

Abstract
The fifth edition of the World Health Organization classification of soft tissue and bone tumors redefined Ewing sarcoma by fusions between EWSR1/FUS and ETS family of transcription factors, and recognized three tumor groups among Ewing‐like sarcoma: CIC‐rearranged sarcoma, sarcoma with BCOR genetic alterations, and round cell sarcoma with EWSR1::non‐ETS fusions. Although this classification underscores the critical role of molecular genetics in the diagnosis of small round cell sarcoma, each entry is recognized as a specific entity not only because they have different genetics but because their phenotypes are distinct and reasonably robust to support the diagnosis. This review focuses on the morphological aspects of Ewing sarcoma and a subset of Ewing‐like sarcomas (CIC‐rearranged sarcoma, BCOR‐associated sarcoma, and EWSR1::NFATC2 sarcoma) for which phenotypic characteristics have been well established. Classic histological findings, uncommon variations, and recurrent diagnostic pitfalls are addressed, along with the utility of recently developed immunohistochemical markers (NKX2.2, PAX7, ETV4, BCOR, CCNB3, and NKX3.1). Phenotypic expertise would significantly expedite the diagnostic process and complement (or sometimes outperform) genetic testing, even in well‐resourced settings. Morphological knowledge plays an even more substantial role in facilities that do not have easy access to molecular testing.
Keywords: diagnosis, Ewing sarcoma, Ewing‐like sarcoma, fusion gene, immunohistochemistry
Abbreviations
- EMC
extraskeletal myxoid chondrosarcoma
- FISH
fluorescence in situ hybridization
- ITD
internal tandem duplication
- NGS
next‐generation sequencing
- PNET
primitive neuroectodermal tumor
- RT‐PCR
reverse transcription polymerase chain reaction
- WHO
World Health Organization
INTRODUCTION
Undifferentiated small round cell sarcomas are a heterogeneous group of high‐grade malignancies in bone and soft tissue. Ewing sarcoma is prototypic, and its clinicopathological characteristics have been well studied. Recent investigations have identified smaller groups of sarcomas that demonstrate a limited overlap with Ewing sarcoma and embraced them in a colloquial label of Ewing‐like sarcoma. The latest World Health Organization (WHO) classification of soft tissue and bone tumors 1 redefined Ewing sarcoma using a specific combination of gene fusion between EWSR1/FUS and the genes encoding the ETS family of transcription factors. The classification additionally recognized three distinct groups among Ewing‐like sarcomas, namely, CIC‐rearranged sarcoma, sarcoma with BCOR genetic alterations, and round cell sarcoma with EWSR1::non‐ETS fusions, although the last category likely does not represent a single entity as it encompasses tumors with widely different fusions and phenotypes. As such, the WHO system emphasizes the critical role of molecular genetics in the classification of small round cell sarcoma, exemplifying a trend toward molecular‐based tumor classification that is now burgeoning across many different organs.
Nevertheless, it is notable that genetic findings were generally not included in the essential diagnostic criteria in the WHO classification, 1 even for molecularly defined tumor types. For example, diagnosing Ewing sarcoma requires histological and immunohistochemical analysis, with the determination of gene fusion status being reserved for select cases. 1 Similarly, CIC‐rearranged sarcoma is diagnosed primarily based on phenotypic characteristics of the tumor, with a demonstration of CIC rearrangement required in select cases. 1 On one hand, these criteria may represent a compromise with the cause that the WHO classification should be useful worldwide, including in communities without ready access to molecular testing. Meanwhile, the criteria also indicate that our knowledge of the tumor phenotypes is robust enough to make a correct diagnosis in many cases.
This review focuses on the morphological aspects of Ewing sarcoma and a subset of Ewing‐like sarcomas (CIC‐rearranged sarcoma, BCOR‐associated sarcoma, and EWSR1::NFATC2 sarcoma), for which studies have established phenotypic characteristics that enable diagnosis with a high degree of confidence. Classic histological findings, uncommon variations, and recurrent diagnostic pitfalls are addressed, along with the utility of recently developed immunohistochemical markers. Key histological characteristics of the diseases discussed in this review are summarized in Table 1. Supporting Information: Table S1 lists examples of staining condition for the select antibodies discussed, which are currently used in our department.
Table 1.
Histological and molecular summary of Ewing and Ewing‐like sarcomas discussed in this review
| Ewing sarcoma | CIC‐rearranged sarcoma | BCOR‐associated sarcoma | EWSR1::NFATC2 sarcoma | |
|---|---|---|---|---|
| Genetics | EWSR1::FLI1, EWSR1::ERG, EWSR1::ETV1, FUS::ERG, etc. | CIC::DUX4, CIC::NUTM1, CIC::FOXO4, etc. | BCOR::CCNB3, BCOR variant fusions, BCOR ITD, YWHAE::NUTM2B | EWSR1::NFATC2 |
| Architecture | Diffuse, rosetted (uncommon), nested (uncommon) | Lobulated or diffuse | Swirling fascicles, dense sheets, or palisades (uncommon) | Cords, nests, or diffuse (rare) |
| Cytology | Uniform; round, spindle (rare), or squamous (rare) | Relatively uniform and minimally pleomorphic; round, epithelioid (uncommon), or spindle (uncommon) | Uniform; oval, spindle, or round | Relatively uniform; round or oval |
| Stroma | Minimal or sclerotic (uncommon) | Sclerotic or minimal, often focally myxoid | Fibrous or myxoid, often hypervascular | Fibrous, hyaline, or myxoid, sometimes with eosinophils |
| Useful IHC | CD99, NKX2.2, PAX7 | ETV4, WT1, NUT (CIC::NUTM1 case only) | BCOR, SATB2, PAX7, CCNB3 (BCOR::CCNB3 case only) | NKX2.2, PAX7, NKX3.1 |
EWING SARCOMA
Definition and diagnostic molecular summary
Ewing sarcoma is defined by fusions between EWSR1 or FUS and one of the ETS family of transcription factor genes, most commonly FLI1 (>90%) or ERG. 1 The fusions can be detected using a variety of methods, including fluorescence in situ hybridization (FISH), reverse transcription polymerase chain reaction (RT‐PCR), and next‐generation sequencing (NGS). The decision of when to seek molecular testing depends on the confidence level of pathologists and access to molecular assays; however, in general, genetic testing would be useful in cases with atypical clinicopathological features in which the pathologists feel uncomfortable rendering definitive diagnoses based on phenotype alone. FISH can be falsely negative in cases with EWSR1::ERG more commonly than in those with EWSR1::FLI1, 2 a frequent reason for diagnostic challenges and consultations in our experience.
Clinical summary
Tumors most commonly occur in children or young adults; however, patients of any age can be affected. Ewing sarcoma is classically reported in bones. However, a significant proportion of cases occur in soft tissues, with a predilection for the older population. 3 In bones, the pelvic girdle is the most commonly involved site, and the chest wall and tubular bones are also common sites. In tubular bones, diaphysis is typically involved, and an ill‐defined permeative mass is associated with an aggressive periosteal reaction (e.g., onion skin) and massive soft tissue mass. Radiology may be mistaken for osteomyelitis, and the pus‐like gross appearance of sarcoma may compound such misinterpretation during surgery. Ewing sarcoma is treated with specific chemotherapy regimens and local surgical control. A favorable prognostic factor is a complete pathological response to chemotherapy (0% viable tumor cells) in resected specimens. 4
Histomorphology
Molecularly defined Ewing sarcoma shows a histological spectrum. However, most examples (~80%) demonstrate classic patterns, 5 , 6 in which small round cells proliferate diffusely (Figure 1a). Tumor cells are highly discohesive and tend to dissociate at the edge of the section/core as floating single cells. Tumor cells are monotonous and have a smooth nuclear membrane, fine chromatin pattern, and one to three small nucleoli. The thin cytoplasm is amphophilic or clear. Deep eosinophilia or rhabdoid forms are exceptional. Degenerated small cells with shrunken chromatin (“dark cells”) are often admixed. Necrosis is frequent, and mitosis is detectable.
Figure 1.
