

Abstract
Cerebral cortical encephalitis (CCE) is a recently described myelin oligodendrocyte glycoprotein antibody‐associated disease (MOGAD) phenotype. In this observational retrospective study, we characterized 19 CCE patients (6.7% of our MOGAD cohort). Headache (n = 15, 79%), seizures (n = 13, 68%), and encephalopathy (n = 12, 63%) were frequent. Magnetic resonance imaging revealed unilateral (n = 12, 63%) or bilateral (n = 7, 37%) cortical T2 hyperintensity and leptomeningeal enhancement (n = 17, 89%). N‐Methyl‐D‐aspartate receptor autoantibodies coexisted in 2 of 15 tested (13%). CCE pathology (n = 2) showed extensive subpial cortical demyelination (n = 2), microglial reactivity (n = 2), and inflammatory infiltrates (perivascular, n = 1; meningeal, n = 1). Most received high‐dose steroids (n = 17, 89%), and all improved, but 3 had CCE relapses. This study highlights the CCE spectrum and provides insight into its pathogenesis. ANN NEUROL 2023;93:297–302
Introduction
Cerebral cortical encephalitis (CCE) is a recently described myelin oligodendrocyte glycoprotein antibody‐associated‐disease (MOGAD) phenotype characterized by cortical T2–fluid‐attenuated inversion recovery (T2‐FLAIR) hyperintensity on brain magnetic resonance imaging (MRI), first reported by Ogawa et al. 1
Although this phenotype is increasingly recognized, the literature is limited to a few case series and case reports, 2 , 3 , 4 , 5 and neuropathology data and details on treatment responses and outcomes are scarce. Moreover, data from US cohorts are lacking. We aim to report the clinical, radiologic, and pathologic features, as well as outcomes of CCE in a MOGAD cohort from the United States.
Patients and Methods
We searched our MOGAD cohort of patients seen at Mayo Clinic between January 1, 2000 and December 31, 2021. A total of 285 (196 adult onset, 89 childhood onset [age < 18 years]) cases were reviewed and MRIs screened (by C.V.‐S. and E.P.F.) for patients meeting the following inclusion criteria: (1) unilateral or bilateral cortical T2‐FLAIR hyperintensity, with or without corresponding leptomeningeal enhancement; (2) serum positivity for myelin oligodendrocyte glycoprotein (MOG)‐IgG; and (3) exclusion of alternative etiologies. 6
MOG‐IgG positivity was assessed by live cell‐based assay (florescence‐activated cell sorting), in fresh or stored serum (IgG1 secondary antibody) and cerebrospinal fluid (CSF; pan‐IgG secondary antibody) samples, at the Mayo Clinic Neuroimmunology Laboratory. An immunoglobulin‐binding index ≥ 2.5 or titer ≥ 1:20 was considered positive, as previously described. 6
We collected demographic, clinical, radiologic, laboratory, treatment, and outcome data. In patients with relapsing CCE, information was collected from the first event. A neuroradiologist (K.N.K.) reviewed brain MRI of included cases for cortical T2 hyperintensity and swelling, white matter T2 hyperintensity or hypointensity, and T1 postgadolinium enhancement. 1 , 2 , 5 Follow‐up MRIs beyond 6 months were evaluated for lesion resolution.
Continuous and categorical variables were reported as median (range) and number (%).
Neuropathological evaluation was undertaken with formalin‐fixed paraffin‐embedded 5μm‐thick sections that were stained with hematoxylin and eosin. Immunohistochemistry was performed with the EnVision FLEX immunohistochemistry system (Dako, Glostrup, Denmark) after steam antigen retrieval with citric acid buffer (pH 6.0, Dako). The following primary antibodies were applied: PLP (1:500; Serotec, Oxford, UK), myelin‐associated glycoprotein (MAG; 1:1,000; Abcam, Cambridge, MA), MOG (1:1,000, Abcam), 2′,3′‐cyclic‐nucleotide 3′‐phosphodiesterase (CNPase; 1:400; BioLegend, San Diego, CA), CD68 KP1 (1:100, Dako), CD4 (1:100, Dako), CD8 (1:50, Dako), CD35 (1:50, Dako), Ki67 (1:400; MilliporeSigma, Burlington, MA), neurofilament protein (1:800, Dako), C9neo (polyclonal, 1:200; from Prof Paul Morgan, Cardiff, UK). The demyelinating activity was defined based on myelin debris within macrophages. 7
The study was approved by the Mayo Clinic institutional review board. All patients, or their parents, consented to the use of their medical records for research purposes. Eight patients were included in prior publications. 8 , 9 , 10 , 11
Results
Frequency, Demographics, and Clinical Features of CCE in MOGAD
A total of 19 patients were included, representing 6.7% of our MOGAD cohort; their demographics and clinical features are outlined in the Table S1. The median age was 14 years (range = 2–47). CCE occurred in 13.5% (12/89) of our MOGAD patients with childhood onset, and in 3.6% (7/196) of patients with adult onset. Twelve patients (63%) were female. Clinical features included headache (n = 15, 79%), seizures (n = 13, 68%), encephalopathy (n = 12, 63%), focal cortical features (n = 10, 53%: aphasia, n = 5; hemiparesis, n = 5), and fever (n = 8, 42%).
