

Abstract
The click reaction between a functionalized trans‐cyclooctene (TCO) and a tetrazine (Tz) is a compelling method for bioorthogonal conjugation in combination with payload releasing capabilities. However, the synthesis of difunctionalized TCOs remains challenging. As a result, these compounds are poorly accessible, which impedes the development of novel applications. In this work, the scalable and accessible synthesis of a new bioorthogonal difunctionalized TCO is reported in only four single selective high yielding steps starting from commercially available compounds. The TCO‐Tz click reaction was assessed and revealed excellent kinetic rates and subsequently payload release was shown with various functionalized derivatives. Tetrazine triggered release of carbonate and carbamate payloads was demonstrated up to 100 % release efficiency and local drug release was shown in a cellular toxicity study which revealed a >20‐fold increase in cytotoxicity.
Keywords: bioorthogonal conjugation, click and release, click chemistry, cycloaddition, TCO, tetrazine, trans-cyclooctene
The synthesis of a novel strained difunctionalized trans‐cyclooctene in only four high‐yielding steps is presented and the releasing capabilities of functionalized derivatives were evaluated upon tetrazine activation. This method presents a major step forward in the ease and efficiency of synthesizing releasing difunctionalized trans‐cyclooctenes.

Introduction
Ever since the visionary publication of the Sharpless group about click chemistry, this field has grown significantly and has had a tremendous impact on drug discovery, bioconjugation and modern chemistry in general.[ 1 , 2 ] Many click reactions are bioorthogonal, a term first used by Bertozzi in 2003, indicating that functional groups are only mutually reactive and do not interfere with biochemical processes occurring in living systems.[ 3 , 4 , 5 , 6 ] Examples of bioorthogonal click reactions include the strain‐promoted azide‐alkyne cycloaddition (SPAAC), the Staudinger ligation and the strain‐promoted oxidation‐controlled cyclooctyne‐1,2‐quinone cycloaddition (SPOQC).[ 7 , 8 , 9 ] In the last decade, bioorthogonal chemistry has successfully been employed in both diagnostic (e. g. radiolabeling)[ 10 , 11 ] and therapeutic applications such as antibody‐drug conjugates (ADCs) [12] and prodrugs.[ 13 , 14 , 15 ]
The inverse‐electron‐demand Diels‐Alder reaction (IEDDA), a reaction between an electron‐rich dienophile and an electron‐poor diene, has been applied for the ligation of trans‐cyclooctenes (TCOs) and tetrazines (Tz). [16] This bioorthogonal ligation usually shows high reaction rates and makes in vivo bioconjugation possible at low concentrations, as demonstrated in numerous studies.[ 17 , 18 , 19 , 20 ] As a next step, in 2013 Versteegen et al. reported on the design and synthesis of TCO derivatives that upon reaction with a tetrazine eliminate a substituent at the allylic position of the TCO. This so‐called click‐to‐release approach was applied to enable site‐specific and traceless drug release upon TCO‐tetrazine ligation.[ 21 , 22 ] The fast reaction kinetics and click and release capability of the TCO‐tetrazine adduct opens up countless possibilities for novel applications in drug release and beyond.[ 23 , 24 ] However, the complex synthesis of TCOs ‐ especially of the releasing ones ‐ prevents the roll‐out of this versatile technology towards novel applications.
In 2016 Rossin et al. reported the synthesis of the first difunctionalized cis‐cyclooctene (CCO) with an allylic moiety in seven steps starting from cis,cis‐1,5‐cyclooctadiene (1,5‐COD, Figure 1A). [17] The authors reported the formation of a mixture of the axial and equatorial difunctionalized TCO derivatives after isomerization with UV‐light and a sensitizer as described by the Fox group (Figure 1B). [25] Currently, the synthetic accessibility of the most commonly used difunctionalized TCOs with releasing properties is rather problematic, mainly due to low yielding steps and a lengthy overall synthesis.[ 17 , 24 ] Ideally, a TCO should have multiple orthogonal handles, a fast click reaction with a tetrazine and a rapid release functionality. In comparison with the difunctionalized TCO of Rossin et al., a shorter and higher yielding synthesis method of a TCO with similar properties is desired.
Figure 1.

A) Difunctionalized CCO synthesis in seven steps as reported by Rossin et al. B) CCO isomerization strategy by the Fox group for the synthesis of the corresponding TCOs. C) Our work featuring a straightforward four‐step synthesis of a difunctional releasing strained TCO and the subsequent click and release studies.
Herein, we present the straightforward synthesis of a strained and allylic functionalized TCO with two orthogonal handles in only four steps starting from readily available 1,5‐COD (Figure 1C). After separation, both the endo‐ and exo‐diastereomers were diastereoselectively functionalized at the allylic position prior to the photochemical isomerization step of the cis‐double bond. A recent continuous flow isomerization method including an aqueous extraction to separate the cis‐ and trans‐cyclooctynes, developed by Blanco et al. was employed in this work, facilitating the scalability of this pathway. [26] Functionalized derivatives of the TCO were evaluated for their releasing capabilities. This method represents a major step forward in the ease and efficiency of the synthesis of releasing TCOs.
