

Abstract
Background
We have previously reported arterial remodelling in patients with idiopathic pulmonary fibrosis (IPF) and suggested that endothelial-to-mesenchymal transition (EndMT) might be central to these changes. This study aims to provide evidence for active EndMT in IPF patients.
Methods
Lung resections from 13 patients with IPF and 15 normal controls (NCs) were immunostained for EndMT biomarkers: vascular endothelial cadherin (VE-cadherin), neural cadherin (N-cadherin), S100A4 and vimentin. Pulmonary arteries were analysed for EndMT markers by using computer- and microscope-assisted image analysis software Image ProPlus7.0. All the analysis was done with observer blinded to subject and diagnosis.
Results
Increased expression of mesenchymal markers N-cadherin (p<0.0001), vimentin (p<0.0001) and S100A4 (p<0.05) was noted with downregulation of junctional endothelial VE-cadherin (p<0.01) in the intimal layer of the arteries from patients with IPF compared to NCs. Cadherin switch was observed in IPF patients, showing increase in endothelial N-cadherin and decrease in VE-cadherin (p<0.01). There was also VE-cadherin shift from junctions to cytoplasm (p<0.01), effecting endothelial cell integrity in patients with IPF. In IPF, individual mesenchymal markers vimentin and N-cadherin negatively correlated with diffusing capacity of the lungs for carbon monoxide (r′= −0.63, p=0.03 and r′= −0.66, p=0.01). Further, N-cadherin positively correlated with arterial thickness (r′=0.58, p=0.03).
Conclusion
This is the first study to demonstrate active EndMT in size-based classified pulmonary arteries from IPF patients and potential role in driving remodelling changes. The mesenchymal markers had a negative impact on the diffusing capacity of the lungs for carbon monoxide. This work also informs early origins of pulmonary hypertension in patients with IPF.
Short abstract
EndMT is an active process in patients with IPF, and contributes to pulmonary arterial remodelling and fibrosis. This work also informs the early origins of pulmonary hypertension in patients with IPF. EndMT is a novel therapeutic target in IPF. https://bit.ly/3Hof1pZ
Introduction
Idiopathic pulmonary fibrosis (IPF) is a highly progressive lung disease with limited therapeutic options [1]. The current IPF pathophysiology emphasises the critical role of alveolar epithelial injury, followed by an aberrant wound healing process resulting in fibrosis and destructive lung scarring, although the causes are unknown [1, 2]. Fibrosis of the lung restricts efficient gas exchange, effectively inducing systemic hypoxic conditions and death due to oxygen deficiency. Similar to the epithelial injury, alterations to pulmonary vasculature also occur, resulting in abnormal vascular remodelling [3]. The modification to the vascular structure includes proliferative intima, thickening of the medial smooth muscle layer, plexiform lesions and complete occlusion or narrowing of the vessels by scarred tissues [1, 3–5]. Such vascular remodelling can lead to pulmonary hypertension (PH), a comorbidity that frequently complicates IPF pathology and worsens prognosis, especially in late stages of the disease [6, 7].
Recent findings suggest the active involvement of several cells in IPF. However, cells vulnerable to transition under pathological conditions, such as airway basal epithelial cells, alveolar type II pneumocytes, macrophages and pulmonary endothelial cells, are crucial contributors to tissue fibrosis [8, 9]. The transition of endothelium occurs through endothelial-to-mesenchymal transition (EndMT), which is a critical mechanism in vascular remodelling that can further progress to fibrotic alterations [10–12]. EndMT-induced vascular changes are reported in IPF [1, 13, 14], intestinal fibrosis [15] and cardiac fibrosis [16]. Our recent study has found that arterial wall thickening, luminal occlusion and disruptive extracellular matrix (ECM) deposition occurs in IPF patients, and EndMT possibly drives these changes [3]. However, the contribution of EndMT to vascular remodelling and its association with lung fibrosis in IPF patients is poorly defined. In EndMT, loss of junctional proteins such as vascular endothelial cadherins (VE-cadherins) and gain of migratory mesenchymal traits occur. The increase in endothelial expression of mesenchymal proteins such as neural cadherin (N-cadherin), S100A4 and vimentin are suggestive of EndMT [17, 18].
This study investigates EndMT as a dynamic process in relation to vascular remodelling in IPF by analysing the EndMT biomarkers. We performed a quantitative assessment of endothelial junctional marker VE-cadherin and mesenchymal markers S100A4, vimentin and N-cadherin protein levels in the classified pulmonary arteries from patients with IPF and compared them with NCs. Furthermore, we analysed the correlations between the mesenchymal markers, arterial remodelling and lung function parameters.
Material and methods
Study population
Surgically resected human lung tissues from 13 patients with IPF were obtained from Alfred Health Biobank Melbourne (ethics ID: 336-13). None of the patients was on any antifibrotic medication, and all had pathologist-verified histopathology reports of usual interstitial pneumonia. Lung tissue specimens from 15 healthy normal control (NC) subjects, comprising small airways and parenchymal areas (ethics ID: H00-50110), were provided by the James Hogg Lung Registry, University of British Columbia. Detailed subject demographics are provided in table 1.
TABLE 1.
Subject demographics and clinical characteristics
| Factor | Normal control (NC) | IPF |
| Total number n | 15 | 13 |
| Age years | 44±20.1 | 64±5.06 |
| Sex (F/M) n | 6/5 | 6/7 |
| Body mass index kg·m−2 | NA | 26.69±3.03 |
| Smoking status: (current smoker/ex-smoker/never-smoker) n | Nonsmokers | 0/7/6 |
| Smoking pack-years | 20.84±23.16 | |
| Lung physiology | ||
| FEV1 L | 1.70 ±0.40 | |
| FEV1 % predicted | 60.17±12.22 | |
| FVC L | 1.97±0.51 | |
| FVC % predicted | 53.5±12.98 | |
| DLCO mmol·min−1·kPa−1 | 5.91±2.92 | |
| DLCO corrected % predicted | 25.85±15.30 | |
Data presented as mean±sd unless otherwise indicated. NA: not available; IPF: idiopathic pulmonary fibrosis; FEV1: forced expiratory volume in 1 s; FVC: forced vital capacity; DLCO: diffusing capacity of the lung for carbon monoxide.
