

Abstract
The tumor suppressor p53 is critical for tumor suppression, but the regulatory role of p53 in alcohol‐induced fatty liver remains unclear. Here, we show a role for p53 in regulating ethanol metabolism via acetaldehyde dehydrogenase 2 (ALDH2), a key enzyme responsible for the oxidization of alcohol. By repressing ethanol oxidization, p53 suppresses intracellular levels of acetyl‐CoA and histone acetylation, leading to the inhibition of the stearoyl‐CoA desaturase‐1 (SCD1) gene expression. Mechanistically, p53 directly binds to ALDH2 and prevents the formation of its active tetramer and indirectly limits the production of pyruvate that promotes the activity of ALDH2. Notably, p53‐deficient mice exhibit increased lipid accumulation, which can be reversed by ALDH2 depletion. Moreover, liver‐specific knockdown of SCD1 alleviates ethanol‐induced hepatic steatosis caused by p53 loss. By contrast, overexpression of SCD1 in liver promotes ethanol‐induced fatty liver development in wild‐type mice, while it has a mild effect on p53 −/− or ALDH2 −/− mice. Overall, our findings reveal a previously unrecognized function of p53 in alcohol‐induced fatty liver and uncover pyruvate as a natural regulator of ALDH2.
Keywords: alcoholic fatty liver, ALDH2, histone acetylation, p53, protein–protein interaction
Subject Categories: Metabolism, Molecular Biology of Disease
The tumor suppressor p53 acts as a direct inhibitor of acetaldehyde dehydrogenase 2 activity, which reduces ethanol‐induced lipid accumulation in the liver.

Introduction
Tumor suppressor p53 controls various biological processes, including apoptosis, cell cycle, DNA repair, and cell proliferation through its target genes (Vousden & Prives, 2009). Emerging studies show that p53 plays an important role in regulating the metabolism of glucose, lipids, and amino acids, among others (Olovnikov et al, 2009; Vousden & Ryan, 2009; Cheung & Vousden, 2010; Floter et al, 2017; Liu et al, 2019; Lahalle et al, 2021; Liu & Gu, 2021). Notably, hepatic p53 has been demonstrated to be a key regulator in different liver diseases, such as hepatic insulin resistance, development of nonalcoholic fatty liver disease (NAFLD) and NAFLD‐hepatitis, hepatocellular carcinoma (HCC) development, and liver regeneration (Krstic et al, 2018). For instance, p53 controls the mevalonate pathway to mediate liver tumor suppression through SREBP2 (Moon et al, 2019). Our recent studies have shown that p53 inhibits NAFLD and NAFLD‐induced HCC through directly transcriptional suppression of SQLE expression (Sun et al, 2021). Alcoholic liver disease is one of the leading causes of mortality worldwide (Lee et al, 2019; Campana et al, 2021). Although the progression of this disease is well characterized, including early fatty liver, steatosis, and steatohepatitis, with some individuals eventually progressing to fibrosis and liver failure (Osna et al, 2017), the exact mechanisms underlying its pathogenesis remain unclear. Therefore, it is crucial to gain insight into the underlying mechanisms of alcohol metabolism and whether p53 plays a role in alcoholic liver disease and alcohol metabolism.
Liver is the primary organ responsible for the detoxification of alcohol. Acetaldehyde dehydrogenase 2 (ALDH2) is the key enzyme responsible for metabolism of the alcohol metabolite acetaldehyde in liver (Wang et al, 2020). ALDH2 catalyzes the reaction of acetaldehyde and NAD+ to form acetate, NADH and H+. It is a tetramer composed of four identical subunits, each containing three functional domains of coenzyme or NAD+ binding, catalysis, and oligomerization (Larson et al, 2005; Adeniji et al, 2018). Approximately 40–50% of Asian people have an inactive ALDH2 mutation that results in acetaldehyde accumulation after alcohol consumption (Larson et al, 2005). Acetaldehyde is considered to be highly toxic, mutagenic, and carcinogenic and plays an important role in the pathogenesis of alcoholic liver disease (Setshedi et al, 2010), but is rapidly converted to acetate by ALDH2. A recent study showed that ALDH2 deficiency promotes alcohol‐related liver cancer through the oxidation of mitochondrial DNA and activation of multiple oncogenic pathways by acetaldehyde (Seo et al, 2019). Interestingly, ALDH2 −/− mice is more susceptible to liver inflammation but more resistant to alcohol‐induced steatosis and blood ALT elevation (Kwon et al, 2014). However, it was reported that ALDH2 +/− mice are susceptible to alcohol‐induced fatty liver because acetaldehyde disrupts the epithelial barrier dysfunction (Chaudhry et al, 2015). Therefore, the role of ALDH2 in alcohol‐induced fatty liver remains still unclear.
In this study, we unexpectedly found that p53 can directly bind to and inhibit ALDH2. Moreover, we discovered that pyruvate has an ability to enhance ALDH2 activity by interacting with ALDH2. Through inhibiting ethanol oxidation, p53 regulates intracellular acetate and acetyl‐CoA levels, thereby impacting SCD1 gene expression through ethanol‐derived histone acetylation. Thus, these findings demonstrate a previously unrecognized function of p53 to inhibit ethanol metabolism and reveal that pyruvate is a natural regulator of ALDH2 activity.
Results
p53 inhibits ethanol metabolism by suppressing the activity of ALDH2
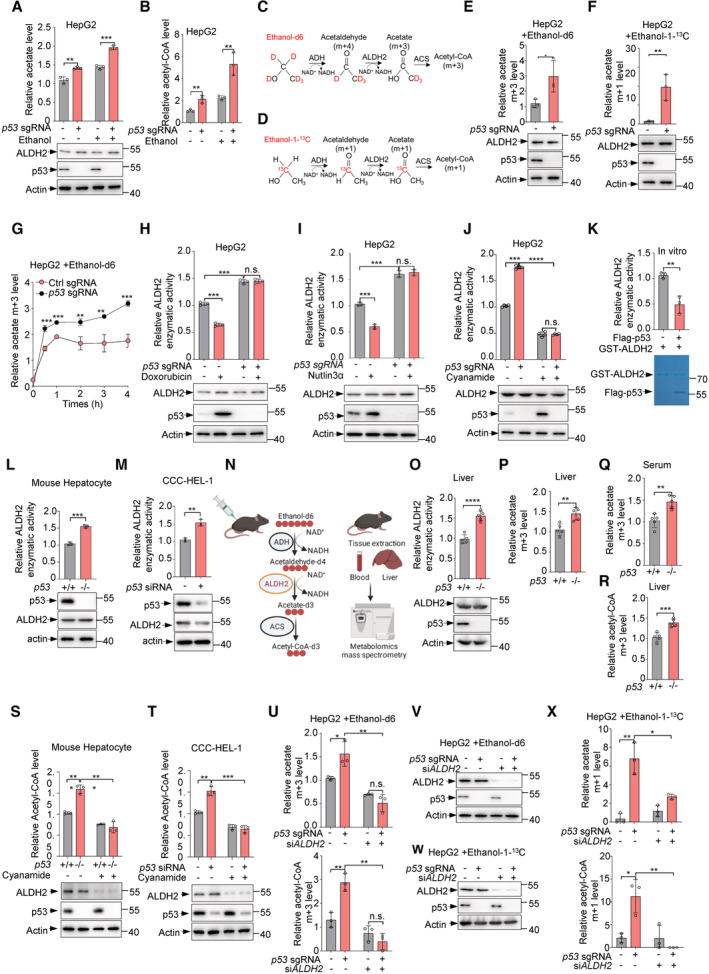
To assess the effect of p53 on ethanol metabolism, we compared the levels of acetate, the end product of ethanol metabolism, in control and p53 knockout HepG2 cells. Knockout of p53 using small guide RNA (sgRNA) resulted in increased levels of acetate (Fig 1A, top panel). Notably, ethanol treatment increased intracellular acetate levels and enhanced the increase in acetate levels caused by p53 loss (Fig 1A, top panel), suggesting a significant effect of p53 on ethanol metabolism. Acetate can be converted to acetyl‐CoA by the enzyme acetyl‐CoA synthetase (ACS; Ingram‐Smith et al, 2006). Consistently, lack of p53 led to an increase in the levels of acetyl‐CoA in ethanol‐treated and ‐untreated cells (Fig 1B), while p53 depletion did not decrease ALDH2 levels (Fig 1A, bottom panel).
Figure 1. p53 inhibits ethanol metabolism through suppressing ALDH2.
-
A, BHepG2 cells infected with lentivirus expressing p53 sgRNA or control sgRNA were treated with ethanol (5 mM) for 0.5 h. Relative levels of acetate (A) and acetyl‐CoA (B) were analyzed. Protein levels of p53 and ALDH2 were examined by western blot (A, bottom panel).
-
C, DSchematic diagram of ethanol metabolism using ethanol‐d6 and ethanol‐1‐13C.
-
E, FHepG2 cells infected with lentivirus expressing p53 sgRNA or control sgRNA were treated with ethanol‐d6 (5 mM) or ethanol‐1‐13C (5 mM) for 0.5 h. Relative acetate m + 3 and acetate m + 1 levels (E, F, top panels) were examined. Protein levels of p53 and ALDH2 (E, F, bottom panels) were analyzed by western blot.
-
GHepG2 cells infected with lentivirus expressing p53 sgRNA or control sgRNA were treated with ethanol‐d6 (5 mM) for 0.5, 1, 2, 3, 4 h and relative acetate m + 3 levels were analyzed.
-
H–JHepG2 cells infected with lentivirus expressing p53 sgRNA or control sgRNA were treated with doxorubicin (2 ng/ml) (H) nutlin3α (10 μM) (I) or cyanamide (1 mM) (J) for 24 h. Relative enzymatic activity of ALDH2 was examined (top panel). Protein expression was determined by western blot analysis (bottom panel).
-
KPurified GST‐ALDH2 proteins were incubated with purified Flag‐p53 proteins or Flag peptides for 0.5 h and relative ALDH2 enzymatic activity was analyzed (top panel). Purified protein was analyzed by SDS‐PAGE followed by Coomassie bright blue staining (bottom panel).
-
LMouse hepatocyte isolated from p53 +/+ and p53 −/− mice and lysates were used to test ALDH2 enzymatic activity. Protein levels of p53 and ALDH2 were analyzed by western blot.
-
MCCC‐HEL‐1 cells were transfected with control siRNA or p53 siRNA and ALDH2 enzymatic activity (top panel) and protein expression (bottom panel) were analyzed.
-
NWork flow of ethanol‐d6 tracing in WT and p53 −/− mouse liver and serum.
-
ORelative enzymatic activities of ALDH2 (top panel) from mouse liver tissues were measured. Protein expression (bottom panel) was examined by western blot.
-
P–Rp53 +/+ and p53 −/− mice were injected intraperitoneally with ethanol‐d6 (2 g/kg) for 0.5 h. Relative acetate m + 3 levels in mouse liver tissues (P) and mouse serum (Q), and relative hepatic acetyl‐CoA (R) were examined.
-
SMouse hepatocyte isolated from p53 +/+ and p53 −/− mice and lysates were treated with or without cyanamide (1 mM). Relative acetyl‐CoA level (top panel) and protein expression (bottom panel) were measured.
-
TCCC‐HEL‐1 cells transfected with control siRNA or p53 siRNA were treated with or without cyanamide (1 mM). Relative acetyl‐CoA level (top panel) and protein expression (bottom panel) were analyzed.
-
U–XHepG2 cells infected with lentivirus expressing p53 sgRNA or control sgRNA were transfected with control siRNA or ALDH2 siRNA and were treated with ethanol‐d6 (5 mM) (U) or ethanol‐1‐13C (5 mM) (X) for 0.5 h. (U) Relative acetate m + 3 and acetyl‐CoA m + 3 levels are shown. (X) Relative acetate m + 1 and acetyl‐CoA m + 3 levels were examined. Protein levels of p53 and ALDH2 were analyzed by western blot (V, W).
Data information: Data in (A, B, E–M, S–U, X) are from n = 3 biological independent samples and (O–R) are from n = 5 mice per group. All western blots are representative of three independent experiments. Data are the mean ± SD. P values were determined by unpaired two‐tailed Student's t tests. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, n.s., not significant.
Source data are available online for this figure.
We next investigated whether the increased acetate and acetyl‐CoA are derived from ethanol induced by p53 loss, we cultured the cells in the presence of ethanol‐d6 or ethanol‐1‐13C and assayed the cellular levels of isotope‐labeled acetate (m + 3) and acetate (m + 1) respectively (Fig 1C and D). Significantly, p53 deficiency let to an increase in the levels of ethanol‐d6‐derived acetate (m + 3) and ethanol‐1‐13C‐derived acetate (m + 1) (Fig 1E and F). Interestingly, when p53 was absent, the levels of ethanol‐d6‐deprived acetate increased rapidly 30 min after the addition of ethanol‐d6 and plateaued after approximately 1 h (Fig 1G).
Given that p53 has little effect on ALDH2 expression (Fig 1A, bottom panel), we next assayed the activity of ALDH2. When p53 was depleted in HepG2 cells with sgRNA, ALDH2 activity increased (Fig 1H and I). Conversely, treatment of p53 +/+ HepG2 cells with doxorubicin (Dox) or nutlin3α to activate p53, resulted in a lower activity of ALDH2 (Fig 1H and I). Expectedly, none of these treatments altered ALDH2 activity in p53‐depleted HepG2 cells (Fig 1H and I). In addition, supplying cells with cyanamide, an inhibitor of ALDH, reduced ALDH2 activity, and minimized the difference in ALDH2 activity between p53‐proficient and ‐deficient cells (Fig 1J). Moreover, an in vitro activity assay using purified proteins showed that the inhibition of ALDH2 by p53 was direct (Fig 1K). Notably, increased ALDH2 activity was observed in primary hepatocytes from p53 −/− mice, but not in those from p53 +/+ mice (Fig 1L). Similar results were obtained in human normal liver CCC‐HEL‐1 cells (Fig 1M).