Seizure semiology included focal motor onset with preserved (n = 2) or impaired awareness (n = 5), focal nonmotor onset with impaired awareness (n = 4), and unknown onset tonic–clonic (n = 2). Four had secondary generalization (1 with status epilepticus).
Encephalopathy was postictal in 3 (1 requiring intubation). In the remaining 9 patients, encephalopathy was mild (n = 4; somnolence, irritability, decreased activity), moderate (n = 4; confusion, disorientation, agitation, lethargy), or severe (n = 1; comatose requiring intubation).
In 1 case, cortical T2‐FLAIR hyperintensity was asymptomatically detected on surveillance MRI and right optic neuritis developed 3 weeks later. CCE was the first attack in 13 (68%), and other MOGAD syndromes (eg, optic neuritis) occurred within 1 month in 12 (63%), with 3 fulfilling acute disseminated encephalomyelitis (ADEM) criteria. Attack details are summarized in the Table S1.
Laboratory Features in CCE
CSF Analysis
CSF (see Table S1) revealed pleocytosis in 16 of 17 (94%, median = 49 white blood cells [WBC]/μl, range = 10–505, normal = 0–5). Oligoclonal bands were absent in all 15 tested. Opening pressure was elevated (>250mmH2O) in 4 of 7 assessed.
Serology
Serum MOG‐IgG median end‐titer (available in 10) was 1:100 (range = 1:40–1:1,000) and CSF MOG‐IgG was positive in 6 of 7 tested. CSF N‐methyl‐D‐aspartate receptor (NMDA‐R) antibodies coexisted in 2 of 15 patients tested (13%). The full details are included in Table S1.
MRI Findings
MRI showed cortical swelling with T2‐FLAIR hyperintensity (unilateral, n = 12, 63%; bilateral, n = 7, 37%) and gadolinium enhancement in 17 (89%; leptomeningeal, n = 17; cortical, n = 5; Figs 1 and 2 and Table S1). Adjacent subcortical T2‐FLAIR hypointensity was observed in all cases. Ten patients (53%) had concomitant cerebral parenchymal T2 hyperintensities (see Table S1). Follow‐up imaging beyond 6 months revealed resolution of the cortical abnormalities in 12 of 13 available.
FIGURE 1.
