

Abstract
Patients with severe COVID‐19 often suffer from lymphopenia, which is linked to T‐cell sequestration, cytokine storm, and mortality. However, it remains largely unknown how severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) induces lymphopenia. Here, we studied the transcriptomic profile and epigenomic alterations involved in cytokine production by SARS‐CoV‐2‐infected cells. We adopted a reverse time‐order gene coexpression network approach to analyze time‐series RNA‐sequencing data, revealing epigenetic modifications at the late stage of viral egress. Furthermore, we identified SARS‐CoV‐2‐activated nuclear factor‐κB (NF‐κB) and interferon regulatory factor 1 (IRF1) pathways contributing to viral infection and COVID‐19 severity through epigenetic analysis of H3K4me3 chromatin immunoprecipitation sequencing. Cross‐referencing our transcriptomic and epigenomic data sets revealed that coupling NF‐κB and IRF1 pathways mediate programmed death ligand‐1 (PD‐L1) immunosuppressive programs. Interestingly, we observed higher PD‐L1 expression in Omicron‐infected cells than SARS‐CoV‐2 infected cells. Blocking PD‐L1 at an early stage of virally‐infected AAV‐hACE2 mice significantly recovered lymphocyte counts and lowered inflammatory cytokine levels. Our findings indicate that targeting the SARS‐CoV‐2‐mediated NF‐κB and IRF1‐PD‐L1 axis may represent an alternative strategy to reduce COVID‐19 severity.
Keywords: ChIP‐sequencing, lymphopenia, PD‐L1, SARS‐CoV‐2, time‐series transcriptomics
1. INTRODUCTION
The emergence of coronavirus disease‐2019 (COVID‐19), caused by severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) that infects and damages multiple organs such as lung, kidney, and gastrointestinal tissues, poses a considerable threat to human health and public safety worldwide. 1 , 2 To understand the disease pathogenesis of SARS‐CoV‐2, various genome‐wide analyses have been conducted to characterize interactions between the virus and host cells, such as African green monkey kidney (Vero E6) cells, 3 , 4 human lung adenocarcinoma (LUAD) Calu‐1 cells, 5 and human colon organoids. 6 Although recent studies have explored host responses to SARS‐CoV‐2 infection by means of transcriptome analyses, the transcriptional dynamics of host cells in response to viral infection, from entry to egress, have remained largely unexplored.
Lymphopenia, a medical condition defined by abnormally low counts of lymphocytes, is a major immunological abnormality in patients with severe COVID‐19. 7 Angiotensin‐converting enzyme‐2 (ACE‐2) has been identified as the receptor for SARS‐CoV‐2 viral entry. 8 Increased expression of ACE2 in lung epithelia may contribute to increased disease susceptibility of COVID‐19 patients. 9 Interestingly, ACE2 is tightly regulated by DNA hypomethylation in lung epithelium, 9 suggesting that epigenetic regulation may involve viral infection. Indeed, emerging evidence suggests that viruses may evolve to hijack the host epigenetic machinery for viral entry, replication, or pathogenesis. 10 For example, SARS‐CoV‐2‐induced DNA hypomethylation of genes encoding transmembrane serine protease 2 (TMPRSS2) and nuclear factor‐κB (NF‐κB) may promote viral entry and excessive immune responses, respectively. 11 , 12 Reversible epigenetic regulatory mechanisms that include DNA methylation, histone modification, and chromatin remodeling play a crucial role in regulating gene expression programs. 13 , 14 Genome‐wide sequencing technologies, such as chromatin immunoprecipitation sequencing (ChIP‐seq), have tremendously advanced our understanding of how global histone modifications control gene expression. 15 For example, histone H3 lysine 4 trimethylation (H3K4me3) at promoter regions has been identified as a hallmark of actively transcribed genes. 16 However, as yet, it remains very few ChIP‐seq studies for COVID‐19 and it is still unclear how SARS‐CoV‐2 commandeers the host epigenetic machinery to affect cellular responses via histone modifications.
Large‐scale retrospective studies have shown that hyperinflammation and uncontrolled immune responses are features of severe COVID‐19. 17 , 18 Overproduction of cytokines that can cause lung damage, such as interleukin‐1β (IL‐1β), IL‐6, interferon‐γ (IFN‐γ), tumor necrosis factor‐α (TNF‐α), or transforming growth factor‐β (TGF‐β), may serve as biomarkers of SARS‐CoV‐2 infection. 19 , 20 , 21 , 22 A recent study reported that programmed cell death protein 1 (PD‐1) and T cell immunoglobulin mucin‐3 (Tim‐3), markers of T cell exhaustion, were highly expressed on the surface of CD4+ and CD8+ T cells in COVID‐19 patients, contributing to SARS‐CoV‐2 induced sepsis and death. 23
Increased levels of the ligand of PD‐1, programmed death ligand‐1 (PD‐L1), have been observed in numerous solid tumors (e.g., lung cancers, head and neck carcinoma, and melanoma), as well as in the white blood cells (e.g., monocytes, basophils, eosinophils, and natural killer cells) of patients suffering severe COVID‐19, indicating that elevated PD‐L1 may be associated with cytokine storm and disease severity. 24 , 25 , 26 , 27 , 28 , 29 Although it has been reported that the interaction of PD‐1 and PD‐L1 upon T cells encountering tumor cells triggers signal transduction to induce T‐cell exhaustion, 30 the underlying mechanisms of SARS‐CoV‐2 upregulate PD‐L1 in cancer cells are still poorly understood.
2. MATERIALS AND METHODS
2.1. Cell culture, stable transfectants, and transfection
Caco‐2, HEK293T (RRID:CVCL_YA60), and A549 cells (RRID:CVCL_0023) were obtained from the American Type Culture Collection, and Vero E6 cell (RRID:CVCL_0574) was received from Dr. Yi‐Ling Lin's Lab (Institute of Biomedical Sciences [IBMS], Academia Sinica). All cells were confirmed by STR genotyping. These cells were grown in DMEM/F12 with 10% fetal bovine serum (FBS). To generate an A549‐hACE2 stably expressing cell line, we infected A549 with lentivirus produced from transfected HEK293T cells with pCMV‐dR8.2 dvpr (RRID:Addgene_8455), pCMV‐VSV‐G (RRID:Addgene_8454), and pLAS2w‐ACE2 plasmids via the calcium phosphate transfection method.
2.2. Virus isolation and infection
Patient‐derived SARS‐CoV‐2 (TCDC#4) was isolated by the Centers for Disease Control (CDC) in Taiwan. The virus was propagated and amplified in Vero E6 cells in MEM supplemented with 2% FBS at 37°C and 5% CO2. Target cells were infected with SARS‐CoV‐2 at an MOI of 0.1. MOI was determined by comparing the cell counts at various viral medium concentrations. Caco‐2, A549‐hACE2, and Vero E6 cells were infected with SARS‐CoV‐2 at the indicated time. All procedures following Taiwan CDC laboratory biosafety guidelines were conducted in a biosafety Level‐3 facility in the IBMS, Academia Sinica.
2.3. RNA‐sequencing read processing and data analysis
Low‐quality bases and reads were removed using Trimmomatic (v.0.39). The processed paired‐end reads were mapped to the human genome (GRCh38) and SARS‐CoV‐2 genome (NC_045512.2) using Hisat2 (v.2.1.0). The expression level (fragments per kilobase of transcript per million mapped fragments [FPKM]) of each gene was estimated using Stringtie (v.2.0.2b). Nuclear protein‐coding genes with FPKM ≥ 1 in at least one sample were selected for further analysis. The upper‐quartile normalization procedure was applied to compare the FPKMs of the chosen genes across samples in a set of transcriptomes. Expression levels of selected genes from all samples were used to run the principal component analysis (PCA) and K‐means clustering using MORPHEUS (https://software.broadinstitute.org/morpheus). NOISeq 31 R package with q = 0.999 (differentially expression probability) was used to identify differentially expressed genes (DEGs) between any pair of conditions. To determine the enriched pathways for upregulated and downregulated DEGs, both the over‐representation analysis and the gene set enrichment analysis (GSEA) were applied. The correlation analysis in STAT1, PD‐L1, and Tim‐3 was analyzed by the Timer2 database (https://pubmed.ncbi.nlm.nih.gov/32442275/). Time‐ordered gene coexpression network (TO‐GCN) was analyzed by previous description. 32 Prediction of infiltration levels for immune cell types was using the ssGSEA 33 implementation in the R package gene set variation analysis (GSVA). 34 ssGSEA is a rank‐based method that computes an overexpression measure for a gene list of interest relative to all other genes in the genome. The normalized RNA‐sequencing dataset was used as input to analyze GSVA enrichment scores.
2.4. Real‐time quantitative polymerase chain reaction (RT‐qPCR)
Total RNA was extracted using Quick‐RNA Miniprep Kit (R1055; Zymo Research). The cDNA was prepared by ToolsQuant II Fast RT Kit (KRT‐BA06‐2; Biotools) according to the manufacturer's protocol. RT‐qPCR was performed using EvaGreen master mix (Bio‐Rad) with cDNA serving as a template. The reactions were carried out in a Bio‐Rad CFX Connect system. The primers used are as follows:
PD‐L1_F: 5ʹ‐ACCTGGCTGCACTAATTGTCT‐3ʹ;
PD‐L1_R: 5ʹ‐GGGAGAGCTGGTCCTTCAAC‐3ʹ;
mouse PD‐L1_F: 5ʹ‐TGCGGACTACAAGCGAATCACG‐3ʹ;
mouse PD‐L1_R: 5ʹ‐CTCAGCTTCTGGATAACCCTCG‐3ʹ.
2.5. ChIP assays
The chromatin samples were prepared from Caco‐2 and A549‐hACE2 cells with a ChIP‐IT Express kit (Active Motif). The chromatin samples (50 µl) were incubated with NF‐κB (39369; Active Motif) or interferon regulatory factor 1 (IRF1) (8478; Cell Signaling) (5 µg) for 12 h at 4°C. Primers used to detect the PD‐L1, TNF‐α, or IFN‐γ promoter regions are as follows:
PD‐L1_F: 5ʹ‐GCTTTATTCCTAGGACACCAACA‐3ʹ;
PD‐L1_R: 5ʹ‐TCCTGACCTTCGGTGAAATC‐3ʹ.
TNF‐α_F: 5ʹ‐CACAGCTTTTCCCTCCAACC‐3ʹ;
TNF‐α_R: 5ʹ‐CTAGAACTGGGAGGGGCTTC‐3ʹ.
IFN‐γ_F: 5ʹ‐CTCCTCTGGCTGCTGGTATT‐3ʹ;
IFN‐γ_R: 5ʹ‐CCCTGCCTATCTGTCACCAT‐3ʹ.
2.6. Library preparation
ChIP DNA Quantification and Qualification were monitored by Agilent 2100 Bioanalyzer. A total amount of 30 ng ChIP DNA (fragments ranging from 100 to 500 bp) per sample was used as input material for the library preparations. Sequencing library was generated using ThruPLEX® DNA‐seq Kit (Catalog #: R400407; Rubicon Genomics) following the manufacturer's recommendations, and index codes were added to each sample. Briefly, DNA fragments were end polished, A‐tailed, and ligated with the full‐length adapter for Illumina sequencing, followed by further PCR amplification. After PCR products were purified (AMPure XP system), libraries were analyzed for size distribution on 1% agarose gels and quantified by real‐time PCR (3 nM). According to the manufacturer's instructions, the clustering of the index‐coded samples was performed on a cBot Cluster Generation System using Novaseq5000/6000 S4 Reagent Kit (Illumina). After cluster generation, the DNA libraries were sequenced on Illumina NovaSeq 6000 platform, and 150 bp paired‐end reads were generated.
