2018). All in all, the concerted activity of RNA‐ and DNA‐dependent strategies offers leakproof repression of repeat expression at every level.
As mentioned before, establishment of such stable silencing typically requires for the retention of repeat transcripts on chromatin. Indeed, studies in budding yeast, and in other model organisms, suggest that slowly processed transcripts can be actively retained on chromatin and then targeted for degradation in an exosome‐dependent manner (Custodio et al, 1999; Zenklusen et al, 2002; de Almeida et al, 2010; Eberle et al, 2010; Kallehauge et al, 2012; Muniz et al, 2021). Many interesting hypotheses were proposed on the role, the control, as well as the mechanisms of action of chromatin‐associated RNAs (Dinger et al, 2009; Deveson et al, 2017; Smurova & De Wulf, 2018; Talbert & Henikoff, 2018; Li & Fu, 2019; Muniz et al, 2021; Trigiante et al, 2021) Inspiring work across species has provided insight into how specificity could be achieved in different contexts (Wilson & Doudna, 2013; Holoch & Moazed, 2015; Martienssen & Moazed, 2015; Duempelmann et al, 2020). In many cases, there is involvement of protein complexes containing members of the Argonaute protein family in association with small RNA molecules that act as specificity factors via base pairing. These processes were best described in Schizosaccharomyces pombe (Holoch & Moazed, 2015; Martienssen & Moazed, 2015; Duempelmann et al, 2020), where double‐stranded RNA precursors, generated from pericentric DNA repeats are processed into small interfering RNAs (siRNAs) primarily by the collaborative action of the RNA‐dependent RNA polymerase complex (RDRC) and the ribonuclease Dicer 1 (Dcr1p; Volpe et al, 2002; Motamedi et al, 2004; Verdel et al, 2004; Martienssen & Moazed, 2015). Thanks to RDRC activity the RNAi response is amplified, and siRNAs are then loaded to an Argonaute‐containing complex, referred to as RNA‐induced transcriptional silencing (RITS) complex. The loaded RITS is responsible for directing and tethering of the H3K9 methylation machinery to proximal nucleosomes (Irvine et al, 2006; Schalch et al, 2009). H3K9 methylation in turn recruits homologs of the vertebrate heterochromatin protein 1 (HP1) and co‐repressor complexes (see Box 3). This allows for histone deacetylation and heterochromatin spreading. Likewise, RDRC and RITS complexes are also capable of recognizing H3K9 methylation marks, hence further sustaining siRNA biogenesis, heterochromatin maintenance and spread (Volpe et al, 2003; Bühler et al, 2006). Similar RNA interference‐dependent (RNAi) pathways have been identified in other model organisms such as Caenorhabditis elegans (Gu et al, 2012; Weiser & Kim, 2019; Duempelmann et al, 2020) and Droshophila melanogaster (Pal‐Bhadra et al, 2004; Bhattacharjee et al, 2019). These systems provide elegant models for studying how RNAs mediate specific targeting of heterochromatin formation to repeat DNA.
Even though RDRC/RITS complexes do not exist in mammals, Dicer can still downregulate the expression of centromeric repeats and transposable elements in mouse embryonic stem cells (mESCs; Kanellopoulou et al, 2005). An alternative class of small RNAs has also evolved to repress TEs in the germline of many organisms including mammals. These so‐called Piwi‐interacting RNAs (piRNAs) are defined by their interaction with the Piwi clade of Argonaute proteins. They are primarily derived from single‐stranded long non‐coding RNAs, from loci which can be described as graveyards of TEs. piRNAs also profit from an RDRC‐independent amplification mechanism that is often referred to as the “ping‐pong” cycle. In mice, it relies on the RNA slicing capacity of the protein called Piwi‐like 2 (PIWIL2 or MILI). Their biogenesis and function in restricting retrotransposons has been extensively reviewed elsewhere (Ernst et al, 2017; Czech et al, 2018; Ozata et al, 2019). Importantly for this review, piRNAs in mammals not only act at the level of PTGS, but are also involved in regulating heterochromatin and TGS of repetitive elements. However, before describing these new discoveries, it is pertinent to briefly introduce why repetitive elements need to be controlled, as well as the canonical RNA‐independent pathways involved in their silencing.
Canonical silencing of repetitive elements in mammals
Both satellite repeats and transposable elements act at the verge of evolution and numerous detailed previous reviews have discussed their role in reshuffling the genome, especially at regulatory elements, and in acting as building blocks for new genes (Goodier & Kazazian, 2008; Cordaux & Batzer, 2009; Rebollo et al, 2012; Elbarbary et al, 2016; Bourque et al, 2018; Platt et al, 2018; Rodriguez‐Terrones & Torres‐Padilla, 2018; Thakur et al, 2021; Fueyo et al, 2022). There are multiple examples of “domestication”, or so‐called exaptation of transposable elements as they acquire new and important regulatory functions (Gerdes et al, 2016). For instance, mouse oocytes express an alternative isoform of Dicer, driven by an intronic LTR retrotransposon, and this specific TE insertion is particularly important for meiosis and female fertility (Flemr et al, 2013). However, many changes to a repetitive element are not well tolerated by the cells and can lead to catastrophic consequences.
Mobilization and aberrant transcription of transposable elements, poses a potentially lethal threat to cells, in particular their genomic integrity and transcriptional regulation (Orr & Zoghbi, 2007; Hancks & Kazazian, 2016). In case of LINE1 elements for example, the very mechanism of transposition itself results in novel insertions and occasionally also small deletions (Moran et al, 1996; Gilbert et al, 2002; Beck et al, 2011; Richardson et al, 2014). Another remarkable example of TE interference with the gene activity is that of Intra‐cisternal A Particles (IAPs). Such elements can act as sites of heterochromatin nucleation and spread into flanking regions, as assessed by chromatin profiling of H3K9me3 and H4K20me3 marks (Rebollo et al, 2011). Though in rare cases, this results in transcriptional silencing of nearby genes, hence potentially jeopardizing cell fitness. TEs can also form new gene regulatory elements. For example, an IAP element inserted up to 100 kb upstream the Agouti gene acts as a cryptic promoter and drives ectopic expression of the latter. As a result, incomplete epigenetic silencing of the element leads to mosaic Agouti expression, and the heritable pleiotropic phenotype of yellow fur, obesity, diabetes, and increased tumor‐susceptibility (Duhl et al, 1994; Morgan et al, 1999). It is now well‐documented that transposable elements are primary sources of mutations (see Box 2) and can interfere with regulation of gene expression at multiple levels. Both features are tightly linked to disease (Hancks & Kazazian, 2016). Therefore, heterochromatin formation and silencing of repetitive elements should in most cases be stable, even though at some developmental stages their de‐repression can provide a source of genetic variability.
Stable repression is particularly important for TEs, which account for the more abundant and active portion of repetitive elements. In almost every somatic tissue, TEs are kept silenced within the heterochromatin compartment, which is established by the concerted action of multiple factors. Apart from the RNA‐mediated mechanisms the canonical RNA‐independent repression plays a vital role. The latter process typically depends on Kruppel‐associated box (KRAB)‐containing zinc finger proteins. This class of TFs can recognize and bind to retroviral DNA sequences resembling the structure of LTRs (see Box 2) and recruit the KRAB domain binding corepressor KAP1/TRIM28. These corepressors in turn can induce heterochromatin formation by recruiting histone deacetylases, the DNA methylation machinery, heterochromatin protein 1 (HP1), and the histone methyltransferase SETDB1/ESET (Friedman et al, 1996; Schultz et al, 2001; Schultz et al, 2002; Sripathy et al, 2006; Wolf & Goff, 2009; Rowe et al, 2010; Quenneville et al, 2012; Turelli et al, 2014; Wolf et al, 2015). Unlike the RNA‐dependent repression, KRAB factors can also target mutated TEs, which are no longer transcribed but retain the necessary sequence motif for zing finger domain binding. Moreover, this canonical pathway can also maintain repression in cellular contexts where heterochromatin would not allow for even low‐level TE expression.
Although various mechanisms contribute to TE silencing at the chromatin level across tissues, their relative importance is context specific. For instance, while the role of H3K9 histone methyltransferases in TE silencing of mouse ESCs is well documented (Hutnick et al, 2010; Matsui et al, 2010; Rowe et al, 2010; Karimi et al, 2011; Bulut‐Karslioglu et al, 2014; He et al, 2015; He et al, 2019), DNA methylation seems to be particularly crucial in maintaining TE silencing in somatic tissues (Walsh et al, 1998; Hutnick et al, 2010; Roulois et al, 2015). Conversely, extensive DNA demethylation in mouse ESCs is well tolerated leading to only a transient TE upregulation, which is subsequently reversed thanks to de novo deposition of H3K27me3 and H3K9me2/3 repressive marks (Walter et al, 2016). In line with these findings, TEs' DNA methylation is partially erased at two developmental stages, that is, pre‐implantation stages and germline development (Greenberg & Bourc'his, 2019), two periods when cells reprogram their epigenome to acquire either parent‐independent or sex‐specific chromatin patterns, respectively. These developmental windows are thus particularly sensitive to de‐repression of transposons. Moreover, how silencing complexes are specifically targeted to TEs in the absence of DNA methylation is an active field of research. One intriguing finding comes from germline development in mice, where Alexei Aravin and others have identified a male‐specific RNA pathway that mediates TE silencing, both in an epigenetic and in a post‐transcriptional manner (Aravin et al, 2006, 2008; Kuramochi‐Miyagawa et al, 2008, 2010; Ernst et al, 2017; Greenberg & Bourc'his, 2019; Ozata et al, 2019). While RNA‐mediated regulation of TE silencing in other organisms had been extensively studied (Kwon & Chung, 2020), its role in mammals and its potential interaction with chromatin repressive mechanisms remained unclear until recently. In the following section, we will discuss recent advances in this field.
RNA‐mediated silencing of repetitive elements in mammals
Chromatin‐associated RNAs
While much is known about how heterochromatin maintains silencing of repetitive elements, the question of how it is targeted in the first place remains more elusive. The KRAB/TRIM28 pathway is an elegant example of how this can be achieved through trans‐acting DNA binding proteins with silencing activity. However, a large proportion of repetitive elements are heterochromatinized independently of TRIM28 (Rowe et al, 2010; Maksakova et al, 2013). Interestingly, nuclear RNAs have recently emerged as alternative mediators of specificity for silencing both in cis and in trans.
In the nucleus, RNAs can be found either in a soluble state in the nucleoplasm, associated with the nuclear matrix or the chromatin itself. Chromatin‐associated RNAs (caRNAs) can be divided into either nascent transcripts that remain at their site of synthesis, acting in cis, or transcripts that are released from the transcription site in order to act at different loci, known as trans‐acting RNAs. Even though caRNAs can be found almost everywhere in the genome, in most cases their function remains elusive. They are generated mainly from genic sequences (~80%, including long ncRNA), primarily intronic, while their targets can be both protein coding and intergenic regions (Li et al, 2017; Zhou et al, 2019; Bonetti et al, 2020). Based on chromatin‐fractionation protocols (see Box 4), a large portion of the caRNAs, are mapped to promoters, enhancers and importantly for this review also repeat elements. For example in a recent study, Xu et al found that more than 50% of the IAPez elements (Intra‐cisternal A Particles), which belong to the family of LTR‐ERV retrotransposons, associate with chromatin at their sites of origin thus highlighting their potential for regulating chromatin state.
Box 4. Studying RNA‐chromatin interactions.
Establishing RNA‐chromatin interactions is not a trivial process as there are limitations in the methods used for their identification. Chromatin fractionation protocols, which are commonly used for detection of ca‐RNAs, retain RNA molecules that are associated not only with chromatin but also the general “nuclear matrix” (Creamer et al, 2021). Moreover, this approach, though unbiased, does not provide information on the mechanism how they are generated nor their mode of action. Other more sophisticated methods, which are based on proximity ligation, can provide further insights on RNA localization on chromatin (Li & Fu, 2019; Mishra & Kanduri, 2019; Kato & Carninci, 2020). The choice of the most suitable technique however requires taking into consideration parameters such as the amount of input material, the required coverage and the mapability of reads. MARGI‐seq (Sridhar et al, 2017), CHAR‐seq (Bell et al, 2018), GRID‐seq (Li et al, 2017; Zhou et al, 2019), RD‐SPRITE (Goronzy et al, 2022) and RADICL‐seq (Bonetti et al, 2020) are the most recent additions to the toolbox for identification of RNA–DNA/chromatin interactions. Out of all, the latter is the one provides a considerable advantage, as it allows for removal of nascent transcripts from the sequenced pool through addition of the RNA Pol II inhibitor actinomycin D in combination with RNAse H treatment.
Despite the highly context‐specific mode of action of caRNAs, certain principles can be described. In naïve mESCs, Li et al (2017) reported that around 63% of caRNAs act in trans and the vast majority of them (~90%) exhibit non sequence‐specific interactions. In fact, most of the regulatory caRNAs are likely to be found in complexes with RNA‐binding proteins (Lunde et al, 2007; Hentze et al, 2018), which can recognize an RNA sequence motif, the structure of the RNA molecule itself (single stranded, double stranded etc), a higher‐order structure, such as a stem‐loops and pseudo‐knots, a specific RNA‐modification or a combination of the above. With regards to their function, traditionally caRNAs include the fraction of the nascent pre‐mRNAs, the small nuclear RNAs (snRNAs) of the splicing machinery, some small nucleolar RNAs (snoRNAs), long non‐coding (lncRNAs), promoter upstream transcripts (PROMTs), enhancer RNAs (eRNAs), stable intronic sequence RNAs (sisRNAs), and the repeat‐derived RNAs which we will focus on in the remainder of this review (Chan & Pek, 2019; Li & Fu, 2019; Khelifi & Hussein, 2020; Agostini et al, 2021).