Results and Discussion
In the design of new TCOs, we were inspired by our previous work on bicyclo[6.1.0]nonyne (BCN), where we demonstrated that the fusion of a cyclopropane with the cyclooctyne induced excellent conjugation rates and kinetics due to the increased ring strain. [8] Cyclopropanated CCOs have also been isomerized by the Fox group to obtain strained TCOs (sTCOs), revealing an enhanced reactivity towards tetrazines as a result of the fusion with the cyclopropane ring.[ 27 , 28 ] Our synthesis commenced with monocyclopropanation of 1,5‐COD (1) by reaction with ethyl diazoacetate in the presence of a catalytic amount of copper(II) acetylacetonate under neat conditions to allow 1,5‐COD recovery after completion. [29] This afforded a mixture of the exo‐ and endo‐diastereomers of 2 (Scheme 1) in a 2 : 1 ratio, respectively (61 % total yield), which were separated by silica gel chromatography. In our experience, substitution reactions on eight‐membered rings have proven to be inefficient, therefore we envisioned that activating 2 with N‐iodosuccinimide (NIS) in the presence of sodium acetate and acetic acid would incorporate both the iodide and acetate in a single step as in the work of Goto and co‐workers. [30] We were pleased to find that this reaction, which proceeds through the corresponding iodiranium ion, leads to highly diastereoselective acetate addition resulting in the acetylated iodohydrin 3 in 83 % yield.
Scheme 1.

Synthesis of the difunctionalized TCO derivatives starting from commercially available cis,cis‐1,5‐cyclooctadiene (1). i. copper(ii) acetylacetonate (0.01 equiv), ethyl diazoacetate (0.17 equiv), 90 °C, 18 h, 61 % (exo/endo 2 : 1). ii. sodium acetate (3.0 equiv), acetic acid, NIS (1.2 equiv), 21 °C, 3 h, 83 %. iii. DBU (3.0 equiv), toluene, 100 °C, 18 h, 89 %. iv. methyl benzoate (2.03 equiv), heptane, MTBE, hv, silver nitrate (2.97 equiv.), 21 °C, 16 h, 5/6 7 : 13, 46 %. v. potassium carbonate (2.0 equiv), ethanol, 21 °C, 18 h, 7: 28 %, 8: 59 %. vi. sodium acetate (3.0 equiv), acetic acid, NIS (1.2 equiv), 21 °C, 72 h, 82 %. vii. DBU (3.0 equiv), toluene, 100 °C, 48 h, 48 %. viii. methyl benzoate (0.4 equiv), heptane, hv, silver nitrate (2.0 equiv), 21 °C, 43 h, 11/12 1 : 1, 31 %. ix. potassium carbonate (2.0 equiv), ethanol, 21 °C, 18 h, 13/14 1 : 1, 43 %.
Subsequently, a dehydroiodination reaction was performed with 1,8‐diazabicyclo[5.4.0]undec‐7‐ene (DBU) in toluene to afford difunctionalized CCO 4 in 89 % yield, presenting the Oac and CHCO2Et groups cis to each other. The stereochemical elucidation of 4 was carried out based on the single‐crystal X‐ray diffraction (Figure 2).
Figure 2.
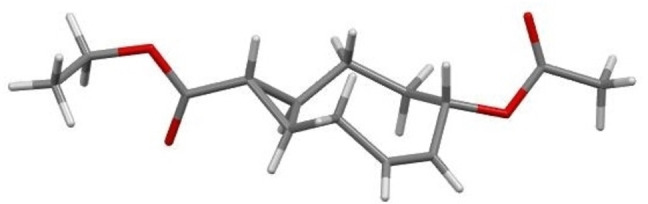
X‐ray crystal structure of compound 4. Deposition Number 2108332 contains the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service.
Compound 4 was then subsequently isomerized into the TCO derivative with UV irradiation at a wavelength of 254 nm in the presence of methyl benzoate (photosensitizer) in a continuous flow setup.[ 25 , 26 ] Selective removal of the TCO with aqueous silver nitrate via liquid‐liquid extraction coupled to the photoflow system gave access to multigram amounts of the desired TCO.14 With the isomerization a new stereoelement was introduced, giving rise to an inseparable mixture of the exo‐diastereomers 5 and 6. Fortunately, the products could be easily separated with silica gel chromatography after ethanolysis of the acetate with potassium carbonate in ethanol to furnish the corresponding hydroxylated TCOs 7 and 8, having the allylic hydroxyl group available for functionalization with a payload.
When the alkene of compound 4 is isomerized, the two planes C ‐C ‐H on both sides of the alkene rotate both clock‐ and counterclockwise to form P‐ and M‐cyclooctenes 5 and 6, respectively.[ 26 , 31 ] In these cases, the resulting TCOs adopt a half‐chair‐chair conformation where the acetoxy group occupies an axial position in the P‐isomer 5 and an equatorial position in the M‐isomer 6 (Scheme 2).
Scheme 2.

Isomerization of CCO 4 into M‐ and P‐TCOs 5 and 6, respectively.
Next to the exo‐derivatives, the endo‐TCO analogues 13 and 14 were synthesized utilizing the same synthetic methodology (Scheme 1, endo‐pathway). The iodohydrin step was performed on 2 and afforded 9 diastereoselectively in 82 % yield. The endo‐TCO‐precursor 10 was obtained after dehydroiodination with DBU and subsequently isomerized into the corresponding endo‐TCOs 11 and 12 in a 1 : 1 ratio. Similar to the exo‐pathway, we obtained an inseparable mixture of the equatorial and axial diastereomers 11 and 12. Unfortunately, after ethanolysis with potassium carbonate an inseparable mixture of the two diastereoisomers 13 and 14 was obtained.