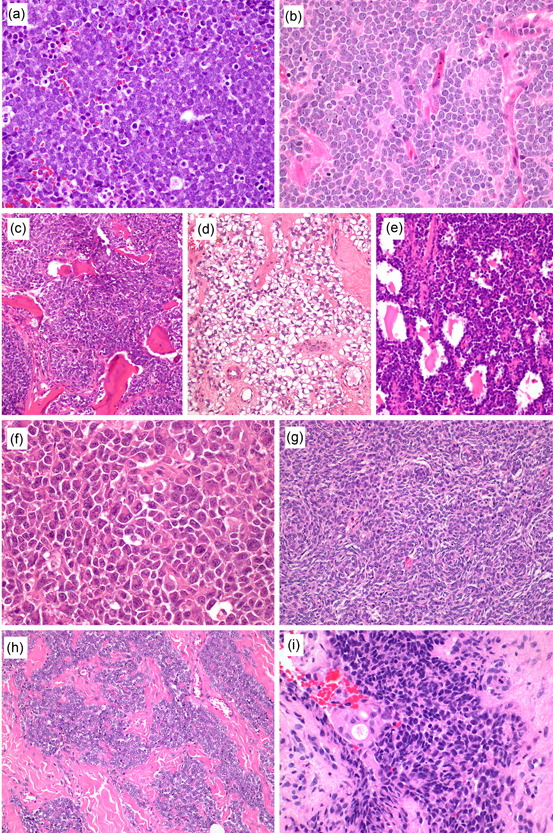
H&E stain findings of Ewing sarcoma. Most Ewing sarcomas demonstrate a classic histology with diffuse sheets of discohesive uniform small round cells (a). Primitive neuroectodermal tumor pattern features Homer Write rosettes, resembling neuroblastoma (b). Other uncommon patterns include reactive bone formation (c), prominent cytoplasmic clearing (d), and alveolar pattern (e). Large cell pattern is characterized by larger nuclear size and prominent nucleoli (f, a case with EWSR1::FLI1). Spindle cell pattern demonstrates swirling fascicles of short spindle cells, simulating synovial sarcoma (g, a case with EWSR1::FLI1). Nested epithelioid pattern is often confused with neuroendocrine tumors because of cohesive nests within the fibrous stroma (h, a case with EWSR1::FLI1). Adamantinoma‐like variant shows basaloid squamous differentiation with peripheral palisading and rare keratinization (i, a case with EWSR1::FLI1).
Nonclassic histology includes the primitive neuroectodermal tumor (PNET) pattern, a relatively common variation, particularly in soft tissue. In this pattern, Homer Wright type rosettes are present, which may cause confusion with neuroblastoma or neuroendocrine tumors (Figure 1b). Reactive bone formation can rarely be seen, and such a case has the risk of being mistaken for small cell osteosarcoma (Figure 1c). Some of these cases may even show a sclerosing radiological appearance, exacerbating confusion. Clear cytoplasmic changes can be observed owing to the accumulation of glycogen, which is rarely prominent (Figure 1d). Alveolar growth pattern is another uncommon variation, mimicking alveolar rhabdomyosarcoma (Figure 1e). The large cell pattern, also known as “atypical Ewing sarcoma,” features large cells with prominent nucleoli, a mild degree of nuclear pleomorphism, and eosinophilic cytoplasm, which may lead to rhabdoid cells (Figure 1f). A rare subset of Ewing sarcoma shows a spindle cell pattern, in which short spindle to oval cells form vague whirling fascicles or a reticular pattern in a myxoid‐to‐hyalinized background (Figure 1g). The nested epithelioid pattern can be found in soft tissue examples and is characterized by cohesive nests and archipelagos of round cells within a sclerotic background (Figure 1h). 3 , 7 , 8 Nested growth is more common in middle‐aged or older adults and is prone to be mistaken for a neuroendocrine tumor, particularly in the thoracic or abdominal cavity. The adamantinoma‐like variant shows exaggerated epithelial differentiation, often toward basaloid squamous morphology (Figure 1i). 9 , 10 , 11 The cells grow with or without peripheral palisading, and abrupt keratinization or keratin pearls are rarely observed. This variant is predilected to the head and neck location but has been reported in the extremities, including the tibial cortex, a classic location of adamantinoma.
Immunohistochemistry
Ewing sarcoma virtually always expresses CD99, 5 , 6 , 12 often in a diffuse, strong, and crisp membranous pattern (Figure 2a) unless compounded by preanalytical problems (e.g., under‐/over‐fixation). Although CD99 reactivity is widely known to be nonspecific in the context of round cell sarcoma, this diffuse strong membranous staining quality is still characteristic, albeit not entirely specific. More specific markers include NKX2.2 (Figure 2b) and PAX7 (Figure 2c), representing targets of the EWSR1::FLI1 and related fusions. 13 , 14 NKX2.2 is a member of the NK2 family of transcription factors that plays a critical role in the development and differentiation of the central nervous system and gastrointestinal/pancreatic endocrine cells. 15 PAX7 is a paired‐box transcription factor required for the developmental specification of adult skeletal muscle stem cells. 16 NKX2.2 and PAX7 are expressed in 90%–93% and 90%–99% of Ewing sarcoma, respectively, 13 , 15 , 17 , 18 , 19 and the combined positivity of CD99, NKX2.2, and PAX7 are highly specific for Ewing sarcoma. 18 , 19 , 20 NKX2.2 has a slightly lower sensitivity than PAX7 in large resection specimens or heavily decalcified samples. 13 , 18 , 19 NKX2.2 is normally expressed in neuroendocrine cells along the gastrointestinal tract and pancreas and weakly in oligodendroglia but not in skeletal muscles or bone. 15 The lack of internal positive control in bone or soft tissue should be considered when interpreting negative staining. In non‐Ewing tumors, NKX2.2 is often positive in olfactory neuroblastoma, mesenchymal chondrosarcoma, small cell carcinoma, neuroendocrine tumors of the gastrointestinal tract and pancreas, and weakly in glioma. 15 PAX7 is normally expressed in satellite cells in the skeletal muscle, sinonasal mucosa, and pituitary gland pars intermediate. 19 There are no PAX7‐positive cells in the bone. Besides Ewing sarcoma, PAX7 is often positive in rhabdomyosarcoma, synovial sarcoma, and round cell sarcoma with BCOR genetic alterations. 13 , 19 In addition, PAX7 can be positive in the invasive stromal front of the tumor in the soft tissue, perhaps representing regenerating/entrapped skeletal muscle satellite cells. 19
Figure 2.
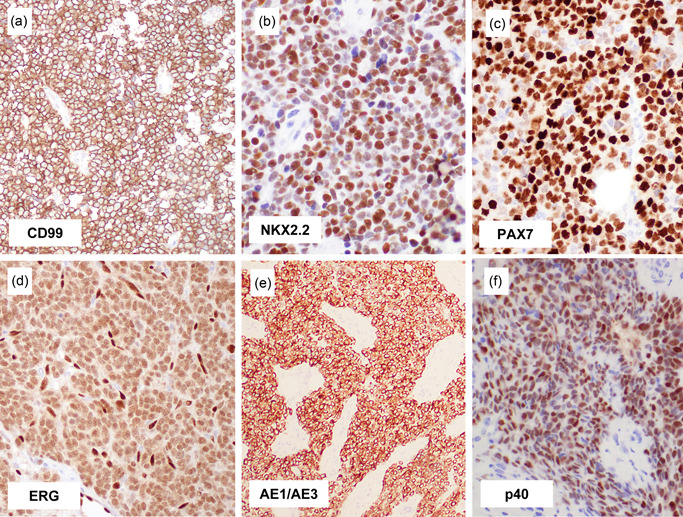
Immunohistochemical findings of Ewing sarcoma. Ewing sarcoma virtually always expresses CD99, often in a diffuse strong, and membranous pattern (a). Most examples are positive for NKX2.2 (b) and PAX7 (c), irrespective of histological pattern and fusion gene variant, and this combination of staining is highly specific for Ewing sarcoma. Diffuse ERG expression indicates the presence of EWSR1::ERG fusion (d). Keratin can be expressed in Ewing sarcoma, which can be rarely diffuse and strong (e), mimicking epithelial tumors. Adamantinoma‐like Ewing sarcoma expresses high‐molecular‐weight cytokeratin and p40 (f), consistent with squamous differentiation.
CD99, NKX2.2, and PAX7 are positive in Ewing sarcoma, irrespective of the histological pattern (including the adamantinoma‐like variant). They are also positive irrespective of fusion gene variant (including EWSR1::FLI1, EWSR1::ERG, EWSR1::ETV1, and FUS::ERG) in our experience. Even in institutions with easy access to molecular testing, such a phenotypic assessment provides a rapid diagnosis in a few days and/or serves a complementary role when these molecular tests fail or provide misleading results. Ewing sarcomas with EWSR1::ERG often (~50%) produce false‐negative EWSR1 FISH results (i.e., no or only equivocally split EWSR1 break‐apart signals), 2 and even in that setting, immunohistochemical assessment along with histomorphology reliably reaches the correct diagnosis. ERG immunostaining may also be helpful, as it produces diffuse nuclear labeling, 21 albeit typically at a lower intensity than vascular endothelial cells (Figure 2d).
Approximately 30% of Ewing sarcoma variably expresses keratin. 5 Keratin expression can be prominent, and when this happens in tumors with the nested epithelioid pattern involving the body cavity of adult patients, it can be easily confused with neuroendocrine tumors (Figure 2e). The expression of neuroendocrine markers is inconsistent. CD56 is often expressed. Synaptophysin can be expressed particularly in cases with a PNET pattern; however, concomitant chromogranin A expression is uncommon, and diffuse strong and uniform synaptophysin and chromogranin A coexpression is exceptional. Epithelial differentiation in Ewing sarcoma culminates in the adamantinoma‐like variant, in which diffuse strong keratin positivity is combined with p40 (or p63) expression (Figure 2f). S100 protein expression is uncommon, whereas desmin expression is rare. Myogenin and myoD1 are consistently negative, and their reactivity should prompt consideration of rhabdomyosarcoma. Ewing sarcoma often expresses c‐kit, cyclin D1, and claudin‐1, 22 but they are unlikely to mislead assessment in an appropriate clinicopathological context.