The inherent characteristics of RNA molecules render them highly potent and versatile candidates for chromatin regulation. First, their ability of base pairing provides an opportunity for locus‐specific interactions. One such well‐studied case is the formation of RNA:DNA hybrids, also known as R loops (Santos‐Pereira & Aguilera, 2015). Typically, R loops are formed when nascent RNAs anneal back to their template, displacing the complementary DNA strand. This results in the formation of a transient three‐strand structure, which predominantly occurs in GC‐skewed regions, often found in regulatory sequences, such as promoters and enhancers, but also repetitive elements, centromeres and highly transcribed genes (Santos‐Pereira & Aguilera, 2015; Niehrs & Luke, 2020). Such structures have recently been identified as important regulators of gene expression, by impeding transcription factor binding at promoters, hence conferring transcriptional silencing, or by excluding repressors and thus promoting transcriptional activation (García‐Muse & Aguilera, 2019). Furthermore, RNAs can be found in association with the DNA in a structure called triplex. This structure is formed when an RNA molecule invades the major groove of poly‐purine double‐stranded DNA, and forms interactions via Hoogsteen base‐pairing (Li & Fu, 2019; Mishra & Kanduri, 2019). However, most commonly RNAs exert their function through protein‐mediated interactions. In such cases, the RNA plays the role of the scaffold for one or more RNA‐binding protein, which is in direct contact with chromatin. One of the best well‐studied example of a caRNA is Xist, a lncRNA recruiting a plethora of transcriptional repressors and chromatin modifiers to inactivate one of the X chromosomes in female placental mammals (Żylicz & Heard, 2020). All in all, caRNAs are a varied class of RNAs that encompass many repeat elements. Such RNAs are closely associated with chromatin and thus have a potential to function as specificity factors initiating heterochromatin formation. Another feature of RNAs molecules, highly prevalent in caRNAs, is their covalent modifications, also known as the epi‐transcriptome. Hitherto, more than 170 modifications have been identified in the MODOMICS database (Boccaletto et al, 2018), with the N‐6 methyl‐adenosine (m6A) being one of the major and one of the most abundant (Zhang et al, 2020). In the sections below, we will discuss the importance of this RNA mark for RNA‐mediated heterochromatic silencing of TEs.
Role of m6A in epigenetic silencing of TEs
Chemical modifications of RNAs have emerged as important mediators of TE silencing at the RNA as well as the chromatin level. Recent technical and methodological advances allowed for the accurate detection of RNA modifications, especially of the most abundant m6A (Wang & Jia, 2020). Similarly to histone modifications, m6A dynamics rely on the concerted action of “writer” and “eraser” complexes. The major m6A methyltransferase complex (MTC) in polyA RNAs consists of the METTL3, which exerts the catalytic activity, the METTL14, which is an allosteric activator of METTL3, and other proteins such as WTAP and ZC3H13, which direct and facilitate interactions of the complex with other molecules (Huang et al, 2020; Kan et al, 2022). The m6A mark deposition occurs co‐transcriptionally (Ke et al, 2017), primarily within the context of the DRm6ACH consensus sequence (D = G, A, or U, R = G or A, H = A, C, or U). In mammals METTL3 is perhaps the sole enzyme responsible for this activity in DRm6ACH motif (Geula et al, 2015; Liu et al, 2020a; preprint: Poh et al, 2021) on the other hand, there are two main demethylases: ALKBH5 and the FTO (Huang et al, 2020).
Various “reader” proteins are also involved in regulating the multifaceted effects of m6A modification. Among the best studied are the YT521‐B homology (YTH) domain‐containing proteins, which include YTHDF1/2/3 and YTHDC1/2 (Shi et al, 2019). So far, m6A modification has been linked to the regulation of transcript splicing, nuclear export, translation, and mRNA decay (Wang et al, 2015; Xiao et al, 2016; Roundtree et al, 2017; Shi et al, 2017). As such, m6A is involved in virtually every physiological process (Huang et al, 2020; Kan et al, 2022), with pluripotency being one of the most pronounced (Batista et al, 2014; Wang et al, 2014; Geula et al, 2015; Wen et al, 2018; Chen et al, 2021; Liu et al, 2021, 2020a). Mettl3‐ko mouse embryos exhibit early embryonic lethality by E8.5 (Geula et al, 2015). Consistently, conversion of mESCs failed to efficiently exit naïve pluripotency and upregulate gastrulation markers. Hence, m6A could play an important role in the execution of specific and highly controlled transcriptional programs. In line with this, m6A has recently attracted much attention with regards to various diseases including cancer, liver steatosis, and heart disease (Huang et al, 2020; Yang et al, 2020; Jiang et al, 2021). In the following paragraphs, we are discussing the mechanistic aspects of how m6A functions in the context of TE repression.
Almost every type of RNA can be modified with m6A and this also includes caRNAs (Huang et al, 2020). Approximately 15–30% of caRNAs contain m6A in mouse ESCs (Liu et al, 2020a). It is noteworthy that m6A methylation on caRNAs is not uniformly distributed among different classes of RNAs, with around 35% of repeat‐derived caRNAs being enriched with m6A (Liu et al, 2020a). According to recent studies, LTR‐ERVK transcripts and especially from IAPez elements, exhibit high enrichment for m6A modification and METTL3 protein occupancy at their 5'UTRs (Chelmicki et al, 2021; Liu et al, 2021; Xu et al, 2021). This is in contrast to most polyA and non‐caRNAs which carry m6A methylation primarily at their stop‐codon regions and 3'UTRs (Dominissini et al, 2012; Meyer et al, 2012; Xu et al, 2021). METTL3 not only binds and modifies RNAs, it can also be detected in proximity to specific genomic regions that are often associated with retrotransposons. In mESCs about 90% of METTL3 peaks on chromatin co‐localize with H3K9me3 and H4K20me3 (Xu et al, 2021), indicating a potential function of m6A in targeting heterochromatin formation. In naïve pluripotent cells, these repressive marks mainly decorate telomeric, satellite, and long terminal repeats (LTRs), such as IAPs and early transposon elements (ETns; Martens et al, 2005; Mikkelsen et al, 2007; Efroni et al, 2008; Wen et al, 2009; Zylicz et al, 2015; Schlesinger & Meshorer, 2019). Indeed METTL3 chromatin‐binding is detected at repeat regions in ESCs but this pattern changes drastically upon differentiation, with METTL3 becoming predominantly bound to promoters of specific active genes (Xu et al, 2021). This is likely related to previous findings that m6A modification regulates the pluripotency network during differentiation (Batista et al, 2014; Wang et al, 2014; Geula et al, 2015; Zhang et al, 2020; Chen et al, 2021; Liu et al, 2021), but also the downregulation of LTR‐ERV retrotransposons upon exit from pluripotency (Martens et al, 2005; Macfarlan et al, 2012; Göke et al, 2015; Grow et al, 2015; Chuong et al, 2017; Chen et al, 2021). Therefore, METTL3 specifically in pluripotent cells, localizes to lowly transcribed repeats/retrotransposons where it might regulate their expression and heterochromatin environment.
The fate of m6A‐modified transcripts associated with chromatin remains elusive. Overall, METTL3 and YTHDC1 seem to bind chromatin‐associated retrotransposon transcripts, particularly the cis‐acting LINE1 (L1) and ERV superfamilies, such as the ERVL and ERVK/IAP. Once bound, METTL3/YTHDC1 down‐regulate TE expression on many levels and in an m6A‐dependent manner (Chelmicki et al, 2021; Liu et al, 2021; Xu et al, 2021). Indeed, the acute auxin‐inducible degradation of both METTL3 and METTL14 in mESC resulted in a strong increase in the stability of IAP transcripts. This was measured by tracking RNA levels upon inhibiting RNAPI and RNAPII‐transcription with actinomycin D (Chelmicki et al, 2021). These elegant experiments indicate an important role of m6A in PTGS at IAP elements. Indeed, further analysis showed that YTHDF2 can bind and downregulate IAP and other ERVK transcripts, in an m6A‐dependent manner (Chelmicki et al, 2021). Similar to these results, YTHDC1 can also recognize m6A‐modified repeat RNAs (Fig 1) and targets them for degradation (Liu et al, 2020a). This function is presumably facilitated by an interaction of m6A readers with core components of the nuclear exosome targeting (NEXT) complex such as RBM7 and ZCCHC8 (Liu et al, 2020a). In contrast, chronic loss of METTL3 in mESC resulted in derepression of IAPez but had no effect on the stability of these transcripts, indicating transcriptional regulation mechanisms to be responsible for their upregulation (Xu et al, 2021).
Figure 1. RNA m6A‐directed formation of heterochromatin in ES cells.
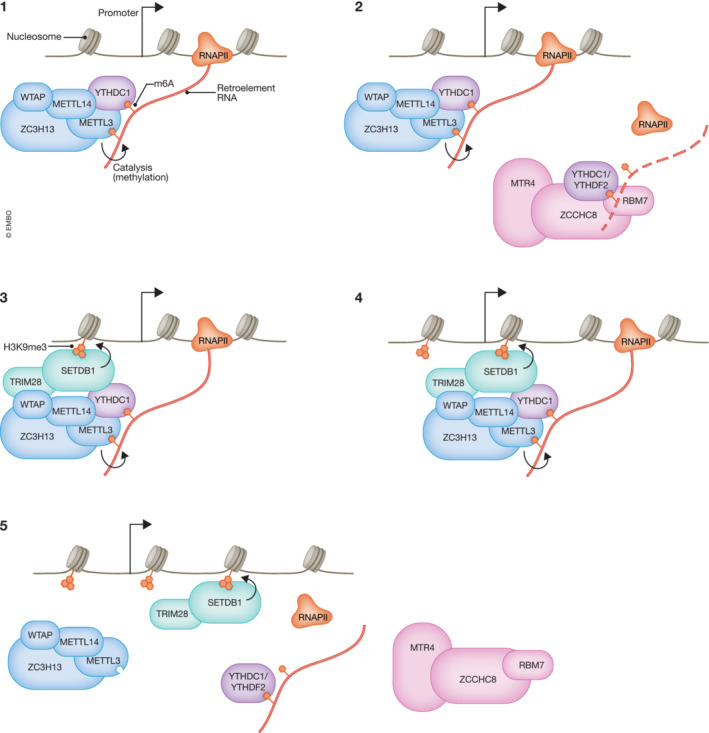
Genomic loci of retrotransposons such as LINE1 and IAP elements are usually decorated by the H3K9me3 and H4K20me3 chromatin marks (Mikkelsen et al, 2007). The former mark is often required for the deposition of the latter, as well as for efficient chromatin silencing (Schotta et al, 2004). N6‐adenine methylation (m6A) on chromatin‐associated retrotransposon RNAs can alter heterochromatic landscape and lead to transcriptional silencing. (1) At first, m6A is deposited on nascent retrotransposon RNAs by the METTL3‐METTL14 complex toward the 5′ end of chromatin‐associated RNAs (Xu et al, 2021). Different m6A readers recognize this modification and mediate various functions, however, the full complexity of their network remains elusive. One such m6A reader YTHDC1, seems to positively regulate METTL3‐METTL14 complex formation. (2) YTHDC1 as well as YTHDF2 are also involved in targeted degradation of the m6A labeled RNAs via the Nuclear Exosome Targeting Complex (NEXT; Liu et al, 2020a; Chelmicki et al, 2021; Chen et al, 2021; Garland et al, 2022). (3) m6A on such RNAs can also lead to METTL3‐dependent recruitment of the histone methyltransferase complex SETDB1‐TRIM28. (4, 5) Subsequent spreading of H3K9me3 mark on proximal nucleosomes results in decreased chromatin accessibility and transcriptional silencing (Liu et al, 2020a; Xu et al, 2021).
Thus, METTL3 plays a dual function in regulating the abundance of m6A‐marked transcripts, both through transcriptional and post‐transcriptional repression (Chelmicki et al, 2021; Liu et al, 2021; Xu et al, 2021). In mESC chromatin accessibility and nascent transcription are dramatically hampered by the catalytic activity of METTL3 and the presence of the m6A reader, YTHDC1 (Liu et al, 2020a). Studies in METTL3 knock‐out cells revealed that this increase in chromatin accessibility is accompanied by a global increase in epigenetic modifications associated with euchromatin, such as H3K4me3 and H3K27ac (Liu et al, 2020a), and depletion of heterochromatin‐ and repeat‐associated marks such as H3K9me2 (Li et al, 2020), H3K9me3 and H4K20me3 (Xu et al, 2021). Interestingly, the expression of the respective epigenetic writers or erasers remained unchanged, which indicated a role of m6A in recruitment of histone modifiers on chromatin. Furthermore, loss of METTL3 resulted in decreased levels of H3K9me3 and H4K20me3 specifically on IAPEz loci, which is furthermore accompanied by a reduction in the histone variant H3.3 and of DNA methylation. Notably, the effect of METTL3‐KO on DNA methylation was not restricted to IAP elements. According to Chelmicki et al (2021), however, the changes of H3K9me3, H3K4me3, and H3K27ac that have been reported in knock‐out cell lines, could not be reproduced when acute depletion of both METTL3:METTL14 proteins was deployed. Nonetheless, transcription upregulation of ERVs was immediate and more pronounced than in chronic loss of function models (Chelmicki et al, 2021). These results highlight the predominant function of m6A on post‐transcriptional regulation of TEs and its secondary and possibly more protracted role in epigenetic regulation of TE loci (Fig 1).