In order to study the click and release kinetics various allylic functionalized TCO derivatives were synthesized. First, we aimed to monitor the click and release reactions with 19F NMR spectroscopy by eliminating a fluorinated payload upon tetrazine activation. The allylic alcohols of the TCOs 7, 8, 13 and 14 were functionalized with a pentafluorophenyl (PFP) group (Scheme 3). The exo‐PFP derivatives were synthesized in one step from 7 and 8 with bis(pentafluorophenyl) carbonate (DPFPC) and N,N‐diisopropylethylamine (DIPEA) in dichloromethane to afford 15 and 16 in 43 and 32 % yield, respectively. Although the deacetylated endo‐TCOs were obtained as a mixture of axial and equatorial diastereoisomers, we still studied the release capabilities. Therefore, the endo‐PFP analogues 17 and 18 were synthesized in a similar fashion with DPFPC and DIPEA in an overall 33 % yield obtained as an inseparable mixture of the axial and equatorial derivatives (ratio 1 : 2.5 respectively).
Scheme 3.

Synthesis of the PFP derivatives 15, 16, 17 and 18 from the corresponding TCOs 7, 8, 13 and 14 with DPFPC (2.75 equiv) and DIPEA (3.0 equiv) in dichloromethane, at room temperature for 18 h. The endo‐derivatives were obtained as an inseparable mixture of the axial (17) and equatorial (18) diastereoisomers in a 1 : 2.5 ratio respectively. PFP=pentafluorophenyl.
Having the new allylic functionalized TCO derivatives in hand, we set out to investigate the click and release kinetics with 19F NMR spectroscopy (Scheme 4). The functionalized TCOs 15, 16, 17 and 18 were dissolved in CDCl3, a solution of 3,6‐di‐2‐pyridyl‐1,2,4,5‐tetrazine (DPTZ, 19) was added and a 19F NMR spectrum was recorded every 48 seconds. In case of the axial‐TCO 15, we observed complete release of the PFP moiety within 20 minutes. Further investigation of the click and release reactions with 15 revealed that at −20 °C solely the click conjugate was observed and no release of PFP was obtained. Only after elevating the temperature to 10 °C, the PFP carbonate was released upon tautomerization from the conjugated product at such a rate that the intermediate tautomeric product could not be observed (see: Figure S8). The equatorial‐TCO 16 clearly showed conjugation with tetrazine 19, but unfortunately no release of the PFP moiety. Instead, a stable tautomerized product with the intact PFP carbonate moiety was formed over time (Scheme 4). This rate is in line with previous studies showing that the equatorial isomer has a significant reactivity decrease because of the steric and possible electronic effects, as again confirmed in this study.[ 21 , 32 ]
Scheme 4.

19F NMR click reaction kinetics study of the axial‐TCOs 15 and 17 and equatorial‐TCOs 16 and 18 by reacting them with DPTZ (19) in CDCl3. TCO 15 revealed complete release of PFP upon the click reaction within 20 minutes. In case of 16, only the click conjugate and the tautomerized products were observed, but no release of PFP. In case of the diastereomic mixture of the endo‐derivatives 17 and 18, minor release of PFP upon the click reaction of the axial isomer with 19 was observed, the equatorial primarily afforded a stable tautomeric product.
In case of the diastereomic mixture of the endo‐derivatives 17 and 18, we observed a significantly slower click reaction under the same conditions than for the exo‐TCOs. Similar to the exo‐derivatives, the reaction was followed via 19F NMR spectroscopy. The different axial and equatorial intermediates and PFP product had an isolated signal which was integrated over time. The endo‐axial isomer 17 showed only minor PFP release within 120 minutes and the equatorial isomer 18 primarily afforded the tautomeric product and no PFP release (see: Figure S9).
Since we observed minor release of PFP with the endo‐TCO derivatives 17 and 18, we focused on the exo‐isomers, also taking into account that the endo‐TCO (13 and 14) was obtained as an inseparable diastereomeric mixture. Besides, the precursor was synthesized in an overall lower yield as compared to the exo‐one and an excess of the exo‐derivative was formed in the cyclopropanation step (2, 2 : 1 ratio). Conversely, we showed that the mixture of isomers in 2 can be isomerized solely into the exo‐isomer with 1 M potassium tert‐butoxide in THF and dry ethanol (data not shown). [33]
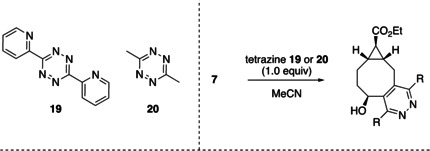
Next, the click kinetics of the axial exo‐TCO 7 were investigated upon reaction with a single equivalent of tetrazine 19 or 20. The second‐order rate constants were determined in acetonitrile at room temperature. [21] For the click reaction of tetrazine 19 with 7 at 188 μM we found a K 2=594.0 M−1 s−1±12.38 and for tetrazine 20 K 2=0.8589 M−1 s−1±0.001, as also expected due to increased electron density of tetrazine 19 (see: Table 1). When the click reaction was carried out at 97 μM we encountered similar values. These results seem to demonstrate that our new TCO outperforms the currently available releasing difunctional TCOs in the literature in terms of click kinetics.
Table 1.
Second‐order rate constants for reaction of TCO 7 with tetrazines 19 and 20. Values were determined using UV/Vis spectroscopy at 540 nm (specific for tetrazine) in acetonitrile at 21 °C.
|
| ||
|---|---|---|
|
tetrazine |
97 μM |
188 μM |
|
19 |
516.2 M−1 s−1±16.78 |
594.0 M−1 s−1±12.38 |
|
20 |
0.9864 M−1 s−1±0.004 |
0.8589 M−1 s−1±0.001 |
Next to the carbonate‐linked PFP TCOs, we wanted to explore more stable carbamate linked derivatives, as these have already been reported in the literature to provide successful release. [22] We synthesized fluorescently caged exo‐TCO derivatives by coupling 7‐amino‐4‐methylcoumarin (AMC) via a carbamate linkage to the allylic alcohol on our TCO. With this approach we could directly asses the release properties of our system with straightforward fluorescence measurements, since the characteristic fluorescence of AMC can easily be quenched by modifying the free amine as also demonstrated in similar TCO‐AMC release studies in the recent literature.[ 34 , 35 ] The best synthesis results for the fluorescent TCO derivatives 21 and 22 were obtained by first forming the isocyanate of AMC with triphosgene and DIPEA in toluene at reflux temperature. Subsequently, the TCO derivative with the free allylic hydroxyl 7 or 8 and 4‐(dimethylamino)pyridine (DMAP) were dissolved in dichloromethane and the AMC‐isocyanate was added. This afforded both the axial and equatorial exo‐TCO derivatives 21 and 22 in 63 and 42 % yield, respectively.