CIC‐REARRANGED SARCOMA
Definition and diagnostic molecular summary
The tumor is defined by the presence of fusion involving CIC gene. Over 90% of tumors have CIC::DUX4 fusion, whereas rare alternative fusion partners include NUTM1, FOXO4, LEUTX, and NUTM2A. 23 , 24 , 25 , 26 , 27 Molecular diagnosis of CIC‐rearranged sarcoma is challenged by a multitude of intraexonic breakpoints, which hampers sensitive detection by multiplex RT‐PCR methods. FISH using CIC break‐apart probes also suffers from moderate sensitivity (~85%). 28 , 29 NGS using fusion detection algorithms commonly filters out CIC::DUX4 fusion, necessitating manual inspection of the fusion reads. 28 , 30 Recently, recurrent ATXN1 or ATXN1L fusions to DUX4, NUTM1, and NUTM2A have been reported in central nervous system sarcomas with overlapping histology, immunophenotype, and methylation profile with CIC::DUX4 sarcoma, suggesting an expansion of the disease concept. 31 , 32 , 33 , 34
Clinical summary
As the most common tumor type among what was previously known as Ewing‐like sarcoma, CIC‐rearranged sarcomas represent 60%–70% of previously unclassifiable small round cell sarcomas. 35 , 36 The tumor has a wide age distribution, affecting children though the elderly, but young adults are most commonly affected. Most tumors arise in the deep soft tissues; however, approximately 10% occur in the viscera, including the kidney, gastrointestinal tract, and central nervous system. Tumors with CIC::NUTM1 fusion are overrepresented in the central nervous system, 23 , 37 although CIC::DUX4 fusion appears more common even in this location based on our experience. Primary bone cases are exceptionally rare, unlike Ewing sarcoma. 38 CIC‐rearranged sarcoma often manifests as a mass, and many patients present with metastatic disease. The disease is highly malignant, with significantly worse survival than that of Ewing sarcoma. 36 , 39 Localized tumors are treated with surgery and Ewing‐type chemotherapy is often considered, with a poor response. No prognostic markers have been identified to date.
Histomorphology
The tumor is grossly large, relatively well‐circumscribed, gray, and soft. Necrosis and hemorrhage are prominent. The histology is distinctive and differs from that of Ewing sarcoma. When the tumor periphery is available for review, it is often lobulated within the sclerotic stroma (Figure 3a), although sclerosis is often lacking in cases in the central nervous system. CIC‐rearranged sarcomas consist of diffuse sheets of small, round cells. Tumor cells often show relatively monotonous but minimally pleomorphic nuclei, including variations in size and shape (Figure 3b), unlike the extremely uniform round cytology of Ewing sarcoma. 36 In addition, CIC‐rearranged sarcoma cells have more ample cytoplasm and a greater degree of intercellular cohesion than Ewing sarcoma. 36 Nuclei tend to display vesicular chromatin and prominent nucleoli, with brisk mitotic figures. Necrosis is prominent. In some cases, the cells become decidedly epithelioid and mimic carcinoma or epithelioid sarcoma (Figure 3c). 40 A characteristic and diagnostically helpful finding is a focal myxoid change observed in half of the cases, in which tumor cells grow in a reticular to corded pattern within the myxoid stroma (Figure 3d). 36 , 41 This pattern can be prominent to the degree of simulating extraskeletal myxoid chondrosarcoma (EMC) or myoepithelial tumors. We have seen several CIC‐rearranged sarcomas that were originally misinterpreted as high‐grade cellular EMC; such a diagnosis, particularly in young patients who are not typically involved by EMC, is worth reconsideration. Myxoid changes are rare in Ewing sarcoma, with the exception of specimens post neoadjuvant therapy. Focal spindle cell component can be present, and it is rarely prominent (Figure 3e). Focal cartilaginous differentiation was reported in a few cases. 36 , 42 The histological findings of tumors with non‐DUX4 fusion and ATXN1 fusion appear to be similar to those of CIC::DUX4 sarcomas. 23 , 24 , 32
Figure 3.
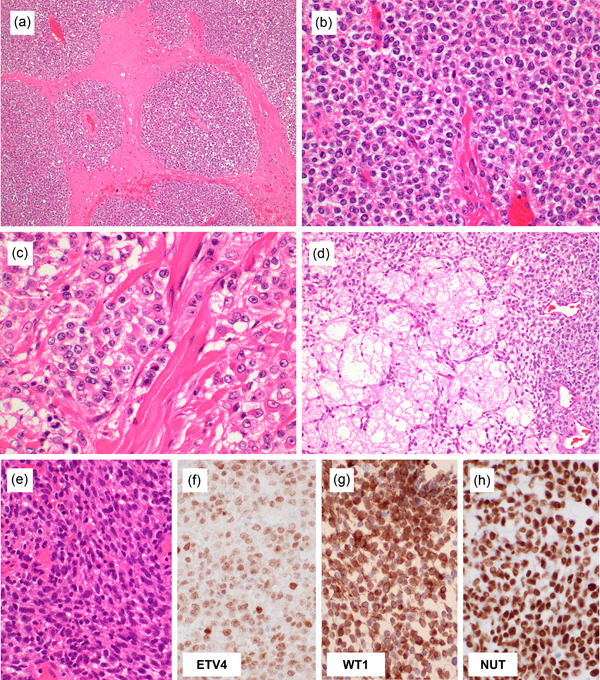
Histological findings of CIC‐rearranged sarcoma. The tumors commonly show peripheral lobulation within the fibrotic stroma (a). The tumor cells are relatively monotonous but consistently show a minimal degree of nuclear size variation and prominent nucleoli (b). The cells are more cohesive with each other and harbor a larger amount of cytoplasm than Ewing sarcoma. Focal areas of epithelioid cells can be present (c). Characteristic myxoid change is observed in half of the cases, at least focally, in which tumor cells are arranged in a reticular pattern, mimicking myoepithelial tumors (d). Spindle cell component is uncommonly admixed (e). CIC‐rearranged sarcomas often co‐express ETV4 (f) and WT1 (g). CIC::NUTM1‐positive sarcomas express NUT diffusely (h).
Immunohistochemistry
Most of the CIC‐rearranged sarcomas are positive for ETV4 (~90%; Figure 3f) and WT1 (70%–95%; Figure 3g). ETV4 provides nuclear staining and is highly sensitive and specific, 29 , 43 , 44 reflecting the characteristic upregulation of PEA3 family genes. 27 Weak or focal ETV4 expression may be observed in Ewing sarcoma and other round cell tumors, and an appropriate cutoff should be internally established. WT1 is slightly less sensitive than ETV4 but has the advantage of wide availability in many laboratories. 36 , 43 , 45 WT1 staining in CIC‐rearranged sarcoma often gives intense cytoplasmic staining in addition to weaker nuclear staining; only nuclear staining should be considered specific. When CIC‐rearranged sarcoma occurs in the body cavity, WT1 expression may be a cause of misdiagnosis as malignant mesothelioma of the small cell type. CIC‐rearranged sarcomas often express calretinin and, sometimes, D2‐40, and such a “mesothelioma panel” needs to be used with caution when dealing with round cell malignancy. 36 Nuclear DUX4 immunoreactivity using C‐terminus antibody can be positive in CIC::DUX4 sarcoma. 46 However, the number of cases tested for this antibody has been relatively small and requires further validation.
CD99 expression is frequent but usually heterogeneous in terms of intensity and extent. Diffuse strong membranous expression of CD99 is rare, unlike in Ewing sarcoma. 36 , 45 CIC‐rearranged sarcoma may show focal keratin staining, usually as a rare, dotted expression in a small minority of cells. Diffuse keratin expression has been exceptionally recorded. 47 S100 protein is negative. Expression of myogenic markers (actin and desmin) is rare and focal at most. Myogenin and myoD1 are negative. CIC‐rearranged sarcoma is negative for NKX2.2 and PAX7, which helps distinguish it from Ewing sarcoma. 19 , 36 CIC::NUTM1 sarcoma shows a positive expression of NUT protein (Figure 3h); speckled expression, characteristic of BRD4/3::NUTM1 carcinoma, is lacking. 23
Many CIC‐rearranged sarcomas express ERG to a variable degree. 41 , 47 A small subset co‐expresses ERG and CD31, thus creating confusion with angiosarcoma. 47 This can be compounded by occasional cleft‐like hemorrhagic pseudovascular spaces and intracytoplasmic vacuoles in CIC‐rearranged sarcomas. ERG and CD31 expression in CIC‐rearranged sarcomas is focal or multifocal and heterogeneous, unlike uniformly diffuse and strong co‐expression in epithelioid angiosarcoma. Furthermore, CIC‐rearranged sarcoma lacks true vasoformative architecture, enabling effective distinctions based on phenotype in most examples. 47
BCOR‐ASSOCIATED SARCOMA
Definition and diagnostic molecular summary
BCOR‐associated sarcomas, or sarcomas with BCOR genetic alterations based on the WHO terminology, 1 encompass at least four molecular subtypes: BCOR::CCNB3 sarcoma, sarcoma with BCOR variant fusions with a non‐CCNB3 partner, sarcomas with internal tandem duplication (ITD) of BCOR exon 15, and YWHAE::NUTM2B sarcoma. Despite different molecular abnormalities and clinical features, combining these four molecular groups into a unifying BCOR‐associated sarcoma is justified by similar histological findings and overlapping RNA expression profiles. 48 BCOR::CCNB3 fusion is created by a paracentric inversion within the X chromosome. 49 As such, its detection by FISH may be difficult using an ordinary BCOR break‐apart design and may require an alternative probe set. 50 Non‐CCNB3 partners to BCOR include MAML3, ZC3H7B, KMT2D, CIITA, and CHD9. 48 , 50 , 51 , 52 BCOR break‐apart FISH may produce false‐negative results for cases with KMT2D::BCOR and CIITA::BCOR. 50 , 51 Demonstration of BCOR ITD can be challenging when using formalin‐fixed, paraffin‐embedded tissues and some targeted NGS platforms.
