

Abstract
Objective:
We evaluated the hypothesis that the development of renal dysfunction and congestive heart failure (CHF) caused by volume overload in rats with angiotensin II (ANG II)-dependent hypertension is associated with altered renal vascular responsiveness to ANG II and to epoxyeicosatrienoic acids (EETs).
Methods:
Ren-2 transgenic rats (TGRs) were used as a model of ANG II-dependent hypertension. CHF was induced by volume overload achieved by the creation of the aorto-caval fistula (ACF). Renal blood flow (RBF) responses were determined to renal arterial administration of ANG II, native 11,12-EET, an analog of 14,15-EETs (EET-A), norepinephrine (NE), acetylcholine (Ach) and bradykinin (Bk) in healthy (i.e., sham-operated) TGR and ACF TGR (5 weeks after ACF creation).
Results:
Selective intrarenal administration of neither vasoactive drug altered mean arterial pressure in any group. Administration of ANG II caused greater decreases in RBF in ACF TGR than in sham-operated TGR, whereas after administration of NE the respective decreases were comparable in the 2 groups. Administration of Ach and Bk elicited significantly higher RBF increases in ACF TGR as compared with sham-operated TGR. In contrast, administration of 11,12-EET and EET-A caused significantly smaller RBF increases in ACF TGR than in sham-operated TGR.
Conclusion:
The findings show that 5 weeks after creation of ACF, the TGR exhibit exaggerated renal vasoconstrictor responses to ANG II and reduced renal vasodilatory responses to EETs, suggesting that both these alterations might play an important role in the development of renal dysfunction in this model of CHF.
Keywords: Congestive heart failure, Hypertension, Aorto-caval fistula, Renal blood flow, Renal dysfunction, Renal vascular reactivity, Angiotensin II, Epoxyeicosatrienoic acid, Norepinephrine, Acetylcholine, Bradykinin
Introduction
Congestive heart failure (CHF) is a major public health problem affecting currently 4% of the adult population in Europe [1]. In spite of an array of therapeutic approaches available, the prognosis is still gloomy: the patients’ survival rate, especially of those with CHF accompanied by impairment of renal hemodynamics, is worse than that in most types of common cancers [2–8]. Notably, it was proposed that hypertension and renal dysfunction are 2 most important independent risk factors for the progression of CHF [9–13]. Unfortunately, current pharmacological regimes usually fail to prevent renal dysfunction in patients with CHF [2, 5, 8, 12]. Evidently, new pharmacological measures targeting renal dysfunction are needed. However, the prerequisite here is a better understanding of the mechanism(s) underlying the development of renal dysfunction in CHF, particularly in originally hypertensive individuals. This is still to come and appropriate focused experimental studies are needed to achieve this goal.
In this context, it should be noted that reduced renal blood flow (RBF) is a very common finding in individuals with CHF, and it is detected at its early stage [12, 14–16]. Adequate and stable perfusion of the kidneys is essential for normal renal function [17, 18], and the RBF decrease is indeed the first warning sign of the development of serious renal dysfunction in CHF [12, 14–16]. Since the activation of the renin-angiotensin-aldosterone system (RAAS) is one of the earliest neurohormonal responses in CHF and has a critical role in its pathophysiology [6, 8, 12, 16, 19–24], the research has been focused on the role of the RAAS in RBF control: there is evidence, even though not entirely conclusive, that impairment of renal hemodynamics in CHF critically depends on intrarenal angiotensin II (ANG II) [25–27]. Perhaps even more likely, altered intrarenal interaction of ANG II with other vasoactive neurohormonal factors might be decisive for RBF reduction and renal dysfunction [7, 9, 22, 23, 28–33]. This notion has been further supported by our recent finding that in the rat model of CHF induced by chronic volume overload due to the creation of the aorto-caval fistula (ACF), the renal vascular responsiveness to ANG II was not enhanced, whereas the rats exhibited augmented renal vascular responsiveness to norepinephrine (NE) [34]. However, this study was performed in CHF rats that were normotensive and before the onset of CHF displayed normal endogenous RAAS activity [34]. Thus, it is possible that in hypertensive rats the renal hemodynamics alterations might still be dependent on intrarenal ANG II actions.
The Ren-2 transgenic rat (TGR) model combines the endogenous activation of the RAAS and hypertension [35, 36], the 2 well-recognized critically important factors promoting the progression of CHF. We found recently that CHF decompensation develops within 5 weeks in ACF TGR but not in normotensive transgene-negative, Hannover Sprague-Dawley (HanSD) rats [37, 38]. In addition, we found that increasing renal tissue concentrations of epoxyeicosatrienoic acids (EETs), renal vasodilatory and natriuretic agents that are produced by cytochrome P-450-dependent epoxygenase pathway of arachidonic acid metabolism, attenuated renal dysfunction and delayed the progression of CHF in ACF TGR but not in ACF HanSD rats [37, 38]. Considering all that knowledge, we hypothesized that the development of renal dysfunction and CHF caused by volume overload in rats with ANG II-dependent hypertension is associated with altered renal vascular responsiveness to ANG II and to EETs.
To test this hypothesis, we compared the renal vascular responsiveness to intrarenal administration of ANG II and EETs in sham-operated (without ACF) TGR and HanSD rats and also in ACF TGR and ACF HanSD rats. To find out if the assumed altered renal vascular responsiveness to ANG II and EETs in ACF TGR is specific or simply represents altered reactivity to diverse endogenous vasoconstrictors and vasodilators, RBF’s responses to NE, acetylcholine (Ach) and bradykinin (Bk) were also examined. Moreover, to gain a better insight whether possible differences in the renal vascular responsiveness to vasoconstrictor and vasodilator agents are not modified by different degree of compensatory activation of neurohormonal systems, plasma concentrations ANG II, NE, ANG-1–7 (ANG 1–7) and aldosterone and kidney concentrations of NE, ANG II and ANG 1–7 were determined in TGR and HanSD rats and also in ACF TGR and ACF HanSD rats.
Methods
Ethical Approval, Animals, CHF Model, Chemicals
The studies were performed in accordance with the guidelines and practices established by the Animal Care and Use Committee of the Institute for Clinical and Experimental Medicine, Prague, which accord with the European Convention on Animal Protection and Guidelines on Research Animal Use and was approved by this committee and consequently by the Ministry of Health of the Czech Republic (the decision number for this project is 26899/2017–3/OZV issued by Ministry of Health of the Czech Republic). All animals used in the present study were bred at the Center of Experimental Medicine of this Institute, which is accredited by the Czech Association for Accreditation of Laboratory Animal Care. Heterozygous TGR were generated by breeding male homozygous TGR with female homozygous transgene-negative normotensive HanSD rats and age-matched HanSD rats served as controls. The animals were kept on a 12-/12-h light/dark cycle. Throughout the experiments rats were fed a normal salt, normal protein diet (0.45% NaCl, 19–21% protein) manufactured by SEMED (Prague, Czech Republic) and had free access to tap water.
Male TGR and HanSD rats, at the initial age of 8 weeks, derived from several litters, were randomly assigned to experimental groups to make sure that the animals from a single litter did not prevail in any group. Rats were anesthetized (tiletamine + zolazepam, Virbac SA, Carros Cedex, France, 8 mg/kg; and xylasine, Spofa, Czech Republic, 4 mg/kg intramuscularly) and CHF was induced by volume overload caused by the ACF created using needle technique as originally described by Garcia and Diebold [39]) and employed and validated by many investigators including our own group [34, 37–46]. After exposure of the abdominal aorta and inferior caval vein between the renal arteries and iliac bifurcation, the aorta was temporarily occluded at this segment for about 40 s. An 18-gauge needle (diameter 1.2 mm) was inserted into the abdominal aorta and advanced across its wall into the inferior caval vein to create ACF. Thereafter, the needle was withdrawn and the puncture site was sealed with cyanoacrylate tissue glue. The successful creation of the ACF was confirmed by the inspection of pulsatile flow of oxygenated blood from the abdominal aorta into the caval vein. Sham-operated rats underwent an identical procedure but without creating ACF. This is a model with many features similar to untreated human CHF; its additional advantage is that the onset of the compensated and decompensated phase of CHF is here precisely defined and consistent [38, 40, 41, 43, 44]. We have recently repeatedly demonstrated that in heterozygous TGR the onset of the decompensated phase of CHF occurs 5 weeks after ACF induction and at this time HanSD rats are still definitely in the phase of compensated CHF [37, 38, 40, 41]. This knowledge provides us the rationale to study the animals (sham or ACF groups) 5 weeks after operation.
ANG II, NE, Ach, and Bk were purchased from the Sigma Aldrich company (Prague, Czech Republic). The native 11,12-EET was purchased from Cayman Pharma (Neratovice, Czech Republic) and the analog 14,15-EET (EET-A) was developed and synthesized by the Professor Falck’s group. The chemical structure of EET-A is described in our previous studies [45, 46].
Preparations for Acute Studies of Renal Vascular Responses to Vasoactive Agents
The rats were anesthetized with thiopental sodium (80 mg/kg i.p.) and placed on a heated surgical table to maintain body temperature at 37 °C. Tracheostomy was performed to maintain patent airways, and the external end of the tracheal cannula was placed inside a small plastic chamber into which humidified 95% oxygen/5% carbon dioxide mixture was continuously delivered. The right jugular vein was catheterized with PE-50 tubing for the infusion of solutions. The right femoral artery was cannulated for arterial blood pressure monitoring. Mean arterial pressure (MAP) was monitored using a pressure transducer and recorded using a computerized data acquisition system (PowerLab, ADInstruments, UK). The left kidney was exposed via a flank incision, isolated from the surrounding tissue and placed in a lucite cup. For selective intrarenal drug administration, a tapered PE-10 catheter was inserted into the aorta via the left femoral artery and passed 1–2 mm down the left renal artery. This catheter was kept patent by a continuous infusion of heparinized saline at a rate of 2 μL/min throughout the experiment. During the surgery, animals received an intravenous infusion of saline containing 6% bovine serum albumin (Sigma Chemical Co., Prague, Czech Republic) at a rate of 20 μL/min. With the surgery completed, saline solution was infused to compensate for fluid losses. An ultrasonic transient-time flow probe (1RB, Transonic Systems, Altron Medical Electronic GmbH, Germany), connected to a Transonic flowmeter, was placed on the left renal artery and RBF was continuously recorded. On completion of surgery, a 45-min equilibration period was allowed. The protocol consisted of evaluating MAP and RBF responses to intrarenal bolus doses of vasoactive agents. Vasoactive agents were loaded in a small volume (20 μL) to a Cheminert valve and then rapidly infused into the animal by a bolus of saline (150 μL) at a rate of 90 μL/min. Two different doses of each vasoactive agent were administrated in random order. The changes in RBF and MAP were expressed as a percent of baseline values and the baseline values were always assessed separately for each administration. This experimental approach was employed and validated by previous studies, including ours [34, 47–49].
