

Abstract
Suppressing the amount of carbon dioxide in the atmosphere is an essential measure toward addressing global warming. Specifically, the photocatalytic CO2 reduction reaction (CRR) is an effective strategy because it affords the conversion of CO2 into useful carbon feedstocks by using sunlight and water. However, the practical application of photocatalyst‐promoting CRR (CRR photocatalysts) requires significant improvement of their conversion efficiency. Accordingly, extensive research is being conducted toward improving semiconductor photocatalysts, as well as cocatalysts that are loaded as active sites on the photocatalysts. In this review, we summarize recent research and development trends in the improvement of cocatalysts, which have a significant impact on the catalytic activity and selectivity of photocatalytic CRR. We expect that the advanced knowledge provided on the improvement of cocatalysts for CRR in this review will serve as a general guideline to accelerate the development of highly efficient CRR photocatalysts.
Keywords: alloying, cocatalysts, CO2 reduction reaction, metal nanoparticles, morphology, photocatalysts
Catalytic CRRiteria: This review summarizes recent research and development trends in the improvement of cocatalysts, which have a significant impact on the catalytic activity and selectivity of photocatalytic carbon dioxide reduction reaction (CRR). It provides advanced knowledge on the improvement of cocatalysts for CRR, serving as a general guide to accelerate the development of highly efficient CRR photocatalysts.

1. Introduction
1.1. Photocatalytic CO2 reduction
Since the Industrial Revolution in the late 18th century, humankind has consumed large quantities of fossil fuels (coal, oil, and natural gas). The use of fossil fuels generates nitrides and sulfides, which are harmful to the ecosystem, cause air pollution, and increase greenhouse gas emissions (e. g., carbon dioxide), which ultimately lead to climate change. Therefore, reducing the use of fossil fuels and thereby suppressing CO2 emissions are pressing issues that need to be addressed to prevent global warming. In addition, given the current situation in which it is not possible to promptly and completely stop the use of fossil fuels and shift to a society that uses alternative greener fuels (e. g., hydrogen, ammonia) as its energy source, it is essential to establish means to reduce CO2 concentration in the atmosphere in parallel with reducing the use of fossil resources. As a strategy to reduce atmospheric CO2 concentration, a technology to capture and bury emitted CO2 underground has been established and is currently being applied. Alternatively, as a more efficient strategy, the emitted CO2 could be recycled (carbon cycle) through its conversion into carbon monoxide (CO) and organic compounds (synthesis gases and hydrocarbon compounds such as raw materials for chemicals, agrochemicals, and pharmaceuticals; Figure 1).
Figure 1.

Schematic illustrations showing the transformation from CO2 in the atmosphere to useful carbon feedstocks.
To reduce CO2 to useful compounds (CO2 reduction reaction (CRR)), photochemical reduction using sunlight and water (H2O) and electrochemical reduction using H2O are attracting attention as clean and sustainable technologies. However, between these two technologies, photochemical reduction, termed artificial photosynthesis, is more advantageous as it does not require complex device design or detailed components and it is therefore less expensive to implement. Consequently, photochemical methods are gaining more interest as an environmentally sustainable means of CRR. However, high conversion efficiencies from sunlight are yet to be achieved for CRR by artificial photosynthesis, [1] for instance, through the fabrication of highly efficient photocatalysts/cocatalysts capable of high energy conversion efficiency from sunlight.
1.2. Cocatalysts for photocatalytic CRR
In CRR, semiconductor photocatalysts absorb light energy and generate excited electrons (e−) with reduction power and excited holes (h+) with oxidizing power (Figure 2A). However, when these semiconductor photocatalysts are used for CRR, a high conversion efficiency cannot be obtained in most cases. This is largely due to: 1) deactivation of the carriers by exciton recombination and thermal relaxation in a relatively short period of time and 2) the small number of active sites on the photocatalyst surface that can serve as reaction sites. Thus, a cocatalyst (active site) consisting of metal and/or metal oxide particles is generally loaded on the photocatalyst (Figure 2A). By loading such cocatalysts, 1) the excited e− or h+ generated within the photocatalyst are transferred to the cocatalyst, thereby extending the charge separation lifetime and 2) surface atoms and electronic states are created that are suitable for adsorption and desorption of the substrate, thereby improving the reactivity and selectivity of CRR. In addition, loading of the cocatalyst is effective in preventing self‐deactivation of the photocatalyst caused by its reducing or oxidizing power, thereby improving the durability of the photocatalyst.
Figure 2.

A) Schematic illustration of photocatalytic CRR: Step 1: light absorption, Step 2: charge separation, and Step 3: surface reactions. B) Principle of CRR using semiconductor photocatalysts.
Currently, the CRR process has the following limitations: 1) the reaction is difficult to proceed; 2) side reactions and reverse reactions are likely to occur; and 3) the selectivity of the reduction products is low. Limitation 1 is related to the fact that CO2 has a large negative standard Gibbs energy of evolution (−394.4 kJ mol−1) and is therefore a very stable molecule. As CO2 is more energetically stable than methanol (CH3OH; −166 kJ mol−1), methane (CH4; −51 kJ mol−1), or ethylene (C2H4; 68 kJ mol−1), the CRR process is an uphill reaction and reduction is thus difficult to proceed. Limitation 2 is related to the fact that the reduction potentials of CO2 and H2O are comparable to each other. This causes a competitive H2 evolution reaction (HER) to occur, in which H2O is reduced to H2, in parallel with CRR, making it difficult to only obtain the desired CO2‐reduced species with a high selectivity (Figure 2B). In addition, as oxygen (O2) and other substances produced in O2 evolution reaction (OER) are present in the system, a reverse reaction to CO2 can also occur upon reaction of these products and substances. Limitation 3 is related to the fact that there are various reducing species of CRR (e. g., CO, formic acid (HCOOH), formaldehyde (HCHO), acetic acid, CH4, and C2H4). Once such mixtures are obtained, their separation requires a great amount of energy. To overcome these challenges and realize artificial photosynthesis with high efficiency, high selectivity, and high durability, it is essential to improve the cocatalyst. [2]
1.3. Purpose and contents of this review
As described above, the improvement of cocatalysts plays an important role in the engineering of highly functional photocatalysts. Therefore, we have conducted extensive research on improving the cocatalysts on photocatalysts. [3] Our work as well as the research conducted by other groups have provided much insights into the selection/fabrication of appropriate cocatalysts for water‐splitting photocatalysts. [4] In contrast, the development of suitable cocatalysts for CRR is still in its infancy. To realize CRR with high efficiency, high selectivity, and high durability, a comprehensive review and summary of the findings and progress achieved to date is necessary. Therefore, in the present review, we summarize representative studies conducted to date on the design and engineering of cocatalysts in CRR using semiconductor photocatalysts.
In Section 2, the mechanism of CRR with semiconductor photocatalysts is described in detail, followed by a discussion on the characteristics and types of semiconductor photocatalysts used in CRR in Section 3. In Section 4, we describe examples of previous studies and findings on controlling the properties of cocatalysts, which is the subject of this review. Section 5 provides a summary of insights gained to date and an outlook on future prospects in this area.
The focus of the present review is on the control of cocatalyst particles in CRR using semiconductor photocatalysts. Therefore, readers interested in improving CRR photocatalysts using surface plasmon resonance, [5] single‐atom metal loading, [6] functional carbon, [7] or metal complex loading [8] are referred to those review articles.
2. Basis of Photocatalytic CRR
2.1. Mechanism
Photocatalytic CRR generally proceeds in H2O or in a vapor atmosphere. CRR proceeds according to the following three major steps (Figure 2A):[ 2 , 9 ] Step 1) the semiconductor photocatalyst absorbs light, which leads to the evolution of excited e− in the conduction band (CB) and excited h+ in the valence band (VB); Step 2) the excited e− and h+ diffuse to the photocatalyst surface and, when a cocatalyst is present, further migrate to the cocatalyst; and Step 3) CRR [Eqs. (1)–(5)] or OER [Eq. (6)] proceeds on the cocatalyst or on the photocatalyst surface.
| (1) |
| (2) |
| (3) |
| (4) |
| (5) |
| (6) |
In a typical CRR, a proton (H+) is included in the reaction system [Eqs. (1)–(5)]. This is because of the high stability of CO2 that requires a large reducing power of −1.9 eV (vs. normal H2 electrode (NHE); at pH 7) for its conversion into an anion radical species. Such a large reduction power is difficult to generate with visible‐light‐driven semiconductor photocatalysts. In contrast, in multi‐electron reactions using H+ [Eqs. (1)–(5)], HCOOH, CO, HCHO, CH3OH, and CH4 are the reaction products. The reduction potential required for these reactions to proceed is much lower than that in one‐electron reactions (reactions via anion radical species). Therefore, most CRR processes that employ semiconductor photocatalysts are designed as multi‐electron reaction systems using H+.
2.2. Reaction system
Theoretically, CRR proceeds when the CB minimum edge (CBM) of the semiconductor photocatalyst is more negative than the reduction potential of CO2. OER proceeds when the VB maximum edge (VBM) of the semiconductor photocatalyst is more positive than the oxidation potential of H2O (Figure 2B). Control of the CBM and VBM positions is relatively easy to achieve for semiconductor photocatalysts of sufficiently large band gaps (BGs; UV‐light‐driven semiconductor photocatalysts). However, as shown in Figure 3, visible light (∼400≤λ≤∼800 nm) accounts for about 43 % of sunlight. Therefore, it is indispensable to make effective use of sunlight using photocatalysts with small BGs, which can absorb visible light (namely, visible‐light‐driven semiconductor photocatalysts), for engineering semiconductor photocatalysts with high efficiencies. However, it is difficult to achieve appropriate CBM and VBM positions simultaneously in such semiconductor photocatalysts. Therefore, only a limited number of one‐step photocatalytic materials (Figure 4A) that can reduce CO2 under visible light have been reported.
Figure 3.
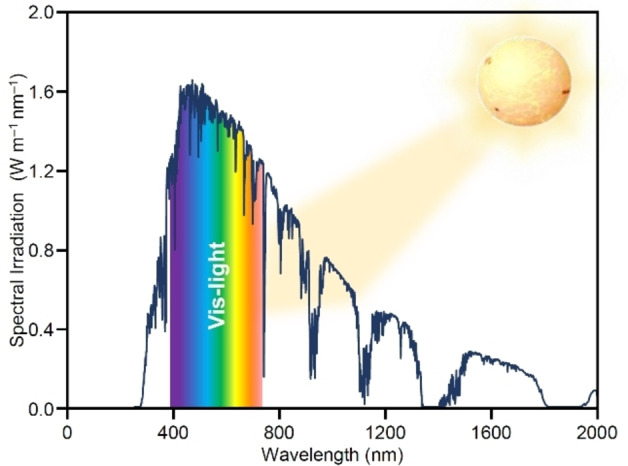
The sunlight spectrum. Visible light (∼400≤λ≤∼800 nm) accounts for about 43 % of sunlight.
Figure 4.
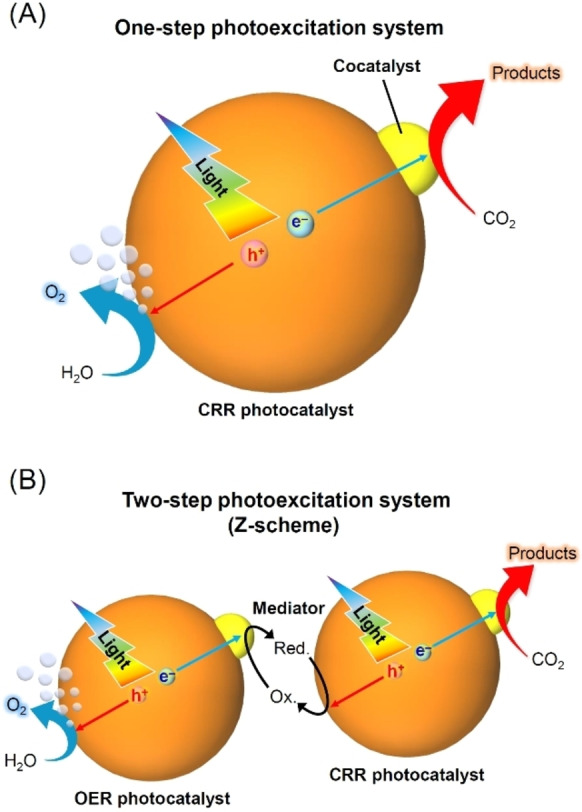
Schematic illustrations of photocatalytic reactions: A) one‐step photoexcitation system for CRR and B) Z‐scheme photoexcitation system for CRR. Red. and Ox. denote reductants and oxidants, respectively.
In contrast, CRR can also proceed according to a two‐step reaction mechanism involving a semiconductor photocatalyst capable of instigating both CRR and OER. These reactions function by combining a photocatalyst, which causes CRR and OER, and a redox couple (mediator), which is responsible for the transfer of excitons (e− and h+; Figure 4B). Such a system that mimics plant photosynthesis is called a Z‐scheme type. When using such Z‐scheme, there are significantly more types of photocatalysts available for study, and it is easier to use longer wavelength light with these photocatalysts than with the one‐step photocatalysts. However, there are some limitations such as the theoretically low conversion efficiency, blocking of light absorption, and photocatalyst inactivation due to side reactions resulting from the use of a mediator.
3. Photocatalysts Used for CRR
Since Honda and colleagues reported using a semiconductor for CRR in 1979, [10] various types of CRR photocatalysts have been subsequently developed. To date, the semiconductor photocatalysts include metal oxides, [11] metal sulfides, [12] metal‐free photocatalysts such as graphitic carbon nitride (g‐C3N4), [13] and metal‐organic frameworks (MOFs).[ 8a , 14 ] These semiconductor photocatalysts have a band structure suitable for CRR (Figure 5).[ 12 , 15 ]
Figure 5.

Band structures of A) UV‐ and B) visible‐light‐driven photocatalysts for CRR. The band position of the photocatalyst is calculated to be at pH 7.[ 12 , 15 ]
Because metal and O2 form a strong bond, metal oxides are highly stable in air and H2O. Therefore, metal oxides are often used as photocatalytic materials for the fabrication of semiconductor photocatalysts. Among the range of metal oxides available, those containing d0 metal elements (transition metal ions such as titanium ion (Ti4+), zirconium ion (Zr4+), niobium ion (Nb5+), tantalum ion (Ta5+), vanadium ion (V5+), tungsten ion (W6+), and cerium ion (Ce4+)) and d10 metal elements (typical metal ions such as zinc ion (Zn2+), indium ion (In3+), gallium ion (Ga3+), germanium ion (Ge4+), tin ion (Sn4+), and antimony ion (Sb5+)) can function as CRR photocatalysts (e. g., TiO2, Ga2O3, WO3, and ZnO).[ 15a , 15b , 15c , 16 ] Photocatalysts such as BaLa4Ti4O15, SrTiO3, and ZnGa2O4, which contain several metal elements, are also used for CRR. [17] Among them, metal oxides such as BiVO4, Bi2WO6, InTaO4, and InVO4 have a narrow BG and thereby absorb visible light.[ 15d , 15g , 15j , 18 ] Visible‐light‐driven photocatalysts can also be obtained by modifying the electronic structure of the UV‐light‐driven metal oxide photocatalysts. Representative approaches of achieving such modification are by: 1) hifting the energy position of the VBM to a negative potential by anion or metal cation substitution; [19] 2) forming impurity levels in the BG by doping; [20] and 3) narrowing the BG through the formation of a solid solution. [21] Accordingly, several visible‐light‐driven metal‐oxide‐based photocatalysts have been developed and reported to date through the application of these modification processes.
Typical examples of metal sulfide semiconductor photocatalysts include CdS and ZnS.[ 12 , 22 ] In contrast to the VB of metal oxides that is composed of O 2p orbitals, the VB of metal sulfides is composed of S 3p orbitals, which are located on the more negative side than the O 2p orbitals. Therefore, the BG of metal sulfides has a suitable width to absorb visible light, and CRR can be performed on metal sulfide photocatalysts under visible light irradiation. However, metal sulfides are readily oxidized by the h+ generated upon light irradiation, resulting in self‐decomposition and reduced durability of the photocatalyst. However, when the reaction is carried out in an aqueous solution containing sulfur species, self‐decomposition is suppressed, and a relatively high activity is achieved.
g‐C3N4 is an organic semiconductor photocatalyst that does not include a metal element.[ 13 , 15h , 23 ] g‐C3N4 can be synthesized by a simple method of thermal polymerization[ 15h , 24 ] using N‐containing precursors (e. g., urea, melamine, and cyanamide). Moreover, these precursors are naturally abundant hence g‐C3N4 can be synthesized at a low cost. g‐C3N4 has a BG of 2.7–2.9 eV, which is suitable for visible light absorption, and the band structure is also suitable for CRR because the CBM is composed of C p z orbitals and the VBM is composed of N p z orbitals. Because of these desirable properties, g‐C3N4 has attracted great attention as a visible‐light‐driven photocatalytic material for CRR.
MOFs, which are composed of metal ions/clusters and organic linker molecules, have also been extensively studied as a photocatalytic material for CRR.[ 8a , 14a , 14b , 25 ] In MOFs, charge separation occurs efficiently because of their large specific surface area and high density of catalytic activity sites. These characteristics make MOFs attractive as a CRR photocatalyst.
4. Cocatalyst Used for Photocatalytic CRR
As described in Section 3, the use of semiconductor photocatalysts with a suitable band structure enables CRR to proceed. However, as discussed in Section 2, the use of semiconductor photocatalysts alone often results in, for instance, low catalytic activity and the simultaneous evolution of multiple by‐products even when the reduction reaction occurs. To overcome these limitations, metal nanoparticles (NPs) are often loaded on the surface of the semiconductor photocatalyst as cocatalysts. Previous studies have reported that metal NPs or metal oxide NPs consisting of platinum, [26] palladium,[ 26l , 26u , 26ab , 26af , 27 ] gold,[ 26l , 26aa , 26ab , 26ac , 26ai , 26aj , 27k , 28 ] rhodium,[ 26z , 26al , 29 ] silver,[ 26l , 26ac , 26ai , 26aj , 26am , 27k , 30 ] ruthenium,[ 26aj , 26ak , 28b , 31 ] copper, –[ 26aj , 27g , 27k , 32 ] nickel,[ 27h , 32n , 33 ] molybdenum, [34] titanium,[ 32x , 35 ] indium, [36] or iridium, [37] can function as CRR cocatalysts. In particular, Pt and PdNPs are used for the hydrogenation reactions because they are favorable for the adsorption and desorption of protons and thereby easy to produce CH4. Cu is used to form a C−C bond whose formation is important for producing C2 products, such as ethane (C2H6) and ethanol (C2H5OH). To improve CRR activity and selectivity, controlling the properties of the cocatalyst is essential. Four examples of cocatalyst property control are discussed subsequently: 1) particle size control (Section 4.1; Figure 6A); [38] 2) chemical composition control (Section 4.2; Figure 6B);[ 38c , 39 ] 3) morphology control (Section 4.3; Figure 6C);[ 39d , 40 ] and 4) surface structure control (Section 4.4; Figure 6D). [41] Types of cocatalyst and photocatalysts, CRR products, and other information in the literatures are summarized in Table 1.
Figure 6.

