

Abstract
Most combinations of chemo‐ and biocatalysis take place in aqueous media or require a solvent change with complex intermediate processing. Using enzymes in the same organic solvent as the chemocatalyst eliminates this need. Here, it was shown that a complete chemoenzymatic cascade to form dioxolanes could be carried out in a purely organic environment. The result, including downstream processing, was compared with a classical mode, shifting solvent. First, a two‐step enzyme cascade starting from aliphatic aldehydes to chiral diols (3,4‐hexanediol and 4,5‐octanediol) was run either in an aqueous buffer or in the potentially biobased solvent cyclopentyl methyl ether. Subsequently, a ruthenium molecular catalyst enabled the conversion to dioxolanes [e. g., (4S,5S)‐dipropyl‐1,3‐dioxolane]. Importantly, the total synthesis of this product was not only highly stereoselective but also based on the combination of biomass, CO2, and hydrogen, thus providing an important example of a bio‐hybrid chemical.
Keywords: asymmetric catalysis, biocatalysis, cascade reaction, chirality, unconventional media
A winning combination: The combination of bio‐ and chemocatalysis is a powerful method to access complex (chiral) molecules. Here, a 2‐step enzymatic cascade is combined in the same organic solvent with a ruthenium‐catalyzed chemical conversion to form dioxolanes from aliphatic aldehydes. The method of combining the two worlds can be applied easily to other hybrid systems.
Introduction
In the light of climate change and the dependency of the chemical industry on fossil resources, production of fine chemicals and liquid energy carriers based on renewable raw materials is gaining traction in the past years. [1] Biocatalysis plays an important role especially in the production of molecules relevant for the pharmaceutical or chemical industry. This includes chiral products, which are based on the inherently high selectivity most enzymes have and their evolution to convert renewable starting materials effectively. [2] Additionally, the mild reaction conditions under which biotransformations take place and their biodegradability render them environmentally benign. During the past decades, using novel molecular biology and engineering tools such as multi‐target screening, modelling, and simulation tools, an increasing number of enzymes with enormous synthesis potential could be obtained, and with this, novel synthesis routes could be realized. [2]
Sequential combinations of unit‐operations using biocatalysis and chemocatalysis amount to a broader spectrum of transformations and high‐value products. [3] A number of Reviews have been dedicated to this subject in the past years, highlighting the importance of this topic. [4] However, the reaction conditions in which both catalyst systems are most active differ substantially. Consequently, incompatibility with respect to pressure, temperature, pH, or reaction solvent limits the envisaged efficient combination. Most enzymes evolved in the aqueous cytosol and thus show highest activities usually in buffered reaction systems. Molecular transition metal catalysts, on the other hand, are often sensitive to water or oxygen and frequently require high pressures or temperatures for reactions with gaseous reactants. Furthermore, water can disrupt hydrogen‐bonding or van der Waals interactions, which are often essential for the activation of substrates. This can also affect the catalytic activity or stereocontrol. [4e] Thus, water is not always the best solvent in hand, and a straightforward combination of both systems in a one‐pot sequential or even simultaneous mode is a challenge. Common solutions are the use of water‐tolerant chemocatalysts or a compartmentalization of both steps in space or time. [5] Additionally, enzyme engineering [6] or enzyme immobilization using encapsulation/entrapment [7] poses possibilities for increasing their stability in non‐natural conditions. Different approaches combine bio‐ and chemocatalysis in deep eutectic solvents (DES) [8] or biphasic systems. [9] However, very few examples of a hybrid system in an organic solvent with enzymes other than hydrolases or proteases are available. [10] Also, if the chemocatalyst operates in organic conditions, mostly a workup of the intermediate is performed in order not to interfere with the subsequently used enzyme. [3c]
Most purified enzymes, except for lipases[ 4a , 4c , 11 ] among others, are very susceptible to organic solvents, and already small quantities can lead to an inactivation. Several techniques improve the enzyme stability in organic solvents. [12] In this work they are formulated as lyophilized whole cells (LWC). [13] This enzyme formulation combines the advantages of being about 90 % cheaper in production in comparison to purified enzymes, [14] and of the cells already containing necessary cofactors for the reaction such as nicotinamide adenine dinucleotide (NADH) for oxidoreductases. Using this formulation, enzymes can be applied in a so‐called micro‐aqueous reaction system, in which only one organic phase is present and the hydration shell around the enzymes is provided by small quantities of buffer. The buffer is then absorbed by the cells, and thus no second aqueous phase forms. Next to advantages regarding substrate load and wastewater reduction, the application of enzymes in non‐aqueous solvent systems can be advantageous for hybrid systems, when bio‐ and chemocatalysts are combined. Many chemocatalysts used for the production of high‐value fine chemicals are susceptible to water [15] or were developed in organic solvents to reach high product concentrations. The use of enzymes in a nonaqueous solvent, such as an organic solvent, obviates the need for such solvent switching between bio‐ and chemocatalysis if a suitable solvent for both can be demonstrated. This offers new opportunities to combine the best available catalysts, regardless of whether they are of biological or chemical origin, into hybrid processes. Consequently, the approach not only optimizes processes economically but also bears high potential for sustainable process development especially when renewable resources should be applied, and a straightforward downstream processing (DSP) is envisaged.