Clinical summary
BCOR::CCNB3 sarcoma is the most common genetic subtype and approximately 80% of the cases affect teenage boys. Bones are more commonly affected than soft tissues, and the sarcomas have a proclivity to involve the pelvic or sacral bones. Acral bones such as the calcaneus can be involved. 53 , 54 , 55 Sarcomas with fusions between BCOR and alternative partners affect both children and adults. 50 , 52 Sarcomas with BCOR ITD and YWHAE::NUTM2B most commonly arise in infants, and infantile BCOR ITD sarcoma corresponds to histology reported as primitive myxoid mesenchymal tumors of infancy. 56 , 57 However, soft tissue sarcoma with BCOR ITD rarely arises in adults. 50 BCOR‐associated sarcoma is a high‐grade malignancy, although its prognosis has not been clearly defined. In one series, the overall survival for BCOR::CCNB3 sarcoma patients was not significantly different from those for head and neck synovial sarcoma patients and Ewing sarcoma patients. 48 The treatment strategy for BCOR‐associated sarcoma has not been established. However, the tumor is commonly treated using Ewing‐like regimens along with local surgical control when feasible.
Histomorphology
Often considered as a member of Ewing‐like sarcoma, BCOR‐associated sarcoma can present as purely round cytology, similar to Ewing sarcoma (Figure 4a). However, in our experience, the tumors commonly manifest as spindle cell sarcoma and many of them can be confused with synovial sarcoma and malignant peripheral nerve sheath tumor. 58 BCOR::CCNB3 sarcoma often consists of a dense proliferation of monotonous spindle or oval cells in a swirling fascicle (Figure 4b). Whorls, curlicues, and nuclear palisading are uncommonly observed (Figure 4c). 53 Nuclear pleomorphism is rare. Some tumors focally display low cellularity, resembling benign fibroblastic tumors (Figure 4d), and such a histology may predominate after chemotherapy. 55 The stroma is edematous to myxoid and variably hypervascular. The vascularity of BCOR‐associated sarcoma is generally higher than that of Ewing sarcoma and CIC‐rearranged sarcoma and often takes the form of branching capillaries. Organoid trabecular structures have been rarely observed (Figure 4e). Focal osteoblastic differentiation has been rarely reported, leading to the misdiagnosis of small cell osteosarcoma. 49 , 55 Uniform tumor cells harbor fine chromatin and smooth nuclear contours. The histological pattern described above is shared by tumors with other BCOR genetic variants. Specific phenotypes associated with BCOR genetic variants include marked nuclear palisading in adult BCOR‐associated sarcomas (Figure 4f) and prominent myxoid stroma in pediatric soft tissue tumors with BCOR ITD (Figure 4g). 50 , 56
Figure 4.
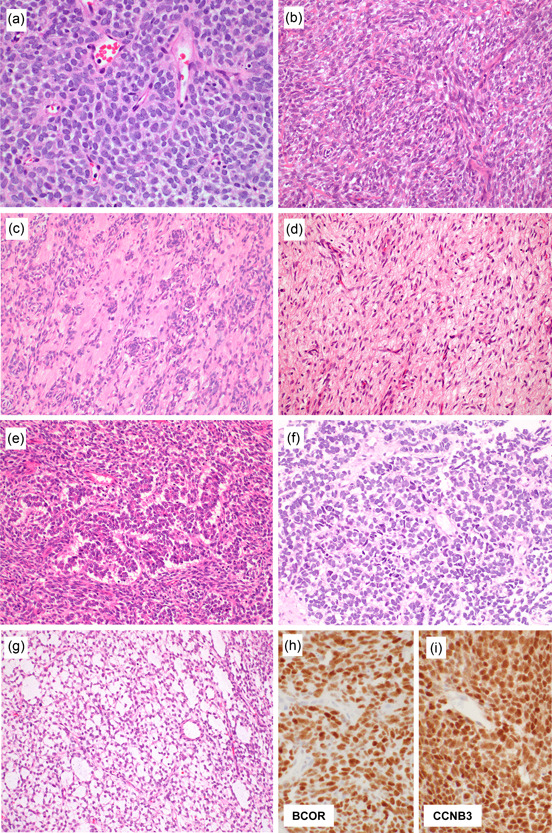
Histological findings of BCOR‐associated sarcoma. BCOR‐associated sarcoma can present with round cell cytology similar to Ewing sarcoma (a). However, it commonly manifests as spindle cell sarcoma, consisting of dense proliferation of monotonous short spindle or oval cells in a swirling fascicle within a variably myxoid and often hypervascular stroma (b). Whorls, curlicue, and nuclear palisading may be seen (c). Some tumors focally display low cellularity mimicking benign lesions (d), and this is particularly common in post chemotherapy specimens. Organoid trabecular structure is rare (e). Nuclear palisading can be present in adult BCOR‐associated sarcomas (f). Prominent myxoid stroma characterizes pediatric soft tissue tumors with BCOR ITD, which corresponds to the primitive myxoid mesenchymal tumor of infancy (g). Over 90% of tumors are positive for BCOR (h). CCNB3 expression is specifically observed in BCOR::CCNB3 sarcoma (i).
Immunohistochemistry
BCOR is a sensitive marker for BCOR‐associated sarcomas, staining >90% of tumors (Figure 4h). 59 However, it has limited specificity and frequent BCOR expression in synovial sarcoma and solitary fibrous tumors, which are two close histological mimics of BCOR‐associated sarcoma, is particularly problematic. 59 , 60 CCNB3 expression is specifically observed in BCOR::CCNB3 sarcoma (Figure 4i), which is very useful for the diagnosis of this molecular variant. 49 , 53 , 54 However, the reactivity may become negative after chemotherapy, 53 , 55 and sarcomas with other BCOR molecular alterations lack CCNB3 expression. SATB2 is also often positive, which may represent a pitfall in bone, as SATB2 is known to be expressed in osteoblastic tumors, including osteosarcoma. 59 , 61 BCOR‐associated sarcoma often over‐expresses NTRK3, which can be detected using the PanTRK antibody, 51 representing a pitfall in NTRK‐rearranged spindle cell tumors. Unlike BCOR‐associated sarcomas, NTRK‐rearranged spindle cell tumors usually do not express BCOR but are commonly positive for CD34 and/or S100 protein. CD99 expression may vary and can be negative. 54 NKX2.2, ETV4, and WT1 are usually negative, while PAX7 expression is often positive. 19 , 53
Relationship with BCOR‐associated visceral tumors
BCOR‐associated sarcomas have parallels in visceral organs such as the kidney, uterus, and brain. BCOR ITD is present in the majority of clear cell sarcomas of the kidney, 62 while BCOR::CCNB3 and YWHAE::NUTM2B are present in a smaller percentage. 63 , 64 YWHAE::NUTM2B, ZC3H7B::BCOR, and BCOR ITD are present in high‐grade endometrial stromal sarcoma. 65 , 66 , 67 BCOR ITD characterizes a subset of high‐grade neuroepithelial tumors. 37 , 68 These various types of tumors show at least partly overlapping histology with their musculoskeletal counterparts, often with similar BCOR expression. However, each tumor type has distinct clinical features and/or a unique immunoprofile and is considered separate from BCOR‐associated sarcomas in bone and soft tissue. For example, high‐grade neuroepithelial tumors with BCOR ITD can express neuroepithelial markers OLIG2 and NeuN. 68
EWSR1::NFATC2 SARCOMA
Definition and diagnostic molecular summary
EWSR1::NFATC2 sarcoma is likely the most common and phenotypically best‐characterized tumor type among round cell sarcoma with the EWSR1::non‐ETS fusion category in the latest WHO classification. Tumors are defined by the presence of a fusion between EWSR1 and NFATC2, often via unbalanced translocation and concomitant amplification. The presence of fusion is inferred by the amplified 5' EWSR1 signals when analyzed using EWSR1 break‐apart FISH, and this finding is very useful for diagnosis. 69 An alternative, non‐amplified, FUS::NFATC2 fusion is present in a small number of cases. EWSR1::NFATC2 sarcoma and FUS::NFATC2 sarcomas are presently described together in the WHO classification. However, the two groups reportedly did not cluster together using RNA expression analysis, and the classification is somewhat controversial. 70 Identical NFATC2 fusions with EWSR1/FUS have been reported in simple bone cysts and intraosseous vascular tumors, but the fusions are not amplified. 71 , 72 , 73
Clinical summary
EWSR1::NFATC2 sarcoma occurs in bone, or less commonly, soft tissue, in adults. 74 , 75 , 76 Bone primary tumors often affect the metaphysis or diaphysis of long bones, such as the femur and humerus. The characteristic radiographic appearance includes a significant extracortical component with saucerization of the cortex associated with cortical buttressing. 77 The cortical involvement and nested histological growth pattern of tumors prompted the original diagnosis of Ewing‐like adamantinoma in some cases. 77 In soft tissues, the extremities, retroperitoneum, chest wall, and head and neck are reported sites. Overall, the tumors are well‐circumscribed and often painful. Growth can be slow with a long preoperative history, reflected by cortical buttressing in bone cases. Most tumors are locally controlled by wide excision; however, a small number of aggressive cases are on record. 75 FUS::NFATC2 sarcoma is also rare and presents as a destructive tumor in the long tubular bones. 70 , 76 , 78 It has a male predilection, and both children and adults can be involved.