Schematic illustrations of photocatalyst functionalization by controlling the A) particle size (Section 4.1), B) chemical composition (Section 4.2), C) morphology (Section 4.3), and D) surface structure (Section 4.4) of the cocatalyst.
Table 1.
Cocatalysts, size of cocatalysts, photocatalysts, CRR products, light sources, selectivities, and references for the selected literature.
|
Section |
Cocatalyst [wt%] |
Size of cocatalyst [nm] |
Photocatalysts |
CRR products |
Light sources |
Selectivity [%] |
Ref. |
|---|---|---|---|---|---|---|---|
|
4.1 (particle size) |
PtNPs (N/A)[a] |
1.04±0.08 |
TiO2 |
CH4 |
400 W Xe lamp, at 250–388 nm |
N/A[a] |
[38a] |
|
|
PtNPs (1.6) |
1.8 |
TiO2‐SiO2(HTSO) |
CO, CH4 |
300 W Xe lamp, AM 1.5 filter[b] |
39.1[c] |
[38b] |
|
4.2 (chemical composition) |
Pd7Cu1 NPs (N/A)[a] |
∼6 |
TiO2 |
CH4 |
300 W Xe lamp, >400 nm |
95.9[c] |
[39a] |
|
|
Pd1Cu2 NPs (1.69) |
8.8 |
g‐C3N4 |
CH4 |
300 W Xe lamp, at 420–780 nm |
96.5[c] |
[39b] |
|
|
Pt0.4Cu0.6 NPs (N/A)[a] |
N/A [a] |
TiO2 |
CH4 |
300 W Xe lamp, at 300–400 nm |
100[d] |
[39c] |
|
4.3 (morphology) |
tetra‐PdNPs (5.8) |
4.9 |
g‐C3N4 |
CO, C2H5OH, CH4 |
300 W Xe lamp, >400 nm |
78.1[e] |
[40a] |
|
|
tetra‐PdNPs (0.39) |
3.2 |
g‐C3N4 |
CH4, CH3OH |
300 W Xe lamp |
N/A[a] |
[40b] |
|
|
concave cube‐PtCuNPs (8.5) |
∼10.0 |
C3N4 |
CO, CH4 |
300 W Xe lamp, at 400–780 nm |
90.6[c] |
[39d] |
|
4.4 (surface structure) |
Cu2O/PtNPs (Cu: 1.70, Pt: 0.92) |
3.1±0.5 |
TiO2 |
CO, CH4 |
200 W Xe lamp, at 320–780 nm |
85.0[f] |
[41a] |
|
|
Cr(OH)3 ⋅ xH2O/AgNPs (Cr: 1.00, Ag: 1.00) |
N/A [a] |
Ga2O3 |
CO |
400 W Hg lamp |
83.8[g] |
[41b] |
|
|
Cr(OH)3 ⋅ xH2O/AgNPs (Cr: 0.25, Ag: 0.25) |
N/A [a] |
Ga2O3 |
CO |
400 W Hg lamp |
83.8[g] |
[41c] |
[a] N/A represent not applicable. [b] AM1.5G represent global standard solar spectrum (AM1.5G). [c] Selectivity in CH4 evolution is evaluated based on the required electrons using the following equation: Selectivity (%)=[8ν(CH4)]/[2ν(CO)+8ν(CH4)+2ν(H2)]×100, where ν(H2), ν(CO) and ν(CH4) stand for the evolution rates for H2, CO and CH4, respectively. [d] Selectivity in CH4 evolution is evaluated based on the required electrons using the following equation: Selectivity (%)=n(CH4)/[n(CO)+n(CH4)]×100, where n(CO) and n(CH4) stand for number of reacted electrons for CO and CH4, respectively. [e] Selectivity in carbon product evolution is evaluated based on the required electrons using the following equation: Selectivity (%)=[2ν(CO)+12ν(C2H5OH)+8ν(CH4)]/[2ν(CO)+12ν(C2H5OH)+8ν(CH4)+2ν(H2)]×100, where ν(H2), ν(CO), ν(CH4) and ν(C2H5OH) stand for the evolution rates for H2, CO, CH4, and C2H5OH, respectively. [f] Selectivity in carbon product evolution is evaluated based on the required electrons using the following equation: Selectivity (%)=[2n(CO)+8n(CH4)]/[2n(CO)+8n(CH4)+2n(H2)]×100, where n(H2), n(CO), and n(CH4) stand for the amounts (moles) of H2, CO, and CH4, respectively. [g] Selectivity in CO evolution is evaluated based on the required electrons using the following equation: Selectivity (%)=[2ν(CO)]/[2ν(CO)+2ν(H2)]×100, where ν(H2), and ν(CO) stand for the evolution rates for H2, and CO, respectively.
4.1. Particle size control
The size of the metal NPs that serve as cocatalysts has a significant effect on CRR activity and selectivity. [38] Biswas and colleagues investigated the effect of PtNP cocatalysts of different particle sizes on CRR activity in 2012. [38a] PtNPs of different particle sizes were formed on TiO2 (PtNPs/TiO2) by changing the deposition time of Pt atoms using a sputtering method (Figure 7A). When Pt was deposited for 20 s, small PtNPs (1.04±0.08 nm) were loaded on the photocatalyst. The resulting photocatalyst showed high activity (1361 μmol g−1 h−1), as assessed by the rate of CH4 evolution, with an estimated quantum yield of 2.41 % (Figure 7B). In comparison, when Pt was deposited for either a shorter time (e. g., 10 s) or a longer time (e. g., ≥30 s), smaller (0.63±0.06 nm) or larger (≥1.34±0.15 nm) PtNPs were loaded. These resulting photocatalysts showed lower activity for CH4 evolution than the photocatalyst loaded with 1.04±0.08 nm PtNPs (Figure 7B). The different CH4 evolution rates displayed by the PtNPs/TiO2 photocatalysts were attributed to changes in the Fermi level of the cocatalyst (Figure 7C). If the PtNPs are too small, electron transfer from the CB of TiO2 to PtNPs is unlikely to occur because the PtNPs have discrete energy bands. In contrast, if the PtNPs are too large, they can easily become the recombination center for e− and h+ owing to their electronic structure, which is comparable to that of the bulk metal. These results show that to improve the catalytic activity of CRR, it is essential to obtain an appropriate electronic structure, which can be achieved by controlling the size of the cocatalyst particles.
Figure 7.

A) Transmission electron microscope (TEM) images of PtNPs/TiO2 prepared by using different Pt deposition times: 20 (a), 30 (b), 45 (c), and 60 s (d). B) CO and CH4 yields obtained during CRR on commercially available TiO2 powder (P25), pristine TiO2 columnar film (TiO2 film), and PtNPs/TiO2 films prepared by using different Pt deposition times (10, 20, 30, 45, and 60 s). C) Schematic diagram of CO2‐photoreduction mechanism with PtNPs/TiO2 nanostructured films. The magnified circle (center) shows that the photogenerated e− can move rapidly within the highly oriented TiO2 single crystals and flow to the Pt deposits, where a redox reaction occurs to convert CO2 into CO and CH4. The energy levels of the PtNPs/TiO2‐CO2 system is also shown. Reproduced with permission from ref. [38a]. Copyright: 2012, American Chemical Society.
Likewise, Zhang and colleagues have reported the importance of controlling the particle size of cocatalysts in improving the catalytic activity of CRR in 2018. [38b] They synthesized PtNPs on porous TiO2‐SiO2 (HTSO) by a simple acid–base‐mediated alcohol reduction method (Figure 8A). They investigated the relation between the size of the PtNPs and CRR activity on the obtained photocatalysts, and revealed that the photocatalysts loaded with smaller PtNPs (i. e., 1.8 nm) showed higher CH4 evolution rate and HER rate than those loaded with larger PtNPs (3.4–7.0 nm; Figure 8B). To rationalize the results obtained, femtosecond transient absorption (fs‐TA) spectroscopy and transient photocurrent response measurements were performed. The results revealed that decreasing the particle size enhanced charge transfer at the metal‐support interface. Specifically, the authors suggested that the Fermi level shifted to the positive side as the particle size became smaller, which facilitated charge transfer at the interface. In contrast, larger PtNPs showed the high selectivity of CH4 evolution. When the size of PtNPs was larger, the ratio of terrace sites, such as Pt(111) planes, increased (Figure 8C). Density functional theory (DFT) calculations indicated that the rate‐determining step of *CO to *COH hydrogenation was more likely to occur on such Pt(111) surfaces (Figure 8D), thus explaining the higher CH4 selectivity displayed by the larger PtNPs. These results indicate that changes in the exposed crystal faces associated with size changes of the cocatalyst also have a significant effect on the selectivity of CRR. The study demonstrates that 1) the smaller particle size of the PtNPs improves the catalytic activity of CRR and HER but 2) the generation of terrace sites is required on the surface of the cocatalyst for improving the selectivity of CH4 evolution.
Figure 8.

A) Schematic illustration of the acid‐base‐mediated alcohol reduction method for controlling the size of PtNPs (a) and HRTEM images of PtNPs of sizes 1.8 (b), 3.4 (c), 4.3 (d), or 7.0 nm (e) loaded on HTSO. B) CH4 yields obtained during CRR on PtNPs/HTSO with different sized PtNPs under different atmospheres. C) Correlations between the selectivity for CH4 and surface site proportion as a function of the size of PtNPs. D) Free energy diagrams for the reduction of CO2 to CH4 using the thermochemical model on Pt(111) surface and Pt55. Reproduced with permission from ref. [38b]. Copyright: 2018. Springer Nature Limited.
4.2. Chemical composition control
Alloying of the cocatalyst improves the adsorption of intermediates and facilitates the progression of multi‐electron reactions, resulting in the improvement of the catalytic activity and selectivity in CRR.[ 38c , 39 ]
Xiong and colleagues successfully obtained alloy cocatalysts with isolated Cu atoms within PdNPs on TiO2 in 2017. [39a] Furthermore, the authors found that the resulting PdCu alloy NPs‐loaded photocatalysts (Pd x Cu1 NPs/TiO2; x=1, 3, 5, 7, 9, or 11) exhibited higher CH4 selectivity than PdNPs/TiO2 photocatalyst in which pure PdNPs were loaded as cocatalysts. The Pd x Cu1 NPs/TiO2 (x=1, 3, 5, 7, 9, or 11) photocatalysts with different alloy ratios were obtained by controlling the concentrations of the Pd and Cu salt precursors. Transmission electron microscopy (TEM) images (Figure 9Aa, b) and high‐angle annular dark‐field scanning TEM (HAADF‐STEM) images (Figure 9Ac, d) showed that Pd x Cu1 alloy cocatalysts were loaded on TiO2 with similar shape and size (average particle size: ∼6 nm) regardless of the Cu content. Among the Pd x Cu1 NPs/TiO2 photocatalysts examined, Pd7Cu1 NPs/TiO2 displayed the highest CH4 evolution rate (19.6 μmol g−1 h−1) and selectivity (95.9 %; Figure 9B). Diffuse reflection infrared Fourier transform spectroscopy (DRIFTS) showed that the evolution of intermediates during CRR was enhanced on the Pd7Cu1 NPs/TiO2 surface relative to the Pd1Cu1 NPs/TiO2 surface. First‐principles simulations also showed that CO2 was more strongly adsorbed on the cocatalyst surface of the Pd−Cu pair when Cu atoms were surrounded by a higher number of Pd atoms (Figure 9C). These two factors were interpreted as the origin of the increase in the CH4 evolution rate and selectivity of the final product when the Cu atoms were isolated in the Pd lattice to form an alloy cocatalyst.
Figure 9.

A) TEM images of Pd1Cu1 NPs/TiO2 (a) and Pd7Cu1 NPs/TiO2 (b) and HAADF‐STEM images of Pd1Cu1 NPs (c) and Pd7Cu1 NPs (d). The insets in (c) and (d) show atomic‐resolution images taken from the regions indicated by the boxes. B) Average evolution rates of CH4 and CO during CRR on different photocatalysts in the presence of H2O. C) Most favorable configurations and adsorption energies of CO2 at an isolated Cu atom (a) and two neighboring Cu atoms (b). Cu: orange, Pd: blue, C: gray, and O: red. Reproduced with permission from ref. [39a]. Copyright: 2017, American Chemical Society.
Bai and colleagues also reported a study on PdCu alloy cocatalysts with similar metal species in 2018. [39b] The authors investigated the CRR activity of PdCu‐ordered alloy cocatalysts (Figure 10A) formed on g‐C3N4 nanosheets. The results demonstrated that the Pd1Cu2 NPs/g‐C3N4 photocatalyst annealed at 375 °C in H2 atmosphere 1) had ordered alloy layers with a body‐centered cubic (bcc) structure (Figure 10A) and 2) had a higher CH4 evolution rate (4.95 μmol h−1 g−1) and selectivity (96.5 %) than the other examined PdCuNPs/g‐C3N4 photocatalysts with different composition ratios (Figure 10B). The authors further found that in the PdCu‐ordered alloy cocatalyst, 1) the electronic interaction between Pd and Cu was enhanced, thus increasing their electron trapping capacity and 2) isolated Cu sites were exposed on the cocatalyst surface (Figure 10A). The origin of the high catalytic activity and selectivity of the Pd1Cu2 NPs/g‐C3N4 photocatalyst was attributed to these two phenomena.
Figure 10.

A) Schematic illustrating enhanced electron‐trapping ability and enhanced number of the isolated Cu sites in ordered PdCu cocatalysts prepared at various Pd/Cu molar ratios and at various annealing temperatures; inset: atomic model of the ordered PdCu intermetallic structure. B) Average evolution rates of H2, CO, and CH4 and CH4 selectivity during CRR on g‐C3N4‐based photocatalysts under visible light irradiation. Reproduced with permission from ref. [39b]. Copyright: 2018, The Royal Society of Chemistry.
In a study by Zhao and colleagues in 2022, the authors investigated the selectivity and mechanism of CRR using photocatalysts loaded with PtCu alloy NPs composed of Pt and Cu. [39c] PtCuNPs/TiO2 were prepared by the H2 reduction method. X‐ray photoelectron spectroscopy measurements, STEM measurements, and energy‐dispersive X‐ray spectroscopy measurements confirmed the loading of the alloy NPs, consisting of Pt and Cu, on TiO2. The results of the catalytic activity study revealed that Pt0.4Cu0.6 NPs/TiO2 did not produce H2, and CH4 was obtained at 100 % selectivity (Figure 11A). Multiple experiments showed that alloying 1) suppressed the recombination of the photogenerated carriers and 2) enhanced charge transfer from the photocatalyst to the cocatalyst. In‐situ Fourier transform infrared spectroscopy (FTIR) and DFT calculations were performed to elucidate the mechanism of the selective evolution of CH4. The results suggested that the selective reduction to CH4 occurred by the following mechanism: 1) CO2 molecules adsorb chemically on the surface of the catalyst and they trap e−, producing the intermediate CO2 − (Figure 11B); 2) CO2 − causes the evolution of *CO, an important intermediate species for the generation of CH4 and 3) activation and hydrogenation of *CO is promoted (Figure 11C). The authors suggested that in this mechanism, desorption of *CO from the PtCu alloy catalyst surface is difficult because *CO and PtCuNPs form a strong bond (Figure 11D), resulting in increased CH4 selectivity and reaction activity.
Figure 11.

A) CO and CH4 yields during CRR over Pt x Cu y NPs/TiO2. B) CO2 in‐situ FTIR spectra of Pt0.4Cu0.6/TiO2. C) CO in‐situ FTIR spectra of Pt0.4Cu0.6 NPs/TiO2. D) Free energy diagrams of CO2 reduction on PtCuNPs. An asterisk denotes the adsorbed intermediate on the substrate. Reproduced with permission from Ref. [39c]. Copyright: 2022, The Royal Society of Chemistry.
4.3. Morphological control
The shape and crystal facets of the metal cocatalyst have significant effects on adsorption of CO2 and thereby the catalytic activity. Therefore, it is important to form cocatalysts with appropriate facets to obtain the desired outcome. Such optimum shape and crystal facets depend on the metal species of the cocatalyst.[ 39d , 40 ]
In 2014, Xiong and colleagues examined two types of photocatalysts loaded with differently shaped PdNP (∼4–6 nm) cocatalysts (cubic; cube‐PdNPs and tetrahedral; tetra‐PdNPs; Figure 12A) to investigate the effect of metal cocatalyst shape on CRR activity. [40a] The PdNPs cocatalysts of different shapes were formed on g‐C3N4 using appropriate capping agents in the liquid phase. As confirmed from the high‐resolution (HR) TEM images in Figure 12B, the NPs in the form of cube‐PdNPs and tetra‐PdNPs were loaded on the photocatalyst (cube‐PdNPs/g‐C3N4 and tetra‐PdNPs/g‐C3N4) and were composed of Pd(100) and Pd(111) planes, respectively. Measurements of the CRR activity revealed that tetra‐PdNPs/g‐C3N4 displayed approximately four times higher carbon product (CH4, C2H5OH, CO) selectivity than cube‐PdNPs/g‐C3N4. The photocurrent and photoluminescence measurements indicated that differences in the cocatalyst shape had no effect on charge transfer from g‐C3N4 to PdNPs. Therefore, simulation calculations were performed, which showed that the Pd(111) facet displayed a higher CO2 adsorption energy (E a; Figure 12C) and a lower activation energy barrier than the Pd(100) facet. These results suggest that loading of tetra‐PdNPs composed of Pd(111) planes is effective in engineering appropriate photocatalysts for CRR using Pd as a cocatalyst element.
Figure 12.