Immunohistochemical staining for EndMT markers
The lung tissue resections were deparaffinised using xylene and ethanol following antigen retrieval using a Decloaking Chamber (Biocare Medical, Pacheco, CA, USA) at 110°C for 15 min with target retrieval citrate buffer pH 6.0 (Dako S2369) and at 95°C for 15 min with retrieval buffer pH 9.0 (Dako S2367), respectively. The tissues were stained with EndMT primary antibodies, polyclonal rabbit anti-human S100A4 (1:1000; Dako A5114), VE-cadherin (CD144) mouse monoclonal (1:150; Thermofisher 14144982), vimentin mouse monoclonal (1:200; Dako M7020) and mouse monoclonal anti-N-cadherin (1:100; Abcam Ab98952) for 60 min; with inflammatory markers, including mouse anti-human CD4 (1:50, Dako M7310), mouse anti-human CD8 (1:50, Leica Biosystems, NCL-CD8-4B11-L-CE), CD68 (1:100, Dako M0814), mouse anti-human neutrophil elastase (1:200, Dako M0752) and mouse monoclonal mast cell chymase (CMA1, 1:100, Abcam ab2377); and with the pericyte marker platelet-derived growth factor receptor-β (PDGFR-β, 1:100, LSBio, LS-C150026); followed by secondary HRP rabbit/mouse antibodies (Dako K5007). The protein markers were visualised as brown with the addition of DAB substrate (Dako K5007). The nucleus was counterstained with haematoxylin (Australian Biostain P/L).
Measurement strategies for pulmonary arteries
Arterial images were acquired from NCs and patients with IPF by using a 4× objective in a vertical uni-direction to avoid overlap. We used a Leica DM 500 microscope with attached Leica IC50W digital camera for analysis. Using measuring tools from the ProPlus 7.0 program, the external length (from one end to the other end of the adventitia) for pulmonary arteries was measured. These measurements were used for arterial classification into six groups: 100–1000 µm (interspaced 100 or 200 µm), similar to the strategy used in our earlier study [3].
VE-cadherin and N-cadherin expression in the basement membrane of pulmonary arteries
We used different magnifications to capture images across all arterial sizes. For example, for smaller arteries measuring 100–199 μm a 63× objective, for 200–399 μm and 400–599 μm a 40× objective and for larger arteries 600–1000 μm a 20× objective was used. Further, for arterial size and subject, five images were randomly selected using an online random number generator for both VE-cadherin and N-cadherin. Firstly, two trace lines were drawn using the imaging software, one at the basement membrane and another one at the outer endothelium as the reference. The mean distance between the trace lines was calculated for basement membrane length using the automated program of tissue image analysis software Image-Pro Plus v7. Further, cells which stained positive (brown colour) for junctional and cytoplasmic VE-cadherin in the reference basement membrane area were counted (supplementary figure S1a). Likewise, cells which stained positive (brown colour) for junctional and cytoplasmic N-cadherin in the reference basement membrane area were counted (supplementary figure S1b). Additionally, total cells in the basement membrane were counted using the imaging software.1
VE-cadherin junctional and cytoplasmic positive cell percentage per average basement membrane was calculated using the following formula:
 |
Similarly, N-cadherin junctional and cytoplasmic positive cell percentage per average basement membrane was calculated using the following formula:
 |
Pulmonary total arterial layer expression for vimentin, N-cadherin and S100A4
Image acquisition and randomisation were followed in the same way as VE-cadherin and N-cadherin cell percentage measurements. For total arterial expression for each mesenchymal marker (vimentin, N-cadherin and S100A4 expression), an area of interest (AOI) from the start of the outer end of the intima facing the lumen to the outer border of adventitia was manually selected using imaging software. Subsequently, mesenchymal staining colour (brown) and total dark objects from the AOI were counted using image ProPlus 7.0 software. A similar strategy was used for individual layers of intima, media and adventitia. For intimal layer mesenchymal marker expression, the outer luminal to the inner elastin layer was selected as the AOI, while for the medial mesenchymal marker expressions the external outline of the inner elastin membrane and internal lining of the external elastin membrane of the atrial smooth muscle layer was considered. The areas bordering the external elastin to the arteries’ outermost connective tissue were considered as adventitia for the mesenchymal marker expression measurements (supplementary figure S1c). Percentage of vimentin, N-cadherin and S100A4 expression was calculated using the following formula:
 |
Correlations between the mesenchymal marker expression and vascular remodelling changes
Our group has previously published the study on morphometric assessment of vascular remodelling changes observed among IPF patients [3, 19]. Here, we used same morphometric assessment data for correlation purposes since the patient's cohort was same. The correlation between each mesenchymal marker expression and arterial remodelling changes was calculated to understand the impact of EndMT.
Statistical analysis
All cross-sectional data were tested for their normal distributions using the D'Agostino–Pearson omnibus normality test. Analysis of variance was performed with ordinary one-way ANOVA using Bonferroni multiple comparison tests, which compared mean and standard deviation across all groups of interest; specific group differences with correction for multiple comparisons were assessed using Dunn's test. Finally, for correlations, we performed regression analyses using Spearman's rank correlation test. All analysis was done using GraphPad prismV9, with a p-value ≤0.05 being considered significant.