On a mechanistic level, the broader MTC complex consists of multiple subunits which regulate and direct its function. METTL3 localization on chromatin depends on its catalytic activity, as well as on METTL14 and other partners of the MTC complex, such as WTAP and ZC3H13 (Xu et al, 2021). Once localized on chromatin however, its catalytic activity is no longer required for deposition of H3K9me3 (Xu et al, 2021), indicating that MTC can function as an m6A‐independent secondary scaffold. Indeed, METTL3 and YTHDC1 physically bind to the TRIM28/SETDB1 histone methyltransferase complex in ESC and potentiate its recruitment specifically to IAPez elements (Liu et al, 2021; Xu et al, 2021). Consistently, SETDB1 loss increases the abundance of IAPez transcripts, which are now able to acquire more m6A methylation. Moreover, YTHDC1 is also found to be enriched at LINE1 and IAPez elements on chromatin level. Its localization relies on METTL3 catalytic activity, as well as its own ability to recognize m6A modification (Xu et al, 2021). Both YTHDC1 and SETDB1 potentiate the localization of METTL3 on chromatin and promote heterochromatin formation, suggesting a positive feedback loop (Xu et al, 2021). All in all, recent findings have uncovered an important role for the m6A machinery in RNA stability and targeting heterochromatin formation to specific retrotransposons. Next, we will discuss another group of RNA‐binding proteins‐ SPOC‐domain containing factors, which can also link transcripts, the m6A machinery and chromatin repressors.
SPOC‐domain containing proteins
Studies related to X‐chromosome inactivation revealed Spen paralogue and orthologue C‐terminal (SPOC) domain proteins as potential factors bridging chromatin associated RNAs with the m6A machinery and chromatin repressors (Shi et al, 2001; McHugh et al, 2015; Moindrot et al, 2015; Dossin et al, 2020). SPEN and SPOCD1 are two such proteins, that both exhibit RNA binding activity and can interact with chromatin remodeling and/or repressor complexes. Depletion of either SPOC‐protein was found to result in upregulation of ERVK, ETn, and LINE1 transposable elements in specific cellular contexts (Carter et al, 2020; Zoch et al, 2020). With regards to SPEN, it is reported to specifically repress the expression of ERVKs and this is accompanied by a loss in H3K9me3 and a substantial gain in chromatin accessibility (Carter et al, 2020; Zoch et al, 2020). As mentioned in the previous chapter similar chromatin effects have been observed upon METTL3 and YTHDC1 knock‐out, which also results in a more active chromatin profile with increased H3K27ac and H3K4me3 levels at repeat elements (Carter et al, 2020; Liu et al, 2020a; Xu et al, 2021). However, the exact mechanism of SPEN‐regulated ERVK transcription remains unknown. Interestingly, SPEN can interact with the m6A RNA methylation machinery, as well as other co‐repressors such as NuRD and NCoR/SMRT (Dossin et al, 2020). Moreover, SPEN's three RNA‐binding domains were reported to directly bind to ERVK RNAs (Carter et al, 2020). Thus, it is tempting to speculate that SPEN localizes to ERVK transcripts retained on chromatin where it can bridge m6A machinery with chromatin repressors. Intriguingly, mESCs lacking SPEN cannot efficiently differentiate, a phenotype consistent with the effects observed in absence of m6A‐related proteins (Carter et al, 2020; Chen et al, 2021; Geula et al, 2015). Nevertheless, Spen‐ko mice can survive up to embryonic day ~E13.5 in contrast to Mettl3‐ko mice, which are aborted earlier (Geula et al, 2015; Kuroda et al, 2003), indicating that additional players must be involved.
Similarly to SPEN, SPOCD1 is also involved in silencing of repeat elements but it does so by linking the piRNA pathway to chromatin regulation. In mouse male germ cells SPOCD1 interacts with MIWI2, a major regulator of the piRNA pathway, which plays distinct functions in the cytoplasm and the nucleus. While extensive work has established a post‐transcriptional regulatory role of cytoplasmic piRNAs, the nuclear MIWI2 is proposed to target DNA methylation to transposable elements (Aravin et al, 2006; Carmell et al, 2007; Aravin et al, 2008; Kuramochi‐Miyagawa et al, 2008; Kuramochi‐Miyagawa et al, 2010; Watanabe et al, 2018). Indeed, Miwi2‐ko mice not only show upregulation of LINE1 elements during spermatogenesis but also their DNA demethylation (Kuramochi‐Miyagawa et al, 2008). A model whereby piRNA‐loaded MIWI2 targets SPOCD1 to specific repetitive element loci has been proposed (Fig 2). At these loci, SPOCD1 can recruit de novo DNA methylation machinery (Fig 2) and subsequent H3K9me2/3‐deposition can take place through G9a, SETDB1, or SUV39H1/2, in a DNA methylation‐dependent or ‐independent manner (Peters et al, 2001; Estève et al, 2006; Tachibana et al, 2007; Fritsch et al, 2010; Quenneville et al, 2012; Pezic et al, 2014; Ernst et al, 2017; Greenberg & Bourc'his, 2019). In line with this model, loss of SPOCD1 results not only in the upregulation of young subfamilies of retrotransposons but also their drastic DNA demethylation, which phenocopies MIWI2‐KO (Zoch et al, 2020). Recent studies have identified DNMT3C as an important player in establishing this piRNA‐dependent DNA methylation of transposable elements in the mouse germline (Barau et al, 2016; Jain et al, 2017). TEX15 is also proposed to indirectly recruit the H3K9me3 machinery (Schöpp et al, 2020). All in all, recent findings suggest that SPOCD1 provides the direct mechanistic link between the piRNA pathway and heterochromatin formation in the male mouse germline.
Figure 2. piRNA‐directed DNA methylation in the developing germline.
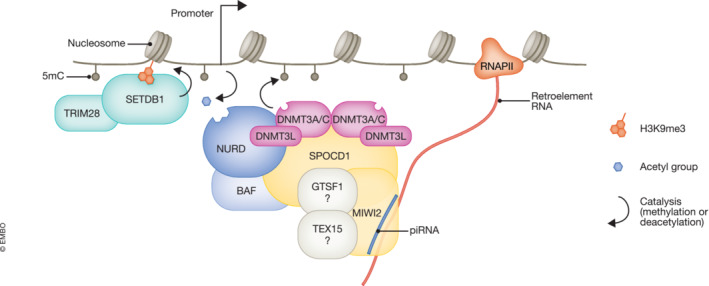
The developing germline undergoes extensive changes on its epigenome. During this period, cells are particularly vulnerable to active retrotransposition (Ernst et al, 2017). To cope with this, the germ cells utilize a small RNA‐mediated silencing pathway, which can act both on transcriptional and post‐transcriptional level. It was recently proposed that these small RNAs, known as piRNAs, may direct the MIWI2‐SPOCD1 complex to nascent retrotransposon transcripts, in a sequence‐specific manner. This complex interacts with several other epigenetic modifiers such as the DNA methylation machinery as well as the NuRD, and the BAF chromatin remodeling complexes. Their recruitment results in the deposition of 5‐methylcytosine (5mC) mark at promoters of specific retrotransposon subfamilies and it can potentially affect other important silencing marks such as H3K9me3, directed by SETDB1 (Greenberg & Bourc'his, 2019) or histone deacetylation, directed by the NuRD complex (Seto & Yoshida, 2014).
Another SPOC domain‐containing protein that recently attracted much attention is the transgene activator suppressor (TASOR/FAM208A/RAP140/C3ORF63/KIAA1105). Together with the M‐phase phosphoprotein 8 (MPHOSPH8/MPP8) and Periphilin 1 (PPHLN1), they constitute a complex known as human silencing hub (HUSH) which bridges TE transcripts to chromatin modifiers (Tchasovnikarova et al, 2015). Initially, TASOR and its partners were identified as targets in screens for suppressors of position effect variegation (PEV), human immunodeficiency retrovirus (HIV) as well as evolutionarily young retro‐transposons in mammalian cells (Yeung et al, 2009; Liu et al, 2011, 2018; Tchasovnikarova et al, 2015; Fukuda et al, 2018; Robbez‐Masson et al, 2018). Importantly for this review, the HUSH complex has also recently been linked to RNA. Indeed, Periphilin 1 can bind transcripts derived from evolutionary young LINE1 TEs (Seczynska et al, 2022). Moreover, the self‐associating properties of the arginine/tyrosine‐rich disordered N‐terminal region of Periphilin 1 seems to be crucial for HUSH assembly, HUSH‐dependent silencing and H3K9me3 deposition (Prigozhin et al, 2020). Based on protein homology to other arginine‐rich disordered domains it has been proposed that the N‐terminal domain of periphilin could self‐assemble into large solid‐like ribonucleoprotein particles on nascent transcripts (Prigozhin et al, 2020). Nevertheless, Periphilins' structure potentially precludes it from inducing the formation of fibers hydrogels or liquid–liquid phase separation typically associated with transcriptional silencing, RNA poll II and splicing machinery exclusion (Strom et al, 2017; Maharana et al, 2018; Gao et al, 2019; Ries et al, 2019; Fu & Zhuang, 2020; Liu et al, 2020b; Cheng et al, 2021).
Another component of the HUSH complex linked to RNA‐mediated silencing is TASOR. Just like other SPOC domain‐containing proteins, it functions as an assembly platform, linking repeat RNAs and RNA‐machinery (RNA Polymerase II) to transcriptional co‐repressors (Castello et al, 2012; Douse et al, 2020; Matkovic et al, 2022). Indeed, the HUSH complex directs H3K9me3 deposition in conjunction with the histone methyltransferase SETDB1, its methylated chaperone ATF7IP (Fig 3; Tchasovnikarova et al, 2015; Timms et al, 2016; Tsusaka et al, 2018), and furthermore interacts with the chromatin remodeler MORC2 (Tchasovnikarova et al, 2017; Liu et al, 2018). The HUSH complex is known to interact with chromatin also by recognizing H3K9me3 and recruitment to retroviral DNA through MPP8 (Fig 3; Zhu et al, 2018). Nevertheless, HUSH binding sites on chromatin comprise only ~1% of SETDB1 binding sites but the majority of those relate to potentially dangerous LINE1 transposable elements located in introns of transcriptionally active genes (Douse et al, 2020; Liu et al, 2018). Notably, such evolutionarily young transcribed LINE1 elements attract dozens of RNA binding proteins, some of which are known to interfere with TE expression (Attig et al, 2018). Seczynska et al (2022) recently reported that HUSH is targeted to newly invaded retroelements by recognizing long intronless transcripts, in a spliceosome‐independent manner, thus highlighting the role of transcription in silencing of TEs. However, the exact molecular mechanisms of HUSH‐dependent transcriptional silencing remain enigmatic. Lastly, the HUSH complex can potentially link epigenetic silencing of TEs and RNA decay in a way reminiscent of the m6A‐dependent pathway and the RNA quality control in S. pombe (Garland et al, 2022; Matkovic et al, 2022). Specifically, it has been reported that MPP8 recruits the nuclear exosome targeting (NEXT) complex on chromatin via an interaction with its MTR4 and ZCCHC8 subunits, and drives RNA degradation of LINE1 and ERV elements in naïve mESCs (Garland et al, 2022). The HUSH complex exhibits remarkable homology in domain organization and functional similarities with RITS, the complex responsible for silencing centromeric repeats in S. pombe (Douse et al, 2020) and it appears that the modes of action for silencing repetitive elements exhibit some conservation across evolutionary distant species.
Figure 3. HUSH‐mediated silencing.
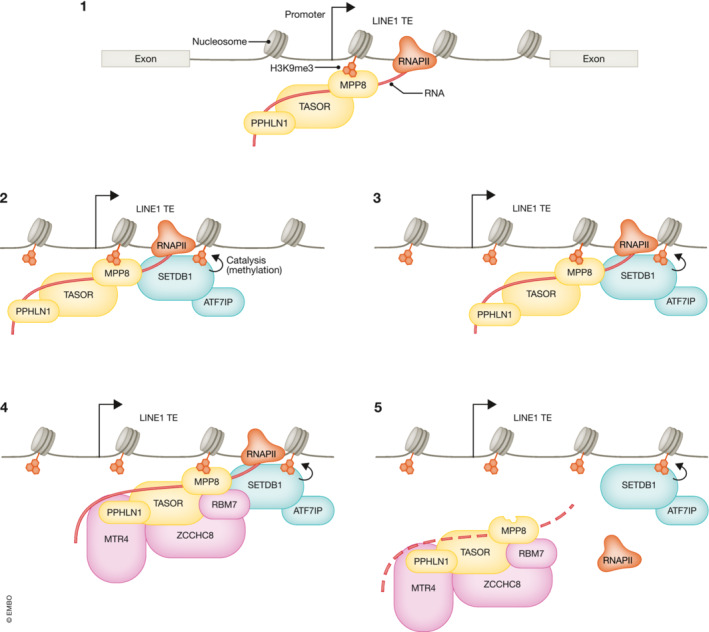
The human silencing complex (HUSH) is known to regulate the expression of evolutionary young TEs, such as LINE1 elements, primarily residing within introns. (1) Core subunits of the complex, namely TASOR and PPHLN1, have intrinsic RNA binding capacity which allows them to bind nascent LINE1 transcripts (Castello et al, 2012; Prigozhin et al, 2020). (2) The complex is known to confer transcriptional silencing by recruiting SETDB1:ATF7IP on chromatin, and guiding H3K9me3 deposition at LINE1 loci in an MPP8‐dependent manner (Tchasovnikarova et al, 2015; Timms et al, 2016; Douse et al, 2020). (3) The MPP8 subunit can also recognize H3K9me3 marks through its chromodomain, hence sustaining LINE1 heterochromatinization (Douse et al, 2020). (4) In parallel, both TASOR and MPP8 subunits directly interact with MTR4 and ZCCHC8 components of the Nuclear Exosome Targeting Complex (NEXT). (5) This allows for the recruitment of NEXT at TE loci allowing for targeted RNA decay (Garland et al, 2022).
