

Abstract
In Denmark, vaccination against Severe Acute Respiratory Syndrome Corona Virus 2 (SARS‐CoV‐2) has been with the Pfizer‐BioNTech (BTN162b2) or the Moderna (mRNA‐1273) mRNA vaccines. Patients with chronic hepatitis C virus (HCV) infection followed in our clinic received mRNA vaccinations according to the Danish roll‐out vaccination plan. To monitor HCV infection, RNA was extracted from patient plasma and RNA sequencing was performed on the Illumina platform. In 10 of 108 HCV patient samples, full‐length or traces of SARS‐CoV‐2 spike mRNA vaccine sequences were found in blood up to 28 days after COVID‐19 vaccination. Detection of mRNA vaccine sequences in blood after vaccination adds important knowledge regarding this technology and should lead to further research into the design of lipid‐nanoparticles and the half‐life of these and mRNA vaccines in humans.
Keywords: Hepatitis C virus, SARS‐CoV‐2, vaccine, blood, mRNA
INTRODUCTION
With the emergency approval by the FDA of two mRNA vaccines in December 2020, and subsequently very large‐scale vaccine production and mass immunization programs, a breakthrough in protective measures against the global pandemic with severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) was achieved. Both the Pfizer‐BioNTech (BTN162b2) and the Moderna (mRNA‐1273) vaccines code for production of the full‐length SARS‐CoV‐2 spike protein. To ensure stability, these vaccines are composed of codon‐optimized modified spike mRNA, have two stabilizing proline substitutions, and the mRNA is encapsulated in lipid nanoparticles (LNPs) [1, 2]. The modified nucleotide sequences allow perfect identification of the vaccine sequences as being different from any coronavirus sequence. Upon intramuscular injection, the vaccine mRNA is taken up by muscle and immune cells, and transported to the regional lymph nodes and concentrated in the spleen [3]. The vaccines consist of nonreplicating mRNA and are expected to naturally decompose both within the cytosol after translation and at the injection site. The half‐life of mRNA translation is estimated to be short, from hours to a day and translation is described to span up to 10 days [4, 5, 6]. The Infectious Diseases Society of America (IDSA) informs that the vaccine mRNA is degraded quickly by normal intracellular processes and states that there is no evidence for long‐term detection of mRNA vaccines in vaccinated individuals by RNA‐seq [1, 7].
In Denmark, the predominant vaccines against Corona Virus Disease 2019 (COVID‐19) have been the two mRNA‐based vaccines from Pfizer‐BioNTech and Moderna. As of July 28, 2022, 80.2% of the population has received two doses and 81.6% at least one dose [8]. At Copenhagen University Hospital, Amager‐Hvidovre, Denmark, patients with chronic hepatitis C virus (HCV) infection are routinely followed at Department of Infectious Diseases, while measurement of HCV viral load and genotyping by whole RNA‐Seq genome sequencing of their HCV RNA, directly from plasma samples, are performed at Department of Clinical Microbiology [9]. In this paper, we describe the unexpected finding of SARS‐CoV‐2 vaccine mRNA sequences in plasma from 10 HCV patient samples up to 28 days after COVID‐19 vaccination. These patients had recently received SARS‐CoV‐2 mRNA vaccinations according to the Danish roll‐out vaccination plan.
METHODS
We analyzed five consecutive sequencing runs, from May 2021 to the end of June 2021, with 108 HCV patients, five negative controls and five HCV‐positive controls consisting of HCV grown in cell culture [10]. Samples were from patients that were HCV positive and came for HCV treatment evaluation with no relation to their vaccination characteristics. The direct RNA sequencing (RNA‐seq) procedure has been described previously [11]. Briefly, RNA was extracted with the ZR viral RNA kit (Zymo Research, Irvine, CA, USA) and depleted for human rRNA with the NEBNext rRNA depletion kit (New England BioLabs, Ipswich. MA, USA). Stranded RNA‐seq libraries were prepared with the NEBNext Ultra II directional RNA library prep kit (New England BioLabs). Sequencing was performed with 2 × 150‐bp paired‐end reads on a NextSeq instrument (Illumina, San Diego, CA, USA).