Moreover, we also synthesized aliphatic carbamate derivatives of the exo‐TCOs to further elucidate the scope of their release properties (Scheme 5). To this extent, we coupled a glycine or sarcosine derivative to the allylic alcohol of the TCO. These compounds were chosen with the work of Carlson and co‐workers in mind, who showed that an unmethylated nitrogen ‐ as in the glycine derivative ‐ can result in formation of a dead‐end product, while an N‐methylated nitrogen as in sarcosine should prevent this. [36] All compounds were generated in a two‐step synthesis, wherein hydroxy TCOs 7 and 8 were first activated with DPFPC, DIPEA and DMAP. In the second step, the amine was added in a basic environment to form the desired carbamate. The glycine derivatives 23 and 24 were obtained in 27 and 42 % yield and the sarcosine derivatives 25 and 26 in 31 and 25 %, respectively.
Scheme 5.

Synthesis of the functionalized exo‐TCO carbamate derivatives from the axial and equatorial isomers 7 and 8. 21/22: 7‐amino‐4‐methylcoumarin (1.25 equiv), triphosgene (1.25 equiv.), DIPEA (4.6 equiv.), DMAP (3.0 equiv.), toluene, DCM, 21 °C, 18 h, 42–63 %. 23/24: i. DPFPC (2.0 equiv.), DIPEA (5.0 equiv.), DMAP (0.1 or 5.0 equiv.), MeCN. ii. glycine methyl ester hydrochloride (5.0‐7.0 equiv.), DIPEA (2.5‐4.5 equiv.), MeCN, 21 °C, 18 h, 27–42 %. 25/26 i. DPFPC (2.0 equiv.), DIPEA (5.0 equiv.), DMAP (0.1 equiv.), MeCN. ii. sarcosine methyl ester hydrochloride (2.0 equiv.), DIPEA (2.5 equiv), MeCN, 21 °C, 18 h, 25–31 %.
Next, we examined the elimination kinetics of the caged fluorescent TCO derivatives 21 and 22. Both derivatives were incubated in phosphate‐buffered saline (PBS, pH=7.4) and subsequently a 2.5‐fold excess of either tetrazine 19 or 20 was added. The characteristic fluorescence of the released AMC (excitation 380 nm, emission 450 nm) was measured as a function over time in a plate reader. As positive control the maximum amount of possible released AMC was also measured, in this way the releasing efficiency could readily be calculated. As expected, and also observed in our 19F NMR experiments, the equatorial‐TCO derivative 22 furnished minimal release of the fluorescent AMC moiety with tetrazine 19 or 20 (<3% in 22 h, Figure 3). Nonetheless, the axial analogue 21 afforded complete release of the fluorescent AMC within 23 h upon ligation with 20, after less than three hours already more than 50 % release was observed (k elim=8.15×10−5 s−1). As anticipated, the elimination reaction was heavily depending on the used tetrazine, as for tetrazine 19 the release was significantly slower (10 % within 23 h, k elim=2.96×10−6 s−1), as also confirmed in the recent literature. [35]
Figure 3.

Release study of the caged fluorescent TCO derivatives 21 and 22 by the click and release reaction with tetrazine 19 or 20. A) The used tetrazines in this study. B) Study conditions, either 21 or 22 was incubated with tetrazine 19 or 20 and the fluorescence (excitation: 380 nm, emission: 450 nm) was measured at certain time points after the addition of the tetrazine. C) The release efficiency plotted as a function over time.
Hereafter, tetrazine 20 was chosen to compare the four allylic carbamate TCO derivatives 23‐26. The derivatives were dissolved in a mixture of deuterated methanol and PBS (1 : 1, v/v) and hereafter 20 was added. 1H NMR spectra were measured over a period of three hours. All derivatives revealed a complete click reaction with 20 within the first five minutes. The axial compounds 23 and 25 showed rapid release of the amino acid payloads (Figure 4, in red) accompanied by a respective decrease in the initial click conjugates (Figure 4, in blue). The respective equatorial derivatives 24 and 26 revealed less release and a more gradual decrease of the click conjugate. Compared to their axial analogues, 38 % of the glycine equatorial isomer 24 and 23 % of the sarcosine equatorial isomer 26 released payload successfully.
Figure 4.

Release study of the allylic amine TCO derivatives 23–26. A) Studied reaction of TCO derivatives 23–26 and 3,6‐dimethyl‐1,2,4,5‐tetrazine (20). B) The signal intensity of the protons measured in the 1H NMR study of the tetrazine methyl‐protons of the intermediate (in blue) and release product (in red) of the axial (•) and equatorial (▪) isomers.
Hereafter, the stability of the releasing axial TCO was investigated in both PBS and mouse serum at 37 °C. The free carboxylic derivative of 7 dissolved in PBS showed no changes in the 1H NMR spectrum after seven days (see: Figure S1). TCO 7 was stable in mouse serum up to several hours and showed no isomerization into the CCO analogue, however, slow degradation of 7 over time was observed (see: Figure S2).