A) TEM and B) HRTEM images of cube‐PdNPs (a) and tetra‐PdNPs (b). C) Optimized configurations of CO2 adsorbed onto Pd(100) (a) or Pd(111) (b) facets; the adsorption energy Ea is also noted. Pd: green and blue, C: gray, and O: red. Reproduced with permission from ref. [40a]. Copyright: 2014, The Royal Society of Chemistry.
The effect of cocatalyst shape on selectivity during CRR was also reported by Yu and colleagues in 2017. [40b] PdNPs of different shapes were synthesized using different capping agents and subsequently loaded onto g‐C3N4 by electrostatic interaction. From the CRR activity measurements, it was found that tetra‐PdNPs/g‐C3N4 displayed 1.42 times higher CH3OH evolution rate than cube‐PdNPs/g‐C3N4 (Figure 13A). In situ FTIR spectrum of tetra‐PdNPs/g‐C3N4 during photoirradiation displayed absorption bands related to the evolution of intermediates (HCOOH and HCHO; Figure 13B). This result suggests that CRR on tetra‐PdNPs/g‐C3N4 surfaces proceeds according to a multistep mechanism involving the evolution of intermediates, such as HCOOH and HCHO, which are subsequently converted into CH4 and CH3OH. In addition, DFT calculations showed that 1) CO2 was more strongly adsorbed on tetra‐PdNPs than on cube‐PdNPs at all sites of the bridge, Top1, and Top2 positions (Figure 13Ca–c) and 2) the product, CH3OH, more readily desorbed from the Pd(111) surface than from the Pd(100) surface (Figure 13Cd). They concluded that tetra‐PdNPs/g‐C3N4 with a Pd(111) facet exhibit high CRR activity due to these factors.
Figure 13.

A) Evolution yields of CH4 and CH3OH during CRR on different photocatalysts – CN: g‐C3N4 only, CN‐1: photocatalyst subjected to the same treatment but without PdNPs, cube: cube‐PdNPs/g‐C3N4, and tetra: tetra‐PdNPs/g‐C3N4. B) In‐situ FTIR spectra of tetra‐PdNPs/g‐C3N4 subjected to different CRR conditions. C) Optimized geometrical structures of CO2 adsorption on Pd(100) facets or Pd(111) facets at the bridge position (a), Top1 position (b), and Top2 position (c), and optimized geometrical structures oof CH3OH adsorption on Pd(100) facets or Pd(111) facets (d). Pd: blue and orange, C: green, O: red, and H: white. Reproduced with permission from ref. [40b]. Copyright: 2017, Elsevier B.V.
This shape dependence was also observed for photocatalysts featuring alloy cocatalysts. In 2017, Bai and colleagues reported that the rate and selectivity of CH4 evolution was dependent on the crystal facet of PtCu alloy cocatalysts. [39d] PtCu alloy cocatalysts were formed on g‐C3N4 by hydrothermal reduction of a solution containing the metal salts (K2PtCl4, CuCl2) and polyvinylpyrrolidone. The shape of the cocatalyst was controlled by adjusting the amount of hydrogen chloride (HCl), which acts as an oxidative etching agent, to obtain cube‐PtCuNPs/g‐C3N4 (Figure 14Aa) and concave cube‐PtCuNPs/g‐C3N4 (with a concave surface; Figure 14Ab). The HRTEM measurements revealed that the (100) and (730) facets were exposed in each cocatalyst (Figure 14Ac, d). The concave cube‐PtCuNPs/g‐C3N4 displayed CO and CH4 evolution rates of 0.046 and 0.112 μmol h−1, respectively. These evolution rates were respectively about two and three times higher than the CO and CH4 evolution rates obtained on cube‐PtCuNPs/g‐C3N4 (Figure 14B). Furthermore, concave cube‐PtCuNPs/g‐C3N4 exhibited a CH4 selectivity of 90.6 %, which was higher than that (85.9 %) displayed by cube‐PtCuNPs/g‐C3N4. The DFT calculations suggested that the adsorption energy of CO2 was higher on the PtCu(730) facet than on the PtCu(100) facet, indicating that the PtCu(730) facet had superior CO2 molecular adsorption capacity (Figure 14C). In addition, there is generally a relatively strong electronic interaction between the Pt atom in the low‐coordinated state and CO2. These results explained the high CH4 evolution activity and selectivity displayed by concave cube‐PtCuNPs/g‐C3N4.
Figure 14.

A) TEM (top) and HRTEM (bottom) images of cube‐PtCu (left) and concave cube‐PtCu (right) NPs. B) Photocatalytic H2, CO, and CH4 evolution rates during CRR on photocatalysts g‐C3N4, cube‐PtCuNPs/g‐C3N4, or concave cube‐PtCuNPs/g‐C3N4. C) Most stable configurations of CO2 adsorbed on PtCu(100) or PtCu(730) facets and associated adsorption energies. Pd: blue, Cu: pink, C: gray, and O: red. Reproduced with permission from ref. [39d]. Copyright: 2017, The Royal Society of Chemistry.
4.4. Surface structure control
To improve the activity and selectivity of CRR, it is also important to suppress the competing HER and reverse reaction that produces CO2. Therefore, several groups have attempted to form cocatalysts with a core–shell structure using two different metal species. [41]
In 2013, Wang and colleagues reported the synthesis of cocatalysts with a core–shell structure composed of Pt and Cu2O and their high selectivity for the evolution of CO and CH4. [41a] A photodeposition method was used to form PtNPs on TiO2 with a particle size of about 3.1 nm. Copper sulfate was then added to form Cu2O/PtNPs/TiO2, in which PtNPs (core) were covered by Cu2O (shell) in a stepwise photodeposition. The thickness of the shell layer composed of Cu2O was controlled by the light irradiation time (x h) (Cu2O/PtNPs/TiO2‐x h). As confirmed by TEM analysis, the Cu shell completely covered the PtNPs after light irradiation for 5 h (Cu2O/PtNPs/TiO2‐5 h; Figure 15A). The evolution of the shell layer after 5 h of light irradiation was also confirmed by high‐sensitivity low‐energy ion scattering (HS‐LEIS) measurements (Figure 15B). Cu2O/PtNPs/TiO2‐5 h, with a Cu content of 3 wt%, showed a CRR selectivity of 85 % (Figure 15C). Within this core–shell configuration, the PtNPs as the core have a high electron capture capacity, while the Cu2O as the shell suppresses the reduction of H2O to H2. Therefore, the core–shell configuration of this Cu2O/PtNPs/TiO2 photocatalyst appears to be conducive to suppressing the competing HER and enabling CRR on the photocatalyst.
Figure 15.

A) HRTEM images of Cu2O/PtNPs/TiO2‐1 h (a), Cu2O/PtNPs/TiO2‐2 h (b), Cu2O/PtNPs/TiO2‐5 h (c), and Cu2O/PtNPs/TiO2‐10 h (d). B) HS‐LEIS spectra of Cu/TiO2 (a), Pt/TiO2 (b), Pt−Cu/TiO2 (c), and Cu2O/PtNPs/TiO2‐5 h (d) photocatalysts recorded at 5 keV 20Ne+. C) Dependence of the photocatalytic behavior on the Cu content in the Cu2O/PtNPs/TiO2‐5 h catalysts for CRR in the presence of H2O. Reproduced with permission from ref. [41a] Copyright: 2013, Wiley‐VCH.
In contrast, in the core–shell‐type cocatalyst reported in 2018 by Tanaka and colleagues, the reverse reaction of CRR was significantly suppressed instead of the HER (Figure 16A). [41b] The core–shell‐type cocatalysts consisting of AgNP cores and chromium hydroxide (Cr(OH)3) shells were formed on Ga2O3 by photodeposition (Cr(OH)3 ⋅ xH2O/AgNPs/Ga2O3). HRTEM analysis confirmed the formation of Cr(OH)3 ⋅ xH2O shell of about 3–5 nm thickness on the AgNP surface (Figure 16B). The obtained Cr(OH)3 ⋅ xH2O/AgNPs/Ga2O3 photocatalyst exhibited an evolution rate of 480 μmol h−1 and a selectivity of 83.8 % for the conversion of CO2 into CO. These values were respectively 2.4 and 2.0 times higher than those of AgNPs/Ga2O3 photocatalyst without a core–shell structure. Cr(OH)3 ⋅ xH2O, which forms the shell layer, is assumed to change to the carbonate compound, Cr2(OH)2m (CO3)(3‐m) ⋅ xH2O, in NaHCO3 solution. This shell layer of carbonate compounds is believed to provide continuous supply of CO2 molecules to the AgNP core, while preventing the approach of O2, thereby suppressing the reverse reaction on Cr(OH)3 ⋅ xH2O/AgNPs/Ga2O3 and leading to high CO selectivity and evolution rate.
Figure 16.

A) Evolution rates of H2 (▴), O2 (▪), CO (•), and CO2 (⧫) of the reverse reaction for the photocatalytic conversion of CO2 in H2O over AgNPs/Ga2O3 (a) and Cr(OH)3 ⋅ xH2O/AgNPs/Ga2O3 (b). Photocatalyst powder: 0.5 g; reaction solution: 1.0 L H2O; flowing rates of gases: 20 mL min−1 CO/Ar mixture gas (5.0 %), 0.64 mL min−1 O2, and 9.4 mL min−1 Ar; solution: 1.0 L H2O; Ag loading amount: 1.0 mol%; Cr loading amount: 1.0 mol%; light source: 400 W high‐pressure Hg lamp. B) TEM (a) and HRTEM (b) images of Cr(OH)3 ⋅ xH2O/AgNPs/Ga2O3. Reproduced with permission from ref. [41b]. Copyright: 2018, The Royal Society of Chemistry.
In 2019, Tanaka and colleagues studied the effect of shell thickness on the photocatalytic activity and selectivity for the same photocatalyst. [41c] The studies revealed that 1) high CO evolution rates were obtained when Cr(OH)3 ⋅ xH2O/AgNPs/Ga2O3 was prepared at Ag : Cr ratios of 1 : 1 and 2) the highest CO evolution rate (525.3 μmol h−1) and selectivity (85.2 %) were obtained for photocatalysts loaded with Ag and Cr both at 0.25 wt%. Examination of the chemical composition and shape of the Cr shell on the photocatalyst revealed that Cr(OH)3 ⋅ xH2O was transformed to Cr(OH) x (CO3) y during the photocatalytic reaction (Figure 17A). Furthermore, the rate of CO evolution was significantly dependent on the thickness of the shell layer (Figure 17B). When a shell layer with appropriate thickness was formed on the surface of the Ag core cocatalyst, CO2 was stably trapped within the active site, allowing CO2 to be preferentially reduced to CO (Figure 17C). However, a thicker shell layer would prevent the approach of carbon species and protons to the Ag core. These results reveal that the formation of a shell layer of appropriate thickness is key to achieve high catalytic activity and selectivity. Such functional shells have been reported to also form using carbon[ 7b , 42 ] and NiO.[ 41d , 41v ]
Figure 17.
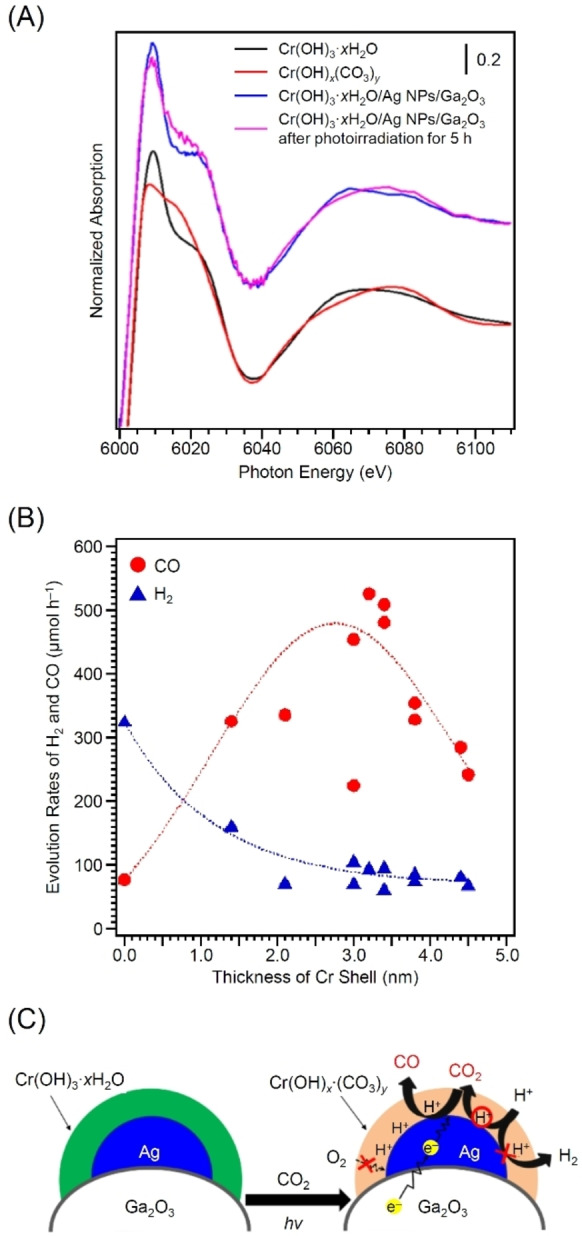
A) Cr K‐edge X‐ray absorption near‐edge structure spectra of Cr(OH)3 ⋅ xH2O (black), Cr(OH) x (CO3) y (red), as‐prepared 1.0 mol% Cr(OH)3 ⋅ xH2O/AgNPs/Ga2O3 (blue), and Cr(OH)3 ⋅ xH2O/AgNPs/Ga2O3 after photoirradiation for 5 h (pink). B) Dependence of the evolution rates of CO (•) and H2 (▴) on the thickness of the Cr(OH)3 ⋅ xH2O shell with various loading amounts of Ag and Cr. Dotted lines represent the data‐fitted curves. C) Schematic illustration of the mechanism of the photocatalytic conversion of CO2 into CO on Cr/Ag/Ga2O3. Reproduced with permission from ref. [41c]. Copyright: 2019, American Chemical Society.
5. Summary and Outlook
The area of CRR with semiconductor photocatalysts is still in its developing stage, with knowledge acquired from extensive studies conducted in the area. To contribute to future advances in engineering CRR photocatalysts with high efficiency, high selectivity, and high durability, this review has provided a summary of the major findings of recent studies conducted in the field, with a focus on the effects of controlling the properties of metal NP cocatalysts on CRR efficiency. The main insights and discussion points are as follows:
A decrease in the size of metal NP cocatalysts affords an increase in the number of active sites owing to an increase in the specific surface area. However, if the size is excessively reduced, the electronic states become discretized. Therefore, electron transfer from the photocatalyst to the cocatalyst is disrupted, potentially leading to a reduction in activity. In addition, changes in particle size lead to changes in the exposed crystal facet. Therefore, to achieve high selectivity and activity, the loading of metal NPs of suitable size is important.
Alloying PdNPs or PtNPs with Cu suppresses recombination of the photogenerated carriers, enhances charge transfer from the photocatalyst to the cocatalyst, and stabilizes the reaction intermediates, enhancing the selectivity and rate of CH4 evolution. If isolated Cu sites and ordered alloying are achieved, the selectivity of the products can be further enhanced.
Control of the exposed crystal facets of the metal NP cocatalysts contributes to the improvement of the activity and selectivity of the CRR. In the PdNP cocatalyst, metal NPs with exposed Pd(111) surfaces are suitable cocatalysts for CRR. In the PtCu alloy cocatalyst, exposure of the PtCu(730) surface leads to highly efficient and selective CH4 evolution.
Suppression of the competing HER or the reverse reaction improves the activity and selectivity of CRR. In particular, the formation of shell layers can be effectively used to suppress these reactions.
Given the knowledge gained to date, further areas remain to be explored, as summarized below, to guide the development of future CRR photocatalysts.
CRR is a multi‐electron reaction that employs several electrons simultaneously, and a longer charge separation lifetime contributes significantly to the activity. It is expected that the carrier lifetimes of various photocatalysts will be elucidated by fluorescence lifetime measurements, TA spectroscopy, [43] and time‐resolved microwave spectroscopy, [44] and that photocatalysts and cocatalysts with high charge separation efficiency will be designed and developed based on the knowledge obtained.[ 26ba , 45 ]
Previous studies have demonstrated that the catalytic activity can be enhanced when cocatalysts are loaded on suitable crystal facets for the reduction and oxidation reactions.[ 28c , 30n , 30o , 46 ] Therefore, it is expected that CRR photocatalysts with improved activity and selectivity will be developed in the future by establishing methods to selectively load cocatalysts that have size, alloy structures, crystal facets, and shell structures suitable for achieving high activity and selectivity on the crystal facets of the photocatalysts where CRR occurs.
It is expected that the geometric structure of highly active and selective cocatalysts will be revealed at the atomic level by aberration‐corrected TEM and STEM [47] measurements, thereby providing a clearer understanding of the effect of surface structure on catalytic activity.
Atomically precise metal nanoclusters (NCs), [48] whose geometric/electronic structures have been revealed by single‐crystal X‐ray structural analysis and DFT calculations have been reported to be highly active cocatalysts for photocatalytic water‐splitting reaction and other catalytic reactions. In addition, the use of atomically precise metal NCs as cocatalysts is also beneficial to obtain deeper understanding of the reaction mechanisms. Indeed, we have succeeded in revealing the reason why Pd doping of AuNC cocatalysts decreases the water‐splitting activity, whereas Pt doping of AuNC cocatalysts increases it by using the atomically precise metal NCs as a cocatalyst. [3d] Moreover, single‐atom (SA) catalysts have been reported to be efficient cocatalysts for photocatalytic CRR, [49] and their use also seems to be beneficial for obtaining deeper understanding of the reaction mechanisms. In future studies, it is expected that deeper understanding will be obtained of the reaction mechanisms in CRR by using the atomically precise metal NCs and SAs as cocatalysts and through performing operando measurements [50] such as in‐situ X‐ray absorption fine structure, in‐situ hard X‐ray photoelectron spectroscopy, and DRIFTS.
DFT calculations are important for understanding the rate‐limiting step of a reaction. [51] However, at present, metal NP cocatalyst structures are often calculated in a simplified manner owing to computational cost issues. In the future, theoretical calculations on actual cocatalysts are expected to be conducted, providing deeper understanding of the reaction mechanisms and thereby providing clear design guidelines for the development of highly active and selective catalysts.
It is expected that researchers in the field of photocatalysis, as well as those in the fields of metal NC chemistry, [52] surface spectroscopic chemistry, [53] and theoretical chemistry [54] will actively engage in the development of materials for application in the CRR, thereby significantly advancing the field of CRR photocatalysts.
Although the number of published papers on the photocatalytic conversion of CO2 increases year by year, the current state of the art is uneven. Namely, of the previous studies, some do not satisfy the requirements of the field of photocatalytic conversion of CO2. The important requirements are as follows: 1) isotope experiments should show the carbon source of the reduction product to be CO2 (not contamination); 2) the competing H2 production must also be analyzed accurately, and the number of excited electrons consumed in the production of the CO2 reduction products must be greater than those in H2 production; 3) the oxidation product (in most cases, O2; the oxidation product of H2O) must be analyzed accurately, and the ratio of excited electrons to holes consumed in the reaction must be 1. In future studies, these requirements are expected to be inevitably satisfied to avoid misunderstanding and thereby establish clear design guidelines for high‐performance CRR photocatalysts.
Conflict of interest
There are no conflicts to declare.
6.
Biographical Information
Tokuhisa Kawawaki is a Junior Associate Professor in the Department of Applied Chemistry at Tokyo University of Science. He received his Ph. D. degree in applied chemistry from the University of Tokyo in 2015. In 2016, he worked as a Japan Society for the Promotion of Science (JSPS) postdoctoral fellow at the University of Melbourne. He then worked as a JSPS super postdoctoral fellow at Kyoto University. In 2019, he moved to his current University. His current research topics include the synthesis of metal nanoparticles and nanoclusters in solution and their application in photoelectrochemistry and photocatalysis.

