

Abstract
The prebiotic generation of sugars in the context of origins of life studies is of considerable interest. Among the important intramolecular processes of sugars are carbonyl migrations and accompanying epimerizations. Herein we describe the carbonyl migration‐epimerization process occurring down the entire carbon chain of chirally pure d‐tetroses sugars under mild conditions. Employing chirally pure 1‐13C‐erythrose, 4‐13C‐erythrose and 1‐13C‐threose, we (1) identify all the species formed as the carbonyl migrates down the four‐carbon chain and (2) assess the rates associated with the production of each of these species. Competing aldol reactions and oxidative fragmentation processes were also observed. Further observations of self‐condensation of glycolaldehyde mainly yielding 2‐keto‐hexoses (sorbose and tagatose) and tetrulose also provides a basis for understanding the effect of carbonyl migrations on the product distribution in plausible prebiotic scenarios.
Keywords: aldol reaction, carbonyl migration, epimerization, formose reaction, tetroses
Prebiotic sugars: An investigation of carbonyl migration and epimerization, proceeding down the entire carbon chain of tetrose sugars (d‐erythrose and d‐threose) in aqueous media, and ultimately leading to a mixture of d‐erythrulose, rac‐erythrulose and rac‐tetroses arising from the above reactions, is presented in this study.

Introduction
Carbohydrates perform several important roles in life, including energy production and storage, structural components of membranes and cell walls, and integral parts of RNA/DNA and glycoproteins. Carbohydrates are complex chiral molecules which can undergo a plethora of acid‐, base‐ and metal‐catalyzed intra‐ and inter‐molecular reactions. Among the important intramolecular processes are carbonyl migration and accompanying epimerization also known as the Lobry de Bruyn‐Alberda van Ekenstein (LdB‐AvE) transformation.[ 1 , 2 , 3 ] Carbonyl migration is also an important mechanism for how biology manages sugar metabolism. For example, enzyme‐catalyzed carbonyl migration occurs in core metabolic pathways, [4] such as isomerization of glyceraldehyde 3‐phosphate to dihydroxyacetone phosphate, fructose 6‐phosphate to glucose 6‐phosphate in gluconeogenesis, and ribulose 5‐phosphate to ribose 5‐phosphate in the pentose phosphate pathway. When it comes to prebiotic carbohydrate synthesis, the postulated autocatalysis step in the formose reaction requires carbonyl migration to isomerize ketotetroses to aldotetroses which subsequently undergo a retroaldol fragmentation to yield two molecules of glycolaldehyde. [5] It is apparent that carbonyl migration plays an important role in biochemistry and it is possible that this fundamental biochemical transformation may trace its origins back to an era that preceded enzymatic catalysis, however, a systematic exploration of carbonyl migration relevant to prebiotic sugar chemistry would be significant.
Scheme 1 illustrates a series of reversible carbonyl migrations and epimerizations of a generalized chiral aldose‐ a 2,3‐dihydroxy aldehyde, both of which are usually reversible processes. The initial carbonyl migration results in a 1,2‐aldo‐keto transformation. The reverse 2,1‐keto‐aldo migration is accompanied by an epimerization at the original C2‐position. Carbonyl migration from the C2‐postion to the C3‐position represents a keto‐keto transformation which also results in epimerization at the C2‐position. The reverse keto‐keto migration from the C3 position to C2 position leads to epimerization at the original C3‐postion. The migration‐epimerization process can, in principle, continue down the chain of longer linear sugar molecules ultimately leading to an aldehyde functionality at the opposite end of the original aldose. Adding another dimension to these processes is the intervention of cyclic hemiacetal species which can interrupt or potentially shut down the migration‐epimerization process. [6]
Scheme 1.
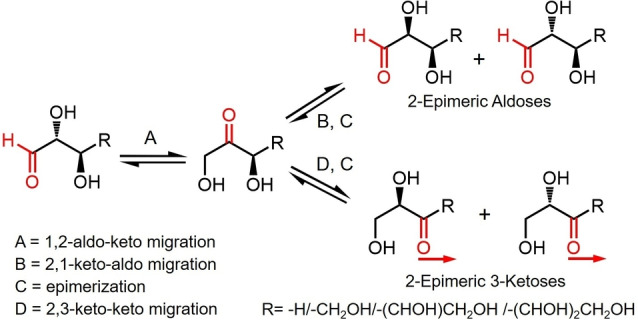
Carbonyl migration and epimerization along sugar carbon chain.
Carbonyl migration/epimerization of monosaccharides usually take place in basic media.[ 7 , 8 , 9 , 10 ] For example, mannose and four keto‐hexoses (fructose, psicose, sorbose, tagatose) were observed in the reaction of glucose in aqueous KOH. [11] The epimerization of glucose affords mannose and carbonyl migration of glucose resulted in the formation of these keto‐hexoses. Moreover, three (d ‐manno‐, d‐gluco‐, and d‐altro‐) 2‐heptuloses were investigated from carbonyl migration and epimerization of d ‐manno‐3‐heptuloses by the action of calcium hydroxide. [8] These reactions depend on the reaction conditions, proceeded via a proton transfer mechanism involving cis‐ and trans‐ enediol intermediates and/or a 1,2‐hydride shift mechanism (Scheme 2).[ 2 , 7 , 12 , 13 ] Hexoses and pentoses labeled with 13C at the C1‐position have been shown to exhibit carbonyl migration/ epimerization processes down the entire carbon chain qualitatively indicating that both aldo‐keto as well as keto‐keto and keto‐aldo migrations are taking place. However, due to the limitations of the analytical tools employed, none of these reports identify all the intermediates as the carbonyl functionality migrates down the chain. It is also important to emphasize that many of these reported studies were conducted under highly basic conditions (pH≥11) where fragmentations (retro‐aldol reactions) followed by recombinations (aldol reactions) were also observed along with the formation of unidentified colored side products‐ processes which complicate the interpretation of the carbonyl migration/epimerization results.
Scheme 2.

a) Carbonyl migration via cis‐ and trans‐ enediol intermediates, and b) carbonyl migration via a 1,2‐hydride shift.