In this work, two two‐step enzymatic cascades are presented, which produce either 3,4‐hexanediol or 4,5‐octanediol from propanal or butanal, respectively (Figure 1). These aldehydes can be produced from biomass by oxidation of the respective, potentially biobased, alcohols and thus display an alternative access to petroleum‐based diols. [16] The resulting diol can subsequently be used as a substrate to produce cyclic acetals. Cyclic acetals are considered as promising alternative platform molecules for the chemical industry with a versatile applicability as bio‐based solvent or monomer for ring‐opening polymerization.[ 15a , 15c , 15d ] In addition, in the Cluster of Excellence “The Fuel Science Center”, [17] these molecules are intensively tested as bio‐hybrid fuels for combustion engines. [18] Cyclic acetals reduce soot and particle emissions due to their higher oxygen content and show promising laminar burning velocities, while exhibiting improved boiling points and energy densities to previous fuel candidates.[ 18 , 19 ] For the recently introduced chemical transformation of diols to dioxolanes not formaldehyde, but hydrogen and carbon dioxide as C1 source could be applied. Here, a further important improvement is presented that allows the acetals to be synthesized directly from formic acid at milder reaction conditions. Formic acid itself can be obtained via the hydrogenation of CO2, [20] resulting in a benign pathway towards the formation of the methylene unit within the dioxolane structures. For this novel transformation based on formic acid, the tailored combination of the transition metal catalyst [Ru(triphos)(tmm)] [triphos: 1,1,1‐tris(diphenylphosphino‐methyl)ethane, (tmm: trimethylene methane)] [21] and a Lewis acid is used. [22] In this way, a hybrid process including bio‐ and chemocatalysis towards cyclic acetals based on renewable raw materials is presented. For the determination of the absolute configuration, non‐commercial stereoisomers of 3,4‐hexanediol and 4,5‐octanediol were either synthesized enzymatically and purified or prepared chemically using Sharpless’ asymmetric dihydroxylation. [23]
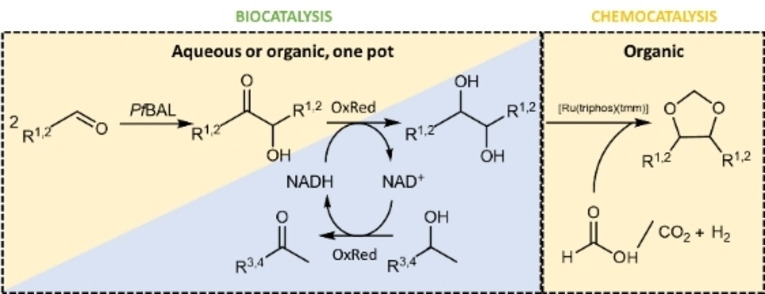
Figure 1.
Reaction scheme for the enzymatic production of diols from aldehydes and the subsequent formation of 1,3‐dioxolanes with a [Ru(triphos)(tmm)] catalyst. The left blue/yellow reaction is possible in both aqueous and organic reaction conditions. R1=ethyl, R2=propyl, R3=methyl, R4=hydroxy ethyl, PfBAL=benzaldehyde lyase from Pseudomonas fluorescens, OxRed=oxidoreductase.
Results and Discussion
Reaction environment selection of buffered and micro‐aqueous reaction system
Two biocatalytic cascades towards 3,4‐hexanediol and 4,5‐octanediol starting with aldehydes were tested both in organic and aqueous reaction conditions (see Figure 2). A micro‐aqueous reaction system (MARS) could be successfully tested for applications with carboligases and alcohol dehydrogenases using cyclopentyl methyl ether (CPME). [24] Although up to now CPME is produced from cylopentene from fossil‐based feedstocks, the atom economy during production is excellent. In the future, there is the potential to produce CPME from sugar. [25] Additionally, CPME is considered to be less toxic than other organic solvents for biocatalysis such as methyl‐tert‐butyl ether (MTBE) or 2‐methyltetrahydrofuran (MTHF). [12] This combined with the potential biogenic origin makes CPME currently defined as a “green solvent”. [25] Since most enzymes need a hydration shell for activity, small amounts of an aqueous buffer are necessary for proper enzyme activity. Here, 1 μL of buffer per mg of lyophilized whole cells proved to be sufficient.[ 13 , 26 ] Previous studies have shown that highly concentrated buffer (1 m) is advantageous over less concentrated buffer or water only. [26] The buffer is completely absorbed by the cells to form the aqueous reaction environment, and thus the solvent system still is monophasic organic.
Figure 2.

Enzymatic synthesis of diols under aqueous and organic reaction conditions. Concentrations were determined using GC analysis. The dashed line indicates the addition of co‐substrate and oxidoreductase. 3,4‐Hexanediol synthesis in (A) organic and (B) aqueous reaction conditions. For (A, B) PfBAL was used for carboligation and LbADH for reduction. 4,5‐Octanediol synthesis in (C) organic and (D) aqueous reaction conditions using PfBAL for carboligation and BlBDH for reduction. The reactions were performed in technical triplicates.
Catalyst screening for enzymatic reactions
Different biocatalysts were screened for the enzymatic reactions for their activity towards the substrates. The stereoselectivity of the enzymes, although very important, was not the main focus of the screening, as the production of optically pure stereoisomers is subject of future research. Especially for the sterically more demanding cascade towards 4,5‐octanediol, the following enzymes were screened: alcohol dehydrogenases from Ralstonia spec (RADH), butanediol dehydrogenases from Bacillus licheniformis (BlBDH), [27] and alcohol dehydrogenase from Lactobacillus brevis (LbADH). [28] For the C−C bond formation of two aldehyde molecules (propanal or butanal) the following ThDP‐dependent enzymes were tested: a variant of the pyruvate decarboxylase from Acetobacter pasteurianus (ApPDCE469G), [29] the benzoylformate decarboxylase from Pseudomonas putida (PpBFD), [30] and benzaldehyde lyase from Pseudomonas fluorescens (PfBAL)[ 30 , 31 ] were screened for activity (see Supporting Information Tables S1 and S2). The best enzyme for the ligation reaction was for both butanal and propanal the PfBAL with 46 % conversion to butyroin and 16 % propioin in MARS, respectively. Subsequently, oxidoreductases were used for the reduction of the carbonyl group to the respective diol. Propioin was reduced with the highest conversions of 88 % by the LbADH to 3,4‐hexanediol in MARS. The LbADH did not accept the sterically more demanding butyroin, whereas the BlBDH could reduce this substrate with a conversion of 27 % in MARS. The final cascades therefore consisted of a carboligation step using PfBAL for both propanal and butanal with a subsequent reduction with either LbADH for propioin or BlBDH for butyroin. The reduction reaction is dependent on the cofactor NADH+H+, which is oxidized to NAD+. For an appropriate cofactor recycling, sacrificial proton donors were used. For the LbADH this was 2‐propanol, which was oxidized to acetone. The BlBDH does not show any activity towards this substrate, since it requires two vicinal hydroxy or carbonyl functions. [27] Thus for the recycling reaction for the cofactor of BlBDH, 1,2 propanediol is used. The BlBDH showed decent activities towards the substrate, and furthermore, it can be produced from renewable resources. [32]
Small‐scale enzymatic reactions
To compare the activity of the enzymes in both organic and aqueous conditions, the production of 3,4‐hexanediol and 4,5‐octanediol was tested using 200 mm of the respective aldehyde as substrate in either CPME or aqueous 50 mm triethanolamine (TEA) buffer. Due to the ligation of two aldehyde molecules in the first step, 100 mm product could be maximally accessed. Every other reaction parameter except for the solvent was identical. The production of the intermediate (propioin and butyroin, respectively) and the product in this two‐step one‐pot reaction is tracked over time by gas chromatography (GC) analysis (Figure 2).