Biographical Information
Yuki Akinaga is a masters student in the Negishi group at Tokyo University of Science. He received his B. Sc. degree in chemistry from Tokyo University of Science in 2021. His main research focuses on the development of high‐performance water‐splitting and CRR photocatalysts using metal clusters and single atoms as cocatalysts.

Biographical Information
Daichi Yazaki is a masters student in the Negishi group at Tokyo University of Science. He received his B. Sc. degree in chemistry from Tokyo University of Science in 2021. His main research interest is the development of high‐performance water‐splitting photocatalysts using atomically precise metal clusters as cocatalysts.

Biographical Information
Hinano Kameko is a masters student in the Negishi group at Tokyo University of Science. She received her B. Sc. degree in chemistry from Tokyo University of Science in 2022. Her research focuses on the development of high‐performance water‐splitting and CRR photocatalysts by using metal clusters and single atoms as cocatalysts.

Biographical Information
Daisuke Hirayama is a masters student in the Negishi group at Tokyo University of Science. He received his B. Sc. degree in chemistry from Tokyo University of Science in 2022. His main research interest is on the development of high‐performance water‐splitting photocatalysts by using atomically precise metal clusters as cocatalysts.

Biographical Information
Yuichi Negishi is a professor in the Department of Applied Chemistry at Tokyo University of Science. He received his Ph. D. degree in chemistry in 2001 under the supervision of Prof. Atsushi Nakajima from Keio University. Before joining Tokyo University of Science in 2008, he was employed as an assistant professor at Keio University and at the Institute for Molecular Science. His current research interests include the precise synthesis of stable and functionalized metal nanoclusters for use in energy‐ and environmental‐related application areas.