The liver is the primary organ responsible for the breakdown of ethanol. Thus, we extended our analysis into animals. p53 +/+ and p53 −/− mice were injected intraperitoneally with ethanol‐d6 for 30 min (Fig 1N). Consistently, deficiency of p53 was associated with a strong increase in ALDH2 activity in mouse liver tissues (Fig 1O, top panel). In line with previous data (Fig 1A), p53 had little effect on the expression of ALDH2 in liver tissues (Fig 1O, bottom panel). Significantly, liquid chromatography with tandem mass spectrometry (LC–MS/MS) analysis in mice revealed higher enrichment of the newly synthesized m + 3 acetate levels in liver and serum from p53 −/− mice than p53 +/+ mice (Fig 1P and Q). Furthermore, higher enrichment of m + 3 acetyl‐CoA in liver tissues was observed in p53 −/− mice compared to p53 +/+ mice (Fig 1R). Together, these findings indicate that p53 may inhibit ALDH2 and ethanol metabolism in vitro and in vivo.
We further determined whether ALDH2 is required for p53‐mediated production of acetate and acetyl‐CoA. In mouse primary hepatocytes and human normal liver CCC‐HEL‐1 cells, p53 depletion increased cellular acetyl‐CoA levels, and this effect could be blocked by cyanamide (Fig 1S and T). Furthermore, knockdown of ALDH2 using small interfering RNA (siRNA) reduced the levels of acetate and acetyl‐CoA synthesized from ethanol, especially in p53‐deficient cells, and minimized the difference between p53‐proficient and p53‐deficient cells tracing with ethanol‐d6 (Fig 1U and V). Similar results were observed when cells were cultured with ethanol‐1‐13C (Fig 1W and X). Taken together, these results suggest that p53 inhibits ethanol metabolism through ALDH2.
p53 interacts with ALDH2 and inhibits its activity
We next investigated the mechanism by which p53 inhibits ALDH2 activity. Since p53 had little effect on ALDH2 expression, we thus examined whether p53 interacts with ALDH2. Coimmunoprecipitation assay showed that HA‐tagged p53 associated with Flag‐tagged ALDH2 (Figs 2A and EV1A). Notably, the interaction between p53 and ALDH2 could occur at the endogenous level in HepG2 cells, mouse primary hepatocytes, and human CCC‐HEL‐1 cells (Figs 2B and EV1B–D). Moreover, the interaction between p53 and ALDH2 was also found in liver tissues from WT, p53 −/− or ALDH2 −/− mice (Fig 2C). The binding between p53 and ALDH2 was direct as evidenced by a pulldown assay with purified recombinant proteins (Fig 2D). In addition, the interaction between p53 and ALDH2 was enhanced when cells were treated with doxorubicin (Dox) or nutlin3 (Fig EV1E), whereas ethanol had no effect (Fig EV1F).
Figure 2. p53 interacts with ALDH2 and inhibits its enzymatic activity.
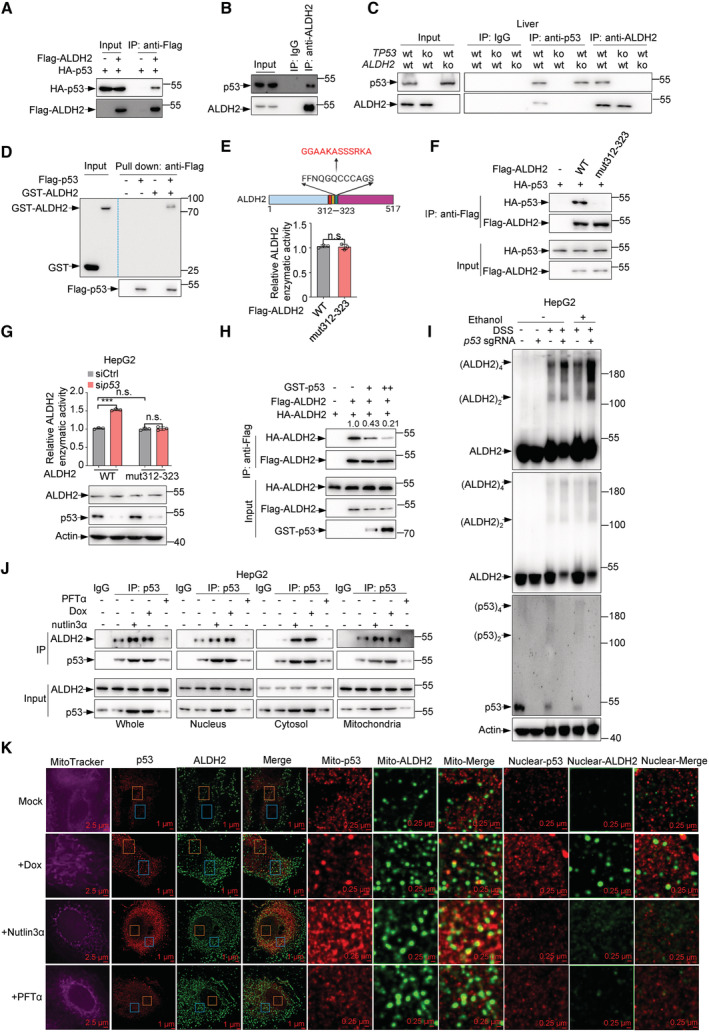
- Lysates from 293T cells transfected with HA‐p53 and Flag‐ALDH2 or vector control (−) as indicated were immunoprecipitated using anti‐Flag M2 affinity gels, and bound proteins were analyzed by western blot.
- Lysates from HepG2 cells were immunoprecipitated using ALDH2 antibody, and bound proteins were analyzed by western blot.
- Lysates from mice liver tissues (WT, p53 −/− and ALDH2 −/−) were immunoprecipitated using p53 or ALDH2 antibody, and bound proteins were analyzed by western blot.
- Purified Flag‐p53 proteins were incubated with purified recombinant GST or GST‐ALDH2 proteins as indicated for 6 h at 4°C, followed by pull down with anti‐Flag M2 affinity gels and immunoblot analysis.
- Relative enzymatic activities of purified Flag‐ALDH2 and Flag‐ALDH2 mut312‐323 proteins were analyzed.
- Lysates from 293T cells transfected with HA‐p53 and Flag‐ALDH2 or Flag‐ALDH2 mut312‐323 or vector control (−) as indicated were immunoprecipitated using anti‐Flag M2 affinity gels, and bound proteins were analyzed by western blot.
- HepG2 cells were infected with lentivirus expressing ALDH2 sgRNA or control sgRNA and overexpressed hygromycin‐resistant ALDH2 mut312‐323 in ALDH2‐knockout cell lines. The cells were then transfected with control siRNA or p53 siRNA for 48 h and treated with ethanol (5 mM) for 0.5 h and relative ALDH2 enzymatic activities were examined. Protein expression was analyzed by western blot.
- Lysates from 293 T cells transfected with HA‐ALDH2, Flag‐ALDH2 and increasing amounts of GST‐p53 or vector control (−) as indicated were immunoprecipitated using anti‐Flag M2 affinity gels, and bound proteins were analyzed by western blot.
- HepG2 cells infected with lentivirus expressing ALDH2 sgRNA or control sgRNA were treated with ethanol (5 mM) for 0.5 h and cell lysates were treated with disuccinimidyl suberate (DSS) or DMSO for 0.5 h and protein levels of p53 and ALDH2 were examined.
- Subcellular fractionation of HepG2 cells treated with doxorubicin (2 ng/ml) or nutlin3α (10 μM) or PFTα (10 μM) for 24 h were immunoprecipitated using p53 antibody, and bound proteins were analyzed by western blot.
- HepG2 cell treated with doxorubicin (2 ng/ml) or nutlin3α (10 μM) or PFTα (10 μM) for 24 h were performed STORM imaging analysis. Scale bar is shown in Figures.
Data information: Data in (A–D, F, and H–K) represent three independent experiments and (E, G) are from n = 3 biological independent samples. Data are the mean ± SD. P values were determined by unpaired two‐tailed Student's t tests. ***P < 0.001, n.s., not significant.
Source data are available online for this figure.
Figure EV1. p53 interacts with ALDH2.

-
ALysates from 293T cells transfected with HA‐p53 and Flag‐ALDH2 or vector control (−) as indicated were immunoprecipitated using anti‐Flag M2 affinity gels, and bound proteins were analyzed by western blot.
-
B–DLysates from HepG2 cells (B), mouse hepatocyte (C) and CCC‐HEL‐1 cells (D) were immunoprecipitated using p53 antibody, and bound proteins were analyzed by western blot.
-
ELysates from 293T cells transfected with HA‐p53 and Flag‐ALDH2, or vector control (−) as indicated and treated with doxorubicin (2 ng/ml) or nutlin3α (10 μM) for 24 h were immunoprecipitated using anti‐Flag M2 affinity gels, and bound proteins were analyzed by western blot.
-
FLysates from 293T cells transfected with HA‐p53 and Flag‐ALDH2, or vector control (−) as indicated and treated with or without ethanol (5 mM) for 24 h were immunoprecipitated using anti‐Flag M2 affinity gels, and bound proteins were analyzed by western blot.
-
GLysates from 293T cells transfected with HA‐ALDH2 and Flag‐p53 wildtype or deletion mutants as indicated were immunoprecipitated using anti‐Flag M2 affinity gels, and bound proteins were analyzed by western blot.
-
HLysates from 293T cells transfected with HA‐p53 and Flag‐ALDH2, Flag‐ALDH2‐Δ284–291, Δ312–323 were immunoprecipitated using anti‐Flag M2 affinity gels, and bound proteins were analyzed by western blot.
-
ILysates from 293T cells transfected with HA‐p53 and Flag‐ALDH2, Flag‐ALDH2‐N186A, E285A, C319A were immunoprecipitated using anti‐Flag antibody, and bound proteins were analyzed by western blot.
-
JHepG2 cells were infected with lentivirus expressing ALDH2 sgRNA or control sgRNA and overexpressed hygromycin‐resistant ALDH2‐mut312‐323 in ALDH2‐knockout cell lines. The cells were then transfected with control siRNA or p53 siRNA for 48 h and treated with ethanol (5 mM) for 0.5 h and relative levels of acetate (J, left panel) and acetyl‐CoA (J, right panel) were examined.
Data information: Data in (A–I) represent three independent experiments and (J) are from n = 3 biological independent samples. Data are the mean ± SD. P values were determined by unpaired two‐tailed Student's t tests. **P < 0.01, ***P < 0.001, n.s., not significant.
Source data are available online for this figure.
To determine the structural determinants of p53 binding to ALDH2, we used a set of p53 domain deletion mutants. Immunoprecipitation assay showed that ALDH2 interacted with the carboxy‐terminal region of p53 (Fig EV1G). By screening a series of ALDH2 deletion mutants, we found that a 12aa‐deletion mutation (Δ312–323) within the catalytic domain of ALDH2 abrogated the interaction between ALDH2 and p53 (Fig EV1H). The residue Asn186 (N186) is predicted to be a transition state stabilizer for ALDH2, and Glu285 (E285) and Cys319 (C319) are critical for its catalytic function (UniProt knowledgebase). Intriguingly, point mutants N186A, E285A, and C319A of ALDH2 were still able to interact with p53 (Fig EV1I). Furthermore, we generated a mutant ALDH2 (ALDH2 mut312‐323) with an enzymatic activity similar to that of wildtype (Fig 2E). Unlike the wild‐type ALDH2, ALDH2 mut312‐323 showed no ability to bind to p53 (Fig 2F). Consistent with this, p53 failed to inhibit the activity of ALDH2 mut312‐323 (Fig 2G), and no inhibitory effect of p53 on cellular acetate and acetyl‐CoA level was observed in ALDH2 mutation‐expressing cells (Fig EV1J).
ALDH2 enzyme requires a tetramer for its functional activity (Yoshida et al, 1984; Li et al, 2006). In transfection experiments, p53 reduced the interaction of two differently tagged ALDH2 proteins (Flag‐ALDH2 and HA‐ALDH2) in a dose‐dependent manner (Fig 2H). Moreover, p53 depletion resulted in an increase in ALDH2 tetramers with a corresponding decrease in ALDH2 monomers (Fig 2I). These results indicate that p53 may disrupt the formation of the tetrameric ALDH2 holoenzyme.
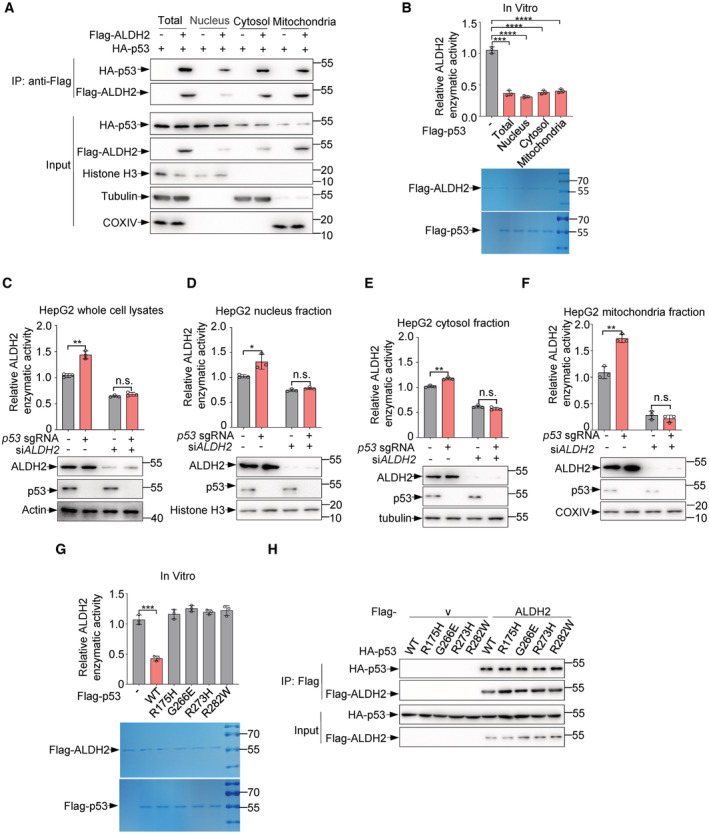
ALDH2 has been reported to be located in mitochondria (Osna et al, 2017), while p53 is present in several subcellular organelles, including mitochondria (O'Brate & Giannakakou, 2003). Consistently, overexpressed ALDH2 interacted with p53 primarily in the mitochondria, with a small amount of binding occurring in the cytoplasm and nucleus (Fig EV2A). In keeping with this, endogenous p53‐ALDH2 interactions occur in the mitochondria, with a small amount of binding occurring in the cytoplasm and nucleus (Fig 2J). This interaction was enhanced when cells were treated with doxorubicin or nutlin3α, but not PFTα, an inhibitor of p53 transcriptional activity (Fig 2J). To assess the inhibitory effect of p53 in different subcellular organelles on ALDH2, we purified p53 protein from these cellular compartments. Interestingly, cytoplasmic, mitochondrial, and nuclear p53 all exhibited inhibitory activity towards ALDH2 (Fig EV2B). Also, the inhibition of ALDH2 by p53 was observed in these subcellular organelles (Fig EV2C–F). In supporting of this, stochastic optical reconstruction microscopy (STORM) analysis revealed that ALDH2‐p53 interaction does occur in the mitochondria, but also in the nucleus (Fig 2K).