Overall, according to the current model, SPOC domain‐containing proteins act as repressor hubs that become functional only when recruited to chromatin through retrotransposon or noncoding RNAs. At these loci, they can associate with the m6A machinery, RNA Polymerase II and a multitude of chromatin co‐repressors. Thus SPOC‐domain proteins are important repressors of retrotransposons. However, alternative RNA‐dependent strategies have evolved to regulate the expression of another class of repeat elements, the centromeric repeats. Regulation of these repetitive elements is crucial for overall genome stability in mammalian cells and will be discussed in further detail in the following section.
Satellite repeat silencing
Transcription of the centromeric and pericentric heterochromatin has an eminent role for the function of the centromere and thus genome stability. The first evidence for the presence of RNA molecules as a component of animal kinetochores dates back to electron micrography images generated in the 1970s (Rieder, 1979). Indeed, the notion that centromere‐related heterochromatin and the repeat elements embedded within remain constantly silent has been disproven. Despite their low abundance, it is clear that centromeric RNAs are transcribed in a cell cycle‐regulated manner by RNAPII, that they are capped and mostly polyadenylated (Lu & Gilbert, 2007; Talbert & Henikoff, 2018; Bury et al, 2020), and are required for centromere‐nucleoprotein assembly and genome integrity (Bouzinba‐Segard et al, 2006; Wong et al, 2007; Talbert & Henikoff, 2018). While multiple studies established that efficient silencing of pericentric satellite repeats is achieved by a positive feedback loop, involving H3K9me3‐dependent SUV39H1 recruitment (Rea et al, 2000; Muchardt et al, 2002; Lehnertz et al, 2003; Peters et al, 2003; Yamamoto & Sonoda, 2003; Fritsch et al, 2010), until recently, the exact mechanism and link to RNA remained elusive.
One hypothesis is that repeat transcripts may be involved in appropriate localization of SUV39H enzymes. Inspired by the findings in fission yeast (Ishida et al, 2012), where small RNAs recruit chromatin repressors, recent studies in mouse and human cells identified the SUV39H1 chromodomain as a broad nucleic acid binding domain. Both in vitro and in vivo, this domain can interact with all types of oligonucleotides (Porro et al, 2014; Scarola et al, 2015; Johnson et al, 2017; Shirai et al, 2017) and its affinity for nucleic acids allows direct binding of RNAs from major satellite and α‐satellite repeats, in mouse and human respectively. Both types of repeat RNAs remain associated with chromatin, likely due to RNA:DNA hybrids formation (Johnson et al, 2017; Shirai et al, 2017; Velazquez Camacho et al, 2017). Notably, SUV39H1's affinity for nucleic acids is independent of its ability to recognize H3K9me3 or to bind HP1 (Fig 4). Moreover, in vitro studies from Velazquez Camacho et al (2017) have showed that association of SUV39H enzymes with native nucleosomes can occur in an RNA‐dependent manner. Indeed both RNAs and H3K9me3/HP1 contribute to pericentric chromatin retention of the enzyme, thereby preventing the spread of heterochromatin and potential position‐effect variegation (Johnson et al, 2017; Shirai et al, 2017). Consistently, knocking‐down major satellite repeats resulted in diffusion of SUV39H1 protein on chromatin, while SUV39H1 loss, leads to upregulation of major satellite repeats (Bulut‐Karslioglu et al, 2014; Shirai et al, 2017; Velazquez Camacho et al, 2017).
Figure 4. RNA–DNA hybrids mediate heterochromatin formation in centromeric repeats in HeLa and mouse ESC.
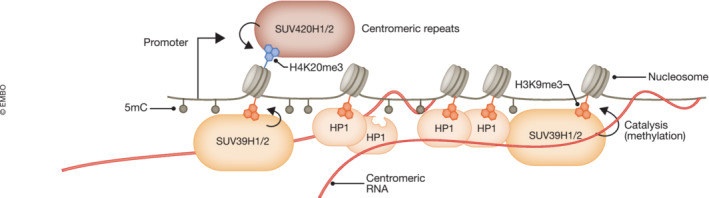
Deposition of H3K9me2/3 is involved in heterochromatin formation and transcriptional repression especially at repetitive elements (Bannister et al, 2001). Chromatin‐associated RNAs produced by centromeric α‐satellite repeats, remain in cis at the transcription site and act as platforms for stable association of SUV39 histone methyltransferase enzymes (Johnson et al, 2017; Velazquez Camacho et al, 2017). Particularly, the chromodomain of SUV39H1, which recognizes H3K9me2/3, has an intrinsic RNA and DNA binding affinity and promotes its centromeric localization (Shirai et al, 2017). Additional chromodomain‐containing proteins such as HP1, which can also bind to RNA, H3K9me2/3 (Muchardt et al, 2002), and SUV39 histone methyltransferases (Yamamoto & Sonoda, 2003) are recruited at the site and mediate chromatin compaction.
However, not all types of repeats are linked to heterochromatin formation, and those which are, appear to function locally. In human cells, RNA‐FISH combined with RNAse treatment revealed that ssRNAs generated from alpha‐satellite repeats, but not beta‐ or Satellite III, colocalize with H3K9me3 enrichment surrounding the centromere/kinetochore center marker HEC1, suggesting specificity for pericentric chromatin (Johnson et al, 2017). These ssRNAs are transcribed primarily by RNAPI and remain associated at their sites of origin. In addition, double knock‐out for SUV39H1 in human DLD1 and HeLa cells, resulted in decreased levels of H3K9me3 as well as HP1 foci, accompanied by an increase in alpha‐satellite repeat transcription. Notably, the transcription of alpha‐satellite did not affect their localization. These results support a model in which repeat transcripts are part of a feedback loop which sustains their local chromatin silencing. Intriguingly, analysis of SUV39H1 mutants lacking either the ability to bind nucleic acids, H3K9me3 or both, showed an additive effect in the double mutant which might indicate an H3K9me3‐independent, ssRNA‐dependent contribution to heterochromatin formation (Johnson et al, 2017). Fluorescence recovery after photobleaching (FRAP) revealed that H3K9me3 deposition is the major determinant of SUV39H1 localization in pericentric chromatin while inhibition of RNA polymerase I increases SUV39H1's mobility (Johnson et al, 2017).
In summary, SUV39H enzymes exhibit both histone‐ and RNA‐ binding activity (Fig 4), mediating proper localization to centromeric and pericentric heterochromatin, primarily in an H3K9me3‐dependent manner (Fig 4). Subsequent interaction with specific repeat‐derived transcripts, retained at their sites of origins, restricts the mobility of the histone modifying enzymes and prevents heterochromatin invasion to euchromatic regions (Fig 4). Intriguingly, a very recent study points out that m6A RNA methylation might also be involved in retention of major satellite repeat RNAs on chromatin, in the form of RNA:DNA hybrids, though the exact mechanism requires further investigation (Duda et al, 2021).
Conclusion
Silencing of repetitive elements is crucial for genome stability and survival of organisms. Cells have developed multiple lines of defense which ensure tight regulation of their expression. In this review, we highlighted the importance of RNA‐dependent mechanisms in mediating the heterochromatinization of repetitive elements primarily in embryonic stem cells and the germline. In these two developmental contexts the repetitive fraction of the genome is partially de‐repressed. There, activation of TEs is important for regulating the expression of germline and pluripotency genes, but also is a direct consequence of global epigenome resetting. At these crucial developmental junctures, RNA‐mediated processes help in de novo heterochromatin establishment and transcriptional repression. Recent evidence supports the notion that transcription of TE is one of their defining functional features which allows for specific targeting and efficient silencing. Thereby even newly transposed, potentially harmful TEs can be repressed just prior to, and shortly after, fertilization. Future studies characterizing the plethora of RNA‐binding proteins should shed light on how stable TE repression is induced and the interplay between RNA‐dependent and independent targeting mechanisms. Finally, in some contexts, such as satellite repeats in non‐transposable repeats remain expressed at low levels. Counterintuitively, these transcripts promote the establishment of well‐defined heterochromatin states, which might play a role in maintaining genome integrity. How wide‐spread this phenomenon is and how it relates to the formation of phase‐separated heterochromatin are questions that should be addressed in future work.
In this review, we have also highlighted some common features of RNA‐mediated silencing pathways that contribute to specificity in de novo heterochromatin formation. Firstly, RNA‐binding domains within chromatin repressor complexes often mediate their recruitment to repeat loci by cis‐acting transcripts. Secondly, there is frequent involvement of the m6A machinery in repressing repeats. Finally, we highlight the use of both transcriptional repression at the chromatin level as well as post‐transcriptional repression of the RNA transcripts themselves. The fact that mammals deploy such intricate and multi‐layered regulatory strategies again stresses the importance of tightly regulating repeat expression, especially in preparation for the successful development.
The last two decades have marked a turning point in unraveling the role that RNA molecules play in maintaining genome stability and controlling chromatin. Major technological advances have allowed us to delve deeper into the intricacies of these complex biological phenomena. The identification of new players in these sophisticated regulatory networks has added layers of complexity to our understanding of how cells and organisms protect their genome from sequence erosion and precisely execute their transcriptional programs. Despite these discoveries it is still unclear how cells initially identify RNAs transcribed from multi‐copy elements and what is the mechanism behind their retention on chromatin. Answering such questions should help in uncovering the molecular requirements for de novo heterochromatin assembly at crucial developmental junctures. Another question that remains to be answered is how widespread the role of low‐level transcription might be in inducing and maintaining heterochromatin formation. Does this mechanism apply to some host genes as well? In line with this, is there a role of low‐level transcription of transposable elements (TEs) utilized as gene regulatory elements? Recent findings also highlighted the importance of the cellular context in which specific repeat repression mechanisms are deployed. However, we still do not fully understand in which contexts RNA‐dependent mechanisms are particularly important. Might they also operate in specific adult or embryonic somatic tissues? We are finally beginning to shed light on the sequence of events that occur during repeat heterochromatinization. All in all, uncovering the multilayered mechanisms mediating repeat repression promises to have a profound impact on our understanding of how genome integrity is maintained in varying cellular and disease contexts.
Author contributions
Nikolaos Stamidis: Writing – original draft; writing – review and editing. Jan Jakub Żylicz: Funding acquisition; writing – review and editing.
Acknowledgements
This work was supported by grants from Novo Nordisk Fonden (NNF) (NNF21CC0073729), Lundbeckfonden (Lundbeck Foundation) (R345‐2020‐1497) and Danmarks Frie Forskningsfond (DFF) (0134‐00031B, 0169‐00031B).