In general, for both cascades acceptable product concentrations could be reached in both organic (63 mm 3,4‐hexanediol; 38 mm 4,5‐octanediol) and buffered media (56 mm 3,4‐hexanediol; 20 mm 4,5‐octanediol) under the conditions applied, which proved both solvent systems as generally appropriate to produce vicinal diols. The PfBAL showed good conversion rates for both propanal (76–85 %) and butanal (66–88 %). For the cascades towards 3,4‐hexanediol and 4,5‐octanediol, the ligation was comparably fast, and 200 mm aldehyde was ligated within 60 min of reaction time. In CPME, the product concentration was increased by 12.5 % for 3,4‐hexanediol and 40 % for the more hydrophobic 4,5‐octanediol. These results show the positive impact of an organic reaction system for hydrophobic substrates such as propanal, and more pronounced with butanal. The mass balance, however, is not completely closed in any of the cases, which is most likely due to the usage of whole cells as a catalyst resulting in a heterogeneous system, in combination with the high reactivity of the substrates. The cells introduce complexity of the system with regard to metabolites, proteins, lipids, or other enzymes, which might react to some extent with the substrate, intermediate, or product. However, no major side products were detected during GC analysis. Furthermore, the distribution between catalyst and liquid phase might not be ideal.
Upscaling of 4,5‐octanediol production
The reactions in Figure 2 were performed in a 1 mL reaction volume. For the further conversion of the diols with chemocatalysis to form a hybrid process, larger quantities of product are needed. As an example, the cascade towards 4,5‐octanediol was performed on a 50 mL scale again both in organic and aqueous conditions for comparison (Figure 3).
Figure 3.
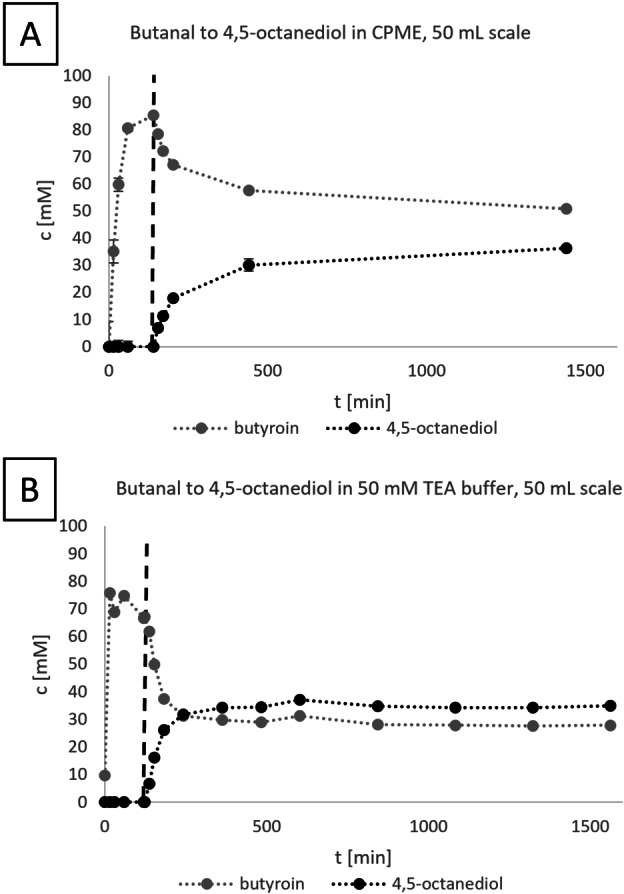
Cascade from butanal to 4,5‐octanediol on a 50 mL scale using the EasyMax® (Mettler Toledo, Columbus, OH, USA) system. Concentrations were determined using GC analysis. The dashed line indicates the addition of BlBDH and 1,2‐propanediol. The measurements were performed for (A) in analytical triplicates for (B) in a single measurement.
In general, the upscaling from the 1 mL reaction system was successful using the Easymax® system for both the aqueous and the MARS system. This system enables fine tuning and monitoring of various process parameters such as pH, temperature, stirring and facilitates automated sampling. The aqueous reaction did proceed faster in the 100 mL reactor than in the 1 mL vial, which can be due to the usage of an overhead stirrer and resulting better mixing.
Efficiency comparison of product separation from buffered and micro‐aqueous reaction systems
The usage of organic media as a reaction medium eases downstream processing. In Table 1, the steps used to purify 4,5‐octanediol from a buffered reaction system and from MARS are presented. The key difference between both is that no extraction of product is necessary for MARS and no proteins or other hydrophilic residues from the whole cells have to be removed since they do not dissolve in the organic reaction environment. However, lipids do dissolve in this environment and must be removed during the purification; here, silica chromatography was used for separation. In general, purifying the product from organic media costs much less energy in comparison to boiling off water, due to the higher enthalpy of evaporation of water in comparison to CPME. [25]
Table 1.