Figure EV2. p53 inhibits ALDH2 enzymatic activity in vivo and in vitro .
-
ASubcellular fractionation of 293T cells transfected with HA‐p53 and Flag‐ALDH2 or vector control (−) as indicated were immunoprecipitated using anti‐Flag M2 affinity gels, and bound proteins were analyzed by western blot. Fractionation controls were COXIV (mitochondria), Tubulin (cytosol), Histone H3 (nucleus).
-
BPurified Flag‐ALDH2 proteins were incubated with purified Flag‐p53, nucleus Flag‐p53, cytosol Flag‐p53 and mitochondria Flag‐p53 proteins or Flag peptides for 0.5 h. Relative enzymatic activities of ALDH2 (top panel) are shown and purified Flag‐ALDH2 and Flag‐p53 proteins were stained by Coomassie bright blue (bottom panel).
-
C–FHepG2 cells infected with lentivirus expressing p53 sgRNA or control sgRNA were transfected with ALDH2 or control siRNA and nucleus, cytosol, and mitochondria fractions of HepG2 cells were isolated. Relative enzymatic activities of ALDH2 (C–F, top panels) were examined and protein levels of p53 and ALDH2 (C–F, bottom panels) were analyzed by western blot.
-
GPurified Flag‐ALDH2 proteins were incubated with purified Flag‐p53, Flag‐p53‐R175H, G266E, R273H, R282W or Flag peptide for 0.5 h and relative enzymatic activities of ALDH2 were examined (top panel). Purified Flag‐ALDH2 and Flag‐p53 proteins were stained by Coomassie bright blue (bottom panel).
-
HLysates from 293T cells transfected with HA‐p53, HA‐p53‐R175H, G266E, R273H, R282W and Flag‐ALDH2 or vector control (−) as indicated were immunoprecipitated using anti‐Flag M2 affinity gels, and bound proteins were analyzed by western blot.
Data information: Data in (A, H) represent three independent experiments and (B–G) are from n = 3 biological independent samples. Data are the mean ± SD. P values were determined by unpaired two‐tailed Student's t tests. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, n.s., not significant.
Source data are available online for this figure.
We next tested whether the tumor‐associated p53 mutants, most of which contain missense mutations, can also inhibit ALDH2 activity. Unlike wild‐type p53, four p53 mutants (R175H, G266E, R273H and R282W) failed to inhibit ALDH2 activity (Fig EV2G), even though all of them still could bind to ALDH2 (Fig EV2H). As tumor‐associated mutations impair the native conformation of p53, the inhibition of ALDH2 by p53 may be due to the native conformation of p53. Collectively, these results indicate that p53 directly binds to and suppresses ALDH2.
Pyruvate binds to ALDH2 and enhances its activity
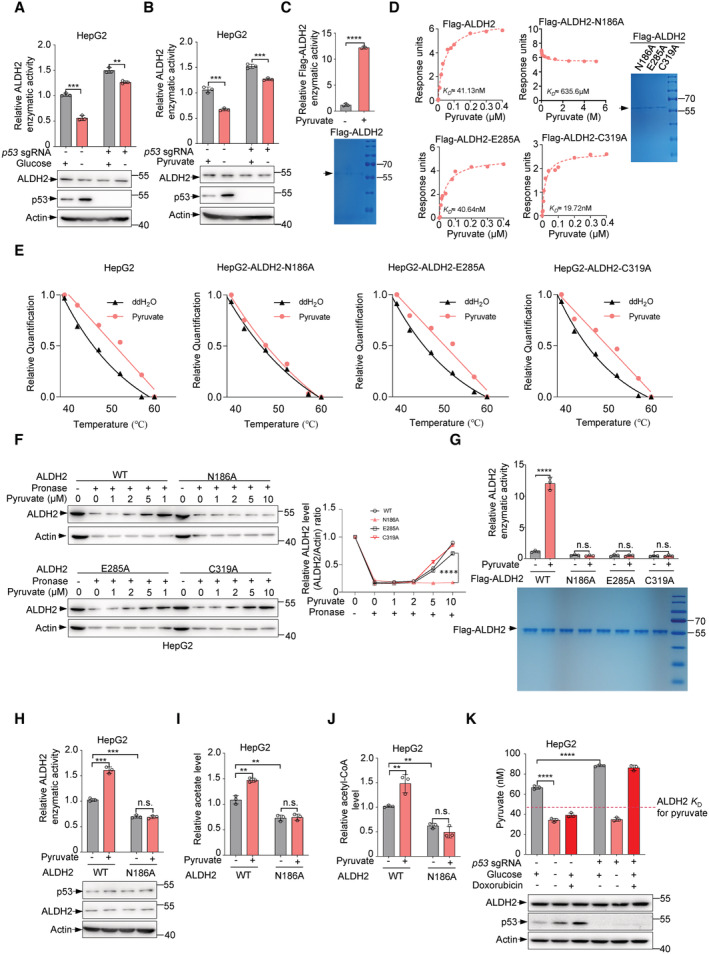
Glucose deprivation can activate p53 (Jones et al, 2005). In keeping with this, the activation of p53 upon glucose starvation resulted in a lower activity of ALDH2 (Fig 3A). Notably, glucose deprivation decreased ALDH2 activity by approximately 45% in p53‐proficient cells (Fig 3A, column 2 vs. column 1) and by approximately 15% in p53‐deficient cells (Fig 3A, column 4 vs. column 3), suggesting that glucose metabolism may be involved in the inhibitory effect of p53 on ALDH2 activity. Pyruvate is the end products of glycolysis and is then transported to the mitochondria where it participates in the tricarboxylic acid (TCA) cycle. Thus, we tested whether pyruvate affects ALDH2 activity. Surprisingly, the activity of ALDH2 was reduced when cells were cultured in media lacking pyruvate (Fig 3B). Moreover, the effect of pyruvate on ALDH2 was stronger in p53 +/+ cells than in p53 −/− cells (Fig 3B), further indicating a role of p53 in pyruvate‐mediated regulation of ALDH2.
Figure 3. Pyruvate directly binds to ALDH2 and promotes its enzymatic activity.
-
A, BHepG2 cells infected with lentivirus expressing p53 sgRNA or control sgRNA under glucose or pyruvate deprivation and cell lysates were used to measure ALDH2 activities (A, B, top panels) and examine p53 and ALDH2 proteins (A, B, bottom panels).
-
CPurified Flag‐ALDH2 protein was incubated with pyruvate (1 mM) for 10 min and relative enzymatic activity of ALDH2 (top panel) was analyzed. Purified Flag‐ALDH2 protein analyzed by SDS‐PAGE was stained by Coomassie bright blue, and indicated by an arrow (bottom panel).
-
DSPR (BIAcore) measurement of the interaction between pyruvate and purified Flag‐ALDH2, Flag‐ALDH2‐N186A, E285K, C319A. The Graphs of equilibrium response units (RU) and compound concentrations are shown (left panels). Purified Flag‐ALDH2‐N186A, E285K and C319A protein were analyzed by SDS‐PAGE followed by Coomassie bright blue staining (D, right panel), indicated by an arrow.
-
ECETSAs exhibit the binding affinity of pyruvate to the N186 amino acid in HepG2 cells. Cell lysates were separated by SDS‐PAGE and analyzed by western blot. Actin was used as an internal control. Relative ALDH2 band intensities were quantified and plotted against corresponding incubation temperatures.
-
FHepG2 cells were infected with lentivirus expressing ALDH2 sgRNA or control sgRNA and overexpressed hygromycin‐resistant ALDH2‐N186A, E285A, C319A in ALDH2‐knockout cell lines. Cells were digested and cell lysates were incubated with the indicated doses of pyruvate for 1 h on ice and 0.5 h at room temperature, followed by pronase digestion. ALDH2 proteins were analyzed by western blot. Relative ALDH2/Actin ratios are shown.
-
GPurified Flg‐ALDH2, Flag‐ALDH2‐N186A, E285A and C319A proteins were incubated with pyruvate (1 mM) for 10 min and relative enzymatic activities of ALDH2 (top panel) were shown. Purified Flg‐ALDH2, Flag‐ALDH2‐N186A, E285A, C319A proteins were analyzed by SDS‐PAGE followed by Coomassie staining of the gels (bottom panel) are shown.
-
H–JHepG2 cells were infected with lentivirus expressing ALDH2 sgRNA or control sgRNA and overexpressed hygromycin‐resistant ALDH2 N186A in ALDH2‐knockout cell lines. The cells were treated with pyruvate (1 mM) for 24 h. Relative ALDH2 activity (H, top panel), acetate (I) and acetyl‐CoA (J) were measured. The expression of p53 and ALDH2 were analyzed by western blot (H, bottom panel).
-
KIntracellular concentration of pyruvate in HepG2 cells infected with lentivirus expressing p53 sgRNA or control sgRNA under glucose deprivation or treated with doxorubicin (2 ng/ml) were measured by liquid chromatography/mass spectrometry (LC/MS) (top panel). The dissociation constant K D of ALDH2 for pyruvate is indicated by the dashed line. Protein expression was determined by western blot (bottom panel).
Data information: Data in (A–C, G–K) are from n = 3 biological independent samples. Data are the mean ± SD. P values were determined by unpaired two‐tailed Student's t tests. **P < 0.01, ***P < 0.001, ****P < 0.0001, n.s., not significant.
Source data are available online for this figure.
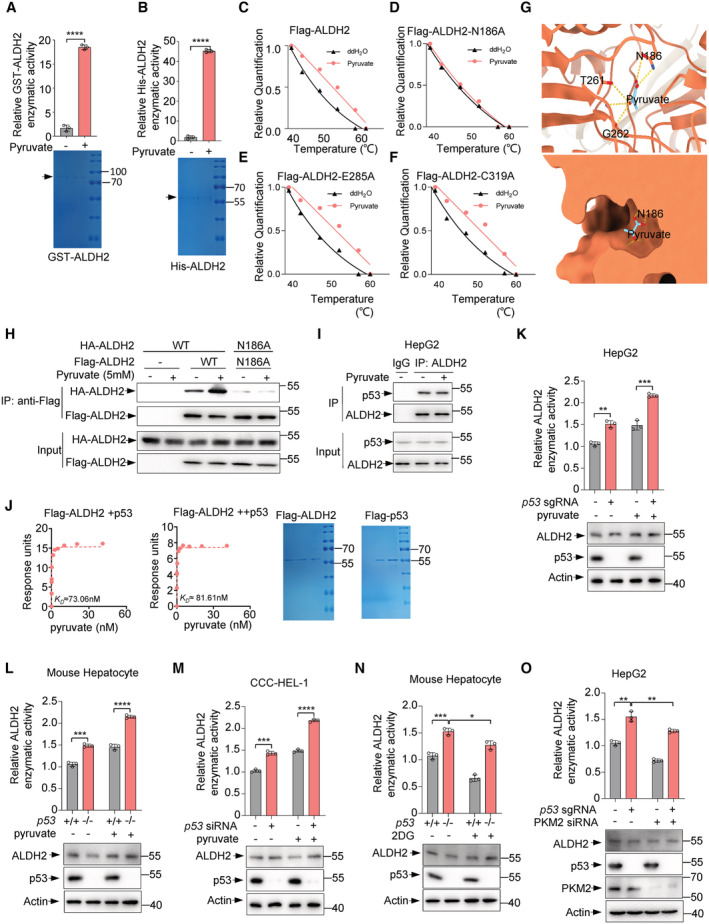
Next, we investigated how pyruvate modulates ALDH2 activity. An in vitro assay showed that pyruvate addition directly increased activity of purified ALDH2 (Figs 3C, and EV3A and B). These findings further provoked us to test whether pyruvate binds to ALDH2. Indeed, a real‐time binding assay using surface plasmon resonance (SPR) showed that pyruvate was able to bind to wildt‐ype ALDH2 with a dissociation constant (K D) of about 41 nM and also to the mutants E285A and C319A, whereas pyruvate showed little affinity towards the mutant N186A (K D ≈ 635 μM; Fig 3D). Consistent with these in vitro SPR data, cellular thermal shift assays (CETSAs) detected obvious shifts in the ALDH2 (including wildtype, mutations E285A and C319A) melting curve in the presence of pyruvate, whereas the melting curve of mutation N186A was not shifted (Figs 3E and EV3C–F). Similarly, drug affinity responsive target stability (DARTS) assays suggest that pyruvate binds to wild‐type ALDH2, E285A and C319A mutants, but not the N186A mutant, and protects them from degradation by pronase in a dose‐dependent manner (Fig 3F). Furthermore, we performed structural analysis of the potential binding site of ALDH2 to pyruvate using AutoDock Vina and found that residue N186 of ALDH2 may form hydrogen bonds with pyruvate (Fig EV3G). In sum, these results suggest that pyruvate binds directly to ALDH2 and that N186 of ALDH2 may be necessary for its binding to pyruvate.
Figure EV3. Pyruvate enhances ALDH2 enzymatic activity.
-
A, BPurified GST‐ALDH2 proteins and His‐ALDH2 proteins were incubated with pyruvate (1 mM) for 10 min and relative enzymatic activities of ALDH2 (top panels) were analyzed. Purified GST‐ALDH2 proteins and His‐ALDH2 proteins were analyzed by SDS‐PAGE followed by Coomassie bright blue staining, and indicated by arrows (bottom panels).
-
C–FLysates from 293T cells transfected with Flag‐ALDH2 wt (C), N186A (D), E285A (E), C319A (F) were used for CETSAs assay. CETSAs exhibit the binding affinity of pyruvate to ALDH2‐N186.
-
GThe predicted structure of ALDH2 binding with pyruvate through AutoDock Vina. Intermolecular interactions between pyruvate and its binding pocket.
-
HLysates from 293T cells transfected with HA‐ALDH2, HA‐ALDH2‐N186A and Flag‐ALDH2, Flag‐ALDH2‐N186A or control vector as indicated in the presence of pyruvate (5 mM) were immunoprecipitated using anti‐Flag M2 affinity gels, and bound proteins were analyzed by western blot.