After we observed that the new TCO gave satisfactory payload release, we decided to synthesize a drug‐conjugated TCO that would release the drug upon tetrazine ligation. This was examined with the release of the chemotherapeutic drug doxorubicin in a cellular environment. [21] The doxorubicin conjugate 27 was synthesized from the 4‐nitrophenyl activated TCO and purified with reversed‐phase HPLC. The effectiveness of doxorubicin release was tested in HeLa cells through an EC50 assay (Figure 5). The cells were incubated for three days at 37 °C with varying concentrations of the indicated compounds (tetrazine 20, doxorubicin, TCO‐Dox (27), or TCO‐Dox (27)+10 equiv. tetrazine 20). Addition of 10 equivalents of tetrazine 20 increased the cytotoxicity of the doxorubicin TCO conjugate 27 with a 21‐fold (75.9±12.08 nM vs. 1.654±0.418 μM) which indicates successful release of doxorubicin (Table 2). This change cannot be attributed to toxicity of the tetrazine, as its EC50 was in the mid‐micromolar range (29.81±5.21 μM). Together, these findings confirmed that conjugation to the TCO reduces doxorubicin's toxicity and that the drug can effectively be released upon tetrazine activation. Besides, HPLC analysis of the same click reaction in solution revealed successful and rapid release of doxorubicin from the conjugate (see: Figure S14).
Figure 5.

Doxorubicin cellular toxicity study of 27 induced by the click reaction with tetrazine 20 in HeLa Cells. The cells were incubated for three days with the indicated compounds at 37 °C after which cellular viability was assessed using a CCK‐8 assay. EC50 curves and values for 3,6‐dimethyl‐1,2,4,5‐tetrazine (Tz (20), in purple), TCO‐doxorubicin (TCO‐DOX (27), in green), TCO‐doxorubicin with 10 equiv. of tetrazine 20 (TCO‐DOX (27)+Tz (20), in red) and doxorubicin (DOX, in blue).
Table 2.
EC50 values for tetrazine 20, TCO‐DOX (27), TCO‐DOX (27)+10 equiv. 20 and doxorubicin from proliferation assays in HeLa cells determined using a CCK‐8 assay.
|
Compound |
EC50 |
95 % CI [a] |
|---|---|---|
|
Tetrazine 20 |
29.81 μM |
24.60–36.40 μM |
|
TCO‐DOX 27 |
1.654 μM |
1.236–2.217 μM |
|
TCO‐DOX 27+10 equiv 20 |
0.076 μM |
0.064–0.096 μM |
|
Doxorubicin |
0.0045 μM |
0.0031–0.0036 μM |
[a] EC50 95 % confidence interval (n=6).
Conclusion
In conclusion, we have developed a straightforward synthesis of a novel difunctionalized strained TCO with cargo‐releasing capabilities in only four high yielding steps. The acetylated iodohydrin intermediate and the subsequent elimination reaction were key steps in this pathway to afford the TCOs in excellent yields. To our knowledge this is the shortest reported synthesis of difunctionalized TCOs with releasing properties allowing improved access to these click reagents for both diagnostic and therapeutic applications. Furthermore, because of the high yields and the continuous photoflow isomerization process our pathway is amenable for larger scale synthesis. The TCOs have two orthogonal handles so that derivatization with different moieties is possible, while the allylic alcohol enables release of a payload upon click reaction with a tetrazine derivative, as demonstrated with carbonate and carbamate linked cargo molecules, even in a cellular environment. Moreover, we identified a TCO that fully retains the allylic moiety upon the click reaction. This product may be valuable for non‐releasable applications requiring stable conjugates with orthogonal handles on the TCO. The broad range applicability of the TCO‐tetrazine click and release system combined with this efficient synthetic accessibility will in our view strongly contribute to new in vitro and in vivo biomedical applications in the near future.
Experimental Section
exo ‐CCO 3: A suspension of cyclooctene 2 (20.4 g, 105 mmol) and sodium acetate (25.8 g, 315 mmol) in acetic acid (240 mL) was stirred and cooled with a water bath until the temperature stabilized to ambient temperature. Next, N‐iodosuccinimide (28.4 g, 126 mmol) was added and the mixture was stirred at ambient temperature for 3 h. After full conversion was observed on TLC, brine (50 mL) and heptane (200 mL) were added to the reaction mixture. The organic layer was collected and washed with brine (250 mL), a mixture of saturated aqueous sodium thiosulfate (50 mL), brine (200 mL) followed by saturated aqueous sodium bicarbonate (200 mL). The aqueous layers were back‐extracted in similar order with two additional portions of heptane (2×200 mL). The combined organic layers were dried with MgSO4 and concentrated to afford the crude product (39 g). This was purified with silica gel column chromatography (25→75 % EtOAc in heptane) to afford exo‐CCO 3 (33.2 g, 83 %) as white crystals. TLC (EtOAc/heptane, 1 : 3 v/v): R F=0.14. 1H NMR (400 MHz, CDCl3) δ 4.86 (ddd, J=7.7, 6.6, 3.5 Hz, 1H), 4.75 (td, J=7.8, 5.3 Hz, 1H), 4.08 (q, J=7.2 Hz, 2H), 2.24–2.17 (m, 2H), 2.15–2.08 (m, 1H), 2.07 (s, 3H), 2.05–1.96 (m, 2H), 1.84–1.70 (m, 1H), 1.54–1.46 (m, 1H), 1.46–1.38 (m, 1H), 1.34–1.25 (m, 2H), 1.23 (t, J=7.1 Hz, 3H), 1.15 (t, J=4.3 Hz, 1H). 13C NMR (101 MHz, CDCl3) δ 173.6, 169.7, 73.3, 60.4, 37.5, 33.7, 33.2, 26.1, 26.1, 26.0, 25.7, 23.6, 21.4, 14.2.