All software was used with default parameters unless specified. Raw reads were trimmed for low‐quality bases with fastp v 0.20.1 with minimum phred quality of 20 and minimum length of 50 [12]. Human depletion was done by mapping the trimmed reads to the human genome hg38 (GenBank accession no. GCA_000001405.27) with Bowtie2 v 2.3.4.1 adding the parameters ‐k 1 ‐X 2000 [13]. Paired reads unmapped to human genome sequences were de novo assembled with VICUNA v 1.3 using minimum contig links of 2 and minimum identify of 90 [14]. As part of our pipeline, contigs above 1000 bp are aligned against NCBI's nt database using BLASTn v2.8.1+ with a minimum value threshold of 1 e‐22 [15]. Contigs that were found to align to coronavirus sequences were subsequently aligned against two assemblies of SARS‐CoV2‐spike‐encoding mRNA vaccines, BNT‐162b2 and mRNA‐1273 using BLASTn for confirmation [16]. Paired reads were also mapped against the two vaccine assemblies using BWA‐mem v 0.7.16 and SAMtools v 1.2 inside the NASP pipeline v 1.1.2 [17, 18, 19, 20]. Stats of number of reads mapping and coverage were calculated only for the SARS‐CoV‐2 spike gene coding region avoiding the 5′ and 3′ UTR as these regions share high percentage of similarity with human gene regions. Coverage was calculated with BEDTools v 2.30.0 and plotted in R v 3.6.1 with ggplot2 [21, 22, 23]. SNP calls were generated with HaplotypeCaller from GATK v 4.2.0.0 and filtered by heterozygous genotype, mapping quality of <30 and symmetric odds ratio of >3 [24]. Consensus sequences were made in GATK and bases covered with less than 10 reads were masked with BEDTools.
The best (highest coverage) spike sequence for the Pfizer‐BioNTech and similarly the two best spike sequences for the Moderna vaccine have been uploaded to NCBI (GenBank accession numbers OK120840‐OK120842). Gaps in the sequences are represented by Ns. Approval by an institutional review board was not required, as this study was performed as an operational activity related to patient management.
RESULTS
De novo assembly of human‐depleted RNA‐seq reads from two patient samples produced contigs of >1000 nt with closest homology to bat coronavirus and 67% homology to the SARS‐CoV‐2 reference genome (NC_045512.2). Translation of the contig nucleotide sequences to amino acid sequences revealed 100% identity to parts of the spike protein of SARS‐CoV‐2. From the literature we found the sequences of the two commercial SARS‐CoV‐2 mRNA vaccines which were used as references to map reads from all the samples [16, 25]. This led to the identification of an additional eight samples with reads that matched the mRNA vaccine sequences. Both mRNA vaccine sequences have been modified and are only ~70% identical to the spike reference genome on a nucleotide level, making them distinct from circulating infectious SARS‐CoV‐2 sequences. Thus, of the 108 patient samples, 10 samples (9.3%) had partial or up to full sequences of the vaccine mRNA sequence (Fig. 1), identified from one to 28 days postvaccination. There was ~100% identity between the detected mRNA nucleotide sequences found in plasma and the specific mRNA vaccine given. The 10 samples had a median of 5.5 million raw read pairs available (see Table S1). Breadth and depth of coverage of the vaccine mRNA sequences ranged from completeness and >20 000, respectively, to short fragments with a depth of coverage of 100 (Fig. 1). None of the negative or the HCV‐positive controls had SARS‐CoV‐2 matching reads.
Fig. 1.