The EMBO Journal (2023) 42: e111717
References
- Agostini F, Zagalak J, Attig J, Ule J, Luscombe NM (2021) Intergenic RNA mainly derives from nascent transcripts of known genes. Genome Biol 22: 136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allshire RC, Madhani HD (2018) Ten principles of heterochromatin formation and function. Nat Rev Mol Cell Biol 19: 229–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aravin A, Gaidatzis D, Pfeffer S, Lagos‐Quintana M, Landgraf P, Iovino N, Morris P, Brownstein MJ, Kuramochi‐Miyagawa S, Nakano T et al (2006) A novel class of small RNAs bind to MILI protein in mouse testes. Nature 442: 203–207 [DOI] [PubMed] [Google Scholar]
- Aravin AA, Sachidanandam R, Bourc'his D, Schaefer C, Pezic D, Toth KF, Bestor T, Hannon GJ (2008) A piRNA pathway primed by individual transposons is linked to De novo DNA methylation in mice. Mol Cell 31: 785–799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attig J, Agostini F, Gooding C, Chakrabarti AM, Singh A, Haberman N, Zagalak JA, Emmett W, Smith CWJ, Luscombe NM et al (2018) Heteromeric RNP assembly at LINEs controls lineage‐specific RNA processing. Cell 174: 1067–1081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannister AJ, Zegerman P, Partridge JF, Miska EA, Thomas JO, Allshire RC, Kouzarides T (2001) Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature 410: 120–124 [DOI] [PubMed] [Google Scholar]
- Barau J, Teissandier A, Zamudio N, Roy S, Nalesso V, Hérault Y, Guillou F, Bourc'his D (2016) The DNA methyltransferase DNMT3C protects male germ cells from transposon activity. Science 354: 909–912 [DOI] [PubMed] [Google Scholar]
- Batista PJ, Molinie B, Wang J, Qu K, Zhang J, Li L, Bouley DM, Lujan E, Haddad B, Daneshvar K et al (2014) m6A RNA modification controls cell fate transition in mammalian embryonic stem cells. Cell Stem Cell 15: 707–719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck CR, Garcia‐Perez JL, Badge RM, Moran JV (2011) LINE‐1 elements in structural variation and disease. Annu Rev Genomics Hum Genet 12: 187–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell JC, Jukam D, Teran NA, Risca VI, Smith OK, Johnson WL, Skotheim JM, Greenleaf WJ, Straight AF (2018) Chromatin‐associated RNA sequencing (ChAR‐seq) maps genome‐wide RNA‐to‐DNA contacts. Elife 7: 1–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharjee S, Roche B, Martienssen RA (2019) RNA‐induced initiation of transcriptional silencing (RITS) complex structure and function. RNA Biol 16: 1133–1146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boccaletto, P. , MacHnicka, M. A. , Purta, E. , Pitkowski, P. , Baginski, B. , Wirecki, T. K. , De Crécy‐Lagard, V. , Ross, R. , Limbach, P. A. , Kotter, A. , Helm, M. , & Bujnicki, J. M. (2018). MODOMICS: a database of RNA modification pathways. 2017 update. Nucleic Acids Res, 46(D1), D303–D307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonetti A, Agostini F, Suzuki AM, Hashimoto K, Pascarella G, Gimenez J, Roos L, Nash AJ, Ghilotti M, Cameron CJF et al (2020) RADICL‐seq identifies general and cell type‐specific principles of genome‐wide RNA‐chromatin interactions. Nat Commun 11: 1018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosch‐Presegué L, Raurell‐Vila H, Thackray JK, González J, Casal C, Kane‐Goldsmith N, Vizoso M, Brown JP, Gómez A, Ausió J et al (2017) Mammalian HP1 isoforms have specific roles in heterochromatin structure and organization. Cell Rep 21: 2048–2057 [DOI] [PubMed] [Google Scholar]
- Bourque G, Burns KH, Gehring M, Gorbunova V, Seluanov A, Hammell M, Imbeault M, Izsvák Z, Levin HL, Macfarlan TS et al (2018) Ten things you should know about transposable elements. Genome Biol 19: 199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouzinba‐Segard H, Guais A, Francastel C (2006) Accumulation of small murine minor satellite transcripts leads to impaired centromeric architecture and function. Proc Natl Acad Sci USA 103: 8709–8714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouha B, Schustak J, Badge RM, Lutz‐Prigge S, Farley AH, Morant JV, Kazazian HH (2003) Hot L1s account for the bulk of retrotransposition in the human population. Proc Natl Acad Sci USA 100: 5280–5285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bühler M, Verdel A, Moazed D (2006) Tethering RITS to a nascent transcript initiates RNAi‐ and heterochromatin‐dependent gene silencing. Cell 125: 873–886 [DOI] [PubMed] [Google Scholar]
- Bulut‐Karslioglu A, DeLaRosa‐Velázquez IA, Ramirez F, Barenboim M, Onishi‐Seebacher M, Arand J, Galán C, Winter GE, Engist B, Gerle B et al (2014) Suv39h‐dependent H3K9me3 Marks intact retrotransposons and silences LINE elements in mouse embryonic stem cells. Mol Cell 55: 277–290 [DOI] [PubMed] [Google Scholar]
- Bury L, Moodie B, Ly J, McKay LS, Miga KHH, Cheeseman IM (2020) Alpha‐satellite rna transcripts are repressed by centromere–nucleolus associations. Elife 9: 1–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmell MA, Girard A, van de Kant HJG, Bourc'his D, Bestor TH, de Rooij DG, Hannon GJ (2007) MIWI2 is essential for spermatogenesis and repression of transposons in the mouse male germline. Dev Cell 12: 503–514 [DOI] [PubMed] [Google Scholar]
- Carter AC, Xu J, Nakamoto MY, Wei Y, Zarnegar BJ, Shi Q, Broughton JP, Ransom RC, Salhotra A, Nagaraja SD et al (2020) Spen links rna‐mediated endogenous retrovirus silencing and x chromosome inactivation. Elife 9: 1–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castello A, Fischer B, Eichelbaum K, Horos R, Beckmann BM, Strein C, Davey NE, Humphreys DT, Preiss T, Steinmetz LM et al (2012) Insights into RNA biology from an atlas of mammalian mRNA‐binding proteins. Cell 149: 1393–1406 [DOI] [PubMed] [Google Scholar]
- Chan SN, Pek JW (2019) Stable Intronic sequence RNAs (sisRNAs): an expanding universe. Trends Biochem Sci 44: 258–272 [DOI] [PubMed] [Google Scholar]
- Chelmicki T, Roger E, Teissandier A, Dura M, Bonneville L, Rucli S, Dossin F, Fouassier C, Lameiras S, Bourc'his D (2021) m6A RNA methylation regulates the fate of endogenous retroviruses. Nature 591: 312–316 [DOI] [PubMed] [Google Scholar]
- Chen C, Liu W, Guo J, Liu Y, Liu X, Liu J, Dou X, Le R, Huang Y, Li C et al (2021) Nuclear m6A reader YTHDC1 regulates the scaffold function of LINE1 RNA in mouse ESCs and early embryos. Protein Cell 12: 455–474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T, Hevi S, Gay F, Tsujimoto N, He T, Zhang B, Ueda Y, Li E (2007) Complete inactivation of DNMT1 leads to mitotic catastrophe in human cancer cells. Nat Genet 39: 391–396 [DOI] [PubMed] [Google Scholar]
- Cheng Y, Xie W, Pickering BF, Chu KL, Savino AM, Yang X, Luo H, Nguyen DT, Mo S, Barin E et al (2021) N6‐Methyladenosine on mRNA facilitates a phase‐separated nuclear body that suppresses myeloid leukemic differentiation. Cancer Cell 39: 958–972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuong EB, Elde NC, Feschotte C (2017) Regulatory activities of transposable elements: from conflicts to benefits. Nat Rev Genet 18: 71–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordaux R, Batzer MA (2009) The impact of retrotransposons on human genome evolution. Nat Rev Genet 10: 691–703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creamer KM, Kolpa HJ, Lawrence JB (2021) Nascent RNA scaffolds contribute to chromosome territory architecture and counter chromatin compaction. Mol Cell 81: 3509–3525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Custodio N, Carmo‐Fonseca M, Geraghty F, Periera HS, Grosveld F, Antoniou M (1999) Inefficient processing impairs release of RNA from the site of transcription. EMBO J 18: 2855–2866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czech B, Munafò M, Ciabrelli F, Eastwood EL, Fabry MH, Kneuss E, Hannon GJ (2018) piRNA‐guided genome defense: from biogenesis to silencing. Annu Rev Genet 52: 131–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Almeida SF, García‐Sacristán A, Custódio N, Carmo‐Fonseca M (2010) A link between nuclear RNA surveillance, the human exosome and RNA polymerase II transcriptional termination. Nucleic Acids Res 38: 8015–8026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deplus R, Brenner C, Burgers WA, Putmans P, Kouzarides T, De Launoit Y, Fuks F (2002) Dnmt3L is a transcriptional repressor that recruits histone deacetylase. Nucleic Acids Res 30: 3831–3838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deveson IW, Hardwick SA, Mercer TR, Mattick JS (2017) The dimensions, dynamics, and relevance of the mammalian noncoding transcriptome. Trends Genet 33: 464–478 [DOI] [PubMed] [Google Scholar]
- Dinger ME, Amaral PP, Mercer TR, Mattick JS (2009) Pervasive transcription of the eukaryotic genome: functional indices and conceptual implications. Brief Funct Genomic Proteomic 8: 407–423 [DOI] [PubMed] [Google Scholar]
- Dominissini D, Moshitch‐Moshkovitz S, Schwartz S, Salmon‐Divon M, Ungar L, Osenberg S, Cesarkas K, Jacob‐Hirsch J, Amariglio N, Kupiec M et al (2012) Topology of the human and mouse m6A RNA methylomes revealed by m6A‐seq. Nature 485: 201–206 [DOI] [PubMed] [Google Scholar]
- Dong KB, Maksakova IA, Mohn F, Leung D, Appanah R, Lee S, Yang HW, Lam LL, Mager DL, Schübeler D et al (2008) DNA methylation in ES cells requires the lysine methyltransferase G9a but not its catalytic activity. EMBO J 27: 2691–2701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dossin F, Pinheiro I, Żylicz JJ, Roensch J, Collombet S, Le Saux A, Chelmicki T, Attia M, Kapoor V, Zhan Y et al (2020) SPEN integrates transcriptional and epigenetic control of X‐inactivation. Nature 578: 455–460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douse CH, Tchasovnikarova IA, Timms RT, Protasio AV, Seczynska M, Prigozhin DM, Albecka A, Wagstaff J, Williamson JC, Freund SMV et al (2020) TASOR is a pseudo‐PARP that directs HUSH complex assembly and epigenetic transposon control. Nat Commun 11: 4940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J, Johnson LM, Jacobsen SE, Patel DJ (2015) DNA methylation pathways and their crosstalk with histone methylation. Nat Rev Mol Cell Biol 16: 519–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duda KJ, Ching RW, Jerabek L, Shukeir N, Erikson G, Engist B, Onishi‐Seebacher M, Perrera V, Richter F, Mittler G et al (2021) m6A RNA methylation of major satellite repeat transcripts facilitates chromatin association and RNA:DNA hybrid formation in mouse heterochromatin. Nucleic Acids Res 49: 5568–5587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duempelmann L, Skribbe M, Bühler M (2020) Small RNAs in the transgenerational inheritance of epigenetic information. Trends Genet 36: 203–214 [DOI] [PubMed] [Google Scholar]
- Duhl DM, Vrieling H, Miller KA, Wolff GL, Barsh GS (1994) Neomorphic agouti mutations in obese yellow mice. Nat Genet 8: 59–65 [DOI] [PubMed] [Google Scholar]
- Eberle AB, Hessle V, Helbig R, Dantoft W, Gimber N, Visa N (2010) Splice‐site mutations cause Rrp6‐mediated nuclear retention of the Unspliced RNAs and transcriptional Down‐regulation of the splicing‐defective genes. PLoS ONE 5: e11540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efroni S, Duttagupta R, Cheng J, Dehghani H, Hoeppner DJ, Dash C, Bazett‐Jones DP, Le Grice S, McKay RDG, Buetow KH et al (2008) Global transcription in pluripotent embryonic stem cells. Cell Stem Cell 2: 437–447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbarbary RA, Lucas BA, Maquat LE (2016) Retrotransposons as regulators of gene expression. Science 351: aac7247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst C, Odom DT, Kutter C (2017) The emergence of piRNAs against transposon invasion to preserve mammalian genome integrity. Nat Commun 8: 1411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estève PO, Hang GC, Smallwood A, Feehery GR, Gangisetty O, Karpf AR, Carey MF, Pradhan S (2006) Direct interaction between DNMT1 and G9a coordinates DNA and histone methylation during replication. Genes Dev 20: 3089–3103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewing AD, Kazazian HH (2010) High‐throughput sequencing reveals extensive variation in human‐specific L1 content in individual human genomes. Genome Res 20: 1262–1270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fachinetti D, Han JS, McMahon MA, Ly P, Abdullah A, Wong AJ, Cleveland DW (2015) DNA sequence‐specific binding of CENP‐B enhances the Fidelity of human centromere function. Dev Cell 33: 314–327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan H, Chu J‐Y (2007) A brief review of short tandem repeat mutation. Genomics Proteomics Bioinformatics 5: 7–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flemr M, Malik R, Franke V, Nejepinska J, Sedlacek R, Vlahovicek K, Svoboda P (2013) A retrotransposon‐driven dicer isoform directs endogenous small interfering RNA production in mouse oocytes. Cell 155: 807–816 [DOI] [PubMed] [Google Scholar]
- Friedman JR, Fredericks WJ, Jensen DE, Speicher DW, Huang XP, Neilson EG, Rauscher FJ (1996) KAP‐1, a novel corepressor for the highly conserved KRAB repression domain. Genes Dev 10: 2067–2078 [DOI] [PubMed] [Google Scholar]
- Fritsch L, Robin P, Mathieu JRR, Souidi M, Hinaux H, Rougeulle C, Harel‐Bellan A, Ameyar‐Zazoua M, Ait‐Si‐Ali S (2010) A subset of the histone H3 lysine 9 methyltransferases Suv39h1, G9a, GLP, and SETDB1 participate in a multimeric complex. Mol Cell 37: 46–56 [DOI] [PubMed] [Google Scholar]