Evaluation of the Indices of the RAAS and Sympathetic Nervous System Activity
Since it is now recognized that plasma and tissue concentrations of ANG II measured under anesthesia are higher than those in conscious rats after decapitation, and there are also marked differences in renin secretion in response to anesthesia and surgery [36, 50–52], in this study ANG II, ANG 1–7, aldosterone and catecholamine concentrations were measured in samples from decapitated animals. The detailed description of the methodology for measurement of the concentration of RAAS components and of catecholamines, as routinely employed in our laboratory, was given previously [52–54].
Experimental Design
The rats were prepared as described above and RBF responses to 2 intrarenal doses of ANG II (2 and 8 ng), NE (6 and 20 ng), Ach (20 and 60 ng), Bk (100 and 300 ng), native 11,12-EET (300 and 600 ng), and EET-A (400 and 800 ng) were determined in the following experimental groups (n = 9 in each group):
Sham-operated HanSD rats 5 weeks after surgery
ACF HanSD rats 5 weeks after ACF creation
Sham-operated TGR 5 weeks after surgery
ACF TGR 5 weeks after ACF creation
Separate groups of animals (n = 7 in each group) were used to evaluate plasma and kidney ANG II, ANG 1–7 and aldosterone levels as the markers of the RAAS along with determination the degree of sympathorenal activation assessed by plasma and kidney concentrations of NE, epinephrine and dopamine.
Statistical Analysis
All values are expressed as mean ± SEM. Statistical analysis of the data was performed using Graph-Pad Prism software (Graph Pad Software, San Diego, CA, USA) employing one-way ANOVA followed by Student-Newman-Keuls test where appropriate. The values exceeding 95% probability limits (p < 0.05) were considered statistically significant.
Results
Figure 1A shows that plasma ANG II levels were significantly higher in sham-operated TGR than corresponding levels in sham-operated HanSD rats. The creation of ACF resulted in significant increases in plasma ANG II concentrations in both groups of rats. Moreover, plasma ANG II concentrations were significantly higher in ACF TGR than corresponding values in ACF HanSD rats.
Fig. 1.
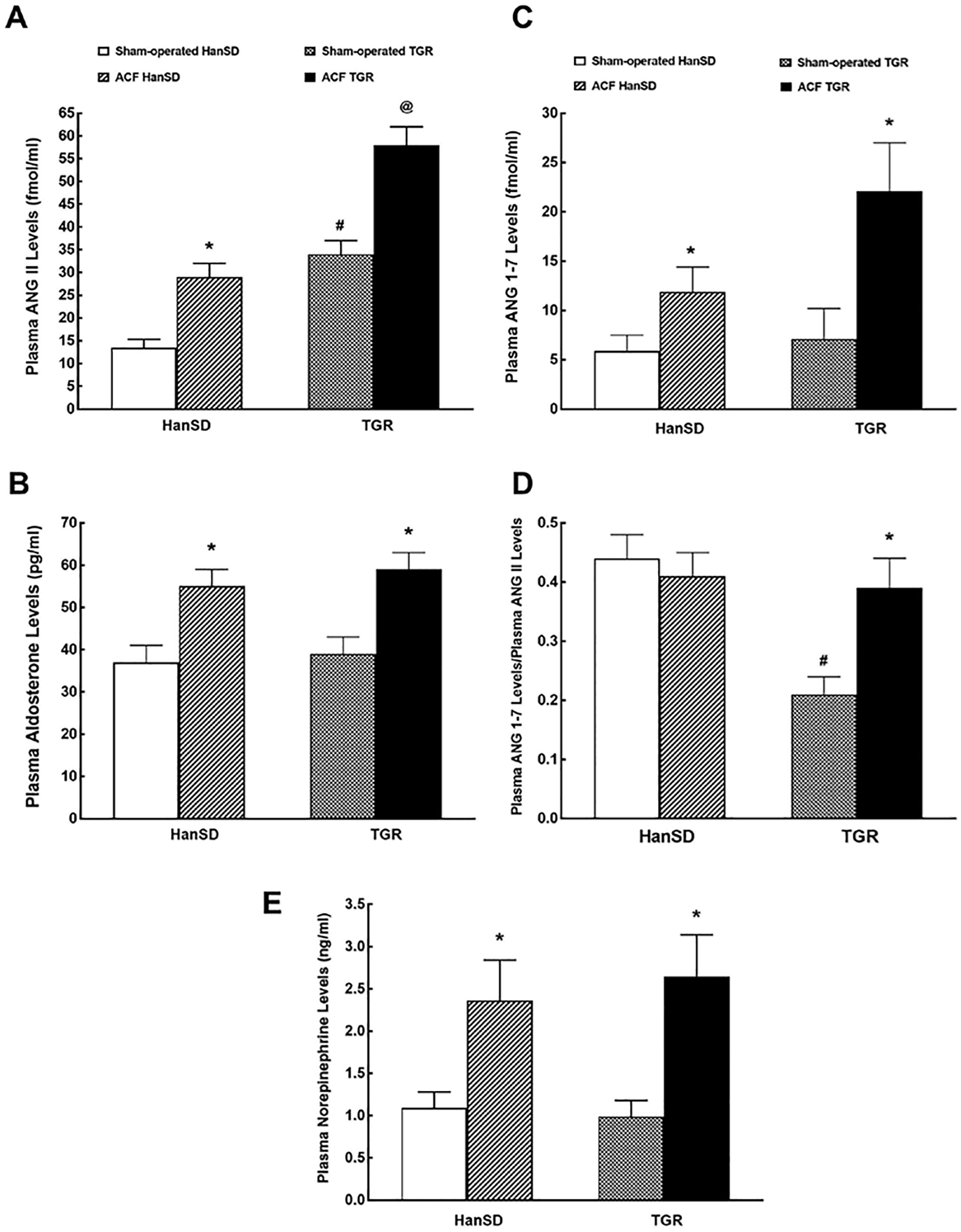
Plasma ANG II (A), plasma aldosterone (B), plasma ANG-1–7 (ANG 1–7; C) levels, plasma ANG 1–7 to ANG II ratio (D) and plasma NE (E) levels in sham-operated transgene-negative HanSD, sham-operated heterozygous Ren-2 TGRs and HanSD and TGR rats with ACF 5 weeks after the creation of ACF or sham-operation. * p < 0.05 versus sham-operated counterparts. # p < 0.05 versus sham-operated HanSD rats. @p < 0.05 versus all other groups. All the values shown are means ± SEM. ACF, aorto-caval fistula; TGR, transgenic rat; HanSD, Hannover Sprague-Dawley; ANG II, angiotensin II.
As shown in Figure 1B, there was no significant difference in plasma aldosterone level between sham-operated HanSD rats and sham-operated TGR. The creation of ACF caused similar significant increases in plasma aldosterone levels in TGR and HanSD rats.
Figure 1C shows that there was no significant difference in plasma ANG 1–7 concentrations between sham-operated HanSD rats and sham-operated TGR. The creation of ACF resulted in significant increases in plasma ANG 1–7 levels in TGR and HanSD rats. While the level tended to be higher in ACF TGR than in ACF HanSD group, the difference did not reach statistical significance.
Data shown in Figure 1D help to assess the state of the systemic balance between the vasodilator and vasoconstrictor axes of the RAAS expressed as the ratio of ANG 1–7 to ANG II. This ratio has been validated in previous studies, including our own [52]. It is shown here that in sham-operated TGR, the ratio is only one half of that in sham-operated HanSD rats. The creation of ACF did not alter the ratio in ACF HanSD rats but resulted in its marked increase in ACF TGR, up to the value observed in sham-operated and ACF HanSD rats.
As shown in Figure 1E, there was no significant difference in plasma NE levels between sham-operated HanSD rats and sham-operated TGR. The creation of ACF resulted in similar increases in plasma NE levels in either group. Plasma concentrations of epinephrine and dopamine showed a similar pattern (data not shown).
As shown in Figure 2A, whole kidney ANG II concentrations were more than twice higher in sham-operated TGR than in sham-operated HanSD rats, and the creation of ACF resulted in marked ANG II increases in both groups, but the concentration was significantly higher in ACF TGR than that in ACF HanSD rats.
Fig. 2.