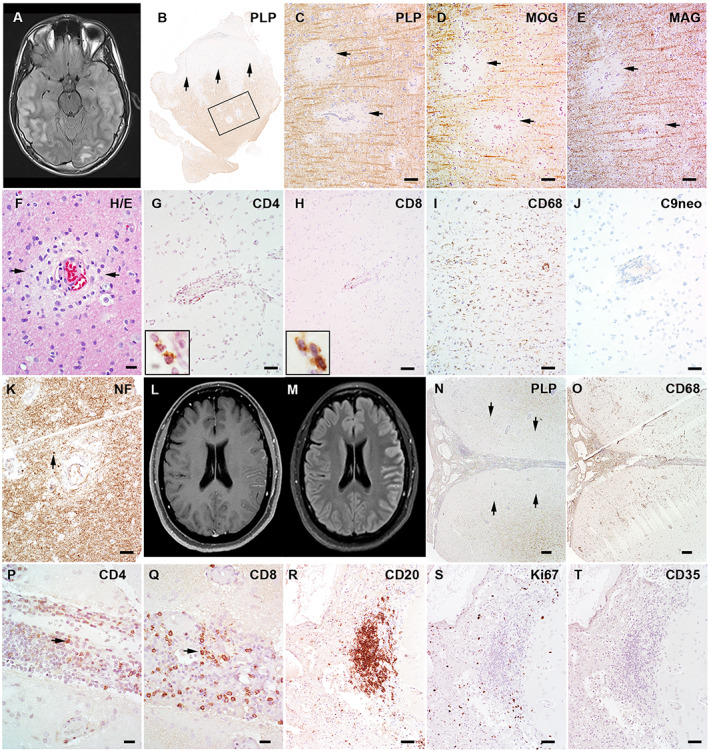
The magnetic resonance imaging (MRI) and pathology of 2 patients with myelin oligodendrocyte glycoprotein (MOG) antibody‐associated disease cerebral cortical encephalitis. (A–K) Patient 12 (Table S1). (A) The axial MRI fluid‐attenuated inversion recovery (FLAIR) image shows swelling and T2 hyperintensity in the right more than left cortex and in subcortical white matter. (B) Cortical biopsy of the right temporal lobe in this patient shows diffuse subpial cortical demyelination (arrows) and small perivascular intracortical demyelination within the frame (proteolipid protein [PLP] immunohistochemistry). (C–E) The enlarged views of framed region in B show perivascular myelin loss to an equal extent in PLP (C), MOG (D), and myelin‐associated glycoprotein (MAG; E). (F) Hematoxylin and eosin (H&E) stain shows mild perivascular infiltration and edema. (G, H) Immunohistochemistry reveals perivascular infiltration of both CD4+ (G) and CD8+ (H) T lymphocytes. (I) Extensive microglial reactions are present beyond the demyelination in the cortex. (J) No C9 neoantigen (terminal complement activation product) is present. (K) Axons are preserved with mild perivascular spheroid formation (indicated with arrow). (L–Q) Patient 18 (Table S1). (L) Axial T1‐weighted postgadolinium MRI shows cortical and leptomeningeal enhancement. (M) Axial FLAIR images reveal cortical T2 hyperintensity and swelling in the left hemispheric cortex. (N) Left hemispheric cortical biopsy using the PLP stain shows extensive subpial cortical demyelination (indicated with arrows) with marked meningeal inflammatory infiltration. (O) CD68 stain highlights extensive macrophage/microglial reactivity in both meninges and subpial lesions. (P, Q) CD4+ (P) and CD8+ (Q) lymphocytes are both present in the subarachnoid space. (R) CD20 immunohistochemistry shows B‐cell abundant aggregates present in the leptomeninges. (S) The staining for the proliferation marker, Ki67, reveals no proliferating activity in the B‐cell aggregates. (T) CD35 immunohistochemistry shows absence of follicle dendritic cells. Scale bars: 100μm (C–E, I, K), 20μm (F, P, Q), 50μm (G, H, J, R‐T), 200μm (N, O). [Color figure can be viewed at www.annalsofneurology.org]
FIGURE 2.
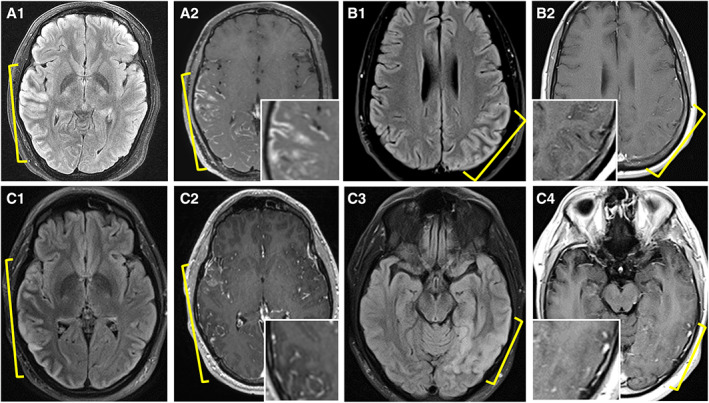
Additional magnetic resonance imaging examples of myelin oligodendrocyte glycoprotein antibody‐associated disease cerebral cortical encephalitis. (A) Axial fluid‐attenuated inversion recovery (FLAIR) image reveals right posterior temporo‐occipital cortical T2 hyperintensity and swelling (A1, yellow bracket), and accompanying T1‐weighted postgadolinium image reveals associated cortical and leptomeningeal enhancement (A2, yellow bracket [inset shows higher magnification]). (B) Axial FLAIR image reveals left parietal cortical T2 hyperintensity and swelling (B1, yellow bracket), and T1‐weighted postgadolinium image reveals concomitant subtle leptomeningeal enhancement (B2 [inset shows higher magnification]). (C) Axial FLAIR image reveals right temporal lobe cortical T2 hyperintensity and swelling (C1, yellow bracket) with T1‐weighted postgadolinium image revealing accompanying leptomeningeal enhancement (C2, yellow bracket [inset shows higher magnification]). Follow‐up imaging 18 months later reveals resolution of right temporal lobe cortical swelling, T2 hyperintensity, and enhancement with new left temporal T2 hyperintensity (C3, yellow bracket) with subtle leptomeningeal enhancement although slightly limited by motion artifact (C4, yellow bracket [inset shows higher magnification]) in a patient with recurrent cerebral cortical encephalitis attacks. [Color figure can be viewed at www.annalsofneurology.org]
Pathology Details