Histomorphology
EWSR1::NFATC2 sarcoma is a grossly yellow to tan and rubbery solid mass. It can be well‐circumscribed, although microscopically infiltrative. EWSR1::NFATC2 sarcomas often consist of cords, nests, or pseudoacini within fibrous, hyaline, or myxoid stroma, thus closely mimicking myoepithelial tumors (Figure 5a–c). 75 , 76 The tumor cells are round to oval and relatively uniform, with variable hyperchromasia. Some cases show focally moderate nuclear pleomorphism, including scattered large cells. Eosinophilic leukocyte infiltration is a notable but inconsistent finding. 76 A small subset of cases is stroma‐poor and demonstrates diffuse sheets of small round cells, similar to Ewing sarcoma (Figure 5d). FUS::NFATC2 sarcoma consists of round or spindle cells. Some tumors have been reported to show a nested pattern or chondroid stroma, but the findings have been inconsistent. 70 , 78
Figure 5.
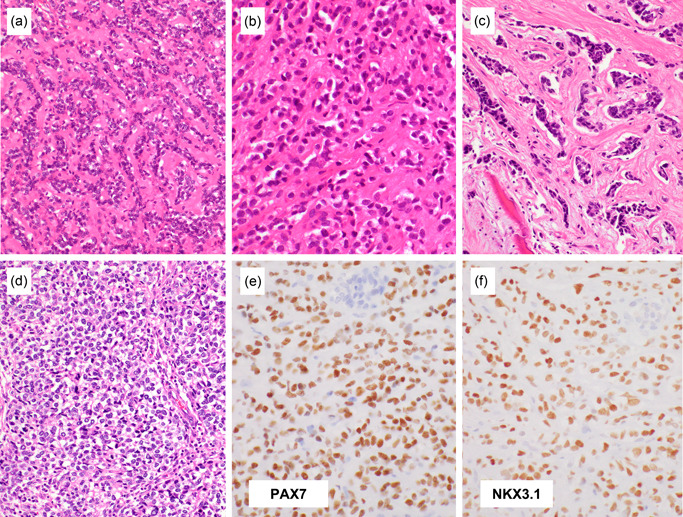
Histological findings of EWSR1::NFATC2 sarcoma. The tumors often consist of cords, nests, or pseudoacini within fibrous, hyaline, or myxoid stroma, thus closely mimicking myoepithelial tumors (a, b) or, rarely, metastatic carcinoma (c). The tumor cells are round to oval and relatively uniform with variable hyperchromasia. Diffuse sheets of small round cells, similar to Ewing sarcoma, are only uncommonly observed (d). EWSR1::NFATC2 sarcoma often variably expresses CD99, NKX2.2, and PAX7 (e), but unlike Ewing sarcoma, ~80% of EWSR1::NFATC2 sarcomas express NKX3.1 (f).
Immunohistochemistry
EWSR1::NFATC2 sarcoma often expresses CD99, NKX2.2, and PAX7 (Figure 5e), 13 , 75 , 76 the same combination as Ewing sarcoma; however, its expression seems more variable than Ewing sarcoma. Focal dot‐like keratin expression is common, which may lead to misinterpretation as metastatic carcinoma. RNA expression profiling identified differential NKX3‐1 overexpression in EWSR1::NFATC2 sarcoma, 70 which was later confirmed by RNA in situ hybridization and immunohistochemistry. 76 , 79 NKX3.1 is immunohistochemically expressed in ~80% of EWSR1::NFATC2 sarcomas (Figure 5f), whereas it is not expressed in many other round cell mimics, including Ewing sarcoma, CIC‐rearranged sarcoma, and BCOR‐associated sarcoma. 76 NKX3.1 (NK3 homeobox 1) is a transcription factor that is essential for prostate development, and it is expressed in the normal prostatic epithelium as well as Sertoli cells and mucous cells in the salivary and bronchial glands. NKX3.1 has been widely used as a prostatic adenocarcinoma marker in surgical pathology in the context of carcinoma of unknown origin. 80 One caveat is that NKX3.1 is also commonly expressed in mesenchymal chondrosarcoma in the primitive noncartilaginous component but not in the well‐differentiated cartilaginous component, and its ancillary diagnostic role is emerging. 76 , 79 , 81 , 82 In mice, Nkx3‐1 and Nkx3‐2 play important roles in chondrogenesis. 83 Because the NKX3.1 expression level in these sarcomas seems lower than in prostatic epithelial cells, 79 appropriate conditioning of the staining method is required, using weak‐moderate staining of Sertoli cells as a positive control.
NKX2.2, PAX7, and NKX3.1 are not expressed in FUS::NFATC2 sarcoma in the tested cases, which is consistent with the differential downregulation of NKX3‐1 in FUS::NFATC2 sarcoma as compared with EWSR1::NFATC2 sarcoma. 70 Sarcomas with EWSR1::NFATC2 and FUS::NFATC2 reportedly express aggrecan, another cartilage‐associated molecule. 78
NKX2.2 and NKX3.1 are not expressed in simple bone cysts and vascular tumors with the same EWSR1::NFATC2 fusions, 71 implicating a different underlying molecular mechanism for each tumor type that incidentally shares gene fusions.
CONCLUSIONS
Undifferentiated round cell sarcomas are increasingly defined by driving molecular genetic changes. However, the tumor phenotype remains at the very center of their diagnosis, as stipulated by the essential diagnostic criteria published in the WHO blue book. Even in a well‐resourced institution, expertise in tumor morphology significantly expedites the diagnostic process. Knowledge of the tumor phenotype also complements genetic testing and can sometimes overcome the errors/difficulties inherent in molecular assays. Phenotypic understanding plays an even more substantial role in facilities that do not have easy access to molecular testing owing to equipment, expertise, and cost limitations. Altogether, phenotypic studies on sarcomas, even those defined on genetic grounds, should significantly improve the quality of care for sarcoma patients. Finally, we are aware that some round cell sarcomas remain, including those harboring EWSR1::PATZ1, 84 , 85 , 86 CRTC1::SS18, 87 , 88 and SS18::POU5F1, 89 , 90 , 91 in which the reported tumor phenotypes are inconsistent and/or the studied cases are few, and, therefore, their diagnosis solely depends on the identification of genetic abnormalities. Further studies should be conducted to determine whether these tumors have a specific recognizable histology.
AUTHOR CONTRIBUTIONS
Akihiko Yoshida wrote the manuscript and prepared the figures.
CONFLICT OF INTEREST
None declared.
Supporting information
Supporting information.
ACKNOWLEDGMENTS
The author is grateful to his colleagues and collaborators who contributed to the research. The author has been announced as the winner of The Japanese Society of Pathology, Pathology Research Award in 2021. This work was partly supported by JSPS KAKENHI (Grant Nos. JP15K19065, JP18K15108, and JP21K06919) and the Rare Cancer Grant of National Cancer Center Hospital (G007).