In contrast, under relatively mild basic conditions, Nagorski and Richard [13] have shown that the carbonyl rearrangement of glyceraldehyde to dihydroxyacetone takes place by either a proton transfer process and/or by a hydride shift pathway depending on the specific reaction conditions. At pD 8.4 in the presence of a pyrophosphate buffer the dominant mechanistic pathway was shown to be the proton transfer process proceeding presumably through enediol intermediates (Scheme 2a). In contrast, when employing 0.01 M deuteroxide approximately 60 % takes place via proton transfer and 40 % by 1,2‐hydride transfer. It was also shown that, in the presence of Zn2+ carbonyl migration proceeded via a hydride transfer process (Scheme 2b, Zn2+ ). Breslow and co‐workers[ 14 , 15 ] have shown that at pH 12 in the presence of Ca2+ (formose reaction conditions) carbonyl migration of glyceraldehyde to dihydroxyacetone proceeded via a 1,2‐hydride transfer mechanism with “a significant tunneling component” (Scheme 2b, Ca2+ ). It would be useful to employ simple linear chirally pure sugars with 13C labels at specific locations thus expanding our knowledge and understanding of carbonyl migration and epimerization.
In order to quantitatively explore both carbonyl migration along the carbon chain of a simple linear sugar and the accompanying epimerization, a series of experiments were conducted employing simple chirally pure C‐4 (tetrose) sugars. We focused on the tetroses since they are postulated to be a central intermediate in many prebiotic scenarios [16] for the higher order sugar synthesis and provide a C‐4 skeleton wherein the carbonyl migration can be studied coupled with its effect on sugar chirality. Herein is presented (1) the effects of temperature, pH and buffer on the rate of reaction of carbonyl migration of d‐erythrose, a simple chirally pure four‐carbon sugar, labeled with 13C in the C1‐position; (2) the accompanying rates of formation of d‐erythrulose, rac‐erythrulose, d‐threose and rac‐tetroses; (3) companion rate experiments involving carbonyl migration and epimerization of erythrose labeled with 13C in the C‐4 position and threose labeled with 13C in the C‐1 position; (4) the incorporation of deuterium into the C‐4 species with respect to time when the carbonyl migration was conducted in D2O, (5) the observation of oxidative fragmentation of erythrose, threose and also when starting with glycolaldehyde during the carbonyl migration and epimerization process.
Results and Discussion
Erythrose is a C‐4 carbon sugar which, in an aqueous environment, exists as a rapidly converting equilibrium mixture of four species‐the open chain aldehyde (1), the corresponding hydrate (2), and α‐ and β‐furanoses (3) and (4), respectively.[ 17 , 18 , 19 ] The 13C NMR spectrum of erythrose shows only the aldehyde hydrate and the two cyclic hemiacetals. Although the free aldehyde is the species which initiates carbonyl migration‐epimerization, it is present in only miniscule amounts. Indeed, even using 13C‐labeled erythrose the free carbonyl carbon is not observed in the 13C NMR spectrum. That carbonyl migrations‐epimerization takes place is only due to the rapid interconversion of the various species through the free aldehyde.
Assuming that the rearrangement of the carbonyl group is capable of taking place along the entire four‐carbon chain of d‐erythrose, the carbonyl‐migrations and epimerizations would lead to a complex mixture of the following structural isomers: d‐erythrulose from 1,2‐(aldo‐keto) carbonyl migration, d‐threose from 2,1‐(keto‐aldo) carbonyl migration, rac‐erythrulose from 2,3‐(keto‐keto) carbonyl migration, and rac‐tetroses from 3,4‐(keto‐aldo) carbonyl migration. It is assumed that the various interconversions proceed via enediol intermediates. This complex set of equilibria are summarized in Scheme 3. In order to facilitate the detection and quantitation of the products formed during carbonyl migration and epimerization of erythrose by 13C NMR, a series of experiments were conducted making use of erythrose labeled with 13C at the C‐1 position. Labeling at only one carbon of erythrose greatly simplifies the analyses since only singlets would be associated with each of the products formed as the carbonyl functionality proceeded down the chain. Thus, following the change in chemical shift of this labeled carbon with respect to time allowed identification of all the new species being formed.
Scheme 3.

Carbonyl migration of d‐erythrose and d‐threose along the carbon chain.
Carbonyl migration of d‐[1‐13C]‐erythrose
A solution containing 80 mM d‐[1‐13C]‐erythrose buffered with 160 mM sodium bicarbonate at pH 8.5 was prepared and monitored over time at 25 °C by 13C NMR spectroscopy. As shown in Figure 1a‐Bottom, the recorded 13C NMR spectrum at t=0 h reveals four major signals assigned to 1‐13C‐β‐d‐erythrofuranose (4, 100.2 ppm, ∼58 %), 1‐13C‐α‐d‐erythrofuranose (3, 94.6 ppm, ∼23 %), 1‐13C‐d‐erythrose hydrate (2, 88.5 ppm, ∼10 %). A small amount of 1‐13C‐d‐erythrulose (5, 65.0 ppm, ∼4 %) was also observed indicating that under these experimental conditions, carbonyl migration had already begun. In addition to d‐erythrulose, the intensity of new peaks assigned to 1‐13C‐α‐d‐threofuranose (10, 101.2 ppm), 1‐13C‐β‐d‐threofuranose (9, 95.7 ppm), 1‐13C‐d‐threose hydrate (8, 88.9 ppm), 4‐13C‐rac‐erythrulose (6, 62.0 ppm), 4‐13C‐rac‐tetrofuranose (12, 72.0, 70.1 and 69.5 ppm) and 4‐13C‐rac‐tetrose hydrate (11, 61.6, 62.4 ppm) were followed over a period of 109 h (Figure 1a‐Top). These resonances were assigned by comparing with the signals of authentic standards under identical conditions The chemical shifts associated with each of the four‐carbon species are listed in Figure 1b‐Bottom. The stacked 13C NMR spectra over this period of time are shown in Figure 2. Figure 1a shows that each of the structures proposed to arise from carbonyl migration and epimerization proceeding down the chain of erythrose in Scheme 3 has been identified. The generation of threose 8–10, the epimer of erythrose, is an indication of a keto‐aldo carbonyl migration from d‐erythrulose 5.