- Fu Y, Zhuang X (2020) m6A‐binding YTHDF proteins promote stress granule formation. Nat Chem Biol 16: 955–963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fueyo R, Judd J, Feschotte C, Wysocka J (2022) Roles of transposable elements in the regulation of mammalian transcription. Nat Rev Mol Cell Biol 24: 19–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuks F, Burgers WA, Godin N, Kasai M, Kouzarides T (2001) Dnmt3a binds deacetylases and is recruited by a sequence‐specific repressor to silence transcription. EMBO J 20: 2536–2544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda K, Okuda A, Yusa K, Shinkai Y (2018) A CRISPR knockout screen identifies SETDB1‐target retroelement silencing factors in embryonic stem cells. Genome Res 28: 846–858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Pei G, Li D, Li R, Shao Y, Zhang QC, Li P (2019) Multivalent m6A motifs promote phase separation of YTHDF proteins. Cell Res 29: 767–769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- García‐Muse T, Aguilera A (2019) R loops: from physiological to pathological roles. Cell 179: 604–618 [DOI] [PubMed] [Google Scholar]
- Garland W, Müller I, Wu M, Schmid M, Imamura K, Rib L, Sandelin A, Helin K, Jensen TH (2022) Chromatin modifier HUSH co‐operates with RNA decay factor NEXT to restrict transposable element expression. Mol Cell 82: 1691–1707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrido‐Ramos M (2017) Satellite DNA: An Evolving Topic. Genes 8: 230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gemayel R, Vinces MD, Legendre M, Verstrepen KJ (2010) Variable tandem repeats accelerate evolution of coding and regulatory sequences. Annu Rev Genet 44: 445–477 [DOI] [PubMed] [Google Scholar]
- Gerdes P, Richardson SR, Mager DL, Faulkner GJ (2016) Transposable elements in the mammalian embryo: pioneers surviving through stealth and service. Genome Biol 17: 1–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geula S, Moshitch‐Moshkovitz S, Dominissini D, Mansour AA, Kol N, Salmon‐Divon M, Hershkovitz V, Peer E, Mor N, Manor YS et al (2015) Stem cells. m6A mRNA methylation facilitates resolution of naïve pluripotency toward differentiation. Science 347: 1002–1006 [DOI] [PubMed] [Google Scholar]
- Gilbert N, Lutz‐Prigge S, Moran JV (2002) Genomic deletions created upon LINE‐1 retrotransposition. Cell 110: 315–325 [DOI] [PubMed] [Google Scholar]
- Göke J, Lu X, Chan YS, Ng HH, Ly LH, Sachs F, Szczerbinska I (2015) Dynamic transcription of distinct classes of endogenous retroviral elements marks specific populations of early human embryonic cells. Cell Stem Cell 16: 135–141 [DOI] [PubMed] [Google Scholar]
- Goodier JL (2016) Restricting retrotransposons: a review. Mob DNA 7: 16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodier JL, Kazazian HH (2008) Retrotransposons revisited: the restraint and rehabilitation of parasites. Cell 135: 23–35 [DOI] [PubMed] [Google Scholar]
- Goronzy IN, Quinodoz SA, Jachowicz JW, Ollikainen N, Bhat P, Guttman M (2022) Simultaneous mapping of 3D structure and nascent RNAs argues against nuclear compartments that preclude transcription. Cell Rep 41: 111730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg MVC, Bourc'his D (2019) The diverse roles of DNA methylation in mammalian development and disease. Nat Rev Mol Cell Biol 20: 590–607 [DOI] [PubMed] [Google Scholar]
- Groh S, Schotta G (2017) Silencing of endogenous retroviruses by heterochromatin. Cell Mol Life Sci 74: 2055–2065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grow EJ, Flynn RA, Chavez SL, Bayless NL, Wossidlo M, Wesche DJ, Martin L, Ware CB, Blish CA, Chang HY et al (2015) Intrinsic retroviral reactivation in human preimplantation embryos and pluripotent cells. Nature 522: 221–246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu SG, Pak J, Guang S, Maniar JM, Kennedy S, Fire A (2012) Amplification of siRNA in Caenorhabditis elegans generates a transgenerational sequence‐targeted histone H3 lysine 9 methylation footprint. Nat Genet 44: 157–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guang S, Bochner AF, Burkhart KB, Burton N, Pavelec DM, Kennedy S (2010) Small regulatory RNAs inhibit RNA polymerase II during the elongation phase of transcription. Nature 465: 1097–1101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guenatri M, Bailly D, Maison C, Almouzni G (2004) Mouse centric and pericentric satellite repeats form distinct functional heterochromatin. J Cell Biol 166: 493–505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hancks DC, Kazazian HH (2016) Roles for retrotransposon insertions in human disease. Mob DNA 7: 9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He J, Fu X, Zhang M, He F, Li W, Abdul MM, Zhou J, Sun L, Chang C, Li Y et al (2019) Transposable elements are regulated by context‐specific patterns of chromatin marks in mouse embryonic stem cells. Nat Commun 10: 34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Q, Kim H, Huang R, Lu W, Tang M, Shi F, Yang D, Zhang X, Huang J, Liu D et al (2015) The Daxx/Atrx complex protects tandem repetitive elements during DNA Hypomethylation by promoting H3K9 Trimethylation. Cell Stem Cell 17: 273–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hentze MW, Castello A, Schwarzl T, Preiss T (2018) A brave new world of RNA‐binding proteins. Nat Rev Mol Cell Biol 19: 327–341 [DOI] [PubMed] [Google Scholar]
- Holoch D, Moazed D (2015) RNA‐mediated epigenetic regulation of gene expression. Nat Rev Genet 16: 71–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Weng H, Chen J (2020) m6A modification in coding and non‐coding RNAs: roles and therapeutic implications in cancer. Cancer Cell 37: 270–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutnick LK, Huang X, Loo TC, Ma Z, Fan G (2010) Repression of retrotransposal elements in mouse embryonic stem cells is primarily mediated by a DNA methylation‐independent mechanism. J Biol Chem 285: 21082–21091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irvine DV, Zaratiegui M, Tolia NH, Goto DB, Chitwood DH, Vaughn MW, Joshua‐Tor L, Martienssen RA (2006) Argonaute slicing is required for heterochromatic silencing and spreading. Science 313: 1134–1137 [DOI] [PubMed] [Google Scholar]
- Ishida M, Shimojo H, Hayashi A, Kawaguchi R, Ohtani Y, Uegaki K, Nishimura Y, Nakayama JI (2012) Intrinsic nucleic acid‐binding activity of Chp1 Chromodomain is required for heterochromatic gene silencing. Mol Cell 47: 228–241 [DOI] [PubMed] [Google Scholar]
- Jackson‐Grusby L, Beard C, Possemato R, Tudor M, Fambrough D, Csankovszki G, Dausman J, Lee P, Wilson C, Lander E et al (2001) Loss of genomic methylation causes p53‐dependent apoptosis and epigenetic deregulation. Nat Genet 27: 31–39 [DOI] [PubMed] [Google Scholar]
- Jain D, Meydan C, Lange J, Claeys Bouuaert C, Lailler N, Mason CE, Anderson KV, Keeney S (2017) Rahu is a mutant allele of Dnmt3c, encoding a DNA methyltransferase homolog required for meiosis and transposon repression in the mouse male germline. PLoS Genet 13: e1006964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssen A, Colmenares SU, Karpen GH (2018) Heterochromatin: Guardian of the genome. Annu Rev Cell Dev Biol 34: 265–288 [DOI] [PubMed] [Google Scholar]
- Jiang X, Liu B, Nie Z, Duan L, Xiong Q, Jin Z, Yang C, Chen Y (2021) The role of m6A modification in the biological functions and diseases. Signal Transduct Target Ther 6: 74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson WL, Yewdell WT, Bell JC, McNulty SM, Duda Z, O'Neill RJ, Sullivan BA, Straight AF (2017) RNA‐dependent stabilization of SUV39H1 at constitutive heterochromatin. Elife 6: 1–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones PL, Veenstra GJ, Wade PA, Vermaak D, Kass SU, Landsberger N, Strouboulis J, Wolffe AP (1998) Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat Genet 19: 187–191 [DOI] [PubMed] [Google Scholar]
- Kallehauge TB, Robert M‐C, Bertrand E, Jensen TH (2012) Nuclear retention prevents premature cytoplasmic appearance of mRNA. Mol Cell 48: 145–152 [DOI] [PubMed] [Google Scholar]
- Kan RL, Chen J, Sallam T (2022) Crosstalk between epitranscriptomic and epigenetic mechanisms in gene regulation. Trends Genet 38: 182–193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanellopoulou C, Muljo SA, Kung AL, Ganesan S, Drapkin R, Jenuwein T, Livingston DM, Rajewsky K (2005) Dicer‐deficient mouse embryonic stem cells are defective in differentiation and centromeric silencing. Genes Dev 19: 489–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapitonov VV, Jurka J (2008) A universal classification of eukaryotic transposable elements implemented in Repbase. Nat Rev Genet 9: 411–412 [DOI] [PubMed] [Google Scholar]
- Karimi MM, Goyal P, Maksakova IA, Bilenky M, Leung D, Tang JX, Shinkai Y, Mager DL, Jones S, Hirst M et al (2011) DNA methylation and SETDB1/H3K9me3 regulate predominantly distinct sets of genes, retroelements, and chimeric transcripts in mescs. Cell Stem Cell 8: 676–687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato M, Carninci P (2020) Genome‐wide technologies to study RNA‐chromatin interactions. Noncoding RNA 6: 20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke S, Pandya‐Jones A, Saito Y, Fak JJ, Vågbø CB, Geula S, Hanna JH, Black DL, Darnell JE, Darnell RB (2017) m6A mRNA modifications are deposited in nascent pre‐mRNA and are not required for splicing but do specify cytoplasmic turnover. Genes Dev 31: 990–1006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khelifi G, Hussein SMI (2020) A new view of genome organization through RNA directed interactions. Front Cell Dev Biol 8: 1–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuramochi‐Miyagawa S, Watanabe T, Gotoh K, Takamatsu K, Chuma S, Kojima‐Kita K, Shiromoto Y, Asada N, Toyoda A, Fujiyama A et al (2010) MVH in piRNA processing and gene silencing of retrotransposons. Genes Dev 24: 887–892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuramochi‐Miyagawa S, Watanabe T, Gotoh K, Totoki Y, Toyoda A, Ikawa M, Asada N, Kojima K, Yamaguchi Y, Ijiri TW et al (2008) DNA methylation of retrotransposon genes is regulated by Piwi family members MILI and MIWI2 in murine fetal testes. Genes Dev 22: 908–917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuroda K, Han H, Tani S, Tanigaki K, Tun T, Furukawa T, Taniguchi Y, Kurooka H, Hamada Y, Toyokuni S et al (2003) Regulation of marginal zone B cell development by MINT, a suppressor of notch/RBP‐J signaling pathway. Immunity 18: 301–312 [DOI] [PubMed] [Google Scholar]
- Kwon Y, Chung YD (2020) RNA‐mediated regulation of chromatin structures. Genes Genomics 42: 609–617 [DOI] [PubMed] [Google Scholar]
- Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W et al (2001) Initial sequencing and analysis of the human genome. Nature 409: 860–921 [DOI] [PubMed] [Google Scholar]
- Lehnertz B, Ueda Y, Derijck AAHA, Braunschweig U, Perez‐Burgos L, Kubicek S, Chen T, Li E, Jenuwein T, Peters AHFM (2003) Suv39h‐mediated histone H3 lysine 9 methylation directs DNA methylation to major satellite repeats at pericentric heterochromatin. Curr Biol 13: 1192–1200 [DOI] [PubMed] [Google Scholar]
- Leung DC, Dong KB, Maksakova IA, Goyal P, Appanah R, Lee S, Tachibana M, Shinkai Y, Lehnertz B, Mager DL et al (2011) Lysine methyltransferase G9a is required for de novo DNA methylation and the establishment, but not the maintenance, of proviral silencing. Proc Natl Acad Sci USA 108: 5718–5723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung DC, Lorincz MC (2012) Silencing of endogenous retroviruses: when and why do histone marks predominate? Trends Biochem Sci 37: 127–133 [DOI] [PubMed] [Google Scholar]
- Li E, Bestor TH, Jaenisch R (1992) Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell 69: 915–926 [DOI] [PubMed] [Google Scholar]
- Li E, Zhang Y (2014) DNA methylation in mammals. Cold Spring Harb Perspect Biol 6: a019133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Fu XD (2019) Chromatin‐associated RNAs as facilitators of functional genomic interactions. Nat Rev Genet 20: 503–519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Zhou B, Chen L, Gou LT, Li H, Fu XD (2017) GRID‐seq reveals the global RNA‐chromatin interactome. Nat Biotechnol 35: 940–950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Xia L, Tan K, Ye X, Zuo Z, Li M, Xiao R, Wang Z, Liu X, Deng M et al (2020) N6‐Methyladenosine co‐transcriptionally directs the demethylation of histone H3K9me2. Nat Genet 52: 870–877 [DOI] [PubMed] [Google Scholar]
- Liu J, Gao M, He J, Wu K, Lin S, Jin L, Chen Y, Liu H, Shi J, Wang X et al (2021) The RNA m6A reader YTHDC1 silences retrotransposons and guards ES cell identity. Nature 591: 322–326 [DOI] [PubMed] [Google Scholar]