Purification of diols from aqueous and organic systems in comparison.
|
Step |
Aqueous |
Organic |
Aim |
|---|---|---|---|
|
centrifugation/filtration |
yes |
yes |
catalyst removal |
|
acid precipitation |
yes |
no |
cell debris removal |
|
organic extraction |
yes |
no |
solvent change for following distillation |
|
Distillation 1 |
yes |
yes |
volume reduction |
|
silica gel chromatography |
yes |
yes |
removal of intermediate and coproducts |
|
Distillation 2 |
yes |
yes |
dry final product |
Using this method, a yield after purification of 50.4 % was achieved for the aqueous system and 90 % for the purification from the MARS. Both products had a purity of about 99 % [nuclear magnetic resonance (NMR) spectroscopy, see Supporting Information Figure S15]. These results show the benefits of a product purification from organic solvents. Still, the recovery of the product was very laborious and time‐consuming, and it should be avoided if possible. In this example, the final diol was used as a substrate for the chemically catalyzed formation of cyclic acetals, using the [Ru(triphos)(tmm)] catalyst.[ 20a , 26 ] When the diol was produced in a nearly anhydrous medium such as the here used CPME‐system, an intermediate workup was not necessary, and the chemical synthesis could take place in the same medium as the biocatalysis. Moreover, the direct contact of the reaction mixture with the catalyst takes advantage of the reduction capability of the catalyst, which also catalyzes the reduction of any remaining intermediate propioin or butyroin to the respective diol. The final dioxolane can be distilled off from the reaction mixture and thereby purified effectively and easily.
Stereoselectivity of enzymatic syntheses
For the determination of the absolute configuration, all stereoisomers of 3,4‐hexanediol and 4,5 octanediol were synthesized (see the Supporting Information). The meso compounds were prepared enzymatically and purified for further analysis. The (R,R)‐ and (S,S)‐stereoisomers were prepared according to the protocol of Denmark and Vogler. [33]
The aim of the enzymatic cascades was the production of diols in high concentrations. For this, the used carboligase PfBAL was chosen, which had previously shown high activity, yet only low stereoselectivity towards the (S)‐propioin and (R)‐butyroin. The BlBDH used for the synthesis of meso‐4,5‐octanediol was highly (S)‐selective resulting in good d. r. values except for the upscaling reaction, where also (4S,5S)‐octanediol was formed as a second stereoisomer. The synthesis of 3,4‐hexanediol resulted mainly in the (3R,4R)‐hexanediol stereoisomer, with the main side product being meso‐3,4‐hexanediol (Table 2). This is due to the higher affinity of the LbADH to the substrate in (R)‐configuration, combined with its (R)‐selectivity.
Table 2.
Enantiomeric ratio (e. r.) and diastereomeric ratio (d. r.) as well as isomeric content of the main product of the synthesis of 3,4‐hexanediol, and 4,5‐octanediol in buffered media and in MARS after 6 h of reaction time.
|
Product |
MARS |
Buffered media |
||
|---|---|---|---|---|
|
e. r. |
d. r. |
e. r. |
d. r. |
|
|
(3R,4R)‐hexanediol |
>99 : 1 |
53 : 47 |
>99 : 1 |
71 : 29 |
|
meso‐4,5‐octanediol (1 mL) |
– |
– |
92 : 8 |
– |
|
meso‐4,5‐octanediol (50 mL) |
– |
– |
95 : 5 (7.3 h) |
– |
Transformation of diols to cyclic acetals using a ruthenium catalyst
The possibility to transform diols to cyclic acetals with carbon dioxide and molecular hydrogen could be realized recently with the development of the molecular catalysts Ru(triphos)(tmm). In these studies, 1,4‐dioxane proved to be the optimal solvent for the synthesis of cyclic acetals. [21] Herein, a further development step should be established by using formic acids as source of the methylene unit in the acetal. In an initial reaction with 3,4‐hexanediol and formic acid, the resulting (4S,5S)‐, (4R,5R)‐, and meso‐4,5‐diethyl‐1,3‐dioxolane were successfully obtained, albeit in a low yield of 9 % (Table 3, entry 1). Both dioxolanes were quantified by multinuclear NMR spectroscopy and analyzed by high‐resolution GC time‐of‐flight (ToF) (see Supporting Information Figures S17 and S22).
Table 3.
Optimization reactions for the synthesis of dioxolanes from racemic 3,4‐hexanediol and 4,5‐octanediol.
|
| |||||
|---|---|---|---|---|---|
|
Entry[a] |
Substrate (x mmol) |
Formic acid [mL] |
TON[d] (RR,SS) |
TON[d] (meso) |
Yield of acetals[d] [%] |
|
1[b] |
3,4‐hexanediol (1.9) |
0.3 |
41 |
10 |
9 |
|
2 |
3,4‐hexanediol (1.9) |
0.3 |
70 |
17 |
15 |
|
3 |
3,4‐hexanediol (1.4) |
0.3 |
59 |
17 |
18 |
|
4 |
3,4‐hexanediol (1.9) |
0.6 |
62 |
17 |
14 |
|
5[c] |
3,4‐hexanediol (1.9) |
2×0.3 |
101 |
32 |
23 |
|
6[c] |
3,4‐hexanediol (1.9) |
3×0.3 |
100 |
26 |
22 |
|
7 |
4,5‐octanediol (1.4) |
0.3 |
41 |
25 |
16 |
|
8[c] |
4,5‐octanediol (1.4) |
2×0.3 |
46 |
27 |
17 |
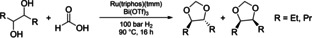
[a] Ru(triphos)(tmm) (0.003 mmol), Bi(OTf)3 (0.032 mmol), formic acid (0.3 mL), CPME (2 mL) 90 °C, 16 h, H2 100 bar. [b] 1,4‐Dioxane (2 mL). [c] Reloaded once/twice with additional formic acid (0.3 mL) after 16 h, respectively, 32/48 h reaction time. [d] TON=turnover number; TON and yield were determined by 1H NMR spectroscopy using mesitylene as internal standard.