Results
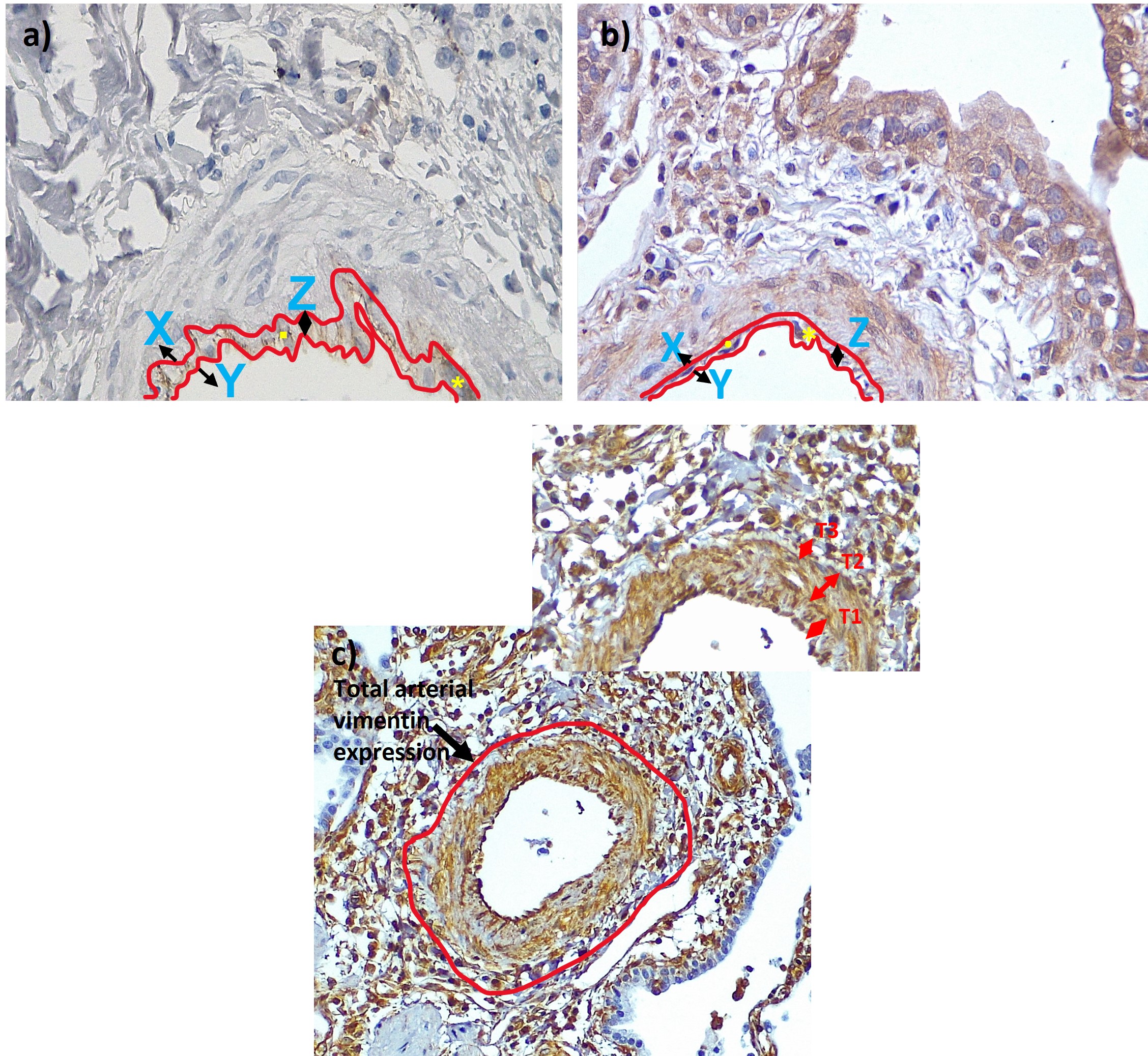
The pulmonary arteries from IPF patients exhibited notable morphological changes such as endothelial proliferation penetrating the lumen, muscular hypertrophy both in intima and medial layers and plexiform lesions compared to arteries from NCs. Cytoplasmic endothelial cell expression was observed for VE-cadherin in tissue sections from patients with IPF, while junctional expression was observed in NCs (figure 1a). Across all arterial sizes, mesenchymal protein N-cadherin expression was observed in all layers of classified arteries in IPF but was not seen in NCs (figure 1b). Strong vimentin and S100A4 staining were seen in all layers in IPF compared to NCs. Vimentin expression was predominantly observed in the smaller and medium-sized arteries (figure 1c). S100A4 staining was predominantly seen in the intima compared to media and adventitia across all arterial ranges in IPF patients. Comparatively there was less S100A4 staining than vimentin and less N-cadherin expression across all arterial sizes in IPF (figure 1d).
FIGURE 1.


a) Slides from normal control (NC) and idiopathic pulmonary fibrosis (IPF) patients of pulmonary arteries immunostained for VE-cadherin measuring 100–199 µm (20× magnification), 200–399 µm (20× magnification), 400–599 µm (20× magnification) and 600–1000 µm (10× magnification). b) Slides from normal control (NC) and idiopathic pulmonary fibrosis (IPF) patients of pulmonary arteries immunostained for N-cadherin measuring 100–199 µm (20× magnification), 200–399 µm (20× magnification), 400–599 µm (20× magnification) and 600–1000 µm (10× magnification). c) Slides from normal control (NC) and idiopathic pulmonary fibrosis (IPF) patients of pulmonary arteries immunostained for vimentin measuring 100–199 µm (20× magnification), 200–399 µm (20× magnification), 400–599 µm (20× magnification) and 600–1000 µm (10× magnification). d) Slides from normal control (NC) and idiopathic pulmonary fibrosis (IPF) patients of pulmonary arteries immunostained for S100A4 measuring 100–199 µm (20× magnification), 200–399 µm (20× magnification), 400–599 µm (20× magnification) and 600–1000 µm (10× magnification).
Endothelial VE-cadherin and N-cadherin cellular expression in IPF
We assessed differential counts for VE- and N-cadherin in the junction and compared them with VE- and N-cadherin cytoplasmic expression, which is presented here as the percentage of total cells in the intimal area. Endothelium of NCs on average consisted of 40–50% cells that expressed junctional VE-cadherin, which drastically reduced to 5–15% in IPF (p<0.01), regardless of the arterial size (figure 2a (a) and b). In addition to the loss of junctional VE-cadherin in IPF patients, we also found an incremental increase in cytoplasmic VE-cadherin, which was fivefold that of cytoplasmic VE-cadherin in NCs across all arterial sizes (p<0.01) (figure 2a (b) and b (b)). A larger difference in cytoplasmic VE-cadherin in IPF and NCs was particularly noted for smaller arteries (range 100–199 µm) (p<0.001) compared to larger arteries. Interestingly, we also identified close to a twofold increase in junctional expression of N-cadherin cells across all arterial sizes (p<0.05) in IPF (figure 2a (c) and b (a)), although the cytoplasmic N-cadherin was only noticeable in IPF patients and was negligible in NCs (figure 2a (d) and b (b)).
