

Abstract
Only a few patients with germline AXIN2 variants and colorectal adenomatous polyposis or cancer have been described, raising questions about the actual contribution of this gene to colorectal cancer (CRC) susceptibility. To assess the clinical relevance for AXIN2 testing in patients suspected of genetic predisposition to CRC, we collected clinical and molecular data from the French Oncogenetics laboratories analyzing AXIN2 in this context. Between 2004 and June 2020, 10 different pathogenic/likely pathogenic AXIN2 variants were identified in 11 unrelated individuals. Eight variants were from a consecutive series of 3322 patients, which represents a frequency of 0.24%. However, loss‐of‐function AXIN2 variants were strongly associated with genetic predisposition to CRC as compared with controls (odds ratio: 11.89, 95% confidence interval: 5.103–28.93). Most of the variants were predicted to produce an AXIN2 protein devoid of the SMAD3‐binding and DIX domains, but preserving the β‐catenin‐binding domain. Ninety‐one percent of the AXIN2 variant carriers who underwent colonoscopy had adenomatous polyposis. Forty percent of the variant carriers developed colorectal or/and other digestive cancer. Multiple tooth agenesis was present in at least 60% of them. Our report provides further evidence for a role of AXIN2 in CRC susceptibility, arguing for AXIN2 testing in patients with colorectal adenomatous polyposis or cancer.
Keywords: adenomatous polyposis, colorectal cancer susceptibility, oligodontia, Wnt signaling pathway
Abbreviations
- CRC
colorectal cancer
- DIX
disheveled and Axin
- Dvl
disheveled
- GSK
glycogen synthase kinase 3β
- P/LP
pathogenic/likely pathogenic
- RGS
regulator of G‐protein signaling
- TBM
tankyrase‐binding motifs
1. INTRODUCTION
Monoallelic germline pathogenic variants in the APC (adenomatous polyposis coli; MIM *611731) and biallelic germline pathogenic variants in MUTYH (human Mut Y homolog; MIM *604933) genes are identified in most patients with classic familial adenomatous polyposis (FAP) or attenuated familial adenomatous polyposis (AFAP). Several other genes including AXIN2 have been associated with risk of developing colorectal adenomatous polyposis and colorectal cancer (CRC). 1 , 2
The AXIN2 gene (MIM* 604025) encodes axis inhibition protein 2, which plays a major role in regulation of the canonical Wnt (Wingless and INT‐1) signaling pathway. 3 , 4 AXIN2 is with APC a key component of the dynamic multiprotein assembly which targets β‐catenin for proteolysis in the absence of a Wnt stimulus. 5 Mutations in AXIN2 have been shown to cause CRC by activating β‐catenin/TCF (T‐cell factor) signaling, 6 leading to transcription of target genes that play a role in control of cell growth and survival. 7 , 8
In patients with colorectal adenomatous polyposis or CRC, only a few pathogenic or likely pathogenic (P/LP) germline variants of AXIN2 have been reported. The first one was described in 2004 in a large Finnish family in which multiple permanent tooth agenesis (oligodontia) and colorectal polyps of various histologies (adenomatous, hyperplastic, and mixed) or CRC segregated with a heterozygous nonsense variant (NM_004655.4:c.1966C>T, p.(Arg656*)). 9 Subsequently, another nonsense AXIN2 variant (c.1989G>A, p.(Trp663*)) was described as segregating with oligodontia, colonic polyposis, early CRC, and other findings including gastric polyps, early breast cancer, and mild ectodermal dysplasia phenotype with sparse hair and eyebrows. 10 Four additional AXIN2 variants were further reported in patients with both oligodontia and colorectal adenomatous polyposis (c.1972del, p.(Ser658Alafs*31)) 11 or CRC (c.1987dup, p.(Trp663Leufs*44)), 12 or with isolated colorectal adenomatous polyposis (c.1994dup, p.(Asn666Glnfs*41)) 13 or CRC (c.254del, p.(Leu85Tyrfs*24)). 14 A variant of uncertain significance (c.1387C>T, p.(Arg463Cys)) was also reported in a patient with isolated colorectal adenomatous polyposis. 15
Given the paucity of available information, many questions remain about the actual contribution of AXIN2 variations to colorectal adenomas and CRC (and other types of cancer) susceptibility, challenging genetic counseling and clinical management. The purpose of this study was to collect and characterize AXIN2 variants identified in French Oncogenetics laboratories, and to report the clinical phenotypes associated with P/LP variants.
2. METHODS
2.1. Patients and controls
We collected AXIN2 P/LP variants identified between 2004 and June 2020 in the French Oncogenetics laboratories from the Genetics and Cancer Group‐Unicancer analyzing AXIN2 in a context of suspected predisposition to CRC.
All the patients but one were originally referred to clinical geneticists because of colorectal polyposis and/or personal or family history of early CRC, leading to analysis of a panel of genes known for their implication in CRC predisposition which included AXIN2 (Figure 1). One patient, a 10‐year‐old child, presented with ectodermal dysplasia, leading to analysis of a panel of genodermatosis‐causing genes, including AXIN2.
FIGURE 1.
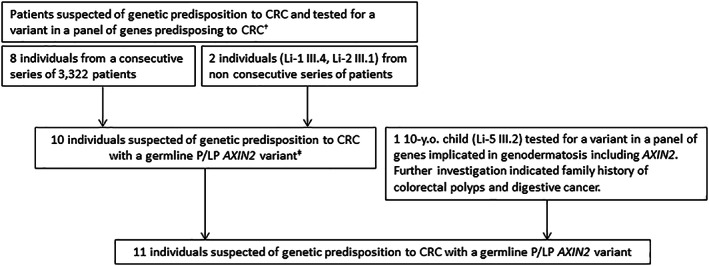
Flowchart indicating the origins of the 11 pathogenic/likely pathogenic (P/LP) AXIN2 variants identified in French Oncogenetics laboratories. †Panel including APC, MUTYH, MLH1, MSH2, MSH6, PMS2, POLE, POLD1, SMAD4, BMPR1A, STK11, PTEN +/‐ NTHL1, GREM1/SCG5, and MSH3. ‡One individual (Re‐3) with AFAP and CRC with co‐occurrence of a P/LP AXIN2 variant and a homozygous pathogenic MUTYH variant.
Information on personal and familial history was obtained during genetic counseling and from medical records. Full informed consent for genetic analyses and the use of clinical data for research purposes was obtained from all patients or legal representatives.
Population control frequencies of AXIN2 variants were obtained from gnomAD v.2.1 database (https://gnomad.broadinstitute.org). Non‐Finnish European, non‐cancer individuals (n = 59 095) were retained for comparisons with patients.
2.2. AXIN2 variant classification
Variants were described according to the HGVS (Human Genome Variation Society) nomenclature guidelines using the reference sequence NM_004655.4 with c.1 corresponding to the first nucleotide of the start codon (www.hgvs.org/varnomen). The pathogenicity assessment was based on the American College of Medical Genetics and Genomics and the Association for Molecular Pathology (ACMG/AMP) criteria. 16 Tools and criteria used for the interpretation are summarized in Figure S1. Allele frequencies in control populations were obtained from gnomAD v.2.1. The impact of missense variants was evaluated using the in silico predictive algorithms PolyPhen‐2 (Polymorphism Phenotyping v.2), SIFT (Sorting Intolerant From Tolerant), and Mutation Taster through Alamut Visual v2.15 (Sophia Genetics). Prediction of the effect on splicing was assessed using SPiP (Splicing Prediction Pipeline, https://github.com/raphaelleman/SPiP) 17 and SpliceAI 18 tools. Prediction of translation initiation sites was performed using ATGpr (Prediction of Translation initiation ATG) 19 and TIS Miner. 20 Using available evidence for their interpretation, variants were classified as benign/likely benign (B/LB), of unknown significance (VUS), and pathogenic/likely pathogenic (P/LP). 16 A consensus in the French Genetics and Cancer Group is to consider allele frequency over 1% in control populations as stand‐alone evidence of benign impact (instead of 5%).
