

Abstract
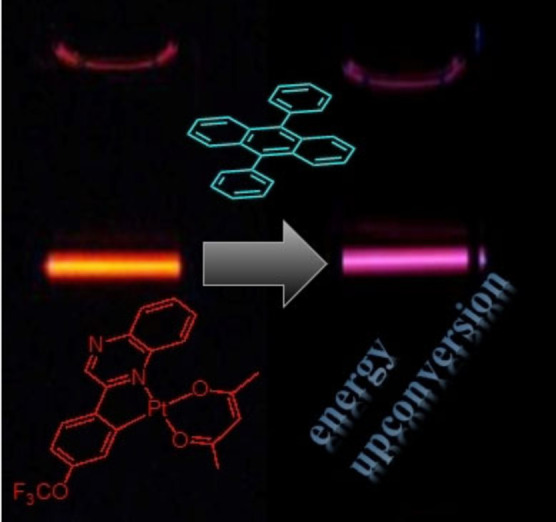
A series of 2‐phenylquinoxaline ligands have been synthesised that introduce either CF3 or OCF3 electron‐withdrawing groups at different positions of the phenyl ring. These ligands were investigated as cyclometalating reagents for platinum(II) to give neutral complexes of the form [Pt(C^N)(acac)] (in which C^N=cyclometalating ligand; acac=acetyl acetonate). X‐ray crystallographic studies on three examples showed that the complexes adopt an approximate square planar geometry. All examples revealed strong Pt−Pt linear contacts of 3.2041(6), 3.2199(3) and 3.2586(2) Å. The highly coloured complexes display efficient visible absorption at 400–500 nm (ϵ ≈5000 M−1 cm−1) and orange red photoluminescent characteristics (λ em=603–620 nm; Φ em ≤37 %), which were subtly tuned by the ligand. Triplet emitting character was confirmed by microsecond luminescence lifetimes and the photogeneration of singlet oxygen with quantum efficiencies up to 57 %. Each complex was investigated as a photosensitiser for triplet–triplet annihilation energy upconversion using 9,10‐diphenylanthracene as the annihilator species: a range of good upconversion efficiencies (Φ UC 5.9–14.1 %) were observed and shown to be strongly influenced by the ligand structure in each case.
Keywords: DFT calculations, platinum complexes, quinoxalines, spectroscopy, upconversion
A series of substituted 2‐phenylquinoxaline ligands have been explored to finely tune the electronic properties of cyclometalated platinum(II) complexes. The complexes were successfully applied as photosensitisers in triplet–triplet annihilation upconversion experiments, demonstrating tuneable upconversion quantum yields of up to 14 %.
Introduction
Luminescent, cyclometalated Pt(II) complexes continue to attract significant attention due to their advantageous application to a wide range of research disciplines. [1] The diamagnetic d8 electronic configuration of Pt(II) implies a square planar coordination geometry and, commonly, mixed ligand complexes can be isolated that have various formulations, including both bidentate and terdentate [2] cyclometalating ligands. As with related cyclometalated Ir(III) complexes, the luminescent properties of Pt(II) species are influenced by the types of ligand in the coordination sphere. The cyclometalating ligand, in particular, can be critical in dictating the luminescent properties of Pt(II) species and a number of studies have investigated effective colour tuning of Pt(II) complexes. This control exerted upon the photophysics of Pt(II) complexes is key to their consideration in light emitting devices, [3] photocatalysis, sensitisers in photooxidation, solar light harvesting, [4] and luminescence bioimaging. [5]
The often highly planar structures of Pt(II) complexes also lends an advantage with respect to applications in the development of novel and functional materials. In particular, responsive sensors [6] based upon Pt(II) complexes have been reported that show vapochromic behaviour. [7] Commonly, the presence of axial Pt⋅⋅⋅Pt interactions [8] provide an additional design feature for supporting stacked structures, and an opportunity to further tune electronics for the consideration of different applications. [9]
Energy upconversion has transformative potential in applications such as photocatalysis and solar cell technologies, both of which could greatly benefit from more efficient harvesting of solar radiation. [10] This is especially important in the case of photocatalysis because it can potentially allow the use of ambient sunlight rather than dedicated light sources such as LEDs, but major technological challenges still remain. [10] Optical bioimaging techniques are also limited by the absorption properties of tissue and biological media; again, exploitation of longer wavelength excitation sources, including the NIR where tissue becomes more transparent, imply that deploying energy upconversion into the design of imaging agents (and theranostic variants) can significantly help to address these challenges. [11]
Triplet–triplet annihilation energy upconversion (TTA‐UC) [12] is a specific class of upconversion that has attracted significant interest because it can demonstrate much improved efficiencies, and thus advantages, compared to other types of upconversion. TTA‐UC requires both a photosensitiser (the component that absorbs the incident light) and an annihilator component (which accepts energy and provides the upconverted light) and in recent years, photoactive metal complexes have proven to be very effective photosensitisers. Attributes such as good molar absorptivity in the visible region (often via charge transfer transitions), and long triplet lifetimes can help drive efficient TTA‐UC. The ease with which photoactive metal complexes can be finely tuned and optimised is thus a distinct advantage in the optimisation of TTA‐UC. A handful of photoactive complexes based on Ru(II), [13] Re(I) [14] and Ir(III) [15] have attracted attention as photosensitisers, and each system has its potential advantages. In particular, we have previously shown that cyclometalated Ir(III) complexes can be successfully utilised to produce TTA‐UC efficiencies up to 39.3 %. [16] More recently an interesting report using a phosphorescent Cr(III) complex has also demonstrated application in TTA‐UC; the viability of earth‐abundant, metal‐based photosensitisers is also an important advance. [17]
Luminescent Pt(II) complexes do have precedent as photosensitisers in TTA‐UC and generally fall into three categories (Scheme 1). Firstly, the well‐known Pt(II) porphyrinates, for example, platinum octaethylporphyrin (PtOEP) [18] which is a deep red emitter that displays efficient absorption in the visible region, high emission quantum yields (>50 %) and long lifetimes (∼90 μs), [19] have been successfully investigated in TTA‐UC in a variety of guises, [20] with a reported TTA‐UC efficiency of 23 %. [21] Secondly, organometallic complexes of Pt(II) have also been investigated; mixed‐ligand species that incorporate chromophore‐functionalised alkynyl ligands (e.g., a naphthalimide derived alkyne, Pt‐1) provide a framework for significantly extending the triplet excited state lifetime of the Pt(II) complex and enabling TTA‐UV efficiencies >20 %. [22] Thirdly, Schiff base ligand complexes, and in particular those that possess a [Pt(N^O)2] coordination sphere, have also shown good applicability as photosensitisers. [23] In a similar design concept to Pt‐1, recent reports have shown that the backbone of the Schiff base ligand can be conjugated with an anthryl chromophore that significantly extends the triplet excited state lifetime (through triplet excited state equilibration) of the complex (Pt‐2) and then yields TTA‐UC efficiencies up to 15 %. [24]
Scheme 1.

Examples of Pt(II) complexes that have been successfully employed as photosensitizers for TTA‐UC in the solution state.
Herein, we describe the synthesis of a series of heteroleptic, cyclometalated Pt(II) complexes that incorporate different organofluorine, CF3 or OCF3, substituted 2‐phenylquinoxaline ligands. The ligand substituents are placed in different positions on the phenyl ring donor to enable subtle modulation of the electronic properties of the complex. The complexes were successfully characterised in both solution and solid state and were shown to possess favourable and efficient photophysical properties that facilitate their use as photosensitisers for TTA‐UC. The variation in complex structures provided a range of TTA‐UC efficiencies up to approximately 14 % for the best performing species.
Results and Discussion
Synthesis of the 2‐phenylquinoxalines
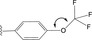
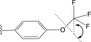
Functionalised quinoxalines represent a useful ligand framework for coordination chemistry studies. [25] Quinoxalines can be synthesized using a range of differing approaches and materials. [26] This has provided quinoxaline‐based ligands that can act as chelators for a variety of different metal ions [27] including a few examples as cyclometalating (C^N) ligands for Ru, [28] Rh, [29] Ir [30] and Pt. [31] The current series of 2‐phenylquinoxaline ligands (HL1–6 ) were designed to allow incorporation of either OCF3 or CF3 substituents onto the different phenyl ring positions. The rationale was that these groups should, firstly, enhance the solubility of the resultant square planar Pt(II) complexes (both OCF3 and CF3 groups are well known to increase lipophilicity [32] ) and secondly, introduce subtle electronic tuning of the excited state nature of the complexes due to their electron withdrawing properties. [33]
From a synthetic perspective, the introduction of OCF3 groups to aryl species is challenging [34] and therefore commercially available OCF3 or CF3 substituted acetophenones (Scheme 2) were chosen as logical entry points to the target ligands. The corresponding 2‐bromoacetophenone species were obtained using dioxane‐dibromide (a solid‐form brominating reagent, that avoids the use of co‐reagents to the reaction). [35] Condensation of the 2‐bromoacetophenone with 1,2‐phenylenediamine (there are alternative ways of forming heterocyclic quinoxalines, but these condensations are generally reproducible) led to the isolation of the six prospective ligands; examples of the NMR spectral data are presented in the Supporting Information (Figures S2–S11) and summarised in the Experimental section.
Scheme 2.

Structures of the isolated 2‐phenyl quinoxaline ligands with synthetic route shown above. Numbering convention for quinoxaline shown top right.
Synthesis and characterisation of the Pt(II) complexes
Different synthetic procedures have been reported to produce cyclometalated Pt(II) species from tetrachloroplatinate(II). Typically, a bimetallic species [Pt(Ln )(μ‐Cl)]2 is formed following addition of the ligand and then the dimer is split using an additional co‐ligand. Initially, a water/2‐ethoxyethanol solvent mixture [36] was employed, however this failed to yield any identifiable Pt(II) dimer product. As an alternative, glacial acetic acid was employed as the solvent and heating tetrachloroplatinate(II) and HLn for three days resulted in the formation of a red‐brown solid that could be isolated by precipitation with water. The resultant crude dimer species were then stirred with a small volume of DMSO to yield the corresponding monometallic [PtCl(Ln )(DMSO)] species (Scheme 3). This synthetic procedure was unsuccessful in the case of HL6 , 2‐(2‐(trifluoromethyl)phenyl)quinoxaline, where the reaction with K2PtCl4 failed to yield product that could be adequately characterised. One could speculate that the likely cyclometalating arrangement of the ligand, which requires the approximate co‐planarity of the phenyl and quinoxaline rings, is inhibited by the steric influence of the ortho CF3 substituent in HL6 .
Scheme 3.

Synthetic route to the Pt(II) complexes using HL3 as an example. Reagents and conditions: i) K2PtCl4, glacial acetic acid, heat, 3 days; ii) DMSO, rt; iii) [Na(acac)], 2‐propanol, rt.
Finally, the [PtCl(Ln )(DMSO)] intermediates were reacted with sodium acetylacetonate to yield the target complexes, [Pt(Ln )(acac)], as highly coloured reddish brown solids. The resultant OCF3/CF3 functionalised Pt(II) complexes possess excellent solubility across a range of common solvents, including non‐polar hexane and toluene. The synthetic details of this approach are described in the Experimental section.