FIGURE 2.

a) Per cent expression of VE-cadherin and N-cadherin positive cells in intimal layer in idiopathic pulmonary fibrosis (IPF) and normal control (NC) across arterial sizes 100–1000 µm: VE-cadherin junctional positive, VE-cadherin cytoplasmic positive, N-cadherin junctional positive and N-cadherin cytoplasmic positive. b) VE-cadherin and N-cadherin positive cells ratio in IPF and NC: VE-cadherin junctional positive versus N-cadherin junctional positive, and VE-cadherin cytoplasmic positive versus N-cadherin cytoplasmic positive. All data are presented as multiple comparisons with ordinary one-way ANOVA; p≤0.05 was considered significant. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001 was considered significant. NS: nonsignificant.
Per cent N-cadherin expression in IPF
There was a significant increase in N-cadherin expression across all arterial sizes in IPF compared to NCs. Total N-cadherin expression was considerably greater (four- to fivefold) in IPF compared to NCs across all arterial sizes (p<0.0001) (figure 3a). Furthermore, intimal and medial N-cadherin % expression revealed a greater fold change compared to that in the adventitial layer across the entire arterial range in IPF (figure 3b, c, d and e). We found that intimal and medial N-cadherin % expression was significantly higher across all classified arterial ranges, i.e., 100–1000 µm (p<0.0001) in IPF (figure 3b, c, d and e). It was greatest in the 200–399 µm and 400–599 µm (p<0.001) (figure 3c, d) compared to the 100–199 µm (p<0.01) and 600–1000 µm (p<0.05) arterial range (figure 3b, e).
FIGURE 3.

N-cadherin expression in normal control (NC) and idiopathic pulmonary fibrosis (IPF): a) total arterial N-cadherin expression across arterial sizes 100–1000 µm, b) 100–199 µm, c) 200–399 µm, d) 400–599 µm and e) 600–1000 µm. All data are presented as multiple comparisons with ordinary one-way ANOVA; p≤0.05 was considered significant. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001 was considered significant.
Vimentin % expression in IPF
The total vimentin percentage significantly increased throughout the arterial ranges in IPF compared to NCs (p<0.0001) (figure 4a). Vimentin expression in intima and media was significantly higher across all classified arterial ranges, i.e., 100–1000 µm (p<0.0001) in IPF (figure 4b, c, d and e). Increase in vimentin expression was also noted in the adventitial layer for all arterial sizes. This was highly significant for the arterial range of 200–599 µm (p<0.001) (figure 4b, c, d, e).
FIGURE 4.

Vimentin expression in normal control (NC) and idiopathic pulmonary fibrosis (IPF): a) total arterial vimentin expression across arterial sizes 100–1000 µm, b) 100–199 µm, c) 200–399 µm, d) 400–599 µm and e) 600–1000 µm. All data are presented as multiple comparisons with ordinary one-way ANOVA; p≤0.05 was considered significant. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001 was considered significant.
S100a4 % expression in IPF
Compared to NC, total percentage of staining for S100A4 was significantly higher in smaller and medium-sized arteries in IPF (100–199 µm (p<0.0001) and 200–399 µm (p<0.001)) (figure 5a). However, in larger arteries S100A4 expression was slightly higher in IPF (10–15%) compared to NCs (8–10%) (figure 5d and e). Intimal expression was significantly higher in smaller arteries measuring 199 µm (p<0.0001; figure 5b) compared to medium-sized arteries of 200–599 µm (p<0.001; figure 5c, d) in IPF. The medial and adventitial S100A4 expression did not change across arterial sizes. Overall, the smaller arteries showed more significant changes in S100A4 expression compared to medium- and large-sized arteries (figure 5b, c, d, f).
FIGURE 5.

S100A4 expression in normal control (NC) and idiopathic pulmonary fibrosis (IPF): a) total arterial vimentin expression across arterial sizes 100–1000 µm, b) 100–199 µm, c) 200–399 µm, d) 400–599 µm and e) 600–1000 µm. All data are presented as multiple comparisons with ordinary one-way ANOVA; p≤0.05 was considered significant. *p<0.05, ***p<0.001, ****p<0.0001 was considered significant. NS: nonsignificant.
Mesenchymal maker expression impacted vascular remodelling and lung physiology
There was a significant positive correlation between increased total N-cadherin and arterial thickening (r′=058, p=0.03) (figure 6a). Total vimentin expression showed a positive association (r′=0.37, p=0.13) with increased arterial thickness (figure 6b). Remarkably, intimal N-cadherin and vimentin expression significantly positively corelated to both intimal (r′=0.64, p=0.01) and adventitial thickness (r′=0.63, p=0.03) (table 2). Total N-cadherin and vimentin expression also showed a significant negative correlation with diffusing capacity of the lung for carbon monoxide (DLCO) % predicted r′= −0.66, p=0.01 and r′= −0.63, p=0.03, respectively (figure 6c, d). We also noted that increases in intimal N-cadherin and vimentin negatively correlated to DLCO % predicted (r′= −0.58, p=0.03; table 2).
FIGURE 6.

Correlations between a) total N-cadherin % expression versus total arterial thickness, b) total vimentin % expression versus total arterial thickness, c) total N-cadherin % expression versus % diffusing capacity of the lung for carbon monoxide (DLCO) and d) total vimentin % expression versus % DLCO.
TABLE 2.
Correlation analysis of mesenchymal markers versus arterial thickness and lung function
| Correlation analysis | Intimal N-cadherin | Intimal vimentin |
| Arterial thickness | ||
| Intima | r′=0.64 | r′=0.60 |
| p=0.01 | p=0.03 | |
| Media | r′=0.12 | r′=0.10 |
| p=0.36 | p=0.38 | |
| Adventitia | r′=0.28 | r′=0.25 |
| p=0.19 | p=0.22 | |
| Lung function | ||
| FEV1 L | r′= −0.40 | r′=0.05 |
| p=0.09 | p=0.44 | |
| FVC L | r′= −0.27 | r′=0.18 |
| p=0.20 | p=0.29 | |
| DLCO % | r′= −0.58 | r′= −0.56 |
| p=0.04 | p=0.03 |
p≤0.05 was considered significant. FEV1: forced expiratory volume in 1 s; FVC: forced vital capacity; DLCO: diffusing capacity of the lung for carbon monoxide.