2.3. Statistical analyses
Fisher's exact test, two‐sided, was used to compare allele frequencies between cases and controls, and to calculate odds ratios (ORs) and confidence intervals (CIs). Results were considered statistically significant when p < 0.05. Statistical tests were carried out using GraphPad Prism 7.00.
3. RESULTS
3.1. AXIN2 variant analysis
Between 2004 and June 2020, 10 different pathogenic/likely pathogenic AXIN2 variants were identified in 11 unrelated individuals (Table 1).
TABLE 1.
Clinical characteristics of index cases and relatives carrying a pathogenic/likely pathogenic AXIN2 variant.
| Family no. | Family member a | Gender | AXIN2 variant | Exon | Age (yrs) at diagnosis | Colorectal polyps (age) | CRC site (age) | Extracolonic digestive tumor (age) | Extradigestive tumor (age) | Tooth agenesis (nb) b | Ectodermal dysplasia | Other manifestations |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Li‐1 | III.4 (IC) | M | c.2092G>T, p.(Glu698*) | 8 | 37 | 85 ad (37); ~60 ad, 2 ssa (38–43) | No | 5 duodenal polyps (37) | No | Yes (12/8) | No | Severe bleeding during polypectomies; retrognathia |
| Li‐2 | III.1 (IC) | F | c.2041C>T, p.(Gln681*) | 8 | 58 | >58 ad, <12 hp (61–72); 88 ad, <10 hp (76–77) | S (58) (pMMR) | 3 fundic gland polyps (75) | Ovary (67) | No (4/0) | No | |
| Li‐3 | II.1 (IC) | F | c.1647T>G, p.(Tyr549*) | 6 | 82 | 1 Ad (77); >20 ad (82) | C (82) (pMMR) | Small bowel cancer (66) | No | ? | No | Severe bleeding during polypectomy; tendancy to gastrointestinal bleeding, anemia; antral angiodysplasia |
| Li‐4 | III.1 (IC) | F | c.2086C>T, p.(Gln696*) | 8 | 62 | 40 ad (62) | S + R (62) | No | Breast (53) | Yes (23/?) | No | Idiopathic myopathy |
| III.2 | F | c.2086C>T, p.(Gln696*) | 8 | 59 | ? | R (59) | No | Yes (23/?) | No | Idiopathic myopathy | ||
| III.3 | F | c.2086C>T, p.(Gln696*) | 8 | 36 | 2 ad (36); 38 ad, 2 ssa, 2 hp (49–57) | No | No (N EGD) | Yes (23/?) | No | Idiopathic myopathy; spina bifida | ||
| Re‐1 | III.6 (IC) | M | c.1928_1941del, p.(Ala643Valfs*59) | 8 | 53 | ≫100 ad (53) | No | No (N EGD) | Melanoma (62) | Yes (32/28) | No | |
| Re‐2 | III.8 (IC) | M | c.2062_2063del, p.(Leu688Aspfs*18) | 8 | 45 | ≥80 ad, ≥4 hp (45) | No | No (N EGD) | No | No | ||
| Re‐3 | IC | F | c.204_214del, p.(Ala69Phefs*68) | 2 | 45 | Multiple ad, nb? (45) | C + S (45) | ? | ? | ? | ? | Co‐occurrence: homozygous MUTYH variant c |
| Ly‐1 | III.5 (IC) | F | c.2062_2063del, p.(Leu688Aspfs*18) | 8 | 52 | 35 ad, 1 hp (52–58) | No | 1 duodenal polyp | No | No | No | |
| Ly‐2 | III.2 (IC) | F | c.1479_1497dup, p.(Leu500Alafs*134) | 6 | 78 | >100 ad (78) | No | No (N EGD) | No | ? | No | |
| Pa‐1 | III.1 (IC) | M | c.1989G > A, p.(Trp663*) | 8 | 68 | ~40 ad (68), 3 hp (70–71) | S (68) | No (N EGD) | No | Yes/Likely d (?) | No | |
| Li‐5 | III.2 (IC) | M | c.‐12_8del, p.0? | 2 | 10 | NA | No | Yes (8/8) | Yes (skin, nails, hair, eyebrows) | |||
| II.4 | M | c.‐12_8del, p.0? | 2 | 47 | 1 ad, 3 hp (47) | No | No, but antral IM (HP‐) | Yes (nb?) | No | |||
| II.1 | M | c.‐12_8del, p.0? | 2 | 52 | ? | No | Vater ampulla cancer (52) | ? | ? | |||
| I.2 | F | c.‐12_8del, p.0? | 2 | −(77) | ? | No | Yes (nb?) | No |
Note: Variant nomenclature follows the Human Genome Variation Society guidelines (https://varnomen.hgvs.org) and is referred for AXIN2 to the NM_004655.4.
Abbreviations: C, caecum; CRC, colorectal cancer; F, female; hp, hyperplastic polyp; HP, Helicobacter pylori; IM, intestinal metaplasia; M, male; N EGD, normal esogastroduodenoscopy; NA, not adapted; pMMR, proficient MMR; R, rectum; S, sigmoid; ssa, sessile serrated adenoma.
Generation and order from Figure 3; IC, index case.
Number of missing permanent teeth/number of missing permanent teeth excluding the third molars; No, absence of tooth agenesis based on clinical examination and questioning; ?, unknown.
NM_001048171.1: c.[1145G>A;1145G>A], p.[Gly382Asp;Gly382Asp].
Dental anomalies justifying a dental apparatus before 45 of age.
All the patients but one were originally referred to clinical geneticists because of colorectal polyposis and/or personal or family history of early CRC, leading to analysis of a panel of genes known for their implication in CRC predisposition which included AXIN2 (Figure 1). Nine different P/LP AXIN2 variants were identified in those 10 unrelated individuals (Figure 1 and Table 1). The frequency of P/LP AXIN2 variants was 0.24%, as assessed from the consecutive series of 3322 patients from four Oncogenetics laboratories (Lille, Rennes, Lyon, Paris; 8/3322 patients). All these variants were protein‐coding loss‐of‐function (LoF) variants (frameshift or stop‐gain). LoF AXIN2 variants were more frequent in the patients suspected of genetic predisposition to CRC compared to controls (0.24% vs. 0.020% [12/59 095], OR = 11.39, 95% CI 5.103–28.93; p < 0.0001) (Table 2). No other P/LP variant was detected in APC, MUTYH or other CRC‐susceptibility genes for 9 out of the 10 patients. Co‐occurrence of a homozygous pathogenic variant in MUTYH (NM_001048171.1: c.1145G>A, p.Gly382Asp) was identified in the last one (Re‐3).
TABLE 2.
Number of LoF AXIN2 variant a carriers in patients suspected of genetic predisposition to CRC and controls.
| Population | Nb of carriers | OR (95% CI) | p Value b |
|---|---|---|---|
| Patients c (n = 3322) | 8 (0.24%) | 11.39 (5.103–28.93) | <0.0001 |
| Controls (non‐cancer, NFE) d (n = 59 095) | 12 (0.020%) |
LoF AXIN2 variants include stop‐gain, frameshift, canonical splice‐site, and start‐loss variants.