The complexes were characterised using a range of spectroscopic and analytical techniques. Firstly, high resolution mass spectrometry data was obtained for [Pt(Ln )(acac)] and revealed the [M+H]+ parent ion in all cases, with the appropriate isotopic distribution at m/z 584, [Pt(L1–3 )(acac)], and m/z 568, [Pt(L4–5 )(acac)]. In four cases higher m/z clusters were also noted (e.g., see Figure S1, Supporting Information) and attributed to the formation of dimerised species, with clusters identified as [M+M+H]+ and [M+M–acac]+.
Solution state characterisation was provided using multinuclear NMR spectroscopy (e.g., Figures S12–26). The unsymmetrical nature of the 2‐phenylquinoxaline ligand induces a number of resonances in the aromatic region which shift upon coordination to Pt(II). For reference, the signature singlet resonance associated with the 3‐position of the quinoxaline ring appears at 8.9–9.3 ppm in the free ligands and is the most downfield signal. Upon coordination to Pt(II) this environment shifts (ca. 9.1–9.7 ppm for the complexes), and typically the most downfield resonance is then associated with a proton on the metalated, organofluorine substituted phenyl ring. The incorporation of the acac ligand was confirmed by the presence of two (induced by the unsymmetrical quinoxaline ligand) methyl groups around 2.1 ppm, and the methine singlet resonance approximately 5.6 ppm. 13C{1H} NMR spectroscopy provided information on the inequivalent acac carbonyl groups with two distinct resonances between 180–190 ppm in all cases. 19F{1H} NMR spectroscopy gave a characteristic singlet resonance of −56.46, −57.95, and −56.93 ppm for the OCF3 species, [Pt(L1–3 )(acac)], and −62.23 and −63.01 ppm for the CF3 complexes, [Pt(L4–5 )(acac)], respectively. These data are indicative of a single fluorine environment in each case, [37] with a more deshielded fluorine environment evident for the OCF3 species. These δF values for the complexes typically represent a subtle deviation from the corresponding free ligands of −57.15, −57.64, −57.67, −62.65 and −62.74 ppm, respectively for HL1–5 .
Solid state IR spectra were also obtained on the complexes and showed a variety of bands that correlate with the presence of the two different ligands. Firstly, features approximately 1600 cm−1 ascribed to ν(C=O) indicated the coordinated acac ligand. IR active CF3 stretching vibrations typically occur between 1100–1200 cm−1 and the symmetric CF3 bending (δ s) modes around 750–800 cm−1. [38] While the CF3 stretches for these complexes fall within a detailed fingerprint region, the spectra do reveal quite strong absorptions around 1000–1200 cm−1. The spectra also showed a sharp feature approximately 760 cm−1 which is tentatively assigned to the δ s CF3 mode.
X‐ray Crystal Structures of [Pt(L1)(acac)], [Pt(L2)(acac)] and [Pt(L3)(acac)]
Single red blade‐shaped, [Pt(L1 )(acac)], both lath‐ and needle‐shaped for [Pt(L2 )(acac)], and red block‐shaped crystals for [Pt(L3 )(acac)] were isolated following slow evaporation of either dichloromethane or chloroform solutions of the complexes.
Data collection parameters, and selected bond lengths and bond angles are shown in Tables S1‐3, Supporting Information. Each of the structures (Figure 1) confirmed the expected formulations, with the 2‐phenylquinoxaline ligand coordinating in a C N cyclometalating mode. [39] In the case of [Pt(L2 )(acac)], where there are two possible positions of Pt−C bond formation, the coordination mode places the OCF3 substituent away from the coordination sphere (i.e., the OCF3 group is trans to the Pt−C bond).
Figure 1.

X‐ray structures of (top to bottom) [Pt(L1 )(acac)], [Pt(L2 )(acac)] at 100 K and [Pt(L3 )(acac)]. Thermal ellipsoids drawn at 50 %.
The complexes adopt an approximate square planar geometry, with significant deviations away from the ideal 90° bond angles. The lowest values were found within the N−Pt−C bond angles, 80.5(3)–81.16(10)°, which are attributed to the 5‐membered chelate ring of the cyclometalating ligand. A comparison with previous structures of [Pt(C^N)(O^O)] complexes with C^N ligands such as 2‐phenylpyridine (ppy), benzo[h]quinoline (bzq), dibenzo[f,h]quinoline (dbq), [40] and 2‐phenylquinoline (pq) [41] reveals that the Pt−C bond lengths (ca. 1.97 Å) are closely comparable with those reported here. The experimental Pt−N distances (ca. 2.04 Å) are slightly longer than for the corresponding ppy, bzq and dbq complexes, but closely comparable to [Pt(pq)(O^O)] which is a ligand structure closely analogous to unsubstituted 2‐phenylquinoxaline. Across the series of complexes, the bond lengths that describe the coordination sphere are indistinguishable and within error, implying that the position of the OCF3 substituent does not strongly influence, either in terms of sterics or electronics, the coordination sphere parameters.
All of the structures (e.g., Figure 2) revealed Pt−Pt contacts at 3.2041(6), 3.2199(3)/3.2020(3) and 3.2586(2) Å for [Pt(L1–3)(acac)], respectively, which are all well within the 2× van der Waals radii of Pt. [42] Typically a Pt−Pt distance of 2.7 Å <d >3.5 Å [43] has been noted for dimers and chains of platinum complexes in the solid state. [44] For example, the structure of the red form of [PtCl2(bipy)], which stacks the square planar complexes in a pseudo linear chain, has a metal‐metal contact of 3.449(1) Å. [45]
Figure 2.

A partial packing diagram obtained from the X‐ray structure of [Pt(L1 )(acac)] showing both the Pt−Pt and the intermolecular π‐π interactions where the centroid‐centroid distance (indicated with arrow) is 3.969(5) Å and the shift distance is 1.755(13) Å.
Perhaps an even closer molecular analogue to the complexes herein is provided by the structure of [Pt(ppy)(acac)] which has a much longer reported Pt−Pt contact at approximately 3.669 Å. [40] Our previous work on the structural characterization of functionalised [Pt(pq)(acac)] complexes revealed a Pt−Pt distance of 3.2365(2) Å, which is more comparable to the complexes here. [46] Analysis of DFT‐optimized (M06|LanL2DZ/6‐31G*) structures by Baders’ quantum theory of atoms in molecules (QTAIM) [47] clearly indicates a bond critical point between the two Pt centres, suggesting a degree of bonding‐type interaction; the bonding situation was further elucidated by natural bonding orbital (NBO) analyses, which suggest the interaction is best described as bilateral d→sd0.3 donor‐acceptor interactions (Figure S27, Supporting Information).
For each structure [Pt(L1–3 )(acac)], the Pt−Pt contact creates Pt dimers within the crystal structure. Closer inspection of these two structures reveals that the Pt−Pt bond appears to be the primary driving force for dimerization. Previous work has noted how intermolecular, ligand‐based π‐stacking can support and promote a strong Pt−Pt interaction, [48] but there are no apparent π‐π stacking interactions evident for [Pt(L1–3 )(acac)] because the 2‐phenylquinoxaline ligands are placed on opposite sides of the molecular pair. However, further packing within the crystal is probably supported by 2‐phenylquinoxaline based π‐stacking interactions that exist between neighbouring dimers (e.g., Figure 2).
In each structure the conformation of the OCF3 group is approximately orthogonal to the plane of the phenyl ring, which previous studies have partly attributed to the preferential positioning of the bulky CF3 group and a nO→σ*C‐F hyperconjugative interaction. [49] Two relevant interactions were identified in supporting NBO calculations (M06|LanL2DZ/6‐31G*) and predicted as nO→π* and sp 2→σ*C‐F interactions. Thus, the NBO calculations imply that one lone pair is described as sp 2 and the other as p in character. The experimental crystal structures reveal that the ∠C−O−C bond angles that describe the OCF3 group's angular relationship to the phenyl ring are ca. 118° (Table 1) which suggests that the oxygen atom approaches sp 2 hybridisation, which is consistent with previous work. [50]
Table 1.
Selected bond angles (°) that describe the conformation of the OCF3 group.
|
Complex |
|
|
|---|---|---|
|
[Pt(L1 )(acac)] |
117.6(7)° |
85(1)° |
|
[Pt(L2 )(acac)] |
113.1(6)/119.6(8)° |
90.5(7)° |
|
[Pt(L3 )(acac)] |
118.3(3)° |
76.7(5)° |
For [Pt(L2 )(acac)], there are two crystal structures. The first crystals (laths) cracked when flash cooled to 100 K and thus a data collection was carried out at 200 K. A second batch of [Pt(L2 )(acac)] was crystallized resulting in a differing morphology (needles) that survived the flash cooling to 100 K; both structures are presented here. The structures are 3‐D similar, and from the data it is also clear that the bond lengths that describe the square planar coordination environments are within experimental error for both samples, although the Pt−Pt distances are slightly different at 3.2199(3) and 3.2020(3) Å. Further work is required to determine whether they are truly polymorphs (the two structures of [Pt(L2 )(acac)] differ in the dimensions and volume of the unit cells – see Table S1, Supporting Information) or that the differences are due to thermal expansion relating to the temperatures at which the data was collected.
Electronic and redox properties of the complexes
Cyclic voltammetry data was recorded (CH2Cl2, 293 K, using 0.1 M [nBu4N][PF6] as the supporting electrolyte) for the complexes. As the Pt2+/3+ couple is typically irreversible (the resultant Pt(III) species are highly susceptible to reaction in solvent [51] ), the primary focus of the electrochemistry was upon the reduction process(es). In all cases a single reversible reduction wave was observed at −1.65, −1.62, −1.62, −1.57 and −1.56 V, for [Pt(L1–5 )(acac)], respectively. Although the values are broadly comparable across the series, the CF3 derivatives may be slightly easier to reduce, which is consistent with a ligand‐based reduction, as noted in related cyclometalated Pt(II) species. [31]
The optical properties of the ligands were described by strong absorption bands in the UV region that can be attributed to the different π→π* transitions originating from the phenyl and quinoxaline rings; the longest wavelength feature is approximately 320–340 nm. Within the series there are generally subtle variations (Figure S28), suggesting that the position of the substituent causes minor perturbation of these transitions, although HL6 showed a comparative hypo‐ and hypsochromic shift which probably relates to the positioning of the CF3 group. The corresponding Pt(II) complexes show more detailed spectra. As well as the ligand‐centred absorptions <400 nm, a new, somewhat broadened feature at 400–500 nm (ϵ ∼5000 M−1 cm−1) was evident in the spectra of the complexes (data collected in Table 2). Previously reported complexes [Pt(C N)(dpm)] (where C N=ppy, bzq, dbq; dpm=dipivolylmethanoate) ascribe the visible band to a spin‐allowed metal‐to‐ligand charge transfer (1MLCT) transition. [40] Comparatively, [Pt(L1–5 )(acac)] therefore show a significant bathochromic shift in the visible absorption wavelength, which is far more reminiscent of previous work on conjugated pyrazine‐based complexes of Pt(II). [31] A very weak shoulder at 500–550 nm was also noted for [Pt(L1–5 )(acac)] and this may be attributed to a spin forbidden 3MLCT transition.
Table 2.