2.7. Pseudovirus infection assay
SARS‐CoV‐2 wild‐type and Omicron variants spike‐pseudotyped lentivirus purchased from National RNAi Core Facility. For pseudovirus infection, The pseudovirus infection assay was conducted as previously described. 35
2.8. Promoter‐luciferase reporter activity of PD‐L1
The reporter gene constructs for PD‐L1 (−373/+328) were generated by PCR amplification. The PCR products were purified with a high PCR product purification kit (GeneJET Plasmid Miniprep kit; Thermo Fisher Scientific) and cloned into the pGL3‐basic firefly luciferase vector (Promega). The sequence of each cloned promoter region was confirmed by sequencing (Applied Biosystems 3730XL system). For reporter assays, cells were cotransfected with the firefly luciferase construct pRL‐SV40 Renilla vector (Promega) and the PD‐L1 promoter construct vector using X‐tremeGENE HP DNA Transfection Reagent (6366236001; Roche). After 6 h, cells were treated with 10 ng/ml TGF‐β (PHG9214; Fisher Scientific), 10 ng/ml IL‐1β (PHC0811; Fisher Scientific), 10 ng/ml IL‐6 (PHC0061; Fisher Scientific), 10 ng/ml EGF (PHG0311, Fisher Scientific), 10 ng/ml IFN‐α (PHC4014, Fisher Scientific), 10 ng/ml IFN‐β (PHC4244, Fisher Scientific), 10 ng/ml IFN‐γ (PHC4031, Fisher Scientific), or 10 ng/ml TNF‐α (PHC3015, Fisher Scientific). Then after 24 h, luciferase activity was measured by using the Dual‐Luciferase Reporter Assay System kit (Promega). A549‐hACE2 cells were transfected with PD‐L1 promoter plasmid expressing luciferase. After that, A549‐hACE2 cells were incubated with SARS‐CoV‐2 wild‐type, and Omicron variants spike‐pseudotyped lentivirus for 48 h followed by reporter assays.
2.9. Western blotting
Cells were lysed with RIPA lysis buffer (50 mM Tris‐HCl, pH 7.6, 150 mM NaCl, 1 mM EDTA, 1% NP‐40, 1% sodium deoxycholate, 0.1% sodium dodecyl sulfate) with protease and phosphatase inhibitors (4693132001; Merck). Western blotting was performed as described previously. 36 , 37 Proteins of interest were studied by hybridizing with α‐tubulin (1:5000; B‐5‐1‐2; Sigma‐Aldrich), or PD‐L1 antibody (1:2000; 104763; GeneTex) overnight incubation at 4°C, then with horseradish peroxidase (HRP)‐conjugated Goat anti‐Mouse IgG (1:2000; ab6789; Abcam), or HRP‐conjugated Goat anti‐Rabbit IgG (1:2000; ab6721; Abcam) secondary antibodies for 1 h. Image acquisition was performed using a UVP BioSpectrum® 500 Imaging System (Ultra‐Violet Products Ltd.).
2.10. Flow cytometry
The PD‐L1 expression in A549 cells was determined by flow cytometry. A549 cells were pretreated with indole‐3‐carbinol (I3C) (S2313; Selleckchem) and Ruxolitinib (S1378; Selleckchem) for 1 h, following by treating with 10 ng/ml TNF‐α and 10 ng/ml IFN‐γ for 48 h. Cells were dispersed and resuspended in FACS buffer followed by incubation with PD‐L1 antibody (104763; GeneTex) for 1 h. After that, PD‐L1 antibody was captured by donkey anti‐Rabbit IgG‐Alexa Fluor 488 (A21206; Thermo Fisher Scientific). The stained cells were analyzed using an LSR II cytometer, and data were processed using FlowJo V10 software.
2.11. Adeno‐associated virus (AAV) infection in mice
AAV6‐mock or AAV6/CB‐hACE2 was produced by AAV core facility in Academia Sinica. Eight to ten weeks old C57BL/6J mice were anesthetized by intraperitoneal injection of a mixture of atropine (0.4 mg/ml)/ketamine (20 mg/ml)/xylazine (0.4%). Mice were then intratracheally injected with 3 × 1011 vg of AAV6‐mock or AAV6/hACE2 in 100 μl saline as previously described. 38 The AAV‐expressing human ACE2 (hACE2) mice were pretreated with anti‐PD‐L1 antibody (100 µg) one day before and after SARS‐CoV‐2 infection. The body weight was measured every day, and lung and PBMC were collected at Day 5 after the SARS‐CoV‐2 challenge.
2.12. Sample preparation for mass cytometry (CyTOF)
Cell lines were trypsinized at a concentration of 3 × 106 cells/ml and stained with a final concentration of 25 μM cisplatin (Catalog #: 201064; Fluidigm) for 1 min at room temperature and subsequently quenched by completed medium (RPMI contained 10% FBS). After fixation with 1.5% paraformaldehyde (Catalog #: 50‐980‐487; Thermo Fisher Scientific [Electron Microscopy Science]) in phosphate‐buffered saline (PBS) for 10 min at room temperature, cells were barcoded by using a Cell‐ID 20‐plex Palladium (Pd) barcoding Kit (Catalog #: 201060; Fluidigm) according to the manufacturer's instructions. For the surface marker staining, barcoded cells were mixed and stained with a surface antibody cocktail for 1 h at room temperature and washed with cell staining media (0.5% bovine serum albumin in PBS). After permeabilization by methanol for 10 min on ice, cells were washed twice with cell staining media and then stained with an intracellular antibody cocktail for 1 h at room temperature. Cells were then fixed overnight with 1.5% paraformaldehyde in PBS containing Cell‐ID™ Intercalator‐Ir. Cells were washed once with cell staining media and twice with ddH2O Before introduction into the CyTOF2 mass cytometer together with EQ™ Four Element Calibration Beads (Catalog #: 201078; Fluidigm). Mass cytometry analyses were performed by the GRC Mass Core Facility of Genomics Research Center, Academia Sinica, Taipei, Taiwan.
2.13. Immunohistochemical staining and histopathological scoring
After mouse blood samples were taken from heart, the left lung of mouse was isolated and fixed in 4% paraformaldehyde. Lung tissue sections were deparaffinized with xylene, rehydrated in graded ethanol, and rinsed in distilled water. For the histological study, tissue sections were stained with hematoxylin and eosin (H&E). The immunohistochemical staining experiments were conducted as previously described. 35 After staining, slides were counterstained in hematoxylin, dehydrated in graded ethanol, clarified in xylene, and coverslipped. Images were taken with an Olympus BX51 microscope (Olympus). The lung section was evaluated with a lung histopathological scoring system. The section was divided into nine areas and numbered. The average scores of these nine areas are used to represent the score of the animal.
2.14. Study subjects
The COVID‐19 study was a secondary analysis of data prospectively collected from The BioRepository of Neurological Disorders at UTHealth, Department of Neurology. A quantitative observational design was used. The samples were collected from subjects admitted to the Memorial Hermann Hospital in Houston, Texas, from June 2018 to September 2020. We recruited 161 patients (72 female, 89 male; average: age 55.3 years, range: 19–101 years) with confirmed COVID‐19. Sixty non‐COVID‐19 patient controls were recruited from the cardiac clinic of the same hospital, including 33 (55.0%) females and 27 males (45.0%), with an average age of 60.5 years (range: 21–96). The plasma samples were collected from controls and COVID‐19 subjects. PD‐L1 levels were measured by ELISA kit (ab214565; Abcam). The human sample collection was approved by the Protection of Human Subjects (CPHS) at the University of Texas Health Science Center at Houston's Institutional Review Board IRB #HSC‐MS‐17‐0452. Experiments and analysis on obtained samples for the current study were performed under IRB #HSC‐MS‐20‐1058.
2.15. Human COVID‐19 lung tissues and immunohistochemistry (IHC)
Lung samples were obtained from tissue blocks of autopsies consented for by the decedent's legal next of kin. Autopsy consents included research permission and were collected under IRB 39881. All the tissue samples were fixed in formalin and embedded in paraffin. The primary antibodies for IHC staining were: anti‐SARS‐CoV‐2 N protein Ab (SinoBiological; 40143‐R019; 1:50); anti‐IFNγ Ab (Novus Biologicals; NBP2‐66900; 1:50); and anti‐PDL1 Ab (Abcam; ab237726; 1:100). As previous study described, 39 we used the color deconvolution plugin for ImageJ analysis to quantify the specific protein expression levels of IHC staining. Each section was divided into three fields. The stained area was divided by the total area in each field. The percentage of the stained area was counted in three fields in each section to obtain the percentage of stained cells by an investigator blinded to the sample condition. Spearman correlation analyses were conducted to compare the results of a total of five lung samples.
2.16. Statistical analysis
Data from individual experiments are presented as mean ± SD (standard deviation) and assessed by one‐way or two‐way analysis of variance with Tukey's post hoc test for multiple comparisons (GraphPad Prism Software Inc.). A p < 0.05 was considered statistically significant.
3. RESULTS
3.1. Time‐series transcriptomics reveals that SARS‐CoV‐2 induces the IFN−STAT1−IRF1 axis to activate gene expression in host cells
To determine the transcriptional dynamics of host cell responses to SARS‐CoV‐2 infection, we performed a time‐series transcriptomic analysis (0–48 h) on human colorectal adenocarcinoma Caco‐2 cells infected with SARS‐CoV‐2 (MOI of 0.1) (Figure 1A). A PCA revealed that SARS‐CoV‐2 infected Caco‐2 cells displayed a unique time‐course transcriptomic profile (Figure 1B). Next, we evaluated the expression levels of viral transcripts in infected Caco‐2 cells according to four viral structural protein‐encoding genes (N, E, M, and S) and seven accessory protein‐encoding genes (ORF1ab, ORF3a, ORF6, ORF7a, ORF7b, ORF8, and ORF10). We found that all of these viral genes were expressed in a highly similar pattern in infected host cells, with a 100‐fold increase being observed at 48 h postinfection (h.p.i.) (representing a log2 expression level difference of ~7). Based on expression levels of SARS‐CoV‐2 transcripts at different periods of viral infection, we defined the time course stages of 1–6, 6–24, and 24–48 h has reflecting viral entry, viral replication, and viral egress, respectively (Figure 1C).
Figure 1.
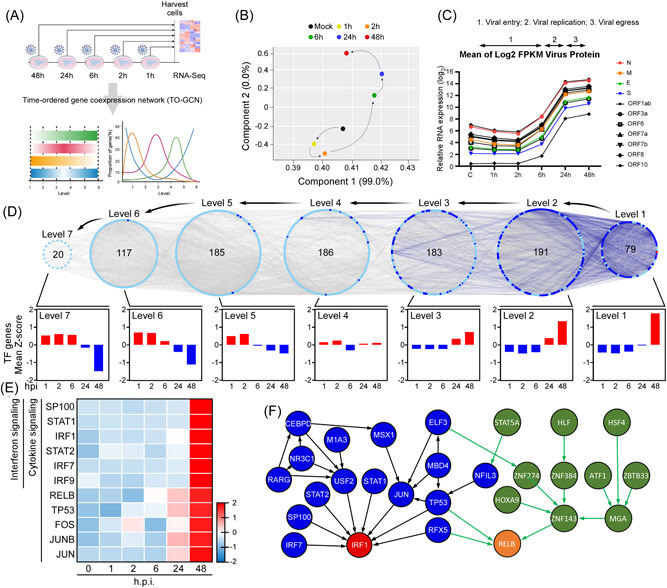
Reverse time‐ordered gene coexpression network (TO‐GCN) revealed IRF1‐ and RELB‐mediated activation of interferons and cytokines signaling. (A) Schematic flowchart of RNA‐sequencing analysis. (B) Principal‐component analysis of RNA‐sequencing results of indicated viral infection time. (C) Expression profiles of viral protein‐coding genes. (D) Reverse TO‐GCN of expressed transcription factor (TF) genes. Each node and edge represent a TF gene and the coexpression relationship between two TF genes, respectively. The numbers in the center of the level represent the number of TF genes in the level. The diagrams of average Z‐scores for TF genes in each level were displayed at the bottom of the figure. (E) Heatmap for expression profiles of TF genes involved in interferon (top six genes) or cytokine signaling pathways. (F) The network analysis for TFs based on TO‐GCN of expressed TF genes. IRF1, interferon regulatory factor 1.