Pathology data were available for 2 patients. The first case (see Fig 1A–K) revealed cortical perivascular edema, hypertrophic astrogliosis, and mild perivascular lymphocytic infiltration. The ratio of infiltrated CD4/CD8 lymphocytes was 2.4 within the Virchow–Robin space without diffuse parenchymal infiltration observed (subarachnoid space was not counted due to meningeal hemorrhage). Perivascular demyelination was present in both the cortex and subcortical white matter, with equal loss of MAG, MOG, and CNPase. The demyelination was inactive in both cortex and white matter characterized by the absence of myelin‐laden macrophages. Terminal complement activation product (C9neo) was absent in meninges, cortex, and white matter. Axons were preserved with scattered spheroids in the perivascular region. The second biopsy (see Fig 1L–Q) showed marked meningeal inflammation, extensive macrophage/microglial reactivity in both meninges and cortex, and extensive subpial cortical inactive demyelination. CD4/CD8 lymphocytic ratio was 0.7 (Virchow–Robin space), 1.1 (subarachnoid space), and 1.4 (diffuse parenchyma). Focal meningeal CD20+ B‐cell abundant aggregates were present (see Fig 1R–T). Ki67 staining showed no active proliferation in the B‐cell abundant aggregates. The immunohistochemistry of follicular dendritic cell marker CD35 showed no immunoreactivity was present. The histology of the B‐cell aggregates in the MOGAD case was different from the ectopic B‐cell follicles found in multiple sclerosis (MS). 12
Initial Diagnosis Assigned
Initially, diagnoses were infectious meningoencephalitis, n = 4; MS, n = 2; vasculitis, n = 2; seizures from suspected cortical dysplasia, n = 1.
Treatments and Outcome
Acute treatments of CCE included one or more of the following: high‐dose steroids, n = 17 (89%); intravenous immunoglobulin, n = 1; or plasma exchange, n = 1. Eleven (58%) patients received antiseizure medications, including 2 who did not receive immunotherapy. Maintenance immunotherapy details are included in the Table S1.
By last follow‐up (median = 3.6 years, range = 0.1–20.3), all experienced improvement, with an Expanded Disability Status Scale of ≤2 in 17 (89%) and of 0 in 12 (63%). Eight patients continued on antiseizure medications, and 1 had drug‐resistant epilepsy. Five patients (26%) had a monophasic course, and 3 had recurrent CCE; attack details are summarized in the Table S1.
Discussion
This study provides new information on CCE frequency in MOGAD, along with extensive clinical, imaging, and pathological analysis in a US cohort. We observed CCE in 6.7% of MOGAD patients, and children predominated. Headaches, seizures, encephalopathy, other focal cortical features, and fever were frequent manifestations, whereas 1 patient was asymptomatic. Initial misdiagnosis occurred in 47%, as isolated CCE did not always prompt consideration of central nervous system demyelination and MOG‐IgG testing, occasionally resulting in unnecessary brain biopsy. Testing CSF NMDA‐R–IgG is important in CCE, given that it coexists in 13% and if positive would prompt a search for teratoma and influence treatment decisions. CCE outcomes were favorable, with brisk steroid response similar to prior reports. 1 , 2 , 3 , 13
CCE is often unilateral and isolated, with concomitant leptomeningeal enhancement but bilateral cortical T2‐FLAIR hyperintensity in one‐third, 14 , 15 and involvement of other brain regions in 53% warrants emphasis. The MOGAD cortical T2‐hyperintense lesions and leptomeningeal enhancement in CCE are more extensive than in MS, although CCE MRI changes can be subtle (see Fig 2B). The frequent cortical T2 lesion resolution also discriminated from MS, 16 and pathologic cortical demyelination excluded seizure‐related signal abnormalities, which can have a similar appearance. 17 Adjacent subcortical T2‐FLAIR hypointensity in CCE was frequent, as noted before, 5 potentially reflecting a transient decrease in interstitial fluid due to cortical dysfunction impeding glymphatic flow into cerebral white matter.