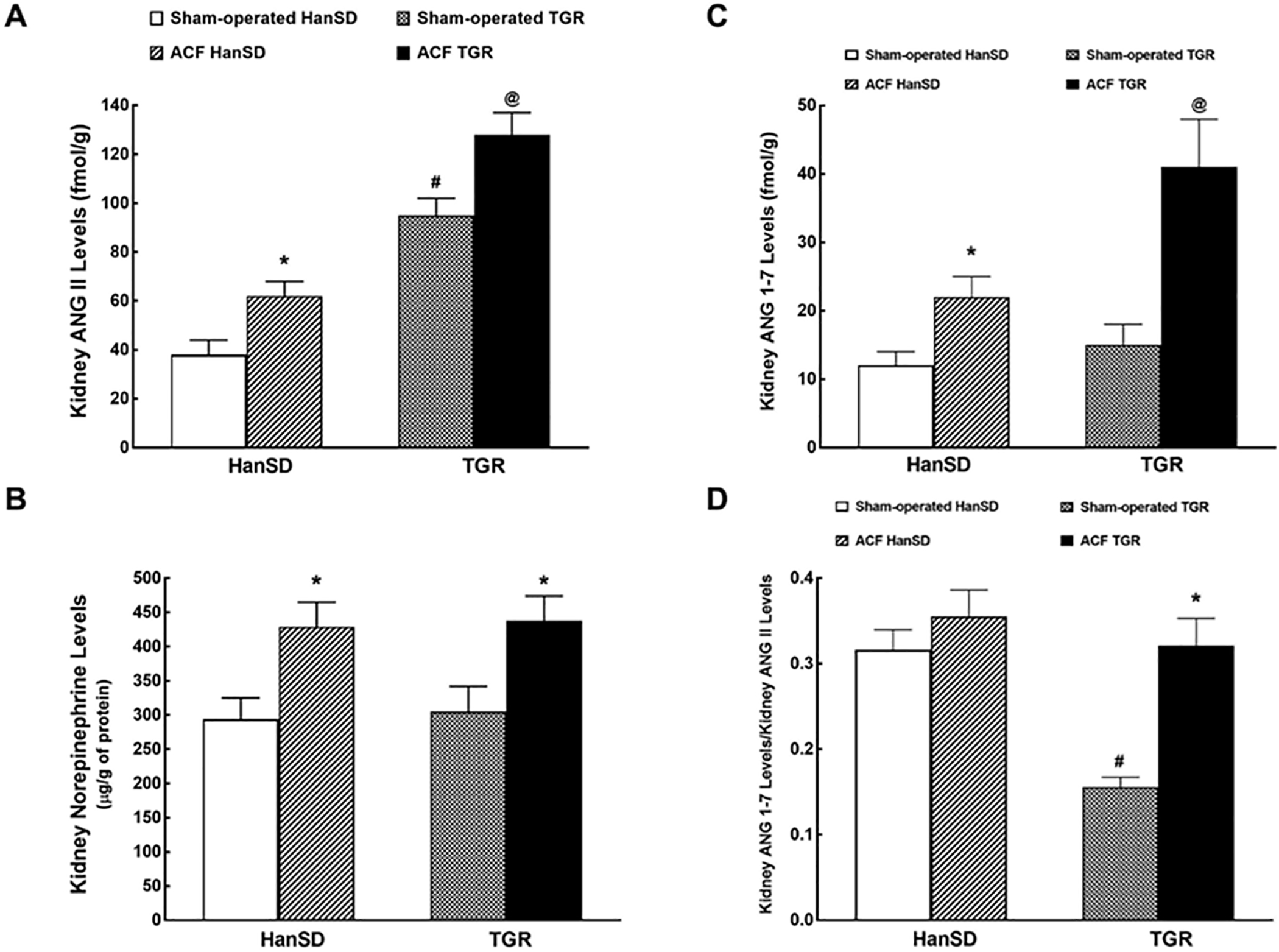
Kidney ANG II (A), kidney NE (B), kidney ANG-1–7 levels (ANG 1–7; C) and kidney ANG 1–7 to ANG II ratio (D) in sham-operated transgene-negative HanSD, sham-operated heterozygous Ren-2 TGRs and HanSD and TGR rats with ACF 5 weeks after the creation of ACF or sham-operation. * p < 0.05 versus sham-operated counterparts. # p < 0.05 versus sham-operated HanSD rats. @ p < 0.05 versus all other groups. All the values shown are means ± SEM. ACF, aorto-caval fistula; TGR, transgenic rat; HanSD, Hannover Sprague-Dawley; ANG II, angiotensin II.
As shown in Figure 2B, there was no significant difference in the renal NE concentration between sham-operated HanSD and TGR. The creation of ACF resulted in similar significant NE increases in TGR and HanSD rats. There were no significant between-group differences in renal concentrations of epinephrine and dopamine (data not shown).
As shown in Figure 2C, there was no significant difference in kidney ANG 1–7 concentrations between sham-operated HanSD rats and sham-operated TGR. The creation of ACF resulted in significant increases in renal ANG 1–7 levels in both groups, but they were more pronounced in ACF TGR than those in ACF HanSD rats.
Figure 2D helps to assess the intrarenal balance between vasodilator and vasoconstrictor axes of the RAAS, again presented as the ratio of ANG 1–7 to ANG II. Similarly as with the circulating peptides, it shows that in sham-operated TGR, the ratio is only half of that in sham-operated HanSD rats. The creation of ACF did not alter this ratio in HanSD rats but resulted in its marked increase in TGR up to the level observed in sham-operated and ACF HanSD rats.
As presented in Table 1, 5 weeks after the creation of ACF, the rats displayed marked cardiac hypertrophy as apparent from increases of left ventricle (LV) weight (LVW, including the interventricular septum) and right ventricle weight compared with sham-operated rats. In addition, ACF rats displayed marked lung congestion as seen from increases of wet lung weight. Notably, cardiac hypertrophy and lung congestion were more pronounced in ACF TGR than that in ACF HanSD rats. Interestingly, the degree of right ventricle hypertrophy was in ACF rats greater than that of the LV as indicated by increases in the right ventricle weight to LVW ratio.
Table 1.
Basal characteristics of body and organ weights in male rats at 5 weeks after the induction of ACF or sham-operation
| Parameter | Group |
|||
|---|---|---|---|---|
| sham-operated HanSD |
sham-operated TGR |
ACF HanSD | ACF TGR | |
|
| ||||
| Body weight, g | 486±9 | 472±11 | 483±8 | 473±8 |
| Heart weight, mg | 1,510±78 | 2,307±81* | 1,759±56# | 2,893±89#, @ |
| LVW, mg | 1,012±71 | 1,344±69* | 1,268±49# | 1,899±59#, @ |
| RVW, mg | 301±27 | 299±29 | 490±37# | 571±29#, @ |
| Lung weight, mg | 1,836±97 | 1,815±99 | 2,239±119# | 3,122±127#, @ |
| Kidney weight, mg | 1,802±99 | 1,795±81 | 1,729±106 | 1,773±113 |
| Liver weight, g | 17.04±0.23 | 16.56±0.28 | 16.99±0.27 | 17.55±0.25 |
| RVW/LVW | 0.297±0.06 | 0.223±0.09 | 0.386±0.07# | 0.301±0.05#, @ |
p < 0.05 sham-operated TGR vs. sham-operated HanSD
p < 0.05 ACF animals vs. sham-operated animals
p < 0.05 ACF TGR vs. ACF HanSD.
Values are means ± SEM.
ACF, aorto-caval fistula; HanSD, transgene-negative normotensive Hannover Sprague-Dawley rats; TGR, Ren-2 renin transgenic hypertensive rats; RVW, right ventricle weight; LVW, left ventricle weight.
Figure 3 illustrates the basal MAP, RBF, and renal vascular resistance values in animals 5 weeks after sham-operation or after creation of ACF. As shown in Figure 3A, sham-operated TGR were markedly hypertensive as compared with sham-operated HanSD rats. A creation of ACF resulted in significant decreases in MAP in TGR as well as in HanSD rats but the pressure remained significantly higher in ACF TGR.
Fig. 3.
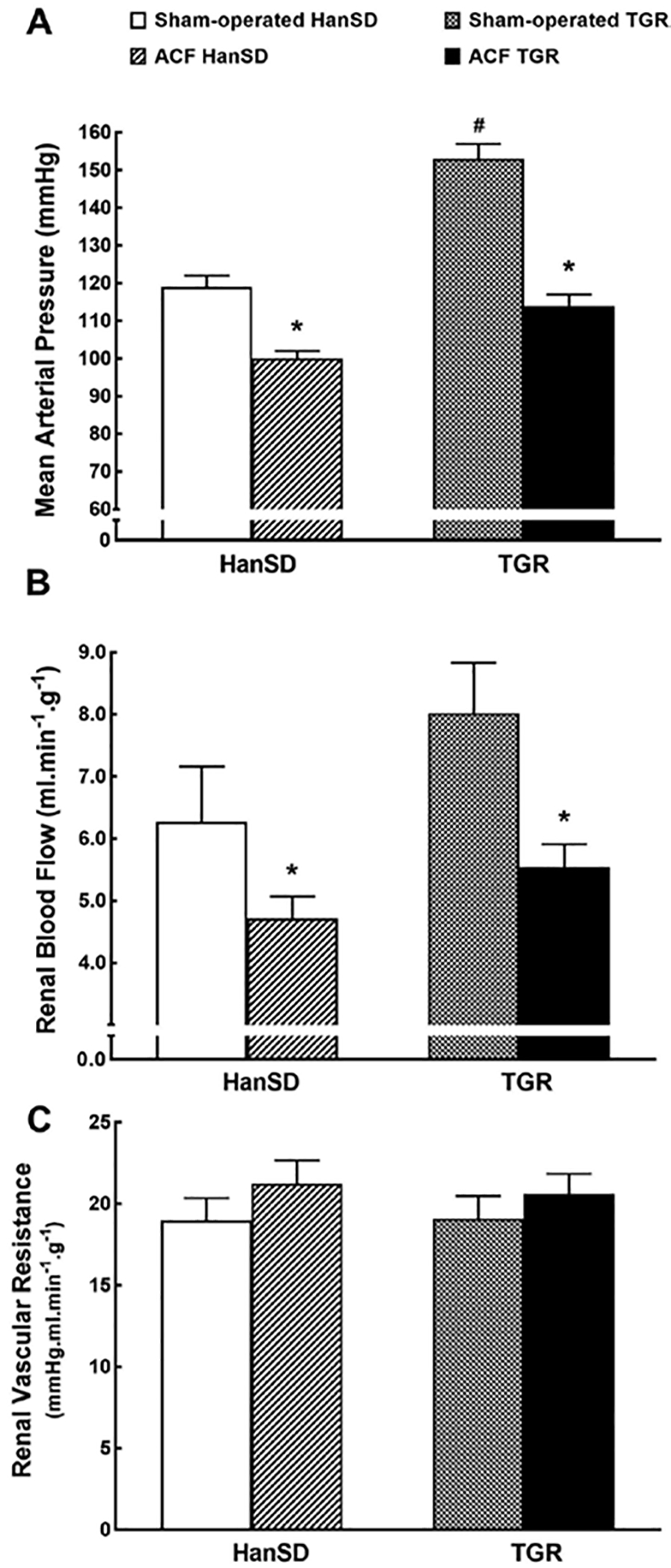
MAP (A), RBF (B), and RVR (C) in sham-operated transgene-negative HanSD, sham-operated heterozygous Ren-2 TGRs and HanSD and TGR rats with ACF 5 weeks after the creation of ACF or sham-operation. * p < 0.05 versus sham-operated counterparts. # p < 0.05 versus all other groups. All the values shown are means ± SEM. ACF, aorto-caval fistula; TGR, transgenic rat; HanSD, Hannover Sprague-Dawley; ANG II, angiotensin II.