Correlation between intimal junctional and cytoplasmic VE-cadherin expression and mesenchymal proteins
We identified an interesting relationship between junctional and cytoplasmic VE-cadherin expression and mesenchymal protein expression in the intimal layer across classified arteries. A negative correlation between VE-cadherin junctional and mesenchymal protein vimentin, N-cadherin and S100A4 was observed between smaller and larger arteries (supplementary figure S2a and d). However, the association between junctional VE-cadherin and intimal vimentin was significant in arteries of size 100–199 µm (r′= −0.85, p=0.001) (supplementary figure S2a), whereas a strong negative relationship was also seen in larger arteries of 600–1000 µm (r′= −0.88, p=0.31) (supplementary figure S2d). Furthermore, the association between junctional VE-cadherin and intimal S100A4 was also negative for all arterial sizes apart from 400–599 µm (supplementary figure S2a–d).
We also observed a consistent positive relationship trend between VE-cadherin cytoplasmic expression and intimal mesenchymal protein vimentin, N-cadherin and S100A4 in smaller and medium-sized arteries (supplementary figure S2a and b). However, we could not identify a comparable pattern in larger arterial sizes (supplementary figure S2c and d), which might be due to the high sample size variability observed between IPF and NCs.
CD4/CD8, mast cells, neutrophil, macrophage and pericyte cell expression in pulmonary arteries from patients with IPF compared to NCs
We found negative expressions of CD4, CD8, neutrophil elastase and CMA1 for mast cells in the arterial wall of patients with IPF. CD68 (macrophage) was the only inflammatory cell type with positive staining, but more so in the lumen of the artery or adventitial layer in IPF. Intimal endothelial cells were negative for CD68. In addition, we observed positive expression for PDGFR-β, which is a marker for pericytes. We found positive staining for pericytes in the intimal layer and the surrounding tissue, but endothelial cells were negative for this marker as shown in the inset in figure 7b. Staining for inflammatory markers and pericytes is shown in figure 7a and b.
FIGURE 7.

a) CD4, CD8 and mast cell chymase (CMA1) staining in arterial layers of normal control (NC) and patients with idiopathic pulmonary fibrosis (IPF). Images were taken at 40× magnification. b) Neutrophil elastase (NE), CD68 and PDGFR-β staining in arterial layers of NC and patients with IPF. Images were taken at 40× magnification, and the insert image was taken at 100× with a bright field.
Discussion
This study, to the best of our knowledge, is the first description of EndMT in the arterial vasculature of patients with IPF. We have provided both qualitative and quantitative data informing EndMT marker expression across different arterial sizes and individual layers. Our study demonstrated downregulation of junctional VE-cadherin and increased cytoplasmic VE-cadherin in patients with IPF compared with NCs. Furthermore, we showed upregulation of the mesenchymal proteins N-cadherin, vimentin and S100A4 across classified arteries in IPF. The mesenchymal expression was most prominent within the intima, suggesting that endothelial transformation via EndMT is an active process in the pulmonary arteries of patients with IPF. Strong co-relations between EndMT and arterial remodelling were observed; these included arterial thickness and ECM deposition. Moreover, we found EndMT to adversely influence lung physiological parameters, especially DLCO. Our findings suggest EndMT as a novel therapeutic target in IPF.
EndMT is a crucial process in developing and progressing fibrotic changes [10–12]. VE-cadherin is an endothelial cell-specific protein [20] that controls permeability and regulates cell motility [21]. Our study noted a significant reduction of VE-cadherin expression across all arterial sizes in IPF and a concurrent increase in cytoplasmic VE-cadherin at the intimal layer across all arterial sizes. Junctional to cytoplasmic VE-cadherin expression shift suggests that endothelial cell integrity is compromised in IPF favouring EndMT. Mammoto et al. [22] cultured human lung microvascular endothelial (L-HMVE) cells in fibronectin-coated polyacrylamide gels with different stiffness to investigate changes in mechanical interactions that could regulate vascular permeability. They observed that L-HMVE cells grown on more flexible substrates appeared round and VE-cadherin-containing cell–cell junctions were disrupted, and this was associated with increased VE-cadherin staining in the cytoplasm [22].
Further, parallel to VE-cadherin expression, we observed an increase in mesenchymal proteins vimentin, S100A4 and N-cadherin in the intimal layer of arteries. Previous IPF studies have detected loss of endothelial phenotypes and acquisition of mesenchymal phenotypes, but these were mainly animal studies [13, 23]. In addition, previous work has suggested that the proliferation of both endothelial and smooth muscle cells that undergo phenotypic changes via EndMT is linked to intimal remodelling and PH pathology [24]. Moreover, augmented mesenchymal protein expression noted in the medial and adventitial layer suggests resident cells may undergo mesenchymal phenotypic transformation.
N-cadherin is also expressed in mesenchymal cells, which promotes motility and invasion [25]. We also discovered the augmented expression of N-cadherin junctional positive and N-cadherin cytoplasmic positive cells in the intimal layer among IPF patients. Previously, upregulated N-cadherin expression in alveolar type II cells has been observed in IPF patients and is considered a potential prognostic indicator based on N-cadherin and Ki-67 expression correlation with histological disease activity [26]. Recently, Ferrell et al. [27] also demonstrated that retention of the N-cadherin prodomain at the cell surface is a potential biomarker of pathological myofibroblasts and is linked with fibrosis of tissues of the heart, lung and liver. Taken together, we believe that increase in cytoplasmic VE-cadherin could lead to the overall decrease in VE-cadherin and increase in N-cadherin indicating the process of EndMT, which drives intimal remodelling changes in IPF [1, 3]. Augmented mesenchymal protein expression noted in the medial and adventitial layer suggests resident cells may undergo mesenchymal phenotypic transformation.