Frequencies between groups were compared using Fisher's exact test. Associated ORs were calculated for each group.
Only patients tested in consecutive series were considered (Figure 1).
Control population data were obtained from gnomAD v2.1 (non‐cancer, non‐Finnish European individuals).
In addition, one patient, a 10‐year‐old child (Li‐5 III.2), presented with ectodermal dysplasia associating ichthyosis, multiple tooth agenesis, sparse dry hair, and recurrent keratitis, leading to analysis of a panel of genodermatosis‐causing genes and identification of a likely pathogenic AXIN2 variant. Medical investigation revealed the existence of family history of CRC and other digestive cancer.
In total, 10 different P/LP AXIN2 variants, of which 8 have never been reported before, were identified in 11 patients (Figure 1; Table 1). Of these 10 variants, 8 were located in the exon 6 (n = 2) or 8 (n = 6), the last 2 being located in exon 2 (first coding exon) (Figure 2). All variants in exons 6 and 8 were predicted to lead to a truncated protein devoid of the C‐terminal DIX (Disheveled and Axin) domain and of all or part of an upstream domain interacting with SMAD3, and to preserve the β‐catenin binding domain. 4 , 25 The c.204_214del variant was predicted to generate an extra‐short protein devoid of all functional domains or to lead to absence of protein production due to nonsense‐mediated mRNA decay (NMD). The last variant was a deletion encompassing the translation initiation site (c.‐12_8del). No alternate in‐frame translation initiation site in close proximity was predicted, suggesting absence of protein synthesis.
FIGURE 2.
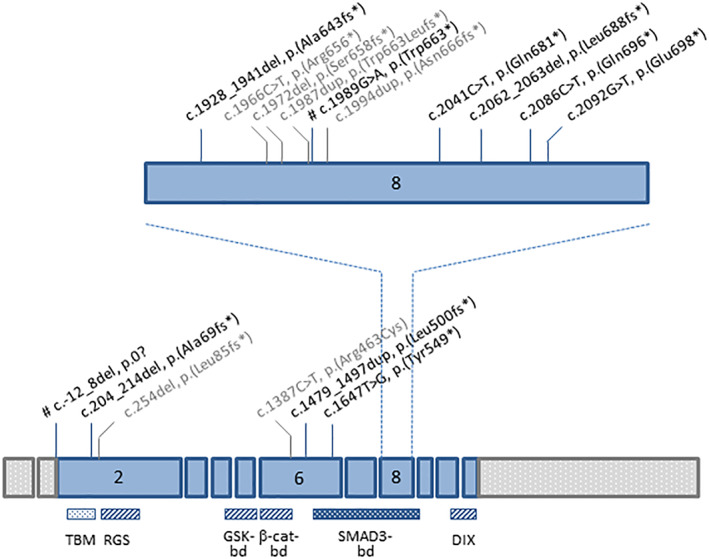
Genomic structure of AXIN2 gene showing the location of variants associated to colorectal adenomatous polyposis or CRC. Boxes represent exons. Pathogenic/likely pathogenic AXIN2 variants described in this study are indicated in black. All were novel, except two (marked by #). 10 , 21 Variants which were described in other studies are indicated in gray. 9 , 11 , 12 , 13 , 14 , 15 Functional domains of AXIN2 protein are shown below. 4 , 22 , 23 , 24 TBM, tankyrase‐binding motifs (role in regulation of AXIN2 stability); 22 , 23 , 24 , 25 RGS, regulator of G‐protein signaling (APC‐binding domain); GSK‐bd, glycogen synthase kinase 3β‐binding domain; β‐cat‐bd, β‐catenin‐binding domain; 4 SMAD3‐bd, SMAD3‐binding domain; 4 , 24 DIX, Disheveled and Axin (Disheveled‐binding domain and Axin homodimerisation). 4
In addition to the P/LP variants, 125 different variants classified as B/LB or as VUS were collected from two laboratories (Lille, Rennes) (Table S1). They consisted in 61 intronic variants and 64 exonic variants, including 60 coding variants (Table S2; Figure S2). Analysis of variant distribution in AXIN2 showed that LoF coding variants were more often located in the region coding the SMAD3‐binding domain in patients suspected of genetic predisposition to CRC compared to controls (7/10 vs. 1/8; p = 0.025) (Table 3). In contrast, non‐LoF coding variants were located all along the coding sequence in both patients and controls, with no enrichment in the SMAD3‐binding domain (23/60 in patients versus 136/427 in controls; p = 0.378) (Table 3).
TABLE 3.
Distribution of LoF a and non‐LoF AXIN2 coding variants in patients suspected of genetic predisposition to CRC and control population.
| Location (aa) 4 , 24 | Functional domain | Exon(s) | Patients | Controls | ||
|---|---|---|---|---|---|---|
| Nb of LoF variants (%) (n = 10) | Nb of non‐LoF variants (n = 60) | Nb of LoF variants (%) (n = 8) | Nb of non‐LoF variants (n = 427) | |||
| 1–22 | – | 2 | 1 (10%) | 1 (1.67%) | 2 (25%) | 8 (1.87%) |
| 23–30 | TBM‐1 | 2 | 1 (0.25%) | |||
| 31–55 | – | 2 | 1 (1.67%) | 11 (2.58%) | ||
| 56–69 | TBM‐2 | 2 | 1 (10%) | 5 (1.17%) | ||
| 70–77 | – | 2 | 1 (1.67%) | 3 (0.70%) | ||
| 78–200 | RGS | 2 | 7 (11.67%) | 39 (9.13%) | ||
| 201–342 | – | 2–4 | 4 (6.67%) | 1 (12.5%) | 59 (13.82%) | |
| 343–396 | GSK3β‐bd | 4–5 | 2 (3.33%) | 29 (6.79%) | ||
| 397–465 | β‐catenin‐bd | 5–6 | 9 (15%) | 2 (25%) | 45 (10.54%) | |
| 466–512 | – | 6 | 1 (10%) | 6 (10%) | 33 (7.73%) | |
| 513–718 | SMAD3‐bd | 6–9 | 7 (70%) | 23 (38.33%) | 1 (12.5%) | 136 (31.85%) |
| 719–785 | – | 9–10 | 3 (5%) | 2 (25%) | 31 (7.25%) | |
| 786–843 | DIX | 10–11 | 3 (5%) | 27 (6.32%) | ||
Abbreviations: aa, amino acid; bd, binding domain; DIX, disheveled and Axin; GSK3, glycogen synthase kinase 3; LoF, loss of function; RGS, regulator of G‐protein signaling; TBM, tankyrase‐binding motif.
LoF coding variants include stop‐gain, frameshift, and start‐loss variants.
3.2. Clinical features associated with AXIN2 P/LP variants
The case Re‐3 was removed from clinical evaluation, since the homozygous MUTYH pathogenic variant is likely to explain at least in part his history of colorectal polyposis and cancer. Clinical data of the 10 remaining index cases and of 5 relatives carrying a P/LP variant in AXIN2 were collected. Only a few family members (n = 10) were tested due to the lack of guidelines regarding the management of individuals carrying a P/LP AXIN2 variant. Clinical features are summarized in Table 1; Figure 3.
FIGURE 3.