exo ‐CCO 4: DBU (39.5 mL, 262 mmol) was added to a stirred solution of CCO 3 (33.2 g, 87.3 mmol) in toluene (279 mL, 2.62 mol). The mixture was stirred at 100 °C for 18 h. The reaction mixture was cooled with an ice bath and filtered to remove the solids. The residue was washed with toluene (2×50 mL). The filtrate was washed with brine (2×250 mL) and the aqueous layers were back‐extracted with toluene (100 mL). The combined organic layers were dried with MgSO4 and concentrated to afford a yellow oil which was purified with silica gel column chromatography (10→20 % EtOAc in heptane) to afford CCO 4 (19.6 g, 89 %). TLC (EtOAc/heptane, 1 : 4 v/v): R F=0.26. 1H NMR (500 MHz, CDCl3) δ 5.81 (dddd, J=11.3, 9.2, 6.9, 2.0 Hz, 1H), 5.62 (dd, J=11.1, 5.3 Hz, 1H), 5.32 (dd, J=10.6, 5.2 Hz, 1H), 4.10 (q, J=7.2 Hz, 2H), 2.44–2.34 (m, 2H), 2.05 (s, 3H), 1.91–1.85 (m, 1H), 1.79–1.63 (m, 3H), 1.44–1.28 (m, 3H), 1.25 (t, J=7.1 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 173.5, 170.1, 133.9, 129.1, 75.3, 60.3, 36.4, 31.2, 28.6, 26.9, 26.4, 25.4, 21.3, 14.2. HRMS (m/z): [M+Na]+ calcd. for C14H20O4Na: 275.1259, found 275.1240.
exo ‐TCOs 5 and 6: A custom‐made long‐necked flask was charged with an aqueous solution of silver nitrate (7.00 g, 41.2 mmol) in water (100 mL). Next, a solution of 4 (3.50 g, 150 mmolar, 13.9 mmol) and methyl benzoate (3.84 g, 3.56 mL, 28.2 mmol) in deoxygenated heptane (160 mL) and MTBE (40 mL) was loaded into a UV irradiation setup (as described in Blanco‐Ania et al.). [26] The continuous process ran for 16 h. Next, the biphasic reaction mixture was loaded into a separation funnel and the aqueous layer was collected. The organic layer was washed with water (100 mL). The combined aqueous layers were washed with heptane (100 mL). Subsequently, 25 % aqueous ammonium hydroxide (25 mL) was added to the aqueous layer before extracting it with EtOAc (100 mL). The combined organic layers were dried with MgSO4 and concentrated to afford an unseparable diastereomeric mixture consisting of a 7 : 13 ratio of the axial and equatorial TCO‐isomers 5 and 6 as a clear oil (1.6 g, 46 %). HRMS (m/z): [M+Na]+ calcd. for C14H20O4Na: 275.1259, found 275.1264. 5 (axial): TLC (EtOAc/heptane, 1 : 1 v/v): R F=0.54. 1H NMR (500 MHz, CDCl3) δ 6.11 (ddd, J=16.8, 10.9, 5.9 Hz, 1H), 5.55 (dd, J=16.9, 3.4 Hz, 1H), 5.12 (q, J=3.1 Hz, 1H), 4.14–4.05 (m, 2H), 2.67 (dt, J=12.6, 6.0 Hz, 1H), 2.51–2.45 (m, 1H), 2.25–2.20 (m, 1H), 2.07 (m, 1H), 2.05 (s, 3H), 2.00–1.95 (m, 1H), 1.80–1.75 (m, 1H), 1.60–1.55 (m, 2H), 1.50–1.45 (m, 1H), 1.25 (m, 3H). 13C NMR (126 MHz, CDCl3) δ 174.9, 170.0, 131.7, 129.5, 70.7, 60.5, 37.2, 31.3, 29.2, 26.7, 21.1, 20.6, 14.2. 6 (equatorial): TLC (EtOAc/heptane, 1 : 1 v/v): R F=0.54. 1H NMR (500 MHz, CDCl3) δ 5.91 (ddd, J=16.0, 9.1, 6.4 Hz, 1H), 5.67 (ddd, J=16.6, 9.7, 1.5 Hz, 1H), 5.01 (td, J=9.8, 5.3 Hz, 1H), 4.14–4.05 (m, 2H), 2.86 (dtt, J=14.7, 9.2, 1.1 Hz, 1H), 2.40–2.35 (m, 1H), 2.38–2.30 (m, 2H), 2.18 (m, 2H), 2.06 (s, 3H) 1.36 (t, J=5.6 Hz, 1H), 1.25 (m, 3H), 1.25‐1.20 (m, 1H), 0.77 (dt, J=15.4, 11.4 Hz, 1H). 13C NMR (126 MHz, CDCl3) δ 174.2, 170.6, 136.1, 131.2, 77.2, 60.4, 37.6, 36.6, 34.6, 30.2, 29.0, 28.4, 21.2, 14.2.