The pathophysiology of CCE in MOGAD remains poorly understood. Prior MOGAD pathology studies showed frequent cortical demyelinating lesions even without a CCE phenotype. 18 , 19 Herein, the 2 CCE cases with pathology showed prominent leptomeningeal inflammation with microglial infiltration along with extensive cortical subpial demyelination. Cortical subpial demyelination was also described in 2 prior CCE cases, 19 and 1 fulminant CCE case showed extensive intracortical demyelination, activated microglia, and macrophages. 18 Conversely, 2 CCE cases had inflammatory changes without demyelination, possibly from early stage biopsy, prior treatment effect, a sampling issue, or seizure‐related changes. 15 , 18 , 20 Meningeal inflammation is more pronounced in MOGAD than MS, showing a higher frequency of meningeal and perivascular inflammatory infiltrates and a higher proportion of CD4+ T cells. B‐cell aggregates found in the second biopsy showed no ectopic lymphoid follicles, which has been reported in MS meninges. 12
The retrospective nature, potential selection bias toward more severe cases, and lack of sufficient tissue to do all stains in both pathology samples were limitations.
To conclude, this study improves our understanding of this characteristic MOGAD phenotype and will help clinicians identify CCE in practice.
Author Contributions
C.V.‐S., Y.G., C.F.L., and E.P.F. contributed to the conception and design of the study. All authors contributed to the acquisition and analysis of data. C.V.‐S., Y.G., C.F.L., and E.P.F. contributed to drafting the text and preparing the figures.
Potential Conflicts of Interest
C.V.‐S., Y.G., E.S., M.M., K.N.K., P.M.E., V.R., J.‐M.T., A.B., C.F.L., report no conflicts of interest. J.J.C. has served as a consultant for Roche and UCB, which have upcoming treatment trials in MOGAD. S.L.‐C. and A.K. have served on an advisory board for Genentech/Roche, which has an upcoming treatment trial in MOGAD. D.D. has consulted for UCB, which has an upcoming treatment trial in MOGAD. S.J.P. reports grants, personal fees, and nonfinancial support from Roche/Genentech, and personal fees for consulting services from UCB, which have upcoming treatment trials in MOGAD. E.P.F. has participated in advisory boards for Roche and UCB who have upcoming treatment trials in MOGAD. E.P.F. has received funding from the NIH (R01NS113828). Mayo Clinic Laboratories offer commercial testing for MOG‐IgG, but none of the authors receive financial compensation for this.
Supporting information
Table S1. Characteristics of 19 Patients with Cerebral Cortical Encephalitis in Myelin Oligodendrocyte Glycoprotein Antibody‐Associated Disease
Acknowledgment
This study was funded by grant funding from the NIH National Institute of Neurological Disorders and Stroke (R01NS113828).
C.F.L. and E.P.F. contributed equally.
Contributor Information
Claudia F. Lucchinetti, Email: clucchinetti@mayo.edu.
Eoin P. Flanagan, Email: flanagan.eoin@mayo.edu.
Data Availability Statement
Anonymized data are available upon reasonable request.