-
ILysates from HepG2 cells treated with or without pyruvate (1 mM) were immunoprecipitated using ALDH2 antibody, and bound proteins were analyzed by western blot.
-
JSPR (BIAcore) measurement of the interaction between pyruvate and purified Flag‐ALDH2 with the increasing amounts of purified p53 proteins. The Graphs of equilibrium response units (RU) and compound concentrations are shown (left panels). Purified Flag‐ALDH2 and Flag‐p53 proteins were analyzed by SDS‐PAGE followed by Coomassie bright blue staining (right panels).
-
KHepG2 cells infected with lentivirus expressing p53 sgRNA or control sgRNA were treated with or without pyruvate (40 nM). ALDH2 enzymatic activity (top panel) and protein expression (bottom panel) were measured.
-
LMouse hepatocyte isolated from p53 +/+ and p53 −/− mice and lysates were treated with or without pyruvate (40 nM). ALDH2 enzymatic activity (top panel) and protein expression (bottom panel) were measured.
-
MCCC‐HEL‐1 cells transfected with control siRNA or p53 siRNA were treated with or without pyruvate (40 nM). ALDH2 enzymatic activity (top panel) and protein expression (bottom panel) were analyzed.
-
NMouse hepatocytes isolated from p53 +/+ and p53 −/− mice and lysates were treated with or without 2DG (1 mM). ALDH2 enzymatic activity (top panel) and protein expression (bottom panel) were measured.
-
OHepG2 cells infected with lentivirus expressing p53 sgRNA or control sgRNA were transfected with control or PKM2 siRNA. ALDH2 enzymatic activity (top panel) and protein expression (bottom panel) were measured.
Data information: Data in (A, B, K–O) are from n = 3 biological independent samples and (C–F, H–J) are representative of three independent experiments. Data are the mean ± SD. P values were determined by unpaired two‐tailed Student's t tests. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, n.s., not significant.
Source data are available online for this figure.
Consistent with the binding data, pyruvate enhanced the activity of wild‐type ALDH2 activity, whereas it had no effect on the activity of ALDH2 N186A mutant in vitro (Fig 3G). Furthermore, pyruvate also failed to enhance the activity of mutant ALDH2 E285A and C319A, even though they still bound to pyruvate, because both mutants lost their catalytic function (Fig 3G). We further investigated the mechanism by which pyruvate enhances ALDH2 activity. Compared to wild‐type ALDH2, the N186A mutant failed to form an oligomeric state (Fig EV3H), and pyruvate increased homologous binding between wild‐type ALDH2, but not the ALDH2 N186A mutant (Fig EV3H). Moreover, treatment with pyruvate‐enhanced ALDH2 activity, while had no effect on ALDH2 N186A mutant in HepG2 cells (Fig 3H). Consistently, the levels of acetate and acetyl‐CoA increased upon pyruvate treatment in the cells carrying wild‐type ALDH2, but not N186A mutation (Fig 3I and J). Taken together, these results suggest that pyruvate enhances ALDH2 activity through directly binding to ALDH2.
We next investigated whether pyruvate influences the binding of p53 to ALDH2. Immunoprecipitation assays showed that pyruvate has no effect on the interaction between p53 and ALDH2 (Fig EV3I). Similarly, p53 did not affect pyruvate‐ALDH2 association (Fig EV3J), indicating that the binding of p53 to ALDH2 is compatible with the binding of pyruvate to ALDH2. Furthermore, we determined the effect of p53 on pyruvate level. Glucose deprivation resulted in a decrease in pyruvate level as expected (Fig 3K). Consistent with the inhibitory effect of p53 on glucose metabolism, knockout of p53 increased pyruvate production, conversely Dox treatment reduced the level of pyruvate which was accompanied by a higher level of p53 expression (Fig 3K). To assess the physiological relevance of these findings, we compared the ALDH2 activity by treating cells with physiological levels of pyruvate (40 nM; Fig EV3K–M). In all cells used, loss of p53 increased ALDH2 activity even in the presence of exogenous pyruvate, and similarly, pyruvate addition still resulted in increased ALDH2 activity in p53‐deficient cells (Figs EV3K–M). Interestingly, reducing intracellular pyruvate levels by supplying mouse primary hepatocytes with a glucose analogue 2DG (2‐Deoxy‐D‐glucose), a competitive glycolytic inhibitor, enlarged the inhibitory effect of p53 on ALDH2 (Fig EV3N, column 3 vs. column 4, and column 1 vs. column 2). Similarly, more inhibition of ALDH2 by p53 was observed in HepG2 cells treated with PKM2 siRNA (Fig EV3O). Thus, it appears that compared to pyruvate, p53 plays a dominant role in the inhibition of ALDH2. Collectively, these results indicate that p53 inhibits ALDH2 activity by directly binding to ALDH2 on the one hand, and by reducing pyruvate production via modulating glucose metabolism on the other.
p53 inhibits alcoholic fatty liver through ALDH2
To determine whether p53 loss contributes to alcohol‐induced fatty liver, we generated p53 −/− mice, and ALDH2 −/− mice, and then crossed p53 −/− mice with ALDH2 −/− mice to establish p53 −/− ALDH2 −/− mice (Fig EV4A and B). Expression of p53 and ALDH2 in these mice was determined by qRT‐PCR analysis (Fig EV4C and D). Mice were placed on a chronic‐binge ethanol diet as shown in Fig 4A (Bertola et al, 2013; Kwon et al, 2014). The body weight of these mice decreased slightly after 2 weeks of ethanol diet feeding (Fig EV4E and F). Ethanol feeding increased liver weight in wild‐type and especially p53 −/− mice, but not in ALDH2 −/− mice (Fig 4B). Remarkably, ALDH2 knockout blocked the increase in liver weight caused by p53 loss (Fig 4B). Liver histology and Oil Red O staining revealed that ethanol feeding led to increased hepatic lipid accumulation in both wildtype and especially in p53 −/− mice, but not in ALDH2 −/− mice (Fig 4C). Notably, p53 deletion enhanced the hepatic inflammatory process induced by ethanol treatment, and this effect was blocked by the ALDH2 loss (Fig 4D–I). Also, ethanol feeding resulted in higher levels hepatic triglyceride (TG) in p53 −/− mice compared to p53 +/+ mice, and depletion of ALDH2 minimized the difference between them (Fig 4J). Moreover, serum concentrations of alanine aminotransferase (ALT) and aspartate aminotransferase (AST), markers for liver inflammation and injury, were increased in ethanol‐fed wild‐type mice and especially in p53 −/− mice, whereas knockout of ALDH2 completely reversed it (Figs 4K and L, and EV4G and H). In addition, in vivo isotope tracing using ethanol‐d6 showed that ALDH2 depletion completely blocked hepatic and serum acetate and hepatic acetyl‐CoA production induced by p53 loss (Fig 4M–O).
Figure EV4. p53 inhibits ethanol‐induced fatty liver through ALDH2.
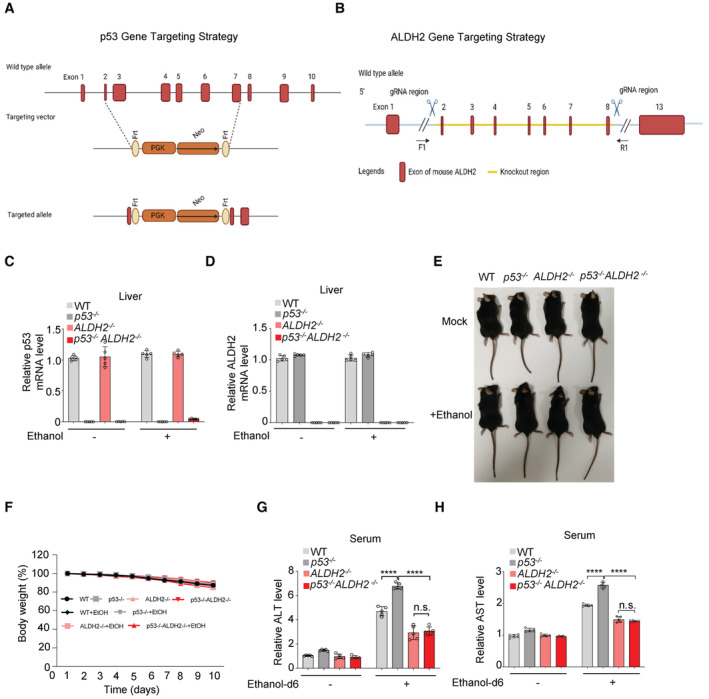
-
A, Bp53 and ALDH2 gene targeting strategies are shown.
-
C–FRelative mRNA level of p53 (C) and ALDH2 (D) in liver tissues of WT, p53 −/−, ALDH2 −/− and p53 −/− ALDH2 −/− C57BL/6 N male mice (related to Fig 4A) were determined by qRT–PCR. Representative mice photos (E) and the changes in body weight (F) are shown.
-
G, HWT, p53 −/−, ALDH2 −/− and p53 −/− ALDH2 −/− C57BL/6 N male mice were intraperitoneally injected with ethanol‐d6 (2 g/kg) or control saline for 0.5 h as shown in Fig 1G. Relative enzymatic activities of serum ALT (G) and AST (H) were measured.
Data information: Data in (C and D) are from n = 5 technical replicates from one of three independent experiments with similar results. Data in (F–H) are from n = 5 mice per group. Data are the mean ± SD. P values were determined by unpaired two‐tailed Student's t tests. ****P < 0.0001, n.s., not significant.
Source data are available online for this figure.
Figure 4. p53 inhibits ethanol‐derived fatty liver through ALDH2.

-
A–L(A) Experiment design for mice model in (A–L). WT, p53 −/−, ALDH2 −/− and p53 −/− ALDH2 −/− C57BL/6 N male mice were treated as described in the methods to establish a mouse model of chronic‐binge ethanol consumption. (B) Relative liver weights were measured. (C, D) H&E (hematoxylin–eosin) staining (C, top) and oil red O staining (C, bottom) and inflammatory score for HE staining (D) for the liver tissues from mice fed with control diet and ethanol diet. (E–I) Relative mRNA levels of IL‐6, IL‐1β, TNF‐α, Cxcl1, Cxcl2, Col1a1 were analyzed. (J) Relative triglyceride level was examined. (K, L) Relative levels of serum ALT (K) and AST (L) were examined.
-
M–OWT, p53 −/−, ALDH2 −/− and p53 −/− ALDH2 −/− C57BL/6 N male mice were injected intraperitoneally with ethanol‐d6 (2 g/kg) for 0.5 h as shown in Fig 1N. Relative acetate and acetate m + 3 levels in mouse liver tissues (M) and mouse serum (N) were examined. (O) Relative acetyl‐CoA and acetyl‐CoA m + 3 levels in mouse liver tissues were measured.
Data information: Scale bar, 100 μm. Data in (B–O) are from n = 5 mice per group. Data are the mean ± SD. P values were determined by unpaired two‐tailed Student's t tests. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, n.s., not significant.
Source data are available online for this figure.
Taken together, these results reveal that p53 alleviates ethanol‐induced fatty liver and ethanol metabolism through ALDH2.
ALDH2 contributes to p53‐mediated histone acetylation
The above findings suggest that p53 inhibits ethanol‐derived acetyl‐CoA, which has been implicated in histone acetylation and gene regulation. Indeed, upon ethanol treatment, p53 deletion increased acetyl CoA levels in the cytoplasm and nucleus and accordingly led to an increase in histone acetylation, which was abolished by ALDH2 silencing (Fig EV5A and B). We next directly investigated whether acetyl groups derived from ethanol are incorporated into histone acetylation. We injected mice intraperitoneally with 2 g/kg ethanol‐d6 or control saline and assessed deuterium incorporation into acetylated histones in liver tissues at 4 h after intraperitoneal injections (Fig 5A). Ethanol‐d6 treatment elevated the incorporation of heavy label into histone acetylation (H3K9, H3K18 and H4) especially in p53 −/− mice, whereas knockout of ALDH2 reduced it and minimized the difference between wildtype and p53 −/− mice (Fig 5B). These in vivo data indicate that p53 inhibits histone acetylation through repressing ALDH2‐mediated ethanol oxidation.
Figure EV5. p53 inhibits acetyl‐CoA and the fraction of monounsaturated fatty acids through ALDH2.

-
AHepG2 cells infected with lentivirus expressing p53 sgRNA or control sgRNA were transfected with ALDH2 siRNA or control siRNA for 48 h and were treated with ethanol for 0.5 h. Then nucleus acetyl‐CoA and cytosol acetyl‐CoA were extracted and analyzed by LC–MS.
-
BHepG2 cells infected with lentivirus expressing p53 sgRNA or control sgRNA were transfected with ALDH2 siRNA or control siRNA for 48 h. Protein levels of histone H3, Ac‐histone H3, p53 and ALDH2 were examined.
-
C, DWT and p53 −/− mice were injected intraperitoneally with ethanol‐d6 (2 g/kg) for 1 h. Chromatin fractions of liver tissues were subjected to ChIP‐seq assay using H3K9ac antibody, H3K27ac antibody. KEGG analysis of the related genes of increased peaks for H3K9ac (C) and H3K27ac (D) in ethanol‐treated p53−/− compared to ethanol‐treated WT mice.
-
EFatty acid (C ≥ 16) distribution within free fatty acid fraction in liver from mice (related to Fig 5H and I).
Data information: Data in (A) are from n = 3 biological independent samples and (B) are representative of three independent experiments. Data are the mean ± SD. P values were determined by unpaired two‐tailed Student's t tests. *P < 0.05, **P < 0.01, ***P < 0.001, n.s., not significant.
Source data are available online for this figure.