Yoshida A. Ewing and Ewing‐like sarcomas: A morphological guide through genetically‐defined entities. Pathol Int. 2023;73:12–26. 10.1111/pin.13293
REFERENCES
- 1. The WHO Classification of Tumours Editorial Board . WHO classification of tumours of soft tissue and bone. 5th ed. Lyon: IARC; 2020. [Google Scholar]
- 2. Chen S, Deniz K, Sung YS, Zhang L, Dry S, Antonescu CR. Ewing sarcoma with ERG gene rearrangements: a molecular study focusing on the prevalence of FUS‐ERG and common pitfalls in detecting EWSR1‐ERG fusions by FISH. Genes Chromosomes Cancer. 2016;55:340–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jahanseir K, Folpe AL, Graham RP, Giannini C, Robinson SI, Sukov W, et al. Ewing sarcoma in older adults: a clinicopathologic study of 50 cases occurring in patients aged >/=40 years, with emphasis on histologic mimics. Int J Surg Pathol. 2020;28:352–60. [DOI] [PubMed] [Google Scholar]
- 4. Albergo JI, Gaston CL, Laitinen M, Darbyshire A, Jeys LM, Sumathi V, et al. Ewing's sarcoma: only patients with 100% of necrosis after chemotherapy should be classified as having a good response. Bone Joint J. 2016;98‐B:1138–44. [DOI] [PubMed] [Google Scholar]
- 5. Folpe AL, Goldblum JR, Rubin BP, Shehata BM, Liu W, Dei Tos AP, et al. Morphologic and immunophenotypic diversity in Ewing family tumors: a study of 66 genetically confirmed cases. Am J Surg Pathol. 2005;29:1025–33. [PubMed] [Google Scholar]
- 6. Llombart‐Bosch A, Machado I, Navarro S, Bertoni F, Bacchini P, Alberghini M, et al. Histological heterogeneity of Ewing's sarcoma/PNET: an immunohistochemical analysis of 415 genetically confirmed cases with clinical support. Virchows Arch. 2009;455:397–411. [DOI] [PubMed] [Google Scholar]
- 7. Kirishima M, Kato I, Hisaoka M, Nakatani Y, Takeda AH, Mizuno K, et al. Solid endobronchial tumor with EWSR1‐FLI1 fusion gene ‐ a diagnostically challenging case of the Ewing sarcoma. Pathol Int. 2021;71:488–90. [DOI] [PubMed] [Google Scholar]
- 8. Rekhi B, Shetty O, Vora T, Gulia A, Bajpai J, Laskar S. Clinicopathologic, immunohistochemical, molecular cytogenetic profile with treatment and outcomes of 34 cases of Ewing sarcoma with epithelial differentiation, including 6 cases with “Adamantinoma‐like” features, diagnosed at a single institution, India. Ann Diagn Pathol. 2020;49:151625. [DOI] [PubMed] [Google Scholar]
- 9. Bridge JA, Fidler ME, Neff JR, Degenhardt J, Wang M, Walker C, et al. Adamantinoma‐like Ewing's sarcoma: genomic confirmation, phenotypic drift. Am J Surg Pathol. 1999;23:159–65. [DOI] [PubMed] [Google Scholar]
- 10. Kikuchi Y, Kishimoto T, Ota S, Kambe M, Yonemori Y, Chazono H, et al. Adamantinoma‐like Ewing family tumor of soft tissue associated with the vagus nerve: a case report and review of the literature. Am J Surg Pathol. 2013;37:772–9. [DOI] [PubMed] [Google Scholar]
- 11. Rooper LM, Bishop JA. Soft tissue special issue: adamantinoma‐like ewing sarcoma of the head and neck: a practical review of a challenging emerging entity. Head Neck Pathol. 2020;14:59–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Machado I, Yoshida A, López‐Guerrero JA, Nieto MG, Navarro S, Picci P, et al. Immunohistochemical analysis of NKX2.2, ETV4, and BCOR in a large series of genetically confirmed Ewing sarcoma family of tumors. Pathol Res Pract. 2017;213:1048–53. [DOI] [PubMed] [Google Scholar]
- 13. Charville GW, Wang WL, Ingram DR, Roy A, Thomas D, Patel RM, et al. EWSR1 fusion proteins mediate PAX7 expression in Ewing sarcoma. Mod Pathol. 2017;30:1312–20. [DOI] [PubMed] [Google Scholar]
- 14. Smith R, Owen LA, Trem DJ, Wong JS, Whangbo JS, Golub TR, et al. Expression profiling of EWS/FLI identifies NKX2.2 as a critical target gene in Ewing's sarcoma. Cancer Cell. 2006;9:405–16. [DOI] [PubMed] [Google Scholar]
- 15. Yoshida A, Sekine S, Tsuta K, Fukayama M, Furuta K, Tsuda H. NKX2.2 is a useful immunohistochemical marker for Ewing sarcoma. Am J Surg Pathol. 2012;36:993–9. [DOI] [PubMed] [Google Scholar]
- 16. Charville GW, Varma S, Forgó E, Dumont SN, Zambrano E, Trent JC, et al. PAX7 expression in rhabdomyosarcoma, related soft tissue tumors, and small round blue cell neoplasms. Am J Surg Pathol. 2016;40:1305–15. [DOI] [PubMed] [Google Scholar]
- 17. Hung YP, Fletcher CDM, Hornick JL. Evaluation of NKX2‐2 expression in round cell sarcomas and other tumors with EWSR1 rearrangement: imperfect specificity for Ewing sarcoma. Mod Pathol. 2016;29:370–80. [DOI] [PubMed] [Google Scholar]
- 18. Machado I, Charville GW, Yoshida A, Navarro S, Righi A, Gambarotti M, et al. Does PAX7 and NKX2.2 immunoreactivity in Ewing sarcoma have prognostic significance? Virchows Arch. 2022;480:909–17. [DOI] [PubMed] [Google Scholar]
- 19. Toki S, Wakai S, Sekimizu M, Mori T, Ichikawa H, Kawai A, et al. PAX7 immunohistochemical evaluation of Ewing sarcoma and other small round cell tumours. Histopathology. 2018;73:645–52. [DOI] [PubMed] [Google Scholar]
- 20. Shibuya R, Matsuyama A, Nakamoto M, Shiba E, Kasai T, Hisaoka M. The combination of CD99 and NKX2.2, a transcriptional target of EWSR1‐FLI1, is highly specific for the diagnosis of Ewing sarcoma. Virchows Arch. 2014;465:599–605. [DOI] [PubMed] [Google Scholar]
- 21. Wang WL, Patel NR, Caragea M, Hogendoorn PC, López‐Terrada D, Hornick JL, et al. Expression of ERG, an Ets family transcription factor, identifies ERG‐rearranged Ewing sarcoma. Mod Pathol. 2012;25:1378–83. [DOI] [PubMed] [Google Scholar]
- 22. Schuetz AN, Rubin BP, Goldblum JR, Shehata B, Weiss SW, Liu W, et al. Intercellular junctions in Ewing sarcoma/primitive neuroectodermal tumor: additional evidence of epithelial differentiation. Mod Pathol. 2005;18:1403–10. [DOI] [PubMed] [Google Scholar]
- 23. Le Loarer F, Pissaloux D, Watson S, Godfraind C, Galmiche‐Rolland L, Silva K, et al. Clinicopathologic features of CIC‐NUTM1 sarcomas, a new molecular variant of the family of CIC‐fused sarcomas. Am J Surg Pathol. 2019;43:268–76. [DOI] [PubMed] [Google Scholar]
- 24. Sugita S, Arai Y, Tonooka A, Hama N, Totoki Y, Fujii T, et al. A novel CIC‐FOXO4 gene fusion in undifferentiated small round cell sarcoma: a genetically distinct variant of Ewing‐like sarcoma. Am J Surg Pathol. 2014;38:1571–6. [DOI] [PubMed] [Google Scholar]
- 25. Sugita S, Arai Y, Aoyama T, Asanuma H, Mukai W, Hama N, et al. NUTM2A‐CIC fusion small round cell sarcoma: a genetically distinct variant of CIC‐rearranged sarcoma. Hum Pathol. 2017;Epub 65:225–30. [DOI] [PubMed] [Google Scholar]
- 26. Song K, Huang Y, Xia CD, Zhu HQ, Wang J. A case of CIC‐rearranged sarcoma with CIC‐LEUTX gene fusion in spinal cord. Neuropathology. 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kawamura‐Saito M, Yamazaki Y, Kaneko K, Kawaguchi N, Kanda H, Mukai H, et al. Fusion between CIC and DUX4 up‐regulates PEA3 family genes in Ewing‐like sarcomas with t(4;19)(q35;q13) translocation. Hum Mol Gen. 2006;15:2125–37. [DOI] [PubMed] [Google Scholar]
- 28. Kao YC, Sung YS, Chen CL, Zhang L, Dickson BC, Swanson D, et al. ETV transcriptional upregulation is more reliable than RNA sequencing algorithms and FISH in diagnosing round cell sarcomas with CIC gene rearrangements. Genes Chromosomes Cancer. 2017;56:501–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yoshida A, Arai Y, Kobayashi E, Yonemori K, Ogura K, Hama N, et al. CIC break‐apart fluorescence in‐situ hybridization misses a subset of CIC‐DUX4 sarcomas: a clinicopathological and molecular study. Histopathology. 2017;71:461–9. [DOI] [PubMed] [Google Scholar]
- 30. Panagopoulos I, Gorunova L, Bjerkehagen B, Heim S. The “grep” command but not FusionMap, FusionFinder or ChimeraScan captures the CIC‐DUX4 fusion gene from whole transcriptome sequencing data on a small round cell tumor with t(4;19)(q35;q13). PLoS One. 2014;9:e99439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pratt D, Kumar‐Sinha C, Cieślik M, Mehra R, Xiao H, Shao L, et al. A novel ATXN1‐DUX4 fusion expands the spectrum of ‘CIC‐rearranged sarcoma' of the CNS to include non‐CIC alterations. Acta Neuropathol. 2021;141:619–22. [DOI] [PubMed] [Google Scholar]