exo ‐TCOs 7 and 8: A solution of the P‐ and M‐isomers 5 and 6 (2.00 g, 7.93 mmol) and potassium carbonate (2.19 g, 15.9 mmol) in ethanol (19.9 mL, 341 mmol) was stirred at 21 °C for 18 h. The flask was shielded from light with aluminum foil. After completion of the reaction was observed with TLC, acetic acid (1.91 mL, 33.3 mmol) in a 1 : 1 water‐brine mixture (50 mL) was added to quench the reaction. Then, EtOAc (25 mL) was added and separated from the aqueous layer, the organic layer was washed with aqueous saturated sodium bicarbonate (25 mL) and then with brine (25 mL). All aqueous layers were back‐extracted with EtOAc (2×25 mL). The combined organic layers were dried with MgSO4, concentrated in vacuo and purified with silica gel column chromatography (10 % acetone in toluene) to afford 7 (470 mg, 28 %) and 8 (980 mg, 59 %). TCO 7 (EtOAc/heptane, 1 : 1 v/v): R F=0.37. 1H NMR (500 MHz, CDCl3) δ 6.26 (ddd, J=16.8, 10.9, 5.9 Hz, 1H), 5.55 (ddd, J=16.8, 3.1, 0.9 Hz, 1H), 4.36 (q, J=3.0 Hz, 1H), 4.08 (qd, J=7.1, 2.1 Hz, 2H), 2.66 (dt, J=12.5, 6.0 Hz, 1H), 2.49–2.42 (m, 1H), 2.25 (dd, J=16.1, 11.9 Hz, 1H), 2.00–1.91 (m, 1H), 1.87 (q, J=8.0 Hz, 1H), 1.79–1.74 (m, 1H), 1.68 (t, J=13.1 Hz, 1H), 1.55–1.48 (m, 2H), 1.46–1.40 (m, 1H), 1.24 (t, J=7.2 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 175.2, 133.5, 130.4, 68.5, 60.4, 39.7, 31.7, 29.1, 27.0, 20.5, 19.3, 14.2. TCO 8 (equatorial): TLC (EtOAc/heptane, 1 : 1 v/v): R F=0.27. 1H NMR (500 MHz, CDCl3) δ 5.76 (ddd, J=15.8, 8.9, 6.5 Hz, 1H), 5.62 (ddd, J=16.5, 9.4, 1.4 Hz, 1H), 4.08 (dd, J=9.8, 5.2 Hz, 1H), 4.05 (q, J=7.1 Hz, 2H), 2.81 (dtt, J=14.8, 9.3, 1.2 Hz, 1H), 2.34–2.29 (m, 1H), 2.28–2.23 (m, 1H), 2.16–2.08 (m, 1H), 2.10–2.05 (m, 1H), 1.43 (dddd, J=13.1, 11.4, 9.7, 0.8 Hz, 1H), 1.32–1.29 (m, 1H), 1.20 (t, J=7.1 Hz, 3H), 1.18–1.13 (m,1H), 0.68 (dt, J=15.4, 11.3 Hz, 1H). 13C NMR (126 MHz, CDCl3) δ 174.5, 140.3, 129.3, 76.1, 60.4, 38.0, 37.7, 30.3, 29.3, 28.6, 28.5, 14.2.
Conflict of interest
The authors declare no conflict of interest.
1.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgments
Rivka Fontijn, Sam Moons, Stephanie Scharmann & Lieke Wouters (Institute for Molecules and Materials, Radboud University) are kindly acknowledged for their synthetic contributions. Dr. Paul Tinnemans (Institute for Molecules and Materials, Radboud University) is kindly thanked for determining the X‐ray crystal structure. This work was supported by LIFT grant 741.018.406 from the Dutch Research Council (NWO) awarded to F.P.J.T.R, T.J.B. and W.L.v.H. and by TTW Demonstrator grant 16929 from the Dutch Research Council (NWO) awarded to D.B.‐A and F.P.J.T.R.
Sondag D., Maartense L., de Jong H., de Kleijne F. F. J., Bonger K. M., Löwik D. W. P. M., Boltje T. J., Dommerholt J., White P. B., Blanco-Ania D., Rutjes F. P. J. T., Chem. Eur. J. 2023, 29, e202203375.
A previous version of this manuscript has been deposited on a preprint server (https://doi.org/10.26434/chemrxiv‐2022‐klj4d).
Data Availability Statement
The data that support the findings of this study are available in the supplementary material of this article.