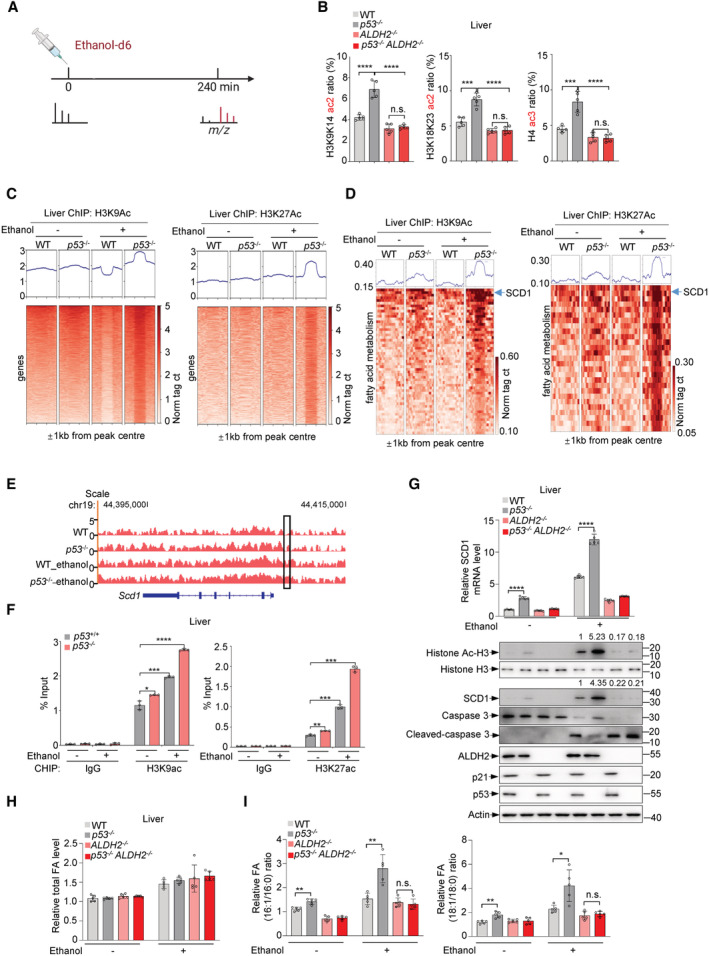
Figure 5. p53 inhibits ethanol‐derived histone acetylation to affect SCD1 expression.
-
AExperimental outline of in vivo ethanol‐d6 mass spectrometry.
-
BWT, p53 −/−, ALDH2 −/− and p53 −/− ALDH2 −/− C57BL/6 N male mice were injected intraperitoneally with ethanol‐d6 (2 g/kg) for 4 h and metabolized heavy ethanol‐d6 is incorporated into histone acetylation in the liver.
-
C, Dp53 +/+ and p53 −/− mice were injected intraperitoneally with ethanol‐d6 (2 g/kg) for 1 h. Chromatin fractions of liver tissues were subjected to ChIP‐seq assay using H3K9ac antibody (left panel), H3K27ac antibody (right panel). Heat map of H3K9ac/H3K27ac binding signal (rows) from −1 to +1 kb surrounding the centre of H3K9ac/H3K27ac‐binding sites for ChIP–seq tags (C). Heatmap of H3K9ac/H3K27ac binding signal (rows) from −1 to +1 kb surrounding the centre of H3K9ac/H3K27ac‐binding sites for ChIP–seq tags of fatty acid metabolism related genes (D).
-
EChIP–seq for H3K9ac in untreated and ethanol‐d6‐treated WT and p53 −/− mice. The genome‐browser track view shows the SCD1 locus.
-
FChIP assays with H3K9ac antibody and H3K27ac antibody or IgG in WT and p53 −/− mice injected intraperitoneally with ethanol‐d6 (2 g/kg) or saline for 1 h. Bound SCD1 response elements (RE) were analyzed by qRT–PCR.
-
G–IWT, p53 −/−, ALDH2 −/− and p53 −/− ALDH2 −/− C57BL/6 N male mice were fed control diet and ethanol diet as shown in Fig 4A. Relative mRNA level of SCD1 (G, top panel) was analyzed. Protein levels of p53, ALDH2, SCD1, Histone H3 and Ac‐H3, caspase 3, cleaved caspase 3, p21 (G, bottom panel) in mice liver tissues were examined. Relative levels of total fatty acids in mice liver tissues (H) and relative ratio of fatty acids (16:1/16:0, 18:1/18:0) (I) were analyzed.
Data information: Data in (B, H, I) are from n = 5 mice per group and (F) are from n = 3 biological independent samples. Data in (G) are from n = 5 technical replicates from one of three independent experiments with similar results. For ChIP‐seq analysis data in (C–E), there were no technical replicates for anti‐H3K9ac or anti‐H3K27ac. Data are the mean ± SD. P values were determined by unpaired two‐tailed Student's t tests. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, n.s., not significant.
Source data are available online for this figure.
Next, we evaluated the functional relevance of ethanol‐derived histone acetylation on p53‐regulated gene expression. ChIP‐seq assays revealed that the enrichment of peaks for K9‐acetylated histone H3 (H3K9ac) was greatly enhanced in ethanol‐treated p53 −/− mice compared to ethanol‐treated wild‐type mice (Fig 5C, left panel). Similar results were observed in ChIP‐seq data using antibodies against H3K27ac (Fig 5C, right panel). Furthermore, KEGG (Kyoto Encyclopedia of Genes and Genomes) pathway enrichment analysis revealed that the related genes of increased peaks in liver from ethanol‐treated p53 −/− mice were mainly enriched in metabolic pathways including fatty acid metabolism (Fig EV5C and D). By further analyzing the promoter regions of fatty acid synthesis‐related genes enriched by histone H3K9 and H3K27 acetylation, we found that the enrichment level of the stearoyl‐CoA desaturase‐1 (SCD1) gene promoter region was most significantly affected by p53 depletion (Fig 5D). Inspection of SCD gene showed that alcohol‐induced histone acetylation at SCD1 gene was induced by p53 depletion (Fig 5E). SCD1 is a central enzyme in lipid metabolism, catalyzing the synthesis of monounsaturated fatty acids (MUFAs), mainly oleate and palmitoleate (Am et al, 2017). Additionally, results of ChIP assays using anti‐H3K9ac and anti‐H3K27ac antibodies showed significantly increased levels of acetylated histones bound to the SCD1 gene in the liver of p53 −/− mice compared to wild‐type mice, particularly in ethanol‐treated p53 −/− mice (Fig 5F). Furthermore, we examined the SCD1 expression in the liver of mice treated with or without ethanol. Loss of p53 resulted in increased SCD1 mRNA and protein expression, particularly in ethanol‐treated mice, which was accompanied by higher levels of histone acetylation (Fig 5G), Notably, when ALDH2 was absent, p53 failed to affect the expression of SCD1, as well as histone acetylation (Fig 5G). In contrast, the expression of the p53 direct transcriptional target gene p21 was not affected by ethanol‐mediated acetylation (Fig 5G). In addition, we determined apoptosis by examining the expression of cleaved caspase 3 in alcoholic fatty liver. As shown in Fig 5G, p53 deficiency abrogated ethanol‐induced hepatic apoptosis. Taken together, these data suggest that p53 regulates SCD1 expression through ethanol‐derived histone acetylation.
p53 loss led to lipid accumulation in the liver of ethanol‐fed mice (Fig 4C). To understand how SCD1 upregulation in p53 −/− mice contributes to alcohol fatty liver progression, we conducted lipidomic analysis of mouse livers. We first measured the abundance of free fatty acids (FFAs) in response to ethanol treatment. Ethanol treatment increased the total FFAs level very little, and there was no difference between wild‐type and p53 −/− mice (Fig 5H). Notably, consistent with the findings that p53 deletion increased SCD1 expression, knockout of p53 resulted in an increased fraction of MUFAs (oleate (18:1) and palmitoleate (16:1); Fig EV5E). Palmitoleate is a major substrate for triglyceride (TG) synthesis in liver. Obviously, ethanol treatment amplified the difference in these metabolites between p53 +/+ and p53 −/− livers, while ALDH2‐depletion reduced it (Fig EV5E). Similarly, the difference in ratio of 16:1/16:0 as well as 18:1/18:0 between wildtype and p53 −/− mice diminished when ALDH2 was knocked out (Fig 5I). Together, these data suggest that p53 may regulate SCD1 through ethanol‐derived histone acetylation, thereby inhibiting ethanol‐induced fatty liver.
p53 inhibits ethanol‐induced fatty liver through SCD1
We next investigated whether SCD1 is required for p53 deficiency‐induced alcoholic fatty liver by generating a mouse model of chronic binge ethanol consumption (Fig 6A). H&E and Oil Red O staining of liver showed increased lipid accumulation in p53 −/− mice after ethanol treatment (Fig 6B). Notably, the increased lipid was blocked by SCD1 depletion (Fig 6B and C). Similar results were obtained when we examined the hepatic triglyceride levels (Fig 6D). Moreover, SCD1 depletion reduced serum ALT and AST levels in ethanol‐fed mice, especially in p53 −/− mice (Fig 6E).
Figure 6. p53 suppresses ethanol‐induced fatty liver through SCD1.
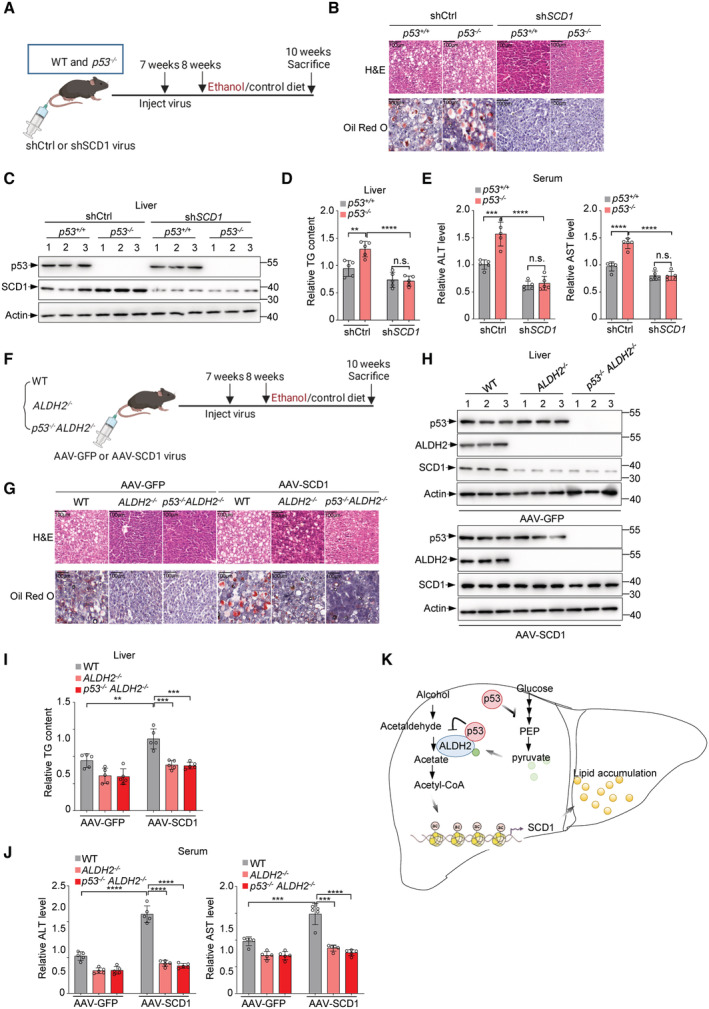
-
A–EWT and p53 −/− mice were injected with adenovirus expressing SCD1 shRNA or control shRNA via tail vein. After 7 days, WT and p53 −/− mice were treated as described in the methods to establish a mouse model of chronic‐binge ethanol consumption. (A) Schematic illustration for experimental design. (B) H&E (hematoxylin–eosin) staining (top panel) and oil red O staining (bottom panel) for the liver tissues from mice fed ethanol diet are shown. (C) Protein levels of p53 and SCD1 were examined by western blot. Relative hepatic triglyceride level (D), relative serum ALT (E, left panel) and AST (E, right panel) level were examined.
-
F–JWT, ALDH2 −/− and p53 −/− ALDH2 −/− mice were injected with adeno‐associated virus expressing AAV‐GFP or AAV‐SCD1 via tail vein. After 7 days, WT, ALDH2 −/− and p53 −/− ALDH2 −/− mice were treated as described in the methods to establish a mouse model of chronic‐binge ethanol consumption. (F) Schematic representation of experimental design. (G) H&E (hematoxylin–eosin) staining (top panel) and oil red O staining (bottom panel) for the liver tissues from mice fed ethanol diet are shown. (H) Protein levels of p53 and SCD1 are shown. Relative hepatic triglyceride level (I), relative serum ALT (J, left panel) and AST (J, right panel) were examined.
-
KSchematic summarizing the role of p53 in regulating ethanol metabolism through ALDH2.
Data information: Scale bar, 100 μm. Data in (D, E, I, J) are from n = 5 mice per group. Data are the mean ± SD. P values were determined by unpaired two‐tailed Student's t tests. **P < 0.01, ***P < 0.001, ****P < 0.0001, n.s., not significant.
Source data are available online for this figure.
To further determine the role of SCD1 in alcoholic fatty liver induced by ALDH2 or p53 loss, we injected AAV particles expressing SCD1 (AAV‐SCD1) or GFP (AAV‐GFP) as a control into the tail vein of mice before feeding these mice a chronic binge ethanol diet (Fig 6F). Again, ethanol feeding resulted in a significant increase in lipid accumulation in wild‐type mice, but had less effect in ALDH2 −/− mice and p53 −/‐ ALDH2 −/− DKO mice (Fig 6G and H). Intriguingly, we noted that although SCD1 overexpression rescued SCD1 expression in ALDH2 −/− mice and p53 −/− ALDH2 −/− DKO mice, it did not increase lipid accumulation in these mice as it did in wild‐type mice (Fig 6G and H), possibly due to a decrease in acetyl‐CoA level caused by ALDH2 deletion. Moreover, SCD1 overexpression increased triglyceride level in wild‐type mice, but weakly restored the reducing triglyceride level in ALDH2 −/− mice and p53 −/‐ ALDH2 −/− DKO mice (Fig 6I), as well as the serum ALT and AST levels (Fig 6J). Taken together, these data suggest that SCD1 is essential for p53 deficiency‐mediated alcoholic steatohepatitis.
Discussion
This study identifies an important role for p53 in inhibiting alcohol‐induced fatty liver. By suppressing ALDH2, p53 exerts a powerful surveillance on the ethanol metabolism. The inhibition of ALDH2 by p53 is achieved by two mechanisms. On the one hand, p53 directly binds to ALDH2 and prevents its dimer or tetramer formation; on the other hand, p53 lowers pyruvate levels to dampen ALDH2 activation (Fig 6K). As a transcription factor, p53 monitors various metabolic pathways by regulating its target genes. Interestingly, p53 is also more directly involved in the regulation of metabolic pathways via protein–protein interaction. For instance, cytoplasmic p53 inhibits pentose phosphate pathway by binding to and inactivating G6PD (Jiang et al, 2011). Here, we found that p53 can bind to and inhibit ALDH2 in mouse liver tissue, as well as in hepatocellular carcinoma cells with or without stress.