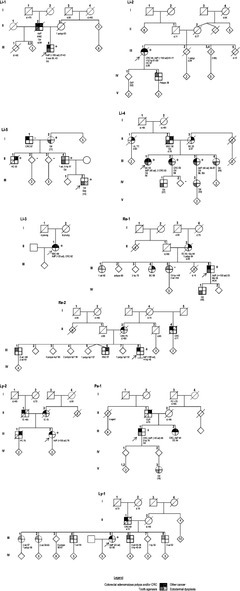
Pedigrees of patients with germline P/LP AXIN2 variants. Filled symbols, affected; +, variant carrier; −, non‐carrier; arrow, index case. Ages at diagnosis or at information gathering (in brackets) or at death (d.) are indicated. For colorectal polyps, the cumulative number from age at first presentation or screening colonoscopy to age at last contact is indicated. AOd, anodontia; AC, ampullary cancer; ad, adenoma; AdP, adenomatous polyposis; BC, breast cancer; CUP, cancer of unknown primary; CRC, colorectal cancer; EDy, ectodermal dysplasia; HNC, head and neck cancer; hp, hyperplastic polyps; IC, intestinal cancer (site not specified); IM, idiopathic myopathy; Lk, leukemia; Me, melanoma; OC, ovarian cancer; Od, oligodontia; PC, prostate cancer; SBC, small bowel cancer; Sbi, spina bifida; SGC, salivary gland cancer; ssa, sessile serrated adenoma; TC, thyroid cancer.
3.2.1. Colorectal polyps and cancer
Of the 15 AXIN2 variant carriers, 11 underwent colonoscopy and 10 had colorectal adenomatous polyposis (≥10 adenomatous polyps) (10/11, 91%). The median age at diagnosis was 55 [range 36–82]. The total number of polyps ranged from more than 20 to several hundred, but was <100 in most cases (7/10, 70%), indicative of attenuated adenomatous polyposis. Polyps were generally distributed throughout the entire colon. Forty adenomas were detected in one of the two relatives who underwent screening colonoscopy. Sessile serrated adenomas and hyperplastic polyps were also reported in several cases.
Five patients (four index cases and one relative, 5/15, 30%) developed CRC. One (Li‐4 III.1) presented with two synchronous colorectal carcinomas. The median age of onset was 62 [range 58–82].
3.2.2. Extracolonic digestive manifestations
Small bowel carcinoma was reported in one patient (Li‐3 II.1) who subsequently developed caecal carcinoma. One patient (Li‐5 II.1) died at age 53 because of an ampullary (papillary) carcinoma of pancreatobiliary histotype. Duodenal polyps (n = 5) were observed in one patient (Li‐1 III.4). No gastric polyposis or cancer was reported, but antral intestinal metaplasia was observed in one case (Li‐5 II.4), which could not be attributed to Helicobacter pylori infection. A few fundic gland polyps (n = 3) were reported in another case (Li‐2 III.1).
3.2.3. Extradigestive manifestations
Ovarian cancer of endometrioid type was observed in one patient (Li‐2 III.1) who had previously developed CRC. A breast cancer and a melanoma were described in two cases.
Multiple tooth agenesis was present in at least nine cases (9/13, 69%). Anodontia (i.e., lack of all permanent teeth) was reported in one case (Re‐1 III.6). Oligodontia (i.e., absence of 6 or more permanent teeth, excluding the third molars) was more frequent as it was reported in eight cases, the number of missing teeth ranging from 8 (12 including the third molars) to 23 permanent teeth. An example is shown in Figure 4.
FIGURE 4.
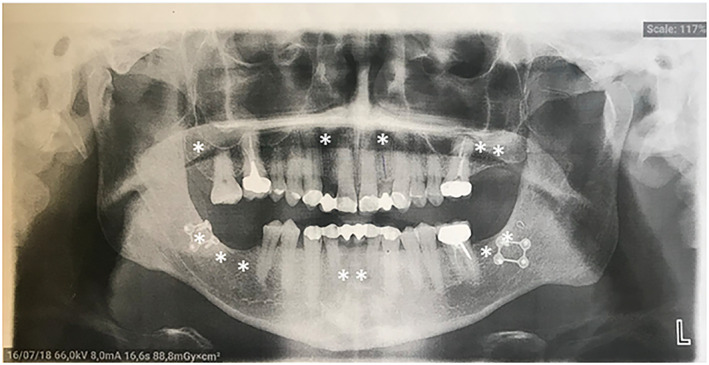
Panoramic radiograph of patient Li‐2 III.1. This radiograph shows oligodontia with absence of 12 permanent teeth (4 incisors [2 maxillary lateral and 2 mandibular central], 1 first molar, 3 second molars, and the 4 third molars). White stars indicate missing permanent teeth.
Additional ectodermal features were observed in one case (Li‐5 III.2) and were the reason for clinical referral. Symptoms associated oligodontia with ichthyosis, sparse dry hair, sparse eyebrows, dysplastic nails, and keratitis. For this patient, co‐occurrence of a heterozygous pathogenic variant in FLG (Filaggrin, MIM *135940) was identified, which could explain cutaneous manifestations, but not the other symptoms, including oligodontia.
A variety of other symptoms were reported. In particular, abnormal severe bleeding during polypectomies was reported in two cases (Li‐1 III.4, Li‐3 II.1), requiring re‐hospitalization in ICU in one case. In one family (Li‐4), idiopathic myopathy was reported in the three variant carriers (III.1, III.2, and III.3).
3.3. Family history of individuals with P/LP AXIN2 variants
Detailed family history data were available for 9 index cases (Figure 3).
Only 10 relatives were tested for AXIN2 variant and 5 of them were found to carry the P/LP variant identified in their family. Nevertheless, adenomatous polyps, CRC or intestinal cancer were reported in a number of relatives. A large variety of other cancers were also observed, including a salivary gland cancer at 50 in one first‐degree relative with oligodontia (Li‐4 II.2), and a head‐and‐neck cancer before 64 in another relative with adenomatous polyposis (Li‐1 II.2).
Tooth agenesis was frequently reported (16 individuals from 5 unrelated families). Notably, oligodontia was reported in seven first‐/second‐degree relatives on three generations in one family (Li‐4).
3.4. Genotype–phenotype correlation
To look for potential genotype–phenotype correlation, P/LP AXIN2 variants identified in our series were classified according to their nature and location in the gene. All the variants identified in exon 6 or 8 were truncating and associated with colorectal adenomatous polyposis. However, the colorectal phenotype was variable, the number of adenomatous polyps ranging from several hundred at 53‐year‐old to less than 50 at age 82.
The c.‐12_8del variant (exon 2), was associated with a particular phenotype, that is, oligodontia and ectodermal dysplasia in the index case, and extracolonic (ampullary) cancer in his paternal uncle (Figure 3, Li‐5). Colorectal findings in this family consisted in only one adenoma with low grade dysplasia in the father, and no evidence of adenomatous polyps in the uncle who developed ampullary cancer. No digestive manifestation was reported in the paternal grandmother (age 77) who was recently shown to carry the variant, but no colonoscopy has yet been performed. Of note, this variant has been reported previously in a patient with breast cancer. 21 No breast cancer was reported in our family.
The presence of multiple tooth agenesis was not associated with variant nature or location.