Acknowledgments
This work was supported by the Japan Society for the Promotion of Science (JSPS) KAKENHI (grants no: 20H02698 and 20H02552), Scientific Research on Innovative Areas “Innovations for Light‐Energy Conversion” (grants no: 18H05178 and 20H05115), Scientific Research on Innovative Areas “Hydrogenomics” (grant no: 21H00027), Scientific Research on Innovative Areas “Aquatic Functional Materials” (grants no: 22H04562), and the JST Adaptable and Seamless Technology Transfer Program through Target‐driven R&D (A‐STEP, grant no: JPMJTM20MS). We also acknowledge funding support from the Yazaki Memorial Foundation for Science and Technology, the Ogasawara Foundation for the Promotion of Science and Engineering, the Kao Foundation for Arts and Sciences, TEPCO Memorial Foundation, the Japan Science Society, the Takahashi Industrial and Economic Research Foundation, and the Kubota Corporation.
Kawawaki T., Akinaga Y., Yazaki D., Kameko H., Hirayama D., Negishi Y., Chem. Eur. J. 2023, 29, e202203387.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1. Lingampalli S. R., Ayyub M. M., Rao C. N. R., ACS Omega 2017, 2, 2740–2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.
- 2a. Li X., Yu J., Jaroniec M., Chen X., Chem. Rev. 2019, 119, 3962–4179; [DOI] [PubMed] [Google Scholar]
- 2b. Wang Q., Pan Z., Nano Res. 2022, 15, 10090–10109. [Google Scholar]
- 3.
- 3a. Kurashige W., Kumazawa R., Ishii D., Hayashi R., Niihori Y., Hossain S., Nair L. V., Takayama T., Iwase A., Yamazoe S., Tsukuda T., Kudo A., Negishi Y., J. Phys. Chem. C 2018, 122, 13669–13681; [Google Scholar]
- 3b. Kurashige W., Mori Y., Ozaki S., Kawachi M., Hossain S., Kawawaki T., Shearer C. J., Iwase A., Metha G. F., Yamazoe S., Kudo A., Negishi Y., Angew. Chem. Int. Ed. 2020, 59, 7076–7082; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 7142–7148; [Google Scholar]
- 3c. Negishi Y., Matsuura Y., Tomizawa R., Kurashige W., Niihori Y., Takayama T., Iwase A., Kudo A., J. Phys. Chem. C 2015, 119, 11224–11232; [Google Scholar]
- 3d. Kurashige W., Hayashi R., Wakamatsu K., Kataoka Y., Hossain S., Iwase A., Kudo A., Yamazoe S., Negishi Y., ACS Appl. Energy Mater. 2019, 2, 4175–4187; [Google Scholar]
- 3e. Kawawaki T., Kataoka Y., Hirata M., Akinaga Y., Takahata R., Wakamatsu K., Fujiki Y., Kataoka M., Kikkawa S., Alotabi A. S., Hossain S., Osborn D. J., Teranishi T., Andersson G. G., Metha G. F., Yamazoe S., Negishi Y., Angew. Chem. Int. Ed. 2021, 60, 21340–21350; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2021, 133, 21510–21520; [Google Scholar]
- 3f. Negishi Y., Mizuno M., Hirayama M., Omatoi M., Takayama T., Iwase A., Kudo A., Nanoscale 2013, 5, 7188–7192. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Kawawaki T., Kawachi M., Yazaki D., Akinaga Y., Hirayama D., Negishi Y., Nanomaterials 2022, 12, 344; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4b. Kawawaki T., Kataoka Y., Ozaki S., Kawachi M., Hirata M., Negishi Y., Chem. Commun. 2021, 57, 417–440; [DOI] [PubMed] [Google Scholar]
- 4c. Kawawaki T., Mori Y., Wakamatsu K., Ozaki S., Kawachi M., Hossain S., Negishi Y., J. Mater. Chem. A 2020, 8, 16081–16113; [Google Scholar]
- 4d. Xiao N., Li S., Li X., Ge L., Gao Y., Li N., Chin. J. Catal. 2020, 41, 642–671; [Google Scholar]
- 4e. Ran J., Zhang J., Yu J., Jaroniec M., Qiao S. Z., Chem. Soc. Rev. 2014, 43, 7787–7812. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Verma R., Belgamwar R., Polshettiwar V., ACS Mater. Lett. 2021, 3, 574–598; [Google Scholar]
- 5b. Vu N.-N., Kaliaguine S., Do T.-O., ChemSusChem 2020, 13, 3967–3991; [DOI] [PubMed] [Google Scholar]
- 5c. Yang J., Guo Y., Lu W., Jiang R., Wang J., Adv. Mater. 2018, 30, 1802227; [DOI] [PubMed] [Google Scholar]
- 5d. Yu S., Wilson A. J., Kumari G., Zhang X., Jain P. K., ACS Energy Lett. 2017, 2, 2058–2070; [Google Scholar]
- 5e. Meng X., Liu L., Ouyang S., Xu H., Wang D., Zhao N., Ye J., Adv. Mater. 2016, 28, 6781–6803. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Wang B., Cai H., Shen S., Small Methods 2019, 3, 1800447; [Google Scholar]
- 6b. Wang L., Chen W., Zhang D., Du Y., Amal R., Qiao S., Wu J., Yin Z., Chem. Soc. Rev. 2019, 48, 5310–5349; [DOI] [PubMed] [Google Scholar]
- 6c. Gao C., Low J., Long R., Kong T., Zhu J., Xiong Y., Chem. Rev. 2020, 120, 12175–12216; [DOI] [PubMed] [Google Scholar]
- 6d. Najafi M., Abednatanzi S., Yousefi A., Ghaedi M., Chem. Eur. J. 2021, 27, 17999–18014; [DOI] [PubMed] [Google Scholar]
- 6e. Hiragond C. B., Powar N. S., Lee J., In S.-I., Small 2022, 18, 2201428; [Google Scholar]
- 6f. Liu L., Li M., Chen F., Huang H., Small Struct. 2022, 2200188; [Google Scholar]
- 6g. Xia T., Long R., Gao C., Xiong Y., Nanoscale 2019, 11, 11064–11070; [DOI] [PubMed] [Google Scholar]
- 6h. Guan X., Mao S. S., Shen S., ChemNanoMat 2021, 7, 873–880; [Google Scholar]
- 6i. Wang L., Wang D., Li Y., Carbon Energy. 2022, 4, 1021–1079; [Google Scholar]
- 6j. Zhang D., Li Y., Li Y., Zhan S., SmartMat. 2022, 3, 417–446; [Google Scholar]
- 6k. Shi X., Cao L. N. Y., Chen M., Huang Y., Chin. Chem. Lett. 2022, 33, 5023–5029. [Google Scholar]
- 7.
- 7a. Wang Y., Bai X., Qin H., Wang F., Li Y., Li X., Kang S., Zuo Y., Cui L., ACS Appl. Mater. Interfaces 2016, 8, 17212–17219; [DOI] [PubMed] [Google Scholar]
- 7b. Yu L., Li G., Zhang X., Ba X., Shi G., Li Y., Wong P. K., Yu J. C., Yu Y., ACS Catal. 2016, 6, 6444–6454; [Google Scholar]
- 7c. Kong X. Y., Tan W. L., Ng B.-J., Chai S.-P., Mohamed A. R., Nano Res. 2017, 10, 1720–1731; [Google Scholar]
- 7d. Bafaqeer A., Tahir M., Amin N. A. S., Chem. Eng. J. 2018, 334, 2142–2153. [Google Scholar]
- 8.
- 8a. Luo Y.-H., Dong L.-Z., Liu J., Li S.-L., Lan Y.-Q., Coord. Chem. Rev. 2019, 390, 86–126; [Google Scholar]
- 8b. Boutin E., Merakeb L., Ma B., Boudy B., Wang M., Bonin J., Anxolabéhère-Mallart E., Robert M., Chem. Soc. Rev. 2020, 49, 5772–5809; [DOI] [PubMed] [Google Scholar]
- 8c. Zou L., Sa R., Lv H., Zhong H., Wang R., ChemSusChem 2020, 13, 6124–6140; [DOI] [PubMed] [Google Scholar]
- 8d. Leung C.-F., Lau T.-C., Energy Fuels 2021, 35, 18888–18899; [Google Scholar]
- 8e. Nikoloudakis E., López-Duarte I., Charalambidis G., Ladomenou K., Ince M., Coutsolelos A. G., Chem. Soc. Rev. 2022, 51, 6965–7045; [DOI] [PubMed] [Google Scholar]
- 8f. Bizzarri C., Eur. J. Org. Chem. 2022, 2022, e202200185. [Google Scholar]
- 9.
- 9a. Zhang Y., Xia B., Ran J., Davey K., Qiao S. Z., Adv. Energy Mater. 2020, 10, 1903879; [Google Scholar]
- 9b. Christoforidis K. C., Fornasiero P., ChemCatChem 2019, 11, 368–382. [Google Scholar]
- 10. Inoue T., Fujishima A., Konishi S., Honda K., Nature 1979, 277, 637–638. [Google Scholar]
- 11.
- 11a. Su T.-M., Qin Z.-Z., Ji H.-B., Jiang Y.-X., Huang G., Environ. Chem. Lett. 2016, 14, 99–112; [Google Scholar]
- 11b. Jia J., Qian C., Dong Y., Li Y. F., Wang H., Ghoussoub M., Butler K. T., Walsh A., Ozin G. A., Chem. Soc. Rev. 2017, 46, 4631–4644. [DOI] [PubMed] [Google Scholar]
- 12. Wang J., Lin S., Tian N., Ma T., Zhang Y., Huang H., Adv. Funct. Mater. 2021, 31, 2008008. [Google Scholar]
- 13.
- 13a. Lu Q., Eid K., Li W., Abdullah A. M., Xu G., Varma R. S., Green Chem. 2021, 23, 5394–5428; [Google Scholar]
- 13b. Aggarwal M., Basu S., Shetti N. P., Nadagouda M. N., Kwon E. E., Park Y.-K., Aminabhavi T. M., Chem. Eng. J. 2021, 425, 131402; [Google Scholar]
- 13c. Lu S., Shen L., Li X., Yu B., Ding J., Gao P., Zhang H., J. Cleaner Prod. 2022, 378, 134589; [Google Scholar]
- 13d. Zhao M., Feng J., Yang W., Song S., Zhang H., ChemCatChem 2021, 13, 1250–1270. [Google Scholar]
- 14.
- 14a. Li X., Zhu Q.-L., EnergyChem 2020, 2, 100033; [Google Scholar]
- 14b. Zhao S.-N., Wang G., Poelman D., Van Der Voort P., Molecules 2018, 23, 2947; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14c. Li D., Kassymova M., Cai X., Zang S.-Q., Jiang H.-L., Coord. Chem. Rev. 2020, 412, 213262; [Google Scholar]
- 14d. Fan W. K., Tahir M., Energy Convers. Manage. 2022, 253, 115180; [Google Scholar]
- 14e. Zhan W., Gao H., Yang Y., Li X., Zhu Q.-L., Adv. Energy Sustainability Res. 2022, 3, 2200004; [Google Scholar]
- 14f. Zhang Y., Xu J., Zhou J., Wang L., Chin. J. Catal. 2022, 43, 971–1000; [Google Scholar]
- 14g. Chen J., Abazari R., Adegoke K. A., Maxakato N. W., Bello O. S., Tahir M., Tasleem S., Sanati S., Kirillov A. M., Zhou Y., Coord. Chem. Rev. 2022, 469, 214664; [Google Scholar]
- 14h. Dao X.-Y., Sun W.-Y., Inorg. Chem. Front. 2021, 8, 3178–3204. [Google Scholar]
- 15.
- 15a. Habisreutinger S. N., Schmidt-Mende L., Stolarczyk J. K., Angew. Chem. Int. Ed. 2013, 52, 7372–7408; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 7516–7557; [Google Scholar]
- 15b. Wu T., Zou L., Han D., Li F., Zhang Q., Niu L., Green Chem. 2014, 16, 2142–2146; [Google Scholar]
- 15c. Park H.-A., Choi J. H., Choi K. M., Lee D. K., Kang J. K., J. Mater. Chem. 2012, 22, 5304–5307; [Google Scholar]
- 15d. Zhu Z., Yang C.-X., Hwang Y.-T., Lin Y.-C., Wu R.-J., Mater. Res. Bull. 2020, 130, 110955; [Google Scholar]
- 15e. Miseki Y., Kato H., Kudo A., Energy Environ. Sci. 2009, 2, 306–314; [Google Scholar]
- 15f. Yan S., Wang J., Gao H., Wang N., Yu H., Li Z., Zhou Y., Zou Z., Adv. Funct. Mater. 2013, 23, 1839–1845; [Google Scholar]
- 15g. Pan P.-W., Chen Y.-W., Catal. Commun. 2007, 8, 1546–1549; [Google Scholar]
- 15h. Mao J., Peng T., Zhang X., Li K., Ye L., Zan L., Catal. Sci. Technol. 2013, 3, 1253–1260; [Google Scholar]
- 15i. Yang J., Hao J., Xu S., Wang Q., Dai J., Zhang A., Pang X., ACS Appl. Mater. Interfaces 2019, 11, 32025–32037; [DOI] [PubMed] [Google Scholar]
- 15j. Zhou Y., Tian Z., Zhao Z., Liu Q., Kou J., Chen X., Gao J., Yan S., Zou Z., ACS Appl. Mater. Interfaces 2011, 3, 3594–3601. [DOI] [PubMed] [Google Scholar]
- 16.
- 16a. Raizada P., Soni V., Kumar A., Singh P., Khan A. A. P., Asiri A. M., Thakur V. K., Nguyen V.-H., J. Materiomics 2021, 7, 388–418; [Google Scholar]
- 16b. Nguyen T. P., Nguyen D. L. T., Nguyen V.-H., Le T.-H., Vo D.-V. N., Trinh Q. T., Bae S.-R., Chae S. Y., Kim S. Y., Le Q. V., Nanomaterials 2020, 10, 337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.
- 17a. Iizuka K., Wato T., Miseki Y., Saito K., Kudo A., J. Am. Chem. Soc. 2011, 133, 20863–20868; [DOI] [PubMed] [Google Scholar]
- 17b. Hemminger J. C., Carr R., Somorjai G. A., Chem. Phys. Lett. 1978, 57, 100–104; [Google Scholar]
- 17c. Yan S. C., Ouyang S. X., Gao J., Yang M., Feng J. Y., Fan X. X., Wan L. J., Li Z. S., Ye J. H., Zhou Y., Zou Z. G., Angew. Chem. Int. Ed. 2010, 49, 6400–6404; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 6544–6548; [Google Scholar]
- 17d. Xie K., Umezawa N., Zhang N., Reunchan P., Zhang Y., Ye J., Energy Environ. Sci. 2011, 4, 4211–4219. [Google Scholar]
- 18.
- 18a. Chen H.-C., Chou H.-C., Wu J. C. S., Lin H.-Y., J. Mater. Res. 2008, 23, 1364–1370; [Google Scholar]
- 18b. Cheng H., Huang B., Liu Y., Wang Z., Qin X., Zhang X., Dai Y., Chem. Commun. 2012, 48, 9729–9731; [DOI] [PubMed] [Google Scholar]
- 18c. Han Q., Bai X., Man Z., He H., Li L., Hu J., Alsaedi A., Hayat T., Yu Z., Zhang W., Wang J., Zhou Y., Zou Z., J. Am. Chem. Soc. 2019, 141, 4209–4213; [DOI] [PubMed] [Google Scholar]
- 18d. Liu Y., Huang B., Dai Y., Zhang X., Qin X., Jiang M., Whangbo M.-H., Catal. Commun. 2009, 11, 210–213. [Google Scholar]
- 19.
- 19a. Zhang N., Ouyang S., Kako T., Ye J., Chem. Commun. 2012, 48, 1269–1271; [DOI] [PubMed] [Google Scholar]
- 19b. Kuriki R., Ichibha T., Hongo K., Lu D., Maezono R., Kageyama H., Ishitani O., Oka K., Maeda K., J. Am. Chem. Soc. 2018, 140, 6648–6655. [DOI] [PubMed] [Google Scholar]
- 20.
- 20a. Olowoyo J. O., Kumar M., Jain S. L., Shen S., Zhou Z., Mao S. S., Vorontsov A. V., Kumar U., Int. J. Hydrogen Energy 2018, 43, 17682–17695; [Google Scholar]
- 20b. Wang Y., Wang F., Chen Y., Zhang D., Li B., Kang S., Li X., Cui L., Appl. Catal. B 2014, 147, 602–609; [Google Scholar]
- 20c. Wang Y., Wang J., Lian W., Liu Y., RSC Adv. 2020, 10, 40117–40126; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20d. Humayun M., Xu L., Zhou L., Zheng Z., Fu Q., Luo W., Nano Res. 2018, 11, 6391–6404. [Google Scholar]
- 21.
- 21a. Gao M., Yang J., Sun T., Zhang Z., Zhang D., Huang H., Lin H., Fang Y., Wang X., Appl. Catal. B 2019, 243, 734–740; [Google Scholar]
- 21b. Kanan D. K., Carter E. A., J. Phys. Chem. C 2012, 116, 9876–9887; [Google Scholar]
- 21c. Liu Q., Xu M., Zhou B., Liu R., Tao F., Mao G., Eur. J. Inorg. Chem. 2017, 2017, 2195–2200. [Google Scholar]
- 22.
- 22a. Fujiwara H., Hosokawa H., Murakoshi K., Wada Y., Yanagida S., Okada T., Kobayashi H., J. Phys. Chem. B 1997, 101, 8270–8278; [Google Scholar]
- 22b. Fujiwara H., Hosokawa H., Murakoshi K., Wada Y., Yanagida S., Langmuir 1998, 14, 5154–5159. [Google Scholar]
- 23.
- 23a. Aggarwal M., Basu S., Shetti N. P., Nadagouda M. N., Kwon E. E., Park Y.-K., Aminabhavi T. M., Chem. Eng. J. 2021, 425, 131402; [Google Scholar]
- 23b. Bhowmik S., Phukan S. J., Sah N. K., Roy M., Garai S., Iyer P. K., ACS Appl. Nano Mater. 2021, 4, 12845–12890. [Google Scholar]
- 24.
- 24a. Li M., Zhang L., Wu M., Du Y., Fan X., Wang M., Zhang L., Kong Q., Shi J., Nano Energy 2016, 19, 145–155; [Google Scholar]
- 24b. Tonda S., Kumar S., Bhardwaj M., Yadav P., Ogale S., ACS Appl. Mater. Interfaces 2018, 10, 2667–2678; [DOI] [PubMed] [Google Scholar]
- 24c. Li M., Zhang L., Fan X., Wu M., Wang M., Cheng R., Zhang L., Yao H., Shi J., Appl. Catal. B 2017, 201, 629–635. [Google Scholar]
- 25.
- 25a. Chen Y., Wang D., Deng X., Li Z., Catal. Sci. Technol. 2017, 7, 4893–4904; [Google Scholar]
- 25b. Guo K., Hussain I., Jie G., Fu Y., Zhang F., Zhu W., J. Environ. Sci. 2023, 125, 290–308. [DOI] [PubMed] [Google Scholar]
- 26.
- 26a. Wang J., Li Y., Zhao J., Xiong Z., Zhang J., Zhao Y., Phys. Chem. Chem. Phys. 2021, 23, 9407–9417; [DOI] [PubMed] [Google Scholar]
- 26b. Mao J., Ye L., Li K., Zhang X., Liu J., Peng T., Zan L., Appl. Catal. B 2014, 144, 855–862; [Google Scholar]
- 26c. Pan Y.-X., Sun Z.-Q., Cong H.-P., Men Y.-L., Xin S., Song J., Yu S.-H., Nano Res. 2016, 9, 1689–1700; [Google Scholar]
- 26d. Tasbihi M., Fresno F., Simon U., Villar-García I. J., Pérez-Dieste V., Escudero C., de la Peña O'Shea V. A., Appl. Catal. B 2018, 239, 68–76; [Google Scholar]
- 26e. Tasbihi M., Schwarze M., Edelmannová M., Spöri C., Strasser P., Schomäcker R., Catal. Today 2019, 328, 8–14; [Google Scholar]
- 26f. Tasbihi M., Fresno F., Álvarez-Prada I., Acharjya A., Thomas A., Escriche L., Romero N., Sala X., de la Peña O'Shea V. A., García-Antón J., J. CO2 Util. 2021, 50, 101574; [Google Scholar]