- 32. Satomi K, Ohno M, Kubo T, Honda‐Kitahara M, Matsushita Y, Ichimura K, et al. Central nervous system sarcoma with ATXN1::DUX4 fusion expands the concept of CIC‐rearranged sarcoma. Genes Chromosomes Cancer. 2022;61:683–8. [DOI] [PubMed] [Google Scholar]
- 33. Siegfried A, Masliah‐Planchon J, Roux FE, Larrieu‐Ciron D, Pierron G, Nicaise Y, et al. Brain tumor with an ATXN1‐NUTM1 fusion gene expands the histologic spectrum of NUTM1‐rearranged neoplasia. Acta Neuropathol Commun. 2019;7:220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Xu F, Viaene AN, Ruiz J, Schubert J, Wu J, Chen J, et al. Novel ATXN1/ATXN1L::NUTM2A fusions identified in aggressive infant sarcomas with gene expression and methylation patterns similar to CIC‐rearranged sarcoma. Acta Neuropathol Commun. 2022;10:102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Italiano A, Sung YS, Zhang L, Singer S, Maki RG, Coindre JM, et al. High prevalence of CIC fusion with double‐homeobox (DUX4) transcription factors in EWSR1‐negative undifferentiated small blue round cell sarcomas. Genes Chromosomes Cancer. 2012;51:207–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yoshida A, Goto K, Kodaira M, Kobayashi E, Kawamoto H, Mori T, et al. CIC‐rearranged sarcomas: a study of 20 cases and comparisons with Ewing sarcomas. Am J Surg Pathol. 2016;40:313–23. [DOI] [PubMed] [Google Scholar]
- 37. Sturm D, Orr BA, Toprak UH, Hovestadt V, Jones DTW, Capper D, et al. New brain tumor entities emerge from molecular classification of CNS‐PNETs. Cell. 2016;164:1060–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gambarotti M, Benini S, Gamberi G, Cocchi S, Palmerini E, Sbaraglia M, et al. CIC‐DUX4 fusion‐positive round‐cell sarcomas of soft tissue and bone: a single‐institution morphological and molecular analysis of seven cases. Histopathology. 2016;69:624–34. [DOI] [PubMed] [Google Scholar]
- 39. Antonescu CR, Owosho AA, Zhang L, Chen S, Deniz K, Huryn JM, et al. Sarcomas with CIC‐rearrangements are a distinct pathologic entity with aggressive outcome: a clinicopathologic and molecular study of 115 cases. Am J Surg Pathol. 2017;41:941–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yamada Y, Kuda M, Kohashi K, Yamamoto H, Takemoto J, Ishii T, et al. Histological and immunohistochemical characteristics of undifferentiated small round cell sarcomas associated with CIC‐DUX4 and BCOR‐CCNB3 fusion genes. Virchows Arch. 2017;470:373–80. [DOI] [PubMed] [Google Scholar]
- 41. Smith SC, Buehler D, Choi EYK, McHugh JB, Rubin BP, Billings SD, et al. CIC‐DUX sarcomas demonstrate frequent MYC amplification and ETS‐family transcription factor expression. Mod Pathol. 2015;28:57–68. [DOI] [PubMed] [Google Scholar]
- 42. Machado I, Yoshida A, Morales MGN, Abrahão‐Machado LF, Navarro S, Cruz J, et al. Review with novel markers facilitates precise categorization of 41 cases of diagnostically challenging, “undifferentiated small round cell tumors”. A clinicopathologic, immunophenotypic and molecular analysis. Ann Diagn Pathol. 2018;34:1–12. [DOI] [PubMed] [Google Scholar]
- 43. Hung YP, Fletcher CD, Hornick JL. Evaluation of ETV4 and WT1 expression in CIC‐rearranged sarcomas and histologic mimics. Mod Pathol. 2016;29:1324–34. [DOI] [PubMed] [Google Scholar]
- 44. Le Guellec S, Velasco V, Pérot G, Watson S, Tirode F, Coindre JM. ETV4 is a useful marker for the diagnosis of CIC‐rearranged undifferentiated round‐cell sarcomas: a study of 127 cases including mimicking lesions. Mod Pathol. 2016;29:1523–31. [DOI] [PubMed] [Google Scholar]
- 45. Specht K, Sung YS, Zhang L, Richter GHS, Fletcher CD, Antonescu CR. Distinct transcriptional signature and immunoprofile of CIC‐DUX4 fusion‐positive round cell tumors compared to EWSR1‐rearranged ewing sarcomas: further evidence toward distinct pathologic entities. Genes Chromosomes Cancer. 2014;53:622–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Siegele B, Roberts J, Black JO, Rudzinski E, Vargas SO, Galambos C. DUX4 immunohistochemistry is a highly sensitive and specific marker for CIC‐DUX4 fusion‐positive round cell tumor. Am J Surg Pathol. 2017;41:423–29. [DOI] [PubMed] [Google Scholar]
- 47. Kojima N, Arai Y, Satomi K, Kubo T, Matsushita Y, Mori T, et al. Co‐expression of ERG and CD31 in a subset of CIC‐rearranged sarcoma: a potential diagnostic pitfall. Mod Pathol. 2022;35:1439–48. [DOI] [PubMed] [Google Scholar]
- 48. Kao YC, Owosho AA, Sung YS, Zhang L, Fujisawa Y, Lee JC, et al. BCOR‐CCNB3 fusion positive sarcomas: a clinicopathologic and molecular analysis of 36 cases with comparison to morphologic spectrum and clinical behavior of other round cell sarcomas. Am J Surg Pathol. 2018;42:604–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Pierron G, Tirode F, Lucchesi C, Reynaud S, Ballet S, Cohen‐Gogo S, et al. A new subtype of bone sarcoma defined by BCOR‐CCNB3 gene fusion. Nature Genet. 2012;44:461–6. [DOI] [PubMed] [Google Scholar]
- 50. Yoshida A, Arai Y, Hama N, Chikuta H, Bando Y, Nakano S, et al. Expanding the clinicopathologic and molecular spectrum of BCOR‐associated sarcomas in adults. Histopathology. 2020;76:509–20. [DOI] [PubMed] [Google Scholar]
- 51. Kao YC, Sung YS, Argani P, Swanson D, Alaggio R, Tap W, et al. NTRK3 overexpression in undifferentiated sarcomas with YWHAE and BCOR genetic alterations. Mod Pathol. 2020;33:1341–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Specht K, Zhang L, Sung YS, Nucci M, Dry S, Vaiyapuri S, et al. Novel BCOR‐MAML3 and ZC3H7B‐BCOR gene fusions in undifferentiated small blue round cell sarcomas. Am J Surg Pathol. 2016;40:433–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Matsuyama A, Shiba E, Umekita Y, Nosaka K, Kamio T, Yanai H, et al. Clinicopathologic diversity of undifferentiated sarcoma with BCOR‐CCNB3 fusion: analysis of 11 cases with a reappraisal of the utility of immunohistochemistry for BCOR and CCNB3. Am J Surg Pathol. 2017;41:1713–21. [DOI] [PubMed] [Google Scholar]
- 54. Shibayama T, Okamoto T, Nakashima Y, Kato T, Sakurai T, Minamiguchi S, et al. Screening of BCOR‐CCNB3 sarcoma using immunohistochemistry for CCNB3: a clinicopathological report of three pediatric cases. Pathol Int. 2015;65:410–4. [DOI] [PubMed] [Google Scholar]
- 55. Puls F, Niblett A, Marland G, Gaston CLL, Douis H, Mangham DC, et al. BCOR‐CCNB3 (Ewing‐like) sarcoma: a clinicopathologic analysis of 10 cases, in comparison with conventional Ewing sarcoma. Am J Surg Pathol. 2014;38:1307–18. [DOI] [PubMed] [Google Scholar]
- 56. Kao YC, Sung YS, Zhang L, Huang SC, Argani P, Chung CT, et al. Recurrent BCOR internal tandem duplication and YWHAE‐NUTM2B fusions in soft tissue undifferentiated round cell sarcoma of infancy: overlapping genetic features with clear cell sarcoma of kidney. Am J Surg Pathol. 2016;40:1009–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Alaggio R, Ninfo V, Rosolen A, Coffin CM. Primitive myxoid mesenchymal tumor of infancy: a clinicopathologic report of 6 cases. Am J Surg Pathol. 2006;30:388–94. [DOI] [PubMed] [Google Scholar]
- 58. Li WS, Liao IC, Wen MC, Lan HHC, Yu SC, Huang HY. BCOR‐CCNB3‐positive soft tissue sarcoma with round‐cell and spindle‐cell histology: a series of four cases highlighting the pitfall of mimicking poorly differentiated synovial sarcoma. Histopathology. 2016;69:792–801. [DOI] [PubMed] [Google Scholar]
- 59. Kao YC, Sung YS, Zhang L, Jungbluth AA, Huang SC, Argani P, et al. BCOR overexpression is a highly sensitive marker in round cell sarcomas with BCOR genetic abnormalities. Am J Surg Pathol. 2016;40:1670–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Argani P, Kao YC, Zhang L, Sung YS, Alaggio R, Swanson D, et al. BCOR Overexpression in renal malignant solitary fibrous tumors: a close mimic of clear cell sarcoma of kidney. Am J Surg Pathol. 2019;43:773–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Conner JR, Hornick JL. SATB2 is a novel marker of osteoblastic differentiation in bone and soft tissue tumours. Histopathology. 2013;63:36–49. [DOI] [PubMed] [Google Scholar]