4. DISCUSSION
AXIN2 is an important negative regulator of the canonical Wnt signaling pathway, which plays a critical role in oncogenesis. 7 , 8 Its key function is to serve as a scaffold, bringing together β‐catenin and GSK3β, thus promoting phosphorylation and subsequent proteasomal degradation of β‐catenin. 3 Notably, the AXIN2 gene is a direct target of the Wnt/β‐catenin pathway and hence also acts as a negative feedback regulator of Wnt signaling. 26 , 27 , 28 AXIN2 also interacts with SMAD3, facilitating its activation by TGFβ receptors and leading to stimulation of TGFβ signaling. 24 , 29 The AXIN2 gene is negatively regulated by TGFβ and mediates cross‐talk between TGFβ and Wnt signaling pathways. 29
AXIN2 somatic mutations or promoter hypermethylation have been reported in a variety of cancers, including CRC, gastric carcinomas, ovarian endometrioid carcinoma, and medulloblastoma. 6 , 30 , 31 , 32 , 33 Mutations in AXIN2 have been reported in approximately 20%–25% of mismatch repair (MMR)‐deficient colorectal tumors and in a few other MMR‐deficient tumors. 6 , 30 , 31 , 32 Almost all mutations correspond to one‐bp deletions/insertions in short mononucleotide repeat sequences located in exon 8, leading to premature protein truncation, 6 , 30 , 31 , 32 and have been shown to activate β‐catenin/TCF signaling. 6
Only a few cases with AXIN2 germline variants and colorectal adenomas or cancer have been described, raising questions about its contribution to CRC (and other types of cancers) susceptibility. The present study allowed characterization of the largest series of patients carrying a P/LP AXIN2 variant identified in a context of CRC predisposition.
Ten different P/LP AXIN2 variants, of which 8 are novel, were identified in 11 patients from unrelated families. Comparison with population‐based controls showed that LoF AXIN2 variants are strongly associated with colorectal adenomatous polyposis and cancer susceptibility. The frequency was estimated to be 0.25%, indicating that AXIN2 variants are a rare cause of colorectal polyposis and cancer. Notably, all P/LP AXIN2 variants identified in our series but two (8/10, 80%) were located in the exon 6 or 8 and predicted to lead to a truncated protein. The pattern of P/LP AXIN2 variants is similar to the few cases previously described and somewhat similar to what is observed in MMR‐deficient tumors. These variants are expected to lead to the synthesis of truncated proteins. Indeed, analysis of a medulloblastoma with a frameshift mutation in AXIN2 exon 6 showed efficient expression of the truncated protein (p.Val506*), as assessed by Western blotting, suggesting no NMD. 33 Moreover, RNA analysis performed in blood and tumor ofone of our patients with a frameshift variant showed expression of the two alleles, also arguing against NMD (Figure S3). However, it cannot be excluded that some variants may be associated with NMD. AXIN2 mutants with a premature stop codon located in exon 6 or 8 (p.Val506*, p.Arg656*, p.Leu688*) have also been shown in vitro to be more stable than the wild‐type protein. 6 , 33 , 34 These variants lead to absence of the DIX domain, which mediates AXIN2 homo‐oligomerisation, as well as binding to other DIX domain‐containing proteins and most notably Disheveled. 35 , 36 , 37 , 38 , 39 , 40 AXIN2 oligomerization and AXIN2‐Disheveled interaction are crucial for regulation of the Wnt/β‐catenin signaling. 35 , 39 , 41 Cell transfection with AXIN2 mutants results in the accumulation of β‐catenin in the cytoplasm and the nuclei and in activation of the Wnt pathway. 6 , 34 These variants also impact the integrity of the SMAD3‐binding domain, which plays a role in TGFβ signaling, with possible impact on the Wnt pathway. 24 , 29 This suggests a major role of the DIX domain and/or of the SMAD3‐binding domain in colorectal tumorigenesis and genetic predisposition to CRC. Conversely, these variants preserve the APC, GSK3β‐, and β‐catenin‐binding domains, suggesting selection for AXIN2 mutants retaining some activity.
Only three AXIN2 truncating variants predicted to be devoid of the β‐catenin domain have been reported in patients suspected of predisposition to CRC. The first one (c.254del, p.(Leu85Tyrfs*84)) was identified in a patient with a rectal cancer but no polyposis at age 79, 14 and another one (c.204_214del, p.(Ala69Phefs*68)) is reported in the present study in a patient with AFAP who also carries a homozygous pathogenic variant in MUTYH, raising the question of their implication in the phenotypes. However, one variant (c.1049del, p.(Pro350Leufs*13)) has been reported recently in a family in which five of the seven variant carriers developed multiple colorectal polyps or/and cancer. 42 Further studies are needed to evaluate the implication of such variants in CRC susceptibility.
Only one of the 10 P/LP variants, that is, c.‐12_8del, was not predicted to lead to a truncated protein, but to lead to absence of protein synthesis. Given the multiple interactions between AXIN2 and other components of the destruction complex, a decrease of the amount of AXIN2 protein is likely to impact regulation of the Wnt/β‐catenin pathway, with consequences depending on the cell type. Indeed, AXIN proteins are thought to be the concentration‐limiting component of the complex. 4 , 43 , 44
AXIN2 variants have been reported in patients with non‐syndromic or syndromic oligodontia without evidence of CRC susceptibility. 9 , 45 , 46 , 47 Notably, most of these variants are missense, suggesting that AXIN2 variants associated with isolated oligodontia differ in nature from those observed in colorectal polyposis and CRC. The only exception corresponds to a de novo frameshift mutation (c.1994dup, p.(Asn666Glnfs*41)) identified in a 13‐year‐old‐patient 9 and a 17‐year‐old‐patient, 45 both with isolated oligodontia. However, given the young age of these patients at genetic diagnosis, predisposition to CRC cannot be excluded. Of note, as our manuscript was being finalized, this variant was reported in a 65‐year‐old patient with colorectal polyposis (57 adenomatous polyps), 48 and in a family with tooth agenesis and variable clinical findings including polyps and CRC, 49 and we recently identified another variant at the same position (c.1994del, p.(Gly665Alafs*24)) in a 66‐year‐old patient presenting with a similar phenotype (46 adenomatous polyps), suggesting once again that truncating variants in exon 8 are associated with colorectal polyposis and cancer susceptibility. Conversely, one missense AXIN2 variant (c.1387C>T, p.(Arg463Cys)) was reported in two siblings with attenuated adenomatous polyposis at young age. 15 However, loss of the mutated allele in polyps argues against its pathogenicity.
In vitro experiments also suggest that AXIN2 truncating and missense variants may have different consequences on the Wnt signaling pathway. Whereas AXIN2 variants p.Arg656*, p.Asn666Glnfs*41 and p.Leu688* have been shown to result in increased levels of β‐catenin and over‐activation of the β‐catenin/TCF signaling, 6 , 34 , 50 the p.His660Tyr variant identified in a family with isolated tooth agenesis resulted in decreased level of β‐catenin and inhibition of the Wnt pathway. 34
In line with previous reports, 10 , 11 , 12 , 13 , 14 , 15 , 42 the colorectal phenotype in the families reported in this study was extremely variable, ranging from several hundred of adenomatous polyps to only one. However, the number of adenomatous polyps was below 100 in most cases and the age of onset of polyps and cancer was relatively late (>35 year‐old), indicating that AXIN2 variants are generally associated with attenuated polyposis. Serrated polyps including sessile serrated adenomas and hyperplastic polyps were also frequent. This phenotype is very similar to the one observed in MUTYH‐associated polyposis. 51 , 52 Regarding digestive extracolonic features, an ampullar carcinoma and a small bowel carcinoma were reported, as well as a few duodenal polyps in one case. Given our observations and previously reported cases, it seems reasonable for patients with a P/LP AXIN2 variant to propose surveillance protocols similar to those offered to patients with germline biallelic variants of MUTYH. 53 , 54 Multiple fundic gland polyps in association with colonic polyposis have been previously reported in a patient carrying a nonsense AXIN2 variant (p.(Trp663*)). 10 However, there was no evidence of gastric polyposis or cancer in our patients, although antral intestinal metaplasia was reported in one case and some fundic gland polyps in another one.