To determine which specific biological processes are modulated by SARS‐CoV‐2 infection, we conducted a pathway enrichment study to analyze DEGs in host cells in response to the three different stages of viral infection. During viral entry (1–6 h), too few DEGs were identified to determine any significant enrichment pathways (data not shown). Consistent with elevated expression of SARS‐CoV‐2 viral transcripts at 6–24 h.p.i. (Figure 1C), we identified significant upregulation of pathways related to viral messenger RNA (mRNA) transcription and translation, eukaryotic translation, and regulation of mitotic cell cycle (Supporting Information: Table S1), supporting that SARS‐CoV‐2 is actively replicating in this timeframe. Interestingly, pathways related to antimicrobial peptides and neutrophil degranulation were significantly downregulated at this stage, indicating that SARS‐CoV‐2 also affects the immune responses of host cells (Supporting Information: Table S1). At the late stage of viral infection (24–48 h.p.i.), we identified induction of pathways associated with the interferon‐stimulated gene 15 (ISG15) antiviral mechanism, cytokine signaling in the immune system, type I interferons (IFN‐α/β) and type II interferon (IFN‐γ), though many other cellular biological processes were suppressed (Supporting Information: Table S1). Thus, SARS‐CoV‐2 may cause severe cellular damage at the stage of viral egress by inducing secretion of type I and type II IFNs and promoting the expression of interferon‐stimulated genes (ISGs).
To understand which biological processes are affected during viral entry (1–6 h), which could not be identified by the DEG analysis, we applied the reverse TO‐GCN method. 32 TO‐GCN allows for the relationships among transcription factor (TF) genes to be arranged into appropriate chronological order according to their coexpression patterns (Figure 1D). All TF genes significantly upregulated at the last time‐point (i.e., 48 h) were designated as the first level (L1). TF genes coexpressed in consecutive levels reflect TF genes upregulated in similar chronological orders, and TF genes at higher levels are upregulated earlier than TF genes in lower levels. 32 Based on this concept, we assigned seven levels (L1–L7) for all TF genes in the TO‐GCN (Figure 1D). Cross‐referencing non‐TF genes coexpressed with the TF genes upregulated from L1–L7, we identified pathways enriched in precisely one level or across multiple levels that represent comprehensive cell responses to SARS‐CoV‐2 infection (Supporting Information: Figure S1A). We found that genes coexpressed in L5–L7 were involved in virus entry (1–6 h), including genes active in cell junction organization, cell–cell communication, cell‐extracellular matrix interaction, basigin interaction, membrane trafficking, and vesicle‐mediated transport (pathway Nos. 2–4, 15, 53, and 54 in Supporting Information: Figure S1B). Consistent with our DEG data for 24–48 h.p.i., interferon and cytokine signaling pathways were also enriched at L1 (pathway Nos. 128 and 131–133 in Supporting Information: Figure S1B). Moreover, genes linked to mRNA processing (pathway Nos. 126, 134, and 139) and senescence (pathway Nos. 136 and 141) were detected in L1 of our TO‐GCN analysis but not in DEGs analysis. Intriguingly, many pathways related to epigenetic regulation were observed for levels L1 and L2 (pathway Nos. 110–112, 118, 123, 127, 129, 137, and 138), indicating that epigenetic modification may be involved in modulating late‐stage SARS‐CoV‐2 infection‐induced cell responses, such as exerted by interferon and cytokine signaling pathways.
To assess which TFs may involve in the modulation of the host immune system in response to SARS‐CoV‐2 infection, we explored the TF genes identified in levels L1 and L2 and noted 11 genes involved in cytokine and interferon signaling (Figure 1E). We constructed a respective regulatory network by integrating the coexpression time‐order and TF target gene pairs from a previously published Encode ChIP‐seq data set 40 (Figure 1F). Our gene regulatory network revealed that STAT1, STAT2, and other TFs may act as the upstream of IRF1 and that the zinc‐finger protein ZNF143 may contribute to activation of RELB in NF‐κB signaling. The NF‐κB family of inducible TFs (which includes RELB) and IRF1 are known to be activated in response to a variety of stimuli, including the TNF protein family and IFNs. 41 Interestingly, we also found that the tumor suppressor p53 (TP53) may mediate cytokine‐ and IFN‐induced cellular signaling by activating RELB and IRF1 (Figure 1F). Notably, pathways related to interferon signaling, hypercytokinemia in the pathogenesis of influenza, and activation of IRF by cytosolic pattern recognition receptors were activated upon SARS‐CoV‐2 infection at 48 h.p.i. (Supporting Information: Figure S1C). Together, these findings indicate that SARS‐CoV‐2 provokes cytokine‐ and IFN‐mediated IRF1 and NF‐κB/RELB signaling via several key TFs at the late stage of the viral lifecycle.
3.2. SARS‐CoV‐2 epigenetically triggers the NF‐κB/IFN‐γ‐mediated STAT1−IRF1−NOS2 axis
Our reverse TO‐GCN results have revealed that epigenetic regulation might be involved in the late stage of SARS‐CoV‐2 infection. To gain insights into the epigenetic activation elicited by SARS‐CoV‐2, we examined the genomic enrichment of H3K4me3, an epigenetic mark often observed at active promoters and that is commonly used to monitor dynamic changes in transcriptional activation. 16 Interestingly, we uncovered a 1.2‐fold increase in H3K4me3 around transcriptional start sites (TSS) at 48 h.p.i. (Figure 2A; Supporting Information: Figure S2A), implying that SARS‐CoV‐2 induces a subset of genes by means of epigenetic activation. More specifically, we identified 8925 regions across the entire genome displaying enhanced H3K4me3 (Supporting Information: Figure S2B). Gene ontology analysis on those regions uncovered the top 10 most enriched pathways (Figure 2B). Consistent with our transcriptomic results (Figure 1), antiviral interferon (IFN‐γ signaling), antiviral mechanism by IFN‐stimulated genes or ISG15, and interleukin‐12 (IL‐12) cytokine signaling were all identified as being induced upon viral infection. Interestingly, SARS‐CoV‐2 induced H3K4me3 enrichment in promoter regions of most TF genes at levels L1 and L2 (Figure 2C). Moreover, as shown in Figure 2D, elevated levels of H3K4me3 were found specifically in the regulatory regions of TP53 and RFX5 upon viral infection. We also examined another activating histone mark “H3K27ac” upon SARS‐CoV‐2 infection for 48 h. In contrast to a significant number of H3K4me3 peaks (8651 peaks, ~50%) was upregulated upon SARS‐CoV‐2 infection, only very few H3K27ac peaks (1280 peaks, <6%) could be found to increase upon SARS‐CoV‐2 infection (Supporting Information: Figure S2B). Consistent with these observations, we could only detect that the expression levels of H3K4 methylase (KMT2E) but not H3K27 acetyltransferase (CITED4 or CBP/P300) 42 were significantly induced after SARS‐CoV‐2 infection (data not shown). This suggests that SARS‐CoV‐2 may mainly mediate H3K4 methylation for gene activation in host cells. In accordance with this view, we mainly focused on H3K4me3 and did not include H3K27ac for SARS‐CoV‐2‐affected gene regulation.
Figure 2.
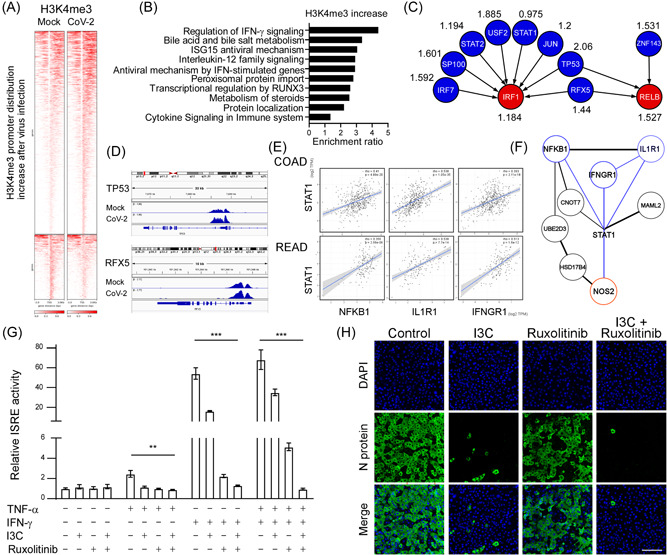
SARS‐CoV‐2 epigenetic activates NF‐κB/IFN‐γ mediated cytokine production and viral infectivity. (A) Heatmap of H3K4me3 ChIP‐sequencing density in a ±3 kb transcription start site region around the summit of each differentially accessible peak in control and virally infected groups in Caco‐2 cell line. (B) Top 10 gene ontology analysis of biological pathways of genes with increased H3K4me3 using WebGestalt (WEB‐based Gene Set Analysis Toolkit) (http://www.webgestalt.org/). (C) The normalized highest H3K4me3 tag intensity in TO‐GCN L1 genes (IRF1 and RELB) and L2 genes (IRF7, SP100, STAT2, USF2, STAT1, JUN, TP53, RFX5, and ZNF143) near promoter regions after being infected with SARS‐CoV‐2 for 48 h compared with mock. The higher normalized value, the more H3K4me3 bind to the promoter region after SARS‐CoV‐2 infection. (D) The Integrative Genomics Viewer browser of H3K4me3 occupancy in TP53, and RFX5 after being infected with SARS‐CoV‐2 for 48 h. (E) Spearman correlation analysis of STAT1 with NFKB1, ILIR1, and IFNGR1 in The Cancer Genome Atlas colon cancer (COAD; n = 460 and rectum cancer data set (READ; n = 171). (F) STRING protein‐protein interaction network showing NFKB1, ILIR1, and IFNGR1 were upstream STAT1/NOS2 axis genes. (G) Luciferase activity fold induction in HEK293T cells transfected with the ISRE reporter plasmid pretreated with 10 μM in I3C, or 4 μM Ruxolitinib for 1 h, then treated with 10 ng/ml human TNF‐α, or 10 ng/ml IFN‐γ for 48 h. Statistical method: two‐way analysis of variance, Tukey post hoc tests, **p < 0.01, ***p < 0.001. (H) Representative immunofluorescence images of the NF‐κB inhibitor (I3C; 10 μM) and the IFN‐γ inhibitor (Ruxolitinib; 4 μM) affect the expression of viral N protein after SARS‐CoV‐2 infection for 48 h in Vero E6 cells. Scale bar = 100 μm. ChIP, chromatin immunoprecipitation; IFN‐γ, interferon‐γ; IRF1, interferon regulatory factor 1; I3C, indole‐3‐carbinol; NF‐κB, nuclear factor‐κB; SARS‐CoV‐2, severe acute respiratory syndrome coronavirus 2; TNF‐α, tumor necrosis factor‐α; TO‐GCN, time‐ordered gene coexpression network.