References
- 1. Ogawa R, Nakashima I, Takahashi T, et al. MOG antibody‐positive, benign, unilateral, cerebral cortical encephalitis with epilepsy. Neurol Neuroimmunol Neuroinflamm 2017;4:e322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Budhram A, Mirian A, Le C, et al. Unilateral cortical FLAIR‐hyperintense lesions in anti‐MOG‐associated encephalitis with seizures (FLAMES): characterization of a distinct clinico‐radiographic syndrome. J Neurol 2019;266:2481–2487. [DOI] [PubMed] [Google Scholar]
- 3. Yao T, Zeng Q, Xie Y, et al. Clinical analysis of adult MOG antibody‐associated cortical encephalitis. Mult Scler Relat Disord 2022;5:103727. [DOI] [PubMed] [Google Scholar]
- 4. Shu H, Ding M, Shang P, et al. Myelin oligodendrocyte glycoprotein antibody associated cerebral cortical encephalitis: case reports and review of literature. Front Hum Neurosci 2021;15:782490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fujimori J, Ogawa R, Murata T, et al. Decreased subcortical T2 FLAIR signal with cortical T2 FLAIR hyperintense lesions in unilateral cerebral cortical encephalitis with myelin oligodendrocyte glycoprotein antibody. Neuroimmunol Rep 2022;2:100096. [Google Scholar]
- 6. López‐Chiriboga AS, Majed M, Fryer J, et al. Association of MOG‐IgG Serostatus with relapse after acute disseminated encephalomyelitis and proposed diagnostic criteria for MOG‐IgG‐associated disorders. JAMA Neurol 2018;75:1355–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Brück W, Porada P, Poser S, et al. Monocyte/macrophage differentiation in early multiple sclerosis lesions. Ann Neurol 1995;38:788–796. [DOI] [PubMed] [Google Scholar]
- 8. Budhram A, Sechi E, Nguyen A, et al. FLAIR‐hyperintense lesions in anti‐MOG‐associated encephalitis with seizures (FLAMES): is immunotherapy always needed to put out the fire? Mult Scler Relat Disord 2020;44:102283. [DOI] [PubMed] [Google Scholar]
- 9. Budhram A, Kunchok AC, Flanagan EP. Unilateral leptomeningeal enhancement in myelin oligodendrocyte glycoprotein immunoglobulin G‐associated disease. JAMA Neurol 2020;77:648–649. [DOI] [PubMed] [Google Scholar]
- 10. Kunchok A, Flanagan EP, Krecke KN, et al. MOG‐IgG1 and co‐existence of neuronal autoantibodies. Mult Scler 2021;27:1175–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhao‐Fleming HH, Valencia Sanchez C, Sechi E, et al. CNS demyelinating attacks requiring Ventilatory support with myelin oligodendrocyte glycoprotein or Aquaporin‐4 antibodies. Neurology 2021;97:e1351–e1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Magliozzi R, Howell O, Vora A, et al. Meningeal B‐cell follicles in secondary progressive multiple sclerosis associate with early onset of disease and severe cortical pathology. Brain 2007;130:1089–1104. [DOI] [PubMed] [Google Scholar]
- 13. Wang L, ZhangBao J, Zhou L, et al. Encephalitis is an important clinical component of myelin oligodendrocyte glycoprotein antibody associated demyelination: a single‐center cohort study in Shanghai. China Eur J Neurol 2019;26:168–174. [DOI] [PubMed] [Google Scholar]
- 14. Fujimori J, Nakamura M, Yagihashi T, Nakashima I. Clinical and radiological features of adult onset bilateral medial frontal cerebral cortical encephalitis with anti‐myelin oligodendrocyte glycoprotein antibody. Front Neurol 2020;11:600169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fujimori J, Takai Y, Nakashima I, et al. Bilateral frontal cortex encephalitis and paraparesis in a patient with anti‐MOG antibodies. J Neurol Neurosurg Psychiatry 2017;88:534–536. [DOI] [PubMed] [Google Scholar]
- 16. Sechi E, Krecke KN, Messina SA, et al. Comparison of MRI lesion evolution in different central nervous system demyelinating disorders. Neurology 2021;97:e1097–e1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cianfoni A, Caulo M, Cerase A, et al. Seizure‐induced brain lesions: a wide spectrum of variably reversible MRI abnormalities. Eur J Radiol 2013;82:1964–1972. [DOI] [PubMed] [Google Scholar]
- 18. Höftberger R, Guo Y, Flanagan EP, et al. The pathology of central nervous system inflammatory demyelinating disease accompanying myelin oligodendrocyte glycoprotein autoantibody. Acta Neuropathol 2020;139:875–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Takai Y, Misu T, Kaneko K, et al. Myelin oligodendrocyte glycoprotein antibody‐associated disease: an immunopathological study. Brain 2020;143:1431–1446. [DOI] [PubMed] [Google Scholar]
- 20. Ikeda T, Yamada K, Ogawa R, et al. The pathological features of MOG antibody‐positive cerebral cortical encephalitis as a new spectrum associated with MOG antibodies: a case report. J Neurol Sci 2018;15:113–115. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Characteristics of 19 Patients with Cerebral Cortical Encephalitis in Myelin Oligodendrocyte Glycoprotein Antibody‐Associated Disease
Data Availability Statement
Anonymized data are available upon reasonable request.