Conversely, although a variety of other cancers have been observed in AXIN2 variant‐carriers, including pancreatobiliary ampullar cancer (the present report), ovarian cancer (the present report and Chan et al. 42 ), breast cancer (the present report and Marvin and colleagues 10 , 42 ), melanoma (the present report and Castiglia et al. 55 ), neuroblastoma, 13 and keratoacanthoma, 42 the link with AXIN2 variants remains unclear and would require larger series to evaluate the actual risk. Particular attention should be paid to the risk of salivary gland cancer and head and neck cancer as they were observed in three relatives, including one with oligodontia and one with adenomatous polyposis.
In line with previous reports 9 , 10 , 11 , 12 oligodontia was frequent in our patients, being observed in 69%. Therefore, association of colorectal polyposis and/or CRC with oligodontia is highly predictive of a germline pathogenic variant in AXIN2. Oligodontia was associated with ectodermal dysplasia phenotype in one case, similar to the description by Marvin et al. 10 Intriguingly, whereas AXIN2 variants are only associated with tooth agenesis, supernumerary teeth together with odontomas and impacted teeth are observed in some patients with FAP caused by inactivating APC variants, indicating that activation of the Wnt pathway can be associated with overproduction as well as failure of tooth development. This underlines the complexity of the role and regulation of the Wnt pathway in tooth development. 56
A variety of other symptoms were reported in our patients, including severe abnormal bleeding during polypectomy in two cases, and antral angiodysplasia and a tendency toward gastrointestinal bleeding in one of them. Although it is not possible to establish a link with AXIN2 variants, this should be taken into account when performing invasive procedures in these patients. In one family, idiopathic myopathy was reported in the three variant carriers. This manifestation may be coincidental but fibromyalgia has been reported in another patient with a truncating AXIN2 variant. 10 Moreover, AXIN2‐dependant Wnt/β‐catenin signaling has been shown to be involved in myotube formation. 57
In conclusion, our study provides additional evidence of a contribution of AXIN2 to CRC susceptibility, arguing for its inclusion in hereditary colorectal polyposis and cancer gene‐panels. This study also extends knowledge on AXIN2 variant spectrum and associated phenotype. However, further collaborative studies are still needed to precise the mechanism of pathogenicity, as well as genotype–phenotype correlation and cancer risk and establish standardized guidelines for counseling, surveillance and management of families with AXIN2‐associated colorectal polyposis or cancer. In the meantime, it may be proposed to follow the protocols adapted to the management of patients with germline biallelic variants of MUTYH.
AUTHOR CONTRIBUTIONS
Julie Leclerc and Marie‐Pierre Buisine had full access to all the data in the study and take responsibility for the integrity of the data and accuracy of the data analysis. Study concept and design: Julie Leclerc, Marie‐Pierre Buisine. Acquisition, analysis, and/or interpretation of molecular data: Julie Leclerc, Marie Beaumont, Stéphane Pinson, Catherine Vermaut, Cathy Flament, Tonio Lovecchio, Lucie Delattre, Christophe Demay, Florence Coulet, Erell Guillerm, Nadim Hamzaoui, Alain Hovnanian, Marie‐Pierre Buisine. Acquisition and analysis of clinical data: Roseline Vibert, Patrick R. Benusiglio, Afane Brahimi, Florence Coulet, Hélène Delhomelle, Sandra Fert‐Ferrer, Clémentine Legrand, Alain Lortholary, David Malka, Florence Petit, Jean‐Christophe Saurin, Sophie Lejeune, and Chrystelle Colas. Drafting of the manuscript: Julie Leclerc, Marie‐Pierre Buisine. Critical revision of the manuscript for important intellectual content: Julie Leclerc, Marie Beaumont, Roseline Vibert, Clémentine Legrand, Florence Petit, and Chrystelle Colas.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
WEB RESOURCES
ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/).
Prediction of Translation initiation ATG (ATGpr) (https://atgpr.dbcls.jp/).
The Genome Aggregation Database (gnomAD) (https://gnomad.broadinstitute.org).
The Human Genome Variation Society (HGVS) (https://varnomen.hgvs.org).
Splicing Prediction Pipeline (SPiP) (https://github.com/raphaelleman/SPiP).
TIS Miner (https://mybiosoftware.com/tag/tis-miner).
Supporting information
FIGURE S1. Flowchart showing the tools and criteria used for classifying AXIN2 variants identified in patients suspected of genetic predisposition to CRC. Criteria and classes are detailed in Richard et al. 16 Criteria: PVS, very strong; PP, supporting; BP, benign supporting; BA, benign stand alone. Classes: P, pathogenic; LP, likely pathogenic; VUS, variant of unknown significance; LB, likely benign; B, benign.
FIGURE S2. Description of non‐pathogenic/likely pathogenic AXIN2 variants identified in patients suspected of genetic predisposition to CRC. The pathogenicity assessment was performed as described in methods and Figure S1.
Abbreviations: P/LP, pathogenic/likely pathogenic; VUS variants of unknown significance; B/LB, benign/likely benign.
FIGURE S3. AXIN2 RT‐PCR analysis. RT‐PCR and Sanger sequencing were performed from blood and tumor RNA of patient Re‐2 III.8 with AXIN2 variant c.2062_2063del. Forward and reverse primers were designed in different exons to avoid amplification of genomic DNA. Electropherograms show the presence of both the wild‐type (WT) and the mutated (Mut) alleles, arguing against NMD.
TABLE S1. Characteristics and classification of AXIN2 variants identified in patients suspected of genetic predisposition to CRC. In addition to the P/LP variants, 125 different variants were collected from two laboratories (Lille, Rennes). These variants were classified as B/LB or VUS according to the ACMG/AMP criteria as described above. Data are summarized in Figure S2.
ACKNOWLEDGMENTS
The authors thank medical and molecular geneticists from the French Genetics and Cancer Group—Unicancer and patients. The authors thank Alice Yvard, Centre Catherine de Sienne, Nantes; Mathilde Warcoin, Department of Genetics, Institut Curie, Paris; Magalie Peysselon, Department of Medical Genetics, CHU Grenoble Alpes, for providing clinical information. We also thank Denis Boidin, Lauriane Briois, and Nora Boucetta, Molecular Oncogenetics, Department of Biochemistry and Molecular Biology, Lille University Hospital; and S. Miskinyte, Department of Genetics, Necker Hospital, Paris, for technical support; Florent Denoual and Marie de Tayrac, Department of Molecular Genetics, CHU Rennes, for bioinformatic support.
Leclerc J, Beaumont M, Vibert R, et al. AXIN2 germline testing in a French cohort validates pathogenic variants as a rare cause of predisposition to colorectal polyposis and cancer. Genes Chromosomes Cancer. 2023;62(4):210‐222. doi: 10.1002/gcc.23112
DATA AVAILABILITY STATEMENT
All P/LP variants presented in this study have been submitted to and are available in ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/, accession numbers SCV001519371 to SCV001519380).