Absorption and emission data of the Pt(II) complexes.[a]
|
Compound |
λabs [b] (ϵ [c])/nm |
λ em [d]/nm |
τ [e]/μs |
τ [f]/μs |
Φ P [g]/% |
|---|---|---|---|---|---|
|
[Pt(L1 )(acac)] |
378 (1.12), 441 (0.46) |
620 |
0.267 |
0.9 (25 %), 2.2 (75 %) |
14.4 |
|
[Pt(L2 )(acac)] |
374 (0.95), 440 (0.58) |
611 |
0.271 |
1.3 (27 %), 2.3 (73 %) |
21.9 |
|
[Pt(L3 )(acac)] |
376 (0.89), 435 (0.51) |
603 |
0.304 |
1.5 (34 %), 2.7 (66 %) |
24.8 |
|
[Pt(L4 )(acac)] |
373 (0.97), 438 (0.57) |
604 |
0.500 |
1.4 (23 %), 2.6 (77 %) |
19.1 |
|
[Pt(L5 )(acac)] |
375 (0.74), 446 (0.42) |
607 |
0.277 |
1.2 (59 %), 1.7 (41 %) |
14.9 |
[a] toluene; [b] maximal UV‐vis absorption wavelength; [c] molar absorption coefficient, in 104 M−1 cm−1; [d] maximal phosphorescence emission wavelength; [e] phosphorescence lifetimes (λex=405 nm in aerated toluene); [f] phosphorescence lifetimes (λex=405 nm in deaerated toluene); [g] phosphorescence quantum yields in N2, λex=440 nm.
Supporting computational studies (density functional theory) were used to calculate the frontier molecular orbitals (Figure 3). These suggest that the highest occupied molecular orbital (HOMO) in these complexes is likely to comprise 5dPt, and π orbitals of the surrounding phenyl and acac ligands. The lowest unoccupied molecular orbital (LUMO) is predicted to predominantly localise on the quinoxaline portion of the cyclometalating ligand. Thus, it may be that MLCT, some intra‐ligand charge transfer (ILCT) and ligand‐to‐ligand charge transfer (LLCT) also contribute to the lowest energy absorption band for these complexes. These calculations also suggest that the position and type of substituent may provide subtle tuning of both the HOMO and LUMO levels. Thus, while the experimental spectra are broadly comparable across the series, there are subtle variations in the position of the longest wavelength band. For example, in the para substituted complexes, the CF3 variant is bathochromically shifted relative to the OCF3 analogue (this is also supported by the smallest computed HOMO‐LUMO energy gap). The triplet state spin density surfaces were calculated at the optimised triplet state geometries and shown to be largely localised on the cyclometalating ligand and Pt centre (Figure 4).
Figure 3.

Selected frontier molecular orbitals and the energy (eV) of the complexes calculated by TDDFT at the B3LYP/GENECP level with Gaussian 09 based on the optimized ground‐state geometry (in vacuum). Isovalue=0.02.
Figure 4.

Isosurfaces of the spin density of the complexes at the optimized triplet state geometries in vacuum. Calculation was performed at the B3LYP/GENECP level with Gaussian 09. Isovalue=0.0004.
Excitation of the complexes (440 nm) into the visible absorption band resulted in observed photoluminescence (data summarised in Table 2) with a broad, modestly structured peak at 604–620 nm (Figure 5): these Pt(II) species are orange‐red emitters and the appearance of the emission spectra are similar to those reported by Williams and Kozhevnikov for conjugated pyrazine‐based ligands on Pt(II). [31] These complex emission features are clearly different to those observed for HL1–6 , which fluoresce approximately 400 nm (Figure S28).
Figure 5.

UV‐Vis absorption spectra of the complexes in toluene. c=1.0×10−5 M, 25 °C. Right: Phosphorescence emission spectra of the compounds in deoxygenated toluene. Optically matched solutions were used (all the solutions show the same absorbance at the excitation wavelength, λ ex=440 nm, A 440 nm=0.1, 25 °C).
The quantum yields of emission in deoxygenated toluene were obtained as 14.4‐24.8 %; this was further increased in deaerated DCM to 21.9–37.1 % (corroborating the increase in integrated intensity of emission from isoabsorbing solutions, Figure S29, Supporting Information), where [Pt(L3 )(acac)] was identified as the most efficient emitter within the series. For the structurally isomeric OCF3 functionalised complexes, [Pt(L1–3 )(acac)], the quantum yield values appear to relate directly to the emission wavelength in accordance with the Energy Gap Law.
The spectral properties of the benchmark complex [Pt(ppy)(acac)] (λ em=486 nm, Φ P=15 % in deoxygenated 2‐MeTHF) [52] contextualise the pronounced red‐shift in emission maximum, and enhancement in quantum yield, induced by the 2‐phenylquinoxaline ligands described here. Furthermore, the very close structural analogue [Pt(pq)(acac)] is less emissive and has a hypsochromically shifted emission (λ em 586 nm, Φ P 9 % in deaerated DCM) [53] relative to [Pt(L1–5 )(acac)].
The good solubility of [Pt(L1–5 )(acac)] allowed comparison of the emission properties in different solvents (Figure 6). The emission spectra of isoabsorbing (λ=440 nm) solutions showed that the emission intensity from the complexes was strongly quenched in polar solvent (MeCN) relative to DCM, toluene and hexane. However, the emission maxima were not strongly solvatochromic implying that there is little change in the dipolarity of the CT excited state versus ground state.
Figure 6.

Phosphorescence emission spectra of (a) [Pt(L1 )(acac)], (b) [Pt(L2 )(acac)], (c) [Pt(L3 )(acac)], (d) [Pt(L4 )(acac)] and (e) [Pt(L5 )(acac)] in different aerated solvents. Optically matched solutions were used (all the solution show the same absorbance at the excitation wavelength, λ ex=440 nm, A 440 nm=0.1, 25 °C).
Time‐resolved measurements yielded sub‐microsecond emission lifetimes on aerated solutions and longer lifetimes (extended to microseconds) on deoxygenated solutions implied a triplet character to the emitting state (see Figures S30‐31, Supporting Information for fitted decay kinetics). The excited state quenching by oxygen was further exemplified by obtaining the photogenerated singlet oxygen (arising from 3Σg −→1Δg) quantum yields for each complex which fell in the range 30–57 %, again indicative of a triplet emitting state. The singlet oxygen production performance was directly comparable to other photoactive species such as [Ru(bipy)3]2+, which is the archetypal 3MLCT emitter.
Additional spectra were obtained using solid state samples and showed that the emission maxima were bathochromically shifted relative to the solution state spectra. Emission wavelengths were noted around 675–700 nm (Figure S32, Supporting Information) and resulted from an excitation wavelength of 450–550 nm; the visually apparent, deep‐red emission was photographically recorded using a crystalline sample of [Pt(L1 )(acac)] under UV irradiation (Figure S32, Supporting Information). The solid state luminescence lifetimes for [Pt(L1–5 )(acac)] were sub‐microsecond in all cases. The emission characteristics of stacked Pt(II) complexes are well known, with both dimerisation and/or aggregation of complexes known to bathochromically shift emission, which is important when considering thin film applications. [54] In luminescent complexes with strong Pt−Pt interactions, solid state photophysical data can imply metal‐metal‐to‐ligand charge transfer (MMLCT) type transitions that originate from a σ* (5dz2/5dz2)→π* transition. [55]
Time‐resolved transient absorption (TA) spectroscopic measurements were also obtained for the five Pt(II) complexes in toluene. As represented in Figures 7 and S33‐35 (Supporting Information), the data shows the change in optical density, following irradiation at 355 nm, as a function of time. The TA spectra generally show three strong positive features in the 300–600 nm region; the broadness and profile of the band at 500–600 nm subtly changes with the type of ligand. The negative band of at ca. 650 nm is due to the phosphorescence of the complex. The decay kinetics of the degassed samples occur over a microsecond timescale which shorten in aerated toluene to which is fully consistent with a triplet transient feature. The triplet state lifetimes were determined as 2.6 μs, 1.5 μs and 2.1 μs, for [Pt(L1 )(acac)], [Pt(L2 )(acac)] and [Pt(L3 )(acac)], respectively. The time domain of these kinetic data are comparable with the photoluminescence lifetimes obtained under similar solvent conditions and therefore implies that the transient absorption spectra probably arise from the same, or closely related, excited state. [56]
Figure 7.

Nanosecond transient absorption spectra of (a) [Pt(L1 )(acac)], (c) [Pt(L2 )(acac)] and (e) [Pt(L3 )(acac)] and the decay trace of (b) [Pt(L1 )(acac)], (d) [Pt(L2 )(acac)] and (f) [Pt(L3 )(acac)] at 540 nm in deaerated toluene excited with nanosecond pulsed laser. λ ex=355 nm. c=3.0×10−5 M, 25 °C.
To assign the ESA bands in the ns‐TA spectra, TDDFT computations were performed to calculate the T1→Tn transitions, based on the optimized T1 state geometry (Supporting Information, Figure S36, S37 and Table S4). For [Pt(L1 )(acac)], the calculated T1→Tn transitions are mainly T1→T28 (3.87 eV, 320 nm), T1→T19 (3.20 eV, 387 nm), T1→T15 (2.76 eV, 450 nm), T1→T14 (2.33 eV, 32 nm) and T1→T13 (2.13 eV, 581 nm) (see Table S4 in the Supporting Information for details). These calculated ESA bands are in agreement with the experimental ns‐TA spectral observations of the ESA bands in the range of 300–350 nm, 350–450 nm, and 500–600 nm (Figure 7a. For the calculated ESA absorption spectra, please refer to Figure S36a). Similar results were obtained for [Pt(L2–5 )(acac)] (Figure S36).
To fully mimic the experimental ns‐TA spectra (difference spectra of the ESA absorption spectrum and the ground state absorption spectrum), we calculated the difference ns‐TA spectra by subtracting the steady state UV‐vis absorption spectrum from the calculated ESA absorption spectrum (note the steady state absorption spectra were multiplied by an arbitrary factor). The results show that three main positive absorption bands in the range of 300–350 nm, 400–500 nm and 500–600 nm were observed (Supporting Information, Figure S37a), which are closely comparable to the experimental results (Figure 7a). Similar results were observed for [Pt(L2–5 )(acac)] (Supporting Information, Figure S37). The absence of the theoretically predicted ESA band >700 nm is reasonable: the PMT detector of the ns‐TA spectrometer usually has a poor sensitivity in the near‐IR region.
Energy upconversion studies
As noted previously, a relatively small number of coordination complexes have been investigated as photosensitisers for TTA‐UC. TTA‐UC studies were therefore undertaken on each of the Pt(II) complexes as photosensitisers. 9,10‐Diphenylanthracene was utilised as the annihilator molecule allowing ease of comparison with other reported systems. Figures 8 and 10 and S38–39 show the emission spectra that result from the addition of DPA to 10−5 M solutions of the different complexes in deoxygenated DCM. In all cases an excitation wavelength of 473 nm was used, which is selective for the visible absorption band of the complexes and avoids direct excitation of the DPA. The DPA triplet excited state is at approximately 700 nm and therefore lies below the triplet emitting level of all of the Pt(II) complexes (we thus estimate a 0.2–0.3 eV triplet state energy gap between the donor and acceptor components). Thus, any emission from DPA in the 400–500 nm range can be attributed to an upconversion process via TTA. Figure 8 describes that upon addition of sequential aliquots of DPA, the emission intensity of [Pt(L1 )(acac)] is concomitantly quenched.