Synergism between IFN‐γ and TNF‐α signaling upon SARS‐CoV‐2 infection promotes inflammatory cell death, that is, PANoptosis (Pyroptosis, Apoptosis, and Necrosis), through the STAT1−IRF1−NOS2 axis, leading to cytokine storm and massive cell death. 43 To investigate if SARS‐CoV‐2 triggers epigenetic regulation to modulate the IFN‐γ and TNF‐α signaling pathways, we conducted STRING protein‐protein interaction network analysis on the H3K4me3‐enriched genes in SARS‐CoV‐2‐infected cells at 48 h.p.i. Interestingly, we uncovered a protein‐protein interaction network that includes IFNGR1, STAT1, NOS2, IL1R1, and NFKB1 (Figure 2E), indicating that SARS‐CoV‐2 may trigger a unique epigenetic activation event to modulate the IFN‐γ and/or TNF‐α signaling pathways. Given that cancer development is strongly influenced by cellular immunity and inflammation, 44 we conducted a pan‐cancer analysis to evaluate if mRNA expression levels of STAT1, NFKB1, IL1R1, and IFNGR1 are coregulated during cancer progression. Intriguingly, we observed that STAT1 gene expression was positively correlated with that of IL1R1, IFN‐γ, and NFKB1 in numerous human cancers, including colorectal cancer (Figure 2F, Supporting Information: Figure S2C), consistent with our findings for SARS‐CoV‐2‐infected Caco‐2 cells.
Notably, we also identified increased H3K4me3 levels at the promoters of NRP1 and BSG, both of which encode SARS‐CoV‐2 spike receptors, and TMPRSS2, with this latter encoding a protease that facilitates SARS‐CoV‐2 cell entry 45 , 46 , 47 (Supporting Information: Figure S2D), indicating that SARS‐CoV‐2 may impair the epigenetic machinery to accelerate the viral infection. Additionally, we observed elevated H3K4me3 levels at the promoter of ISG15 (an upstream stimulator of IFN‐γ), and at CSN5 (COP9 signalosome 5, a downstream target of NF‐κB) (Supporting Information: Figure S2E), 48 , 49 further confirming potential roles for the epigenetic regulatory machinery in SARS‐CoV‐2 modulated IFN‐γ and TNF‐α signaling pathways.
The upregulation ISGs was known to be involved in patients suffering severe COVID‐19, 50 and the synergism of IFN‐γ and TNF‐α may induce lethal cytokine shock. 43 Next, we investigated if a combination of IFN‐γ and TNF‐α can augment ISG induction in primate kidney cell lines, as the kidney is one of the SARS‐CoV‐2 vulnerable tissues. Moreover, cytokine‐induced ISG promoter activation was dramatically diminished by administering Ruxolitinib (an IFN‐γ pathway inhibitor that targets JAK1/2) and I3C (an NF‐κB inhibitor) (Figure 2G). Previous reports have shown that the treatment of I3C (inhibiting NF‐κB signaling) or Ruxolitinib (targeting JAK1/2) inhibits viral replication and cytokine storm in COVID‐19 patients. 51 , 52 , 53 , 54 Based on our transcriptomic/epigenetic analyses and biochemical studies, we hypothesize that SARS‐CoV‐2 may transcriptionally and epigenetically induce IFN‐IRF1 and NF‐κB regulatory axes that contribute to viral replication and cytokine storm. Thus, inhibition of IFN‐γ and TNF‐α/NF‐κB signaling pathways may prevent cytokine overproduction in host cells upon SARS‐CoV‐2 infection. Supporting our hypothesis, we observed that blockage of the IFN‐γ and TNF‐α/NF‐κB signaling pathways by Ruxolitinib and I3C impaired SARS‐CoV‐2 infectivity in African green monkey kidney (Vero E6) cells (Figure 2H). These findings are similar to previous studies showing that SARS‐CoV‐2 induces the IFN‐γ and/or TNF‐α/NF‐κB pathways in COVID‐19 patients, which may serve as potential therapeutic targets for anti‐SARS‐CoV‐2 therapy. 51 , 55 , 56 Altogether, our data reveal an epigenetic regulatory mechanism potentially induced by SARS‐CoV‐2 infection, which triggers the IFN‐γ and TNF‐α/NF‐κB signaling axes.
3.3. SARS‐CoV‐2 mediates immunosuppression and T‐cell exhaustion by upregulating PD‐L1 gene expression
Increased PD‐L1 expression is associated with the cytokine storm and CD8+ T‐cell exhaustion displayed by patients suffering severe COVID‐19. 29 Our transcriptomic and epigenomic analyses have shown that the STAT1/IRF1/NF‐κB regulatory axis might transmit intracellular signals upon IFN‐γ and TNF‐α induction in response to SARS‐CoV‐2 infection. To investigate if the STAT1, IRF1, and NF‐κB regulatory pathways contribute to T‐cell exhaustion and immunosuppression, we sought correlations between these signaling factors and the immunosuppressive ligand PD‐L1 (a PD‐1 ligand), as well as Galectin‐9 and CEACAM1 (Tim‐3 ligands), in cancer samples from The Cancer Genome Atlas (TCGA) database. 57 Our pan‐cancer analysis indicated that mRNA expression levels of STAT1, IRF1, and RELA were positively correlated with mRNAs of PD‐L1, Galectin‐9, and CEACAM1 (Figure 3A and Supporting Information: Figure S3A,B), with the PD‐L1 gene displaying the most pronounced correlation with those TFs. Accordingly, we focused on exploring the possible mechanism by which SARS‐CoV‐2 perturbs PD‐L1 gene regulation.
Figure 3.
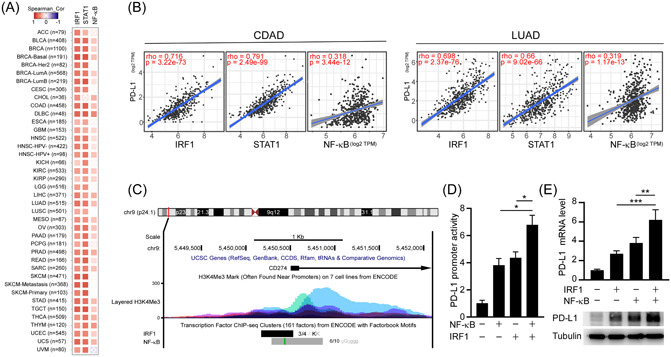
NF‐κB/IRF1 axis upregulates PD‐L1 expression. (A) Spearman correlation analysis of PD‐L1 with IRF1, STAT1, and RELA (NF‐κB) in the TCGA pan‐cancers. (B) Spearman correlation analysis of PD‐L1 with IRF1 and STAT1 in the TCGA colon cancer (COAD; n = 460) and lung cancer data set (LUAD; n = 585). (C) ENCODE data displayed ChIP‐sequencing signals for the occupancy of IRF1, RELA, and histone H3K4me3 around the transcription start sites of PD‐L1. (D) Promoter‐luciferase reporter activity of PD‐L1 (−373/+328) by overexpression of NF‐κB and IRF1. Statistical method: one‐way ANOVA, Tukey post hoc tests, *p < 0.05. (E) Effects of IRF1 and NF‐κB on the mRNA expression (upper panel) and protein expression (lower panel) of PD‐L1 in A549 cells were analyzed by RT‐qPCR and western blotting, respectively. Statistical method: one‐way ANOVA, Tukey post hoc tests, **p < 0.01, ***p < 0.001. ANOVA, analysis of variance; ChIP, chromatin immunoprecipitation; IRF1, interferon regulatory factor 1; LUAD, lung adenocarcinoma; mRNA, messenger RNA; NF‐κB, nuclear factor‐κB; PD‐L1, programmed death ligand‐1; RT‐qPCR, real‐time quantitative polymerase chain reaction; TCGA, The Cancer Genome Atlas.
First, we analyzed mRNA expression levels of PD‐L1 in SARS‐CoV‐2‐susceptible cancer cell lines, such as colon and lung cancers in the TCGA database. We noted that PD‐L1 mRNA levels were significantly and positively correlated with those of STAT1, IRF1, and RELA in colon adenocarcinoma and LUAD (Figure 3B). Moreover, we examined ChIP‐seq signals for genomic occupancies of the IRF1 and RELA (an NF‐κB family member) TFs as well as H3K4me3 modifications according to the Encyclopedia of DNA Elements (ENCODE) database, which revealed enrichment of IRF1 and RELA, as well as the H3K4me3 modification, at the TSS of PD‐L1 (Figure 3C). Hence, IRF1 and RELA appear to coordinately activate PD‐L1 expression via promoter regulation. To validate that hypothesis, we expressed IRF1 and/or RELA in human LUAD A549 cells and assessed endogenous expression levels of PD‐L1 through RT‐qPCR and immunoblotting, as well as PD‐L1 promoter activity through luciferase reporter assays. We found that either IRF1 or RELA upregulated PD‐L1 promoter activity and PD‐L1 expression at mRNA and protein levels. Moreover, coexpression of both TFs further induced PD‐L1 expression in cells (Figure 3D,E). ChIP‐qPCR analyses confirmed that SARS‐CoV‐2 induced binding of IRF1 and NF‐κB/RELA to the promoter region of PD‐L1 in A549 cells (Supporting Information: Figure S3C). These data indicate that SARS‐CoV‐2 may trigger cellular signaling to activate IRF1‐ and NF‐κB‐dependent PD‐L1 gene expression.
Since SARS‐CoV‐2 activates several signaling pathways, including p38, AKT, JAK/STAT, and NF‐κB, 58 we were interested in determining which pathways might induce transcriptional activation of PD‐L1. To assess if SARS‐CoV‐2 induces PD‐L1 expression in lung tissue, we expressed ACE2 in A549 and Calu‐1 cells, followed by infection with an RFP‐hosting SARS‐CoV‐2 pseudovirus, and then distinguished pseudoviral‐infected from noninfected cells based on the RFP reporter. We observed that expression of PD‐L1, but not PD‐L2, was elevated in the infected cells, suggesting that SARS‐CoV‐2 induces PD‐L1 expression (Figure 4A). In addition, major histocompatibility antigens, HLA‐ABC, were also elevated in pseudovirus‐infected cells (Figure 4A). Moreover, T‐cell effector and cytotoxicity signatures (GZMB, PRF1, and IFNG) were inversely associated with SARS‐CoV‐2 cellular entry receptors (ACE2 and TMPRSS2) in the TCGA normal lung tissues and lung cancers database, suggesting that T‐cell exhaustion may occur in vulnerable tissue (Supporting Information: Figure S3D). To further investigate if the STAT1/IRF1/NF‐κB regulatory axis transmits intracellular signals from cytokines to induce transcriptional activation of PD‐L1, we tested the impacts of IFN‐γ, TNF‐α, and other cytokines or growth factors on PD‐L1 gene regulation by means of luciferase reporter assays. We observed that IFN‐γ and TNF‐α significantly induced PD‐L1 promoter activity relative to IL‐1β, IL‐6, TGF‐β, EGF, IFN‐α, and IFN‐β. Interestingly, a combination of IFN‐γ and TNF‐α synergistically boosted transcriptional activation of PD‐L1, whereas combining IL‐1β with IFN‐γ or TNF‐α did not exert the same effect (Supporting Information: Figure S3E). Next, we sought clinical evidence linking PD‐L1 to COVID‐19. We enrolled 161 patients (72 female, 89 male; average age: 55.3 years, range: 19–101 years) with confirmed COVID‐19 at Memorial Hermann Hospital in Houston, Texas, USA. Sixty non‐COVID‐19 patient controls were recruited from the cardiac clinic of the same hospital, including 33 (55.0%) females and 27 males (45.0%), with an average age of 60.5 years (range: 21–96). Demographic and clinical details of all subjects were obtained (Table 1). Subjects with severe COVID‐19 disease showed higher PD‐L1 expression (Figure 4B). The previous report indicates that PD‐L1 can be highly expressed on tumor cell‐derived small extracellular vesicles surface and can be secreted as circulating exosomes into plasma. 59 The soluble PD‐L1 secretion into plasma is due to cleavage of the extracellular domain of PD‐L1, and the concentration of the secreted soluble PD‐L1 protein in plasma has been shown to correlate with PD‐L1 expression in cancer tissues. 60 , 61 Therefore, we detected PD‐L1 expression levels in COVID‐19 patients' plasma samples. Existing therapeutic antibodies in clinical use may lose efficacy against the Omicron variant. 62 To determine if Omicron‐induced infected cells exhibit higher PD‐L1 expression to evade immune surveillance, we accessed PD‐L1 by flow cytometry in A549‐hACE2 cells upon SARS‐CoV‐2 and Omicron pseudovirus infection. We found that Omicron did induce higher PD‐L1 expression (Figure 4C). Importantly, this was due to upregulation of PD‐L1 promoter activity, suggesting that Omicron induces PD‐L1 expression by activating its transcriptional level (Figure 4D). Furthermore, our ChIP assays also demonstrated that SARS‐CoV‐2 induced H3K4me3 and H3K27ac binding to the promoter region of TNF‐α, IFN‐γ in A549‐hACE2 cells (Figure 4E,F). To evaluate the function of SARS‐CoV‐2 in PD‐L1 upregulation in vivo, we infected a nonpathogenic AAV‐hACE2 mouse model susceptible to SARS‐CoV‐2, thereby closely mimicking human COVID‐19 conditions. 63 SARS‐CoV‐2 treatment induced a dramatic increase in TNF‐α, IFN‐γ, and PD‐L1 expression at both mRNA and protein levels in the lung tissues of the AAV‐hACE2 mice (Supporting Information: Figure S3F–H and Figure 4G). Thus, our results indicate that SARS‐CoV‐2 may hijack cellular signaling pathways, such as the IFN−IRF1 and NF‐κB axes, to promote upregulation of PD‐L1 in host cells and AAV‐hACE2 model, which may elicit immune suppression. Accordingly, inhibiting those signaling pathways by means of specific inhibitors (e.g., Ruxolitinib for IFN and I3C for NF‐κB) may reverse PD‐L1 upregulation and, consequently, PD‐L1‐mediated immunosuppression (Figure 4H,I).