REFERENCES
- 1. Stoffel EM, Boland CR. Genetics and genetic testing in hereditary colorectal cancer. Gastroenterology. 2015;149(5):1191‐1203.e2. [DOI] [PubMed] [Google Scholar]
- 2. Heald B, Hampel H, Church J, et al. Collaborative Group of the Americas on Inherited Gastrointestinal Cancer Position statement on multigene panel testing for patients with colorectal cancer and/or polyposis. Fam Cancer. 2020;19(3):223‐239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Behrens J, Jerchow BA, Würtele M, et al. Functional interaction of an axin homolog, conductin, with beta‐catenin, APC, and GSK3β. Science. 1998;280(5363):596‐599. [DOI] [PubMed] [Google Scholar]
- 4. Mai M, Qian C, Yokomizo A, Smith DI, Liu W. Cloning of the human homolog of conductin (AXIN2), a gene mapping to chromosome 17q23‐q24. Genomics. 1999;55(3):341‐344. [DOI] [PubMed] [Google Scholar]
- 5. Stamos J, Weis W. The β‐catenin destruction complex. Cold Spring Harb Perspect Biol. 2013;5(1):a007898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Liu W, Dong X, Mai M, et al. Mutations in AXIN2 cause colorectal cancer with defective mismatch repair by activating β‐catenin/TCF signalling. Nat Genet. 2000;26(2):146‐147. [DOI] [PubMed] [Google Scholar]
- 7. Lustig B, Behrens J. The Wnt signaling pathway and its role in tumor development. J Cancer Res Clin Oncol. 2003;129:199‐221. [DOI] [PubMed] [Google Scholar]
- 8. Clevers H. Wnt/β‐catenin signaling in development and disease. Cell. 2006;127(3):469‐480. [DOI] [PubMed] [Google Scholar]
- 9. Lammi L, Arte S, Somer M, et al. Mutations in AXIN2 cause familial tooth agenesis and predispose to colorectal cancer. Am J Hum Genet. 2004;74(5):1043‐1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Marvin ML, Mazzoni SM, Herron CM, Edwards S, Gruber SB, Petty EM. AXIN2‐associated autosomal dominant ectodermal dysplasia and neoplastic syndrome. Am J Med Genet A. 2011;155A(4):898‐902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Beard C, Purvis R, Winship IM, Macrae FA, Buchanan DD. Phenotypic confirmation of oligodontia, colorectal polyposis and cancer in a family carrying an exon 7 nonsense variant in the AXIN2 gene. Fam Cancer. 2019;18(3):311‐315. [DOI] [PubMed] [Google Scholar]
- 12. Hansen MF, Johansen J, Sylvander AE, et al. Use of multigene‐panel identifies pathogenic variants in several CRC‐predisposing genes in patients previously tested for lynch syndrome. Clin Genet. 2017;92(4):405‐414. [DOI] [PubMed] [Google Scholar]
- 13. Macklin‐Mantia SK, Riegert‐Johnson DL. An American patient with polyposis carrying a Scandinavian AXIN2 pathogenic variant. Hered Cancer Clin Pract. 2020;18:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rohlin A, Rambech E, Kvist A, et al. Expanding the genotype‐phenotype spectrum in hereditary colorectal cancer by gene panel testing. Fam Cancer. 2017;16(2):195‐203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rivera B, Perea J, Sánchez E, et al. A novel AXIN2 germline variant associated with attenuated FAP without signs of oligondontia or ectodermal dysplasia. Eur J Hum Genet. 2014;22(3):423‐426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Richards S, Aziz N, Bale S, et al. ACMG laboratory quality assurance committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405‐424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Leman R, Parfait B, Vidaud D, et al. SPiP: Splicing Prediction Pipeline, a machine learning tool for massive detection of exonic and intronic variant effects on mRNA splicing. Hum Mutat. 2022; 43(12):2308‐2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jaganathan K, Kyriazopoulou Panagiotopoulou S, McRae JF, et al. Predicting splicing from primary sequence with deep learning. Cell. 2019;176(3):535‐548.e24. [DOI] [PubMed] [Google Scholar]
- 19. Salamov AA, Nishikawa T, Swindells MB. Assessing protein coding region integrity in cDNA sequencing projects. Bioinformatics. 1998;14(5):384‐390. [DOI] [PubMed] [Google Scholar]
- 20. Liu H, Wong L. Data mining tools for biological sequences. J Bioinform Comput Biol. 2003;1:139‐167. [DOI] [PubMed] [Google Scholar]
- 21. Susswein LR, Marshall ML, Nusbaum R, et al. Pathogenic and likely pathogenic variant prevalence among the first 10,000 patients referred for next‐generation cancer panel testing. Genet Med. 2016;18:823‐832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Huang SM, Mishina YM, Liu S, et al. Tankyrase inhibition stabilizes axin and antagonizes Wnt signalling. Nature. 2009;461(7264):614‐620. [DOI] [PubMed] [Google Scholar]
- 23. Morrone S, Cheng Z, Moon RT, Cong F, Xu W. Crystal structure of a Tankyrase‐Axin complex and its implications for Axin turnover and Tankyrase substrate recruitment. Proc Natl Acad Sci U S A. 2012;109(5):1500‐1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Furuhashi M, Yagi K, Yamamoto H, et al. Axin facilitates Smad3 activation in the transforming growth factor β signaling pathway. Mol Cell Biol. 2001;21(15):5132‐5141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mariotti L, Pollock K, Guettler S. Regulation of Wnt/β‐catenin signalling by tankyrase‐dependent poly(ADP‐ribosyl)ation and scaffolding. Br J Pharmacol. 2017;174(24):4611‐4636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Leung JY, Kolligs FT, Wu R, et al. Activation of AXIN2 expression by β‐catenin‐T cell factor. A feedback repressor pathway regulating Wnt signaling. J Biol Chem. 2002;277(24):21657‐21665. [DOI] [PubMed] [Google Scholar]
- 27. Jho EH, Zhang T, Domon C, Joo CK, Freund JN, Costantini F. Wnt/β‐catenin/Tcf signaling induces the transcription of Axin2, a negative regulator of the signaling pathway. Mol Cell Biol. 2002;22(4):1172‐1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lustig B, Jerchow B, Sachs M, et al. Negative feedback loop of Wnt signaling through upregulation of conductin/axin2 in colorectal and liver tumors. Mol Cell Biol. 2002;129(4):199‐221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dao DY, Yang X, Chen D, Zuscik M, O'Keefe RJ. Axin1 and Axin2 are regulated by TGF‐β and mediate cross‐talk between TGF‐β and Wnt signaling pathways. Ann N Y Acad Sci. 2007;1116:82‐99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Domingo E, Espín E, Armengol M, et al. Activated BRAF targets proximal colon tumors with mismatch repair deficiency and MLH1 inactivation. Genes Chrom Cancer. 2004;39(2):138‐142. [DOI] [PubMed] [Google Scholar]
- 31. Kim MS, Kim SS, Ahn CH, Yoo NJ, Lee SH. Frameshift mutations of Wnt pathway genes AXIN2 and TCF7L2 in gastric carcinomas with high microsatellite instability. Hum Pathol. 2009;40(1):58‐64. [DOI] [PubMed] [Google Scholar]
- 32. Wu R, Zhai Y, Fearon ER, Cho KR. Diverse mechanisms of beta‐catenin deregulation in ovarian endometrioid adenocarcinomas. Cancer Res. 2001;61(22):8247‐8255. [PubMed] [Google Scholar]
- 33. Koch A, Hrychyk A, Hartmann W, et al. Mutations of the Wnt antagonist AXIN2 (Conductin) result in TCF‐dependent transcription in medulloblastomas. Int J Cancer. 2007;121(2):284‐291. [DOI] [PubMed] [Google Scholar]
- 34. Yue H, Liang J, Yang K, Hua B, Bian Z. Functional analysis of a novel missense mutation in AXIN2 associated with non‐syndromic tooth agenesis. Eur J Oral Sci. 2016;124(3):228‐233. [DOI] [PubMed] [Google Scholar]