The replacement of 1,4‐dioxane by CPME, the established solvent for the enzymatic formation of the diols, would offer the possibility to use a single solvent in both the enzymatic and the chemically catalyzed reaction without intermediate product isolation steps. Surprisingly, in comparison to the reaction with 1,4‐dioxane, the use of CPME increased the yield to 15 % (entry 2). Furthermore, decreasing the substrate concentration increased the yield to 18 % while the catalyst activity decreased only slightly from an overall turnover number of 87 to 76 (entry 3). At the established reaction conditions, a decomposition of formic acid during the transformation could limit the product formation. Thus, a reaction with enhanced formic acid loading was performed. However, increasing the amount of formic acid at the beginning of the reaction from 0.3 to 0.6 mL decreased the yield to 14 % (entry 4), possibly due to the too strong decrease in pH. Therefore, a reaction should be started with 0.3 mL of formic acid and reloaded with the same amount during the reaction. Thus, after 16 h reaction time, the autoclave was cooled and vented to an argon atmosphere. Subsequently, additional 0.3 mL of formic acid were added to the reaction mixture, repressurized with 100 bar of hydrogen and heated for another 16 h at 90 °C. This sequential procedure raised the yield to 23 % (entry 5), but further reloading steps proved not to be beneficial (entry 6). Transferring the established reaction conditions to the substrate 4,5‐octanediol resulted in the formation of (4S,5S)‐, (4R,5R)‐ and meso‐4,5‐dipropyl‐1,3‐dioxolanes in a yield of 16 % (entry 7). Finally, reloading the reaction with formic acid improved the yield further to 17 % (entry 8).
After these promising reactions with the pure starting materials, the 4,5‐octanediol produced by the enzymatic transformation was directly used as substrate in the reaction with the molecular transition metal catalyst (Table 4).
Table 4.
Utilization of 4,5‐octanediol from the enzymatic cascades as substrate for the synthesis of 4,5‐dipropyl‐1,3‐dioxolane.
|
| ||||
|---|---|---|---|---|
|
Entry[a] |
Substrate [x mmol] |
Formic acid [mL] |
Product ratio SS/meso |
Yield acetals[d] [%] |
|
1 |
4,5‐octanediol >99 % meso (1.4) |
0.3 |
0 : 100 |
11 |
|
2 |
4,5‐octanediol 93 % meso (1.4) |
0.3 |
34 : 66 |
13 |
|
3 |
4,5‐octanediol >99 % S,S (1.4) |
0.3 |
100:0 |
18 |
|
4[b] |
4,5‐octanediol >99 % S,S (1.4) |
2×0.3 |
100:0 |
31 |
|
5[c] |
4,5‐octanediol (1.3) without purification 89 % meso |
0.3 |
40 : 60 |
10 |

[a] Ru(triphos)(tmm) (0.003 mmol), Bi(OTf)3 (0.032 mmol), formic acid (0.3 mL), CPME (2 mL) 90 °C, 16 h, H2 (100 bar). [b] Reloaded with formic acid (0.3 mL), Ru(triphos)(tmm) (0.0025 mmol), Bi(OTf)3 (0.017 mmol), CPME (1 mL) after 16 h, 32 h reaction time. [c] 4,5‐Octanediol (89 % meso), butyroin (1.4 mmol). [d] TON and yield were determined by 1H NMR spectroscopy using mesitylene as internal standard.
Initially, meso‐4,5‐octanediol (>99 %) was employed in the catalytic experiment using the established reaction conditions. As a result, meso‐4,5‐dipropyl‐dioxolane was obtained as product leading to a yield of the acetals of 11 % (Table 4, entry 1). Using a sample containing 93 % of meso‐4,5‐octanediol and 7 % of (4S,5S)‐octanediol resulted in a comparable yield of 13 % (entry 2). Employing (4S,5S)‐octanediol in 99 % purity resulted in the formation of (4S,5S)‐dipropyl‐dioxolane in a yield of 18 % (entry 3). The yield of this reaction was significantly higher compared to the transformation with the enriched meso‐sample, hinting towards an increased reactivity of the (4S,5S)‐octanediol substrate in the acetal formation. Moreover, the yield could be even further enhanced to 31 % with a stereoselectivity of >99 % by reloading the reaction with additional formic acid, Ru‐catalyst and the Lewis acid (entry 4). Consequently, deactivation of the catalyst system may currently limit the turnover but could in future be circumvented by a switch to a continuous system. In a concluding experiment, a sample directly from the enzymatic cascade was employed. Initially, the sample volume was slightly reduced under vacuum and filtered over a silica plug. Before the reaction, 4,5‐octanediol (89 % meso), butyroin, and traces of 1,2‐propanediol were present in the mixture. During the reaction, the butyroin was completely hydrogenated to 4,5‐octanediol and 4,5‐dipropyl‐1,3‐dioxolane was obtained in 10.0 % yield (entry 5). This result showed that the catalytic system was highly tolerant to impurities and intermediates of previous reaction steps, which could not be removed by simple filtration. The shifting SS/meso product ratio at low yields compared to the employed SS/meso substrate ratio in entry 2 and 5 result from the increased reactivity of the (4S,5S)‐octanediol compared to the meso‐octanediol. The reactions of pure meso‐ or (4S,5S)‐octanediol prove that racemization is not taking place (entries 1, 3, and 5). Finally, the direct use of CO2 as C1 carbon source was investigated (Figure 4).
Figure 4.

Synthesis of dioxolanes from bioderived diols using CO2 as C1 source.
For these reactions, the challenges associated with carbon dioxide activation and subsequent hydrogenation to formic acid required the use of higher catalyst loadings in combination with HNTf2 as acid additive. Astonishingly, 4,5‐diethyl‐dioxolane could be obtained in a yield of 18 %, and 4,5 dipropyl‐dioxolane showed an increased yield of 25 %. The results clearly demonstrate the possibility to catalytically transform diols from the biocatalytic reaction directly to the respective acetals with carbon dioxide as renewable resource. Moreover, the catalytic system is tolerant to remaining intermediates and impurities after the enzymatic cascade, and the use of the same solvent CPME for all reaction steps enables a straightforward combination of bio‐ and chemocatalytic unit operations.