- Liu J, Dou X, Chen C, Chen C, Liu C, Michelle XM, Zhao S, Shen B, Gao Y, Han D et al (2020a) N6‐methyladenosine of chromosome‐associated regulatory RNA regulates chromatin state and transcription. Science 367: 580–586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Oliveira NMM, Cheney KM, Pade C, Dreja H, Bergin A‐MH, Borgdorff V, Beach DH, Bishop CL, Dittmar MT et al (2011) A whole genome screen for HIV restriction factors. Retrovirology 8: 94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu N, Lee CH, Swigut T, Grow E, Gu B, Bassik MC, Wysocka J (2018) Selective silencing of euchromatic L1s revealed by genome‐wide screens for L1 regulators. Nature 553: 228–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Brind'Amour J, Karimi MM, Shirane K, Bogutz A, Lefebvre L, Sasaki H, Shinkai Y, Lorincz MC (2014) Setdb1 is required for germline development and silencing of H3K9me3‐marked endogenous retroviruses in primordial germ cells. Genes Dev 28: 2041–2055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S‐Y, Feng Y, Wu J‐J, Zou M‐L, Sun Z‐L, Li X, Yuan F (2020b) M 6 a facilitates YTHDF‐independent phase separation. J Cell Mol Med 24: 2070–2072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- López‐Flores I, Garrido‐Ramos MA (2012) The repetitive DNA content of eukaryotic genomes. Genome Dyn 7: 1–28 [DOI] [PubMed] [Google Scholar]
- Lu J, Gilbert DM (2007) Proliferation‐dependent and cell cycle–regulated transcription of mouse pericentric heterochromatin. J Cell Biol 179: 411–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunde BM, Moore C, Varani G (2007) RNA‐binding proteins: modular design for efficient function. Nat Rev Mol Cell Biol 8: 479–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macfarlan TS, Gifford WD, Driscoll S, Lettieri K, Rowe HM, Bonanomi D, Firth A, Singer O, Trono D, Pfaff SL (2012) Embryonic stem cell potency fluctuates with endogenous retrovirus activity. Nature 487: 57–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madireddy A, Gerhardt J (2017) Replication through repetitive DNA elements and their role in human diseases. Adv Exp Med Biol 1042: 549–581 [DOI] [PubMed] [Google Scholar]
- Maharana S, Wang J, Papadopoulos DK, Richter D, Pozniakovsky A, Poser I, Bickle M, Rizk S, Guillén‐Boixet J, Franzmann TM et al (2018) RNA buffers the phase separation behavior of prion‐like RNA binding proteins. Science 360: 918–921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maksakova IA, Romanish MT, Gagnier L, Dunn CA, Van De Lagemaat LN, Mager DL (2006) Retroviral elements and their hosts: insertional mutagenesis in the mouse germ line. PLoS Genet 2: 1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maksakova IA, Thompson PJ, Goyal P, Jones SJM, Singh PB, Karimi MM, Lorincz MC (2013) Distinct roles of KAP1, HP1 and G9a/GLP in silencing of the two‐cell‐specific retrotransposon MERVL in mouse ES cells. Epigenetics Chromatin 6: 15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martens JHA, O'Sullivan RJ, Braunschweig U, Opravil S, Radolf M, Steinlein P, Jenuwein T (2005) The profile of repeat‐associated histone lysine methylation states in the mouse epigenome. EMBO J 24: 800–812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martienssen R, Moazed D (2015) RNAi and heterochromatin assembly. Cold Spring Harb Perspect Biol 7: a019323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matkovic R, Morel M, Lanciano S, Larrous P, Martin B, Bejjani F, Vauthier V, Hansen MMK, Emiliani S, Cristofari G et al (2022) TASOR epigenetic repressor cooperates with a CNOT1 RNA degradation pathway to repress HIV. Nat Commun 13: 1–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui T, Leung D, Miyashita H, Maksakova IA, Miyachi H, Kimura H, Tachibana M, Lorincz MC, Shinkai Y (2010) Proviral silencing in embryonic stem cells requires the histone methyltransferase ESET. Nature 464: 927–931 [DOI] [PubMed] [Google Scholar]
- McHugh CA, Chen CK, Chow A, Surka CF, Tran C, McDonel P, Pandya‐Jones A, Blanco M, Burghard C, Moradian A et al (2015) The Xist lncRNA interacts directly with SHARP to silence transcription through HDAC3. Nature 521: 232–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, Jaffrey SR (2012) Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell 149: 1635–1646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikkelsen TS, Ku M, Jaffe DB, Issac B, Lieberman E, Giannoukos G, Alvarez P, Brockman W, Kim TK, Koche RP et al (2007) Genome‐wide maps of chromatin state in pluripotent and lineage‐committed cells. Nature 448: 553–560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra K, Kanduri C (2019) Understanding long noncoding RNA and chromatin interactions: what we know so far. Noncoding RNA 5: 54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moindrot B, Cerase A, Coker H, Masui O, Grijzenhout A, Pintacuda G, Schermelleh L, Nesterova TB, Brockdorff N (2015) A pooled shRNA screen identifies Rbm15, Spen, and Wtap as factors required for Xist RNA‐mediated silencing. Cell Rep 12: 562–572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran JV, Holmes SE, Naas TP, DeBerardinis RJ, Boeke JD, Kazazian HH (1996) High frequency Retrotransposition in cultured mammalian cells. Cell 87: 917–927 [DOI] [PubMed] [Google Scholar]
- Morgan HD, Sutherland HGE, Martin DIK, Whitelaw E (1999) Epigenetic inheritance at the agouti locus in the mouse. Nat Genet 23: 314–318 [DOI] [PubMed] [Google Scholar]
- Motamedi MR, Verdel A, Colmenares SU, Gerber SA, Gygi SP, Moazed D (2004) Two RNAi complexes, RITS and RDRC, physically interact and localize to noncoding Centromeric RNAs. Cell 119: 789–802 [DOI] [PubMed] [Google Scholar]
- Muchardt C, Guillemé M, Seeler JS, Trouche D, Dejean A, Yaniv M (2002) Coordinated methyl and RNA binding is required for heterochromatin localization of mammalian HP1α. EMBO Rep 3: 975–981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muniz L, Nicolas E, Trouche D (2021) RNA polymerase II speed: a key player in controlling and adapting transcriptome composition. EMBO J 40: 1–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nan X, Ng H, Johnson CA, Laherty CD, Turner BM, Eisenman RN, Bird A (1998) Transcriptional repression by the methyl‐CpG‐binding protein MeCP2 involves a histone deacetylase complex. Nature 393: 386–389 [DOI] [PubMed] [Google Scholar]
- Niehrs C, Luke B (2020) Regulatory R‐loops as facilitators of gene expression and genome stability. Nat Rev Mol Cell Biol 21: 167–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr HT, Zoghbi HY (2007) Trinucleotide repeat disorders. Annu Rev Neurosci 30: 575–621 [DOI] [PubMed] [Google Scholar]
- Otake K, Ohzeki J, Shono N, Kugou K, Okazaki K, Nagase T, Yamakawa H, Kouprina N, Larionov V, Kimura H et al (2020) CENP‐B creates alternative epigenetic chromatin states permissive for CENP‐A or heterochromatin assembly. J Cell Sci 133: 1–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozata DM, Gainetdinov I, Zoch A, O'Carroll D, Zamore PD (2019) PIWI‐interacting RNAs: small RNAs with big functions. Nat Rev Genet 20: 89–108 [DOI] [PubMed] [Google Scholar]
- Padeken J, Methot SP, Gasser SM (2022) Establishment of H3K9‐methylated heterochromatin and its functions in tissue differentiation and maintenance. Nat Rev Mol Cell Biol 23: 623–640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal‐Bhadra M, Leibovitch BA, Gandhi SG, Rao M, Bhadra U, Birchler JA, Elgin SCR (2004) Heterochromatic silencing and HP1 localization in Drosophila are dependent on the RNAi machinery. Science 303: 669–672 [DOI] [PubMed] [Google Scholar]
- Peters AHFM, Kubicek S, Mechtler K, O'Sullivan RJ, Derijck AAHA, Perez‐Burgos L, Kohlmaier A, Opravil S, Tachibana M, Shinkai Y et al (2003) Partitioning and plasticity of repressive histone methylation states in mammalian chromatin. Mol Cell 12: 1577–1589 [DOI] [PubMed] [Google Scholar]
- Peters AHFM, O'Carroll D, Scherthan H, Mechtler K, Sauer S, Schöfer C, Weipoltshammer K, Pagani M, Lachner M, Kohlmaier A et al (2001) Loss of the Suv39h histone methyltransferases impairs mammalian heterochromatin and genome stability. Cell 107: 323–337 [DOI] [PubMed] [Google Scholar]
- Pezic D, Manakov SA, Sachidanandam R, Aravin AA (2014) piRNA pathway targets active LINE1 elements to establish the repressive H3K9me3 mark in germ cells. Genes Dev 28: 1410–1428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platt RN, Vandewege MW, Ray DA (2018) Mammalian transposable elements and their impacts on genome evolution. Chromosome Res 26: 25–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poh HX, Mirza AH, Pickering BF, Jaffrey SR (2021) Understanding the source of METTL3‐independent m6A in mRNA. BioRxiv 10.1101/2021.12.15.472866 [PREPRINT] [DOI] [Google Scholar]
- Porro A, Feuerhahn S, Delafontaine J, Riethman H, Rougemont J, Lingner J (2014) Functional characterization of the TERRA transcriptome at damaged telomeres. Nat Commun 5: 5379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prigozhin DM, Douse CH, Farleigh LE, Albecka A, Tchasovnikarova IA, Timms RT, Oda SI, Adolf F, Freund SMV, Maslen S et al (2020) Periphilin self‐association underpins epigenetic silencing by the HUSH complex. Nucleic Acids Res 48: 10313–10328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quenneville S, Turelli P, Bojkowska K, Raclot C, Offner S, Kapopoulou A, Trono D (2012) The KRAB‐ZFP/KAP1 system contributes to the early embryonic establishment of site‐specific DNA methylation patterns maintained during development. Cell Rep 2: 766–773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rea S, Eisenhaber F, O'Carroll D, Strahl BD, Sun ZW, Schmid M, Opravil S, Mechtler K, Ponting CP, Allis CD et al (2000) Regulation of chromatin structure by site‐specific histone H3 methyltransferases. Nature 406: 593–599 [DOI] [PubMed] [Google Scholar]
- Rebollo R, Karimi MM, Bilenky M, Gagnier L, Miceli‐Royer K, Zhang Y, Goyal P, Keane TM, Jones S, Hirst M et al (2011) Retrotransposon‐induced heterochromatin spreading in the mouse revealed by insertional polymorphisms. PLoS Genet 7: e1002301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebollo R, Romanish MT, Mager DL (2012) Transposable elements: an abundant and natural source of regulatory sequences for host genes. Annu Rev Genet 46: 21–42 [DOI] [PubMed] [Google Scholar]
- Richardson SR, Morell S, Faulkner GJ (2014) L1 retrotransposons and somatic mosaicism in the brain. Annu Rev Genet 48: 1–27 [DOI] [PubMed] [Google Scholar]
- Rieder CL (1979) Localization of ribonucleoprotein in the trilaminar kinetochore of PtK1. J Ultrastruct Res 66: 109–119 [DOI] [PubMed] [Google Scholar]
- Ries RJ, Zaccara S, Klein P, Olarerin‐George A, Namkoong S, Pickering BF, Patil DP, Kwak H, Lee JH, Jaffrey SR (2019) m6A enhances the phase separation potential of mRNA. Nature 571: 424–428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbez‐Masson L, Tie CHC, Conde L, Tunbak H, Husovsky C, Tchasovnikarova IA, Timms RT, Herrero J, Lehner PJ, Rowe HM (2018) The HUSH complex cooperates with TRIM28 to repress young retrotransposons and new genes. Genome Res 28: 836–845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez‐Terrones D, Torres‐Padilla ME (2018) Nimble and ready to mingle: transposon outbursts of early development. Trends Genet 34: 806–820 [DOI] [PubMed] [Google Scholar]
- Roulois D, Loo Yau H, Singhania R, Wang Y, Danesh A, Shen SY, Han H, Liang G, Jones PA, Pugh TJ et al (2015) DNA‐Demethylating agents target colorectal cancer cells by inducing viral mimicry by endogenous transcripts. Cell 162: 961–973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roundtree IA, Luo GZ, Zhang Z, Wang X, Zhou T, Cui Y, Sha J, Huang X, Guerrero L, Xie P et al (2017) YTHDC1 mediates nuclear export of N6‐methyladenosine methylated mRNAs. Elife 6: 1–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe HM, Jakobsson J, Mesnard D, Rougemont J, Reynard S, Aktas T, Maillard PV, Layard‐Liesching H, Verp S, Marquis J et al (2010) KAP1 controls endogenous retroviruses in embryonic stem cells. Nature 463: 237–240 [DOI] [PubMed] [Google Scholar]
- Rowe HM, Trono D (2011) Dynamic control of endogenous retroviruses during development. Virology 411: 273–287 [DOI] [PubMed] [Google Scholar]
- Santos‐Pereira JM, Aguilera A (2015) R loops: new modulators of genome dynamics and function. Nat Rev Genet 16: 583–597 [DOI] [PubMed] [Google Scholar]
- Sassaman DM, Dombroski BA, Moran JV, Kimberland ML, Naas TP, DeBerardinis RJ, Gabriel A, Swergold GD, Kazazian HH (1997) Many human L1 elements are capable of retrotransposition. Nat Genet 16: 37–43 [DOI] [PubMed] [Google Scholar]
- Scarola M, Comisso E, Pascolo R, Chiaradia R, Maria Marion R, Schneider C, Blasco MA, Schoeftner S, Benetti R (2015) Epigenetic silencing of Oct4 by a complex containing SUV39H1 and Oct4 pseudogene lncRNA. Nat Commun 6: 7631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schalch T, Job G, Noffsinger VJ, Shanker S, Kuscu C, Joshua‐Tor L, Partridge JF (2009) High‐affinity binding of Chp1 Chromodomain to K9 methylated histone H3 is required to establish Centromeric heterochromatin. Mol Cell 34: 36–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlesinger S, Meshorer E (2019) Open chromatin, epigenetic plasticity, and nuclear Organization in Pluripotency. Dev Cell 48: 135–150 [DOI] [PubMed] [Google Scholar]
