

Abstract
Objective
Obesity is a significant public health concern across the globe. Research investigating epigenetic mechanisms related to obesity and obesity‐associated conditions has identified differences that may contribute to cellular dysregulation that accelerates the development of disease. However, few studies include Black women, who experience the highest incidence of obesity and early onset of cardiometabolic disorders.
Methods
The association of BMI with epigenome‐wide DNA methylation (DNAm) was examined using the 850K Illumina EPIC BeadChip in two Black populations (Intergenerational Impact of Genetic and Psychological Factors on Blood Pressure [InterGEN], n = 239; and The Genetic Epidemiology Network of Arteriopathy [GENOA] study, n = 961) using linear mixed‐effects regression models adjusted for batch effects, cell type heterogeneity, population stratification, and confounding factors.
Results
Cross‐sectional analysis of the InterGEN discovery cohort identified 28 DNAm sites significantly associated with BMI, 24 of which had not been previously reported. Of these, 17 were replicated using the GENOA study. In addition, a meta‐analysis, including both the InterGEN and GENOA cohorts, identified 658 DNAm sites associated with BMI with false discovery rate < 0.05. In a meta‐analysis of Black women, we identified 628 DNAm sites significantly associated with BMI. Using a more stringent significance threshold of Bonferroni‐corrected p value 0.05, 65 and 61 DNAm sites associated with BMI were identified from the combined sex and female‐only meta‐analyses, respectively.
Conclusions
This study suggests that BMI is associated with differences in DNAm among women that can be identified with DNA extracted from salivary (discovery) and peripheral blood (replication) samples among Black populations across two cohorts.
Study Importance.
What is already known?
Despite existing research on the effects of obesity on DNA methylation among some populations, there is an immediate need for more research among Black/African American (AA) women.
Black/AA populations have a younger age of mortality and onset of cardiometabolic disorders than other ethnic groups.
The incidence and prevalence of obesity have grown to pandemic proportions, with approximately 1.5 billion people worldwide exceeding obesity or overweight criteria.
What does this study add?
Twenty‐five CpG sites were found to be hypomethylated with higher BMI, and three were found hypermethylated.
Of the 28 CpG sites from our discovery cohort of all Black/AA women, we identified 12 novel CpG sites not previously associated with BMI in previous BMI analyses and replicated them among a second all Black/AA cohort.
Some of the sites identified may be unique to Black/AA populations, specifically women.
How might these results change the direction of research or the focus of clinical practice?
Epigenomic studies can be used to develop individual health care plans and identify needs for the individual and community.
INTRODUCTION
The incidence and prevalence of obesity have grown to pandemic proportions, with approximately 1.5 billion people worldwide exceeding obesity or overweight criteria [1, 2, 3]. By 2030, adults with obesity or overweight are expected to account for approximately 58% of the world's population [4]. Body mass index (BMI) is an important quantitative measure of adiposity [1, 5, 6, 7, 8, 9]. Lifestyle, behavior, and environmental influences on the development of obesity include overconsumption of calories, lack of physical activity, medications, and quality of food available [5, 8]. Health consequences due to obesity include hypertension, dyslipidemia, type 2 diabetes, coronary heart disease, stroke, and mortality [5, 7, 8]. However, beyond external lifestyle and environmental factors, genome‐wide association studies have shown significant associations of genes with adiposity, particularly among minority populations [4, 7, 10]. However, literature is lacking on epigenome‐wide association studies (EWAS) among Black/African American (AA) populations, who are disproportionately affected by obesity.
Black/AA populations have a younger age of mortality and onset of cardiometabolic disorders than other ethnic groups [11, 12]. The age of mortality and risk for cardiometabolic disorders can be strongly influenced by environmental factors [11, 12]. Some of these factors include discrimination and socioeconomic status (SES), which can contribute to poor mental and physical health [11, 13]. Over time, environmental factors such as experiencing discrimination can contribute to differences in DNA methylation (DNAm) patterns [14]. DNAm is an epigenetic mark that is involved in the regulation of gene expression [15]. Environmental (i.e., climate) and lifestyle factors (i.e., diet/obesity), as well as time (i.e., aging), are some of the known influences on DNAm patterns and gene expression [4, 5, 10, 15]. Obesity has been shown to be associated with DNAm, particularly at genes involved in physiologic pathways such as inflammation, which can trigger damaging immune and oxidative stress responses [6, 16]. Some changes in DNAm have been shown to causally influence obesity, whereas others are more likely to be a consequence, or biomarker, of the disease process [1, 17, 18]. However, alterations in DNAm resulting from various exposures may be reversible and amenable to interventions [10].