Conclusion
Biocatalysis in combination with a downstream reaction based on chemocatalysis enables effective access to promising products. However, both kinds of catalysts usually reveal their highest activity at very different conditions, and combining them in a one‐pot cascade is challenging. [4c] Herein, we could show that a switch of the formulation of the biocatalysts and the usage of cyclopentyl methyl ether (CPME) as a solvent enables a two‐step cascade in organic conditions. This approach eliminates the need for a subsequent workup when combined with a ruthenium transition metal catalyst reaction to yield cyclic acetals from formic and molecular hydrogen. Interestingly, CPME, which can be produced from renewable raw materials, turned out to be a solvent that is not only suitable for the biocatalysts but is also advantageous for the activity of the molecular ruthenium catalyst. This benefit of organic media already holds true for the transformation, but in the downstream processing, the advantages of the micro‐aqueous reaction system (MARS) become even clearer. [34] This system can be used in the future for further combination of chemo‐enzymatic cascades incorporating, for example, CO2 fixation. Additionally, we could show that (4S,5S)‐octanediol and (4S,5S)‐dipropyl‐1,3‐dioxolane can be obtained with high stereoselectivity, corroborating the effective interplay of the two catalytic systems. Conclusively, the high inherent selectivity of enzyme catalysis can be maintained under the chosen reaction conditions also in the hybrid process. For a subsequent process, enzymes with stereo‐complementary activity can be selected and combined in a modular fashion to access a platform of diols and dioxolanes with different chirality. An increase of the synthesis yield is target of current research. Additionally, suitable alcohol dehydrogenases could be used and improved to access the respective aldehydes from the biogenic alcohols. Also, since most metal catalysts can operate under organic conditions and a number of enzyme classes can be applied as lyophilized whole cells in MARS, [12] numerous potential chemo‐enzymatic cascades can be envisaged. In follow‐up experiments, the chemocatalyst will be optimized for continuous flow reactions with the aim to further increase the yield of the reaction. Overall, this cascade combines biodegradable, non‐toxic biocatalysts in a potentially biobased, green solvent with metal catalysts that can introduce CO2 or CO2‐derived formic acid to yield high‐value dioxolanes, thus combining many factors of sustainable processes.
Experimental Section
The analytical methods are described in the Supporting Information.
Biocatalyst preparation
For the production of the biocatalyst formulated as lyophilized whole cells, the following protocol was used: PfBAL (GenBank AY007242.1), LbADH (GenBank CAD66648.1), BlBDH [27] were produced in E. coli Bl21 (DE3) using autoinduction medium at 37 °C for 2 h followed by 48 h incubation at 20 °C with 90 rpm. The cell suspension was centrifuged for 20 min at 4000×g at 4 °C. The resulting cell pellet was subsequently frozen at −20 °C before it was lyophilized at −54 °C and 0.01 mbar using a Christ LT‐105 freeze drier (Martin Christ, Germany). The dried pellets were crushed using a mortar and stored at −20 °C.
Enzymatic reaction in a MARS
30 mg of lyophilized whole cells as catalyst was weighed in 1.5 mL glass vials, and CPME and 200 mm of aldehyde were added to a final volume of 1 mL. To start the reaction, 30 μL of 1 m TEA buffer pH 9 with 2.5 mm MgCl2 and 0.1 mm thiamine diphosphate was added, and the mixture was stirred at 1000 rpm at 30 °C in an Eppendorf shaker. For reactions containing LbADH for the reduction step, 1 m isopropanol was added for cofactor regeneration; for reaction containing the BlBDH, 600 mm 1,2‐propanediol was used.
Enzymatic reactions in buffered media
Identical to the setup of enzymatic reactions in MARS, but instead of CPME 50 mm TEA buffer pH 9 with 2.5 mm MgCl2 and 0.1 mm thiamine diphosphate was used.
Upscaling of enzymatic reaction
For upscaling in a 50 mL scale, an EasyMax® 402 system with a 100 mL reactor vessel purchased from MettlerToledo® was used. The composition of the solvent system was identical to the 1 mL scale described above. The stirring speed was 350 rpm and an EasySampler (Mettler Toledo, Columbus, OH, USA) was used for automated sampling.
Purification of 4,5‐octanediol
Product purification was performed after production of 4,5‐octanediol on a 50 mL scale in both organic and buffered conditions. The cells were removed by centrifugation at 4000×g for 20 min at room temperature followed by a protein precipitation step for the buffered medium to avoid interphase formation during organic extraction in the next step. HCl was added to lower the pH to 2 until a white precipitate formed. This precipitate was then removed via centrifugation at 4000×g for 20 min at room temperature. The product was extracted from the supernatant into an organic phase by mixing it once with an equal volume of ethyl acetate in a separating funnel. The organic phase was separated using a separation funnel and dried with MgSO4. The next steps were performed with both the organic reaction solution and the organic phase used for extraction of the buffered media. Distillation at 40 °C and 100 mbar reduced the final volume to approximately 4 mL and the solution was added to the top of a silica gel column, which was prepared with a mixture of petrol ether and ethyl acetate (5 : 1). 300 mL of the solvent mixture was used for the elution of the intermediate before 300 mL of a 1 : 1 mixture of petroleum ether and ethyl acetate was used for the fractionated elution of the product. Fractions containing only product were combined and distilled to remove the remaining solvents at 40 °C and 40 mbar until a white, dry powder remained.
Chemical synthesis of stereoisomers of diols
(3R,4R)‐Hexanediol, (3S,4S)‐hexanediol, (4R,5R)‐octanediol, and (4S,5S)‐octanediol were synthesized using the protocol of Denmark and Vogler. [33] The meso‐diols were prepared enzymatically using the above‐mentioned protocol and purified. Details on the synthesis, purification, and properties are given in the Supporting Information.
General procedure for the synthesis of cyclic acetals from diols and formic acid using a ruthenium and acid catalyst
All reactions were conducted in 20 mL stainless‐steel high‐pressure autoclaves. The catalyst [Ru(triphos)(tmm)], the Lewis acid Bi(OTf)3, and the substrate were weighed into a glass inlet with stir bar inside a glovebox. The inlet was placed into the autoclave, which was sealed and repeatedly evacuated and filled with argon for 5 cycles. The solvent CPME and formic acid were added to the autoclave in an argon counter stream. Then, the autoclave was pressurized at room temperature with H2 and heated to reaction temperature using a preheated aluminum cone. After completing the reaction time, the autoclave was cooled in an ice bath for 1 h and subsequently vented to atmosphere. The reaction solution was analyzed by 1H and 13C NMR spectroscopy, and the yields and TONs of the product were determined using mesitylene as internal standard.
Conflict of interest
The authors declare no conflict of interest.