- 62. Ueno‐Yokohata H, Okita H, Nakasato K, Akimoto S, Hata J, Koshinaga T, et al. Consistent in‐frame internal tandem duplications of BCOR characterize clear cell sarcoma of the kidney. Nature Genet. 2015;47:861–3. [DOI] [PubMed] [Google Scholar]
- 63. O'Meara E, Stack D, Lee CH, Garvin AJ, Morris T, Argani P, et al. Characterization of the chromosomal translocation t(10;17)(q22;p13) in clear cell sarcoma of kidney: t(10:17)(q22;p13) in renal clear cell sarcoma. J Pathol. 2012;227:72–80. [DOI] [PubMed] [Google Scholar]
- 64. Wong MK, Ng CCY, Kuick CH, Aw SJ, Rajasegaran V, Lim JQ, et al. Clear cell sarcomas of the kidney are characterised by BCOR gene abnormalities, including exon 15 internal tandem duplications and BCOR‐CCNB3 gene fusion. Histopathology. 2018;72:320–9. [DOI] [PubMed] [Google Scholar]
- 65. Lewis N, Soslow RA, Delair DF, Park KJ, Murali R, Hollmann TJ, et al. ZC3H7B‐BCOR high‐grade endometrial stromal sarcomas: a report of 17 cases of a newly defined entity. Mod Pathol. 2018;31:674–84. [DOI] [PubMed] [Google Scholar]
- 66. Mariño‐Enriquez A, Lauria A, Przybyl J, Ng TL, Kowalewska M, Debiec‐Rychter M, et al. BCOR internal tandem duplication in high‐grade uterine sarcomas. Am J Surg Pathol. 2018;42:335–41. [DOI] [PubMed] [Google Scholar]
- 67. Lee CH, Mariño‐Enriquez A, Ou W, Zhu M, Ali RH, Chiang S, et al. The clinicopathologic features of YWHAE‐FAM22 endometrial stromal sarcomas: a histologically high‐grade and clinically aggressive tumor. Am J Surg Pathol. 2012;36:641–53. [DOI] [PubMed] [Google Scholar]
- 68. Yoshida Y, Nobusawa S, Nakata S, Nakada M, Arakawa Y, Mineharu Y, et al. CNS high‐grade neuroepithelial tumor with BCOR internal tandem duplication: a comparison with its counterparts in the kidney and soft tissue. Brain Pathol. 2018;28:710–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Szuhai K, Ijszenga M, de Jong D, Karseladze A, Tanke HJ, Hogendoorn PCW. The NFATc2 gene is involved in a novel cloned translocation in a Ewing sarcoma variant that couples its function in immunology to oncology. Clin Cancer Res. 2009;15:2259–68. [DOI] [PubMed] [Google Scholar]
- 70. Watson S, Perrin V, Guillemot D, Reynaud S, Coindre JM, Karanian M, et al. Transcriptomic definition of molecular subgroups of small round cell sarcomas. J Pathol. 2018;245:29–40. [DOI] [PubMed] [Google Scholar]
- 71. Hung YP, Fisch AS, Diaz‐Perez JA, Iafrate AJ, Lennerz JK, Nardi V, et al. Identification of EWSR1‐NFATC2 fusion in simple bone cysts. Histopathology. 2021;78:849–56. [DOI] [PubMed] [Google Scholar]
- 72. Ong SLM, Lam SW, van den Akker BEWM, Kroon HM, Briaire‐de Bruijn IH, Cleven AHG, et al. Expanding the spectrum of EWSR1‐NFATC2‐rearranged benign tumors: a common genomic abnormality in vascular malformation/hemangioma and simple bone cyst. Am J Surg Pathol. 2021;45:1669–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Pižem J, Šekoranja D, Zupan A, Boštjančič E, Matjašič A, Mavčič B, et al. FUS‐NFATC2 or EWSR1‐NFATC2 fusions are present in a large proportion of simple bone cysts. Am J Surg Pathol. 2020;44:1623–34. [DOI] [PubMed] [Google Scholar]
- 74. Diaz‐Perez JA, Nielsen GP, Antonescu C, Taylor MS, Lozano‐Calderon SA, Rosenberg AE. EWSR1/FUS ‐ NFATc2 rearranged round cell sarcoma: clinicopathological series of four cases and literature review. Hum Pathol. 2019;90:45–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Wang GY, Thomas DG, Davis JL, Ng T, Patel RM, Harms PW, et al. EWSR1‐NFATC2 translocation‐associated sarcoma clinicopathologic findings in a rare aggressive primary bone or soft tissue tumor. Am J Surg Pathol. 2019;43:1112–22. [DOI] [PubMed] [Google Scholar]
- 76. Yoshida K, Machado I, Motoi T, Parafioriti A, Lacambra M, Ichikawa H, et al. NKX3‐1 Is a useful immunohistochemical marker of EWSR1‐NFATC2 sarcoma and mesenchymal chondrosarcoma. Am J Surg Pathol. 2020;44:719–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Makise N, Yoshida KI, Iijima T, Yoshida A, Ushiku T, Ishida T. Skeletal EWSR1‐NFATC2 sarcoma previously diagnosed as Ewing‐like adamantinoma: a case report and literature review emphasizing its unique radiological features. Pathol Int. 2021;71:614–20. [DOI] [PubMed] [Google Scholar]
- 78. Perret R, Escuriol J, Velasco V, Mayeur L, Soubeyran I, Delfour C, et al. NFATc2‐rearranged sarcomas: clinicopathologic, molecular, and cytogenetic study of 7 cases with evidence of AGGRECAN as a novel diagnostic marker. Mod Pathol. 2020;33:1930–44. [DOI] [PubMed] [Google Scholar]
- 79. Yoshida A, Hashimoto T, Ryo E, Yoshida K, Motoi T, Yatabe Y, et al. Confirmation of NKX3‐1 expression in EWSR1‐NFATC2 sarcoma and mesenchymal chondrosarcoma using monoclonal antibody immunohistochemistry, RT‐PCR, and RNA in situ hybridization. Am J Surg Pathol. 2021;45:578–82. [DOI] [PubMed] [Google Scholar]
- 80. Gurel B, Ali TZ, Montgomery EA, Begum S, Hicks J, Goggins M, et al. NKX3.1 as a marker of prostatic origin in metastatic tumors. Am J Surg Pathol. 2010;34:1097–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Ramanayake N, Vargas AC, Talbot J, Bonar F, Wong DD, Wong D, et al. NKX3.1 immunohistochemistry is highly specific for the diagnosis of mesenchymal chondrosarcomas: experience in the Australian population. Pathology. 2021;53:705–12. [DOI] [PubMed] [Google Scholar]
- 82. Syed M, Mushtaq S, Loya A, Hassan U. NKX3.1 a useful marker for mesenchymal chondrosarcoma: an immunohistochemical study. Ann Diagn Pathol. 2021;50:151660. [DOI] [PubMed] [Google Scholar]
- 83. Lefebvre V, Smits P. Transcriptional control of chondrocyte fate and differentiation. Birth Defects Res C Embryo Today. 2005;75:200–12. [DOI] [PubMed] [Google Scholar]
- 84. Bridge JA, Sumegi J, Druta M, Bui MM, Henderson‐Jackson E, Linos K, et al. Clinical, pathological, and genomic features of EWSR1‐PATZ1 fusion sarcoma. Mod Pathol. 2019;32:1593–604. [DOI] [PubMed] [Google Scholar]
- 85. Chougule A, Taylor MS, Nardi V, Chebib I, Cote GM, Choy E, et al. Spindle and round cell sarcoma with EWSR1‐PATZ1 gene fusion: a sarcoma with polyphenotypic differentiation. Am J Surg Pathol. 2019;43:220–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Michal M, Rubin BP, Agaimy A, Kosemehmetoglu K, Rudzinski ER, Linos K, et al. EWSR1‐PATZ1‐rearranged sarcoma: a report of nine cases of spindle and round cell neoplasms with predilection for thoracoabdominal soft tissues and frequent expression of neural and skeletal muscle markers. Mod Pathol. 2021;34:770–85. [DOI] [PubMed] [Google Scholar]
- 87. Alholle A, Karanian M, Brini AT, Morris MR, Kannappan V, Niada S, et al. Genetic analyses of undifferentiated small round cell sarcoma identifies a novel sarcoma subtype with a recurrent CRTC1‐SS18 gene fusion. J Pathol. 2018;245:186–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Pan R, Wang Z, Wang X, Fang R, Xia Q, Rao Q. CRTC1‐SS18 fusion sarcoma with aberrant anaplastic lymphoma kinase expression. Int J Surg Pathol. 2022;30:99–105. [DOI] [PubMed] [Google Scholar]
- 89. Antonescu CR, Agaram NP, Sung YS, Zhang L, Dickson BC. Undifferentiated round cell sarcomas with novel SS18‐POU5F1 fusions. Genes Chromosomes Cancer. 2020;59:620–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Argani P, Matoso A, Gross JM, Zhang Y, SoRelle JA, Gagan J, et al. Primary renal sarcoma with SS18::POU5F1 gene fusion. Genes Chromosomes Cancer. 2022;61:572–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Shenoy A, Newsom K, Gray B, Zhang Y, Lagmay JP, Islam S, et al. Malignant round cell tumor with SS18‐POU5F1 fusion: is it a myoepithelial neoplasm, a synovial sarcoma or a new entity? Histopathology. 2020;77:681–84. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information.