Figure 8.

TTA upconversion with [Pt(L2 )(acac)] as the photosensitizer and DPA (9,10‐diphenylanthracene) as the acceptor in deoxygenated DCM. c [Pt(L2)]=1.0×10−5 M, 25 °C. (a) Upconversion emission spectra. The asterisks indicate the scattered laser. Excited with a 473 nm cw‐laser with a power density of 80 mW cm−2. (b) Upconverted DPA emission intensity at 410 nm as a function of the incident laser power density. The solid red and green lines are fitting results with slopes of 2.0 and 1.0 in the low‐ and high‐density regions, respectively. c [DPA]=3.0×10−5 M. (c) Upconversion emission spectra as a function of increasing DPA concentration. (d) Photographs of [Pt(L2 )(acac)] alone and the upconversion. (e) Photographs of upconversion solutions observed with band‐pass filter (transparent in the range 380–520 nm). Excited with a 473 nm cw‐laser with a power density of 80 mW cm−2. c [PSs]=1.0×10−5 M, c [DPA]=3.0×10−5 M, 25 °C.
Figure 10.

(a) TTA upconversion emission spectra with [Pt(L4 )(acac)] as the photosensitizer and DPA as the acceptor in deaerated DCM. The asterisks indicate the scattered laser. (b) Photographs of [Pt(L4 )(acac)] alone and the upconversion. (c) Photographs of upconversion solutions observed with band‐pass filter (transparent in the range 380–520 nm). Excited with a 473 nm cw‐laser with a power density of 80 mW cm−2. c [PSs]=1.0×10−5 M, c [DPA]=3.0×10−5 M, 25 °C.
Figure 8 also presents the plot obtained from the intensity of the upconverted fluorescence from DPA as a function of incident power density. A double‐logarithm plot was noted with a gradient change which implies a quadratic dependence at low power density and a linear dependence at high power density. [21] Therefore an optimised laser power density of 80 mW cm−2 was chosen for the TTA‐UC quantum yield measurements.
The data (Table 3) shows that each complex behaves as a viable photosensitiser for TTA‐UC with efficiencies (Φ UC) ranging between 5.9–14.1 %. These values compare very favourably with the relatively small number of examples of organometallic Pt(II) complexes investigated in TTA‐UC.
Table 3.
Emission, singlet oxygen generation and energy upconversion data of the Pt(II) complexes recorded in deaerated DCM.
|
Compound |
λ em [a]/nm |
Φ P [b]/% |
Φ Δ [c]/% |
Φ UC [d]/% |
|---|---|---|---|---|
|
[Pt(L1)(acac)] |
623 |
21.9 |
38 |
5.9 |
|
[Pt(L2)(acac)] |
614 |
32.1 |
54 |
12.1 |
|
[Pt(L3)(acac)] |
608 |
37.1 |
39 |
14.1 |
|
[Pt(L4)(acac)] |
606 |
34.3 |
30 |
9.5 |
|
[Pt(L5)(acac)] |
610 |
27.1 |
58 |
8.5 |
[a] maximal phosphorescence emission wavelength; [b] phosphorescence quantum yields in N2, λ ex=440 nm; [c] Singlet oxygen quantum yield. [Ru(bpy)3]2+ (Φ Δ= 0.57 in DCM) as the standard, λ ex=440 nm. [d] TTA upconversion quantum yield in deoxygenated DCM. [Ru(dmb)3]2+ (Φ P=0.073 in deoxygenated acetonitrile; dmb=4,4′‐dimethyl‐2,2′‐bipyridine) as the reference, λ ex=473 nm.
For example, the best performing sensitiser, [Pt(L3 )(acac)], is directly comparable to Pt‐2 (see Introduction) which features a much longer triplet lifetime. It is therefore noteworthy that although the complexes presented here do not possess comparatively long triplet state lifetimes, they are sufficient to act as efficient sensitisers for TTA‐UC. The data in Table 3 clearly shows the trend, especially within the OCF3 substituted complexes, that the TTA‐UC efficiencies correlate with the overall emissivity of the complexes which, in turn, broadly relates to the emission energy maximum. The variation in TTA‐UC performance therefore demonstrates the importance of the subtle structural alterations of the ligand upon the photosensitiser. The visualised energy upconversion in these systems is shown in Figures 9–10 (one example each of a OCF3 and CF3 functionalised complex) and S11–12 where photographs visually illustrate the effect of adding the DPA annihilator to the different Pt(II) sensitiser solutions.
Figure 9.

(a) TTA upconversion emission spectra with [Pt(L3 )(acac)] as the photosensitizer and DPA as the acceptor in deaerated DCM. The asterisks indicate the scattered laser. (b) Photographs of [Pt(L3 )(acac)] alone and the upconversion. (c) Photographs of upconversion solutions observed with band‐pass filter (transparent in the range 380–520 nm). Excited with a 473 nm cw‐laser with a power density of 80 mW cm−2. c [PSs]=1.0×10−5 M, c [DPA]=3.0×10−5 M, 25 °C.
Conclusion
The design and synthesis of substituted 2‐phenylquinoxaline ligands allows tuning of the physical properties of cyclometalated Pt(II) complexes. As well as enhancing solubilities, particularly in planar complexes, the use of organofluorine substituents provides subtle tuning of the electronic properties and thus photophysical attributes of the resultant Pt(II) complexes. The electronic influence of the OCF3 groups was investigated in detail using a combination of computational methods and solid‐state structures. The delicate tuning of the excited state properties has a strong influence upon their application as photosensitisers for TTA‐UC, with resultant efficiencies in the range of 5.9–14.1 %. The upper range of these values is competitive with the best performing Pt(II) photosensitisers and has been achieved without significant manipulation of the triplet lifetimeexcited state character. The data suggests that the 2‐phenylquinoxaline framework is therefore an excellent basis for further optimisation; additional structural modifications may further enhance their efficiencies in TTA‐UC.
Experimental Section
X‐ray crystallography: Suitable crystals for [Pt(L1 )(acac)], [Pt(L2 )(acac)] and [Pt(L3 )(acac)] were obtained and mounted on a MITIGEN holder in oil on either a Rigaku 007HF diffractometer equipped with HF Varimax confocal mirrors and an AFC11 goniometer and HyPix 6000HE detector ([Pt(L1 )(acac)]), or a Rigaku FRE+ diffractometer equipped with Arc)Sec VHF Varimax confocal mirrors and either an AFC12 goniometer and HyPix 6000 detector ([Pt(L2 )(acac)]_200 K) or with a UG2 goniometer and HyPix 6000HE detector ([Pt(L2 )(acac)]_100 K and [Pt(L3 )(acac)]). [57] Using Olex2 [58] the structure was solved with the ShelXT [59] structure solution program, using the Intrinsic Phasing solution method. The model was refined with version 2014/7 of ShelXL [60] using Least Squares minimisation. All non‐H atoms were refined anisotropically and difference Fourier syntheses were employed in positioning idealized hydrogen atoms and were allowed to ride on their parent C‐atoms.
Deposition Number(s) 2210432 (for [Pt(L 1 )(acac)]), 2210433 (for [Pt(L 2 )(acac)]_200 K), 2210434 (for [Pt(L 2 )(acac)]_100 K), 2210435 (for [Pt(L 3 )(acac)]) contain(s) the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service.
Computational methods: The geometries and spin density surfaces of the compounds were optimized using density functional theory (DFT) with B3LYP functional and GENECP basis set. There were no imaginary frequencies for all optimized structures. All these calculations were performed with Gaussian 09. [61]
NBO calculations were performed using NBO 6.0 [62] and QTAIM calculations with the AIMAll package. [63]
Triplet‐triplet annihilation upconversion: Transient absorption measurements were carried out using a LP980 laser flash photolysis spectrometer (Edinburgh Instruments, UK). All spectra were collected using a pump wavelength of 440 nm (third harmonic of a Continuum Surelite l‐10 Nd:YAG‐laser‐system with Surelite OPO Plus). The probe light for these measurements was a xenon lamp, affording spectral generation between 300<λ<900 nm. Lifetime data was generated using a photomultiplier to collect time resolved signals. The lifetime data was obtained using L900 software package. All compound solutions in flash photolysis experiments were deaerated with N2 for ca. 10 min and the gas was maintained during the measurement.
Associated luminescence spectra were recorded on Shimadzu RF‐5301PC spectrofluorometer. The fluorescence and phosphorescence lifetimes were measured on an OB920 fluorescence/phosphorescence lifetime instrument (Edinburgh, U.K.) with an EPL picosecond pulsed diode laser (405 nm±10 nm, pulse width: 119.9 ps, maximum average power: 5 mW.
Continuous laser (473 nm) was used for upconversion and the power of the laser beam was 4.8 mW. The spot area of the 473 nm laser was ca. 6 mm2. The mixed solution (with different triplet sensitizers and acceptor) was deaerated for 10 min before experiment, and the gas flow was kept during the measurement. The upconverted fluorescence was recorded with a RF 5301PC spectrofluorometer. In order to repress the laser scattering, a small black box was put behind the fluorescent cuvette as beam dump to trap the laser.
The upconversion quantum yields (ΦUC) of all the complexes in toluene were using the [Ru(dmb)3]2+ (ΦP=0.073 in deoxygenated acetonitrile the standard to be determined. The upconversion quantum yield was using the following equation (Eq. (1)) to calculate, where Φ, A, I and η represent the quantum yield, absorbance, integrated photoluminescence intensity and the refractive index, respectively. Symbols with ‘std’ and ′sam′ are the corresponding parameters for the standard used in the measurement of quantum yield and samples to be measured.
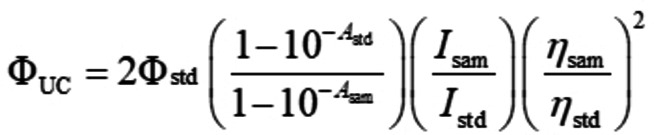
General experimental details: All reactions were performed with the use of vacuum line and Schlenk techniques. Reagents were commercial grade and were used without further purification. 1H, 13C{1H} and 19F{1H} NMR spectra were recorded on a Bruker Avance dpx 400 MHz or Avance dpx 500 MHz spectrometers and were recorded as CDCl3 solutions. Mass spectra were obtained by the staff at Cardiff University. IR spectra were recorded on an ATR equipped Shimadzu IRAffinity‐1 spectrophotometer. UV‐vis data were recorded as solutions on a Perkin Elmer Lamda20 spectrophotometer. Cyclic voltammetry was performed using a PalmSens4 potentiostat. Experiments were performed using HPLC grade CH2Cl2 with an analyte concentration of 1 mM at 293 K, using triply recrystallised [nBu4N][PF6] as the supporting electrolyte at 0.1 M concentration. A three‐electrode setup was used, consisting of a platinum disc working electrode, a platinum wire counter‐electrode and a silver wire pseudo‐reference. Solutions were spared for 10 minutes with CH2Cl2 saturated stream of nitrogen gas. Voltammograms were referenced to the ferrocene/ferrocenium redox couple (set to 0 V) measured using the same conditions.