- 26g. Peng Y., Kang S., Hu Z., ACS Appl. Nano Mater. 2020, 3, 8632–8639; [Google Scholar]
- 26h. Zhao X., Guan J., Li J., Li X., Wang H., Huo P., Yan Y., Appl. Surf. Sci. 2021, 537, 147891; [Google Scholar]
- 26i. Xu J., Liu X., Zhou Z., Deng L., Liu L., Xu M., Energy Fuels 2021, 35, 10820–10831; [Google Scholar]
- 26j. Yoon H. J., Yang J. H., Park S. J., Rhee C. K., Sohn Y., Appl. Surf. Sci. 2021, 536, 147753; [Google Scholar]
- 26k. Li X., He W., Li C., Song B., Liu S., Appl. Catal. B 2021, 287, 119934; [Google Scholar]
- 26l. Jin L., Shaaban E., Bamonte S., Cintron D., Shuster S., Zhang L., Li G., He J., ACS Appl. Mater. Interfaces 2021, 13, 38595–38603; [DOI] [PubMed] [Google Scholar]
- 26m. Chen Y.-X., Xu Y.-F., Wang X.-D., Chen H.-Y., Kuang D.-B., Sustain. Energy Fuels 2020, 4, 2249–2255; [Google Scholar]
- 26n. Rangappa A. P., Kumar D. P., Hong Y., Jeong S., Reddy D. A., Song J. K., Kim T. K., ACS Appl. Energ. Mater. 2020, 3, 10533–10540; [Google Scholar]
- 26o. Han C., Wang B., Wu N., Shen S., Wang Y., Appl. Surf. Sci. 2020, 515, 145952; [Google Scholar]
- 26p. He J., Lv P., Zhu J., Li H., RSC Adv. 2020, 10, 22460–22467; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26q. Chong R., Su C., Du Y., Fan Y., Ling Z., Chang Z., Li D., J. Catal. 2018, 363, 92–101; [Google Scholar]
- 26r. Chong R., Fan Y., Du Y., Liu L., Chang Z., Li D., Int. J. Hydrogen Energy 2018, 43, 22329–22339; [Google Scholar]
- 26s. Sorcar S., Hwang Y., Grimes C. A., In S.-I., Mater. Today 2017, 20, 507–515; [Google Scholar]
- 26t. Zhang X., Wang L., Du Q., Wang Z., Ma S., Yu M., J. Colloid Interface Sci. 2016, 464, 89–95; [DOI] [PubMed] [Google Scholar]
- 26u. Jia W., Liu T., Li Q., Yang J., Catal. Today 2019, 335, 221–227; [Google Scholar]
- 26v. Bera S., Lee J. E., Rawal S. B., Lee W. I., Appl. Catal. B 2016, 199, 55–63; [Google Scholar]
- 26w. Umezawa N., Kristoffersen H. H., Vilhelmsen L. B., Hammer B., J. Phys. Chem. C 2016, 120, 9160–9164; [Google Scholar]
- 26x. Liu G., Xie S., Zhang Q., Tian Z., Wang Y., Chem. Commun. 2015, 51, 13654–13657; [DOI] [PubMed] [Google Scholar]
- 26y. Yu J., Wang K., Xiao W., Cheng B., Phys. Chem. Chem. Phys. 2014, 16, 11492–11501; [DOI] [PubMed] [Google Scholar]
- 26z. Pang H., Liu L., Ouyang S., Xu H., Li Y., Wang D., Int. J. Photoenergy 2014, 2014, 894396; [Google Scholar]
- 26aa. Zhang Z., Wang Z., Cao S.-W., Xue C., J. Phys. Chem. C 2013, 117, 25939–25947; [Google Scholar]
- 26ab. Katsumata K.-I., Sakai K., Ikeda K., Carja G., Matsushita N., Okada K., Mater. Lett. 2013, 107, 138–140; [Google Scholar]
- 26ac. Li X., Zhuang Z., Li W., Pan H., Appl. Catal. A 2012, 429 (430), 31–38; [Google Scholar]
- 26ad. Li X., Li W., Zhuang Z., Zhong Y., Li Q., Wang L., J. Phys. Chem. C 2012, 116, 16047–16053; [Google Scholar]
- 26ae. Varghese O. K., Paulose M., LaTempa T. J., Grimes C. A., Nano Lett. 2009, 9, 731–737; [DOI] [PubMed] [Google Scholar]
- 26af. Blommaerts N., Hoeven N., Esteban D. A., Campos R., Mertens M., Borah R., Glisenti A., De Wael K., Bals S., Lenaerts S., Verbruggen S. W., Cool P., Chem. Eng. J. 2021, 410, 128234; [Google Scholar]
- 26ag. Huang Y., Li K., Zhou J., Guan J., Zhu F., Wang K., Liu M., Chen W., Li N., Chem. Eng. J. 2022, 439, 135744; [Google Scholar]
- 26ah. Xiong Z., Lei Z., Kuang C.-C., Chen X., Gong B., Zhao Y., Zhang J., Zheng C., Wu J. C. S., Appl. Catal. B 2017, 202, 695–703; [Google Scholar]
- 26ai. Conte F., Villa A., Prati L., Pirola C., Bennici S., Ramis G., Rossetti I., Ind. Eng. Chem. Res. 2022, 61, 2963–2972; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26aj. Wang J., Huang C., Chen X., Zhang H., Li Z., Zou Z., Appl. Surf. Sci. 2015, 358, 463–467; [Google Scholar]
- 26ak. Zhang Y., Li P., Tang L.-Q., Li Y.-Q., Zhou Y., Liu J.-M., Zou Z.-G., Dalton Trans. 2017, 46, 10564–10568; [DOI] [PubMed] [Google Scholar]
- 26al. AlOtaibi B., Fan S., Wang D., Ye J., Mi Z., ACS Catal. 2015, 5, 5342–5348; [Google Scholar]
- 26am. Lyulyukin M. N., Kurenkova A. Y., Bukhtiyarov A. V., Kozlova E. A., Mendeleev Commun. 2020, 30, 192–194; [Google Scholar]
- 26an. Wang Y., Lai Q., Zhang F., Shen X., Fan M., He Y., Ren S., RSC Adv. 2014, 4, 44442–44451; [Google Scholar]
- 26ao. Pan B., Luo S., Su W., Wang X., Appl. Catal. B 2015, 168 (169), 458–464; [Google Scholar]
- 26ap. Jiao J., Wei Y., Chi K., Zhao Z., Duan A., Liu J., Jiang G., Wang Y., Wang X., Han C., Zheng P., Energy Technol. 2017, 5, 877–883; [Google Scholar]
- 26aq. Li A., Cao Q., Zhou G., Schmidt B. V. K. J., Zhu W., Yuan X., Huo H., Gong J., Antonietti M., Angew. Chem. Int. Ed. 2019, 58, 14549–14555; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 14691–14697; [Google Scholar]
- 26ar. Wang H., Zhang L., Zhou Y., Qiao S., Liu X., Wang W., Appl. Catal. B 2020, 263, 118331; [Google Scholar]
- 26as. Pan Y.-X., You Y., Xin S., Li Y., Fu G., Cui Z., Men Y.-L., Cao F.-F., Yu S.-H., Goodenough J. B., J. Am. Chem. Soc. 2017, 139, 4123–4129; [DOI] [PubMed] [Google Scholar]
- 26at. Nabil S., Shalaby E. A., Elkady M. F., Matsushita Y., El-Shazly A. H., Catal. Lett. 2022, 152, 3243–3258; [Google Scholar]
- 26au. Xiao J., Chen C., Chen S., Liu H., Peng T., Chem. Eng. J. 2021, 425, 131627; [Google Scholar]
- 26av. Chen C., Hu J., Yang X., Yang T., Qu J., Guo C., Li C. M., ACS Appl. Mater. Interfaces 2021, 13, 20162–20173; [DOI] [PubMed] [Google Scholar]
- 26aw. Jiang Y., Chen H.-Y., Li J.-Y., Liao J.-F., Zhang H.-H., Wang X.-D., Kuang D.-B., Adv. Funct. Mater. 2020, 30, 2004293; [Google Scholar]
- 26ax. Kočí K., Dang Van H., Edelmannová M., Reli M., Wu J. C. S., Appl. Surf. Sci. 2020, 503, 144426; [Google Scholar]
- 26ay. Tasbihi M., Kočí K., Edelmannová M., Troppová I., Reli M., Schomäcker R., J. Photochem. Photobiol. A 2018, 366, 72–80; [Google Scholar]
- 26az. Xie S., Wang Y., Zhang Q., Fan W., Deng W., Wang Y., Chem. Commun. 2013, 49, 2451–2453; [DOI] [PubMed] [Google Scholar]
- 26ba. Dong C., Xing M., Zhang J., Mater. Horiz. 2016, 3, 608–612. [Google Scholar]
- 27.
- 27a. Yui T., Kan A., Saitoh C., Koike K., Ibusuki T., Ishitani O., ACS Appl. Mater. Interfaces 2011, 3, 2594–2600; [DOI] [PubMed] [Google Scholar]
- 27b. Raja K. S., Smith Y. R., Kondamudi N., Manivannan A., Misra M., Subramanian V., Electrochem. Solid-State Lett. 2011, 14, F5–F8; [Google Scholar]
- 27c. Zhu Y., Gao C., Bai S., Chen S., Long R., Song L., Li Z., Xiong Y., Nano Res. 2017, 10, 3396–3406; [Google Scholar]
- 27d. Su K.-Y., Chen C.-Y., Wu R.-J., J. Inst. Chem. 2019, 96, 409–418; [Google Scholar]
- 27e. Zhan Z., Wang H., Huang Q., Li S., Yi X., Tang Q., Wang J., Tan B., Small 2022, 18, 2105083; [DOI] [PubMed] [Google Scholar]
- 27f. Huang Z., Wu J., Ma M., Wang J., Wu S., Hu X., Yuan C., Zhou Y., New J. Chem. 2022, 46, 16889–16898; [Google Scholar]
- 27g. Li D., Zhou C., Xie Z., Chen D., Zhou Y., Shi X., Jiang D., Chen M., Shi W., Solar RRL 2021, 5, 2000813; [Google Scholar]
- 27h. Lan D., Pang F., Ge J., ACS Appl. Energ. Mater. 2021, 4, 6324–6332; [Google Scholar]
- 27i. Zhang R., Huang Z., Li C., Zuo Y., Zhou Y., Appl. Surf. Sci. 2019, 475, 953–960; [Google Scholar]
- 27j. Li N., Zou X., Liu M., Wei L., Shen Q., Bibi R., Xu C., Ma Q., Zhou J., J. Phys. Chem. C 2017, 121, 25795–25804; [Google Scholar]
- 27k. Chen W., Wang Y., Shangguan W., Int. J. Hydrogen Energy 2019, 44, 4123–4132; [Google Scholar]
- 27l. Yang J.-J., Zhang Y., Xie X.-Y., Fang W.-H., Cui G., ACS Catal. 2022, 12, 8558–8571; [Google Scholar]
- 27m. Shen L., Xie Z., Hou L., Yang J., Li Q., Energy Fuels 2022, 36, 11515–11523; [Google Scholar]
- 27n. Li D., Zhou C., Shi X., Zhang Q., Song Q., Zhou Y., Jiang D., J. Mol. Catal. 2022, 526, 112382. [Google Scholar]
- 28.
- 28a. Chen W., Wang Y., Shangguan W., Mater. Lett. 2019, 238, 74–76; [Google Scholar]
- 28b. Dong C., Hu S., Xing M., Zhang J., Nanotechnology 2018, 29, 154005; [DOI] [PubMed] [Google Scholar]
- 28c. Bai Y., Ye L., Wang L., Shi X., Wang P., Bai W., Environ. Sci.-Nano 2016, 3, 902–909; [Google Scholar]
- 28d. Mei B., Pougin A., Strunk J., J. Catal. 2013, 306, 184–189; [Google Scholar]
- 28e. Wang R., Shen J., Sun K., Tang H., Liu Q., Appl. Surf. Sci. 2019, 493, 1142–1149; [Google Scholar]
- 28f. Li F., Zhou H., Fan J., Xiang Q., J. Colloid Interface Sci. 2020, 570, 11–19; [DOI] [PubMed] [Google Scholar]
- 28g. Cai S., Chen J., Li Q., Jia H., ACS Appl. Mater. Interfaces 2021, 13, 14221–14229; [DOI] [PubMed] [Google Scholar]
- 28h. Zhu S., Liao W., Zhang M., Liang S., Chem. Eng. J. 2019, 361, 461–469; [Google Scholar]
- 28i. Chen S., Pan B., Zeng L., Luo S., Wang X., Su W., RSC Adv. 2017, 7, 14186–14191. [Google Scholar]
- 29. Solymosi F., Tombácz I., Catal. Lett. 1994, 27, 61–65. [Google Scholar]
- 30.
- 30a. Collado L., Jana P., Sierra B., Coronado J. M., Pizarro P., Serrano D. P., de la Peña O'Shea V. A., Chem. Eng. J. 2013, 224, 128–135; [Google Scholar]
- 30b. Yamamoto M., Yoshida T., Yamamoto N., Nomoto T., Yamamoto Y., Yagi S., Yoshida H., J. Mater. Chem. A 2015, 3, 16810–16816; [Google Scholar]
- 30c. Li H., Wu X., Wang J., Gao Y., Li L., Shih K., Int. J. Hydrogen Energy 2016, 41, 8479–8488; [Google Scholar]
- 30d. Anzai A., Fukuo N., Yamamoto A., Yoshida H., Catal. Commun. 2017, 100, 134–138; [Google Scholar]
- 30e. Zhu Z., Qin J., Jiang M., Ding Z., Hou Y., Appl. Surf. Sci. 2017, 391, 572–579; [Google Scholar]
- 30f. Zhu Z., Han Y., Chen C., Ding Z., Long J., Hou Y., ChemCatChem 2018, 10, 1627–1634; [Google Scholar]
- 30g. Kawaguchi Y., Akatsuka M., Yamamoto M., Yoshioka K., Ozawa A., Kato Y., Yoshida T., J. Photochem. Photobiol. A 2018, 358, 459–464; [Google Scholar]
- 30h. Yoshida H., Sato M., Fukuo N., Zhang L., Yoshida T., Yamamoto Y., Morikawa T., Kajino T., Sakano M., Sekito T., Matsumoto S., Hirata H., Catal. Today 2018, 303, 296–304; [Google Scholar]
- 30i. Takayama T., Nakanishi H., Matsui M., Iwase A., Kudo A., J. Photochem. Photobiol. A 2018, 358, 416–421; [Google Scholar]
- 30j. Zhu X., Yamamoto A., Imai S., Tanaka A., Kominami H., Yoshida H., Chem. Commun. 2019, 55, 13514–13517; [DOI] [PubMed] [Google Scholar]
- 30k. Zhu X., Anzai A., Yamamoto A., Yoshida H., Appl. Catal. B 2019, 243, 47–56; [Google Scholar]
- 30l. Ishii T., Anzai A., Yamamoto A., Yoshida H., Appl. Catal. B 2020, 277, 119192; [Google Scholar]
- 30m. Hammad A., Anzai A., Zhu X., Yamamoto A., Ootsuki D., Yoshida T., El-Shazly A., Elkady M., Yoshida H., Catal. Lett. 2020, 150, 1081–1088; [Google Scholar]
- 30n. Zhu X., Yamamoto A., Imai S., Tanaka A., Kominami H., Yoshida H., Appl. Catal. B 2020, 274, 119085; [Google Scholar]
- 30o. Wang S., Teramura K., Hisatomi T., Domen K., Asakura H., Hosokawa S., Tanaka T., ACS Sustainable Chem. Eng. 2021, 9, 9327–9335; [Google Scholar]
- 30p. Tao X., Wang Y., Qu J., Zhao Y., Li R., Li C., J. Mater. Chem. A 2021, 9, 19631–19636; [Google Scholar]
- 30q. Sonoda K., Yamamoto M., Tanabe T., Yoshida T., Appl. Surf. Sci. 2021, 542, 148680; [Google Scholar]
- 30r. Fresno F., Galdón S., Barawi M., Alfonso-González E., Escudero C., Pérez-Dieste V., Huck-Iriart C., de la Peña O'Shea V. A., Catal. Today 2021, 361, 85–93; [Google Scholar]
- 30s. Reñones P., Fresno F., Oropeza F. E., de la Peña O'Shea V. A., Materials 2022, 15, 843; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30t. Liu E., Kang L., Wu F., Sun T., Hu X., Yang Y., Liu H., Fan J., Plasmonics 2014, 9, 61–70; [Google Scholar]
- 30u. Takayama T., Iwase A., Kudo A., Bull. Chem. Soc. Jpn. 2015, 88, 538–543; [Google Scholar]
- 30v. Huang Z., Teramura K., Asakura H., Hosokawa S., Tanaka T., J. Mater. Chem. A 2017, 5, 19351–19357; [Google Scholar]
- 30w. Tahir M., Amin N. A. S., Int. J. Hydrogen Energy 2017, 42, 15507–15522; [Google Scholar]
- 30x. Yoshida T., Yamamoto N., Mizutani T., Yamamoto M., Ogawa S., Yagi S., Nameki H., Yoshida H., Catal. Today 2018, 303, 320–326; [Google Scholar]
- 30y. Yamamoto M., Yagi S., Yoshida T., Catal. Today 2018, 303, 334–340; [Google Scholar]
- 30z. Tahir B., Tahir M., Amin N. A. S., Appl. Surf. Sci. 2019, 493, 18–31; [Google Scholar]
- 30aa. Wang Y., Liu M., Chen W., Mao L., Shangguan W., J. Alloys Compd. 2019, 786, 149–154; [Google Scholar]
- 30ab. Basaleh A. S., Mohamed R. M., Appl. Nanosci. 2020, 10, 3865–3874; [Google Scholar]
- 30ac. Zhang F., Li Y.-H., Qi M.-Y., Tang Z.-R., Xu Y.-J., Appl. Catal. B 2020, 268, 118380; [Google Scholar]
- 30ad. Wang S., Teramura K., Hisatomi T., Domen K., Asakura H., Hosokawa S., Tanaka T., ACS Appl. Energ. Mater. 2020, 3, 1468–1475; [Google Scholar]
- 30ae. Soltani T., Zhu X., Yamamoto A., Singh S. P., Fudo E., Tanaka A., Kominami H., Yoshida H., Appl. Catal. B 2021, 286, 119899; [Google Scholar]
- 30af. Wang S., Teramura K., Asakura H., Hosokawa S., Tanaka T., J. Phys. Chem. C 2021, 125, 1304–1312; [Google Scholar]
- 30ag. Wei S., Heng Q., Wu Y., Chen W., Li X., Shangguan W., Appl. Surf. Sci. 2021, 563, 150273; [Google Scholar]
- 30ah. Xiao J., Chen S., Jin J., Li R., Zhang J., Peng T., Langmuir 2021, 37, 12487–12500; [DOI] [PubMed] [Google Scholar]
- 30ai. Sun Y., Li G., Gong Y., Sun Z., Yao H., Zhou X., J. Hazard. Mater. 2021, 403, 124019. [DOI] [PubMed] [Google Scholar]
- 31.
- 31a. Baran T., Wojtyła S., Dibenedetto A., Aresta M., Macyk W., Appl. Catal. B 2015, 178, 170–176; [Google Scholar]
- 31b. Li M., Li P., Chang K., Wang T., Liu L., Kang Q., Ouyang S., Ye J., Chem. Commun. 2015, 51, 7645–7648; [DOI] [PubMed] [Google Scholar]
- 31c. Guo K., Zhu X., Peng L., Fu Y., Ma R., Lu X., Zhang F., Zhu W., Fan M., Chem. Eng. J. 2021, 405, 127011; [Google Scholar]
- 31d. Cabrero-Antonino M., Ferrer B., Baldoví H. G., Navalón S., Chem. Eng. J. 2022, 445, 136426; [Google Scholar]
- 31e. Xie Z., Xu W., Fang F., Zhang K., Yu X., Chang K., Chem. Eng. J. 2022, 429, 132364; [Google Scholar]
- 31f. Pastor E., Pesci F. M., Reynal A., Handoko A. D., Guo M., An X., Cowan A. J., Klug D. R., Durrant J. R., Tang J., Phys. Chem. Chem. Phys. 2014, 16, 5922–5926; [DOI] [PubMed] [Google Scholar]
- 31g. Roldán L., Marco Y., García-Bordejé E., ChemCatChem 2015, 7, 1347–1356. [Google Scholar]
- 32.
- 32a. Yin G., Nishikawa M., Nosaka Y., Srinivasan N., Atarashi D., Sakai E., Miyauchi M., ACS Nano 2015, 9, 2111–2119; [DOI] [PubMed] [Google Scholar]
- 32b. Shoji S., Yin G., Nishikawa M., Atarashi D., Sakai E., Miyauchi M., Chem. Phys. Lett. 2016, 658, 309–314; [Google Scholar]
- 32c. Gusain R., Kumar P., Sharma O. P., Jain S. L., Khatri O. P., Appl. Catal. B 2016, 181, 352–362; [Google Scholar]
- 32d. Jiang M., Gao Y., Wang Z., Ding Z., Appl. Catal. B 2016, 198, 180–188; [Google Scholar]