References
- 1. Kolb H. C., Finn M. G., Sharpless K. B., Angew. Chem. Int. Ed. 2001, 40, 2004–2021; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2001, 113, 2056–2075. [Google Scholar]
- 2. Moses J. E., Moorhouse A. D., Chem. Soc. Rev. 2007, 36, 1249–1262. [DOI] [PubMed] [Google Scholar]
- 3. Hang H. C., Yu C., Kato D. L., Bertozzi C. R., Proc. Natl. Acad. Sci. USA 2003, 100, 14846–14851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sletten E. M., Bertozzi C. R., Acc. Chem. Res. 2011, 44, 666–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sletten E. M., Bertozzi C. R., Angew. Chem. Int. Ed. 2009, 48, 6974–6998; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 7108–7133. [Google Scholar]
- 6. Agarwal P., Bertozzi C. R., Bioconjugate Chem. 2015, 26, 176–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Agard N. J., Prescher J. A., Bertozzi C. R., J. Am. Chem. Soc. 2004, 126, 15046–15047. [DOI] [PubMed] [Google Scholar]
- 8. Dommerholt J., Schmidt S., Temming R., Hendriks L. J. A., Rutjes F. P. J. T., van Hest J. C. M., Lefeber D. J., Friedl P., van Delft F. L., Angew. Chem. Int. Ed. 2010, 49, 9422–9425; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 9612–9615. [Google Scholar]
- 9. Dommerholt J., Rutjes F. P. J. T., van Delft F. L., Cycloadd. in Bioorth. Chem. 2016, 57–76. [Google Scholar]
- 10. Rossin R., Robillard M. S., Curr. Opin. Chem. Biol. 2014, 21, 161–169. [DOI] [PubMed] [Google Scholar]
- 11. Rossin R., Renart Verkerk P., van den Bosch S. M., Vulders R. C. M., Verel I., Lub J., Robillard M. S., Angew. Chem. Int. Ed. 2010, 49, 3475–3478. [DOI] [PubMed] [Google Scholar]
- 12. Rossin R., Versteegen R. M., Wu J., Khasanov A., Wessels H. J., Steenbergen E. J., Ten Hoeve W., Janssen H. M., van Onzen A. H. A. M., Hudson P. J., Nat. Commun. 2018, 9, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Beck A., Goetsch L., Dumontet C., Corvaïa N., Nat. Rev. Drug Discovery 2017, 16, 315–337. [DOI] [PubMed] [Google Scholar]
- 14. Fang Y., Judkins J. C., Boyd S. J., Am Ende C. W., Rohlfing K., Huang Z., Xie Y., Johnson D. S., Fox J. M., Tetrahedron 2019, 75, 4307–4317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Liu L., Liu Y., Zhang G., Ge Y., Fan X., Lin F., Wang J., Zheng H., Xie X., Zeng X., Biochemistry 2018, 57, 446–450. [DOI] [PubMed] [Google Scholar]
- 16. Blackman M. L., Royzen M., Fox J. M., J. Am. Chem. Soc. 2008, 130, 13518–13519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rossin R., van Duijnhoven S. M. J., Ten Hoeve W., Janssen H. M., Kleijn L. H. J., Hoeben F. J. M., Versteegen R. M., Robillard M. S., Bioconjugate Chem. 2016, 27, 1697–1706. [DOI] [PubMed] [Google Scholar]
- 18. Li H., Conde J., Guerreiro A., Bernardes G. J. L., Angew. Chem. Int. Ed. 2020, 59, 16023–16032; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 16157–16166. [Google Scholar]
- 19. Davies S., Oliveira B. L., Bernardes G. J. L., Org. Biomol. Chem. 2019, 17, 5725–5730. [DOI] [PubMed] [Google Scholar]
- 20. Wang M., Svatunek D., Rohlfing K., Liu Y., Wang H., Giglio B., Yuan H., Wu Z., Li Z., Fox J., Theranostics 2016, 6, 887–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Versteegen R. M., Rossin R., ten Hoeve W., Janssen H. M., Robillard M. S., Angew. Chem. Int. Ed. 2013, 52, 14112–14116; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 14362–14366. [Google Scholar]
- 22. Versteegen R. M., Ten Hoeve W., Rossin R., de Geus M. A. R., Janssen H. M., Robillard M. S., Angew. Chem. Int. Ed. 2018, 57, 10494–10499; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 10654–10659. [Google Scholar]
- 23. Wilkovitsch M., Haider M., Sohr B., Herrmann B., Klubnick J., Weissleder R., Carlson J. C. T., Mikula H., J. Am. Chem. Soc. 2020, 142, 19132–19141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. de Geus M. A. R., Maurits E., Sarris A. J. C., Hansen T., Kloet M. S., Kamphorst K., Ten Hoeve W., Robillard M. S., Pannwitz A., Bonnet S. A., Chem. Eur. J. 2020, 26, 9900–9904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Royzen M., Yap G. P. A., Fox J. M., J. Am. Chem. Soc. 2008, 130, 3760–3761. [DOI] [PubMed] [Google Scholar]
- 26. Blanco-Ania D., Maartense L., Rutjes F. P. J. T., ChemPhotoChem 2018, 2, 898–905. [Google Scholar]
- 27. Darko A., Wallace S., Dmitrenko O., Machovina M. M., Mehl R. A., Chin J. W., Fox J. M., Chem. Sci. 2014, 5, 3770–3776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Darko A., Boyd S. J., Fox J. M., Synthesis 2018, 50, 4875–4882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gential G. P. P., Ho N. I., Chiodo F., Meeuwenoord N., Ossendorp F., Overkleeft H. S., van der Marel G. A., Filippov D. V., Bioorg. Med. Chem. Lett. 2016, 26, 3641–3645. [DOI] [PubMed] [Google Scholar]
- 30. Okamoto K., Kondo T., Goto T., Bull. Chem. Soc. Jpn. 1987, 60, 631–636. [Google Scholar]
- 31. Favre H. A., Powell W. H., Nomenclature of Organic Chemistry: IUPAC Recommendations and Preferred Names 2013, Royal Society of Chemistry, 2013. [Google Scholar]
- 32. Wagner J. A., Mercadante D., Nikić I., Lemke E. A., Gräter F., Chem. Eur. J. 2015, 21, 12431–12435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. O'Brien J. G. K., Chintala S. R., Fox J. M., J. Org. Chem. 2017, 83, 7500–7503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fan X., Ge Y., Lin F., Yang Y., Zhang G., Ngai W. S. C., Lin Z., Zheng S., Wang J., Zhao J., Angew. Chem. Int. Ed. 2016, 128, 14252–14256. [Google Scholar]
- 35. Sarris A. J. C., Hansen T., de Geus M. A. R., Maurits E., Doelman W., Overkleeft H. S., Codée J. D. C., Filippov D. V., van Kasteren S. I., Chem. Eur. J. 2018, 24, 18075–18081. [DOI] [PubMed] [Google Scholar]
- 36. Carlson J. C. T., Mikula H., Weissleder R., J. Am. Chem. Soc. 2018, 140, 3603–3612. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Data Availability Statement
The data that support the findings of this study are available in the supplementary material of this article.