The remodelling changes in medial and adventitial layers could be due to mesenchymal transformation of the smooth muscle cells and the adventitial fibroblast [28, 29]. N-cadherin was found to be abundantly expressed in both media and adventitia in IPF compared with the healthy control group. Moreover, the correlation between N-cadherin expression and vascular remodelling changes such as arterial thickness confirms increased N-cadherin expression is associated with increased individual layer thickness, especially intimal thickness. Thus, we suggest that targeting N-cadherin could be a potential strategy to reduce vascular remodelling, as suggested by earlier studies [30, 31].
Vimentin, a cytoskeletal protein that is prevalent in cells of mesenchymal origin, and its overexpression has been linked to increased invasiveness and excessive scarring [32]. Intriguingly, our study also discovered the augmented expression of vimentin at intimal, medial and adventitial layers for different arterial sizes in IPF. Several studies have noted vimentin overexpression in IPF lungs [33–37] with high levels of vimentin in IPF lung tissue related to disease progression [38]. Moreover, a positive correlation between total vimentin expression and arterial thickness suggests they play a role in arterial remodelling in IPF. Additionally, we observed that intimal vimentin expression influenced individual layer thickness, especially the intimal thickness.
S100A4 belongs to the small Ca2+-binding protein family, and it modulates cellular biological functions, such as cell motility, proliferation and metastasis [39–41]. S100A4 causes lung fibrosis by promoting the proliferation and activation of fibroblasts [42–44], and increased S100A4 has been proposed to contribute to the development of IPF [45–48]. S100A4 expression was significantly higher in the intimal layer across all arterial sizes; however, we did not find such a significant expression in medial and adventitial layers.
We next investigated the relationship between VE-cadherin junctional and cytoplasmic positive expression and mesenchymal protein expression, which suggested that upregulated VE-cadherin cytoplasmic expression is associated with increased mesenchymal protein expression, especially in case of smaller and medium-sized arteries. However, larger arteries did not show similar changes, possibly due to the smaller number of larger arteries observed in fibrotic IPF lung tissue. This trend suggests increased cytoplasmic VE-cadherin expression in endothelial cells could indicate active endothelial proliferation and transition.
In assessing the impact of EndMT on lung function, a correlation between expression of mesenchymal markers and lung function parameter DLCO was performed. The correlations suggested that the increase in N-cadherin and vimentin expression could negatively impact lung gas exchange capacity (DLCO) in IPF patients, indicating PH complications. Furthermore, the reduced DLCO in IPF patients has been considered as an indication of concomitant PH to the underlying pulmonary fibrosis [49]. Our findings fit with this theory and suggested pathology.
To further confirm our EndMT findings and cellular expression of mesenchymal markers, we also stained for inflammatory cell populations and pericytes. We found that pulmonary arteries were negative for mast cells, CD4/CD8 and neutrophils. Some positivity was observed for macrophages (CD68+), but mainly in the lumen or adventitial tissue. Endothelial cells were negative for CD68. We also found positive staining for pericytes, but intimal endothelial cells were negative for PDGFR-β. This shows that endothelial cells staining for mesenchymal markers are not inflammatory cells but in fact the cells undergoing EndMT. Pericytes could also be contributing to arterial remodelling in IPF in addition to endothelial cells, but this warrants further work.
One limitation of this study is that the number of IPF and NC samples varied for size-based comparison, as some arterial sizes were not observed in IPF. Another limitation is the unavailability of cardiac function data or a clinical diagnosis of PH for the IPF patients studied; hence, the associations between EndMT and PH could not be established. However, the positive correlation of EndMT with vascular remodelling and negative correlation with DLCO indicates possible PH in IPF patients. Further work is needed to confirm these associations. Future studies are needed to further explore these histopathological findings with relationships to transcriptomics [50].
In summary, endothelial cells acquire mesenchymal traits through EndMT in IPF patients. Significant increased mesenchymal expression in arterial layers potentially contributes to remodelling of muscular arteries and drives physiological changes, such as deficient oxygen absorption. Hence, our study data validated the crucial role of EndMT in IPF pathology and suggest EndMT as a novel therapeutic target for preventing PH and lung fibrosis in general.
Supplementary material
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Figure S1 00487-2022.SUPPLEMENT (1.1MB, jpg)
{kind=link}
Figure S2 00487-2022.SUPPLEMENT2 (660.1KB, jpg)
{kind=link}
Footnotes
Provenance: Submitted article, peer reviewed.
Conflict of interest: S.S. Sohal reports personal fees for lectures from Chiesi, travel support from Chiesi and AstraZeneca, and a research grant from Boehringer Ingelheim outside the submitted work. S.S. Sohal is on the Small Airway Advisory Board for Chiesi Australia.
Conflict of interest: All the other authors have no conflict of interest to declare.
Support statement: This work was supported by research grants from Clifford Craig Foundation Launceston General Hospital and Lung Foundation Australia. Funding information for this article has been deposited with the Crossref Funder Registry.