Synthesis of bromoacetyl precursors
Synthesis of 2‐bromo‐1‐(2‐(trifluoromethoxy)phenyl)ethan‐1‐one: Slow addition of the dioxane dibromide (0.668 g, 2.69 mmol) in a solution of 1,4‐dioxane (20 mL) and diethyl ether (20 mL) into 2’‐(trifluoromethoxy)acetophenone (0.500 g, 2.45 mmol) in the same solvent mixture took place. This mixture was allowed to stir at room temperature for 2 h. The pale yellow solution was added to water and extracted with diethyl ether. The combined ether layers were dried over MgSO4. The solvent was removed in vacuo to give the product as a yellow oil. Yield=0.570 g, 82 %. 1H NMR (300 MHz, CDCl3) δH=7.78 (dd, 3 J HH=7.8, 1.7 Hz, 1H), 7.63–7.53 (m, 1H), 7.44–7.28 (m, 2H), 4.46 (s, 2H) ppm.
Synthesis of 2‐bromo‐1‐(3‐(trifluoromethoxy)phenyl)ethan‐1‐one: The title compound was prepared similarly from 3’‐(trifluoromethoxy)acetophenone (0.250 g, 1.23 mmol) and dioxane dibromide (0.334 g, 1.35 mmol) to give the product as a yellow oil. Yield=0.335 g, 97 %. 1H NMR (500 MHz, CDCl3) δH=7.93–7.90 (m, 1H), 7.85–7.82 (m, 1H), 7.59–7.53 (m, J=11.9, 4.1 Hz, 1H), 7.48–7.45 (m, 1H), 4.43 (s, 2H) ppm.
Synthesis of 2‐bromo‐1‐(4‐(trifluoromethoxy)phenyl)ethan‐1‐one: The title compound was prepared similarly from 4’‐(trifluoromethoxy)acetophenone (0.250 g, 1.23 mmol) and dioxane dibromide (0.334 g, 1.35 mmol) to give the product as yellow crystals. Yield=0.335 g, 97 %. 1H NMR (500 MHz, CDCl3) δH=8.07–8.03 (m, 2H), 7.34–7.29 (m, 2H), 4.42 (s, 2H) ppm.
Synthesis of 2‐bromo‐1‐(2‐(trifluoromethyl)phenyl)ethan‐1‐one: The title compound was prepared similarly from 2’‐(trifluoromethyl)acetophenone (1.500 g, 7.97 mmol) and dioxane dibromide (2.174 g, 8.77 mmol) to give the product as a yellow oil. Yield=1.947 g, 92 %. 1H NMR (500 MHz, CDCl3) δH=7.76–7.73 (m, 1H), 7.67–7.60 (m, 2H), 7.53–7.49 (m, 1H), 4.37 (s, 2H) ppm.
Synthesis of 2‐bromo‐1‐(3‐(trifluoromethyl)phenyl)ethan‐1‐one: The title compound was prepared similarly from 3’‐(trifluoromethyl)acetophenone (2.500 g, 13.29 mmol) and dioxane dibromide (3.623 g, 14.62 mmol) to give the product as a colourless oil. Yield=3.083 g, 87 %. 1H NMR (500 MHz, CDCl3) δH=8.24 (s, 1H), 8.17 (d, 3 J HH=7.8 Hz, 1H), 7.87 (d, 3 J HH=8.2 Hz, 1H), 7.65 (app.t, 3 J HH=7.8 Hz, 1H), 4.46 (s, 2H) ppm.
Synthesis of 2‐bromo‐1‐(4‐(trifluoromethyl)phenyl)ethan‐1‐one: [64] The title compound was prepared similarly from 4’‐(trifluoromethyl)acetophenone (2.500 g, 13.29 mmol) and dioxane dibromide (3.623 g, 14.62 mmol) to give the product as white crystals. Yield=3.107 g, 88 %. 1H NMR (400 MHz, CDCl3) δH=8.06 (d, 3 J HH=8.1 Hz, 2H), 7.71 (d, 3 J HH=8.2 Hz, 2H), 4.45 (s, 1H) ppm.
Synthesis of the substituted 2‐phenylquinoxaline ligands
Synthesis of 2‐(2‐(trifluoromethoxy)phenyl)quinoxaline (HL1): 2‐bromo‐1‐(2‐(trifluoromethoxy)phenyl)ethan‐1‐one (1.302 g, 4.60 mmol) and 1,2‐phenylenediamine (0.547 g, 5.06 mmol) were heated to reflux in ethanol, whilst exposed to air, for 24 h. Once the reaction had cooled, the solvent was removed in vacuo and the product was purified by column chromatography (SiO2, CH2Cl2), where the product eluted as a yellow band with CH2Cl2. The solvent was removed in vacuo to give the product as yellow crystals. Yield=0.515 g, 39 %. 1H NMR (300 MHz, CDCl3) δH=9.21 (s, 1H), 8.19–8.13 (m, 2H), 7.96 (dd, J HH=7.7, 1.8 Hz, 1H), 7.82–7.76 (m, 2H), 7.54 (ddd, J HH=8.1, 7.5, 1.9 Hz, 1H), 7.49 (app. td, J HH=7.5, 1.3 Hz, 1H), 7.45–7.42 (m, 1H) ppm. 13C{1H} NMR (126 MHz, CDCl3) δC=150.5, 147.1 (q, J CF=1.8 Hz), 146.0, 142.6, 141.5, 132.3, 131.4, 131.1, 130.4, 130.3, 129.8, 129.4, 127.8, 121.5, 120.5 (q, 1 JCF =259.0 Hz) ppm. 19F{1H} NMR (376 MHz, CDCl3) δF=−57.15 (s) ppm. MS (ES) m/z calcd for C15H10F3N2O 291.0745; found 291.0754.
Synthesis of 2‐(3‐(trifluoromethoxy)phenyl)quinoxaline (HL2): The ligand was prepared similarly from 2‐bromo‐1‐(3‐(trifluoromethoxy)phenyl)ethan‐1‐one (0.303 g, 1.07 mmol) and 1,2‐phenylenediamine (0.127 g, 1.18 mmol) to give the product as a pale yellow solid. Yield=0.068 g, 24 %.1H NMR (300 MHz, CDCl3) δH=9.31 (s, 1H), 8.19–8.12 (m, 2H), 8.13–8.10 (m, 2H), 7.84–7.74 (m, 2H), 7.59 (app. t, J HH=8.2 Hz, 1H), 7.41–7.35 (m, 1H) ppm. 13C{1H} NMR (126 MHz, CDCl3) δC=150.2, 150.2–150.1 (m), 143.0, 142.3, 142.0, 139.0, 130.7, 130.7, 130.2, 129.9, 129.3, 125.8, 120.7 (q, 1 J CF=257.7 Hz), 122.6, 120.3 ppm. 19F{1H} NMR (376 MHz, CDCl3) δF=−57.64 (s) ppm. MS (ES) m/z calcd for C15H10F3N2O 291.0743; found 291.0745.
Synthesis of 2‐(4‐(trifluoromethoxy)phenyl)quinoxaline (HL3): [65] The ligand was prepared similarly from 2‐bromo‐1‐(4‐(trifluoromethoxy)phenyl)ethan‐1‐one (0.693 g, 2.45 mmol) and 1,2‐phenylenediamine (0.291 g, 2.69 mmol) to give the product as pale yellow crystals. Yield=0.184 g, 26 %. 1H NMR (500 MHz, CDCl3) δH=9.25 (s), 8.22–8.16 (m, 2H), 8.12–8.06 (m, 2H), 7.78–7.69 (m, 2H), 7.37 (d, 3 J HH=8.0 Hz, 2H) ppm. 19F{1H} NMR (376 MHz, CDCl3) δF=−57.67 (s) ppm.
Synthesis of 2‐(3‐(trifluoromethyl)phenyl)quinoxaline (HL4): [66] The ligand was prepared similarly from 2‐bromo‐1‐(3‐(trifluoromethyl)phenyl)ethan‐1‐one (3.376 g, 12.64 mmol) and 1,2‐phenylenediamine (1.641 g, 15.17 mmol) but without the need for purification, as the product precipitated from cooled ethanol to give the product as an off‐white solid. Yield=0.383 g, 11 %. 1H NMR (500 MHz, CDCl3) δH=9.36 (s, 1H), 8.52 (s, 1H), 8.39 (d, 3 J HH=7.9 Hz, 1H), 8.22–8.14 (m, 2H), 7.86–7.76 (m, 3H), 7.71 (app. t, J HH =7.7 Hz, 1H) ppm. 19F{1H} NMR (376 MHz, CDCl3) δF=−62.65 (s) ppm.
Synthesis of 2‐(4‐(trifluoromethyl)phenyl)quinoxaline (HL5): [67] The ligand was prepared similarly from 2‐bromo‐1‐(4‐(trifluoromethyl)phenyl)ethan‐1‐one (3.107 g, 11.64 mmol) and 1,2‐phenylenediamine (1.510 g, 14.0 mmol), but without the need for purification, as the product precipitated from cooled ethanol as off‐white crystals. Yield=0.786 g, 25 %. 1H NMR (500 MHz, CDCl3) δH=9.35 (s, 1H), 8.33 (d, 3 J HH=8.1 Hz, 2H), 8.20–8.13 (m, 2H), 7.83 (d, 3 J HH=8.2 Hz, 2H), 7.85–7.78 (m, 2H) ppm. 19F{1H} NMR (376 MHz, CDCl3) δF=−62.74 (s) ppm.
Synthesis of 2‐(2‐(trifluoromethyl)phenyl)quinoxaline (HL6): [68] The ligand was prepared similarly from 2‐bromo‐1‐(2‐(trifluoromethyl)phenyl)ethan‐1‐one (2.181 g, 8.17 mmol) and 1,2‐phenylenediamine (1.060 g, 9.80 mmol) to give the product as a yellow oil. Yield=0.907 g, 41 %. Yield=0.383 g, 11 %. 1H NMR (500 MHz, CDCl3) δH=8.99 (s, 1H), 8.21–8.12 (m, 2H), 7.86 (d, 3 J HH=7.8 Hz, 1H), 7.85–7.81 (m, 2H), 7.72 (d, 3 J HH=7.6 Hz, 1H), 7.67–7.61 (m, 2H) ppm. 19F{1H} NMR (376 MHz, CDCl3) δF=−56.55 (s) ppm.
General procedure for the synthesis of platinum dimers, [Pt(L)(μ‐Cl)]2 : Based on a modified literature methodology.69 A solution of potassium tetrachloroplatinate(II) (1.0 equiv.) in water (2 mL) was added to a stirring solution of HL (1.0 equiv.) in glacial acetic acid (10 mL) under dinitrogen. The solution was heated to 130 °C for 3 days. Once cooled, the reaction mixture was filtered, washed with AcOH and EtOH and dried to give the product as a solid which was used without further purification.
Synthesis of [Pt(L1)(μ‐Cl)]2 : KPtCl4 (0.172 g, 0.413 mmol) in H2O (∼0.5 mL) was added to a stirred solution of HL1 (0.120 g, 0.413 mmol) in glacial acetic acid (∼10 mL). Yield=0.061 g, 28 %.
Synthesis of [Pt(L2)(μ‐Cl)]2 : The product was prepared similarly with KPtCl4 (0.150 g, 0.36 mmol) and HL2 (0.070 g, 0.24 mmol) to give rise to the product as a brown solid. Yield=0.096 g, 76 %.
Synthesis of [Pt(L3)(μ‐Cl)]2 : The product was prepared similarly with KPtCl4 (0.146 g, 0.35 mmol) and HL3 (0.068 g, 0.23 mmol) to give rise to the product as a brown solid. Yield=0.129 g, 91 %.