1.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgments
This work is supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy – Exzellenzcluster 2186 “The Fuel Science Center” ID: 390919832. We thank Prof. Petra Siegert (University of Applied Sciences Aachen) for providing us with the plasmids for the BlBDH. Open Access funding enabled and organized by Projekt DEAL.
Spöring J.-D., Wiesenthal J., Pfennig V. S., Gätgens J., Beydoun K., Bolm C., Klankermayer J., Rother D., ChemSusChem 2023, 16, e202201981.
Contributor Information
Prof. Dr. Jürgen Klankermayer, Email: jklankermayer@itmc.rwth-aachen.de.
Prof. Dr. Dörte Rother, Email: do.rother@fz-juelich.de.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1.
- 1a. Marr A. C., Liu S., Trends Biotechnol. 2011, 29, 199–204; [DOI] [PubMed] [Google Scholar]
- 1b. Sheldon R. A., Brady D., ChemSusChem 2019, 12, 2859–2881. [DOI] [PubMed] [Google Scholar]
- 2. Turner N. J., Kumar R., Curr. Opin. Chem. Biol. 2018, 43, A1-A3. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Löwe J., Dietz K. J., Gröger H., Adv. Sci. 2020, 7, 1902973; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3b. Zhang X., King-Smith E., Renata H., Angew. Chem. Int. Ed. 2018, 57, 5037–5041; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 5131–5135; [Google Scholar]
- 3c. Gu X., Zhao J., Chen L., Li Y., Yu B., Tian X., Min Z., Xu S., Gu H., Sun J., J. Org. Chem. 2020, 85, 6844–6853. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Huang X., Cao M., Zhao H., Curr. Opin. Chem. Biol. 2020, 55, 161–170; [DOI] [PubMed] [Google Scholar]
- 4b. Hönig M., Sondermann P., Turner N. J., Carreira E. M., Angew. Chem. Int. Ed. 2017, 56, 8942–8973; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 9068–9100; [Google Scholar]
- 4c. Rudroff F., Mihovilovic M. D., Gröger H., Snajdrova R., Iding H., Bornscheuer U. T., Nat. Catal. 2018, 1, 12–22; [Google Scholar]
- 4d. Wang Y., Zao H., Catalysts 2016, 6, 194; [Google Scholar]
- 4e. Ascaso-Alegre C., Mangas-Sanchez J., Eur. J. Org. Chem. 2022, 2022, e202200093. [Google Scholar]
- 5.
- 5a. Liu Y., Liu P., Gao S., Wang Z., Luan P., González-Sabín J., Jiang Y., Chem. Eng. J. 2021, 420, 127659; [Google Scholar]
- 5b. Sato H., Hummel W., Gröger H., Angew. Chem. Int. Ed. 2015, 54, 4488–4492; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 4570–4574; [Google Scholar]
- 5c. Clayton A. D., Labes R., Blacker A. J., Curr. Opin. Green Sustain. Chem. 2020, 26, 100378; [Google Scholar]
- 5d. Cortes-Clerget M., Akporji N., Zhou J., Gao F., Guo P., Parmentier M., Gallou F., Berthon J.-Y., Lipshutz B. H., Nat. Commun. 2019, 10, 2169; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5e. Schmidt S., Castiglione K., Kourist R., Eur. J. Chem. 2018, 24, 1755–1768. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Cao Y., Li X., Ge J., Trends Biotechnol. 2021, 39, 1173–1183; [DOI] [PubMed] [Google Scholar]
- 6b. Devine P. N., Howard R. M., Kumar R., Thompson M. P., Truppo M. D., Turner N. J., Nat. Chem. Rev. 2018, 2, 409–421. [Google Scholar]
- 7. Bai S., Wu C., Gawlitza K., von Klitzing R., Ansorge-Schumacher M. B., Wang D., Langmuir 2010, 26, 12980–12987. [DOI] [PubMed] [Google Scholar]
- 8. Cicco L., Ríos-Lombardía N., Rodríguez-Álvarez M. J., Morís F., Perna F. M., Capriati V., García-Álvarez J., González-Sabín J., Green Chem. 2018, 20, 3468–3475. [Google Scholar]
- 9.
- 9a. Schaaf P., Bayer T., Koley M., Schnürch M., Bornscheuer U. T., Rudroff F., Mihovilovic M. D., Chem. Commun. 2018, 54, 12978–12981; [DOI] [PubMed] [Google Scholar]
- 9b. Gauchot V., Kroutil W., Schmitzer A. R., Eur. J. Chem. 2010, 16, 6748–6751. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. Gómez Baraibar Á., Reichert D., Mügge C., Seger S., Gröger H., Kourist R., Angew. Chem. Int. Ed. 2016, 55, 14823–14827; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 15043–15047; [Google Scholar]
- 10b. Bering L., Craven E. J., Sowerby Thomas S. A., Shepherd S. A., Micklefield J., Nat. Commun. 2022, 13, 380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wang Y., Zhang N., Zhang E., Han Y., Qi Z., Ansorge-Schumacher M. B., Ge Y., Wu C., Eur. J. Chem. 2019, 25, 1716–1721. [DOI] [PubMed] [Google Scholar]
- 12. van Schie M. M., Spöring J.-D., Bocola M., de María P. Domínguez, Rother D., Green Chem. 2021, 23, 3191–3206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jakoblinnert A., Mladenov R., Paul A., Sibilla F., Schwaneberg U., Ansorge-Schumacher M. B., de María P. Domínguez, Chem. Commun. 2011, 47, 12230–12232. [DOI] [PubMed] [Google Scholar]
- 14. Tufvesson P. R., Lima-Ramos J., Nordblad M., Woodley J. M., Org. Process Res. Dev. 2011, 15, 266–274. [Google Scholar]
- 15.