As shown in Figure 3B, there were no significant differences in RBF between sham-operated TGR and sham-operated HanSD rats; there was only a tendency for a higher RBF in the former group. The creation of ACF resulted in significant decreases in RBF in both groups to similar levels. Figure 3C shows that there were no significant differences in renal vascular resistance between experimental groups.
Intrarenal administration of ANG II, NE, Ach, Bk, 11,12-EET, and EET-A did not alter MAP in any experimental group. The percent changes induced varied from −0.9 ± 0.8 to 1.1 ± 1.1, which was not significantly different from MAP responses to intrarenal bolus administration of saline.
Figure 4A shows the maximum decreases in RBF induced by the intrarenal administration of 2 and 8 ng of ANG II. The response was substantially greater in sham-operated TGR as compared with the response of sham-operated HanSD rats. ANG II caused almost identical dose-dependent decreases in RBF in ACF HanSD rats and in sham-operated HanSD rats. In contrast, ANG II caused greater RBF decreases in ACF TGR as compared with sham-operated TGR.
Fig. 4.
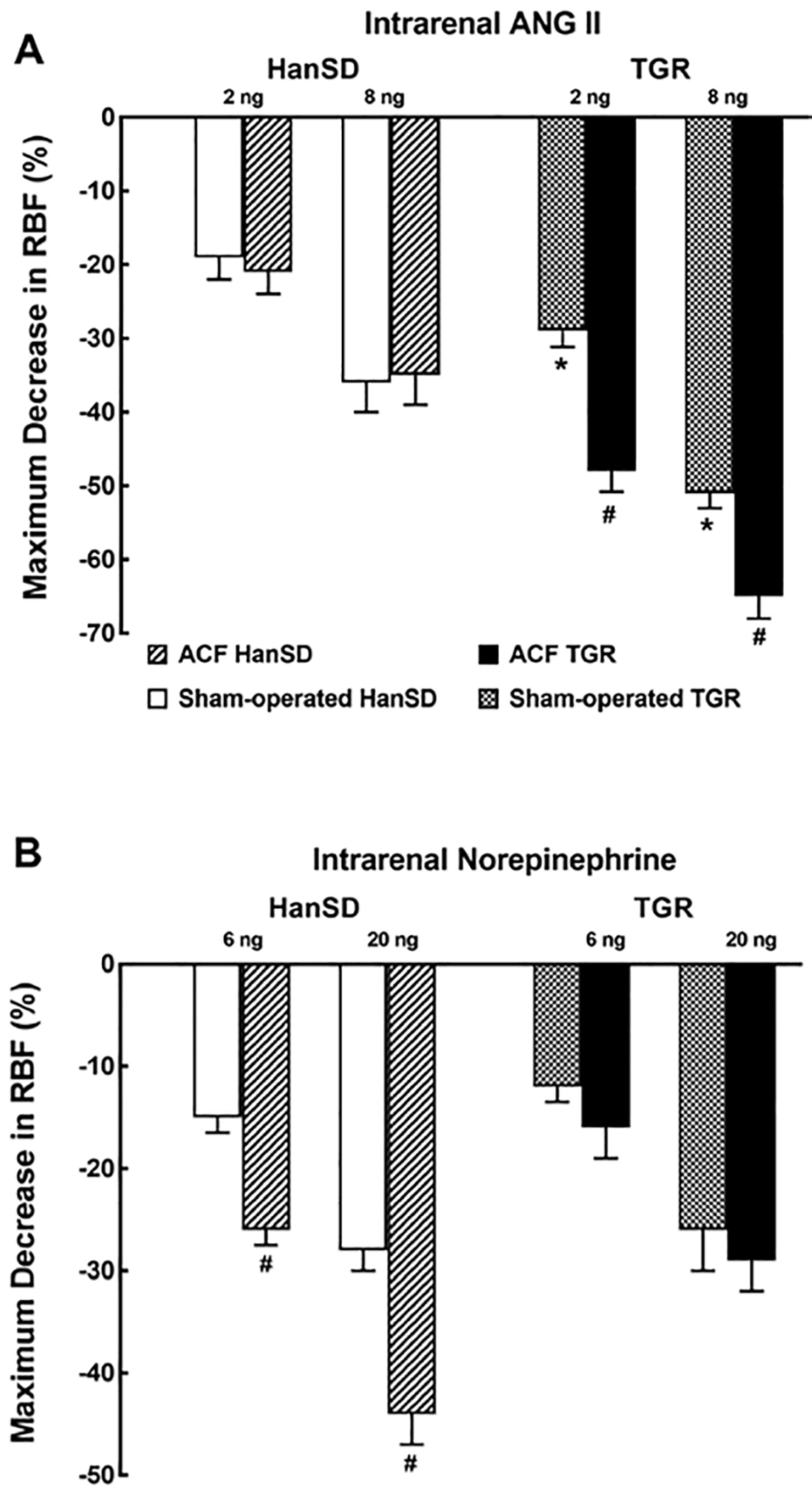
Maximum change in RBF induced by selective intrarenal bolus administration of ANG II (2 and 8 ng, A) and NE (6 and 20 ng, B) in sham-operated transgene-negative HanSD, sham-operated heterozygous Ren-2 TGRs and HanSD and TGR rats with ACF 5 weeks after the creation of ACF or sham-operation. * p < 0.05 versus sham-operated HanSD rats for the same dose of ANG II. # p < 0.05 versus sham-operated counterparts for the same dose. All the values shown are means ± SEM. RBF, Renal blood flow; ACF, aorto-caval fistula; TGR, transgenic rat; HanSD, Hannover Sprague-Dawley; ANG II, angiotensin II.
Figure 4B shows that the intrarenal administration of NE at the doses of 6 and 20 ng elicited similar dose-dependent RBF decreases in sham-operated TGR and HanSD rats. However, NE caused markedly higher decreases in RBF in ACF HanSD rats as compared with sham-operated HanSD rats. Dissimilarly, it elicited comparable decreases in RBF in both TGR groups.
Figure 5 presents RBF responses to intrarenal administration Ach and Bk, the endothelium-dependent vasodilators. As shown in Figure 5A, Ach at the doses of 20 and 60 ng elicited similar RBF increases in sham-operated TGR and HanSD rats. However, it caused significantly greater RBF increases in ACF TGR and ACF HanSD rats than those in their sham-operated counterparts. As shown in Figure 5B, intrarenal administration of 100 and 300 ng of Bk elicited RBF increases with a pattern quite similar to that observed with Ach.
Fig. 5.
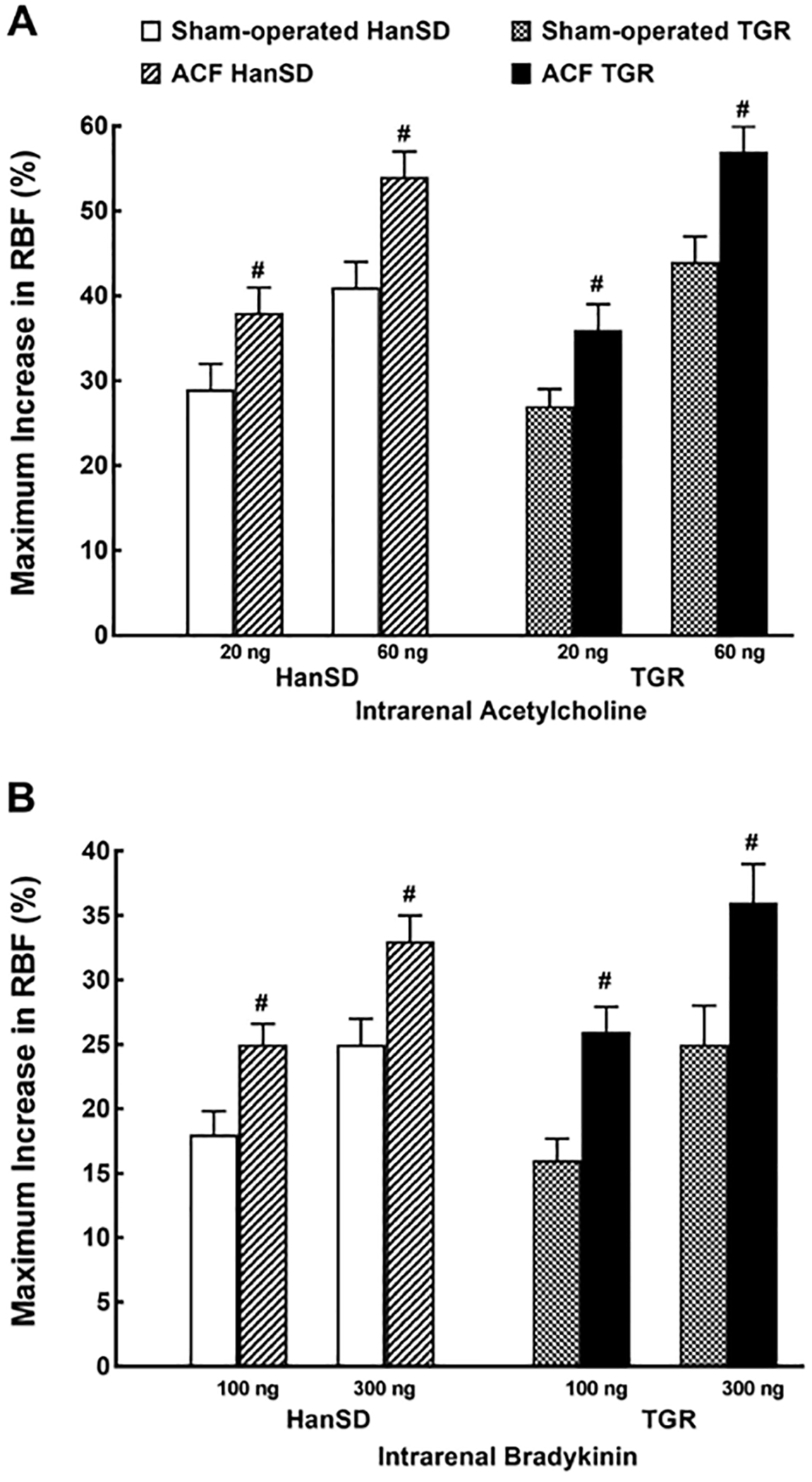
Maximum change in RBF induced by selective intrarenal bolus administration of Ach (20 and 60 ng, A) or Bk (100 and 300 ng, B) in sham-operated transgene-negative HanSD, sham-operated heterozygous Ren-2 TGRs and HanSD and TGR rats with ACF 5 weeks after the creation of ACF or sham-operation. # p < 0.05 versus sham-operated counterparts for the same dose. All the values shown are means ± SEM. RBF, Renal blood flow; ACF, aorto-caval fistula; TGR, transgenic rat; HanSD, Hannover Sprague-Dawley.