- 32e. Núñez J., de la Peña O'Shea V. A., Jana P., Coronado J. M., Serrano D. P., Catal. Today 2013, 209, 21–27; [Google Scholar]
- 32f. Liu L., Gao F., Zhao H., Li Y., Appl. Catal. B 2013, 134–135, 349–358; [Google Scholar]
- 32g. Zhang Q., Gao T., Andino J. M., Li Y., Appl. Catal. B 2012, 123–124, 257–264; [Google Scholar]
- 32h. Shown I., Hsu H.-C., Chang Y.-C., Lin C.-H., Roy P. K., Ganguly A., Wang C.-H., Chang J.-K., Wu C.-I., Chen L.-C., Chen K.-H., Nano Lett. 2014, 14, 6097–6103; [DOI] [PubMed] [Google Scholar]
- 32i. Zhu S., Liang S., Tong Y., An X., Long J., Fu X., Wang X., Phys. Chem. Chem. Phys. 2015, 17, 9761–9770; [DOI] [PubMed] [Google Scholar]
- 32j. Li H., Li C., Han L., Li C., Zhang S., Energy Sources Part A 2016, 38, 420–426; [Google Scholar]
- 32k. Zhang T., Low J., Huang X., Al-Sharab J. F., Yu J., Asefa T., ChemCatChem 2017, 9, 3054–3062; [Google Scholar]
- 32l. Jiang H., Katsumata K.-I., Hong J., Yamaguchi A., Nakata K., Terashima C., Matsushita N., Miyauchi M., Fujishima A., Appl. Catal. B 2018, 224, 783–790; [Google Scholar]
- 32m. Jeong S., Kim G.-M., Kang G.-S., Kim C., Lee H., Kim W.-J., Lee Y. K., Lee S., Kim H., Lim H. K., Lee D. C., J. Phys. Chem. C 2019, 123, 29184–29191; [Google Scholar]
- 32n. Wang Z., Zhou W., Wang X., Zhang X., Chen H., Hu H., Liu L., Ye J., Wang D., Catalysts 2020, 10, 654; [Google Scholar]
- 32o. Hu Z., Liu W., ACS Appl. Mater. Interfaces 2020, 12, 51366–51373; [DOI] [PubMed] [Google Scholar]
- 32p. Duan Z., Feng X., Chen L., J. Solid State Chem. 2020, 286, 121298; [Google Scholar]
- 32q. Ge H., Zhang B., Liang H., Zhang M., Fang K., Chen Y., Qin Y., Appl. Catal. B 2020, 263, 118133; [Google Scholar]
- 32r. Ibarra-Rodríguez L. I., Huerta-Flores A. M., Torres-Martínez L. M., Mater. Res. Bull. 2020, 122, 110679; [Google Scholar]
- 32s. Wang S.-Q., Zhang X.-Y., Dao X.-Y., Cheng X.-M., Sun W.-Y., ACS Appl. Nano Mater. 2020, 3, 10437–10445; [Google Scholar]
- 32t. Sun Z., Fang W., Zhao L., Wang H., Appl. Surf. Sci. 2020, 504, 144347; [Google Scholar]
- 32u. Faria A. L. A., Centurion H. A., Torres J. A., Gonçalves R. V., Ribeiro L. S., Riberio C., da Cruz J. C., Nogueira F. G. E., J. CO2 Util. 2021, 53, 101739; [Google Scholar]
- 32v. Qian X., Zhang L., Lin Y., Wang M., Wang X., Su W., Appl. Surf. Sci. 2021, 568, 150985; [Google Scholar]
- 32w. Garay-Rodríguez L. F., Yoshida H., Torres-Martínez L. M., Juárez-Ramírez I., Int. J. Hydrogen Energy 2021, 46, 32490–32502; [Google Scholar]
- 32x. Xiao Y., Men C., Chu B., Qin Z., Ji H., Chen J., Su T., Chem. Eng. J. 2022, 446, 137028; [Google Scholar]
- 32y. Xi H., Xu Y., Zou W., Ji J., Cai Y., Wan H., Dong L., J. CO2 Util. 2022, 55, 101825; [Google Scholar]
- 32z. Li P., Zhang Y., Zang R., Liu P., Yuan X., Wang X., Energy Fuels 2022, 36, 11654–11659; [Google Scholar]
- 32aa. Wang W., Deng C., Xie S., Li Y., Zhang W., Sheng H., Chen C., Zhao J., J. Am. Chem. Soc. 2021, 143, 2984–2993; [DOI] [PubMed] [Google Scholar]
- 32ab. Yu H., Xuan Y., Zhang K., Chang K., Chem. Eng. J. 2022, 430, 132940; [Google Scholar]
- 32ac. Hurtado L., Natividad R., García H., Catal. Commun. 2016, 84, 30–35. [Google Scholar]
- 33.
- 33a. Lee D.-S., Chen H.-J., Chen Y.-W., J. Phys. Chem. Solids 2012, 73, 661–669; [Google Scholar]
- 33b. Lv X.-J., Fu W.-F., Hu C.-Y., Chen Y., Zhou W.-B., RSC Adv. 2013, 3, 1753–1757; [Google Scholar]
- 33c. Lee D.-S., Chen Y.-W., J. CO2 Util. 2015, 10, 1–6; [Google Scholar]
- 33d. Meng A., Wu S., Cheng B., Yu J., Xu J., J. Mater. Chem. A 2018, 6, 4729–4736; [Google Scholar]
- 33e. Tahir M., Tahir B., Zakaria Z. Y., Muhammad A., J. Cleaner Prod. 2019, 213, 451–461; [Google Scholar]
- 33f. Ye J., He J., Wang S., Zhou X., Zhang Y., Liu G., Yang Y., Sep. Purif. Technol. 2019, 220, 8–15; [Google Scholar]
- 33g. Deng H., Xu F., Cheng B., Yu J., Ho W., Nanoscale 2020, 12, 7206–7213; [DOI] [PubMed] [Google Scholar]
- 33h. Kozlova E. A., Lyulyukin M. N., Markovskaya D. V., Bukhtiyarov A. V., Prosvirin I. P., Cherepanova S. V., Kozlov D. V., Top. Catal. 2020, 63, 121–129; [Google Scholar]
- 33i. Zhao T., Niu Q., Huang G., Chen Q., Gao Y., Bi J., Wu L., J. Colloid Interface Sci. 2021, 602, 23–31; [DOI] [PubMed] [Google Scholar]
- 33j. Liao W., Lu S., Chen W., Zhu S., Xia Y., Yang M.-Q., Liang S., Green Chem. Eng. 2022, 3, 240–249; [Google Scholar]
- 33k. Wang L., Dong Y., Zhang J., Tao F., Xu J., J. Solid State Chem. 2022, 308, 122878; [Google Scholar]
- 33l. Li Q., Feng W., Liu Y., Chen D., Wu Z., Wang H., J. Mater. Chem. A 2022, 10, 15752–15765; [Google Scholar]
- 33m. Singhal N., Goyal R., Kumar U., Energy Fuels 2017, 31, 12434–12438; [Google Scholar]
- 33n. Qin H., Guo R.-T., Liu X.-Y., Shi X., Wang Z.-Y., Tang J.-Y., Pan W.-G., Colloids Surf. A 2020, 600, 124912. [Google Scholar]
- 34.
- 34a. Tu W., Li Y., Kuai L., Zhou Y., Xu Q., Li H., Wang X., Xiao M., Zou Z., Nanoscale 2017, 9, 9065–9070; [DOI] [PubMed] [Google Scholar]
- 34b. Zhang W., Zhou T., Hong J., Xu R., J. Energy Chem. 2016, 25, 500–506; [Google Scholar]
- 34c. Xu F., Zhu B., Cheng B., Yu J., Xu J., Adv. Opt. Mater. 2018, 6, 1800911; [Google Scholar]
- 34d. Tang J.-Y., Yang D., Zhou W.-G., Guo R.-T., Pan W.-G., Huang C.-Y., J. Catal. 2019, 370, 79–87; [Google Scholar]
- 34e. Huang S., Yi H., Zhang L., Jin Z., Long Y., Zhang Y., Liao Q., Na J., Cui H., Ruan S., Yamauchi Y., Wakihara T., Kaneti Y. V., Zeng Y.-J., J. Hazard. Mater. 2020, 393, 122324; [DOI] [PubMed] [Google Scholar]
- 34f. Li H. J. W., Zhou H., Chen K., Liu K., Li S., Jiang K., Zhang W., Xie Y., Cao Z., Li H., Liu H., Xu X., Pan H., Hu J., Tang D., Qiu X., Fu J., Liu M., Sol. RRL 2020, 4, 1900416; [Google Scholar]
- 34g. Jung H., Cho K. M., Kim K. H., Yoo H.-W., Al-Saggaf A., Gereige I., Jung H.-T., ACS Sustainable Chem. Eng. 2018, 6, 5718–5724. [Google Scholar]
- 35.
- 35a. Low J., Zhang L., Tong T., Shen B., Yu J., J. Catal. 2018, 361, 255–266; [Google Scholar]
- 35b. Hu J., Ding J., Zhong Q., J. Colloid Interface Sci. 2021, 582, 647–657; [DOI] [PubMed] [Google Scholar]
- 35c. Zhang J., Shi J., Tao S., Wu L., Lu J., Appl. Surf. Sci. 2021, 542, 148685; [Google Scholar]
- 35d. Ye M., Wang X., Liu E., Ye J., Wang D., ChemSusChem 2018, 11, 1606–1611; [DOI] [PubMed] [Google Scholar]
- 35e. Li J., Wang Z., Chen H., Zhang Q., Hu H., Liu L., Ye J., Wang D., Catal. Sci. Technol. 2021, 11, 4953–4961; [Google Scholar]
- 35f. He F., Zhu B., Cheng B., Yu J., Ho W., Macyk W., Appl. Catal. B 2020, 272, 119006; [Google Scholar]
- 35g. Hong L.-F., Guo R.-T., Yuan Y., Ji X.-Y., Lin Z.-D., Yin X.-F., Pan W.-G., Colloids Surf. A 2022, 639, 128358; [Google Scholar]
- 35h. Wang H., Tang Q., Wu Z., ACS Sustainable Chem. Eng. 2021, 9, 8425–8434; [Google Scholar]
- 35i. Shi Q., Zhang X., Yang Y., Huang J., Fu X., Wang T., Liu X., Sun A., Ge J., Shen J., Zhou Y., Liu Z., J. Energy Chem. 2021, 59, 9–18; [Google Scholar]
- 35j. Saeed A., Chen W., Shah A. H., Zhang Y., Mehmood I., Liu Y., J. CO2 Util. 2021, 47, 101501; [Google Scholar]
- 35k. Chen L., Huang K., Xie Q., Lam S. M., Sin J. C., Su T., Ji H., Qin Z., Catal. Sci. Technol. 2021, 11, 1602–1614; [Google Scholar]
- 35l. Cao S., Shen B., Tong T., Fu J., Yu J., Adv. Funct. Mater. 2018, 28, 1800136; [Google Scholar]
- 35m. Tang Q., Sun Z., Deng S., Wang H., Wu Z., J. Colloid Interface Sci. 2020, 564, 406–417; [DOI] [PubMed] [Google Scholar]
- 35n. Khan A. A., Tahir M., Bafaqeer A., Energy Fuels 2020, 34, 9810–9828. [Google Scholar]
- 36.
- 36a. Hsieh M.-C., Wu G.-C., Liu W.-G., W. A. Goddard III , Yang C.-M., Angew. Chem. Int. Ed. 2014, 53, 14216–14220; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 14440–14444; [Google Scholar]
- 36b. Nguyen N. T., Xia M., Duchesne P. N., Wang L., Mao C., Jelle A. A., Yan T., Li P., Lu Z.-H., Ozin G. A., Nano Lett. 2021, 21, 1311–1319. [DOI] [PubMed] [Google Scholar]
- 37.
- 37a. Tang K., Wang Z., Zou W., Guo H., Wu Y., Pu Y., Tong Q., Wan H., Gu X., Dong L., Rong J., Chen Y.-W., ACS Appl. Mater. Interfaces 2021, 13, 6219–6228; [DOI] [PubMed] [Google Scholar]
- 37b. Shen C., Sun K., Zhang Z., Rui N., Jia X., Mei D., Liu C.-j., ACS Catal. 2021, 11, 4036–4046; [Google Scholar]
- 37c. Kim W., Yuan G., McClure B. A., Frei H., J. Am. Chem. Soc. 2014, 136, 11034–11042. [DOI] [PubMed] [Google Scholar]
- 38.
- 38a. Wang W.-N., An W.-J., Ramalingam B., Mukherjee S., Niedzwiedzki D. M., Gangopadhyay S., Biswas P., J. Am. Chem. Soc. 2012, 134, 11276–11281; [DOI] [PubMed] [Google Scholar]
- 38b. Dong C., Lian C., Hu S., Deng Z., Gong J., Li M., Liu H., Xing M., Zhang J., Nat. Commun. 2018, 9, 1252; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38c. Lee S., Jeong S., Kim W. D., Lee S., Lee K., Bae W. K., Moon J. H., Lee S., Lee D. C., Nanoscale 2016, 8, 10043–10048; [DOI] [PubMed] [Google Scholar]
- 38d. Lee D.-E., Kim D. J., Devthade V., Jo W.-K., Tonda S., Appl. Surf. Sci. 2022, 584, 152532; [Google Scholar]
- 38e. Yang P., Li L., Zhao Z.-J., Gong J., Chin. J. Catal. 2021, 42, 817–823; [Google Scholar]
- 38f. Xie S., Wang Y., Zhang Q., Deng W., Wang Y., ACS Catal. 2014, 4, 3644–3653; [Google Scholar]
- 38g. Zhu Y., Xu Z., Jiang W., Zhong S., Zhao L., Bai S., J. Mater. Chem. A 2017, 5, 2619–2628; [Google Scholar]
- 38h. Mu Q., Zhu W., Yan G., Lian Y., Yao Y., Li Q., Tian Y., Zhang P., Deng Z., Peng Y., J. Mater. Chem. A 2018, 6, 21110–21119; [Google Scholar]
- 38i. Xiong Z., Lei Z., Chen X., Gong B., Zhao Y., Zhang J., Zheng C., Wu J. C. S., Catal. Commun. 2017, 96, 1–5; [Google Scholar]
- 38j. Yoshioka K., Yamamoto M., Tanabe T., Yoshida T., e-J. Surf. Sci. Nanotechnol. 2020, 18, 168–174. [Google Scholar]
- 39.
- 39a. Long R., Li Y., Liu Y., Chen S., Zheng X., Gao C., He C., Chen N., Qi Z., Song L., Jiang J., Zhu J., Xiong Y., J. Am. Chem. Soc. 2017, 139, 4486–4492; [DOI] [PubMed] [Google Scholar]
- 39b. Cai X., Wang A., Wang J., Wang R., Zhong S., Zhao Y., Wu L., Chen J., Bai S., J. Mater. Chem. A 2018, 6, 17444–17456; [Google Scholar]
- 39c. Wang J., Li Y., Zhao J., Xiong Z., Zhao Y., Zhang J., Catal. Sci. Technol. 2022, 12, 3454–3463; [Google Scholar]
- 39d. Lang Q., Yang Y., Zhu Y., Hu W., Jiang W., Zhong S., Gong P., Teng B., Zhao L., Bai S., J. Mater. Chem. A 2017, 5, 6686–6694; [Google Scholar]
- 39e. Jiang D., Zhou Y., Zhang Q., Song Q., Zhou C., Shi X., Li D., ACS Appl. Mater. Interfaces 2021, 13, 46772–46782; [DOI] [PubMed] [Google Scholar]
- 39f. Tan D., Zhang J., Shi J., Li S., Zhang B., Tan X., Zhang F., Liu L., Shao D., Han B., ACS Appl. Mater. Interfaces 2018, 10, 24516–24522; [DOI] [PubMed] [Google Scholar]
- 39g. Zhao L., Ye F., Wang D., Cai X., Meng C., Xie H., Zhang J., Bai S., ChemSusChem 2018, 11, 3524–3533; [DOI] [PubMed] [Google Scholar]
- 39h. Liu Q., Chen Q., Li T., Ren Q., Zhong S., Zhao Y., Bai S., J. Mater. Chem. A 2019, 7, 27007–27015; [Google Scholar]
- 39i. Kar P., Farsinezhad S., Mahdi N., Zhang Y., Obuekwe U., Sharma H., Shen J., Semagina N., Shankar K., Nano Res. 2016, 9, 3478–3493; [Google Scholar]
- 39j. Kang Q., Wang T., Li P., Liu L., Chang K., Li M., Ye J., Angew. Chem. Int. Ed. 2015, 54, 841–845; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 855–859; [Google Scholar]
- 39k. Jiao J., Wei Y., Zhao Y., Zhao Z., Duan A., Liu J., Pang Y., Li J., Jiang G., Wang Y., Appl. Catal. B 2017, 209, 228–239; [Google Scholar]
- 39l. Yang L., Li L., Xia P., Li H., Yang J., Li X., Zeng X., Zhang X., Xiao C., Xie Y., Chem. Commun. 2021, 57, 11629–11632; [DOI] [PubMed] [Google Scholar]
- 39m. Ovcharov M. L., Shvalagin V. V., Granchak V. M., Theor. Exp. Chem. 2014, 50, 53–58; [Google Scholar]
- 39n. Yin G., Abe H., Kodiyath R., Ueda S., Srinivasan N., Yamaguchi A., Miyauchi M., J. Mater. Chem. A 2017, 5, 12113–12119; [Google Scholar]
- 39o. Li A., Wang T., Chang X., Zhao Z.-J., Li C., Huang Z., Yang P., Zhou G., Gong J., Chem. Sci. 2018, 9, 5334–5340; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39p. Yu Y., Dong X., Chen P., Geng Q., Wang H., Li J., Zhou Y., Dong F., ACS Nano 2021, 15, 14453–14464; [DOI] [PubMed] [Google Scholar]
- 39q. Reñones P., Collado L., Iglesias-Juez A., Oropeza F. E., Fresno F., de la Peña O'Shea V. A., Ind. Eng. Chem. Res. 2020, 59, 9440–9450; [Google Scholar]
- 39r. Wang Z., Lee H., Chen J., Wu M., Leung D. Y. C., Grimes C. A., Lu Z., Xu Z., Feng S.-P., J. Power Sources 2020, 466, 228306; [Google Scholar]
- 39s. Sorokina L., Savitskiy A., Shtyka O., Maniecki T., Szynkowska-Jozwik M., Trifonov A., Pershina E., Mikhaylov I., Dubkov S., Gromov D., J. Alloys Compd. 2022, 904, 164012; [Google Scholar]
- 39t. Mankidy B. D., Joseph B., Gupta V. K., Nanotechnology 2013, 24, 405402; [DOI] [PubMed] [Google Scholar]
- 39u. Cai X., Wang J., Wang R., Wang A., Zhong S., Chen J., Bai S., J. Mater. Chem. A 2019, 7, 5266–5276; [Google Scholar]
- 39v. Chen Q., Chen X., Fang M., Chen J., Li Y., Xie Z., Kuang Q., Zheng L., J. Mater. Chem. A 2019, 7, 1334–1340; [Google Scholar]
- 39w. He J., Lyu P., Jiang B., Chang S., Du H., Zhu J., Li H., Appl. Catal. B 2021, 298, 120603; [Google Scholar]
- 39x. Neaţu Ş., Maciá-Agulló J. A., Concepción P., Garcia H., J. Am. Chem. Soc. 2014, 136, 15969–15976; [DOI] [PubMed] [Google Scholar]
- 39y. Li D., Zhou C., Shi X., Zhang Q., Song Q., Zhou Y., Jiang D., Appl. Surf. Sci. 2022, 598, 153843; [Google Scholar]
- 39z. Liu J., Liu M., Yang X., Chen H., Liu S. F., Yan J., ACS Sustainable Chem. Eng. 2020, 8, 6055–6064; [Google Scholar]
- 39aa. Li F., Huang Y., Gao C., Wu X., Mater. Res. Bull. 2021, 144, 111488; [Google Scholar]
- 39ab. Zhou H., Luo Y., Xu J., Deng L., Wang Z., He H., Chem. Eur. J. 2022, 28, e202202044. [DOI] [PubMed] [Google Scholar]
- 40.
- 40a. Bai S., Wang X., Hu C., Xie M., Jiang J., Xiong Y., Chem. Commun. 2014, 50, 6094–6097; [DOI] [PubMed] [Google Scholar]
- 40b. Cao S., Li Y., Zhu B., Jaroniec M., Yu J., J. Catal. 2017, 349, 208–217; [Google Scholar]
- 40c. Ye F., Wang F., Meng C., Bai L., Li J., Xing P., Teng B., Zhao L., Bai S., Appl. Catal. B 2018, 230, 145–153; [Google Scholar]
- 40d. Cho H., Dong Kim W., Yu J., Lee S., Lee D. C., ChemCatChem 2018, 10, 5679–5688; [Google Scholar]
- 40e. Lang Q., Hu W., Zhou P., Huang T., Zhong S., Yang L., Chen J., Bai S., Nanotechnology 2017, 28, 484003; [DOI] [PubMed] [Google Scholar]
- 40f. Huang H., Zhao J., Weng B., Lai F., Zhang M., Hofkens J., Roeffaers M. B. J., Steele J. A., Long J., Angew. Chem. Int. Ed. 2022, 61, e202204563. [DOI] [PubMed] [Google Scholar]
- 41.
- 41a. Zhai Q., Xie S., Fan W., Zhang Q., Wang Y., Deng W., Wang Y., Angew. Chem. Int. Ed. 2013, 52, 5776–5779; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 5888–5891; [Google Scholar]