- 15a. Abel B. A., Snyder R. L., Coates G. W., Science 2021, 373, 783–789; [DOI] [PubMed] [Google Scholar]
- 15b. Gröger H., Hummel W., Curr. Opin. Chem. Biol. 2014, 19, 171–179; [DOI] [PubMed] [Google Scholar]
- 15c. Moity L., Benazzouz A., Molinier V., Nardello-Rataj V., Elmkaddem M. K., De Caro P., Thiébaud-Roux S., Gerbaud V., Marion P., Aubry J.-M., Green Chem. 2015, 17, 1779–1792; [Google Scholar]
- 15d. Snyder R. L., Choo Y., Gao K. W., Halat D. M., Abel B. A., Sundararaman S., Prendergast D., Reimer J. A., Balsara N. P., Coates G. W., ACS Energy Lett. 2021, 6, 1886–1891. [Google Scholar]
- 16.
- 16a. Walther T., François J. M., Biotechnol. Adv. 2016, 34, 984–996; [DOI] [PubMed] [Google Scholar]
- 16b. Green E. M., Curr. Opin. Biotechnol. 2011, 22, 337–343; [DOI] [PubMed] [Google Scholar]
- 16c. Mondal S., Santra S., Rakshit S., Kumar Halder S., Hossain M., Chandra Mondal K., Bioresour. Technol. 2022, 343, 126093. [DOI] [PubMed] [Google Scholar]
- 17.The Fuel Science Center, RWTH Aachen University, https://www.fuelcenter.rwth-aachen.de/cms/~siul/Fuelcenter/?lidx=1, 2019.
- 18. Schieweck B. G., Klankermayer J., Angew. Chem. Int. Ed. 2017, 56, 10854–10857; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 10994–10997. [Google Scholar]
- 19.
- 19a. Beydoun K., Thenert K., Wiesenthal J., Hoppe C., Klankermayer J., ChemCatChem 2020, 12, 1944–1947; [Google Scholar]
- 19b. Omari A., Heuser B., Pischinger S., Rüdinger C., Appl. Energy 2019, 239, 1242–1249; [Google Scholar]
- 19c. vom Lehn F., Cai L., Cáceres B. C., Pitsch H., Combust. Flame 2021, 232, 111525; [Google Scholar]
- 19d. Wildenberg A., Fenard Y., Carbonnier M., Kéromnès A., Lefort B., Serinyel Z., Dayma G., Le Moyne L., Dagaut P., Heufer K. A., Proc. Combust. Inst. 2021, 38, 543–553; [Google Scholar]
- 19e. Westbrook C., Heufer K. A., Wildenberg A., Energy Fuels 2021, 35, 15339–15359. [Google Scholar]
- 20.
- 20a. Mura M. G., Luca L. D., Giacomelli G., Porcheddu A., Adv. Synth. Catal. 2012, 354, 3180–3186; [Google Scholar]
- 20b. Supronowicz W., Ignatyev I., Lolli G., Wolf A., Zhao L., Mleczko L., Green Chem. 2015, 17, 2904–2911; [Google Scholar]
- 20c. Sordakis K., Tang C., Vogt L. K., Junge H., Dyson P. J., Beller M., Laurenczy G., Chem. Rev. 2018, 118, 372–433; [DOI] [PubMed] [Google Scholar]
- 20d. Savourey S., Lefèvre G., Berthet J.-C., Cantat T., Chem. Commun. 2014, 50, 14033–14036; [DOI] [PubMed] [Google Scholar]
- 20e. Inoue Y., Izumida H., Sasaki Y., Hashimoto H., Chem. Lett. 1976, 5, 863–864; [Google Scholar]
- 20f. Graf E., Leitner W., J. Chem. Soc., Chem. Commun. 1992, 623–624; [Google Scholar]
- 20g. Gassner F., Leitner W., J. Chem. Soc., Chem. Commun. 1993, 1465–1466. [Google Scholar]
- 21. Beydoun K., Klankermayer J., Chem. Eur. J. 2019, 25, 11412–11415. [DOI] [PubMed] [Google Scholar]
- 22. Beydoun K., Klankermayer J., ChemSusChem 2020, 13, 488–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.
- 23a. Sharpless K. B., Amberg W., Bennani Y. L., Crispino G. A., Hartung J., Jeong K. S., Kwong H. L., Morikawa K., Wang Z. M., J. Org. Chem. 1992, 57, 2768–2771; [Google Scholar]
- 23b. Kolb H. C., VanNieuwenhze M. S., Sharpless K. B., Chem. Rev. 1994, 94, 2483–2547. [Google Scholar]
- 24. Oeggl R., Maßmann T., Jupke A., Rother D., ACS Sustainable Chem. Eng. 2018, 6, 11819–11826. [Google Scholar]
- 25. de Gonzalo G., Alcántara A. R., Domínguez de María P., ChemSusChem 2019, 12, 2083–2097. [DOI] [PubMed] [Google Scholar]
- 26. Jakoblinnert A., Rother D., Green Chem. 2014, 16, 3472–3482. [Google Scholar]
- 27. Muschallik L., Kipp C. R., Recker I., Bongaerts J., Pohl M., Gellissen M., Schöning M. J., Selmer T., Siegert P., J. Biotechnol. 2020, 324, 61–70. [DOI] [PubMed] [Google Scholar]
- 28. Leuchs S., Greiner L., Chem. Biochem. Eng. Q. 2011, 25, 267–281. [Google Scholar]
- 29. Rother D., Kolter G., Gerhards T., Berthold C. L., Gauchenova E., Knoll M., Pleiss J., Müller M., Schneider G., Pohl M., ChemCatChem 2011, 3, 1587–1596. [Google Scholar]
- 30. Domínguez de María P., Pohl M., Gocke D., Gröger H., Trauthwein H., Stillger T., Walter L., Müller M., Eur. J. Org. Chem. 2007, 2007, 2940–2944. [Google Scholar]
- 31. Hailes H. C., Rother D., Müller M., Westphal R., Ward J. M., Pleiss J., Vogel C., Pohl M., FEBS J. 2013, 280, 6374–6394. [DOI] [PubMed] [Google Scholar]
- 32. Mondal S., Arifa A. A., Biswas P., Catal. Lett. 2017, 147, 2783–2798. [Google Scholar]
- 33. Denmark S. E., Vogler T., Chem. Eur. J. 2009, 15, 11737–11745. [DOI] [PubMed] [Google Scholar]
- 34. Spöring J.-D., Graf von Westarp W., Kipp C. R., Jupke A., Rother D., Org. Process Res. Dev. 2022. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.