Figure 6 shows RBF responses to EET vasodilators (native 11,12-EET or EET-A, a synthetic analog of 14,15 EET). As seen in Figure 6A, intrarenal administration of 11,12-EET at the doses of 300 and 600 ng induced similar RBF increases in sham-operated TGR and sham-operated HanSD rats. 11,12-EET also elicited similar RBF increases in ACF HanSD rats and in sham-operated HanSD rats, whereas RBF increases in ACF TGR were significantly smaller than those in sham-operated TGR. The intrarenal administration of EET-A at the doses of 400 and 800 ng elicited RBF responses that were similar in pattern with those observed with 11,12-EET (Fig. 6B).
Fig. 6.
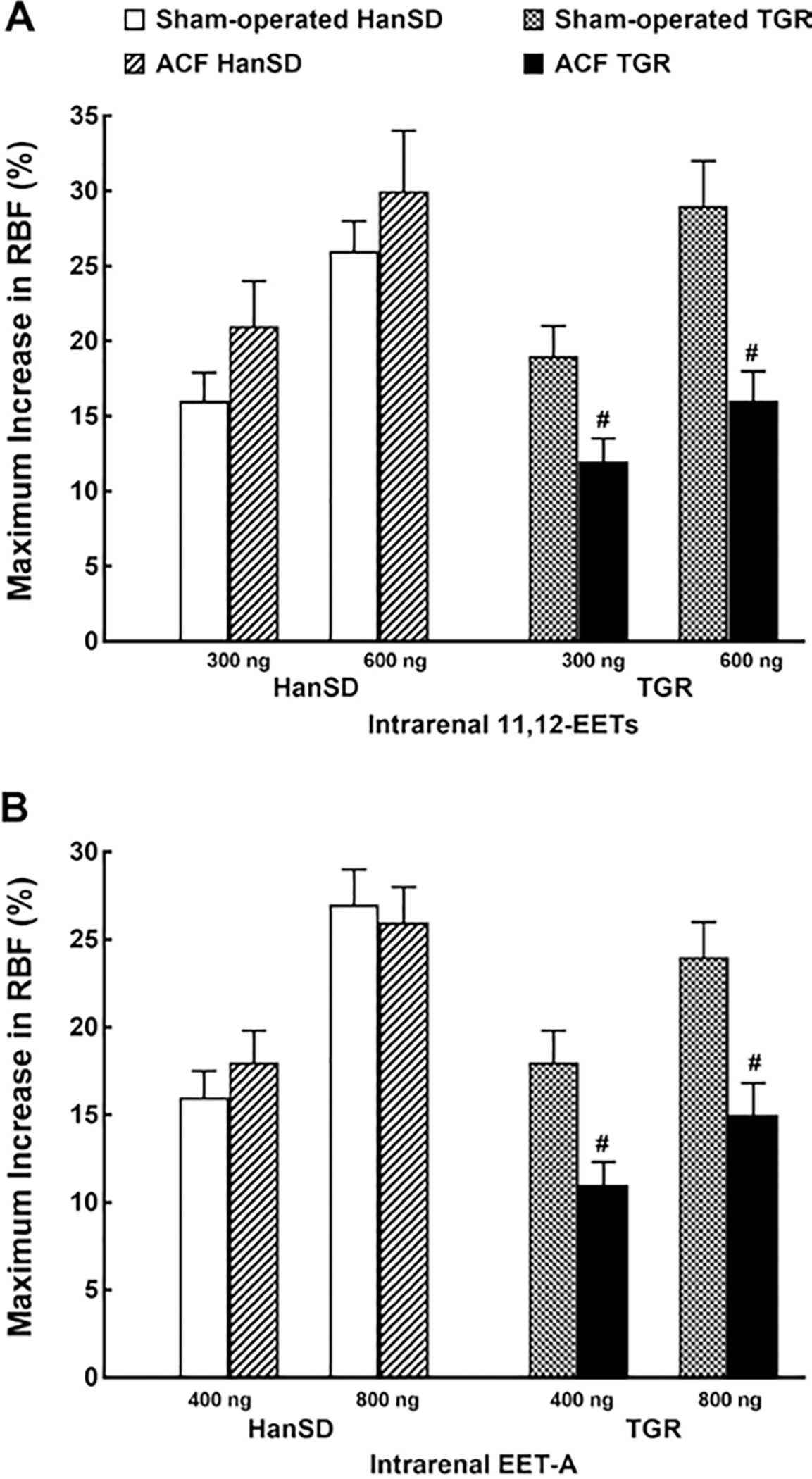
Maximum change in RBF induced by selective intrarenal bolus administration of native 11,12- EETs (11,12-EET, 300 and 600 ng, A) and the analog of 14,15- EET-A (400 and 800 ng, B) in sham-operated transgene-negative HanSD, sham-operated heterozygous Ren-2 TGRs and HanSD and TGR rats with ACF 5 weeks after the creation of ACF or sham-operation. # p < 0.05 versus sham-operated counterparts for the same dose. All the values shown are means ± SEM. EETs, epoxyeicosatrienoic acids; RBF, Renal blood flow; ACF, aorto-caval fistula; TGR, transgenic rat; HanSD, Hannover Sprague-Dawley.
Discussion
The most important finding of the present study is that intrarenal administration of ANG II, at doses that do not alter MAP, elicited substantially greater decreases in RBF in ACF TGR than those in ACF HanSD rats and also than those in the sham-operated TGR counterparts. In contrast, data show that intrarenal administration of the native 11,12-EET as well as of the EET-A, an analog of 14, 15-EET, induced distinctly smaller RBF increases in ACF TGR than those in ACF HanSD rats and, even more so, in sham-operated TGR. For comparison, intrarenal NE, Ach and Bk elicited comparable RBF responses in ACF TGR and ACF HanSD rats. Furthermore, RBF responses to intrarenal endothelium-dependent vasodilators Ach and Bk were enhanced in ACF TGR as compared with sham-operated TGR. On the whole, present data indicate that ACF TGR exhibit enhanced renal vasoconstrictor responses to ANG II and attenuated renal vasodilatory responses to EETs. These findings are in agreement with our hypothesis that selectively exaggerated renal vascular responsiveness to ANG II combined with selectively impaired renal vasodilator responsiveness to EETs are both present at the onset of decompensated phase of CHF and critically contribute to the development of renal dysfunction in ANG II-dependent hypertension. This in turn may accelerate the progression of CHF decompensation and lead to a fatal end.
However, before the formulation of the ultimate conclusion on the importance of our present findings for the pathophysiology of progression of CHF and the prospective development of new therapeutic measures, several issues should be considered.
The first issue is to what extent the present findings are pertinent to the assumed systemic and intrarenal activation of the RAAS and sympathorenal axis. In accordance with the early evidence [53] and later own studies [49, 51, 52], sham-operated TGR exhibit markedly higher plasma and intrarenal ANG II concentrations as compared with sham-operated HanSD rats; however, there is no increase in plasma aldosterone levels. Our present finding of normal plasma aldosterone levels in sham-operated TGR is in agreement with that of previous studies [55, 56], which indicated that the adrenal gland is the major site of Ren-2 renin gene expression; notably, aldosterone production is altered only locally, without affecting circulating aldosterone levels. Moreover, our finding that circulating and intrarenal NE levels are not elevated in sham-operated TGR as compared with sham-operated HanSD rats again supports the previous notion that increased sympathetic nervous system (SNS) activity is not a primary cause of hypertension in the models obtained by the insertion of the mouse Ren-2 renin gene into the rat genome, that is, either TGR or inducible Cyp1a1 Ren-2 TGRs [56–58]. Our novel finding is that even though plasma and kidney ANG 1–7 levels are similar in sham-operated TGR and sham-operated HanSD rats, the circulating and intrarenal balance between the vasodilator axis and vasoconstrictor axis of the RAAS (expressed as ANG 1–7 to ANG II ratio) is shifted toward the latter. Considering that ANG 1–7 is the biologically active peptide of the vasodilatory/natriuretic axis of the RAAS and that the physiological role of this axis is to counteract deleterious actions of the vasoconstrictor/sodium retaining axis, especially under conditions of inappropriately activated RAAS [52, 59], it is conceivable that deficient activity of the vasodilatory/natriuretic axis might contribute to the pathophysiology of hypertension in TGR. Taken together, our present findings confirm again that TGR presents a unique ANG II-dependent model of hypertension with increased activation of the RAAS, wherein increased activity of its vasoconstrictor axis plays a critical hypertensiogenic role.