Figure 1.

a) 13C NMR spectrum for [1‐13C]‐erythrose carbonyl migration at 0 and 109 h at 25 °C, pH 8.5. G1: glycolate, G2: glycerate; b) 13C chemical shifts (ppm) of the labeled carbons associated with each of the species.
Figure 2.

13C NMR spectra of 80 mM d‐[1‐13C]‐erythrose 1 with 160 mM NaHCO3 buffer at pH 8.5 under 25 °C over time. d‐Erythrose (hydrate 2 and cyclic ring 3, 4), d‐erythrulose 5, rac‐erythrulose 6, d‐threose (hydrate 8 and cyclic ring 9, 10), rac‐tetroses (hydrate 11 and cyclic ring 12 and diastereomeric C8 species (14 and 15) were observed in these spectra over time and their peak intensity detected in each spectrum are recorded in Table S1.
Not unexpectedly, diastereomeric octuloses (14, doublets in the 69.5–70.0 and 72.5–73.5 ppm and 15, singlets in the 74.4–75.1 ppm regions) were also observed, and attributed to aldol reactions between the various aldose and ketose stereoisomers present in the reaction solution. [20] The aldol reactions leading to a variety of 13C‐labeled diastereomeric octuloses, are summarized in Scheme S1. In addition, three new peaks assigned to formate (170.1 ppm), glycerate (63.2 ppm) and glycolate (60.4 ppm) suggested that some oxidative fragmentation, promoted by oxygen in the air, has accompanied the carbonyl migrations‐epimerizations and aldol reactions. This oxidation process will be discussed further in a later section.
To quantify the species produced during the carbonyl migration and epimerization of 1‐13C‐d‐erythrose, the intensity of each 13C NMR peak obtained from the spectra displayed in Figure 2 is listed in Supporting Information Table S1. It is important to note that the sum total intensity for all the peaks in the 13C NMR spectra at each of the time intervals remain relatively constant suggesting a good mass balance. The rates of formation and disappearance of the species in Scheme 3 and Scheme S1 over a period of 109 h is graphically displayed in Figure 3. For convenience, in describing the progression of the migration of the carbonyl group along the four‐carbon chain, the 13C‐labeled carbon is designated as C1. Over a period of 18 h the intensity of the peak of erythrose decreases by approximately 60 % (red curve). In this analysis erythrose is the sum of the peak intensities of the aldehydic form (1), the hydrate (2), and the α‐ and β‐ furanose forms (3 and 4). This decrease is accompanied by an increase in the peaks attributed to d‐erythrulose (5) resulting from an aldo‐keto carbonyl migration (black curve). It should be noted that this reaction is reversible; the formation of d‐threose (the epimer of d‐erythrose) via keto‐aldo carbonyl migration is observed (pink curve). As in the case of erythrose, the amount of threose is the sum of the peak attributed to the aldehydic form (7), the hydrate (8), and α‐ and β‐ furanose forms (9 and 10). Keto‐keto migration of the carbonyl group from C2 to C3 produces rac‐erythrulose (green curve). Keto‐aldo carbonyl migration from C3 to C4 completes the full migration over the entire four carbon system producing rac‐tetroses (grey curve) which is a mixture of the linear hydrate and the cyclic hemiacetals of rac‐erythrose and rac‐threose. After reaction for 109 h, the carbonyl migrations‐epimerizations lead to a mixture of ∼10 % d‐erythrose together with ∼32 % d‐erythrulose, ∼18 % rac‐erythrulose, ∼4 % rac‐tetroses and ∼10 % d‐threose.
Figure 3.
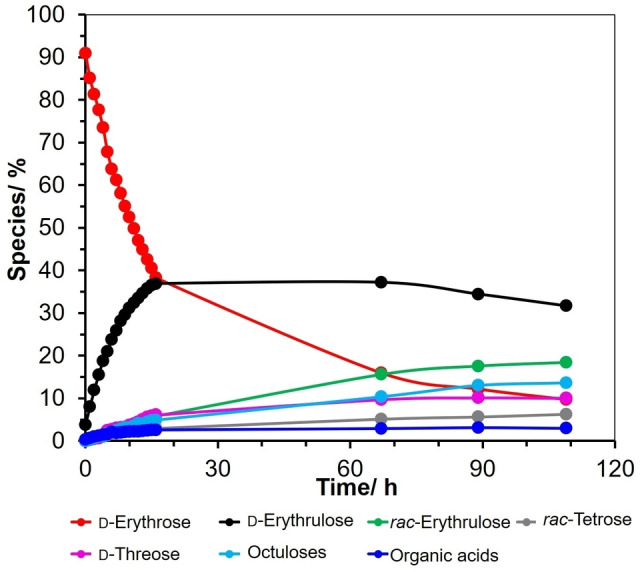
Reaction progress of the d‐erythrose and the formation of d‐erythrulose, rac‐erythrulose, rac‐tetroses, d‐threose, diastereomeric octuloses and organic acids (formate, glycolate and glycerate) as determined by NMR data shown in Figure 2. This observation is consistent with DFT calculations. The difference in the free energies of reaction for erythrulose undergoing a keto‐aldo migration to form either threose or erythrose is only 0.71 kcal/mol.
Scheme S1a indicates that with the initial formation of d‐erythrulose, a base promoted bimolecular aldol reaction with d‐erythrose takes place leading to the formation of diastereomeric octuloses. Any d‐threose produced from the keto‐aldo carbonyl migration from d‐erythrulose can also undergo aldol reactions with d‐erythrulose to form diastereomeric octuloses. In both cases one would expect to observe doublets in the 13C NMR spectra with peaks in the 69 to 76 ppm range. Indeed, significant doublets are observed at 69.5 to 70.0 ppm and 72.5 ppm to 73.5 ppm (14, Figure 1) along with smaller doublets. It should be emphasized that at t=0 h, there are no peaks within this range. As the carbonyl migration proceeds down the chain to produce rac‐erythrulose, subsequently rac‐tetroses, similar aldol reactions produce additional diastereomeric octuloses where the labeled carbons appear as singlets in the 13C NMR spectra within the 74 to 76 ppm (internal ‐CHOH‐) and 60 to 62 ppm (terminal ‐CH2OH) ranges (Scheme S1b, S1c, S1d). The sum total of these aldol reactions results in approximately 14 % octuloses after 109 h. It is expected, however, that the octuloses will continually increase at reaction times greater than 109 h.