Synthesis of [Pt(L4)(μ‐Cl)]2 : The product was prepared similarly with KPtCl4 (0.171 g, 0.41 mmol) and 2‐(3‐(trifluoromethyl)phenyl)quinoxaline (HL4) (0.112 g, 0.41 mmol) to give rise to the product as a brown solid. Yield=0.204 g, 98 %. The product was used in the next step without further purification.
Synthesis of [Pt(L5)(μ‐Cl)]2 : The product was prepared similarly with KPtCl4 (0.182 g, 0.44 mmol) and 2‐(3‐(trifluoromethyl)phenyl)quinoxaline (HL5) (0.120 g, 0.44 mmol) to give rise to the product as a dark red solid. Yield=0.21 g, 95 %. The product was used in the next step without further purification.
General procedure for splitting platinum dimers: Based on a modified literature methodology. [70] Crude [Pt(L)(μ‐Cl)]2 was dissolved in a minimum volume of DMSO before being precipitated with brine (10 mL), filtered on a sinter and washed with water (2×20 mL) and dried.
Synthesis of [PtCl(L1)(DMSO)]: [Pt(L1)(μ‐Cl)]2 (0.061 g, 0.06 mmol) was dissolved in the minimum amount of DMSO (<5 mL). Brine (saturated solution, aq.) was added (∼10 mL) and the mixture was stirred at room temperature overnight. The resultant precipitate was filtered and washed with distilled water, giving rise to an orange/red solid. Yield=0.044 g, 63 %.
Synthesis of [PtCl(L2)(DMSO)]: The product was prepared similarly with [Pt(L2)(μ‐Cl)]2 (0.093 g, 0.09 mmol). The product was formed as a brown solid. Yield=0.084 g, 79 %.
Synthesis of [PtCl(L3)(DMSO)]: The product was prepared similarly with [Pt(L3)(μ‐Cl)]2 (0.124 g, 0.12 mmol). The product was formed as a brown solid. Yield=0.099 g, 69 %.
Synthesis of [PtCl(L4)(DMSO)]: The product was prepared similarly with [Pt(L4)(μ‐Cl)]2 (0.189 g, 0.19 mmol). The product was formed as a brown solid. Yield=0.065 g, 30 %.
Synthesis of [PtCl(L5)(DMSO)]: The product was prepared similarly with [Pt(L6)(μ‐Cl)]2 (0.209 g, 0.21 mmol). The product was formed as an orange solid. Yield=0.075 g, 31 %
General procedure for coordinating β‐diketonates to platinum complexes: Based on a modified literature methodology. [71] [PtCl(L)(DMSO)] (1 equiv.) was dissolved in 3‐pentanone (5 mL), to which, the β‐diketonate (1–10 equiv.) was added. The reaction was stirred at room temperature for 16 h under dinitrogen. The solvent was removed in vacuo and the crude product dissolved in dichloromethane (10 mL) and filtered to remove any insoluble salts and concentrated to dryness in vacuo. The crude product was purified by column chromatography (silica, dichloromethane) wherein elution of the first yellow band with dichloromethane gave the desired product, [Pt(L)(acac)].
Synthesis of [Pt(L1)(acac)]: [PtCl(L1)(DMSO)] (0.045 g, 0.075 mmol) and sodium acetylacetonate (0.105 g, 0.753 mmol) gave rise to the pure product as an orange solid. Yield=0.023 g, 52 %. 1H NMR (500 MHz, CDCl3) δH=9.73 (s, 1H), 9.59 (ddd, J HH=8.9, 1.2, 0.5 Hz, 1H), 8.13 (dd, J HH=8.3, 1.5 Hz, 1H), 7.85 (ddd, J HH=8.7, 6.9, 1.6 Hz, 1H), 7.80–7.75 (m, 2H), 7.30 (app. t, J HH=7.9 Hz, 1H), 7.12‐7.08 (m, 1H), 5.61 (s, 1H), 2.09 (overlapping s, 6H) ppm. 13C{1H} NMR (126 MHz, CDCl3) δC=186.33, 184.61, 144.65, 144.29, 143.15, 140.27, 131.34, 130.58, 130.24, 129.36, 128.53, 126.49, 115.99, 102.19, 28.38, 27.30 ppm. 19F {1H} NMR (376 MHz, CDCl3) δF=−56.46 (s) ppm. MS ES m/z calcd for C20H16F3N2O3 194Pt 583.0740; found: 583.0736. FTIR (solid, cm−1) (ATR) v max: 2951, 2922, 2852, 1587, 1571, 1560, 1522, 1472, 1432, 1387, 1363, 1340, 1319, 1233, 1197, 1156, 1049, 963, 782, 761, 653, 642, 621.
Synthesis of [Pt(L2)(acac)]: [PtCl(L2)(DMSO)] (0.084 g, 0.14 mmol) and sodium acetylacetonate (0.197 g, 1.41 mmol) gave the product as dark red crystals. Yield=0.039 g, 47 %. 1H NMR (500 MHz, CDCl3) δH=9.72 (dd, J HH=8.9, 0.9 Hz, 1H), 9.16 (s, 1H), 8.13 (dd, J HH=8.3, 1.4 Hz, 1H), 7.85 (ddd, J HH=8.7, 6.9, 1.6 Hz, 1H), 7.78 (d, 3 JHH =8.5 Hz, 1H), 7.76 (ddd, J HH=8.2, 6.9, 1.2 Hz, 1H), 7.58‐7.56 (m, 1H), 7.21‐7.17 (m, 1H), 5.61 (s, 1H), 2.10 (s, 3H), 2.07(s, 3H) ppm. 13C{1H} NMR (126 MHz, CDCl3) δC=186.02, 184.29, 161.82, 146.55, 144.65, 143.70, 143.27, 141.62, 139.83, 131.74, 131.41, 130.06, 129.63, 126.07, 123.27, 117.76, 102.13, 28.47, 27.29 ppm. 19F {1H} NMR (376 MHz, CDCl3) δF=−57.95 (s) ppm. HR MS (ES) m/z calcd for C20H16F3N2O3 194Pt 583.0740; found 583.0743 [M+H]+, 1067.1075 [M+M–acac]+ and 1167.1697 [M+M+H]+. UV‐vis (CHCl3): λmax (ϵ/L mol−1 cm−1) 254 (44192), 295 (32012), 356 (13691), 373 (15503), 436 (8557) nm. FTIR (solid, cm−1) (ATR) v max: 2953, 2921, 2852, 1561, 1535, 1522, 1459, 1427, 1390, 1366, 1347, 1329, 1252, 1213, 1161, 1077, 1045, 1025, 993, 956, 948, 937, 906, 887, 863, 816, 796, 762, 727, 695, 660.
Synthesis of [Pt(L3)(acac)]: [PtCl(L3)(DMSO)] (0.099 g, 0.17 mmol) and sodium acetylacetonate (0.231 g, 1.65 mmol) gave the product as red crystals. Yield=48 mg, 50 %. 1H NMR (500 MHz, CDCl3) δH=9.70 (dd, J HH=8.8, 0.8 Hz, 1H), 9.13 (s, 1H), 8.09 (dd, J HH=8.2, 1.3 Hz, 1H), 7.82 (ddd, J HH=8.7, 6.9,1.6 Hz, 1H), 7.73 (ddd, J HH=8.2, 6.9, 1.3 Hz, 1H), 7.72 (d, J HH=8.6 Hz, 1H), 7.57‐7.50 (m, 1H), 7.06‐7.02 (m, 1H), 5.59 (s, 1H, acac), 2.09 (s, 3H), 2.07 (s, 3H) ppm. 13C{1H} NMR (126 MHz, CDCl3) δC=185.99, 184.44, 161.89, 144.36, 143.39, 143.27, 142.10, 141.89, 131.68, 129.77, 129.60, 126.54, 125.88, 121.22, 116.14, 102.15, 28.44, 27.30 ppm. 19F {1H} NMR (376 MHz, CDCl3) δF=−56.93 (s) ppm. HR MS (ES) m/z calcd for C20H16F3N2O3 194Pt 583.0740; found 583.0738 [M+H]+, 1067.1064 [M+M–acac]+ and 1167.1707 [M+M+H]+. UV‐vis (CHCl3): λmax (ϵ/L mol−1 cm−1) 252 (28886), 295 (22927), 358 (8888), 374 (10908), 431 (5959) nm. FTIR (solid, cm−1) (ATR) v max: 2953, 2921, 2852, 1580, 1561, 1537, 1511, 1476, 1459, 1438, 1422, 1381, 1366, 1338, 1254, 1211, 1183, 1155, 1137, 1075, 1042, 1029, 898, 816, 770, 676.
Synthesis of [Pt(L4)(acac)]: [PtCl(L4)(DMSO)] (0.058 g, 0.10 mmol) and sodium acetylacetonate (0.139 g, 1.00 mmol). Additional purification was achieved by recrystallisation (DCM/diethyl ether) to obtain the product as a red solid. Yield=13 mg, 24 %. 1H NMR (500 MHz, CDCl3) δH=9.73 (ddd, J HH=8.9, 1.3, 0.5 Hz, 1H), 9.24 (s, 1H), 8.14 (dd, J HH=8.2, 1.3 Hz, 1H), 7.93‐7.91 (m, 1H), 7.88 (d, J HH=8.1 Hz, 1H), 7.85 (ddd, J HH=8.6, 6.9, 1.6 Hz, 1H), 7.77 (ddd, J HH=8.2, 6.9, 1.3 Hz, 1H), 7.50–7.44 (m, 1H), 5.62 (s, 1H, acac), 2.11 (s, 3H), 2.09 (s, 3H) ppm. 13C{1H} NMR (126 MHz, CDCl3) δC=186.15, 184.49, 144.14, 143.79, 141.64, 131.76, 130.73, 130.08, 129.70, 126.06, 102.16, 28.43, 27.31 ppm.19F{1H} NMR (376 MHz, CDCl3) δF=−62.23 (s) ppm. HR MS (ES+) m/z calcd for C20H16F3N2O2 195Pt 568.0812; found 568.0814, [M+H]+, 1035.1005 [M+M–acac]+ and 1135.1511 [M+M+H]+. UV‐vis (CHCl3): λmax (ϵ/L mol−1 cm−1) 260 (43027), 293 (30199), 355 (12928), 372 (15049), 434 (8080) nm. FTIR (solid, cm−1) (ATR) v max: 2954, 2923, 2850, 1572, 1561, 1536, 1522, 1459, 1433, 1422, 1390, 1362, 1332, 1314, 1295, 1278, 1258, 1209, 1170, 1142, 1135, 1109, 1083, 1073, 1025, 982, 900, 837, 805, 773, 760, 732, 714, 680.