Pyruvate is the end product of glycolysis and is then transported to the mitochondria to support the TCA cycle. Our findings reveal a regulatory role for pyruvate in modulating ALDH2 activity. Mechanistically, pyruvate increases ALDH2 activity through promoting ALDH2 oligomer formation. Interestingly, pyruvate can directly bind to N186 of ALDH2. N186 is predicted to be a transition state stabilizer for ALDH2. Mutant N186A of ALDH2 shows less affinity to pyruvate. In line with this, mutant N186A has less ALDH2 activity. Based on these findings, pyruvate may act as an endogenous regulator of ALDH2 activity.
Although p53 is known for its function in tumor suppression, p53 also has a role in different liver diseases including NAFLD, steatohepatitis, and HCC development (Krstic et al, 2018). The inhibitory effect of p53 on ethanol metabolism that we found in this study represents a previously unrecognized model of p53 regulation of alcoholic fatty liver development. In addition, by regulating ethanol metabolism, p53 regulates cellular acetyl CoA production and alters histone acetylation. Thus, our findings reveal a role of p53 in controlling gene expression via affecting cellular epigenetic state. In addition to the SCD1 gene, it would be also of interest to investigate which genes are regulated by p53‐mediated histone acetylation and to determine their physiological or pathological functions.
ALDH2 is a key enzyme in the alcohol oxidation pathway and has been reported to be a mitochondrial enzyme. Surprisingly, we find that, at least in the cells we used in this study, ALDH2 also localizes in cytoplasm and nucleus, where it is similarly suppressed by p53. The localization of ALDH2 in the nucleus may be an interesting finding. We speculate that ALDH2 may, like many other metabolic enzymes found in the nucleus (Boukouris et al, 2016; Bian et al, 2022), perform noncanonical functions in the nucleus. While we found that p53 regulates the level of acetyl‐CoA and in turn histone acetylation, we have not determined the absolute contribution of p53 binding to nuclear ALDH2 to acetyl CoA production, and the function of ALDH2 in the nucleus needs further investigation. Furthermore, inhibition of ALDH2 may lead to an increase in acetaldehyde in cells and liver tissue, and high levels of acetaldehyde may lead to the susceptibility to many types of cancer. However, in unstressed cells, the levels of p53 are kept low due to its rapid degradation in the proteasome, so that low amount of p53 in normal cells or tissues do not significant increase in acetaldehyde and susceptibility to cancer.
The role of ALDH2 deficiency in the pathogenesis of alcoholic liver injury remains still obscure. Although pharmacological activation of ALDH2 or global overexpression of ALDH2 ameliorates chronic alcohol‐induced hepatic steatosis (Guo et al, 2015; Zhong et al, 2015), improved alcoholic fatty liver in ALDH2 deficient mice has also been also reported (Kwon et al, 2014; Chaudhry et al, 2015). Nevertheless, currently we could speculate that different degrees of variation in ALDH2 expression levels might have different effects on fatty liver, which may result from a combination of complex mechanisms. However, deficiency of ALDH2 in humans leads to the accumulation of acetaldehyde in the blood, but there are few reports on its association with steatosis.
Similar to the effect of ALDH2 on fatty liver, ALDH2 has a controversial function in cancer. ALDH2 polymorphism or mutations is correlated to an increased risk of alcohol‐related cancers including hepatocellular carcinoma (HCC; Brooks et al, 2009; Seo et al, 2019). However, ALDH2 rs671 mutation (Glu to Lys mutation at 487) was also found to be significantly associated with low risk of HCC related to alcohol drinking (Liu et al, 2016) and also plays a protective role in ovarian cancer unrelated to alcohol consumption (Ugai et al, 2018). Thus, the regulatory effects of ALDH2 on tumors are complex, may be tissue type‐specific and are also likely to be context‐dependent, such as whether alcohol is present and under what types of signaling stimulus. Given the lack of a direct mechanistic link between fatty liver and HCC, we currently do not know whether the regulation of ALDH2 by p53 directly leads to HCC and further mechanistic studies are needed.
Recently, a role of p53 in mediating lipid metabolism has emerged, with intriguing metabolic roles in regulating cholesterol homeostasis and lipid droplet formation. Genetically or pharmacologically activated p53 is capable of regulating genes involved in multiple aspects of lipid metabolism, including intracellular regulators of ceramide and fatty acids, systemic lipid uptake and lipoprotein metabolism (Goldstein et al, 2012). However, whether hepatic p53 plays a role in alcohol metabolism remains unclear. In this work, we identify a key role for the p53‐ALDH2‐SCD1 axis in regulating alcohol metabolism and its associated fatty liver disease, adding a new chapter to the understanding of the function of hepatic p53 in regulating lipid metabolism.
Materials and Methods
Regents and antibodies
Cyanamide (catalogue no. 187364, Sigma), ethanol (catalogue no. 51976, Sigma), ethanol‐d6 (catalogue no. 186414, Sigma), ethanol‐1‐13C (catalogue no. 324523, Sigma), 13C6‐glucose (catalogue no. CLM‐1396, Cambridge Isotope Laboratories), Flag M2 affinity gels (catalogue no. A2220, Sigma), HA beads (catalogue no. 88836, ThermoFisher Scientific), GST beads (catalogue no. 17075601, GE Healthcare), doxorubicin (catalogue no. S1208, Selleck), nutlin3α (catalogue no. S8059, Selleck), PFTα, TRIzol (catalogue no. 15596‐018, Invitrogen), Flag peptide (catalogue no. F4799, Sigma), MitoTracker (catalogue no. M22426, ThermoFisher Scientific), Lipofectamine™ 3000 (catalogue no. L3000075, ThermoFisher Scientific), disuccinimidyl suberate (DSS; catalogue no. 21655, ThermoFisher Scientific), pyruvate (catalogue no. P5280, Sigma), CM5 sensor chip (catalogue no. 29104988, Cytiva), pronase (catalogue no. 10165921001, Roche), alanine aminotransferase (ALT) enzymatic activity kit (catalogue no. K752, Biovison), aspartate aminotransferase (AST) enzymatic activity kit (catalogue no. K753, Biovison), ALDH2 enzymatic activity assay kit (catalogue no. ab115348, Abcam), SimpleChIP® Plus Enzymatic Chromatin IP Kit (catalogue no. 9005, Cell Signaling Technology), SYBR Green Q‐PCR Mix (catalogue no. A301‐01, GenStar), RevertAid First Strand cDNA Synthesis Kit (catalogue no. K16225, ThermoFisher Scientific); actin (catalogue no. 66009, 1:5,000, Proteintech), p53 (DO‐1, 1:2,000, Santa Cruz Biotechnology), p53 (catalogue no. 2524, 1:100, Cell Signaling Technology), ALDH2 (catalogue no. MA5‐17029, 1:1,000, ThermoFisher Scientific), ALDH2 (catalogue no. 15310‐1‐AP, 1:1,000, Proteintech), H3 (catalogue no. 4499, 1:1,000, Cell Signaling Technology), anti‐acetyl‐histone H3 Antibody (catalogue no. 06‐599, Merck Millipore), Flag–horseradish peroxidase (Flag–HRP; catalogue no. A8592, 1:3,000, Sigma), haemagglutinin (HA)–HRP (catalogue no. 12013819001, 1:3,000, Sigma), GST (catalogue no. AT0027, 1:3,000, CMCTAG), and SCD1 (catalogue no. 2794, 1:1,000, Cell Signaling Technology).
siRNAs, shRNAs and sgRNAs
siRNA for p53 was purchased from Invitrogen. 5′‐CCGCCUGAGGUUGGCUCUGACUGUA‐3′ (p53) and 5′‐CCCAACAUCAUCAUGUCAGAU‐3′ (ALDH2). siRNAs were transfected into cells using Lipofectamine 3000 Transfection Agent. sgRNA for human p53 was 5′‐CACCGTCCATTGCTTGGGACGGCAA‐3′. shRNA for mouse SCD1 was 5′‐CACCGCCTACGACAAGAACATTCAATCTCGAGATTGAATGTTCTTGTCGTAGG‐3′.
Generation of p53 knockout HepG2 cells
p53‐knocknot HepG2 cells was generated as described previously (Li et al, 2019). Briefly, a lentiviral CRISPR/Cas9 plasmid targeting p53 was created by cloning the annealed sgRNA into pLenti‐CRISPRv2 vector. The sgRNAs sequences targeting human p53 were 5′‐CACCGTCCATTGCTTGGGACGGCAA‐3′ and 5′‐AAACTTGCCGTCCCAAGCAATGGAC‐3′. 293T cells were cotransfected with pLenti‐CRISPRv2, pVSVg and psPAX2 to make lentiviruses. After infection, HepG2 cells were selected by 2 μg/ml puromycin for a week.
Quantitative RT–PCR analysis
RNA extraction and quantitative PCR analysis were carried out as described previously (Zhao et al, 2022). RNA was extracted by TRIzol according to the manufacturer's instructions. Then 2 μg of RNA for each sample was reversed to complementary DNA using a RevertAid First Strand cDNA Synthesis Kit (ThermoFisher Scientific), and 0.2 μg of cDNA was used as a template to perform real‐time quantitative PCR. The primer pairs were: Mouse actin, 5′‐CATTGCTGACAGGATGCAGAAGG‐3′ and 5′‐TGCTGGAAGGTGGACAGTGAGG‐3′; Mouse SCD1, 5′‐GCAAGCTCTACACCTGCCTCTT‐3′ and 5′‐CGTGCCTTGTAAGTTCTGTGGC‐3′; Mouse p53, 5′‐GAAGTCCTTTGCCCTGAAC‐3′ and 5′‐CTAGCAGTTTGGGCTTTCC‐3′.
Mice
p53 −/− and ALDH2 −/− mice on a C57BL/6 background were purchased from Biocytogen Pharmaceuticals (Beijing) and Cyagen Biosciences (Guangzhou), respectively. p53 −/− mice were crossed with ALDH2 −/− mice to obtain p53 −/− ALDH2 −/− mice. All animals were used at 7–8 weeks of age, and only male mice were used. All animal experiments were undertaken in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals, with the approval of the Scientific Investigation Board of the School of Life Sciences, Tsinghua University, Beijing, China. The primers for genotyping were p53‐p1: 5′‐AGTTCTGCCACGTGGTTGGT‐3′; p53‐p2: 5′‐GTCTCCTGGCTCAGAGGGAG‐3′; p53‐p3: 5′‐CAGAGGCCACTTGTGTAGCG‐3′; ALDH2‐F1: 5′‐GACCTATAGAATTCAGCAAACGGA‐3′; ALDH2‐R1: 5′‐GAAGACTGTCAACTGTGAAAGACAG‐3′; ALDH2‐R2: 5′‐GCTACAGAATGAGACTCCACC‐3′.
Mouse model of chronic‐plus‐single‐binge ethanol consumption
The mouse model of chronic‐binge ethanol consumption was described previously (Bertola et al, 2013; Kwon et al, 2014). Briefly, 8‐week‐old male WT, p53 −/−, ALDH2 −/− and p53 −/− ALDH2 −/− C57BL/6J mice were fed a nutritionally adequate, control liquid diet for 1 day, followed by a gradual increase in ethanol concentration from 1 to 4% (v/v) from day 2 to day 5. Next mice were fed a liquid diet containing 5% ethanol for 10 days, and control groups were pair‐fed a control diet for 10 days. At day 11, the mice in the ethanol groups were gavaged with a single dose of ethanol (5 g/kg body weight, 30% ethanol), whereas the mice in the control groups were gavaged with isocaloric dextrin maltose. All mice were sacrificed 9 h after gavage and liver tissue parts and serum of mice were collected.
Adenovirus construction and preparation
Adenoviruses expressing shRNA targeting mouse SCD1 (shSCD1) were cloned into pENTR/U6 and recombined into the BLOCK‐iT Adenoviral RNAi Expression System (catalogue no. K494500, ThermoFisher Scientific) according to according to the manufacturer's instructions. As a control, the LacZ shRNA was similarly cloned into the pAD/Block‐iT‐DEST system. All the adenoviruses were packaged and propagated in HEK293A cells, purified by cesium chloride gradient centrifugation at 107,200 g for 22 h, and desalted by PD‐10 desalting column (catalogue no. 17085101, GE Healthcare).
Adeno‐associated virus construction and preparation
Mouse SCD1 and the GFP gene were cloned into the pAAV2/8 system. The adeno‐associated virus (AAV) was packaged in HEK293A cells (Invitrogen, catalog no. R70507) and purified via discontinuous iodixanol gradient. The quality of AAV preparation was monitored by Coomassie brilliant blue (CBB) G‐250 staining and quantified by real‐time qPCR.
Acetic acid analysis
The Dionex Ultimate 3000 UPLC system was coupled to a TSQ Quantiva Ultra triple‐quadrupole mass spectrometer (Thermo Fisher, CA), equipped with a heated electrospray ionization (HESI) probe in negative ion mode. Extracts from HepG2 cells or mice liver tissues were separated by a BEH C18 column (2.1 × 100 mm, 1.7 μm, Waters). A binary solvent system was used, in which mobile phase A consisted of 100% H2O, and mobile phase B of 100% acetonitrile. A 15‐min gradient with flow rate of 280 μl/min was used as follows: 0–1.5 min at 5% B; 1.5–2.5 min, 5–18% B; 2.5–10 min, 18–48% B;10–10.1 min, 48–98% B; 10.1–12.2 min, 98% B and 12.2–15 min, 5% B. Column chamber and sample tray were held at 55 and 10°C, respectively. The mass spectrometer is operated in selective reaction monitoring mode, and the following mass transitions are monitored over the duration of the chromatographic run: 194 > 137 m/z (derivatized acetic acid) Both of precursor and fragment ion were collected with resolution of 0.7 FWHM, respectively. The source parameters are as follows: spray voltage: 2,500 V; ion transfer tube temperature: 350°C; vaporizer temperature: 300°C; sheath gas flow rate: 35 Arb; auxiliary gas flow rate: 15 Arb. CID gas: 1.5 mTorr. Data analysis and quantitation were performed by the software Xcalibur 3.0.63 (Thermo Fisher, CA).