References
- 1.Gaikwad AV, Eapen MS, McAlinden KD, et al. Endothelial to mesenchymal transition (EndMT) and vascular remodeling in pulmonary hypertension and idiopathic pulmonary fibrosis. Expert Rev Respir Med 2020; 14: 1027–1043. doi: 10.1080/17476348.2020.1795832 [DOI] [PubMed] [Google Scholar]
- 2.Richeldi L, Collard HR, Jones MG. Idiopathic pulmonary fibrosis. Lancet 2017; 389: 1941–1952. doi: 10.1016/S0140-6736(17)30866-8 [DOI] [PubMed] [Google Scholar]
- 3.Gaikwad AV, Lu W, Dey S, et al. Vascular remodelling in IPF patients and its detrimental effect on lung physiology: potential role of endothelial to mesenchymal transition (EndMT). ERJ Open Res 2022; 8: 00571-02021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nathan SD, Shlobin OA, Ahmad S, et al. Serial development of pulmonary hypertension in patients with idiopathic pulmonary fibrosis. Respiration 2008; 76: 288–294. doi: 10.1159/000114246 [DOI] [PubMed] [Google Scholar]
- 5.Farkas L, Gauldie J, Voelkel NF, et al. Pulmonary hypertension and idiopathic pulmonary fibrosis: a tale of angiogenesis, apoptosis, and growth factors. Am J Respir Cell Mol Biol 2011; 45: 1–15. doi: 10.1165/rcmb.2010-0365TR [DOI] [PubMed] [Google Scholar]
- 6.Nathan SD, Barbera JA, Gaine SP, et al. Pulmonary hypertension in chronic lung disease and hypoxia. Eur Respir J 2019; 53: 1801914. doi: 10.1183/13993003.01914-2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lettieri CJ, Nathan SD, Barnett SD, et al. Prevalence and outcomes of pulmonary arterial hypertension in advanced idiopathic pulmonary fibrosis. Chest 2006; 129: 746–752. doi: 10.1378/chest.129.3.746 [DOI] [PubMed] [Google Scholar]
- 8.Kinoshita T, Goto T. Molecular mechanisms of pulmonary fibrogenesis and its progression to lung cancer: a review. Int J Mol Sci 2019; 20: 1461. doi: 10.3390/ijms20061461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wynn TA. Integrating mechanisms of pulmonary fibrosis. J Exp Med 2011; 208: 1339–1350. doi: 10.1084/jem.20110551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ursoli Ferreira F, Eduardo Botelho Souza L, Hassibe Thomé C, et al. Endothelial cells tissue-specific origins affects their responsiveness to TGF-β2 during endothelial-to-mesenchymal transition. Int J Mol Sci 2019; 20: 458. doi: 10.3390/ijms20030458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lv Z, Wang Y, Liu Y-J, et al. NLRP3 inflammasome activation contributes to mechanical stretch–induced endothelial-mesenchymal transition and pulmonary fibrosis. Crit Care Med 2018; 46: e49–e58. doi: 10.1097/CCM.0000000000002799 [DOI] [PubMed] [Google Scholar]
- 12.Piera-Velazquez S, Mendoza FA, Jimenez SA. Endothelial to mesenchymal transition (EndoMT) in the pathogenesis of human fibrotic diseases. J Clin Med 2016; 5: 45. doi: 10.3390/jcm5040045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hashimoto N, Phan SH, Imaizumi K, et al. Endothelial–mesenchymal transition in bleomycin-induced pulmonary fibrosis. Am J Respir Cell Mol Biol 2010; 43: 161–172. doi: 10.1165/rcmb.2009-0031OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Choi SH, Hong ZY, Nam JK, et al. A hypoxia-induced vascular endothelial-to-mesenchymal transition in development of radiation-induced pulmonary fibrosis. Clin Cancer Res 2015; 21: 3716–3726. doi: 10.1158/1078-0432.CCR-14-3193 [DOI] [PubMed] [Google Scholar]
- 15.Rieder F, Kessler SP, West GA, et al. Inflammation-induced endothelial-to-mesenchymal transition: a novel mechanism of intestinal fibrosis. Am J Pathol 2011; 179: 2660–2673. doi: 10.1016/j.ajpath.2011.07.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yoshimatsu Y, Watabe T. Roles of TGF-β signals in endothelial-mesenchymal transition during cardiac fibrosis. Int J Inflam 2011; 2011: 724080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eapen MS, Lu W, Gaikwad AV, et al. Endothelial to mesenchymal transition: a precursor to post-COVID-19 interstitial pulmonary fibrosis and vascular obliteration? Eur Respir J 2020; 56: 2003167. doi: 10.1183/13993003.03167-2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sanchez-Duffhues G, Orlova V, ten Dijke P. In Brief: Endothelial-to-mesenchymal transition. J Pathol 2016; 238: 378–380. doi: 10.1002/path.4653 [DOI] [PubMed] [Google Scholar]
- 19.Bhattarai P, Lu W, Gaikwad AV, et al. Arterial remodelling in smokers and in patients with small airway disease and COPD: implications for lung physiology and early origins of pulmonary hypertension. ERJ Open Res 2022; 8: 00254-2022. doi: 10.1183/23120541.00254-2022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ampugnani MG, Resnati M, Raiteri M, et al. A novel endothelial-specific membrane protein is a marker of cell-cell contacts. J Cell Biol 1992; 118: 1511–1522. doi: 10.1083/jcb.118.6.1511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vestweber D, Winderlich M, Cagna G, et al. Cell adhesion dynamics at endothelial junctions: VE-cadherin as a major player. Trends Cell Biol 2009; 19: 8–15. doi: 10.1016/j.tcb.2008.10.001 [DOI] [PubMed] [Google Scholar]
- 22.Mammoto A, Mammoto T, Kanapathipillai M, et al. Control of lung vascular permeability and endotoxin-induced pulmonary oedema by changes in extracellular matrix mechanics. Nat Commun 2013; 4: 1759. doi: 10.1038/ncomms2774 [DOI] [PubMed] [Google Scholar]
- 23.Jia W, Wang Z, Gao C, et al. Trajectory modeling of endothelial-to-mesenchymal transition reveals galectin-3 as a mediator in pulmonary fibrosis. Cell Death Dis 2021; 12: 327. doi: 10.1038/s41419-021-03603-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ghigna M-R, Dorfmüller P. Pulmonary vascular disease and pulmonary hypertension. Diagn Histopathol 2019; 25: 304–312. doi: 10.1016/j.mpdhp.2019.05.002 [DOI] [Google Scholar]
- 25.Wheelock MJ, Johnson KR. Cadherins as modulators of cellular phenotype. Annu Rev Cell Dev Biol 2003; 19: 207–235. doi: 10.1146/annurev.cellbio.19.011102.111135 [DOI] [PubMed] [Google Scholar]
- 26.Lomas NJ, Watts KL, Akram KM, et al. Idiopathic pulmonary fibrosis: immunohistochemical analysis provides fresh insights into lung tissue remodelling with implications for novel prognostic markers. Int J Clin Exp Pathol 2012; 5: 58–71. [PMC free article] [PubMed] [Google Scholar]