The carbonyl migration experiments reported thus far were conducted at pH 8.5. In order to investigate the effect of pH on carbonyl migration and epimerization, a solution of d‐[1‐13C]‐erythrose was subjected to reaction in buffered aqueous media at pH 5, 7, 8.5 and 10 at 40 °C where the various buffers employed were all at the same concentration (160 mM). The comparative rates of disappearance of d‐[1‐13C]‐erythrose (carbonyl migration and aldol reaction) under these different pH conditions were followed employing 13C NMR analyses. The rate profiles for the disappearance of erythrose is shown in Figure 4a and the corresponding rate profiles for the formation of d‐erythrulose and the diastereomeric octuloses are shown in Figure 4b. For further information the stacked 13C NMR spectra along with the experimental details are summarized in Supporting Information: Figures S1–S4, S19–S22 and the listing of the peak intensities are summarized in Supporting Information: Tables S2–S5.
Figure 4.
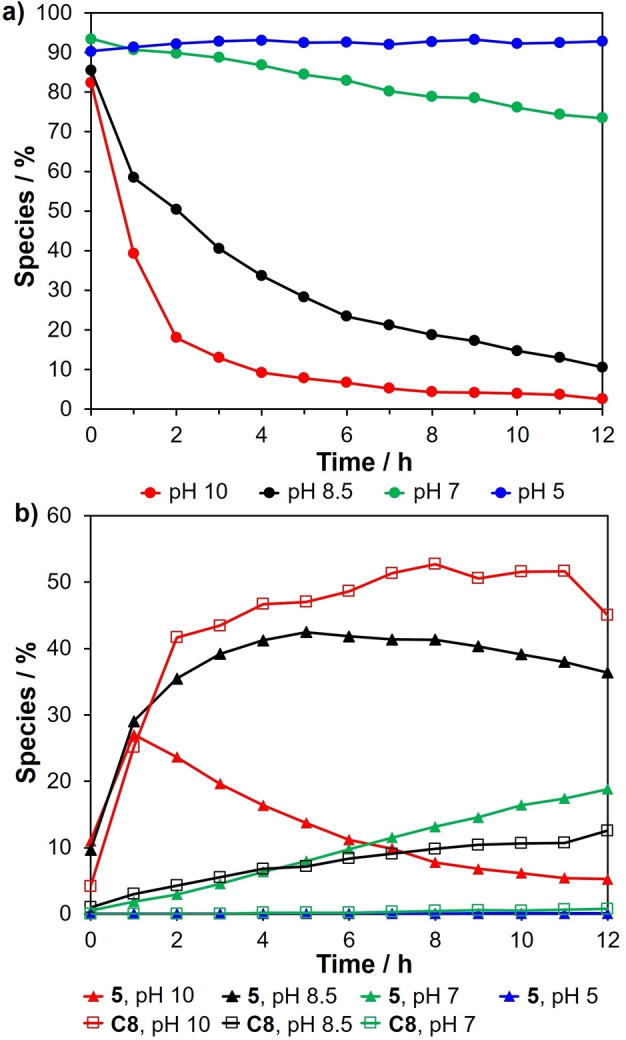
(a) Reaction progress of the d‐erythrose at pH 5, 7, 8.5 and 10 over a 12‐hour period; (b) The rates of the formation of d‐erythrulose 5, and diastereomeric octuloses (C8) at pH 5, 7, 8.5 and 10 over a 12‐hour period. The compositions were measured per hour.
It is clear that there is a dramatic difference in the rate of disappearance of erythrose over the pH range investigated. As shown in Figure 4a, the fastest rates of reaction occur in basic media (pH 10 and pH 8.5‐bicarbonate buffers). Within a period of 12 h most of the erythrose has reacted. In contrast, in slightly acidic media (pH 5‐acetate buffer) negligible reaction takes place over the same period of time. At neutral pH (pH 7‐phosphate buffer), the reaction is relatively slow compared to the reactions at both pH 8.5 and 10. At pH 7, approximately 10 % of the erythrose has reacted after 12 h. Figure 4b indicates that at pH 8.5 and 10 the formation of the diastereomeric octuloses is extremely fast. This is consistent with rapid carbonyl migration of erythrose to erythrulose followed by a rapid aldol process. At pH 7, the formation of the octuloses is relatively slow while at pH 5 these aldol products are not detected within the 12‐hour time period.
Changing the buffer and the buffer concentration at a constant pH provided greater insight to the carbonyl migration and epimerization process. The rates of disappearance of erythrose and the rates of formation of d‐erythrulose at pH 7 as a function of buffer and buffer concentration was explored. Phosphate buffers at concentrations of 160 and 480 mM (Supporting Information: Figure S2 and S5) and a cacodylate buffer at a concentration of 160 mM (Supporting Information: Figure S6) were employed. The results are graphically summarized in Figure 5. Increasing the concentration of phosphate buffer results in a modest increase in the rate of disappearance of d‐erythrose and the rate of formation of d‐erythrulose. Changing the nature of the buffer by employing cacodylate, a weaker base, at the same buffer concentration as phosphate (160 mM) results in a modest decrease in the rate of disappearance of erythrose. These results suggest general base catalysis and are consistent with reports dealing with the isomerization of aldo‐pentoses into keto‐pentoses [21] and the interconversion of glucose to fructose [22] at neutral pH.
Figure 5.