- 35. Kishida S, Yamamoto H, Hino S, Ikeda S, Kishida M, Kikuchi A. DIX domains of Dvl and axin are necessary for protein interactions and their ability to regulate β‐catenin stability. Mol Cell Biol. 1999;19(6):4414‐4422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Fagotto F, Jho E, Zeng L, et al. Domains of axin involved in protein‐protein interactions, Wnt pathway inhibition, and intracellular localization. J Cell Biol. 1999;145(4):741‐756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Julius MA, Schelbert B, Hsu W, et al. Domains of axin and disheveled required for interaction and function in Wnt signaling. Biochem Biophys Res Commun. 2000;276(3):1162‐1169. [DOI] [PubMed] [Google Scholar]
- 38. Schwarz‐Romond T, Fiedler M, Shibata N, et al. The DIX domain of Dishevelled confers Wnt signaling by dynamic polymerization. Nat Struct Mol Biol. 2007;14(6):484‐492. [DOI] [PubMed] [Google Scholar]
- 39. Fiedler M, Mendoza‐Topaz C, Rutherford TJ, Mieszczanek J, Bienz M. Dishevelled interacts with the DIX domain polymerization interface of Axin to interfere with its function in down‐regulating β‐catenin. Proc Natl Acad Sci U S A. 2011;108(5):1937‐1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Liu YT, Dan QJ, Wang J, et al. Molecular basis of Wnt activation via the DIX domain protein Ccd1. J Biol Chem. 2011;286(10):8597‐8608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. MacDonald BT, He X. Frizzled and LRP5/6 receptors for Wnt/β‐catenin signaling. Cold Spring Harb Perspect Biol. 2012;4(12):a007880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chan JM, Clendenning M, Joseland S, et al. Rare germline variants in the AXIN2 gene in families with colonic polyposis and colorectal cancer. Fam Cancer. 2022;21(4):399‐413. [DOI] [PubMed] [Google Scholar]
- 43. Salic A, Lee E, Mayer L, Kirschner MW. Control of beta‐catenin stability: reconstitution of the cytoplasmic steps of the wnt pathway in Xenopus egg extracts. Mol Cell. 2000;5(3):523‐532. [DOI] [PubMed] [Google Scholar]
- 44. Lee E, Salic A, Krüger R, Heinrich R, Kirschner MW. The roles of APC and Axin derived from experimental and theoretical analysis of the Wnt pathway. PLoS Biol. 2003;1(1):e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bergendal B, Klar J, Stecksén‐Blicks C, Norderyd J, Dahl N. Isolated oligodontia associated with mutations in EDARADD, AXIN2, MSX1, and PAX9 genes. Am J Med Genet A. 2011;155A(7):1616‐1622. [DOI] [PubMed] [Google Scholar]
- 46. Wong S, Liu H, Bai B, et al. Novel missense mutations in the AXIN2 gene associated with non‐syndromic oligodontia. Arch Oral Biol. 2014;59(3):349‐353. [DOI] [PubMed] [Google Scholar]
- 47. Fournier BP, Bruneau MH, Toupenay S, et al. Patterns of dental agenesis highlight the nature of the causative mutated genes. J Dent Res. 2018;97(12):1306‐1316. [DOI] [PubMed] [Google Scholar]
- 48. Macklin‐Mantia SK, Hines SL, Chaichana KL, et al. Case report expanding the germline AXIN2‐related phenotype to include olfactory neuroblastoma and gastric adenoma. BMC Med Genet. 2020;21(1):161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Jensen JM, Skakkebæk A, Gaustadness M, et al. Familial colorectal cancer and tooth agenesis caused by an AXIN2 variant: how do we detect families with rare cancer predisposition syndromes? Fam Cancer. 2022;21(3):325‐332. [DOI] [PubMed] [Google Scholar]
- 50. Peterlongo P, Howe LR, Radice P, et al. Germline mutations of AXIN2 are not associated with nonsyndromic colorectal cancer. Hum Mutat. 2005;25(5):498‐500. [DOI] [PubMed] [Google Scholar]
- 51. Wang L, Baudhuin LM, Boardman LA, et al. MYH mutations in patients with attenuated and classic polyposis and with young‐onset colorectal cancer without polyps. Gastroenterology. 2004;127(1):9‐16. [DOI] [PubMed] [Google Scholar]
- 52. Nielsen M, Joerink‐van de Beld MC, Jones N, et al. Analysis of MUTYH genotypes and colorectal phenotypes in patients with MUTYH‐associated polyposis. Gastroenterology. 2009;136(2):471‐476. [DOI] [PubMed] [Google Scholar]
- 53. Vangala DB, Cauchin E, Balmaña J, et al. Screening and surveillance in hereditary gastrointestinal cancers: recommendations from the European Society of Digestive Oncology (ESDO) expert discussion at the 20th European Society for Medical Oncology (ESMO)/world congress on gastrointestinal cancer, Barcelona, June 2018. Eur J Cancer. 2018;104:91‐103. [DOI] [PubMed] [Google Scholar]
- 54. Colas C, Bonadona V, Baert‐Desurmont S, et al. MUTYH‐associated polyposis: review and update of the French recommendations established in 2012 under the auspices of the National Cancer institute (INCa). Eur J Med Genet. 2020;63(12):104078. [DOI] [PubMed] [Google Scholar]
- 55. Castiglia D, Bernardini S, Alvino E, et al. Concomitant activation of Wnt pathway and loss of mismatch repair function in human melanoma. Genes Chrom Cancer. 2008;47(7):614‐624. [DOI] [PubMed] [Google Scholar]
- 56. Järvinen E, Salazar‐Ciudad I, Birchmeier W, Taketo MM, Jernvall J, Thesleff I. Continuous tooth generation in mouse is induced by activated epithelial Wnt/beta‐catenin signaling. Proc Natl Acad Sci U S A. 2006;103(49):18627‐18632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Huraskin D, Eiber N, Reichel M, et al. Wnt/β‐catenin signaling via Axin2 is required for myogenesis and, together with YAP/Taz and Tead1, active in IIa/IIx muscle fibers. Development. 2016;143(17):3128‐3142. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
FIGURE S1. Flowchart showing the tools and criteria used for classifying AXIN2 variants identified in patients suspected of genetic predisposition to CRC. Criteria and classes are detailed in Richard et al. 16 Criteria: PVS, very strong; PP, supporting; BP, benign supporting; BA, benign stand alone. Classes: P, pathogenic; LP, likely pathogenic; VUS, variant of unknown significance; LB, likely benign; B, benign.
FIGURE S2. Description of non‐pathogenic/likely pathogenic AXIN2 variants identified in patients suspected of genetic predisposition to CRC. The pathogenicity assessment was performed as described in methods and Figure S1.
Abbreviations: P/LP, pathogenic/likely pathogenic; VUS variants of unknown significance; B/LB, benign/likely benign.
FIGURE S3. AXIN2 RT‐PCR analysis. RT‐PCR and Sanger sequencing were performed from blood and tumor RNA of patient Re‐2 III.8 with AXIN2 variant c.2062_2063del. Forward and reverse primers were designed in different exons to avoid amplification of genomic DNA. Electropherograms show the presence of both the wild‐type (WT) and the mutated (Mut) alleles, arguing against NMD.
TABLE S1. Characteristics and classification of AXIN2 variants identified in patients suspected of genetic predisposition to CRC. In addition to the P/LP variants, 125 different variants were collected from two laboratories (Lille, Rennes). These variants were classified as B/LB or VUS according to the ACMG/AMP criteria as described above. Data are summarized in Figure S2.
Data Availability Statement
All P/LP variants presented in this study have been submitted to and are available in ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/, accession numbers SCV001519371 to SCV001519380).