Measurements of fatty acids
For fatty acid analysis, samples from mice liver tissues were re‐suspended in 120 μl of dichloromethane (CH2Cl2)/Methanol (MeOH) (v:v = 1:1). The UPLC system was coupled to a Q‐Exactive HF orbitrap mass spectrometer (Thermo Fisher, CA) equipped with a heated electrospray ionization (HESI) probe. Lipid extracts were separated by a CORTECS C18 (100 × 2.1 mm, 2.7 μm) column (Waters, USA). A binary solvent system was used, in which mobile phase A consisted of ACN:H2O (60:40), 10 mM Ammonium acetate, and mobile phase B of IPA:ACN (90:10). A 18‐min gradient with flow rate of 250 μl/min was used. Linear gradient was as follows: 0 min, 30% B; 2.5 min, 30% B; 8 min, 50% B; 10 min, 98% B; 15 min, 98% B; 15.1 min, 30% B; 18 min, 30% B. Column chamber and sample tray were held at 40 and 10°C, respectively. Data with mass ranges of m/z 150–600 was acquired at negative ion mode. The full scan was collected with resolution of 70,000. The source parameters are as follows: spray voltage: 3,000 V; capillary temperature: 320°C; heater temperature: 300°C; sheath gas flow rate: 35 Arb; auxiliary gas flow rate: 10 Arb. Data analysis and lipid identification were performed by the tracefinder (Thermo Fisher, CA) according to endogenous MS database by accurate masses.
Measurements of triglyceride (TG)
The UPLC system was coupled to a Q‐Exactive HFX orbitrap mass spectrometer (Thermo Fisher, CA) equipped with a heated electrospray ionization (HESI) probe. Lipid extracts from mice liver tissues were separated by a Cortecs C18 100 × 2.1 mm column (Waters). A binary solvent system was used, in which mobile phase A consisted of ACN:H2O (60:40), 10 mM ammonium acetate, and mobile phase B of IPA:ACN (90:10), 10 mM ammonium acetate. A 25‐min gradient with flow rate of 250 μl/min was used, as shown in Table 1. Column chamber and sample tray were held at 40 and 10°C, respectively. Data with mass ranges of m/z 300–2,000 was acquired at positive ion mode with data dependent MSMS acquisition. The full scan and fragment spectra were collected with resolution of 60,000 and 15,000, respectively. The source parameters are as follows: spray voltage: 3,000 V; capillary temperature: 320°C; heater temperature: 300°C; sheath gas flow rate: 35 Arb; auxiliary gas flow rate: 10 Arb. Data analysis and lipid identification were performed by the software lipidsearch (Thermo Fisher, CA). All molecular identifications were based on MS2, with a MS1 mass error of < 5 ppm and MS2 mass error of < 8 ppm.
Table 1.
In the assay for Triglyceride measurement, a 25‐min gradient with a flow rate of 250 μl/min was used.
| Time (min) | Flow (ml/min) | %B |
|---|---|---|
| 3 | 0.25 | 30 |
| 19 | 0.25 | 98 |
| 22 | 0.25 | 98 |
| 22.1 | 0.25 | 30 |
| 25 | 0.25 | 30 |
STORM imaging
Briefly, cultured HepG2 cells (ATCC, catalogue no. HB‐8065) were fixed with 4% PFA and 0.1% glutaraldehyde in PBS (pH 7.4) for 10 min, followed by washing off excess PFA and reducing unreacted aldehyde groups with 0.1% sodium borohydride (NaBH4) in PBS. Cells were blocked and permeabilized in blocking buffer (3% w/v BSA, 0.2% v/v Triton X‐100 in PBS) for 1 h at room temperature. After washing, cells were postfixed for 10 min with 4% PFA and 0.1% glutaraldehyde in PBS used for STORM imaging. p53 (/no. 15310‐1‐AP, 1:100, Proteintech) antibodies were used for STORM assay.
Surface plasmon resonance analysis
SPR was carried out as described (Yao et al, 2017). SPR analysis was performed with a BIAcore 8 K biomolecular interaction analysis system (GE Healthcare) according to the manufacturer's instructions. Briefly, purified Flag‐tagged ALDH2 and ALDH2 mutations were immobilized on the surface of a CM5 sensor chip in 10 mM sodium acetate buffer (pH 4.0) and functioned as the stationary phase. Pyruvate was dissolved in PBS, diluted with gradient concentrations and run across each sensor surface in a running buffer of PBS at a flow rate of 30 μl/min for 120 s as a contact phase, followed by 150 s in running buffer as a dissociation phase.
Immunoprecipitation
293T cells (ATCC, catalogue no. CRL‐3216) were cotransfected with Flag‐ALDH2 or mutations and HA‐p53 for 48 h, and then cells were lysed in IP lysis buffer for 1 h. Cells were tested negative for mycoplasma contamination. Anti‐Flag M2 affinity gels were adding to supernatants and incubated for 8 h. After incubation, beads were washed three times with lysis buffer and eluted by elution buffer. Protein samples were boiled in 5× loading buffer for 10 min.
Protein purification and pull‐down assays
GST and GST‐p53 proteins were purified from 293T cells and eluted by GSH. GST or GST‐p53 was incubated with Flag‐ALDH2 bound with Flag M2 affinity gels for 8 h. Then washed the beads with IP lysis buffer for three times and eluted the beads with acid buffer and boiled in 5× loading buffer for 10 min.
Western blotting
Cells (liver tissues were broken by tissue blender) were washed and incubated in IP lysis buffer for 1 h on ice, and boiled in 5× SDS loading buffer. Protein samples were resolved by SDS–PAGE and transferred to a nitrocellulose membrane. The membrane was then blocked in 5% skimmed milk in TBST and probed with the indicated antibodies. After incubation with HRP‐labelled secondary antibody, the membrane was incubated with ECL western blotting detection reagent (catalogue no. 32132, ThermoFisher Scientific) and analysed using a Tanon 5200 chemiluminescence imaging system (Tanon Science & Technology).
H&E staining
Briefly, mouse liver tissues were immediately collected and fixed in 4% PFA for 48 h. The liver slices were then dehydrated and embedded in paraffin, sectioned into 5 mm sections, and stained with hematoxylin–eosin (H&E) to assess hepatic steatosis. The inflammatory scores of H&E stained sections were based on the histological scoring system to assess the liver pathology (Kleiner et al, 2005; Mridha et al, 2017).
Oil red O staining
Briefly, 0.5 g Oil Red O was dissolved in 100 ml isopropanol and filtered using a 0.22‐μm filter. Working solution was generated by mixing six parts stock with four parts H2O, incubating for 30 min at room temperature and then filtering through a 0.22‐μm PES filter. Liver tissues were washed with PBS and fixed with 4% paraformaldehyde for 1 h at room temperature, then sectioned into 5 mm sections and were stained with working solution for 30 min at 37°C. After staining, cells were washed three times in PBS and subjected to image capture.
Alanine aminotransferase (ALT) and aspartate aminotransferase (AST) activity
ALT enzymatic activity was measured using mouse serum according to according to the manufacturer's instructions. Briefly, serum samples were diluted in the assay buffer and prepare 20 μl/well test samples with assay buffer in a 96‐well plate. Then added reaction mix containing ALT assay buffer, OxiRed, ALT enzyme mixture and ALT substrate and read OD 570 nm (A1) at T1 (T1 > 10 min) then again (A2) at T2 after incubating the reaction at 37°C for 60 min, protect from light. The OD of the color generated by oxidation of pyruvate is ∆A570 nm = A2 − A1.
AST enzymatic activity was measured using mouse serum according to according to the manufacturer's instructions. Briefly, serum samples were diluted in the assay buffer and prepare 50 μl/well test samples with assay buffer in a 96‐well plate. Then added reaction mix containing AST assay buffer, AST enzyme mixture, developer and AST substrate and read OD 450 nm (A1) at T1 (T1 > 10 min) then again (A2) at T2 after incubating the reaction at 37°C for 60 min, protect from light. The OD of the color generated by oxidation of pyruvate is ∆A450 nm = A2 − A1.
ALDH2 enzymatic activity
ALDH2 enzymatic activity was examined using HepG2 cell lysates, mouse primary hepatocytes (8‐week‐old male C57BL/6J mice) or liver tissue lysates according to according to the manufacturer's instructions. Briefly, extraction buffer supplemented with phosphatase inhibitors, PMSF and protease inhibitor cocktail prior to use was used to solubilize HepG2 cell pellet, mouse primary hepatocytes or liver tissue pellet (liver tissues were broken by tissue blender) for 20 min on ice and centrifuge at 16,000 × g at 4°C for 20 min. Transfer the supernatants into clean tubes and add all the regents and cover/seal the plate and incubate for 3 h at room temperature. Wash three times and gently add 200 μl 1× activity solution and read OD 450 nm.
Subcellular fractionation assay
Cell fractionation was carried out as described (Jiang et al, 2013). Cells were homogenized in 20 mM HEPES‐KOH buffer, pH 7.5, 10 mM KCL, 1.5 mM MgCl2, 1 mM sodium EDTA buffer, 1 mM sodium EGTA buffer and 1 mM dithiothreitol in the presence of 250 mM sucrose and protease inhibitor cocktail. Homogenates were centrifuged at 500 g for 5 min at 4°C, and the supernatant was collected and centrifuged again at 10,000 g for 20 min to obtain cytosolic and mitochondrial fractions. Resuspend the cells by pipetting them up and down in ice‐cold hypotonic buffer (20 mM Tris–HCl pH 7.4, 10 mM KCl, 2 mM MgCl2, 1 mM EGTA, 0.5 mM DTT, 0.5 mM PMSF) and incubate on ice for 3 min. Add NP‐40 to a final concentration of 0.1%, and incubate on ice for 3 min. Centrifuge the cell suspension at 1,000 rcf at 4°C for 5 min to separate the nuclei (pellet). Resuspend the nuclei by pipetting them up and down in an isotonic buffer (20 mM Tris–HCl pH 7.4, 150 mM KCl, 2 mM MgCl2, 1 mM EGTA, 0.5 mM DTT, 0.5 mM PMSF) containing 0.1% NP‐40 and incubate on ice for 10 min. Centrifuge the nuclei at 1,000 rcf at 4°C for 3 min.
Cellular thermal shift assay
A CETSA was performed to determine the direct binding between pyruvate and ALDH2 in HepG2 cells. Briefly, HepG2 cells were pretreated with 5 mM pyruvate for 16 h before being subjected to the CETSA protocol. Cells were chilled on ice, washed with PBS plus protease inhibitor cocktail and then transferred into 200 μl PCR tubes. The cells were heat shocked in a Bio‐Rad T100 thermal cycler at the indicated temperatures for 3 min to denature the proteins and cooled at room temperature for 3 min. All samples were then subjected to three freeze–thaw cycles using dry ice and a thermal cycler at the indicated temperatures to lyse cells, sequentially centrifuged at 14,000 g for 10 min at 4°C and the cell debris discarded. The supernatants were boiled with SDS buffer for western blot analysis. Relative ALDH2 band intensities were quantified using Image J software and plotted against the corresponding incubation temperatures.
Drug‐affinity responsive target stability assays
DARTS was performed as described previously with minor modifications. Briefly, HepG2 cells were harvested in lysis buffer M‐PER (catalogue no. 78501, ThermoFisher Scientific) with protease inhibitor cocktail, then centrifuged at 16,200 g for 10 min. The supernatants were collected and incubated with varying concentrations of pyruvate or PBS (vehicle) for 1 h on ice and then kept at room temperature for another 30 min. Samples were digested with pronase (12.5 μg/ml) for 20 min at room temperature followed by addition of SDS loading buffer to stop the reaction. Actin was used as a negative control.
ChIP‐seq assay and ChIP‐qPCR analysis
ChIP assay was performed using SimpleChIP® enzymatic chromatin IP kit (catalog no. 9005, Cell Signaling Technology) according to according to the manufacturer's instructions. Briefly, liver tissues were homogenated by tissue blender and subsequently cross‐linked with 1.5% formaldehyde solution at room temperature for 20 min. Stop cross‐linking by adding 0.125 M glycine and mix for 5 min at room temperature. Centrifuge tissue and remove supernatant and wash one time with PBS. Then resuspend cells in buffer A and incubate on ice for 10 min. Centrifuge and resuspend pellet in buffer B and add 0.5 μl of micrococcal nuclease, mix and incubate for 20 min at 37°C to digest DNA. Stop digest by adding 0.5 M EDTA and pellet nuclei by centrifugation at 16,000 g for 1 min at 4°C and remove supernatant. Resuspend nuclear pellet in ChIP buffer and incubate on ice for 10 min. Sonicate lysate with several pulses to break nuclear membrane and incubate samples for 30 s on wet ice between pulses. Centrifuge at 9,400 g for 10 min at 4°C and cells were subjected to immunoprecipitation with indicated antibodies against Ac‐H3K9, Ac‐H3K27 overnight, and samples were then washed with low‐salt buffer, high‐salt buffer. Histone–DNA complexes were eluted using elution buffer (1% SDS and 100 mM NaHCO3), incubated at 65°C overnight in the presence of 0.54 M NaCl. Then DNase‐free RNase (10 mg/ml) was added into samples and incubated for 1 h at 37°C. Samples were then incubated with proteinase K for another 1 h at 55°C. Finally, DNA was purified using Qiaex II beads (Qiagen). The primer pairs were: H3K9ac‐SCD1‐RE, 5′‐CTGGTCTACAAGGTGAGTTCC‐3′ and 5′‐GGAACTATGTAGAGTAGGCTGC‐3′; H3K27ac‐SCD1‐RE, 5′‐CCTCCCTTTCCCAACAAGATG‐3′ and 5′‐CCCTCCCTTCCTTTAGCTCTC‐3′.
ChIP‐seq data processing
ChIP‐seq data processing was performed as described previously (Huang et al, 2020). Briefly, FastQC (v0.11.8) was used for quality control and filtering of raw readings. The sequencing results from ChIP‐seq data for H3K9ac and H3K27ac were aligned to the mm10 genome using bowtie2 (v2.3.5.1). Peaks were called using MACS2 (v2.2.6) for H3K9ac and H3K27ac. The significance threshold of 10–5 was used for all data sets. Different peaks were generated using bdgdiff. The ChIP‐seq pileup heatmaps were generated using plotHeatmap. Visualization was realized by IGV (v2.8.9). GO/KEGG enrichment analysis was performed using the R package clusterProfiler (v4.0).