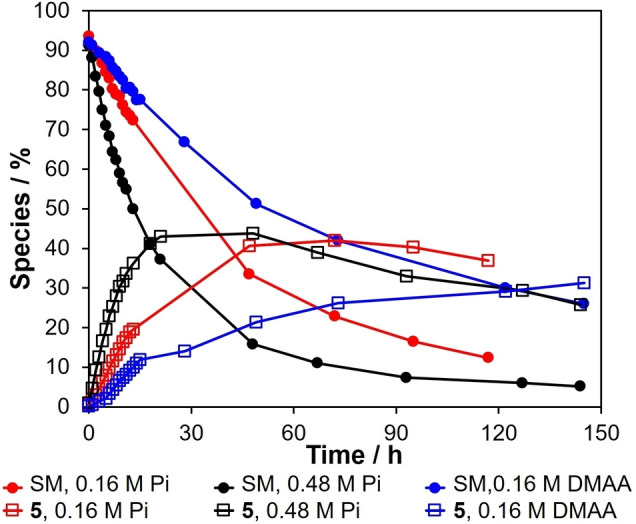
Reaction progress of the d‐erythrose (SM) and the formation of d‐erythrulose 5 at pH 7 in 160 mM, 480 mM Pi and 160 mM cacodylate (DMAA) buffer.
Carbonyl migration of d‐[4‐13C]‐erythrose
In order to provide additional evidence for the interpretation of the experimental results shown in Figure 1, carbonyl migration experiments employing erythrose labeled with 13C in the C4‐position was investigated at pH 8.5 at 40 °C and compared with the results of the experiments conducted with erythrose labeled in the C1‐position. As shown in Figure 6, three major resonances were recorded at 0 h, assigned to be 4‐13C‐α,β‐d‐erythrose furanose 3’ and 4’ (70.1 ppm), 4‐13C‐d‐erythrose hydrate 2’ (61.6 ppm) as well as 4‐13C‐d‐erythrulose 5’ (62.0 ppm), respectively. New resonances correspond to 1‐13C‐rac‐tetrose furanose 12’ (94.5–101.8 ppm), 1‐13C‐rac‐tetrose hydrate 11’ (88.4–89.2 ppm), 4‐13C‐α‐d‐threose furanose 10’ (72.0 ppm), 4‐13C‐β‐d‐threose furanose 9’ (69.5 ppm), 1‐13C‐rac‐erythrulose 6’ (65.0 ppm), 4‐13C‐d‐threose hydrate 8’ (62.4 ppm), were also observed to grow over time. The peak intensities for each of the species as a function of time are summarized in Supporting Information Table S8. These observations indicate that all of the carbonyl migration species determined in d‐[1‐13C]‐erythrose solution were consistent with those observed in the d‐[4‐13C]‐erythrose. In comparison to the use of d‐[1‐13C]‐erythrose, the rac‐tetroses in the spectra of d‐[4‐13C]‐erythrose were more easily observed within the 88–102 ppm region. Further analysis of the 13C NMR spectra shown in Figure S25 reveals that the migration rates of carbonyl group from C1 to C4 achieved in d‐[4‐13C]‐erythrose are essentially the same as those in d‐[1‐13C]‐erythrose under similar conditions (Figure S21).
Figure 6.

a) 13C NMR spectrum for [4‐13C]‐erythrose carbonyl migration at 0 and 36 h at 40 °C, pH 8.5. G2: glycerate; b) 13C chemical shifts (ppm) of the labeled carbons associated with each of the species.
It should be noted that, if erythrose itself would undergo retro‐aldol under these reaction conditions, then the aldol reactions of resulting glycolaldehyde could lead to other recombination products, thus affecting the product distribution and interfere with the carbonyl‐migration analysis. To test whether such retro‐aldol reactions are occurring and are reflected in the products formed, a control experiment employing a 1 : 1 mixture of d‐[1‐13C]‐erythrose and d‐[4‐13C]‐erythrose was carried out at pH 8 and 40 °C. Analysis by 13C NMR and MS‐spectra (Figure S8 and S18), however, showed product distribution that was similar to those obtained from d‐[1‐13C]‐erythrose alone or from d‐[4‐13C]‐erythrose alone under identical conditions. No doubly 13C‐labeled erythrose (or erythrulose) was detected in the mixed reaction of d‐[1‐13C]‐erythrose and d‐[4‐13C]‐erythrose (Figure S18d), which suggests that a retro‐aldol of the individual erythrose were not occurring at the detectable limits of these analytical techniques. The only doubly 13C‐labeled products observed were peaks corresponding to octuloses consistent with aldol reaction between erythrose/erythrulose combinations. These observations, under the conditions where carbonyl migrations are taking place, suggests that the retro‐aldol reaction of erythrose/erythrulose are not interfering with the product distribution.
Carbonyl migration of d‐[1‐13C]‐threose
Threose, like its diastereomer erythrose, exists as a rapidly converting equilibrium mixture of four species in an aqueous medium: the linear aldehyde, the corresponding hydrate, and the α‐ and β‐ cyclic hemiacetals (Scheme 3). 1‐13C Labeled threose was subjected to the identical reaction conditions at pH 8.5 as applied to 1‐13C‐erythrose. It was observed, however, that carbonyl migration of threose at 25 °C was significantly slower than the 1‐13C‐erythrose. As a consequence, in order to make a direct comparison between 1‐13C‐threose and 1‐13C‐erythrose the reaction of each aldose was each conducted at 40 °C. The comparative results are shown in Figure 7b and the stacked 13C NMR spectra for the reaction of 1‐13C‐threose is shown in Figure 7a and the chemical shifts and peak intensities for each of the species in the equilibria are tabulated in Supporting Information Table S9.
Figure 7.

a) 13C NMR spectrum for [1‐13C]‐threose carbonyl migration at 0 and 13 h at 40 °C, pH 8.5. G1: glycolate, D: dihydroxyacetone, G2: glycerate. b) Left: 1‐13C‐d‐Threose carbonyl migration: Reaction progress of d‐threose and the formation of d‐erythrulose, rac‐erythrulose, rac‐tetrose, d‐erythrose and diastereomeric octuloses at pH 8.5 over a 13‐hour time period at 40 °C. Right: 1‐13C‐d‐Erythrose carbonyl migration: Reaction progress of d‐erythrose and the formation of d‐erythrulose, rac‐erythrulose, rac‐tetroses, d‐threose and diastereomeric octuloses at pH 8.5 over a 12‐hour time period at 40 °C.