Figure 4.
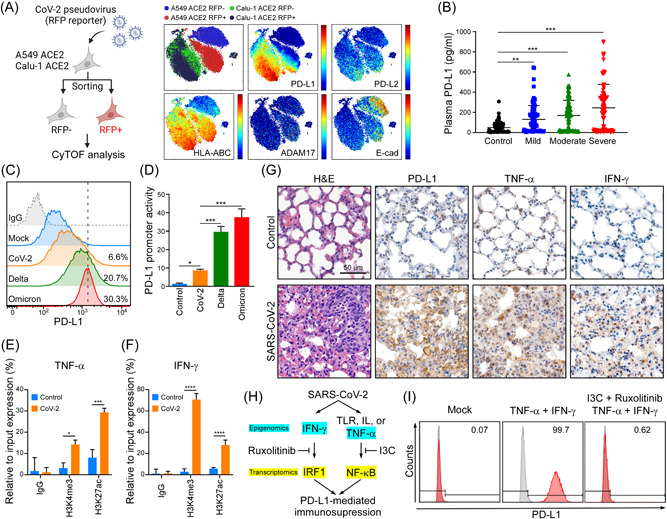
SARS‐CoV‐2 induces PD‐L1 through TNF‐α/IFN mediated NF‐κB/IRF1 axis. (A) CyTOF analysis of PD‐L1, PD‐L2, HLA‐ABC, ADAM17, and E‐cad in A549‐hACE2 and Calu‐1 ACE2 upon SARS‐CoV‐2 pseudovirus infection. (B) ELISA detection of the indicated group's plasma samples binding to precoated PD‐1. Data shown are means ± SD from control (n = 60), mild (n = 48), moderate (n = 59), and severe (n = 54) patients. **p < 0.01, ***p < 0.001 (one‐way ANOVA). (C) Flow cytometry analysis of PD‐L1 in A549‐hACE2 cells upon SARS‐CoV‐2, and Omicron pseudovirus infection. (D) Promoter‐luciferase reporter activity of PD‐L1 (−373/+328) by infection of SARS‐CoV‐2, and Omicron pseudovirus. Statistical method: one‐way ANOVA, Tukey post hoc tests, *p < 0.05, ***p < 0.001. (E, F) ChIP analysis of enrichment of H3K4me3 and H3K27ac modification in TNF‐α (E) and IFN‐γ (F) promoters after SARS‐CoV‐2 infection in A549‐hACE2 cells for 1 day. The enrichment levels were analyzed by RT‐qPCR and shown as the percentage of input. Statistical method: two‐way ANOVA, Tukey post hoc tests, *p < 0.05, ***p < 0.001, ****p < 0.0001 (n = 3 with mean ± SD shown). (G) Representative data of immunohistochemistry staining of TNF‐α and IFN‐γ in lung tissues of SARS‐CoV‐2 infected AAV‐hACE2 mice at 5 dpi. Scale bar = 50 µm. (H) Schematic diagram of SARS‐CoV‐2 induces PD‐L1‐mediated immunosuppression through NF‐κB/IRF1 axis. (I) Flow cytometry was performed to detect surface PD‐L1 protein expression. A549 cells were pretreated with I3C and Ruxolitinib for 1 h, following by treating with 10 ng/ml TNF‐α and 10 ng/ml IFN‐γ for 48 h. AAV‐hACE2, adeno associated virus‐human angiotensin‐converting enzyme 2; ANOVA, analysis of variance; IFN‐γ, interferon‐γ; IRF1, interferon regulatory factor 1; NF‐κB, nuclear factor‐κB; PD‐1, programmed cell death protein 1; PD‐L1, programmed death ligand‐1; SARS‐CoV‐2, severe acute respiratory syndrome coronavirus 2; RT‐qPCR, real‐time quantitative polymerase chain reaction; TNF‐α, tumor necrosis factor‐α.
Table 1.
Demographic and characteristics of the healthy controls and COVID‐19 subjects (n = 221)
| Control | Mild | Moderate | Severe | |
|---|---|---|---|---|
| Total subjects, n | 60 | 48 | 59 | 54 |
| Sex, male, n (%) | 27 (45.0%) | 25 (52.1%) | 28 (47.5%) | 36 (66.7%) |
| Age, years, mean ± SD | 60.5 ± 17.8 | 50.7 ± 16.6 | 52.8 ± 16.4 | 62.1 ± 18.0 |
| Race/ethnicity, n (%) | ||||
| White | 41 (68.3%) | 4 (8.3%) | 2 (3.6%) | 5 (9.8%) |
| African American | 13 (21.7%) | 14 (29.2%) | 13 (23.2%) | 8 (15.7%) |
| Hispanic | 3 (5.0%) | 28 (58.3%) | 37 (66.1%) | 35 (68.6%) |
| Asian | 3 (5%) | 2 (4.2%) | 3 (5.4%) | 2 (3.9%) |
| Other | 0 (0%) | 0 (0%) | 1 (1.8%) | 1 (2.0%) |
3.4. Blockage of PD‐L1 alleviates SARS‐CoV‐2‐induced lymphopenia in mice
Lymphopenia is observed in almost 80% of SARS‐CoV‐2‐infected patients admitted to the hospital, contributing to severe COVID‐19. 20 , 64 Although we found that Ruxolitinib and/or I3C treatment impaired SARS‐CoV‐2 infectivity and abrogated abnormal cytokine production by suppressing the IFN‐γ and TNF‐α signaling axes, these signaling inhibitors might not be appropriate therapeutic choices because they can elicit serious adverse effects. For instance, Ruxolitinib can cause anemia, neutropenia, and thrombocytopenia. 65 Similarly, I3C treatment may lead to skin rashes, tremors, nausea, and loss of balance. 66 I3C has also been shown to increase liver enzyme activity in rats, as well as possible drug–drug interactions in humans. 67 To avoid unfavorable side‐effects, we targeted PD‐L1 downstream of the signaling axes by means of neutralizing anti‐PD‐L1 antibody treatment.
From a clinical standpoint, we sought to determine the clinical relevance of targeting PD‐L1 using a neutralizing anti‐PD‐L1 antibody (B7‐H1) in terms of preventing SARS‐CoV‐2 infectivity and lymphopenia in AAV‐hACE2 mice. First, we demonstrated that expression of hACE2 was detectable in mouse lungs and that hACE2 expression was unaffected by B7‐H1 treatment (Figure 5A). AAV‐hACE2 mice were pretreated with anti‐PD‐L1 or IgG antibodies (as control) before challenging them with SARS‐CoV‐2. Notably, administration of anti‐PD‐L1 antibody in SARS‐CoV‐2‐infected AAV‐hACE2 mice remarkably prevented weight loss relative to the control group (Figure 5B), confirming a protective role for the anti‐PD‐L1 antibody upon encountering SARS‐CoV‐2 infection. Supporting these findings, we found that anti‐PD‐L1 antibody treatment significantly reduced viral infectivity and abolished viral replication in mouse lungs (Figure 5C,D). Since an elevated neutrophil‐to‐lymphocyte ratio (NLR) is a hematologic biomarker for severe COVID‐19, 20 we used flow cytometry to count CD45+ immune cells in the peripheral blood of AAV‐hACE2 mice. Administration of anti‐PD‐L1 antibody in SARS‐CoV‐2‐infected AAV‐hACE2 mice greatly reduced the NLR, as well as amounts of SARS‐CoV‐2 nucleocapsid (N) protein and SARS‐CoV‐2‐dependent production of IFN‐γ significantly diminishing COVID‐19 disease severity (Figure 5E–I). The inflammation and lung damage between the IgG and anti‐PD‐L1 groups were assessed using histopathological scores on the lung tissue. Moderate to severe histopathology was observed in the lungs of IgG‐treated mice following the viral challenge (Figure 5J), which is consistent with the previous study. 68 The pneumonia was reduced in anti‐PD‐L1‐treated mice upon SARS‐CoV‐2 infection. Consistently, anti‐PD‐L1 groups showed less IFN‐γ positive cells in the infected lung tissues (Figure 5K). Interestingly, PD‐L1 and IFN‐γ expressions were also observed from COVID‐19 lung biopsy tissues (Figure 6A). Furthermore, we found that the levels of N protein expression were positively correlated with PD‐L1 expression levels in COVID‐19 lung tissues (Figure 6B,C). Thus, our results strongly support that targeting PD‐L1 could be a novel SARS‐CoV‐2 antiviral strategy to curtail COVID‐19 severity.
Figure 5.