Stable isotope labelling of liver histone acetylation
Stable isotope labeling assay was carried out as described previously (Mews et al, 2019). Ethanol‐d6 was intraperitoneally injected into mice (2 g/kg) and deuterium integrated into acetylated histones at baseline, as well as at 4 h after intraperitoneal injections were assessed. Liver tissues were broken by tissue blender. For acid extraction, broken tissue pellets were lysed in hypotonic lysis buffer containing 10 mM HEPES, 10 mM KCl, 1.5 mM MgCl2, 0.5 mM dithiothreitol and protease inhibitors. H2SO4 was then added to a final concentration of 0.2 M and incubated for 30 min at 4°C. After spinning down, proteins in supernatants were precipitated in 33% trichloroacetic acid, washed with acetone and resuspended in ddH2O. Protein extracts were mixed with 15 μl derivatization mix, consisting of propionic anhydride and acetonitrile in a 1:3 ratio (v/v), and this was immediately followed by the addition of 7.5 μl ammonium hydroxide to maintain pH 8.0. The sample was incubated for 15 min at 37°C, dried and the derivatization procedure was repeated one more time. Samples were then resuspended in 50 mM NH4HCO3 and incubated with trypsin (enzyme:sample ratio of 1:20) overnight at room temperature. After digestion, the derivatization reaction was performed again twice to derivatize the N termini of the peptides. Samples were desalted using C18 stage tips before quantitative liquid chromatography–mass spectrometry (LC–MS) analysis. LC–MS was used to quantify the relative abundance of isotopically labelled histone acetylation in the liver protein extracts.
Statistical analysis
All experiments were performed at least three times independently. No statistical methods were used to predetermine sample size. For animal experiments, real‐time PCR and LC–MS analysis, the investigators were blinded to group allocation during data collection and/or analysis. The authors were not blinded to all other experiments. Results are shown as means ± s.d. All statistical methods used are specified in the figure legends. Statistical analysis was performed, and P values were obtained using GraphPad Prism v.7.0 (GraphPad Software).
Author contributions
Wenjing Du: Supervision; writing – original draft; writing – review and editing. Peng Jiang: Conceptualization; resources. Pengbo Yao: Data curation; formal analysis; investigation; visualization; methodology. Hongchao Liu: Methodology. Wei Li: Data curation. Zhenxi Zhang: Methodology.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Source Data for Expanded View
PDF+
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Acknowledgements
We thank D. Shi, Y. Mao, L. Zhang, F. Wu and Z. Yang for their technical help. We thank X. Wang, Y. Wang, L. Xu, Y. Jiao and X. Liu from the Metabolomics Facility at Technology Center for Protein Sciences at Tsinghua University for their flux and metabolic profiling. We are grateful to N. Wang for helping with the protein structural analysis. This work was supported by the National Key Research and Development Program of China (2022YFA0806302), CAMS Innovation Fund for Medical Sciences (CIFMS) (2021‐I2M‐1‐016), Haihe Laboratory of Cell Ecosystem Innovation Fund (HH22KYZX0011), CAMS Basic Research Fund (2019‐RC‐HL‐007) and State Key Laboratory Special Fund (2060204) to WD.
The EMBO Journal (2023) 42: e112304
Contributor Information
Peng Jiang, Email: pengjiang@tsinghua.edu.cn.
Wenjing Du, Email: wenjingdu@ibms.pumc.edu.cn.
Data availability
The CHIP‐seq data have been deposited in the Gene Expression Omnibus under accession GSE222743 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE222743). The remaining data supporting the findings of this study are available from the correspondence upon reasonable request. Correspondence and requests for materials should be addressed to Dr. Wenjing Du (wenjingdu@ibms.pumc.edu.cn).
References
- Adeniji EA, Olotu FA, Soliman MES (2018) Alcohol metabolic inefficiency: structural characterization of polymorphism‐induced ALDH2 dysfunctionality and allosteric site identification for Design of Potential Wildtype Reactivators. Protein J 37: 216–222 [DOI] [PubMed] [Google Scholar]
- Am AL, Syed DN, Ntambi JM (2017) Insights into Stearoyl‐CoA Desaturase‐1 regulation of systemic metabolism. Trends Endocrinol Metab 28: 831–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertola A, Mathews S, Ki SH, Wang H, Gao B (2013) Mouse model of chronic and binge ethanol feeding (the NIAAA model). Nat Protoc 8: 627–637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bian X, Jiang H, Meng Y, Li YP, Fang J, Lu Z (2022) Regulation of gene expression by glycolytic and gluconeogenic enzymes. Trends Cell Biol 32: 786–799 [DOI] [PubMed] [Google Scholar]
- Boukouris AE, Zervopoulos SD, Michelakis ED (2016) Metabolic enzymes moonlighting in the nucleus: metabolic regulation of gene transcription. Trends Biochem Sci 41: 712–730 [DOI] [PubMed] [Google Scholar]
- Brooks PJ, Enoch MA, Goldman D, Li TK, Yokoyama A (2009) The alcohol flushing response: an unrecognized risk factor for esophageal cancer from alcohol consumption. PLoS Med 6: e50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campana L, Esser H, Huch M, Forbes S (2021) Liver regeneration and inflammation: from fundamental science to clinical applications. Nat Rev Mol Cell Biol 22: 608–624 [DOI] [PubMed] [Google Scholar]
- Chaudhry KK, Samak G, Shukla PK, Mir H, Gangwar R, Manda B, Isse T, Kawamoto T, Salaspuro M, Kaihovaara P et al (2015) ALDH2 deficiency promotes ethanol‐induced gut barrier dysfunction and fatty liver in mice. Alcohol Clin Exp Res 39: 1465–1475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung EC, Vousden KH (2010) The role of p53 in glucose metabolism. Curr Opin Cell Biol 22: 186–191 [DOI] [PubMed] [Google Scholar]
- Floter J, Kaymak I, Schulze A (2017) Regulation of metabolic activity by p53. Metabolites 7: 21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein I, Ezra O, Rivlin N, Molchadsky A, Madar S, Goldfinger N, Rotter V (2012) p53, a novel regulator of lipid metabolism pathways. J Hepatol 56: 656–662 [DOI] [PubMed] [Google Scholar]
- Guo R, Xu X, Babcock SA, Zhang Y, Ren J (2015) Aldehyde dedydrogenase‐2 plays a beneficial role in ameliorating chronic alcohol‐induced hepatic steatosis and inflammation through regulation of autophagy. J Hepatol 62: 647–656 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Huang Y, Zhang H, Wang L, Tang C, Qin X, Wu X, Pan M, Tang Y, Yang Z, Babarinde IA et al (2020) JMJD3 acts in tandem with KLF4 to facilitate reprogramming to pluripotency. Nat Commun 11: 5061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingram‐Smith C, Woods BI, Smith KS (2006) Characterization of the acyl substrate binding pocket of acetyl‐CoA synthetase. Biochemistry 45: 11482–11490 [DOI] [PubMed] [Google Scholar]
- Jiang P, Du W, Wang X, Mancuso A, Gao X, Wu M, Yang X (2011) p53 regulates biosynthesis through direct inactivation of glucose‐6‐phosphate dehydrogenase. Nat Cell Biol 13: 310–316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang P, Du W, Mancuso A, Wellen KE, Yang X (2013) Reciprocal regulation of p53 and malic enzymes modulates metabolism and senescence. Nature 493: 689–693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones RG, Plas DR, Kubek S, Buzzai M, Mu J, Xu Y, Birnbaum MJ, Thompson CB (2005) AMP‐activated protein kinase induces a p53‐dependent metabolic checkpoint. Mol Cell 18: 283–293 [DOI] [PubMed] [Google Scholar]
- Kleiner DE, Brunt EM, Van Natta M, Behling C, Contos MJ, Cummings OW, Ferrell LD, Liu YC, Torbenson MS, Unalp‐Arida A et al (2005) Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 41: 1313–1321 [DOI] [PubMed] [Google Scholar]
- Krstic J, Galhuber M, Schulz TJ, Schupp M, Prokesch A (2018) p53 as a dichotomous regulator of liver disease: the dose makes the medicine. Int J Mol Sci 19: 921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon HJ, Won YS, Park O, Chang B, Duryee MJ, Thiele GE, Matsumoto A, Singh S, Abdelmegeed MA, Song BJ et al (2014) Aldehyde dehydrogenase 2 deficiency ameliorates alcoholic fatty liver but worsens liver inflammation and fibrosis in mice. Hepatology 60: 146–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahalle A, Lacroix M, De Blasio C, Cisse MY, Linares LK, Le Cam L (2021) The p53 pathway and metabolism: the tree that hides the Forest. Cancers (Basel) 13: 133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson HN, Weiner H, Hurley TD (2005) Disruption of the coenzyme binding site and dimer interface revealed in the crystal structure of mitochondrial aldehyde dehydrogenase “Asian” variant. J Biol Chem 280: 30550–30556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee BP, Vittinghoff E, Dodge JL, Cullaro G, Terrault NA (2019) National Trends and long‐term outcomes of liver transplant for alcohol‐associated liver disease in the United States. JAMA Intern Med 179: 340–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Zhang D, Jin W, Shao C, Yan P, Xu C, Sheng H, Liu Y, Yu J, Xie Y et al (2006) Mitochondrial aldehyde dehydrogenase‐2 (ALDH2) Glu504Lys polymorphism contributes to the variation in efficacy of sublingual nitroglycerin. J Clin Invest 116: 506–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Mao Y, Zhao L, Li L, Wu J, Zhao M, Du W, Yu L, Jiang P (2019) p53 regulation of ammonia metabolism through urea cycle controls polyamine biosynthesis. Nature 567: 253–256 [DOI] [PubMed] [Google Scholar]
- Liu Y, Gu W (2021) The complexity of p53‐mediated metabolic regulation in tumor suppression. Semin Cancer Biol 85: 4–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Yang HI, Lee MH, Jen CL, Hu HH, Lu SN, Wang LY, You SL, Huang YT, Chen CJ (2016) Alcohol drinking mediates the association between polymorphisms of ADH1B and ALDH2 and hepatitis B‐related hepatocellular carcinoma. Cancer Epidemiol Biomarkers Prev 25: 693–699 [DOI] [PubMed] [Google Scholar]
- Liu J, Zhang C, Hu W, Feng Z (2019) Tumor suppressor p53 and metabolism. J Mol Cell Biol 11: 284–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mews P, Egervari G, Nativio R, Sidoli S, Donahue G, Lombroso SI, Alexander DC, Riesche SL, Heller EA, Nestler EJ et al (2019) Alcohol metabolism contributes to brain histone acetylation. Nature 574: 717–721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon SH, Huang CH, Houlihan SL, Regunath K, Freed‐Pastor WA, Morris JP, Tschaharganeh DF, Kastenhuber ER, Barsotti AM, Culp‐Hill R et al (2019) p53 represses the mevalonate pathway to mediate tumor suppression. Cell 176: 564–580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mridha AR, Haczeyni F, Yeh MM, Haigh WG, Ioannou GN, Barn V, Ajamieh H, Adams L, Hamdorf JM, Teoh NC et al (2017) TLR9 is up‐regulated in human and murine NASH: pivotal role in inflammatory recruitment and cell survival. Clin Sci (Lond) 131: 2145–2159 [DOI] [PubMed] [Google Scholar]
- O'Brate A, Giannakakou P (2003) The importance of p53 location: nuclear or cytoplasmic zip code? Drug Resist Updat 6: 313–322 [DOI] [PubMed] [Google Scholar]
- Olovnikov IA, Kravchenko JE, Chumakov PM (2009) Homeostatic functions of the p53 tumor suppressor: regulation of energy metabolism and antioxidant defense. Semin Cancer Biol 19: 32–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osna NA, Donohue TM Jr, Kharbanda KK (2017) Alcoholic liver disease: pathogenesis and current management. Alcohol Res 38: 147–161 [PMC free article] [PubMed] [Google Scholar]
- Seo W, Gao Y, He Y, Sun J, Xu H, Feng D, Park SH, Cho YE, Guillot A, Ren T et al (2019) ALDH2 deficiency promotes alcohol‐associated liver cancer by activating oncogenic pathways via oxidized DNA‐enriched extracellular vesicles. J Hepatol 71: 1000–1011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Setshedi M, Wands JR, Monte SM (2010) Acetaldehyde adducts in alcoholic liver disease. Oxid Med Cell Longev 3: 178–185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun H, Li L, Li W, Yang F, Zhang Z, Liu Z, Du W (2021) p53 transcriptionally regulates SQLE to repress cholesterol synthesis and tumor growth. EMBO Rep 22: e52537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ugai T, Kelemen LE, Mizuno M, Ong JS, Webb PM, Chenevix‐Trench G, Australian Ovarian Cancer Study Group , Wicklund KG, Doherty JA, Rossing MA et al (2018) Ovarian cancer risk, ALDH2 polymorphism and alcohol drinking: Asian data from the ovarian cancer association consortium. Cancer Sci 109: 435–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vousden KH, Prives C (2009) Blinded by the light: the growing complexity of p53. Cell 137: 413–431 [DOI] [PubMed] [Google Scholar]
- Vousden KH, Ryan KM (2009) p53 and metabolism. Nat Rev Cancer 9: 691–700 [DOI] [PubMed] [Google Scholar]
- Wang W, Wang C, Xu H, Gao Y (2020) Aldehyde dehydrogenase, liver disease and cancer. Int J Biol Sci 16: 921–934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao P, Sun H, Xu C, Chen T, Zou B, Jiang P, Du W (2017) Evidence for a direct cross‐talk between malic enzyme and the pentose phosphate pathway via structural interactions. J Biol Chem 292: 17113–17120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida A, Huang IY, Ikawa M (1984) Molecular abnormality of an inactive aldehyde dehydrogenase variant commonly found in Orientals. Proc Natl Acad Sci USA 81: 258–261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao M, Yao P, Mao Y, Wu J, Wang W, Geng C, Cheng J, Du W, Jiang P (2022) Malic enzyme 2 maintains protein stability of mutant p53 through 2‐hydroxyglutarate. Nat Metab 4: 225–238 [DOI] [PubMed] [Google Scholar]
- Zhong W, Zhang W, Li Q, Xie G, Sun Q, Sun X, Tan X, Sun X, Jia W, Zhou Z (2015) Pharmacological activation of aldehyde dehydrogenase 2 by Alda‐1 reverses alcohol‐induced hepatic steatosis and cell death in mice. J Hepatol 62: 1375–1381 [DOI] [PMC free article] [PubMed] [Google Scholar]