The 13C NMR spectrum at 0 h reveals three major signals assigned to 1‐13C‐α‐d‐threofuranose (10, 101.2 ppm, ∼50 %), 1‐13C‐β‐d‐threofuranose (9, 95.7 ppm, ∼37 %), and 1‐13C‐d‐threose hydrate (8, 88.9 ppm, ∼9 %), as well as a small signal of 1‐13C‐d‐erythrulose (5, 65.0 ppm, ∼1 %). Immediately after heating to 40 °C the signal for d‐erythrulose (5) was observed to increase in intensity while the signals for threose decreased proportionally. In addition to the d‐erythrose, the new resonances assigned to 1‐13C‐β‐d‐erythrofuranose (4, 100.2 ppm), 1‐13C‐d‐erythrose hydrate (2, 88.5 ppm), 4‐13C‐rac‐erythrulose (6, 62.1 ppm), 4‐13C‐rac‐tetrofuranose (12, 72.0 ppm), and diastereomeric C8 species (14, doublets in the 66.5–67.5 and 68.5–69.5 ppm, and 15, singlets in the 74.4–76.0 ppm regions) also appeared over a period of 13 h. The corresponding information for 1‐13C‐erythrose at 40 °C is shown in Supporting Information Figure S3 and Table S4. The comparative results in Figure 7b show that erythrose (2 h, pH 8.5, 40 °C) has an obvious shorter half‐life than threose (more than 12 h) under identical reaction conditions. The reaction products for 1‐13C‐erythrose are essentially the same as those found at 25 °C; only difference is that the rate of reaction is faster at 40 °C.
Proposed mechanistic pathways for carbonyl migration
As previously discussed, two major mechanisms have been reported to explain the migration of a carbonyl functional group along the chain of a linear sugar molecules: (1) proton abstraction with the formation of an enediol (enediolate) intermediate (Scheme 2a) and (2) an intramolecular (concerted) 1,2‐ hydride shift (Scheme 2b). There is evidence that the carbonyl migration described in this report proceed via the former pathway. In the carbonyl migration of erythrose and threose at pH 8.5, it was observed that almost immediately after the formation of erythrulose (carbonyl migration from C1 to C2) the diastereomeric octuloses were produced (Figure 8a). This observation strongly suggests the formation of an erythrulose enolate intermediate via proton abstraction leading to a classical aldol process.
Figure 8.

The evidence support the proposed mechanism that carbonyl migration occurs predominantly through enediol formation. a) Proposed aldol pathway for the formation of the diastereomeric octuloses. b) MS spectra of d‐erythrose in D2O at pH 8.5 from m/z 143–148 over time.
In order to determine if enolate (enediol) intermediates were involved in the carbonyl migration process, a series of experiments with unlabeled threose was conducted in D2O and followed by mass spectrometry. These experiments provided a means of following the incorporation of deuterium into the four‐carbon sugar as the carbonyl migration proceeded. Note that deuterium incorporation into the sugar chain should only be observed if carbonyl migration proceeds by enediol (enediolate) intermediates while no deuterium incorporation should be observed if carbonyl migration takes place via and intramolecular 1,2‐hydride shift (Scheme S2a). The results, over a period of 12 h, are shown in Figure 8b. It is clear that as the carbonyl migration progressed, deuterium incorporation increased. After 12 h, 5 deuterium atoms were incorporated into the four‐carbon sugar. The results are consistent with the deprotonation pathway involving enediol intermediates (Figure 8a). The detail of deuterium incorporation into threose carbon chain is shown in Scheme S2b. It is concluded that a major pathway for the carbonyl migration process involves a base promoted deprotonation‐protonation process with an enediol intermediate. It must be emphasized that these experiments do not negate the possibility that the intramolecular 1,2‐hydride shift can take place. This pathway may still be a minor component in the carbonyl migration process.
Oxidative fragmentation of erythrose and threose
It has been demonstrated that subjecting 1‐13C‐erythrose and 1‐13C‐threose to mildly basic conditions in an aqueous environment resulted in both carbonyl migration along the 4‐carbon chain and aldol reactions producing a variety of diastereomeric octuloses. Since these experiments were conducted in an air environment, it was not surprising that a variety of oxidative fragmentation products [23] were also observed in neutral and basic media. The specific oxidation products identified included formate, glycerate and glycolate (see Supporting Information Figures 1, S1–S6 and S8; Tables S1–S7 and S9). When threose solutions were enriched with air, the pathways leading to the formation of formate, glycerate and glycolate took place over a much shorter period of time (Supporting Information Figure S11, S12 and S26b). Similar experiments, performed with degassed solutions (sparged with N2), produced much reduced amounts of these oxidation products (Supporting Information Figure S10 and S26a). The proposed mechanistic pathways for the formation of formate, glycerate and glycolate are shown in Scheme 4. The enolate derived from a tetrose or tetrulose reacts with oxygen to form a peroxyanion intermediate followed by formation of a dioxetane which subsequently fragments into either formate and glycerate or two molecules of glycolate. This suggested pathway is consistent with the mechanistic pathway suggested by Dubourg and Naffa.[ 24 , 25 ]
Scheme 4.

Proposed reaction pathways for the formation of formate, glycerate and glycolate from tetroses or tetruloses with oxygen.