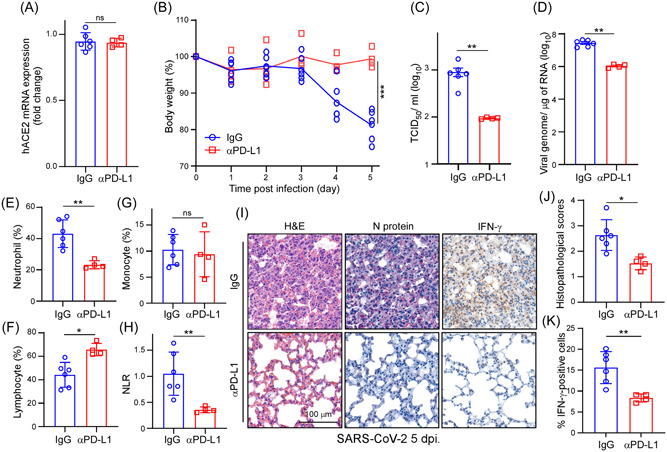
Blockage of PD‐L1 recovers the severity of mice. (A) RT‐qPCR analysis of hACE2 mRNA expression in lung tissues of SARS‐CoV‐2 infected AAV‐hACE2 mice treated with IgG (n = 6) or anti‐PD‐L1 antibody (n = 4). (B) The weight loss curve of AAV‐hACE2 mice infected with SARS‐CoV‐2 (n = 6). Statistic method: Mann–Whitney U test, ***p < 0.001. (C) Tissue culture infection dose 50% (TCID50) was determined in the IgG or anti‐PD‐L1 antibody treatment AAV‐hACE2 mice lung tissues. Results are mean values ± SD of three independent experiments (n = 6 with mean ± SD shown). Statistic method: Mann–Whitney U test, **p < 0.01. (D) RT‐qPCR was performed to analyze viral RNA expression in lung tissues of SARS‐CoV‐2 infected AAV‐hACE2 mice treated with IgG or anti‐PD‐L1 antibody (n = 6). Statistic method: Mann–Whitney U test, **p < 0.01. (E–H) The percentage of neutrophils, lymphocytes, monocytes, and NLR (neutrophil‐to‐lymphocytes ratio) in the peripheral blood of SARS‐CoV‐2 infected AAV‐hACE2 mice treated with IgG or anti‐PD‐L1 antibody (n = 6). *p < 0.05, **p < 0.01. (I) Representative microscopic images of N protein and IFN‐γ expression in lung tissues of SARS‐CoV‐2 infected AAV‐hACE2 mice. Scale bar = 100 µm. (J) Histopathology score of AAV‐hACE2 mouse lungs after SARS‐CoV‐2 infection. (n = 6). *p < 0.05. (K) Quantification of the percentage of IFN‐γ‐positive cells in the lung tissue of SARS‐CoV‐2‐infected AAV‐hACE2 mice treated with IgG or anti‐PD‐L1 antibody. **p < 0.01, (n = 6). AAV‐hACE2, adeno associated virus‐human angiotensin‐converting enzyme 2; IFN‐γ, interferon‐γ; mRNA, messenger RNA; PD‐L1, programmed death ligand‐1; SARS‐CoV‐2, severe acute respiratory syndrome coronavirus 2; RT‐qPCR, real‐time quantitative polymerase chain reaction.
Figure 6.
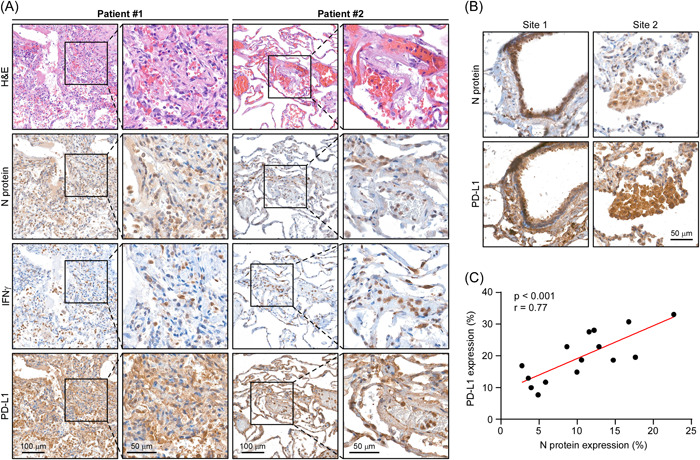
PD‐L1 correlates with N protein in human COVID‐19 lung tissues. (A) Representative microscopic images of N protein, IFN‐γ, and PD‐L1 expression in human COVID‐19 lung tissues. (B) Representative microscopic images of N protein and PD‐L1 expression in human COVID‐19 lung tissues. (C) Spearman's rank correlation analysis between the protein expression levels of N protein and PD‐L1 in (B). (3 fields per lung tissue; total 15 fields). IFN‐γ, interferon‐γ; PD‐L1, programmed death ligand‐1.
4. DISCUSSION
The dynamic host/virus gene regulatory network uncovered in this study reveals the potential etiology and biological mechanisms underlying COVID‐19. We employed transcriptomics and epigenomics to identify key regulators involved in SARS‐CoV‐2 pathogenesis. Using a TO‐GCN approach to analyze our transcriptomic data, we identified STAT1 and IRF1 as crucial TFs involved in interferon and cytokine signaling pathways (at the L1 level), and those pathways are controlled by epigenetic regulation (at the L2 level). SARS‐CoV‐2 infection exerts global and gene‐specific effects on host chromatin, ultimately upregulating IFN‐stimulating and cytokine‐regulatory genes. 43 Importantly, suppressing chromatin structure by inhibiting topoisomerase can alleviate excessive inflammatory gene production and cytokine storms. 69 Our epigenomic data also indicate that TNF‐α and IFN‐γ trigger PANopotosis via the STAT1/NOS2 signaling axis, as reported by others, 43 which may be due to enriched H3K4me3‐mediated epigenetic activation. Furthermore, we also observed that SARS‐CoV‐2 promoted H3K4me3 histone marking of pathogenic factors such as NRP1, BSG, and TMPRSS2.
Since TNF‐α and IFN‐γ secretion cannot be measured at the in vitro system because the P3 disinfection procedure would disrupt protein conformation in the medium, we performed indirect methods, such as epigenomic and transcriptomic studies, and revealed that the IFN‐related genes are highly enriched by H3K4me3 in SARS‐CoV‐2 infected Caco‐2 cells (Figure 2B). In addition, transcriptomic analyses showed that mRNA expression levels of IFN downstream TFs, such as IRF1 and NF‐κB, were highly elevated following viral infection (Figure 1E). In our transcriptomic data, the strongest differences in genetic changes occurred at 24–48 h.p.i. At this later stage of viral infection, viral replication only caused 14% of cell death (p = 0.2776, no significant difference, data not shown), thus possibly excluding the indirectly changing in the IFN‐IRF1 and NF‐κB signaling.
We found that antiviral interferon and cytokine signalings—such as by ISG15, IFN‐γ, and IL‐12 were enriched in Caco‐2 cells upon SARS‐CoV‐2 infection. The IFN‐γ pathway is known to be highly activated after SARS‐CoV‐2 infection. 43 IL‐12 induces transcription of IFN‐γ by triggering the downstream JAK2 and TYK2 signaling cascades in Th1 cells. 70 An expression quantitative trait locus (eQTL) analysis to uncover differential TYK2 expression and explore its association with COVID‐19 revealed rs11085727 to be the most significant cis‐eQTL (p = 9.47 × 10–6) for TYK2 (data not shown) in the cultured fibroblast data set of the GTEx portal. Furthermore, rs11085727 T allele replacement was associated with elevated TYK2 expression and a more severe COVID‐19 phenotype (data not shown). These findings provide clinical support to the notion that the TYK2 rs11085727 T allele may contribute to COVID‐19 severity.
Growth factors and inflammatory cytokines such as EGF, IL‐6, IFN‐γ, TNF‐α, and TGF‐β can induce PD‐L1 expression. 71 , 72 , 73 , 74 , 75 Several studies have demonstrated that SARS‐CoV‐2 infection stimulates IFN‐γ, TNF‐α, EGF, IL‐6, or TGF‐β production. 76 , 77 , 78 , 79 Here, we have shown that a combination of IFN‐γ and TNF‐α additively induces PD‐L1 expression at the mRNA and protein levels (Figure 3E). Moreover, the TNF‐α/NF‐κB axis induces CSN5‐mediated deubiquitination and stabilization of PD‐L1. 49 , 80 We also observed elevated H3K4me3 marking in the regulatory regions of CSN5 upon viral infection, indicating that epigenetic regulation of CSN5 also contributes to PD‐L1 stability. The combination of I3C (an NF‐κB inhibitor) and Ruxolitinib (targets JAK1/2) directly inhibits viral replication and cytokine storm. 51 , 52 , 53 , 54 By blocking viral infection and cytokine secretion, we could reduce virus‐induced RELA and IRF1‐mediated PD‐L1 upregulation in vitro models. Consistently, TNF‐α and IFN‐γ blockade diminished PD‐L1 expression in lung tissue in the AAV‐hACE2 mice model (data not shown). We also report the IRF1 and RELA TFs involved in PD‐L1 expression, as highlighted elsewhere. 73 , 81 In addition, blocking NF‐κB downregulated glycosylation‐mediated ACE2‐spike interaction. 35 , 82 Thus, targeting the NF‐κB and IRF1 pathways could be beneficial for treating COVID‐19, 51 , 83 with combinatorial anti‐NF‐κB and anti‐IRF treatment representing an alternative strategy for impairing viral infectivity and preventing systemic inflammation.
Cancer patients are more susceptible to SARS‐CoV‐2 and are more likely to develop severe COVID‐19. 84 , 85 It remains unclear if anti‐PD‐1/PD‐L1 treatment would be beneficial for COVID‐19 patients. Immune checkpoint inhibitors (ICI) may enhance the cytokine storm associated with higher COVID‐19 morbidity and mortality. 86 , 87 A few clinical cases have reported that patients with metastatic squamous head and neck cancer, 28 lung cancer, 24 , 27 or Merkel cell carcinoma 88 who received ICI treatment could be at greater risk of recall immune‐mediated pneumonitis upon SARS‐CoV‐2 infection. In contrast, melanoma patients suffering from COVID‐19 displayed better outcomes upon ICI co‐treatment. 89 , 90 The incidence rates of ICI‐induced pneumonitis (mimicking COVID‐19 infection) and anti‐PD‐1/PD‐L1‐related pneumonitis range from 2.5% to 5%, 26 , 91 potentially explaining somewhat why COVID‐19 patients receiving anti‐PD‐1/PD‐L1 agents develop a greater risk of interstitial pneumonitis. 92 Importantly, ICI treatment does not appear to significantly increase the risk of serious adverse events compared with chemotherapy. 93
Upon SARS‐CoV‐2 infection, we noticed that anti‐PD‐L1 treatment reduced NLR but had no effect on monocyte numbers (Figure 5E–H). The decreased NLR may be due to both decreased neutrophil numbers and increased lymphocyte numbers after anti‐PD‐L1 treatment (Figure 5E,F). Anti‐PD‐L1 therapy has been associated with reduced NLR values and is beneficial in the overall survival of renal cell carcinoma and nonsmall‐cell lung carcinoma patients. 94 Therefore, anti‐PD‐Li therapy may also benefit to SARS‐CoV‐2 infected patients. Nevertheless, further studies are needed to dissect how anti‐PD‐L1 therapy affects neutrophils and lymphocytes following SARS‐CoV‐2 infection.
5. CONCLUSIONS
Our data demonstrate that anti‐PD‐L1 antibody pretreatment promotes lymphocyte numbers and abrogates the production of inflammatory cytokines in an AAV‐hACE2 mouse model (Figure 5F,I). Those outcomes indicate that anti‐PD‐L1 treatment may limit T‐cell exhaustion and block virus infectivity at the early stage of virus entry. However, COVID‐19 patients may not be suitable for ICI treatment since T cells are associated with viral clearance and they dampen overactive innate immune responses. 95 Hence, although PD‐1/PD‐L1 checkpoint blockade benefits melanoma patients suffering from COVID‐19, whether it represents an appropriate therapeutic target for other cancer patients battling COVID‐19 requires further research.
AUTHOR CONTRIBUTIONS
Hsiang‐Chi Huang and Shih‐Han Wang designed and performed the experiments, analyzed data, and wrote the manuscript; Guo‐Chen Fang, Chun‐Che Liao, Cheng‐Pu Sun, Jia‐Tsrong Jan, Hsiu‐Hua Ma, Hui‐Ying Ko, Yi‐An Ko, Ming‐Tsai Chiang, Jian‐Jong Liang, Chun‐Tse Kuo, and Te‐An Lee performed experiments and analyzed data; Shih‐Yu Chen analyzed data, provided scientific input, and wrote the manuscript; Wen‐Cheng Chou analyzed data and wrote the manuscript; Diego Morales‐Scheihing, Lu Cui, Gerlinde Wernig and Louise D. McCullough provided resources and analyzed data; Chen‐Yang Shen, Yi‐Ling Lin, and Mi‐Hua Tao provided scientific input. Yao‐Ming Chang, Shu‐Ping Wang, Yun‐Ju Lai, and Chia‐Wei Li supervised the entire project, designed the experiments, analyzed data, and wrote the manuscript.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
All animal procedures approved by Institutional Animal Care and Use Committee (IACUC) Academia Sinica. Human samples were collected under IRB #HSC‐MS‐17‐0452 for The Biorepository of Neurological Disorders. Experiments and analysis on selected samples for the current study were performed under IRB #HSC‐MS‐20‐1058. This work was funded by the Huffington Foundation to Dr. Louise McCullough and NIMH grant R01MH127856, "Sex specific immune response to SARS‐CoV‐2 leads to chronic neurologic symptoms". Human COVID‐19 lung tissues were provided by Dr. Gerlinde Wernig at Stanford University, and Stanford Hospital Autopsy service under IRB #39881.