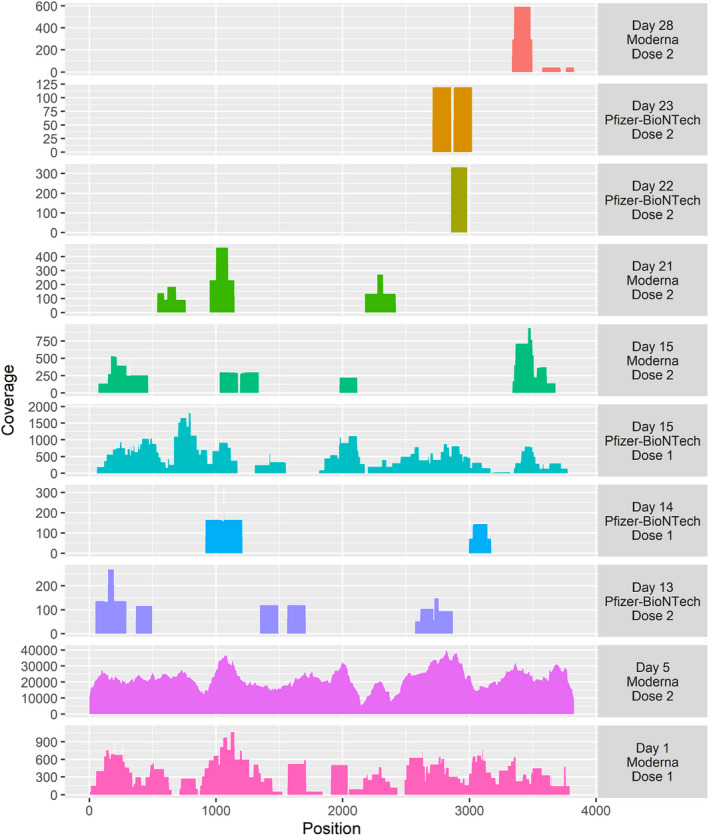
Mapping of trimmed and filtered reads to the coding regions of the spike protein of the Moderna mRNA vaccine or Pfizer BioNTech vaccine. Each colored plot is a single sample. The right‐hand column shows the day postvaccination, the vaccine given and found in blood and if sample was drawn after 1. vaccination or revaccination. The two bottom samples are from the same patient after first and second vaccination with the Moderna vaccine.
DISCUSSION
We surprisingly found fragments of COVID‐19 vaccine mRNA up to 28 days postvaccination in blood from chronic HCV patients vaccinated with mRNA vaccines from both Pfizer‐BioNTech and Moderna.
Analysis of mRNA vaccine function has focused on the immune response and on protection of vaccinated individuals from SARS‐CoV‐2 induced severe COVID‐19 [26, 27]. The LNPs have been reported to be rapidly cleared by immune cells and mRNA is degraded by exonucleases in tissue and blood [28, 29, 30]. A recent study did not detect the vaccine mRNA by quantitative PCR in human milk after 4–48 h postdose 1 or 2 with BNT162b2 or mRNA‐1273 [31].
RNA‐seq is widely used in biological and medical research. We routinely use direct total RNA‐seq to obtain full length HCV RNA sequences and from these infer their genotype. Our pipeline includes taxonomic analysis of contigs of more than 1000 nt which led to the discovery of coronavirus sequences that when first blasted had closest identity to a bat coronavirus.
We expect that vaccine mRNA detected in plasma is contained within LNPs and that the LNPs in plasma have been slowly released from the injection site either directly to the blood or through the lymph system. Without the LNPs protecting the mRNA, the mRNA would rapidly degrade. This allows prolonged spike protein production giving an advantage for a continuous immune response in some persons. Current studies on half‐life of mRNA vaccines could have underestimated the half‐life of the LNPs, primarily using results from half‐life of studies of mRNA in the cytosol of human cells.
In samples where we observed only fragments of vaccine mRNA, this could indicate that the concentration of LNPs in plasma is low. This is in accordance with our findings that HCV viral load should be higher than 10 000 IU/mL to obtain a full‐length HCV RNA genome (size ~9600 nt) as genome coverage correlates with viral load. The number of SARS‐CoV‐2 vaccine mRNA molecules is therefore likely lower than ~4000 IU/mL, explaining the partial read coverage randomly spread over the coronavirus vaccine sequence.