It was initially postulated that the source of formate was from the oxidation of formaldehyde derived from a retro‐aldol reaction of tetrulose. In control experiments it was observed that formate was not detected when 13C labeled formaldehyde was submitted to the identical reaction conditions applied to erythrose or threose at pH 8.5 thus eliminating formaldehyde as the formate precursor (Supporting Information Figure S13). It is interesting to note that the oxidation of hexoses in alkaline solution leads to the almost quantitative production of formate under high oxygen pressure. [26]
Aldol condensation of glycolaldehyde and accompanying oxidative fragmentation
Based on the observations in the erythrose and threose carbonyl migration studies, the question naturally arose as to the products that would be observed accompanying the formation of C4 sugars derived from the aldol reaction of glycolaldehyde, as this is an important step in the formose‐type reaction scenarios. In order to probe this question, the reaction of an aqueous solution of unlabeled glycolaldehyde at room temperature and pH 8.5 (HCO3 − buffer) was investigated, first, in a reaction flask open to air environment. The experimental protocol and observations along with the corresponding 1H and 13C NMR spectra for the reactions conducted in 0.5 M and 1.0 M HCO3 − are shown in Supporting Information Figures S14 and S15. The reactions were observed to proceed at a faster rate at the higher buffer concentration. As expected, tetrulose was produced, presumably via the initially formed tetroses from the condensation of two glycolaldehyde units. The signals corresponding to tetroses were not observed in the NMR spectrum suggesting a fast aldose to ketose conversion. In addition, many of the oxidation products observed in the carbonyl migration studies of erythrose and threose (Scheme 4) were also present. These include formate, glycerate, glycolate and glyoxal hydrate; the retro‐aldol product formaldehyde hydrate derived from tetrose/tetrulose was also observed. The proposed mechanisms for the formation of glyoxal hydrate and formaldehyde hydrate are shown in Scheme 5a and 5b, respectively. The enolate derived from a glycolaldehyde reacts with oxygen to afford a peroxyanion intermediate followed by generation of a hydroperoxide which subsequently fragments into H2O2 and glyoxal. The C2 peroxyanion intermediate can also lead to a dioxetane which can fragment into two molecules of formate.
Scheme 5.

Proposed reaction pathways for a) the formation of glyoxal hydrate (Path a) and an additional pathway for the formation of formate (Path b). b) Retro‐aldol processes describing the formation of formaldehyde hydrate, glycolaldehyde and dihydroxyacetone. c) aldol reaction of glycolaldehyde yielding erythrulose and 2‐keto‐hexoses (tagatose and sorbose) via carbonyl migration.
When the presence of oxygen was minimized by employing degassed solvents and running the reaction under N2 atmosphere, tetrulose was formed as the main product. The formation of the oxidation products glycerate, formate, glycolate and glyoxyal hydrate proceeded much more slowly (over a period of days). The retro‐aldol product formaldehyde hydrate was also detected. The comparative 13C NMR spectra of the aldol reaction of glycolaldehyde under a nitrogen atmosphere followed by the continuation of the reaction in an air environment are displayed in Supporting Information S16. At 40 °C and pH 7.0 (phosphate buffer), in addition to tetrose/tetrulose formation, formate, glycerate, glyoxal hydrate were also observed (Supporting Information S17).
The studies of carbonyl migration and aldol reactions under relatively mild basic conditions herein may provide insight into how carbonyl migration guides the product distribution under various prebiotic conditions for sugars synthesis.[ 27 , 28 , 29 ] For example, In addition to formaldehyde addition, glycolaldehyde addition reaction is also an alternative chain‐growth mechanism in the formose reaction. Self‐condensation of glycolaldehyde yields tetroses (erythrose, threose). A further aldol reaction with another glycolaldehyde would be expected to afford aldohexoses, however, the 13C NMR spectra in S16 revealed no obvious aldohexoses. Instead, 2‐keto‐hexoses (sorbose and tagatose) were observed as the major C6 sugars together with erythrulose. This result suggests that most of the newly formed tetroses were converted to tetruloses via carbonyl migration, consistent with the observation in Figure 3, followed by aldol reaction with glycolaldehyde to yield linear 3‐keto hexoses and further carbonyl migration of 3‐keto hexoses to afford 2‐keto‐hexoses (Scheme 5c). It should be noted that, only miniscule amounts of formaldehyde, glycolaldehyde, and dihydroxyacetone were present in the above 13C‐labeled tetrose studies, indicating that retrol‐aldol reaction of tetroses to smaller sugars rarely occurs if at all ‐ at least under the above conditions in the absence of Ca2+. This result is also consistent with the lack of pentoses observed in the aldol reaction of glycolaldehyde under similar conditions.
Moreover, the carbonyl migration mechanism of the formose reaction is still under some debate as to whether the primary mechanism involves formation of enediol intermediates or 1,2‐hydride transfers. Our results have shown that, in absence of Ca2+, and with high concentration of buffer anion, carbonyl migration of tetroses proceeds predominantly through enediol intermediates, consistent with the evidence reported by Benner. [30] When carbonyl migration takes place in the presence of Ca2+ with hydroxide, the dominant mechanistic pathway was shown to proceed through a 1,2‐hydride transfer. [14] Indeed, the concentration of Ca2+ has been found to influence the product distribution in the formose reaction, [31] and appears to be an important factor in the carbonyl migration mechanism.
Conclusion
Labeled chirally pure sugars 1‐13C‐d‐erythrose, 4‐13C‐d‐erythrose and 1‐13C‐d‐threose undergo carbonyl migration with epimerization along the entire four‐carbon chain under mild conditions. The rates of disappearance of the starting sugar and the rates of appearance of each of the species in the carbonyl migration (d‐erythrulose, rac‐erythrulose, d‐threose and rac‐tetroses) was found to be dependent on temperature, the pH of the aqueous media and the buffer nature and the buffer concentration. When the carbonyl migration experiments were conducted in an air environment, oxidative fragmentations take place producing formate, glycerate and glycolate. Similar oxidation products were observed in the aldol reaction of glycolaldehyde under conditions comparable to those employed in the carbonyl migration studies. Mass spectral analysis of the reaction of unlabeled threose at pH 8.5 in D2O resulted in incorporation of deuterium atoms along the four‐carbon chain as carbonyl migration progressed. This fact indicates that in high concentration of buffer anion without Ca2+, the major pathway of carbonyl migration is via a proton transfer mechanism, consistent with previously reported result [30] in the formose reaction. The observation of sorbose and tagatose together with erythrulose, as the major product, in a base promoted aldol reaction of glycolaldehyde supports the notion that carbonyl migration also has a significant effect on the nature of the product distribution under plausible prebiotic scenarios (such as formose reaction).