Additionally, also in agreement with recent findings, we observed that ACF rats exhibit increased plasma ANG II, aldosterone and NE levels and also elevated intrarenal concentrations of ANG II and NE [31, 34, 38, 42]. This confirms that the activation of the vasoconstrictor/sodium retaining axis of the RAAS along with the activation of the sympathorenal axis is present in this model of CHF. This further supports the notion that CHF is not only a hemodynamic disorder but also a neurohormonal syndrome, with activation of the RAAS and SNS contributing to CHF progression [16, 20, 21, 23, 28–30, 60].
In this regard, our important novel finding is that ACF creation in TGR results in a further increase of already elevated plasma and kidney ANG II levels: indeed, plasma and intrarenal concentrations of ANG II in ACF TGR were almost twice higher than those in ACF HanSD rats. This indicates that ACF-induced CHF in this ANG II-dependent model of hypertension is accompanied by a more pronounced activation of the vasoconstrictor axis of the RAAS than observed in normotensive animals. On the other hand, we found that plasma aldosterone levels in ACF TGR and ACF HanSD rats were elevated to similar levels. Of particular interest is our finding that the vasodilatory/natriuretic axis of the RAAS was more activated in ACF TGR than that in ACF HanSD rats (based on the ANG 1–7 to ANG II ratio in the plasma and in the kidney), which indicates a more pronounced compensatory activation of this axis, possibly counteracting the prominent activation of the vasoconstrictor/sodium retaining axis of the RAAS in ACF TGR. Nevertheless, we propose that pronounced hyperactivity of the vasoconstrictor/sodium retaining axis of the RAAS is an important determinant of an earlier onset of the decompensation phase of CHF in ACF TGR.
Another set of the present findings to be considered here is the organ morphometric data. In agreement with previous studies, including our own [31, 37, 38, 40–45], 5 weeks after creation of ACF, all the animals displayed striking cardiac hypertrophy with significant lung congestion but without any increase in liver weight, indicating LV failure. The novel finding is that the degree of these changes was discernibly higher in ACF TGR than that in ACF HanSD rats. Specifically, the percent increases in LVW and lung weight were markedly higher in ACF TGR than those in ACF HanSD rats (41 ± 3 vs. 25 ± 2 and 72 ± 4 vs. 22 ± 3%, respectively, p < 0.05 in both cases). Even if organ weight may be thought an “unsophisticated” marker, previous studies evaluating the relationship between organ morphometry and cardiac function assessed by echocardiography and by direct hemodynamic pressure-volume analyses have demonstrated that the organ weights are a reliable predictor of the onset of cardiac decompensation in experimental models of HF and particularly in the volume overload model [38, 43, 44, 61–63]. Collectively, the present organ weight data suggest that ACF TGR were in the stage of advanced HF and rapidly progressed to the decompensation phase. This clearly indicates that the present findings are relevant to the pathophysiology of the onset of HF decompensation, at least in the model employed here.
Of considerable interest are also our findings pertaining to RBF responses to Ach and Bk, the endothelium-dependent vasodilators, as observed in sham-operated as well as in ACF animals. In agreement with observations made by Jacinto et al. [48] and Kopkan et al. [49], we showed that sham-operated TGR exhibited exaggerated renal vasodilator responses to these agents as compared with sham-operated HanSD rats. It means that augmented renal vascular responsiveness to ANG II cannot be explained by an impaired vascular responsiveness to endogenous vasodilators. Instead, our findings suggest that enhanced renal vascular responsiveness to endothelium-dependent vasodilators may buffer the renal vasoconstrictor actions of ANG II and thus attenuate the decreases in RBF in ACF animals. It is noteworthy that previous studies in patients and animals with HF demonstrated impaired endothelium-dependent renal vasodilatory responses that have been proposed to play a role in the development of renal dysfunction in HF and the progression of this disease [31, 32, 64–68]. In an attempt to reconcile our findings with these previous studies, it is important to point out that the majority of studies in HF subjects evaluated RBF responses to systemic and not intrarenal administration of vasoactive agents. Such procedure can cause significant changes in MAP [31, 32, 64–67], which are known to activate compensatory mechanism(s) that alter RBF responses. Therefore, it is now recommended that any conclusions concerning renal vascular reactivity should be based on direct intrarenal arterial administration of vasoactive agents [48, 49, 69–72].
It has to be admitted that the mechanism(s) responsible for the altered renal vascular responsiveness to ANG II and to EETs, as observed in ACF TGR, remain unclear and the present study was not designed to address them. However, considering several potentially relevant issues can provide some insight in the background of the altered responses.
First, it is possible that the enhanced renal vascular responsiveness to ANG II might be secondary to increased number and/or affinity of vascular ANG II type 1 (AT1) and type 2 (AT2) receptors. It was previously reported that in ANG II-infused hypertensive rats [73] and in TGR [49] the kidney AT1 receptor protein levels were not significantly different from those in their normotensive counterparts, despite markedly elevated circulating and intrarenal ANG II concentrations. However, it cannot be excluded that the enhanced vascular responsiveness to ANG II reflects increased sensitivity of the AT1 signaling pathway in the vascular smooth muscle of ACF TGR. Regarding the role of AT2 receptors, it seems unlikely because the AT2 receptors protein level in ANG II-infused hypertensive rats and TGR is extremely low and not different from the values observed in normotensive controls [49, 73, 74]. Nevertheless, little information is available regarding ANG II receptor levels and signaling in the vascular smooth muscle of renal resistance vessels in ACF rats, and there is no such information for ACF TGR. Additional studies are required to address this issue.
Second, the interpretation of the reduced renal vasodilatory responses to EETs in ACF TGR is even more difficult compared to ANG II because EETs act as endothelial-derived hyperpolarizing factors and their vasoactive actions are substantially modified by interactions with hormonal and paracrine systems, such as RAAS, SNS, as well as purinergic and endothelin systems [75]. Considering that the activities of RAAS, SNS, and endothelin systems are markedly activated in CHF [12, 16, 20–23, 28–31, 75, 76], it is conceivable that the differential compensatory activation of these systems in ACF TGR and ACF HanSD rats might also alter the renal vascular responsiveness to the administered native or synthetic EETs. Future studies would be needed to dissect such complex interrelationships.
In the context of the above-discussed issues, 2 limitations of the present study should be mentioned. The first one relates to the technique employed in the evaluation of renal vascular responses to vasoactive agents in vivo. Even though the technique is in common use [25–27, 34, 37, 38, 47–49], one should be aware that experiments are performed in anesthetized, surgically stressed animals, and involve fairly invasive manipulation of the kidney. Thus, it is possible that ACF TGR and ACF HanSD rats respond differently to those experimental conditions, which might to some extent influence the outcome of such renal in vivo studies.
Another limitation relates to the models of hypertension and CHF. Admittedly, TGR represent an artificial model of hypertension, wherein the insertion of the mouse renin gene (Ren-2) into the genome of normotensive rats is associated with striking activation of the RAAS [35, 36]. The ACF model of CHF is also specific due to its primary “high-output” feature: in the majority of patients with CHF, the initial insult is different. Nevertheless, the model still has many features similar to untreated human CHF, especially regarding neurohormonal activation and the development of renal dysfunction [31, 34, 37, 38, 40–44, 62–65]. It should be mentioned here that in our original study evaluating the sequels of ACF creation in TGR [37], we found that after an initial profound drop in MAP below the level observed in sham-operated HanSD rats, MAP returned to the values observed in sham-operated HanSD rats within 56 h. Thus, these MAP values were within the range of renal autoregulatory capacity and adequate for normal renal function, providing appropriate baseline conditions for studies in ACF TGR. Therefore, we suggest that ACF TGR is suitable to be employed in studies evaluating the role of hypertension and RAAS in the progression of CHF, in particular, its form associated with renal dysfunction.
Conclusion
On the whole, while admitting the limitations discussed above, we can still conclude that our present results show that ACF TGR studied 5 weeks after creation of ACF exhibit exaggerated renal vascular responsiveness to ANG II and reduced renal vasodilatory responses to EETs. This suggests that such combined alterations play an important role in the development of renal dysfunction and can accelerate the onset of decompensated phase of CHF in this hypertensive model. As discussed above, we believe that our novel findings regarding the pathophysiology of CHF should be considered in attempts to develop new pharmacological strategies for the treatment of renal dysfunction in CHF individuals, especially those displaying hypertension before the onset of CHF.
Acknowledgments
This study was primarily supported by the Ministry of Health of the Czech Republic grant no. NV-18-02-00053 to L.Č. All rights reserved.
Footnotes
Disclosure Statement
Authors declare that they have no conflicts of interest to disclose.