Supporting information
Supplementary information.
ACKNOWLEDGMENTS
This work was funded in part by the following: Ministry of Science and Technology (MOST 109‐2314‐B‐001‐002‐MY3 and MOST 109‐2314‐B‐001‐008‐MY3 to Chun‐Che Liao; MOST 108‐3114‐Y‐001‐002 to Mi‐Hua Tao; MOST 108‐2320‐B‐001‐034‐MY2 to Shih‐Yu Chen) MOST 108‐2320‐B‐001‐007‐MY2 and MOST 110‐2628‐B‐001‐017 to Shu‐Ping Wang); Academia Sinica (AS‐SUMMIT‐109, AS‐KPQ‐109‐BioMed to Mi‐Hua Tao; AS‐CDA‐110‐L09, AS‐GC‐110‐05, and AS‐KPQ‐110‐EIMD to Shih‐Yu Chen); University of Massachusetts Lowell (Faculty start‐up D50210000000022 to Yun‐Ju Lai). The authors thank Taiwan CDC for providing SARS‐CoV‐2 TCDC#4 (hCoV‐19/Taiwan/4/2020) and funding support from Academia Sinica for IBMS P3 facility (AS‐CFII‐108‐102) and the Ministry of Science and Technology, Taiwan for COVID‐19 study (MOST 109‐3114‐Y‐001‐001). Enrollment of COVID‐19 and control subjects for The Biorepositoritory of Neurological discorders were funded by the Huffington Foundation and NIMH grant R01MH127856, "Sex specific immune response to SARS‐CoV‐2 leads to chronic neurologic symptoms" to Dr. Louise McCullough. We also thank Biosafety Level 3 Facility in Genomic Research Centers (Grant AS‐CFII‐108‐101), Biosafety Level 3 Facility in Institute of Biomedical Sciences (Grant AS‐CFII‐108‐102), AAV Core Facility (Grant AS‐CFII109‐103), Light Microscopy Core Facility, Flow Cytometry Core Facility (Grant AS‐CFII108‐111), Pathology Core Laboratory, and DNA Sequencing Core Facility (AS‐CFII‐108‐115) at Academia Sinica. We also thank Dr. Lu Cui and Dr. Gerlinde Wernig for providing human COVID‐19 lung tissues at Stanford University, and Stanford Hospital Autopsy service.
Huang H‐C, Wang S‐H, Fang G‐C, et al. Upregulation of PD‐L1 by SARS‐CoV‐2 promotes immune evasion. J Med Virol. 2023;95:e28478. 10.1002/jmv.28478
Contributor Information
Yao‐Ming Chang, Email: petitming@ibms.sinica.edu.tw.
Shu‐Ping Wang, Email: spwang@ibms.sinica.edu.tw.
Yun‐Ju Lai, Email: yunju_lai@uml.edu.
Chia‐Wei Li, Email: cwli@ibms.sinica.edu.tw.
DATA AVAILABILITY STATEMENT
Data deposition: The Illumina reads reported in this paper have been deposited in the NCBI Sequence Read Archive, https://www.ncbi.nlm.nih.gov/sra (accession no. PRJNA744316).
REFERENCES
- 1. Cheung KS, Hung IFN, Chan PPY, et al. Gastrointestinal manifestations of SARS‐CoV‐2 infection and virus load in fecal samples from a Hong Kong cohort: systematic review and meta‐analysis. Gastroenterology. 2020;159(1):81‐95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Synowiec A, Szczepański A, Barreto‐Duran E, Lie LK, Pyrc K. Severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2): a systemic infection. Clin Microbiol Rev. 2021;34(2):e00133‐20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Davidson AD, Williamson MK, Lewis S, et al. Characterisation of the transcriptome and proteome of SARS‐CoV‐2 reveals a cell passage induced in‐frame deletion of the furin‐like cleavage site from the spike glycoprotein. Genome Med. 2020;12(1):68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kim D, Lee JY, Yang JS, Kim JW, Kim VN, Chang H. The architecture of SARS‐CoV‐2 transcriptome. Cell. 2020;181(4):914‐921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sun J, Ye F, Wu A, et al. Comparative transcriptome analysis reveals the intensive early stage responses of host cells to SARS‐CoV‐2 infection. Front Microbiol. 2020;11:593857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Triana S, Metz‐Zumaran C, Ramirez C, et al. Single‐cell analyses reveal SARS‐CoV‐2 interference with intrinsic immune response in the human gut. Mol Syst Biol. 2021;17(4):e10232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jafarzadeh A, Jafarzadeh S, Nozari P, Mokhtari P, Nemati M. Lymphopenia an important immunological abnormality in patients with COVID‐19: possible mechanisms. Scand J Immunol. 2021;93(2):e12967. [DOI] [PubMed] [Google Scholar]
- 8. Gheblawi M, Wang K, Viveiros A, et al. Angiotensin‐converting enzyme 2: SARS‐CoV‐2 receptor and regulator of the renin‐angiotensin system: celebrating the 20th anniversary of the discovery of ACE2. Circ Res. 2020;126(10):1456‐1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pinto BGG, Oliveira AER, Singh Y, et al. ACE2 expression is increased in the lungs of patients with comorbidities associated with severe COVID‐19. J Infect Dis. 2020;222(4):556‐563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Saksena N, Bonam SR, Miranda‐Saksena M. Epigenetic lens to visualize the severe acute respiratory syndrome coronavirus‐2 (SARS‐CoV‐2) infection in COVID‐19 pandemic. Front Genet. 2021;12:581726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sawalha AH, Zhao M, Coit P, Lu Q. Epigenetic dysregulation of ACE2 and interferon‐regulated genes might suggest increased COVID‐19 susceptibility and severity in lupus patients. Clin Immunol. 2020;215:108410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Aliee H, Massip F, Qi C, et alDeterminants of SARS‐CoV‐2 receptor gene expression in upper and lower airways. MedRxiv. 2020.
- 13. Sharma S, Kelly TK, Jones PA. Epigenetics in cancer. Carcinogenesis. 2010;31(1):27‐36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Atlante S, Mongelli A, Barbi V, Martelli F, Farsetti A, Gaetano C. The epigenetic implication in coronavirus infection and therapy. Clin Epigenetics. 2020;12(1):156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Park PJ. ChIP‐seq: advantages and challenges of a maturing technology. Nat Rev Genet. 2009;10(10):669‐680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hyun K, Jeon J, Park K, Kim J. Writing, erasing and reading histone lysine methylations. Exp Mol Med. 2017;49(4):e324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bhaskar S, Sinha A, Banach M, et al. Cytokine storm in COVID‐19—immunopathological mechanisms, clinical considerations, and therapeutic approaches: the REPROGRAM consortium position paper. Front Immunol. 2020;11:1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kim JS, Lee JY, Yang JW, et al. Immunopathogenesis and treatment of cytokine storm in COVID‐19. Theranostics. 2021;11(1):316‐329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jose RJ, Manuel A. COVID‐19 cytokine storm: the interplay between inflammation and coagulation. Lancet Respir Med. 2020;8(6):e46‐e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Liu J, Li S, Liu J, et al. Longitudinal characteristics of lymphocyte responses and cytokine profiles in the peripheral blood of SARS‐CoV‐2 infected patients. EBioMedicine. 2020;55:102763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hamidi SH, Kadamboor Veethil S, Hamidi SH. Role of pirfenidone in TGF‐β pathways and other inflammatory pathways in acute respiratory syndrome coronavirus 2 (SARS‐Cov‐2) infection: a theoretical perspective. Pharmacol Rep. 2021;73:712‐727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Costela‐Ruiz VJ, Illescas‐Montes R, Puerta‐Puerta JM, Ruiz C, Melguizo‐Rodríguez L. SARS‐CoV‐2 infection: the role of cytokines in COVID‐19 disease. Cytokine Growth Factor Rev. 2020;54:62‐75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Diao B, Wang C, Tan Y, et al. Reduction and functional exhaustion of T cells in patients with coronavirus disease 2019 (COVID‐19). Front Immunol. 2020;11:827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Di Noia V, D'Aveni A, Squadroni M, Beretta GD, Ceresoli GL. Immune checkpoint inhibitors in SARS‐CoV‐2 infected cancer patients: the spark that ignites the fire? Lung Cancer. 2020;145:208‐210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Vitte J, Diallo AB, Boumaza A, et al. A granulocytic signature identifies COVID‐19 and its severity. J Infect Dis. 2020;222(12):1985‐1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chang HL, Wei PJ, Wu KL, Huang HL, Yang CJ. Checkpoint inhibitor pneumonitis mimicking COVID‐19 infection during the COVID‐19 pandemic. Lung Cancer. 2020;146:376‐377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Robilotti EV, Babady NE, Mead PA, et al. Determinants of COVID‐19 disease severity in patients with cancer. Nature Med. 2020;26(8):1218‐1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dipasquale A, Persico P, Lorenzi E, Rahal D, Santoro A, Simonelli M. COVID‐19 lung injury as a primer for immune checkpoint inhibitors (ICIs)‐related pneumonia in a patient affected by squamous head and neck carcinoma treated with PD‐L1 blockade: a case report. J Immunother Cancer. 2021;9(2):e001870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chen J, Vitetta L. Increased PD‐L1 expression may be associated with the cytokine storm and CD8+ T‐cell exhaustion in severe COVID‐19. J Infect Dis. 2021;223(9):1659‐1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. McDermott DF, Atkins MB. PD‐1 as a potential target in cancer therapy. Cancer Med. 2013;2(5):662‐673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tarazona S, García‐Alcalde F, Dopazo J, Ferrer A, Conesa A. Differential expression in RNA‐seq: a matter of depth. Genome Res. 2011;21(12):2213‐2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chang YM, Lin HH, Liu WY, et al. Comparative transcriptomics method to infer gene coexpression networks and its applications to maize and rice leaf transcriptomes. Proc Natl Acad Sci USA. 2019;116(8):3091‐3099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Barbie DA, Tamayo P, Boehm JS, et al. Systematic RNA interference reveals that oncogenic KRAS‐driven cancers require TBK1. Nature. 2009;462(7269):108‐112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hänzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA‐seq data. BMC Bioinform. 2013;14:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Huang HC, Lai YJ, Liao CC, et al. Targeting conserved N‐glycosylation blocks SARS‐CoV‐2 variant infection in vitro. EBioMedicine. 2021;74:103712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sun LL, Yang RY, Li CW, et al. Inhibition of ATR downregulates PD‐L1 and sensitizes tumor cells to T cell‐mediated killing. Am J Cancer Res. 2018;8(7):1307‐1316. [PMC free article] [PubMed] [Google Scholar]
- 37. Han Y, Chen MK, Wang HL, et al. Synergism of PARP inhibitor fluzoparib (HS10160) and MET inhibitor HS10241 in breast and ovarian cancer cells. Am J Cancer Res. 2019;9(3):608‐618. [PMC free article] [PubMed] [Google Scholar]
- 38. Santry LA, Ingrao JC, Yu DL, et al. AAV vector distribution in the mouse respiratory tract following four different methods of administration. BMC Biotechnol. 2017;17(1):43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ruifrok AC, Johnston DA. Quantification of histochemical staining by color deconvolution. Anal Quant Cytol Histol. 2001;23(4):291‐299. [PubMed] [Google Scholar]