To our knowledge, our study is the first to detect Pfizer‐BioNTech and Moderna COVID‐19 mRNA vaccine sequences in blood after vaccination, and therefore provides new knowledge regarding the timeframe in which the mRNA can be detected. This study examined a cohort of HCV‐positive patients with presumed functional immune systems, as most of the patients can be cured, with negative HCV RNA 12 weeks after end of treatment with direct‐acting antivirals. A future prospective study to establish the half‐life of mRNA vaccines in vaccine recipients could be performed using mRNA vaccine‐specific PCRs. These findings are interesting and should lead to further research into the design of LNPs and the half‐life of LNPs and mRNA vaccines, but it should be emphasized that our data does not in any way change the conclusion that both mRNA vaccines are safe and effective.
CONFLICT OF INTEREST
Authors have no conflicts of interest to declare.
Supporting information
Table S1. Sequencing and mapping report of the 10 patient samples. Mapping and coverage statistics are calculated for only the SARS‐CoV‐2 spike gene coding region of the vaccine.
References
- 1. Nance KD, Meier JL. Modifications in an emergency: the role of N1‐Methylpseudouridine in COVID‐19 vaccines. ACS Cent Sci. 2021;7:748–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Schoenmaker L, Witzigmann D, Kulkarni JA, Verbeke R, Kersten G, Jiskoot W, et al. mRNA‐lipid nanoparticle COVID‐19 vaccines: structure and stability. Int J Pharm. 2021;601:120586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tombácz I, Laczkó D, Shahnawaz H, Muramatsu H, Natesan A, Yadegari A, et al. Highly efficient CD4+ T cell targeting and genetic recombination using engineered CD4+ cell‐homing mRNA‐LNP. Mol Ther. 2021;29(11):3293–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wayment‐Steele HK, Kim DS, Choe CA, Nicol JJ, Wellington‐Oguri R, Watkins AM, et al. Theoretical basis for stabilizing messenger RNA through secondary structure design. Nucleic Acids Res. 2021;49(18):10604–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lindsay KE, Bhosle SM, Zurla C, Beyersdorf J, Rogers KA, Vanover D, et al. Visualization of early events in mRNA vaccine delivery in non‐human primates via PET‐CT and near‐infrared imaging. Nat Biomed Eng. 2019;3:371–80. [DOI] [PubMed] [Google Scholar]
- 6. Pardi N, Tuyishime S, Muramatsu H, Kariko K, Mui BL, Tam YK, et al. Expression kinetics of nucleoside‐modified mRNA delivered in lipid nanoparticles to mice by various routes. J Control Release. 2015;217:345–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Vaccines FAQ. [Internet]; 2021. https://www.idsociety.org/covid‐19‐real‐time‐learning‐network/vaccines/mrna‐vaccines/#overviewandmechanism
- 8. Danish vaccination data for covid‐19. [Internet]; 2022. https://experience.arcgis.com/experience/9824b03b114244348ef0b10f69f490b4/page/Kommunalt
- 9. Schønning K, Pedersen MS, Johansen K, Landt B, Nielsen LG, Weis N, et al. Analytical and clinical performance of the Hologic Aptima HCV quant dx assay for the quantification of HCV RNA in plasma samples. J Virol Methods. 2017;248:159–65. [DOI] [PubMed] [Google Scholar]
- 10. Ramirez S, Bukh J. Current status and future development of infectious cell‐culture models for the major genotypes of hepatitis C virus: essential tools in testing of antivirals and emerging vaccine strategies. Antiviral Res. 2018;158:264–87. [DOI] [PubMed] [Google Scholar]