- Schöpp T, Zoch A, Berrens RV, Auchynnikava T, Kabayama Y, Vasiliauskaitė L, Rappsilber J, Allshire RC, O'Carroll D (2020) TEX15 is an essential executor of MIWI2‐directed transposon DNA methylation and silencing. Nat Commun 11: 3739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schotta G, Lachner M, Sarma K, Ebert A, Sengupta R, Reuter G, Reinberg D, Jenuwein T (2004) A silencing pathway to induce H3‐K9 and H4‐K20 trimethylation at constitutive heterochromatin. Genes Dev 18: 1251–1262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz DC, Friedman JR, Rauscher FJ (2001) Targeting histone deacetylase complexes via KRAB‐zinc finger proteins: the PHD and bromodomains of KAP‐1 form a cooperative unit that recruits a novel isoform of the mi‐2alpha subunit of NuRD. Genes Dev 15: 428–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz DC, Ayyanathan K, Negorev D, Maul GG, Rauscher FJ (2002) SETDB1: a novel KAP‐1‐associated histone H3, lysine 9‐specific methyltransferase that contributes to HP1‐mediated silencing of euchromatic genes by KRAB zinc‐finger proteins. Genes Dev 16: 919–932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seczynska M, Bloor S, Cuesta SM, Lehner PJ (2022) Genome surveillance by HUSH‐mediated silencing of intronless mobile elements. Nature 601: 440–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seto E, Yoshida M (2014) Erasers of histone acetylation: the histone deacetylase enzymes. Cold Spring Harb Perspect Biol 6: a018713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi H, Wang X, Lu Z, Zhao BS, Ma H, Hsu PJ, Liu C, He C (2017) YTHDF3 facilitates translation and decay of N6‐methyladenosine‐modified RNA. Cell Res 27: 315–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi H, Wei J, He C (2019) Where, when, and how: context‐dependent functions of RNA methylation writers, readers, and erasers. Mol Cell 74: 640–650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Downes M, Xie W, Kao HY, Ordentlich P, Tsai CC, Hon M, Evans RM (2001) Sharp, an inducible cofactor that integrates nuclear receptor repression and activation. Genes Dev 15: 1140–1151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirai A, Kawaguchi T, Shimojo H, Muramatsu D, Ishida‐Yonetani M, Nishimura Y, Kimura H, Nakayama JI, Shinkai Y (2017) Impact of nucleic acid and methylated H3K9 binding activities of Suv39h1 on its heterochromatin assembly. Elife 6: e25317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smit AFA, Hubley R, Green P (n.d.) RepeatMasker Open‐4.0. http://www.repeatmasker.org
- Smith ZD, Meissner A (2013) DNA methylation: roles in mammalian development. Nat Rev Genet 14: 204–220 [DOI] [PubMed] [Google Scholar]
- Smurova K, De Wulf P (2018) Centromere and Pericentromere transcription: roles and regulation … in sickness and in health. Front Genet 9: 1–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sridhar B, Rivas‐Astroza M, Nguyen TC, Chen W, Yan Z, Cao X, Hebert L, Zhong S (2017) Systematic mapping of RNA‐chromatin interactions In vivo. Curr Biol 27: 602–609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sripathy SP, Stevens J, Schultz DC (2006) The KAP1 corepressor functions to coordinate the assembly of de novo HP1‐demarcated microenvironments of heterochromatin required for KRAB zinc finger protein‐mediated transcriptional repression. Mol Cell Biol 26: 8623–8638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strom AR, Emelyanov AV, Mir M, Fyodorov DV, Darzacq X, Karpen GH (2017) Phase separation drives heterochromatin domain formation. Nature 547: 241–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sultana T, Zamborlini A, Cristofari G, Lesage P (2017) Integration site selection by retroviruses and transposable elements in eukaryotes. Nat Rev Genet 18: 292–308 [DOI] [PubMed] [Google Scholar]
- Tachibana M, Nozaki M, Takeda N, Shinkai Y (2007) Functional dynamics of H3K9 methylation during meiotic prophase progression. EMBO J 26: 3346–3359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talbert PB, Henikoff S (2018) Transcribing centromeres: noncoding RNAs and kinetochore assembly. Trends Genet 34: 587–599 [DOI] [PubMed] [Google Scholar]
- Tchasovnikarova IA, Timms RT, Douse CH, Roberts RC, Dougan G, Kingston RE, Modis Y, Lehner PJ (2017) Hyperactivation of HUSH complex function by Charcot–Marie–tooth disease mutation in MORC2. Nat Genet 49: 1035–1044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tchasovnikarova IA, Timms RT, Matheson NJ, Wals K, Antrobus R, Göttgens B, Dougan G, Dawson MA, Lehner PJ (2015) Epigenetic silencing by the HUSH complex mediates position‐effect variegation in human cells. Science 348: 1481–1485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakur J, Packiaraj J, Henikoff S (2021) Sequence, chromatin and evolution of satellite DNA. Int J Mol Sci 22: 4309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timms RT, Tchasovnikarova IA, Antrobus R, Dougan G, Lehner PJ (2016) ATF7IP‐mediated stabilization of the histone methyltransferase SETDB1 is essential for heterochromatin formation by the HUSH complex. Cell Rep 17: 653–659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trigiante G, Blanes Ruiz N, Cerase A (2021) Emerging roles of repetitive and repeat‐containing RNA in nuclear and chromatin organization and gene expression. Front Cell Dev Biol 9: 415–429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsusaka T, Kikuchi M, Shimazu T, Suzuki T, Sohtome Y, Akakabe M, Sodeoka M, Dohmae N, Umehara T, Shinkai Y (2018) Tri‐methylation of ATF7IP by G9a/GLP recruits the chromodomain protein. Epigenetics Chromatin 11: 1–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turelli P, Castro‐Diaz N, Marzetta F, Kapopoulou A, Raclot C, Duc J, Tieng V, Quenneville S, Trono D (2014) Interplay of TRIM28 and DNA methylation in controlling human endogenous retroelements. Genome Res 24: 1260–1270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velazquez Camacho O, Galan C, Swist‐Rosowska K, Ching R, Gamalinda M, Karabiber F, De La Rosa‐Velazquez I, Engist B, Koschorz B, Shukeir N et al (2017) Major satellite repeat RNA stabilize heterochromatin retention of Suv39h enzymes by RNA‐nucleosome association and RNA:DNA hybrid formation. Elife 6: 1–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdel A, Jia S, Gerber S, Sugiyama T, Gygi S, Grewal SIS, Moazed D (2004) RNAi‐mediated targeting of heterochromatin by the RITS complex. Science 303: 672–676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volpe TA, Kidner C, Hall IM, Teng G, Grewal SIS, Martienssen RA (2002) Regulation of heterochromatic silencing and histone H3 Lysine‐9 methylation by RNAi. Science 297: 1833–1837 [DOI] [PubMed] [Google Scholar]
- Volpe T, Schramke V, Hamilton GL, White SA, Teng G, Martienssen RA, Allshire RC (2003) RNA interference is required for normal centromere function in fission yeast. Chromosome Res 11: 137–146 [DOI] [PubMed] [Google Scholar]
- Walsh CP, Chaillet JR, Bestor TH (1998) Transcription of IAP endogenous retroviruses is constrained by cytosine methylation. Nat Genet 20: 116–117 [DOI] [PubMed] [Google Scholar]
- Walter M, Teissandier A, Pérez‐Palacios R, Bourc'his D (2016) An epigenetic switch ensures transposon repression upon dynamic loss of DNA methylation in embryonic stem cells. Elife 5: 1–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Zhao BS, Roundtree IA, Lu Z, Han D, Ma H, Weng X, Chen K, Shi H, He C (2015) N6‐methyladenosine modulates messenger RNA translation efficiency. Cell 161: 1388–1399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Li Y, Toth JI, Petroski MD, Zhang Z, Zhao JC (2014) N6‐methyladenosine modification destabilizes developmental regulators in embryonic stem cells. Nat Cell Biol 16: 191–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Jia G (2020) Detection methods of epitranscriptomic mark N6‐methyladenosine. Essays Biochem 64: 967–979 [DOI] [PubMed] [Google Scholar]
- Watanabe T, Cui X, Yuan Z, Qi H, Lin H (2018) MIWI2 targets RNAs transcribed from piRNA‐dependent regions to drive DNA methylation in mouse prospermatogonia. EMBO J 37: 1–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiser NE, Kim JK (2019) Multigenerational regulation of the Caenorhabditis elegans chromatin landscape by germline small RNAs. Annu Rev Genet 53: 289–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen B, Wu H, Shinkai Y, Irizarry RA, Feinberg AP (2009) Large histone H3 lysine 9 dimethylated chromatin blocks distinguish differentiated from embryonic stem cells. Nat Genet 41: 246–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen J, Lv R, Ma H, Shen H, He C, Wang J, Jiao F, Liu H, Yang P, Tan L et al (2018) Zc3h13 regulates nuclear RNA m6A methylation and mouse embryonic stem cell self‐renewal. Mol Cell 69: 1028–1038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wicker T, Sabot F, Hua‐Van A, Bennetzen JL, Capy P, Chalhoub B, Flavell A, Leroy P, Morgante M, Panaud O et al (2007) A unified classification system for eukaryotic transposable elements. Nat Rev Genet 8: 973–982 [DOI] [PubMed] [Google Scholar]
- Wicker T, Sabot F, Hua‐Van A, Bennetzen JL, Capy P, Chalhoub B, Flavell A, Leroy P, Morgante M, Panaud O et al (2009a) Reply: a unified classification system for eukaryotic transposable elements should reflect their phylogeny. Nat Rev Genet 10: 276 [DOI] [PubMed] [Google Scholar]
- Wicker T, Sabot F, Hua‐Van A, Bennetzen JL, Capy P, Chalhoub B, Flavell A, Leroy P, Morgante M, Panaud O et al (2009b) A unified classification system for eukaryotic transposable elements should reflect their phylogeny. Nat Rev Genet 10: 276 [DOI] [PubMed] [Google Scholar]
- Wilson RC, Doudna JA (2013) Molecular mechanisms of RNA interference. Annu Rev Biophys 42: 217–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf D, Goff SP (2009) Embryonic stem cells use ZFP809 to silence retroviral DNAs. Nature 458: 1201–1204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf G, de Iaco A, Sun MA, Bruno M, Tinkham M, Hoang D, Mitra A, Ralls S, Trono D, Macfarlan TS (2020) Krab‐zinc finger protein gene expansion in response to active retrotransposons in the murine lineage. Elife 9: 1–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf G, Yang P, Füchtbauer AC, Füchtbauer E‐M, Silva AM, Park C, Wu W, Nielsen AL, Pedersen FS, Macfarlan TS (2015) The KRAB zinc finger protein ZFP809 is required to initiate epigenetic silencing of endogenous retroviruses. Genes Dev 29: 538–554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong LH, Brettingham‐Moore KH, Chan L, Quach JM, Anderson MA, Northrop EL, Hannan R, Saffery R, Shaw ML, Williams E et al (2007) Centromere RNA is a key component for the assembly of nucleoproteins at the nucleolus and centromere. Genome Res 17: 1146–1160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao W, Adhikari S, Dahal U, Chen Y‐S, Hao Y‐J, Sun B‐F, Sun H‐Y, Li A, Ping X‐L, Lai W‐Y et al (2016) Nuclear m(6)a reader YTHDC1 regulates mRNA splicing. Mol Cell 61: 507–519 [DOI] [PubMed] [Google Scholar]
- Xing J, Zhang Y, Han K, Salem AH, Sen SK, Huff CD, Zhou Q, Kirkness EF, Levy S, Batzer MA et al (2009) Mobile elements create structural variation: analysis of a complete human genome. Genome Res 19: 1516–1526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Li J, He C, Wen J, Ma H, Rong B, Diao J, Wang L, Wang J, Wu F et al (2021) METTL3 regulates heterochromatin in mouse embryonic stem cells. Nature 591: 317–321 [DOI] [PubMed] [Google Scholar]
- Yamamoto K, Sonoda M (2003) Self‐interaction of heterochromatin protein 1 is required for direct binding to histone methyltransferase, SUV39H1. Biochem Biophys Res Commun 301: 287–292 [DOI] [PubMed] [Google Scholar]
- Yang C, Hu Y, Zhou B, Bao Y, Li Z, Gong C, Yang H, Wang S, Xiao Y (2020) The role of m6A modification in physiology and disease. Cell Death Dis 11: 960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yap KL, Zhou M‐M (2010) Keeping it in the family: diverse histone recognition by conserved structural folds. Crit Rev Biochem Mol Biol 45: 488–505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yap KL, Zhou M‐M (2011) Structure and mechanisms of lysine methylation recognition by the chromodomain in gene transcription. Biochemistry 50: 1966–1980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung ML, Houzet L, Yedavalli VSRK, Jeang K‐T (2009) A genome‐wide short hairpin RNA screening of Jurkat T‐cells for human proteins contributing to productive HIV‐1 replication. J Biol Chem 284: 19463–19473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng Y, Chen T (2019) DNA methylation reprogramming during mammalian development. Genes 10: 257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zenklusen D, Vinciguerra P, Wyss J‐C, Stutz F (2002) Stable mRNP formation and export require Cotranscriptional recruitment of the mRNA export factors Yra1p and Sub2p by Hpr1p. Mol Cell Biol 22: 8241–8253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Zhai Y, Zhang S, Dai X, Li Z (2020) Roles of N6‐Methyladenosine (m6A) in stem cell fate decisions and early embryonic development in mammals. Front Cell Dev Biol 8: 1–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou B, Li X, Luo D, Lim DH, Zhou Y, Fu XD (2019) GRID‐seq for comprehensive analysis of global RNA–chromatin interactions. Nat Protoc 14: 2036–2068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Wang GZ, Cingöz O, Goff SP (2018) NP220 mediates silencing of unintegrated retroviral DNA. Nature 564: 278–282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoch A, Auchynnikava T, Berrens RV, Kabayama Y, Schöpp T, Heep M, Vasiliauskaitė L, Pérez‐Rico YA, Cook AG, Shkumatava A et al (2020) SPOCD1 is an essential executor of piRNA‐directed de novo DNA methylation. Nature 584: 635–639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zylicz JJ, Dietmann S, Günesdogan U, Hackett JA, Cougot D, Lee C, Surani MA (2015) Chromatin dynamics and the role of G9a in gene regulation and enhancer silencing during early mouse development. Elife 4: 1–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Żylicz JJ, Heard E (2020) Annual review of biochemistry. Annu Rev Biochem 89: 255–282 [DOI] [PubMed] [Google Scholar]