- 40. Abascal F, Acosta R, Addleman NJ, et al. Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature. 2020;583(7818):699‐710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Feng H, Zhang YB, Gui JF, Lemon SM, Yamane D. Interferon regulatory factor 1 (IRF1) and anti‐pathogen innate immune responses. PLoS Pathog. 2021;17(1):e1009220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Beacon TH, Delcuve GP, López C, et al. The dynamic broad epigenetic (H3K4me3, H3K27ac) domain as a mark of essential genes. Clin Epigenetics. 2021;13(1):138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Karki R, Sharma BR, Tuladhar S, et al. Synergism of TNF‐α and IFN‐γ triggers inflammatory cell death, tissue damage, and mortality in SARS‐CoV‐2 infection and cytokine shock syndromes. Cell. 2021;184(1):149‐168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Shalapour S, Karin M. Immunity, inflammation, and cancer: an eternal fight between good and evil. J Clin Invest. 2015;125(9):3347‐3355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Cantuti‐Castelvetri L, Ojha R, Pedro LD, et al. Neuropilin‐1 facilitates SARS‐CoV‐2 cell entry and infectivity. Science. 2020;370(6518):856‐860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wang K, Chen W, Zhang Z, et al. CD147‐spike protein is a novel route for SARS‐CoV‐2 infection to host cells. Signal Transduct Target Ther. 2020;5(1):283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hoffmann M, Kleine‐Weber H, Schroeder S, et al. SARS‐CoV‐2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181(2):271‐280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Perng YC, Lenschow DJ. ISG15 in antiviral immunity and beyond. Nat Rev Microbiol. 2018;16(7):423‐439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lim SO, Li CW, Xia W, et al. Deubiquitination and stabilization of PD‐L1 by CSN5. Cancer Cell. 2016;30(6):925‐939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zhou Z, Ren L, Zhang L, et al. Heightened innate immune responses in the respiratory tract of COVID‐19 patients. Cell Host Microbe. 2020;27(6):883‐890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kircheis R, Haasbach E, Lueftenegger D, Heyken WT, Ocker M, Planz O. NF‐κB pathway as a potential target for treatment of critical stage COVID‐19 patients. Front Immunol. 2020;11:598444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hariharan A, Hakeem AR, Radhakrishnan S, Reddy MS, Rela M. The role and therapeutic potential of NF‐kappa‐B pathway in severe COVID‐19 patients. Inflammopharmacology. 2021;29(1):91‐100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Nilsson‐Payant BE, Uhl S, Grimont A, et al. The NF‐κB transcriptional footprint is essential for SARS‐CoV‐2 replication. J Virol. 2021;95(23):e0125721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bagca BG, Avci CB. The potential of JAK/STAT pathway inhibition by ruxolitinib in the treatment of COVID‐19. Cytokine Growth Factor Rev. 2020;54:51‐62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hariharan A, Hakeem AR, Radhakrishnan S, Reddy MS, Rela M. The role and therapeutic potential of NF‐kappa‐B pathway in severe COVID‐19 patients. Inflammopharmacology. 2021;29:91‐100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Heuberger J, Trimpert J, Vladimirova D, et al. Epithelial response to IFN‐γ promotes SARS‐CoV‐2 infection. EMBO Mol Med. 2021;13(4):e13191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Wolf Y, Anderson AC, Kuchroo VK. TIM3 comes of age as an inhibitory receptor. Nat Rev Immunol. 2020;20(3):173‐185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Farahani M, Niknam Z, Mohammadi Amirabad L, et al. Molecular pathways involved in COVID‐19 and potential pathway‐based therapeutic targets. Biomed Pharmacother. 2022;145:112420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Yu ZL, Liu JY, Chen G. Small extracellular vesicle PD‐L1 in cancer: the knowns and unknowns. NPJ Precis Oncol. 2022;6(1):42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Gu D, Ao X, Yang Y, Chen Z, Xu X. Soluble immune checkpoints in cancer: production, function and biological significance. J Immunother Cancer. 2018;6(1):132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Shiraishi T, Toyozumi T, Sakata H, et al. Soluble PD‐L1 concentration is proportional to the expression of PD‐L1 in tissue and is associated with a poor prognosis in esophageal squamous cell carcinoma. Oncology. 2022;100(1):39‐47. [DOI] [PubMed] [Google Scholar]
- 62. Supasa P, Zhou D, Dejnirattisai W, et al. Reduced neutralization of SARS‐CoV‐2 B.1.1.7 variant by convalescent and vaccine sera. Cell. 2021;184(8):2201‐2211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Israelow B, Song E, Mao T, et al. Mouse model of SARS‐CoV‐2 reveals inflammatory role of type I interferon signaling. J Exp Med. 2020;217(12):e20201241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Guan W, Ni Z, Hu Y, et al. Clinical characteristics of coronavirus disease 2019 in China. N Engl J Med. 2020;382(18):1708‐1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Tefferi A, Pardanani A. Serious adverse events during ruxolitinib treatment discontinuation in patients with myelofibrosis. Mayo Clin Proc. 2011;86(12):1188‐1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Auborn K, Abramson A, Bradlow HL, Sepkovic D, Mullooly V. Estrogen metabolism and laryngeal papillomatosis: a pilot study on dietary prevention. Anticancer Res. 1998;18(6b):4569‐4573. [PubMed] [Google Scholar]
- 67. Leibelt DA. Evaluation of chronic dietary exposure to indole‐3‐carbinol and absorption‐enhanced 3,3'‐diindolylmethane in sprague‐dawley rats. Toxicol Sci. 2003;74(1):10‐21. [DOI] [PubMed] [Google Scholar]
- 68. Sun CP, Jan JT, Wang IH, et al. Rapid generation of mouse model for emerging infectious disease with the case of severe COVID‐19. PLoS Pathog. 2021;17(8):e1009758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Ho JSY, Mok BWY, Campisi L, et al. TOP1 inhibition therapy protects against SARS‐CoV‐2‐induced lethal inflammation. Cell. 2021;184(10):2618‐2632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Constantine GM, Lionakis MS. Recent advances in understanding inherited deficiencies in immunity to infections. F1000Research. 2020;9:243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Li CW, Lim SO, Chung EM, et al. Eradication of triple‐negative breast cancer cells by targeting glycosylated PD‐L1. Cancer Cell. 2018;33(2):187‐201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Ju X, Zhang H, Zhou Z, Wang Q. Regulation of PD‐L1 expression in cancer and clinical implications in immunotherapy. Am J Cancer Res. 2020;10(1):1‐11. [PMC free article] [PubMed] [Google Scholar]
- 73. Antonangeli F, Natalini A, Garassino MC, Sica A, Santoni A, Di Rosa F. Regulation of PD‐L1 expression by NF‐κB in cancer. Front Immunol. 2020;11:584626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Zhang W, Liu Y, Yan Z, et al. IL‐6 promotes PD‐L1 expression in monocytes and macrophages by decreasing protein tyrosine phosphatase receptor type O expression in human hepatocellular carcinoma. J Immunother Cancer. 2020;8(1):e000285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Song S, Yuan P, Wu H, et al. Dendritic cells with an increased PD‐L1 by TGF‐β induce T cell anergy for the cytotoxicity of hepatocellular carcinoma cells. Int Immunopharmacol. 2014;20(1):117‐123. [DOI] [PubMed] [Google Scholar]
- 76. Hondermarck H, Bartlett NW, Nurcombe V. The role of growth factor receptors in viral infections: an opportunity for drug repurposing against emerging viral diseases such as COVID‐19? FASEB Bioadv. 2020;2(5):296‐303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Gubernatorova EO, Gorshkova EA, Polinova AI, Drutskaya MS. IL‐6: relevance for immunopathology of SARS‐CoV‐2. Cytokine Growth Factor Rev. 2020;53:13‐24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Ferreira‐Gomes M, Kruglov A, Durek P, et al. SARS‐CoV‐2 in severe COVID‐19 induces a TGF‐β‐dominated chronic immune response that does not target itself. Nat Commun. 2021;12(1):1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Bouadma L, Wiedemann A, Patrier J, et al. Immune alterations in a patient with SARS‐CoV‐2‐Related acute respiratory distress syndrome. J Clin Immunol. 2020;40(8):1082‐1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Gou Q, Dong C, Xu H, et al. PD‐L1 degradation pathway and immunotherapy for cancer. Cell Death Dis. 2020;11(11):955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Garcia‐Diaz A, Shin DS, Moreno BH, et al. Interferon receptor signaling pathways regulating PD‐L1 and PD‐L2 expression. Cell Rep. 2017;19(6):1189‐1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Huang HC, Liao CC, Wang SH, et al. Hyperglycosylated spike of SARS‐CoV‐2 gamma variant induces breast cancer metastasis. Am J Cancer Res. 2021;11(10):4994‐5005. [PMC free article] [PubMed] [Google Scholar]
- 83. Bagheri A, Moezzi SMI, Mosaddeghi P, et al. Interferon‐inducer antivirals: potential candidates to combat COVID‐19. Int Immunopharmacol. 2021;91:107245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Ahmadi M, Saffarzadeh N, Habibi MA, Hajiesmaeili F, Rezaei NColon Cancercancer and SARS‐CoV‐2: Impactimpact of ACE2 Expressionexpression in Susceptibilitysusceptibility to COVID‐19. bioRxiv. 2020.
- 85. Sidaway P. COVID‐19 and cancer: what we know so far. Nat Rev Clin Oncol. 2020;17(6):336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Gillessen S, Powles T. Advice regarding systemic therapy in patients with urological cancers during the COVID‐19 pandemic. Eur Urol. 2020;77(6):667‐668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Postow MA, Sidlow R, Hellmann MD. Immune‐related adverse events associated with immune checkpoint blockade. N Engl J Med. 2018;378(2):158‐168. [DOI] [PubMed] [Google Scholar]
- 88. da Costa CM, de Souza ZS, Real Salgues AC, et al. COVID‐19 in a patient with advanced Merkel cell carcinoma receiving immunotherapy. Immunotherapy. 2020;12(15):1133‐1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Moritz RKC, Gutzmer R, Zimmer L, et al. SARS‐CoV‐2 infections in melanoma patients treated with PD‐1 inhibitors: a survey of the German ADOREG melanoma registry. Eur J Cancer. 2021;144:382‐385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Isgrò MA, Vitale MG, Celentano E, et al. Immunotherapy may protect cancer patients from SARS‐CoV‐2 infection: a single‐center retrospective analysis. J Transl Med. 2021;19(1):132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Choi J, Lee SY. Clinical characteristics and treatment of immune‐related adverse events of immune checkpoint inhibitors. Immune Netw. 2020;20(1):e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Bersanelli M. Controversies about COVID‐19 and anticancer treatment with immune checkpoint inhibitors. Immunotherapy. 2020;12(5):269‐273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Mandala M, Lorigan P, De Luca M, et al. SARS‐CoV‐2 infection and adverse events in patients with cancer receiving immune checkpoint inhibitors: an observational prospective study. J Immunother Cancer. 2021;9(2):e001694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Faget J, Peters S, Quantin X, Meylan E, Bonnefoy N. Neutrophils in the era of immune checkpoint blockade. J Immunother Cancer. 2021;9(7):e002242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. de Candia P, Prattichizzo F, Garavelli S, Matarese G. T cells: warriors of SARS‐CoV‐2 infection. Trends Immunol. 2021;42(1):18‐30. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary information.
Data Availability Statement
Data deposition: The Illumina reads reported in this paper have been deposited in the NCBI Sequence Read Archive, https://www.ncbi.nlm.nih.gov/sra (accession no. PRJNA744316).