- 11. Pedersen MS, Mollerup S, Nielsen LG, Jenssen H, Bukh J, Schønning K. Genome sequence of an unknown subtype of hepatitis C virus genotype 6: another piece for the taxonomic puzzle. Microbiol Resour Announc. 2019;8(42):e01030–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chen S, Zhou Y, Chen Y, Gu J. Fastp: an ultra‐fast all‐in‐one FASTQ preprocessor. Bioinformatics. 2018;34(17):i884–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Langmead B, Salzberg S. Fast gapped‐read alignment with bowtie 2. Nat Methods. 2012;9(4):357–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yang X, Charlebois P, Gnerre S, Coole MG, Lennon NJ, Levin JZ, et al. De novo assembly of highly diverse viral populations. BMC Genomics. 2012;13:475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, et al. BLAST+: architecture and applications. BMC Bioinformatics. 2009;10:421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Assemblies‐of‐putative‐SARS‐CoV2‐spike‐encoding‐mRNA‐sequences‐for‐vaccines‐BNT‐162b2‐and‐mRNA‐1273. [Internet]; 2021. https://github.com/NAalytics/Assemblies‐of‐putative‐SARS‐CoV2‐spike‐encoding‐mRNA‐sequences‐for‐vaccines‐BNT‐162b2‐and‐mRNA‐1273
- 17. Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25(16):2078–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bonfield JK, Marshall J, Danecek P, Li H, Ohan V, Whitwham A, et al. HTSlib – C library for reading/writing high‐throughput sequencing data. Gigascience. 2021;10(2):giab007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA‐MEM. arXiv. 2013;1303.3997v2 [q‐bio.GN].
- 20. Sahl JW, Lemmer D, Travis J, Schupp JM, Gillece JD, Aziz M, et al. NASP: an accurate, rapid method for the identification of SNPs in WGS datasets that supports flexible input and output formats. Microbial Genomics. 2016;2(8):e000074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26(6):841–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. R Core Team . R: a language and environment for statistical computing [internet]. Vienna, Austria: R Foundation for Statistical Computing; 2016. https://www.R‐project.org/ [Google Scholar]
- 23. Wickham H. ggplot2: elegant graphics for data analysis [internet]. New York: Springer‐Verlag; 2016. https://ggplot2.tidyverse.org [Google Scholar]
- 24. Poplin R, Ruano‐Rubio V, DePristo MA, Fennell TJ, Carneiro MO, Van der Auwera GA, et al. Scaling accurate genetic variant discovery to tens of thousands of samples. bioRxiv. 2017;201178. [Google Scholar]
- 25. World Health Organization . Messenger RNA encoding the full‐length SARS‐CoV‐2 spike glycoprotein [Internet]; 2021. https://berthub.eu/articles/11889.doc
- 26. Ong DSY, Fragkou PC, Schweitzer VA, Chemaly RF, Moschopoulos CD, Skevaki C, et al. How to interpret and use COVID‐19 serology and immunology tests. Clin Microbiol Infect. 2021;27(7):981–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Science Brief: COVID‐19 Vaccines and Vaccination. [Internet]; 2021. https://www.cdc.gov/coronavirus/2019‐ncov/science/science‐briefs/fully‐vaccinated‐people.html
- 28. Delehedde C, Even L, Midoux P, Pichon C, Perche F. Intracellular routing and recognition of lipid‐based mRNA nanoparticles. Pharmaceutics. 2021;13(7):945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Houseley J, Tollervey D. The many pathways of RNA degradation. Cell. 2009;136(4):763–76. [DOI] [PubMed] [Google Scholar]
- 30. Hou X, Zaks T, Langer R, Dong Y. Lipid nanoparticles for mRNA delivery. Nat Rev Mater. 2021;6(12):1078–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Golan Y, Prahl M, Cassidy A, Lin CY, Ahituv N, Flaherman VJ, et al. Evaluation of messenger RNA from COVID‐19 BTN162b2 and mRNA‐1273 vaccines in human milk. JAMA Pediatr. 2021;175(10):1069–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Sequencing and mapping report of the 10 patient samples. Mapping and coverage statistics are calculated for only the SARS‐CoV‐2 spike gene coding region of the vaccine.