- 41b. Pang R., Teramura K., Tatsumi H., Asakura H., Hosokawa S., Tanaka T., Chem. Commun. 2018, 54, 1053–1056; [DOI] [PubMed] [Google Scholar]
- 41c. Pang R., Teramura K., Asakura H., Hosokawa S., Tanaka T., ACS Sustainable Chem. Eng. 2019, 7, 2083–2090; [Google Scholar]
- 41d. Tsai C.-W., Chen H. M., Liu R.-S., Asakura K., Chan T.-S., J. Phys. Chem. C 2011, 115, 10180–10186; [Google Scholar]
- 41e. Pang R., Teramura K., Asakura H., Hosokawa S., Tanaka T., J. Phys. Chem. C 2019, 123, 2894–2899; [Google Scholar]
- 41f. Guo Q., Xu J., Luo Y., Yang Y., Wang Z., He H., J. Phys. Chem. C 2021, 125, 26389–26397; [Google Scholar]
- 41g. Liu Q., Wang S., Mo W., Zheng Y., Xu Y., Yang G., Zhong S., Ma J., Liu D., Bai S., Small 2022, 18, 2104681; [DOI] [PubMed] [Google Scholar]
- 41h. Lee D.-E., Moru S., Bhosale R., Jo W.-K., Tonda S., Appl. Surf. Sci. 2022, 599, 153973; [Google Scholar]
- 41i. Kikkawa S., Teramura K., Asakura H., Hosokawa S., Tanaka T., J. Phys. Chem. C 2018, 122, 21132–21139; [Google Scholar]
- 41j. Wang Y., Lai Q., He Y., Fan M., Catal. Commun. 2018, 108, 98–102; [Google Scholar]
- 41k. Soltani T., Yamamoto A., Singh S. P., Anzai A., Fudo E., Tanaka A., Kominami H., Yoshida H., ACS Appl. Energ. Mater. 2021, 4, 6500–6510; [Google Scholar]
- 41l. Chen Q., Mo W., Yang G., Zhong S., Lin H., Chen J., Bai S., Small 2021, 17, 2102105; [DOI] [PubMed] [Google Scholar]
- 41m. Xi Y., Zhang Y., Cai X., Fan Z., Wang K., Dong W., Shen Y., Zhong S., Yang L., Bai S., Appl. Catal. B 2022, 305, 121069; [Google Scholar]
- 41n. Wei Y., Jiao J., Zhao Z., Zhong W., Li J., Liu J., Jiang G., Duan A., J. Mater. Chem. A 2015, 3, 11074–11085; [Google Scholar]
- 41o. Wei Y., Jiao J., Zhao Z., Liu J., Li J., Jiang G., Wang Y., Duan A., Appl. Catal. B 2015, 179, 422–432; [Google Scholar]
- 41p. Wu H.-K., Li Y.-H., Qi M.-Y., Lin Q., Xu Y.-J., Appl. Catal. B 2020, 278, 119267; [Google Scholar]
- 41q. Zhang M., Wu M., Wang Z., Cheng R., Leung D. Y. C., Lu Z., Feng S. P., Mater. Sci. Energy Technol. 2020, 3, 734–741; [Google Scholar]
- 41r. Pang R., Teramura K., Morishita M., Asakura H., Hosokawa S., Tanaka T., Commun. Chem. 2020, 3, 137; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41s. He W., Wei Y., Xiong J., Tang Z., Wang Y., Wang X., Deng J., Yu X., Zhang X., Zhao Z., Sep. Purif. Technol. 2022, 300, 121850; [Google Scholar]
- 41t. Xu X., Asakura H., Hosokawa S., Tanaka T., Teramura K., Appl. Catal. B 2023, 320, 121885; [Google Scholar]
- 41u. Zhang Y., Wu Y., Wan L., Ding H., Li H., Wang X., Zhang W., Appl. Catal. B 2022, 311, 121255; [Google Scholar]
- 41v. Han C., Zhang R., Ye Y., Wang L., Ma Z., Su F., Xie H., Zhou Y., Wong P. K., Ye L., J. Mater. Chem. A 2019, 7, 9726–9735. [Google Scholar]
- 42. Li H., Zhang X., MacFarlane D. R., Adv. Energy Mater. 2015, 5, 1401077. [Google Scholar]
- 43.
- 43a. Yamakata A., Kawaguchi M., Nishimura N., Minegishi T., Kubota J., Domen K., J. Phys. Chem. C 2014, 118, 23897–23906; [Google Scholar]
- 43b. Yamakata A., Kawaguchi M., Murachi R., Okawa M., Kamiya I., J. Phys. Chem. C 2016, 120, 7997–8004; [Google Scholar]
- 43c. Vequizo J. J. M., Matsunaga H., Ishiku T., Kamimura S., Ohno T., Yamakata A., ACS Catal. 2017, 7, 2644–2651; [Google Scholar]
- 43d. Yamakata A., Vequizo J. J. M., J. Photochem. Photobiol. C 2019, 40, 234–243; [Google Scholar]
- 43e. Yabuta M., Takeda A., Sugimoto T., Watanabe K., Kudo A., Matsumoto Y., J. Phys. Chem. C 2017, 121, 22060–22066. [Google Scholar]
- 44. Nakada A., Saeki A., Higashi M., Kageyama H., Abe R., J. Mater. Chem. A 2018, 6, 10909–10917. [Google Scholar]
- 45. Di J., Zhu C., Ji M., Duan M., Long R., Yan C., Gu K., Xiong J., She Y., Xia J., Li H., Liu Z., Angew. Chem. Int. Ed. 2018, 57, 14847–14851; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 15063–15067. [Google Scholar]
- 46.
- 46a. Akple M. S., Low J., Liu S., Cheng B., Yu J., Ho W., J. CO2 Util. 2016, 16, 442–449; [Google Scholar]
- 46b. Wang S., Teramura K., Hisatomi T., Domen K., Asakura H., Hosokawa S., Tanaka T., Chem. Sci. 2021, 12, 4940–4948; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46c. Yu X., Ordomsky V. V., Khodakov A. Y., ChemCatChem 2020, 12, 740–749. [Google Scholar]
- 47.
- 47a. Sinkler W., Sanchez S. I., Bradley S. A., Wen J., Mishra B., Kelly S. D., Bare S. R., ChemCatChem 2015, 7, 3779–3787; [Google Scholar]
- 47b. Rasouli S., Myers D., Kariuki N., Higashida K., Nakashima N., Ferreira P., Nano Lett. 2019, 19, 46–53. [DOI] [PubMed] [Google Scholar]
- 48.
- 48a. Kurashige W., Yamazoe S., Yamaguchi M., Nishido K., Nobusada K., Tsukuda T., Negishi Y., J. Phys. Chem. Lett. 2014, 5, 2072–2076; [DOI] [PubMed] [Google Scholar]
- 48b. Niihori Y., Eguro M., Kato A., Sharma S., Kumar B., Kurashige W., Nobusada K., Negishi Y., J. Phys. Chem. C 2016, 120, 14301–14309; [Google Scholar]
- 48c. Niihori Y., Hossain S., Kumar B., Nair L. V., Kurashige W., Negishi Y., APL Mater. 2017, 5, 053201; [Google Scholar]
- 48d. Nair L. V., Hossain S., Takagi S., Imai Y., Hu G., Wakayama S., Kumar B., Kurashige W., Jiang D.-E., Negishi Y., Nanoscale 2018, 10, 18969–18979; [DOI] [PubMed] [Google Scholar]
- 48e. Hossain S., Imai Y., Suzuki D., Choi W., Chen Z., Suzuki T., Yoshioka M., Kawawaki T., Lee D., Negishi Y., Nanoscale 2019, 11, 22089–22098; [DOI] [PubMed] [Google Scholar]
- 48f. Hossain S., Imai Y., Motohashi Y., Chen Z., Suzuki D., Suzuki T., Kataoka Y., Hirata M., Ono T., Kurashige W., Kawawaki T., Yamamoto T., Negishi Y., Mater. Horiz. 2020, 7, 796–803; [Google Scholar]
- 48g. Hossain S., Ono T., Yoshioka M., Hu G., Hosoi M., Chen Z., Nair L. V., Niihori Y., Kurashige W., Jiang D.-E., Negishi Y., J. Phys. Chem. Lett. 2018, 9, 2590–2594; [DOI] [PubMed] [Google Scholar]
- 48h. Kumar B., Kawawaki T., Shimizu N., Imai Y., Suzuki D., Hossain S., Nair L. V., Negishi Y., Nanoscale 2020, 12, 9969–9979; [DOI] [PubMed] [Google Scholar]
- 48i. Negishi Y., Hashimoto S., Ebina A., Hamada K., Hossain S., Kawawaki T., Nanoscale 2020, 12, 8017–8039; [DOI] [PubMed] [Google Scholar]
- 48j. Hossain S., Miyajima S., Iwasa T., Kaneko R., Sekine T., Ikeda A., Kawawaki T., Taketsugu T., Negishi Y., J. Chem. Phys. 2021, 155, 024302; [DOI] [PubMed] [Google Scholar]
- 48k. Hossain S., Niihori Y., Nair L. V., Kumar B., Kurashige W., Negishi Y., Acc. Chem. Res. 2018, 51, 3114–3124; [DOI] [PubMed] [Google Scholar]
- 48l. Negishi Y., Nakazaki T., Malola S., Takano S., Niihori Y., Kurashige W., Yamazoe S., Tsukuda T., Häkkinen H., J. Am. Chem. Soc. 2015, 137, 1206–1212; [DOI] [PubMed] [Google Scholar]
- 48m. Negishi Y., Munakata K., Ohgake W., Nobusada K., J. Phys. Chem. Lett. 2012, 3, 2209–2214; [DOI] [PubMed] [Google Scholar]
- 48n. Negishi Y., Sakamoto C., Ohyama T., Tsukuda T., J. Phys. Chem. Lett. 2012, 3, 1624–1628; [DOI] [PubMed] [Google Scholar]
- 48o. Kurashige W., Yamaguchi M., Nobusada K., Negishi Y., J. Phys. Chem. Lett. 2012, 3, 2649–2652; [DOI] [PubMed] [Google Scholar]
- 48p. Niihori Y., Matsuzaki M., Pradeep T., Negishi Y., J. Am. Chem. Soc. 2013, 135, 4946–4949; [DOI] [PubMed] [Google Scholar]
- 48q. Kurashige W., Niihori Y., Sharma S., Negishi Y., J. Phys. Chem. Lett. 2014, 5, 4134–4142; [DOI] [PubMed] [Google Scholar]
- 48r. Niihori Y., Kurashige W., Matsuzaki M., Negishi Y., Nanoscale 2013, 5, 508–512; [DOI] [PubMed] [Google Scholar]
- 48s. Negishi Y., Kurashige W., Niihori Y., Nobusada K., Phys. Chem. Chem. Phys. 2013, 15, 18736–18751; [DOI] [PubMed] [Google Scholar]
- 48t. Kurashige W., Munakata K., Nobusada K., Negishi Y., Chem. Commun. 2013, 49, 5447–5449; [DOI] [PubMed] [Google Scholar]
- 48u. Niihori Y., Kikuchi Y., Kato A., Matsuzaki M., Negishi Y., ACS Nano 2015, 9, 9347–9356; [DOI] [PubMed] [Google Scholar]
- 48v. Kurashige W., Yamazoe S., Kanehira K., Tsukuda T., Negishi Y., J. Phys. Chem. Lett. 2013, 4, 3181–3185; [Google Scholar]
- 48w. Niihori Y., Uchida C., Kurashige W., Negishi Y., Phys. Chem. Chem. Phys. 2016, 18, 4251–4265; [DOI] [PubMed] [Google Scholar]
- 48x. Niihori Y., Matsuzaki M., Uchida C., Negishi Y., Nanoscale 2014, 6, 7889–7896; [DOI] [PubMed] [Google Scholar]
- 48y. Niihori Y., Koyama Y., Watanabe S., Hashimoto S., Hossain S., Nair L. V., Kumar B., Kurashige W., Negishi Y., J. Phys. Chem. Lett. 2018, 9, 4930–4934; [DOI] [PubMed] [Google Scholar]
- 48z. Hossain S., Kurashige W., Wakayama S., Kumar B., Nair L. V., Niihori Y., Negishi Y., J. Phys. Chem. C 2016, 120, 25861–25869; [Google Scholar]
- 48aa. Niihori Y., Shima D., Yoshida K., Hamada K., Nair L. V., Hossain S., Kurashige W., Negishi Y., Nanoscale 2018, 10, 1641–1649; [DOI] [PubMed] [Google Scholar]
- 48ab. Niihori Y., Hashimoto S., Koyama Y., Hossain S., Kurashige W., Negishi Y., J. Phys. Chem. C 2019, 123, 13324–13329; [Google Scholar]
- 48ac. Hossain S., Suzuki D., Iwasa T., Kaneko R., Suzuki T., Miyajima S., Iwamatsu Y., Pollitt S., Kawawaki T., Barrabés N., Rupprechter G., Negishi Y., J. Phys. Chem. C 2020, 124, 22304–22313; [Google Scholar]
- 48ad. Nair L. V., Hossain S., Wakayama S., Takagi S., Yoshioka M., Maekawa J., Harasawa A., Kumar B., Niihori Y., Kurashige W., Negishi Y., J. Phys. Chem. C 2017, 121, 11002–11009. [Google Scholar]
- 49. Ou H., Li G., Ren W., Pan B., Luo G., Hu Z., Wang D., Li Y., J. Am. Chem. Soc. 2022, 144, 22075–22082. [DOI] [PubMed] [Google Scholar]
- 50.
- 50a. Harada M., Einaga H., Langmuir 2007, 23, 6536–6543; [DOI] [PubMed] [Google Scholar]
- 50b. Ma J., Zou Y., Jiang Z., Huang W., Li J., Wu G., Huang Y., Xu H., Phys. Chem. Chem. Phys. 2013, 15, 11904–11908; [DOI] [PubMed] [Google Scholar]
- 50c. Tada M., Murata S., Asakoka T., Hiroshima K., Okumura K., Tanida H., Uruga T., Nakanishi H., Matsumoto S.-I., Inada Y., Nomura M., Iwasawa Y., Angew. Chem. Int. Ed. 2007, 46, 4310–4315; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 4388–4393; [Google Scholar]
- 50d. Takagi Y., Wang H., Uemura Y., Nakamura T., Yu L., Sekizawa O., Uruga T., Tada M., Samjeské G., Iwasawa Y., Yokoyama T., Phys. Chem. Chem. Phys. 2017, 19, 6013–6021; [DOI] [PubMed] [Google Scholar]
- 50e. Wang X., Shi H., Kwak J. H., Szanyi J., ACS Catal. 2015, 5, 6337–6349; [Google Scholar]
- 50f. Hu H., Cai S., Li H., Huang L., Shi L., Zhang D., J. Phys. Chem. C 2015, 119, 22924–22933. [Google Scholar]
- 51.
- 51a. Wu J., Xiong L., Hu Y., Yang Y., Zhang X., Wang T., Tang Z., Sun A., Zhou Y., Shen J., Zou Z., Appl. Catal. B 2021, 295, 120277; [Google Scholar]
- 51b. Gao G., Jiao Y., Waclawik E. R., Du A., J. Am. Chem. Soc. 2016, 138, 6292–6297; [DOI] [PubMed] [Google Scholar]
- 51c. Zhang Z., Zhang L., Yao S., Song X., Huang W., Hülsey M. J., Yan N., J. Catal. 2019, 376, 57–67. [Google Scholar]
- 52.
- 52a. Agrachev M., Ruzzi M., Venzo A., Maran F., Acc. Chem. Res. 2019, 52, 44–52; [DOI] [PubMed] [Google Scholar]
- 52b. Kwak K., Lee D., Acc. Chem. Res. 2019, 52, 12–22; [DOI] [PubMed] [Google Scholar]
- 52c. Nieto-Ortega B., Bürgi T., Acc. Chem. Res. 2018, 51, 2811–2819; [DOI] [PubMed] [Google Scholar]
- 52d. Sakthivel N. A., Dass A., Acc. Chem. Res. 2018, 51, 1774–1783; [DOI] [PubMed] [Google Scholar]
- 52e. Whetten R. L., Weissker H.-C., Pelayo J. J., Mullins S. M., López-Lozano X., Garzón I. L., Acc. Chem. Res. 2019, 52, 34–43; [DOI] [PubMed] [Google Scholar]
- 52f. Bhattarai B., Zaker Y., Atnagulov A., Yoon B., Landman U., Bigioni T. P., Acc. Chem. Res. 2018, 51, 3104–3113; [DOI] [PubMed] [Google Scholar]
- 52g. Gan Z., Xia N., Wu Z., Acc. Chem. Res. 2018, 51, 2774–2783; [DOI] [PubMed] [Google Scholar]
- 52h. Ghosh A., Mohammed O. F., Bakr O. M., Acc. Chem. Res. 2018, 51, 3094–3103; [DOI] [PubMed] [Google Scholar]
- 52i. Yan J., Teo B. K., Zheng N., Acc. Chem. Res. 2018, 51, 3084–3093; [DOI] [PubMed] [Google Scholar]
- 52j. Tian S., Liao L., Yuan J., Yao C., Chen J., Yang J., Wu Z., Chem. Commun. 2016, 52, 9873–9876; [DOI] [PubMed] [Google Scholar]
- 52k. Yamazoe S., Kurashige W., Nobusada K., Negishi Y., Tsukuda T., J. Phys. Chem. C 2014, 118, 25284–25290; [DOI] [PubMed] [Google Scholar]
- 52l. Kumara C., Aikens C. M., Dass A., J. Phys. Chem. Lett. 2014, 5, 461–466; [DOI] [PubMed] [Google Scholar]
- 52m. Yao C., Lin Y.-j., Yuan J., Liao L., Zhu M., Weng L.-H., Yang J., Wu Z., J. Am. Chem. Soc. 2015, 137, 15350–15353; [DOI] [PubMed] [Google Scholar]
- 52n. Fei W., Antonello S., Dainese T., Dolmella A., Lahtinen M., Rissanen K., Venzo A., Maran F., J. Am. Chem. Soc. 2019, 141, 16033–16045; [DOI] [PubMed] [Google Scholar]
- 52o. Sun C., Mammen N., Kaappa S., Yuan P., Deng G., Zhao C., Yan J., Malola S., Honkala K., Häkkinen H., Teo B. K., Zheng N., ACS Nano 2019, 13, 5975–5986; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52p. Lee S., Bootharaju M. S., Deng G., Malola S., Baek W., Häkkinen H., Zheng N., Hyeon T., J. Am. Chem. Soc. 2020, 142, 13974–13981; [DOI] [PubMed] [Google Scholar]
- 52q. Zhu M., Aikens C. M., Hollander F. J., Schatz G. C., Jin R., J. Am. Chem. Soc. 2008, 130, 5883–5885; [DOI] [PubMed] [Google Scholar]
- 52r. Joshi C. P., Bootharaju M. S., Alhilaly M. J., Bakr O. M., J. Am. Chem. Soc. 2015, 137, 11578–11581; [DOI] [PubMed] [Google Scholar]
- 52s. Yang H., Wang Y., Huang H., Gell L., Lehtovaara L., Malola S., Häkkinen H., Zheng N., Nat. Commun. 2013, 4, 2422; [DOI] [PubMed] [Google Scholar]
- 52t. Kawawaki T., Imai Y., Suzuki D., Kato S., Kobayashi I., Suzuki T., Kaneko R., Hossain S., Negishi Y., Chem. Eur. J. 2020, 26, 16150–16193; [DOI] [PubMed] [Google Scholar]
- 52u. Bao Y., Wu X., Gao H., Zhou M., Chen S., Jin S., Yu H., Zhu M., Dalton Trans. 2020, 49, 17164–17168; [DOI] [PubMed] [Google Scholar]
- 52v. Bhat S., Baksi A., Mudedla S. K., Natarajan G., Subramanian V., Pradeep T., J. Phys. Chem. Lett. 2017, 8, 2787–2793; [DOI] [PubMed] [Google Scholar]
- 52w. Jadzinsky P. D., Calero G., Ackerson C. J., Bushnell D. A., Kornberg R. D., Science 2007, 318, 430–433; [DOI] [PubMed] [Google Scholar]
- 52x. Qian H., Zhu M., Wu Z., Jin R., Acc. Chem. Res. 2012, 45, 1470–1479; [DOI] [PubMed] [Google Scholar]
- 52y. Tsukuda T., Bull. Chem. Soc. Jpn. 2012, 85, 151–168. [Google Scholar]
- 53.
- 53a. Mitsui M., Nagaoka S., Matsumoto T., Nakajima A., J. Phys. Chem. B 2006, 110, 2968–2971; [DOI] [PubMed] [Google Scholar]
- 53b. Nakaya M., Iwasa T., Tsunoyama H., Eguchi T., Nakajima A., Nanoscale 2014, 6, 14702–14707; [DOI] [PubMed] [Google Scholar]
- 53c. Minamitani E., Takagi N., Arafune R., Frederiksen T., Komeda T., Ueba H., Watanabe S., Prog. Surf. Sci. 2018, 93, 131–145. [Google Scholar]
- 54. Iida K., Noda M., Ishimura K., Nobusada K., J. Phys. Chem. A 2014, 118, 11317–11322. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.