Synthesis of [Pt(L5)(acac)]: [PtCl(L5)(DMSO)] (0.064 g, 0.11 mmol) and sodium acetylacetonate (0.154 g, 1.10 mmol). Additional purification was achieved by recrystallisation (DCM/diethyl ether) to obtain the product as a red solid. Yield=38 mg, 62 %. 1H NMR (500 MHz, CDCl3) δH=9.74 (dd, J HH=8.9, 1.1 Hz, 1H), 9.22 (s, 1H), 8.13 (dd, J HH=8.2, 1.5 Hz, 1H), 8.03‐7.94 (m, 1H), 7.85 (ddd, J HH=8.8, 6.9, 1.6 Hz, 1H), 7.80–7.75 (m, 2H), 7.44‐7.40 (m, 1H), 5.60 (s, 1H, acac), 2.10 (s, 3H), 2.08 (s, 3H) ppm. 13C{1H} NMR (126 MHz, CDCl3) δC=186.06, 184.55, 143.96, 143.37, 141.89, 131.80, 130.26, 129.66, 126.62, 126.22, 124.91, 120.93, 102.15, 28.43, 27.30 ppm. 19F{1H} NMR (376 MHz, CDCl3) δF=−63.01 ppm. HR MS (ES) m/z calcd for C20H16F3N2O2 195Pt 568.0812; found 568.0814 [M+H]+, 1035.1066 [M+M–acac]+ and 1135.1519 [M+M+H]+. UV‐vis (CHCl3): λmax (ϵ/L mol−1 cm−1) 260 (43027), 293 (30199), 355 (12928), 372 (15049), 434 (8080) nm. FTIR (solid, cm−1) (ATR) v max: 2917, 2850, 1578, 1561, 1542, 1520, 1476, 1461, 1392, 1319, 1276, 1258, 1163, 1153, 1138, 1131, 1112, 1075, 1032, 1017, 975, 961, 911, 820, 775, 760, 714, 671.
Notes
Deposition Number(s) 2210432, 2210433, 2210434, 2210435 contain(s) the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service.
Conflict of interest
The authors declare no conflict of interest.
1.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Acknowledgments
Cardiff University (Knowledge Economy Skills Scholarship to SAF) and STG Aerospace (Dr Andrew Hallett and Dr Sean O'Kell) are thanked for financial support. The Leverhulme Trust is also thanked for funding (RPG‐2021‐003). We thank the staff of the Engineering and Physical Sciences Research Council (EPSRC) UK National Crystallographic Service at the University of Southampton.
Fitzgerald S. A., Xiao X., Zhao J., Horton P. N., Coles S. J., Knighton R. C., Ward B. D., Pope S. J. A., Chem. Eur. J. 2023, 29, e202203241.
Contributor Information
Prof. Jianzhang Zhao, Email: zhaojzh@dlut.edu.cn.
Prof. Simon J. A. Pope, Email: popesj@cardiff.ac.uk.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1.For example: Archer S., Weinstein J. A., Coord. Chem. Rev. 2012, 256, 2530; [Google Scholar]; Rausch A. F., Murphy L., Williams J. A. G., Yersin H., Inorg. Chem. 2012, 51, 312; [DOI] [PubMed] [Google Scholar]; Fuertes S., Chueca A. J., Perálvarez M., Borja P., Torrell M., Carreras J., Sicilia V., ACS Appl. Mater. Interfaces 2016, 8, 16160; [DOI] [PubMed] [Google Scholar]; Chi Y., Chou P.-T., Chem. Soc. Rev. 2010, 39, 638; [DOI] [PubMed] [Google Scholar]; Chakraborty A., Yarnell J. E., Sommer R. D., Roy S., Castellano F. N., Inorg. Chem. 2018, 57, 1298; [DOI] [PubMed] [Google Scholar]; Huo S., Carroll J., Vezzu D. A. K., Asian J. Org. Chem. 2015, 4, 1210. [Google Scholar]
- 2.For example: Zhao D., Krause J. A., Connick W. B., Inorg. Chem. 2015, 54, 8339; [DOI] [PubMed] [Google Scholar]; Chan M. H.-Y., Wong H.-L., Yam V. W.-W., Inorg. Chem. 2016, 55, 5570; [DOI] [PubMed] [Google Scholar]; Ortiz R. J., Braun J. D., Williams J. A. G., Hubert D. E., Inorg. Chem. 2021, 60, 16881. [DOI] [PubMed] [Google Scholar]
- 3. Chi Y., Chou P.-T., Chem. Soc. Rev. 2010, 39, 638; [DOI] [PubMed] [Google Scholar]; Visbal R., Gimeno M. C., Chem. Soc. Rev. 2014, 43, 3551. [DOI] [PubMed] [Google Scholar]
- 4. Stewart S. F., Refson K., Bennett R. D., Best J., Mel'nikov M. Y., Weinstein J. A., Inorg. Chem. 2012, 51, 9748. [DOI] [PubMed] [Google Scholar]
- 5. Botchway S. W., Charnley M., Haycock J. W., Parker A. W., Rochester D. L., Weinstein J. A., Williams J. A. G., Proc. Natl. Acad. Sci. USA 2008, 105, 16071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kobayashi A., Kato M., Eur. J. Inorg. Chem. 2014, 2014, 4469. [Google Scholar]
- 7. Wenger O. S., Chem. Rev. 2013, 113, 3686; [DOI] [PubMed] [Google Scholar]; Bryant M. J., Skelton J. M., Hatcher L. E., Stubbs C., Madrid E., Pallipurath A. R., Thomas L. H., Woodall C. H., Christensen J., Fuertes S., Robinson T. P., Beavers C. M., Teat S. J., Warren M. R., Pradaux-Caggiano F., Walsh A., Marken F., Carbery D. R., Parker S. C., McKeown N. B., Malpass-Evans R., Carta M., Raithby P. R., Nat. Commun. 2017, 8, 1800; [DOI] [PMC free article] [PubMed] [Google Scholar]; Lien C.-Y., Hsu Y.-F., Liu Y.-H., Peng S.-M., Shinmyozu T., Yang J.-S., Inorg. Chem. 2020, 59, 11584. [DOI] [PubMed] [Google Scholar]
- 8. Nazari M., Shahsavari H. R., Appl. Organomet. Chem. 2019, 33, e5020. [Google Scholar]
- 9. Zheng X., Chan M. H.-Y., Chan A. K.-W., Cao S., Ng M., Sheong F. K., Li C., Goonetilleke E. C., Lam W. W. Y., Lau T.-C., Huang X., Yam V. W.-W., Proc. Nat. Acad. Sci. 2022, 119, e2116543119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Richards B. S., Hudry D., Busko D., Turshatov A., Howard I. A., Chem. Rev. 2021, 121, 9165. [DOI] [PubMed] [Google Scholar]
- 11.For example: Liu Q., Yang T., Feng W., Li F., J. Am. Chem. Soc. 2012, 134, 5390. [DOI] [PubMed] [Google Scholar]
- 12. Bharmoria P., Bildirir H., Moth-Poulsen K., Chem. Soc. Rev. 2020, 49, 6529. [DOI] [PubMed] [Google Scholar]
- 13. Kerzig C., Wenger O. S., Chem. Sci. 2018, 9, 6670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yi X., Zhao J., Sun J., Guo S., Zhang H., Dalton Trans. 2013, 42, 2062. [DOI] [PubMed] [Google Scholar]
- 15. Lu Y., Wang J., McGoldrick N., Cui X., Zhao J., Caverly C., Twamley B., Maille G. M. O., Irwin B., Conway-Kenny R., Draper S. M., Angew. Chem. Int. Ed. 2016, 55, 14688. [DOI] [PubMed] [Google Scholar]
- 16. Phillips K. A., Stonelake T. M., Chen K., Hou Y., Zhao J., Coles S. J., Horton P. N., Keane S. J., Stokes E. C., Fallis I. A., Hallett A. J., O′Kell S. P., Beames J. M., Pope S. J. A., Chem. Eur. J. 2018, 24, 8577. [DOI] [PubMed] [Google Scholar]
- 17. Wang C., Reichenauer F., Kitzmann W. R., Kerzig C., Heinze K., Resch-Genger U., Angew. Chem. Int. Ed. 2022, 134, e202202238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Eastwood D., Gouterman M., J. Mol. Spectrosc. 1970, 35, 359. [Google Scholar]
- 19. Ji S., Wu W., Wu Y., Zhao T., Zhou F., Yang Y., Zhang X., Liang X., Wu W., Chi L., Wang Z., Zhao J., Analyst 2009, 134, 958. [DOI] [PubMed] [Google Scholar]
- 20. Islangulov R., Lott J., Weder C., Castellano F. N., J. Am. Chem. Soc. 2007, 129, 12652; [DOI] [PubMed] [Google Scholar]; Tanaka K., Ohashi W., Inafuka K., Shiotsu S., Chujo Y., Dyes Pigm. 2020, 172, 107821; [Google Scholar]; Kim J.-H., Kim J.-H., J. Am. Chem. Soc. 2012, 134, 17478; [DOI] [PubMed] [Google Scholar]; Goudarzi H., Keivanidis P. E., J. Phys. Chem. C 2014, 118, 14256; [Google Scholar]; Haefele A., Blumhoff J., Khnayzer R. S., Castellano F. N., J. Phys. Chem. Lett. 2012, 3, 299; [Google Scholar]; Kamada K., Sakagami Y., Mizokuro T., Fujiwara Y., Kobayashi K., Narushima K., Hirata S., Vacha M., Mater. Horiz. 2017, 4, 83; [Google Scholar]; Hwang S.-Y., Song D., Seo E.-J., Hollmann F., You Y., Park J.-B., Sci. Rep. 2022, 12, 9397; [DOI] [PMC free article] [PubMed] [Google Scholar]; Duan P., Yanai N., Kimizuka N., J. Am. Chem. Soc. 2013, 135, 19056. [DOI] [PubMed] [Google Scholar]
- 21. Xu K., Zhao J., Escudero D., Mahmoud Z., Jacquemin D., J. Phys. Chem. 2015, 119, 23801. [Google Scholar]
- 22. Zhong F., Zhao J., Eur. J. Inorg. Chem. 2017, 5196. [Google Scholar]
- 23. Wu W., Sun J., Ji S., Wu W., Zhao J., Guo H., Dalton Trans. 2011, 40, 11550. [DOI] [PubMed] [Google Scholar]
- 24. Chen K., Hussain M., Razi S. S., Hou Y., Yildiz E. A., Zhao J., Yaglioglu H. G., Di Donato M., Inorg. Chem. 2020, 59, 14731. [DOI] [PubMed] [Google Scholar]
- 25. Wu X., Gorden A. E. V., A S., Tonks, Vilseck J. Z., J. Org. Chem. 2007, 72, 8691. [DOI] [PubMed] [Google Scholar]
- 26. Zhang R., Qin Y., Zhang L., Luo S., Org. Lett. 2017, 19, 5629; [DOI] [PubMed] [Google Scholar]; Shee S., Panja D., Kundu S., J. Org. Chem. 2020, 85, 2775; [DOI] [PubMed] [Google Scholar]; Qi C., Jiang H., Huang L., Chen Z., Chen H., Synthesis 2011, 387; [Google Scholar]; Yuan H., Li K., Chen Y., Wang Y., Cui J., Chen B., Synlett 2013, 24, 2315; [Google Scholar]; Chen T., Chen X., Wei J., Lin D., Xie Y., Zeng W., Org. Lett. 2016, 18, 2078; [DOI] [PubMed] [Google Scholar]; Shen J., Wang X., Yang Z., Cheng G., Cui X., Org. Lett. 2016, 18, 1378; [DOI] [PubMed] [Google Scholar]; Cho Y.-H., Kim K.-H., Cheon C.-H., J. Org. Chem. 2014, 79, 901; [DOI] [PubMed] [Google Scholar]; Bandaru S. S. M., Bhilare S., Chrysochos N., Gayakhe V., Trentin I., Schulzke C., Kapdi A. R., Org. Lett. 2018, 20, 473. [DOI] [PubMed] [Google Scholar]