Experimental Section
Please see supporting Information for general procedures for carbonyl migration experiments of d‐[1‐13C]‐erythrose, d‐[4‐13C]‐erythrose and d‐[1‐13C]‐threose as well as aldol reaction of glycolaldehyde. Relevant NMR spectra for carbonyl migration experiments of d‐[1‐13C]‐erythrose, d‐[4‐13C]‐erythrose and d‐[1‐13C]‐threose as well as aldol reaction of glycolaldehyde, and MS spectra are also provided in supporting Information.
Conflict of interest
The authors declare no conflict of interest.
1.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgments
This work was supported by the NASA Exobiology Program NNH20ZA001N‐EXO Grant 20‐EXO‐0006 and jointly by National Science Foundation and the NASA Astrobiology Program under the Center for Chemical Evolution grant no. CHE‐1504217. This work was also supported in part by the Astrobiology Center Program of National Institutes of Natural Sciences (NINS) (Grant Number AB031017), Japan Society for the Promotion of Science (JSPS) Grant‐in‐aid 21K14029 and by the WPI‐funded Earth‐Life Science institute at Tokyo institute of Technology. The authors would like to acknowledge Christopher Butch and Marcos A. Amezeua, Jr. for performing preliminary experiments.
Yi R., Kern R., Pollet P., Lin H., Krishnamurthy R., Liotta C. L., Chem. Eur. J. 2023, 29, e202202816.
Contributor Information
Dr. Ruiqin Yi, Email: yiruiqin@elsi.jp.
Prof. Ramanarayanan Krishnamurthy, Email: rkrishna@scripps.edu.
Prof. Charles L. Liotta, Email: charles.liotta@chemistry.gatech.edu.
Data Availability Statement
The data that support the findings of this study are available in the supplementary material of this article.
References
- 1.J. C. Speck Jr, in Adv. Carbohydr. Chem., Elsevier, 1958, pp. 63–103. [DOI] [PubMed]
- 2.S. J. Angyal, in Glycoscience, Springer, 2001, pp. 1–14.
- 3. Delidovich I., Palkovits R., ChemSusChem 2016, 9, 547–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Romano A. H., Conway T., Res. Microbiol. 1996, 147, 448–455. [DOI] [PubMed] [Google Scholar]
- 5. Breslow R., Tetrahedron Lett. 1959, 1, 22–26. [Google Scholar]
- 6. Serianni A. S., Pierce J., Huang S. G., Barker R., J. Am. Chem. Soc. 1982, 104, 4037–4044. [Google Scholar]
- 7. Isbell H. S., Linek K., K. E. Hepner Jr , Carbohydr. Res. 1971, 19, 319–327. [Google Scholar]
- 8. Schaffer R., J. Org. Chem. 1964, 29, 1473–1475. [Google Scholar]
- 9. Sowden J. C., Thompson R. R., J. Am. Chem. Soc. 1958, 80, 1435–1437. [Google Scholar]
- 10. King-Morris M. J., Serianni A. S., Carbohydr. Res. 1986, 154, 29–36. [DOI] [PubMed] [Google Scholar]
- 11. El Khadem H. S., Ennifar S., Isbell H. S., Carbohydr. Res. 1987, 169, 13–21. [Google Scholar]
- 12. El Khadem H. S., Ennifar S., Isbell H. S., Carbohydr. Res. 1989, 185, 51–59. [Google Scholar]
- 13. Nagorski R. W., Richard J. P., J. Am. Chem. Soc. 2001, 123, 794–802. [DOI] [PubMed] [Google Scholar]
- 14. Appayee C., Breslow R., J. Am. Chem. Soc. 2014, 136, 3720–3723. [DOI] [PubMed] [Google Scholar]
- 15. Cheng L., Doubleday C., Breslow R., Proc. Nat. Acad. Sci. 2015, 112, 4218–4220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kitadai N., Maruyama S., Geosci. Front. 2018, 9, 1117–1153. [Google Scholar]
- 17. Alkorta I., Popelier P. L. A., Carbohydr. Res. 2011, 346, 2933–2939. [DOI] [PubMed] [Google Scholar]
- 18. Azofra L. M., Alkorta I., Elguero J., Popelier P. L. A., Carbohydr. Res. 2012, 358, 96–105. [DOI] [PubMed] [Google Scholar]
- 19. Azofra L. M., Alkorta I., Elguero J., Carbohydr. Res. 2013, 372, 1–8. [DOI] [PubMed] [Google Scholar]
- 20. Westerlund E., Carbohydr. Res. 1981, 91, 21–30. [Google Scholar]
- 21. Delidovich I., Gyngazova M. S., Sánchez-Bastardo N., Wohland J. P., Hoppe C., Drabo P., Green Chem. 2018, 20, 724–734. [Google Scholar]
- 22. Delidovich I., Palkovits R., Green Chem. 2016, 18, 5822–5830. [Google Scholar]
- 23. Gleason W. B., Barker R., Can. J. Chem. 1971, 49, 1425–1432. [Google Scholar]
- 24.H. S. ISBELL, in Carbohydrates Solut., ACS Publications, 1973, pp. 70–87.
- 25. Shalaby M. A., Isbell H. S., El Khadem H. S., J. Carbohydr. Chem. 1995, 14, 429–437. [Google Scholar]
- 26. Bamford C. H., Collins J. R., Proc. R. Soc. London Ser. A 1950, 204, 62–84. [Google Scholar]
- 27. Sagi V. N., Punna V., Hu F., Meher G., Krishnamurthy R., J. Am. Chem. Soc. 2012, 134, 3577–3589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Colón-Santos S., Cooper G. J. T., Cronin L., ChemSystemsChem 2019, 1, e1900014. [Google Scholar]
- 29. Haas M., Lamour S., Christ S. B., Trapp O., Commun. Chem. 2020, 3, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kim H.-J., Ricardo A., Illangkoon H. I., Kim M. J., Carrigan M. A., Frye F., Benner S. A., J. Am. Chem. Soc. 2011, 133, 9457–9468. [DOI] [PubMed] [Google Scholar]
- 31. Robinson W. E., Daines E., van Duppen P., de Jong T., Huck W. T. S., Nat. Chem. 2022, 1–9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Data Availability Statement
The data that support the findings of this study are available in the supplementary material of this article.