- 27.Ferrell PD, Oristian KM, Cockrell E, et al. Pathologic proteolytic processing of N-cadherin as a marker of human fibrotic disease. Cells 2022; 11: 156. doi: 10.3390/cells11010156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kuwabara JT, Tallquist MD. Tracking adventitial fibroblast contribution to disease: a review of current methods to identify resident fibroblasts. Arterioscler Thromb Vasc Biol 2017; 37: 1598–1607. doi: 10.1161/ATVBAHA.117.308199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stenmark KR, Davie N, Frid M, et al. Role of the adventitia in pulmonary vascular remodeling. Physiology (Bethesda) 2006; 21: 134–145. [DOI] [PubMed] [Google Scholar]
- 30.Jones M, Sabatini PJB, Lee FSH, et al. N-Cadherin upregulation and function in response of smooth muscle cells to arterial injury. Arterioscler Thromb Vasc Biol 2002; 22: 1972–1977. doi: 10.1161/01.ATV.0000036416.14084.5A [DOI] [PubMed] [Google Scholar]
- 31.Lyon CA, Koutsouki E, Aguilera CM, et al. Inhibition of N-cadherin retards smooth muscle cell migration and intimal thickening via induction of apoptosis. J Vasc Surg 2010; 52: 1301–1309. doi: 10.1016/j.jvs.2010.05.096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li H, Chang L, Du WW, et al. Anti-microRNA-378a enhances wound healing process by upregulating integrin beta-3 and vimentin. Mol Ther 2014; 22: 1839–1850. doi: 10.1038/mt.2014.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang Y, Fujita J, Bandoh S, et al. Detection of antivimentin antibody in sera of patients with idiopathic pulmonary fibrosis and non-specific interstitial pneumonia. Clin Exp Immunol 2002; 128: 169–174. doi: 10.1046/j.1365-2249.2002.01811.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Felton VM, Borok Z, Willis BC. N-acetylcysteine inhibits alveolar epithelial-mesenchymal transition. Am J Physiol Lung Cell Mol Physiol 2009; 297: L805–L812. doi: 10.1152/ajplung.00009.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pandit KV, Corcoran D, Yousef H, et al. Inhibition and role of let-7d in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2010; 182: 220–229. doi: 10.1164/rccm.200911-1698OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu G, Philp AM, Corte T, et al. Therapeutic targets in lung tissue remodelling and fibrosis. Pharmacol Ther 2021; 225: 107839. doi: 10.1016/j.pharmthera.2021.107839 [DOI] [PubMed] [Google Scholar]
- 37.Zhang D, Zhuang R, Li J, et al. MicroSPECT imaging-guided treatment of idiopathic pulmonary fibrosis in mice with a vimentin-targeting 99mTc-labeled N-acetylglucosamine-polyethyleneimine. Mol Pharm 2021; 18: 4140–4147. doi: 10.1021/acs.molpharmaceut.1c00545 [DOI] [PubMed] [Google Scholar]
- 38.Surolia R, Li FJ, Wang Z, et al. Vimentin intermediate filament assembly regulates fibroblast invasion in fibrogenic lung injury. JCI Insight 2019; 4: e123253. doi: 10.1172/jci.insight.123253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Grigorian M, Andresen S, Tulchinsky E, et al. Tumor suppressor p53 protein is a new target for the metastasis-associated Mts1/S100A4 protein: functional consequences of their interaction. J Biol Chem 2001; 276: 22699–22708. doi: 10.1074/jbc.M010231200 [DOI] [PubMed] [Google Scholar]
- 40.Boye K, Mælandsmo GM. S100A4 and metastasis: a small actor playing many roles. Am J Pathol 2010; 176: 528–535. doi: 10.2353/ajpath.2010.090526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li Z-H, Bresnick AR. The S100A4 metastasis factor regulates cellular motility via a direct interaction with myosin-IIA. Cancer Res 2006; 66: 5173–5180. doi: 10.1158/0008-5472.CAN-05-3087 [DOI] [PubMed] [Google Scholar]
- 42.Xia H, Gilbertsen A, Herrera J, et al. Calcium-binding protein S100A4 confers mesenchymal progenitor cell fibrogenicity in idiopathic pulmonary fibrosis. J Clin Invest 2017; 127: 2586–2597. doi: 10.1172/JCI90832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li Y, Bao J, Bian Y, et al. S100A4(+) macrophages are necessary for pulmonary fibrosis by activating lung fibroblasts. Front Immunol 2018; 9: 1776. doi: 10.3389/fimmu.2018.01776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang W, Ohno S, Steer B, et al. S100a4 is secreted by alternatively activated alveolar macrophages and promotes activation of lung fibroblasts in pulmonary fibrosis. Front Immunol 2018; 9: 1216. doi: 10.3389/fimmu.2018.01216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee J-U, Chang HS, Shim E-Y, et al. The S100 calcium-binding protein A4 level is elevated in the lungs of patients with idiopathic pulmonary fibrosis. Respir Med 2020; 171: 105945. doi: 10.1016/j.rmed.2020.105945 [DOI] [PubMed] [Google Scholar]
- 46.Li Z, Li Y, Liu S, et al. Extracellular S100A4 as a key player in fibrotic diseases. J Cell Mol Med 2020; 24: 5973–5983. doi: 10.1111/jcmm.15259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kagimoto A, Tsutani Y, Kushitani K, et al. Serum S100 calcium-binding protein A4 as a novel predictive marker of acute exacerbation of interstitial pneumonia after surgery for lung cancer. BMC Pulm Med 2021; 21: 186. doi: 10.1186/s12890-021-01554-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Qin H-Y, Li M-D, Xie G-F, et al. Associations among S100A4, Sphingosine-1–Phosphate, and pulmonary function in patients with chronic obstructive pulmonary disease. Oxid Med Cell Longev 2022; 2022: 6041471. doi: 10.1155/2022/6041471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ruffenach G, Hong J, Vaillancourt M, et al. Pulmonary hypertension secondary to pulmonary fibrosis: clinical data, histopathology and molecular insights. Respir Res 2020; 21: 303. doi: 10.1186/s12931-020-01570-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ackermann M, Stark H, Neubert L, et al. Morphomolecular motifs of pulmonary neoangiogenesis in interstitial lung diseases. Eur Respir J 2020; 55: 1900933. doi: 10.1183/13993003.00933-2019 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Figure S1 00487-2022.SUPPLEMENT (1.1MB, jpg)
Figure S2 00487-2022.SUPPLEMENT2 (660.1KB, jpg)