- 27.For example, Dhanaraj C. J., Johnson J., J. Photochem. Photobiol. B 2016, 161, 108; [DOI] [PubMed] [Google Scholar]; Dhanaraj C. J., Johnson J., Joseph J., Joseyphus R. S., J. Coord. Chem. 2013, 66, 1416; [Google Scholar]; Cummings S. D., Eisenberg R., Inorg. Chem. 1995, 34, 2007; [Google Scholar]; Zhang S.-M., Hu T.-L., Li J.-R., Du J.-L., Bu X.-H., CrystEngComm 2008, 10, 1595; [Google Scholar]; Andrews M., Laye R. H., Harding L. P., Pope S. J. A., Polyhedron 2008, 27, 2365. [Google Scholar]
- 28. Clark A. M., Rickard C. E. F., Roper W. R., Wright L. J., J. Organomet. Chem. 2000, 598, 262. [Google Scholar]
- 29. Guo B.-B., Azam M., Al Resayes S. I., Lin Y.-J., Jin G.-X., Chem. Eur. J. 2020, 26, 558. [DOI] [PubMed] [Google Scholar]
- 30. Hwang F.-M., Chen H.-Y., Chen P.-S., Liu C.-S., Chi Y., Shu C.-F., Wu F., Chou P.-T., Peng S.-M., Lee G.-H., Inorg. Chem. 2005, 44, 1344; [DOI] [PubMed] [Google Scholar]; Sengottuvelan N., Yun S.-J., Kim D.-Y., Hwang I.-H., Kang S. K., Kim Y.-I., Bull. Korean Chem. Soc. 2013, 34, 167; [Google Scholar]; Sengottuvelan N., Yun S.-J., Kang S. K., Kim Y.-I., Bull. Korean Chem. Soc. 2011, 32, 4321; [Google Scholar]; Otaif H. Y., Adams S. J., Horton P. N., Coles S. J., Beames J. M., Pope S. J. A., RSC Adv. 2021, 11, 39718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Culham S., Lanoe P.-H., Whittle V. L., Durrant M. C., Williams J. A. G., Kozhevnikov V. N., Inorg. Chem. 2013, 52, 10992. [DOI] [PubMed] [Google Scholar]
- 32. Leroux F. R., Manteau B., Vors J.-P., Pazenok S., Beilstein J. Org. Chem. 2008, 4, 10.3762/bjoc.4.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.For example with emissive Ir(III) complexes: Groves L. M., Schotten C., Beames J. M., Platts J. A., Coles S. J., Horton P. N., Browne D. L., Pope S. J. A., Chem. Eur. J. 2017, 23, 9407. [DOI] [PubMed] [Google Scholar]
- 34. Zhang Q.-W., Hartwig J. F., Chem. Commun. 2018, 54, 10124. [DOI] [PubMed] [Google Scholar]
- 35. Chaudhuri S. K., Roy S., Bhar S., Beilstein J. Org. Chem. 2012, 8, 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cho J. Y., Suponitsky K. Y., Li J., Tirnofeeva T. V., Barlow S., Marder S. R., J. Organomet. Chem. 2005, 690, 4090. [Google Scholar]
- 37. Cobb S. L., Murphy C. D., J. Fluorine Chem. 2009, 130, 132. [Google Scholar]
- 38. Burger H., Pawelke G., Haas A., Willner H., Downs A. J., Spectrochim. Acta 1978, 34 A, 287; [Google Scholar]; Brown J. D., Dobbie R. C., Straughan B. P., J. Chem. Soc. Dalton Trans. 1973, 1691. [Google Scholar]
- 39. Tamayo A. B., Alleyne B. D., Djurovich P. I., Lamansky S., Tsyba I., Ho N. M., Bau R., Thompson M. E., J. Am. Chem. Soc. 2003, 125, 7377; [DOI] [PubMed] [Google Scholar]; Lamansky S., Djurovich P., Murphy D., Abdel-Razzaq F., Kwong R., Tsyba I., Bortz M., Mui B., Bau R., Thompson M. E., Inorg. Chem. 2001, 40, 1704; [DOI] [PubMed] [Google Scholar]; Colombo M. C., Brunold T. C., Riedener T., Gudel H. U., Fortsch M., Burgi H.-B., Inorg. Chem. 1994, 33, 545. [Google Scholar]
- 40. Bossi A., Rausch A. F., Leitl M. J., Czerwieniec R., Whited M. T., Djurovich P. I., Yersin H., Thompson M. E., Inorg. Chem. 2013, 52, 12403. [DOI] [PubMed] [Google Scholar]
- 41. Stacey O. J., Platts J. A., Coles S. J., Horton P. N., Pope S. J. A., Inorg. Chem. 2015, 54, 6528. [DOI] [PubMed] [Google Scholar]
- 42. Alvarez S., Dalton Trans. 2013, 42, 8617. [DOI] [PubMed] [Google Scholar]
- 43. Novoa J. J., Aullón G., Alemany P., Álvarez S., J. Am. Chem. Soc. 1995, 117, 7169. [Google Scholar]
- 44. Komeda S., Lutz M., Spek A. L., Yamanaka Y., Sato T., Chikuma M., Reedijk J., J. Am. Chem. Soc. 2002, 124, 4738. [DOI] [PubMed] [Google Scholar]
- 45. Connick W. B., Henling L. M., Marsh R. E., Gray H. B., Inorg. Chem. 1996, 35, 6261; [Google Scholar]; Tzeng B.-C., Lee G.-H., Peng S.-M., Inorg. Chem. Commun. 2003, 6, 1341. [Google Scholar]
- 46. Stacey O. J., Ward B. D., Coles S. J., Horton P. N., Pope S. J. A., Dalton Trans. 2016, 45, 10297; [DOI] [PubMed] [Google Scholar]; Lowe J. A., Stacey O. J., Horton P. N., Coles S. J., Pope S. J. A., J. Organomet. Chem. 2016, 805, 87; [Google Scholar]; Stacey O. J., Amoroso A. J., Platts J. A., Horton P. N., Coles S. J., Lloyd D., Wiliams C. F., Hayes A. J., Dunsford J. J., Pope S. J. A., Chem. Commun. 2015, 51, 12305. [DOI] [PubMed] [Google Scholar]
- 47. Kumar P. S. V., Raghavendra V., Subramanian V., J. Chem. Sci. 2016, 128, 1527. [Google Scholar]
- 48. Poater A., Moradell S., Pinilla E., Poater J., Sola M., Martinez M. A., Llobet A., Dalton Trans. 2006, 1188. [DOI] [PubMed] [Google Scholar]
- 49. Lee K. N., Lee J. W., Ngai M.-Y., Tetrahedron 2018, 74, 7127; [DOI] [PMC free article] [PubMed] [Google Scholar]; Federsel D., Herrmann A., Christen D., Sander S., Willner H., Oberhammer H., J. Mol. Struct. 2001, 567, 127. [Google Scholar]
- 50. Serfaty I. W., Hodgins T., McBee E. T., J. Org. Chem. 1972, 37, 2651; [Google Scholar]; Manteau B., Genix P., Brelot L., Vors J.-P., Pazenok S., Giornal F., Leuenberger C., Leroux F. R., Eur. J. Org. Chem. 2010, 2010, 6043A. [Google Scholar]
- 51. Kvam P. I., Puzyk M. V., Balashev K. P., Songstad J., Acta Chem. Scand. 1995, 49, 335. [Google Scholar]
- 52. Brooks J., Babayan Y., Lamansky S., Djurovich P. I., Tsyba I., Bau R., Thompson M. E., Inorg. Chem. 2002, 41, 3055. [DOI] [PubMed] [Google Scholar]
- 53. Lui C., Song X., Wang Z., Qiu J., ChemPlusChem 2014, 79, 1472. [Google Scholar]
- 54. Kim J., Batagoda T., Lee J., Sylvinson D., Ding K., Saris P. J. G., Kaipa U., Oswald I. W. H., Omary M. A., Thompson M. E., Forrest S. R., Adv. Mater. 2019, 31, 1900921. [DOI] [PubMed] [Google Scholar]
- 55.For example: Xue M., Lam T.-L., Cheng G., Liu W., Low K.-H., Du L., Xu S., Hung F.-F., Phillips D. L., Che C.-M., Adv. Opt. Mater. 2022, 10.1002/adom.202200741; [DOI] [Google Scholar]; Pinter P., Soellner J., Strassner T., Eur. J. Inorg. Chem. 2021, 2021, 3104. [Google Scholar]
- 56. Wilson G. J., Launikonis A., Sasse W. H. F., Mau A. W.-H., J. Phys. Chem. A 1997, 101, 4860. [Google Scholar]
- 57. Coles S. J., Gale P. A., Chem. Sci. 2012, 3, 683. [Google Scholar]
- 58. Dolomanov O. V., Bourhis L. J., Gildea R. J., Howard J. A. K., Puschmann H., J. Appl. Crystallogr. 2009, 42, 339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Sheldrick G. M., Acta Crystallogr. Sect. A 2015, 71, 3. [Google Scholar]
- 60. Sheldrick G. M., Acta Crystallogr. Sect. C 2015, 27, 3. [Google Scholar]
- 61.Gaussian 09, Revision C.01, M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman, and D. J. Fox, Gaussian, Inc., Wallingford CT, 2016.
- 62.E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter, J. E. Bohmann, C. M. Morales, C. R. Landis, F. Weinhold, NBO 6.0, Theoretical Chemistry Institute, University of Wisconsin, Madison, 2013.
- 63.T. A. Keith, AIMAll, TK Gristmill Software, Overland Park KS, USA, 2019.
- 64. Chen Z.-W., Ye D.-N., Ye M., Zhou Z.-G., Li S.-H., Liu L.-X., Tet. Lett. 2014, 55, 1373. [Google Scholar]
- 65. Wang X.-T., Song J.-L., Zhong M., Kang H.-J., Xie H., Che T., Shu B., Peng D., Zhang L., Zhang S.-S., Eur. J. Org. Chem. 2020, 2020, 3635. [Google Scholar]
- 66. Song S., Shi X., Zhu Y., Ren Q., Zhou P., Zhou J., Li J., J. Org. Chem. 2022, 87, 4764. [DOI] [PubMed] [Google Scholar]
- 67. Liu K. M., Liao L. Y., Duan X. F., Chem. Commun. 2015, 51, 1124. [DOI] [PubMed] [Google Scholar]
- 68. Lou S.-J., Xu D.-Q., Xia A.-B., Wang Y.-F., Liu Y.-K., Du X.-H., Xu Z.-Y., Chem. Commun. 2013, 49, 6218. [DOI] [PubMed] [Google Scholar]
- 69. Godbert N., Pugliese T., Aiello I., Bellusci A., Crispini A., Ghedini M., Eur. J. Inorg. Chem. 2007, 5105. [Google Scholar]
- 70. Black D. S., Deacon G. B., Edwards G. L., Aust. J. Chem. 1994, 47, 217. [Google Scholar]
- 71. Santoro A., Whitwood A. C., Williams J. A. G., Kozhevnikov V. N., Bruce D. W., Chem. Mater. 2009, 21, 3871. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.