References
- 1.Maggioni AP. Epidemiology of heart failure in Europe. Heart Fail Clin. 2015. Oct;11(4):625–35. [DOI] [PubMed] [Google Scholar]
- 2.Ponikowski P, Voors AA, Anker SD, Bueno H, Cleland JG, Coats AJ, et al. ; ESC Scientific Document Group. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: the Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC)Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur Heart J. 2016. Jul;37(27):2129–200. [DOI] [PubMed] [Google Scholar]
- 3.Stewart S, MacIntyre K, Hole DJ, Capewell S, McMurray JJ. More ‘malignant’ than cancer? Five-year survival following a first admission for heart failure. Eur J Heart Fail. 2001. Jun;3(3):315–22. [DOI] [PubMed] [Google Scholar]
- 4.Ronco C, Haapio M, House AA, Anavekar N, Bellomo R. Cardiorenal syndrome. J Am Coll Cardiol. 2008. Nov;52(19):1527–39. [DOI] [PubMed] [Google Scholar]
- 5.Braunwald E The war against heart failure: the Lancet lecture. Lancet. 2015. Feb;385(9970):812–24. [DOI] [PubMed] [Google Scholar]
- 6.Beldhuis IE, Streng KW, Ter Maaten JM, Voors AA, van der Meer P, Rossignol P, et al. Renin-angiotensin system inhibition, worsening renal function, and outcome in heart failure patients with reduced and preserved ejection fraction. Circ Heart Fail. 2017. Feb;10(2):e003588. [DOI] [PubMed] [Google Scholar]
- 7.Di Nicolò P The dark side of the kidney in cardio-renal syndrome: renal venous hypertension and congestive kidney failure. Heart Fail Rev. 2018. Mar;23(2):291–302. [DOI] [PubMed] [Google Scholar]
- 8.Kassi M, Hannawi B, Trachtenberg B. Recent advances in heart failure. Curr Opin Cardiol. 2018. Mar;33(2):249–56. [DOI] [PubMed] [Google Scholar]
- 9.Braam B, Joles JA, Danishwar AH, Gaillard CA. Cardiorenal syndrome—current understanding and future perspectives. Nat Rev Nephrol. 2014. Jan;10(1):48–55. [DOI] [PubMed] [Google Scholar]
- 10.Re RN. A reassessment of the pathophysiology of progressive cardiorenal disorders. Med Clin North Am. 2017. Jan;101(1):103–15. [DOI] [PubMed] [Google Scholar]
- 11.Mann JF, Böhm M. Dual renin-angiotensin system blockade and outcome benefits in hypertension: a narrative review. Curr Opin Cardiol. 2015. Jul;30(4):373–7. [DOI] [PubMed] [Google Scholar]
- 12.Mullens W, Verbrugge FH, Nijst P, Tang WH. Renal sodium avidity in heart failure: from pathophysiology to treatment strategies. Eur Heart J. 2017. Jun;38(24):1872–82. [DOI] [PubMed] [Google Scholar]
- 13.Heywood JT, Fonarow GC, Costanzo MR, Mathur VS, Wigneswaran JR, Wynne J; ADHERE Scientific Advisory Committee and Investigators. High prevalence of renal dysfunction and its impact on outcome in 118,465 patients hospitalized with acute decompensated heart failure: a report from the ADHERE database. J Card Fail. 2007. Aug;13(6):422–30. [DOI] [PubMed] [Google Scholar]
- 14.Barger AC, Muldowney FP, Liebowitz MR. Role of the kidney in the pathogenesis of congestive heart failure. Circulation. 1959. Aug;20(2):273–85. [DOI] [PubMed] [Google Scholar]
- 15.Packer M, Lee WH, Kessler PD. Preservation of glomerular filtration rate in human heart failure by activation of the renin-angiotensin system. Circulation. 1986. Oct;74(4):766–74. [DOI] [PubMed] [Google Scholar]
- 16.Packer M, McMurray JJ. Importance of endogenous compensatory vasoactive peptides in broadening the effects of inhibitors of the renin-angiotensin system for the treatment of heart failure. Lancet. 2017. May;389(10081):1831–40. [DOI] [PubMed] [Google Scholar]
- 17.Navar LG. Renal autoregulation: perspectives from whole kidney and single nephron studies. Am J Physiol. 1978. May;234(5):F357–70. [DOI] [PubMed] [Google Scholar]
- 18.Carlström M, Wilcox CS, Arendshorst WJ. Renal autoregulation in health and disease. Physiol Rev. 2015. Apr;95(2):405–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pfeffer MA, Pfeffer JM, Steinberg C, Finn P. Survival after an experimental myocardial infarction: beneficial effects of long-term therapy with captopril. Circulation. 1985. Aug;72(2):406–12. [DOI] [PubMed] [Google Scholar]
- 20.Dube P, Weber KT, Weber KT. Congestive heart failure: pathophysiologic consequences of neurohormonal activation and the potential for recovery: part I. Am J Med Sci. 2011. Nov;342(5):348–51. [DOI] [PubMed] [Google Scholar]
- 21.Packer M The neurohormonal hypothesis: a theory to explain the mechanism of disease progression in heart failure. J Am Coll Cardiol. 1992. Jul;20(1):248–54. [DOI] [PubMed] [Google Scholar]
- 22.Patel VB, Zhong JC, Grant MB, Oudit GY. Role of the ACE2/angiotensin 1–7 axis of the renin-angiotensin system in heart failure. Circ Res. 2016. Apr;118(8):1313–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rossi F, Mascolo A, Mollace V. The pathophysiological role of natriuretic peptide-RAAS cross talk in heart failure. Int J Cardiol. 2017. Jan;226:121–5. [DOI] [PubMed] [Google Scholar]
- 24.Orsborne C, Chaggar PS, Shaw SM, Williams SG. The renin-angiotensin-aldosterone system in heart failure for the non-specialist: the past, the present and the future. Postgrad Med J. 2017. Jan;93(1095):29–37. [DOI] [PubMed] [Google Scholar]
- 25.Ichikawa I, Pfeffer JM, Pfeffer MA, Hostetter TH, Brenner BM. Role of angiotensin II in the altered renal function of congestive heart failure. Circ Res. 1984. Nov;55(5):669–75. [DOI] [PubMed] [Google Scholar]
- 26.Hostetter TH, Pfeffer JM, Pfeffer MA, Dworkin LD, Braunwald E, Brenner BM. Cardiorenal hemodynamics and sodium excretion in rats with myocardial infarction. Am J Physiol. 1983. Jul;245(1):H98–103. [DOI] [PubMed] [Google Scholar]
- 27.Stanton RC, Brenner BM. Role of the kidney in congestive heart failure. Acta Med Scand Suppl. 1986;707 S707:21–5. [DOI] [PubMed] [Google Scholar]
- 28.Antoine S, Vaidya G, Imam H, Villarreal D. Pathophysiologic mechanisms in heart failure: role of the sympathetic nervous system. Am J Med Sci. 2017. Jan;353(1):27–30. [DOI] [PubMed] [Google Scholar]
- 29.Polhemus DJ, Trivedi RK, Gao J, Li Z, Scarborough AL, Goodchild TT, et al. Renal sympathetic denervation protects the failing heart via inhibition of neprilysin activity in the kidney. J Am Coll Cardiol. 2017. Oct;70(17):2139–53. [DOI] [PubMed] [Google Scholar]
- 30.Jönsson S, Agic MB, Narfström F, Melville JM, Hultström M. Renal neurohormonal regulation in heart failure decompensation. Am J Physiol Regul Integr Comp Physiol. 2014. Sep;307(5):R493–7. [DOI] [PubMed] [Google Scholar]
- 31.Abassi Z, Goltsman I, Karram T, Winaver J, Hoffman A. Aortocaval fistula in rat: a unique model of volume-overload congestive heart failure and cardiac hypertrophy. J Biomed Biotechnol. 2011;2011:729497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Duggan DJ, Tabrizchi R. Angiotensin II control of regional haemodynamics in rats with aortocaval fistula. Exp Physiol. 2016. Sep;101(9):1192–205. [DOI] [PubMed] [Google Scholar]
- 33.Szczepanska-Sadowska E, Czarzasta K, Cudnoch-Jedrzejewska A. Dysregulation of the renin-angiotensin system and the vasopressinergic system interactions in cardiovascular disorders. Curr Hypertens Rep. 2018. Mar;20(3):19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kratky V, Kopkan L, Kikerlova S, Huskova Z, Taborsky M, Sadowski J, et al. The role of renal vascular reactivity in the development of renal dysfunction in compensated and decompensated congestive heart failure. Kidney Blood Press Res. 2018;43(6):1730–41. [DOI] [PubMed] [Google Scholar]
- 35.Mullins JJ, Peters J, Ganten D. Fulminant hypertension in transgenic rats harbouring the mouse Ren-2 gene. Nature. 1990. Apr;344(6266):541–4. [DOI] [PubMed] [Google Scholar]
- 36.Husková Z, Kramer HJ, Vanourková Z, Červenka L. Effects of changes in sodium balance on plasma and kidney angiotensin II levels in anesthetized and conscious Ren-2 transgenic rats. J Hypertens. 2006. Mar;24(3):517–27. [DOI] [PubMed] [Google Scholar]
- 37.Červenka L, Melenovský V, Husková Z, Škaroupková P, Nishiyama A, Sadowski J. Inhibition of soluble epoxide hydrolase counteracts the development of renal dysfunction and progression of congestive heart failure in Ren-2 transgenic hypertensive rats with aorto-caval fistula. Clin Exp Pharmacol Physiol. 2015. Jul;42(7):795–807. [DOI] [PubMed] [Google Scholar]
- 38.Červenka L, Melenovský V, Husková Z, Sporková A, Bürgelová M, Škaroupková P, et al. Inhibition of soluble epoxide hydrolase does not improve the course of congestive heart failure and the development of renal dysfunction in rats with volume overload induced by aorto-caval fistula. Physiol Res. 2015;64(6):857–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Garcia R, Diebold S. Simple, rapid, and effective method of producing aortocaval shunts in the rat. Cardiovasc Res. 1990. May;24(5):430–2. [DOI] [PubMed] [Google Scholar]
- 40.Červenka L, Škaroupková P, Kompanowska-Jezierska E, Sadowski J. Sex-linked differences in the course of chronic kidney disease and congestive heart failure: a study in 5/6 nephrectomized Ren-2 transgenic hypertensive rats with volume overload induced using aorto-caval fistula. Clin Exp Pharmacol Physiol. 2016. Oct;43(10):883–95. [DOI] [PubMed] [Google Scholar]
- 41.Melenovsky V, Skaroupkova P, Benes J, Torresova V, Kopkan L, Cervenka L. The course of heart failure development and mortality in rats with volume overload due to aorto-caval fistula. Kidney Blood Press Res. 2012;35(3):167–73. [DOI] [PubMed] [Google Scholar]
- 42.Cohen-Segev R, Francis B, Abu-Saleh N, Awad H, Lazarovich A, Kabala A, et al. Cardiac and renal distribution of ACE and ACE-2 in rats with heart failure. Acta Histochem. 2014. Oct;116(8):1342–9. [DOI] [PubMed] [Google Scholar]
- 43.Oliver-Dussault C, Ascah A, Marcil M, Matas J, Picard S, Pibarot P, et al. Early predictors of cardiac decompensation in experimental volume overload. Mol Cell Biochem. 2010. May;338(1–2):271–82. [DOI] [PubMed] [Google Scholar]
- 44.Hutchinson KR, Guggilam A, Cismowski MJ, Galantowicz ML, West TA, Stewart JA Jr, et al. Temporal pattern of left ventricular structural and functional remodeling following reversal of volume overload heart failure. J Appl Physiol (1985). 2011. Dec;111(6):1778–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sporková A, Husková Z, Škaroupková P, Rami Reddy N, Falck JR, Sadowski J, et al. Vasodilatory responses of renal interlobular arteries to epoxyeicosatrienoic acids analog are not enhanced in Ren-2 transgenic hypertensive rats: evidence against a role of direct vascular effects of epoxyeicosatrienoic acids in progression of experimental heart failure. Physiol Res. 2017. Mar;66(1):29–39. [DOI] [PubMed] [Google Scholar]
- 46.Červenka L, Husková Z, Kopkan L, Kikerlová S, Sedláková L, Vaňourková Z, et al. Two pharmacological epoxyeicosatrienoic acid-enhancing therapies are effectively antihypertensive and reduce the severity of ischemic arrhythmias in rats with angiotensin II-dependent hypertension. J Hypertens. 2018. Jun;36(6):1326–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chatziantoniou C, Daniels FH, Arendshorst WJ. Exaggerated renal vascular reactivity to angiotensin and thromboxane in young genetically hypertensive rats. Am J Physiol. 1990. Aug;259(2 Pt 2):F372–82. [DOI] [PubMed] [Google Scholar]
- 48.Jacinto SM, Mullins JJ, Mitchell KD. Enhanced renal vascular responsiveness to angiotensin II in hypertensive ren-2 transgenic rats. Am J Physiol. 1999. Feb;276(2):F315–22. [DOI] [PubMed] [Google Scholar]
- 49.Kopkan L, Kramer HJ, Husková Z, Vanourková Z, Skaroupková P, Thurmová M, et al. The role of intrarenal angiotensin II in the development of hypertension in Ren-2 transgenic rats. J Hypertens. 2005. Aug;23(8):1531–9. [DOI] [PubMed] [Google Scholar]
- 50.Fox J, Guan S, Hymel AA, Navar LG. Dietary Na and ACE inhibition effects on renal tissue angiotensin I and II and ACE activity in rats. Am J Physiol. 1992. May;262(5 Pt 2):F902–9. [DOI] [PubMed] [Google Scholar]
- 51.Husková Z, Kramer HJ, Thumová M, Vanourková Z, Bürgelová M, Teplan V, et al. Effects of anesthesia on plasma and kidney ANG II levels in normotensive and ANG II-dependent hypertensive rats. Kidney Blood Press Res. 2006;29(2):74–83. [DOI] [PubMed] [Google Scholar]
- 52.Husková Z, Kopkan L, Červenková L, Doleželová Š, Vaňourková Z, Škaroupková P, et al. Intrarenal alterations of the angiotensin-converting enzyme type 2/angiotensin 1–7 complex of the renin-angiotensin system do not alter the course of malignant hypertension in Cyp1a1-Ren-2 transgenic rats. Clin Exp Pharmacol Physiol. 2016. Apr;43(4):438–49. [DOI] [PubMed] [Google Scholar]
- 53.Mitchell KD, Jacinto SM, Mullins JJ. Proximal tubular fluid, kidney, and plasma levels of angiotensin II in hypertensive ren-2 transgenic rats. Am J Physiol. 1997. Aug;273(2 Pt 2):F246–53. [DOI] [PubMed] [Google Scholar]
- 54.Čertíková Chábová V, Kujal P, Škaroupková P, Varňourková Z, Vacková Š, Husková Z, et al. Combined inhibition of soluble epoxide hydrolase and renin-angiotensin system exhibits superior renoprotection to renin-angiotensin system blockade in 5/6 nephrectomized Ren-2 transgenic hypertensive rats with established chronic kidney disease. Kidney Blood Press Res. 2018;43(2):329–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Peters J, Ganten D. Adrenal renin expression and its role in ren-2 transgenic rats TGR(mREN2)27. Horm Metab Res. 1998. Jun-Jul;30(6–7):350–4. [DOI] [PubMed] [Google Scholar]
- 56.Lee MA, Böhm M, Paul M, Bader M, Ganten U, Ganten D. Physiological characterization of the hypertensive transgenic rat TGR(mREN2)27. Am J Physiol. 1996. Jun;270(6 Pt 1):E919–29. [DOI] [PubMed] [Google Scholar]
- 57.Zicha J, Dobešová Z, Behuliak M, Pintérová M, Kuneš J, Vaněčková I. Nifedipine-sensitive blood pressure component in hypertensive models characterized by high activity of either sympathetic nervous system or renin-angiotensin system. Physiol Res. 2014;63(1):13–26. [DOI] [PubMed] [Google Scholar]
- 58.Zicha J, Hojná S, Kopkan L, Červenka L, Vaněčková I. The absence of sympathoexcitation during the development of hypertension in Cyp1a1 Ren-2 transgenic rats. Physiol Res. 2019. Apr;68(2):329–34. [DOI] [PubMed] [Google Scholar]
- 59.Santos RA, Sampaio WO, Alzamora AC, Motta-Santos D, Alenina N, Bader M, et al. The ACE2/angiotensin-(1–7)/MAS axis of the renin-angiotensin system: focus on angiotensin-(1–7). Physiol Rev. 2018. Jan;98(1):505–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Goldsmith SR, Sobotka PA, Bart BA. The sympathorenal axis in hypertension and heart failure. J Card Fail. 2010. May;16(5):369–73. [DOI] [PubMed] [Google Scholar]
- 61.Brower GL, Henegar JR, Janicki JS. Temporal evaluation of left ventricular remodeling and function in rats with chronic volume overload. Am J Physiol. 1996. Nov;271(5 Pt 2):H2071–8. [DOI] [PubMed] [Google Scholar]
- 62.Melenovsky V, Benes J, Skaroupkova P, Sedmera D, Strnad H, Kolar M, et al. Metabolic characterization of volume overload heart failure due to aorto-caval fistula in rats. Mol Cell Biochem. 2011. Aug;354(1–2):83–96. [DOI] [PubMed] [Google Scholar]
- 63.Brower GL, Levick SP, Janicki JS. Differential effects of prevention and reversal treatment with Lisinopril on left ventricular remodeling in a rat model of heart failure. Heart Lung Circ. 2015. Sep;24(9):919–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Abassi ZA, Gurbanov K, Mulroney SE, Potlog C, Opgenorth TJ, Hoffman A, et al. Impaired nitric oxide-mediated renal vasodilation in rats with experimental heart failure: role of angiotensin II. Circulation. 1997. Nov;96(10):3655–64. [DOI] [PubMed] [Google Scholar]
- 65.Abassi Z, Gurbanov K, Rubinstein I, Better OS, Hoffman A, Winaver J. Regulation of intrarenal blood flow in experimental heart failure: role of endothelin and nitric oxide. Am J Physiol. 1998. Apr;274(4):F766–74. [DOI] [PubMed] [Google Scholar]
- 66.Kubo SH, Rector TS, Bank AJ, Williams RE, Heifetz SM. Endothelium-dependent vasodilation is attenuated in patients with heart failure. Circulation. 1991. Oct;84(4):1589–96. [DOI] [PubMed] [Google Scholar]
- 67.Katz SD, Schwarz M, Yuen J, LeJemtel TH. Impaired acetylcholine-mediated vasodilation in patients with congestive heart failure. Role of endothelium-derived vasodilating and vasoconstricting factors. Circulation. 1993. Jul;88(1):55–61. [DOI] [PubMed] [Google Scholar]
- 68.Katz SD, Hryniewicz K, Hriljac I, Balidemaj K, Dimayuga C, Hudaihed A, et al. Vascular endothelial dysfunction and mortality risk in patients with chronic heart failure. Circulation. 2005. Jan;111(3):310–4. [DOI] [PubMed] [Google Scholar]
- 69.Johns EJ, Kopp UC, DiBona GF. Neural control of renal function. Compr Physiol. 2011. Apr;1(2):731–67. [DOI] [PubMed] [Google Scholar]
- 70.Cervenka L, Wang CT, Navar LG. Effects of acute AT1 receptor blockade by candesartan on arterial pressure and renal function in rats. Am J Physiol. 1998. May;274(5):F940–5. [DOI] [PubMed] [Google Scholar]
- 71.Majid DS, Navar LG. Nitric oxide in the control of renal hemodynamics and excretory function. Am J Hypertens. 2001. Jun;14(6 Pt 2):74S–82S. [DOI] [PubMed] [Google Scholar]
- 72.Forrester SJ, Booz GW, Sigmund CD, Coffman TM, Kawai T, Rizzo V, et al. Angiotensin II signaling transduction: an update on mechanisms of physiology and pathophysiology. Physiol Rev. 2018. Jul;98(3):1627–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Harrison-Bernard LM, Zhuo J, Kobori H, Ohishi M, Navar LG. Intrarenal AT1 receptor and ACE binding in ANG II-induced hypertensive rats. Am J Physiol. 2002;281:F19–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhuo J, Ohishi M, Mendelsohn FA. Roles of AT1 and AT2 receptors in the hypertensive Ren-2 gene transgenic rat kidney. Hypertension. 1999. Jan;33(1 Pt 2):347–53. [DOI] [PubMed] [Google Scholar]
- 75.Imig JD. Epoxyeicosatrienoic acids and 20-hydroxyeicosatetraenoic acid on endothelial and vascular function. Adv Pharmacol. 2016;77:105–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Miyauchi T, Sakai S. Endothelin and the heart in health and diseases. Peptides. 2019. Jan;111:77–88. [DOI] [PubMed] [Google Scholar]