Despite existing research on the effects of obesity on DNAm among some populations, there is an immediate need for more research among Black/AA women [7, 16]. Ancestry and sex are two known influences of obesity related changes in DNAm [17]. Previous EWASs focused on evaluating DNAm related to BMI and obesity have been conducted primarily among European and South Asian populations, with limited representation of populations with African ancestry [1, 7, 16, 17, 18, 19, 20]. Because women tend to have a higher BMI than men, and Black/AA women have among the highest incidence of obesity [5], it is important to better understand the influence of obesity on features that can alter physiology, such as DNAm, in order to better address this health disparity and attenuate the development of cardiometabolic disorders. The purpose of this study is to conduct an EWAS among Black/AA women in the well characterized Intergenerational Impact of Genetic and Psychological Factors on Blood Pressure (InterGEN) cohort and replicate findings in the Genetic Epidemiology Network of Arteriopathy (GENOA) cohort to identify differences in DNAm associated with obesity.
METHODS
Discovery cohort
The InterGEN study is a longitudinal cohort study in Connecticut that examined the effects of genetic, epigenetic, and psychological factors on blood pressure (BP) in mother/child dyads. Recruitment began in April 2015 and follow‐up was completed in 2019. Eligibility criteria included the following: mothers (≥21 years old) who self‐identify as Black/AA, speak English, and have no mental illness that could interfere with psychological measurements. Women enrolled with a biological child (3‐5 years old) who lived with them most of the time and could provide a saliva sample. More information on the cohort and psychological measures can be found in previous reports [9, 21, 22, 23, 24]. During the baseline visit, study personnel collected clinical measurements of BP, height, weight, percent body fat, percent body water (mothers only), and saliva for DNA analysis from both mother and child. Demographic information, health history, and psychological measures (including parenting, experiences of perceived racism and discrimination, and depression) were collected through mother's report using Audio Computer‐Assisted Self‐Interview software. The present study included only InterGEN mothers (Table 1). Yale University and Columbia University's Institutional Review Boards (IRB) approved the study procedures.
TABLE 1.
Participant characteristics, InterGEN study (n = 239) and GENOA study (N = 961)
| InterGEN, | GENOA | ||
|---|---|---|---|
| Women (n = 239) | Total sample (N = 961) | Women (n = 685) | |
| Age (y), mean (SD) ** | 31.3 (5.8) | 57.5 (10.3) | 57.1 (10.4) |
| Education, n (%) ** | |||
| High school or less | 100 (41.8) | 587 (61.1) | 421 (65.5) |
| Some college or associate's degree | 105 (44) | 157 (16.3) | 111 (16.2) |
| Bachelor's degree or higher | 34 (14.2) | 217 (22.6) | 153 (22.3) |
| BMI category, n (%)** | |||
| Underweight (<18.5 kg/m2) | 13 (5.4) | 4 (0.4) | 3 (0.4) |
| Normal (18.5‐24.9 kg/m2) | 58 (24.3) | 140 (14.6) | 79 (11.5) |
| Overweight (25‐29.9 kg/m2) | 59 (24.7) | 319 (33.2) | 197 (28.8) |
| Obesity (≥30 kg/m2) | 109 (45.6) | 498 (51.8) | 406 (59.3) |
| BMI, mean (SD) * | 29.7 (8.3) | 31.3 (6.5) | 32.3 (6.8) |
| Current smoker, n (%) * | |||
| Yes | 52 (21.8) | 161 (16.8) | 92 (13.4) |
| No | 187 (78.2) | 800 (83.2) | 593 (86.6) |
Note: Participants were given the option not to respond to any of the questions, so all numbers may not add up to total N. P values from either t test for continuous variables or χ2 test for categorical variables.
Abbreviations: GENOA, Genetic Epidemiology Network of Arteriopathy; InterGEN, Intergenerational Impact of Genetic and Psychological Factors on Blood Pressure.
p < 0.01,
p < 0.001.
DNAm
Researchers collected saliva samples for DNA using the Oragene (OG)‐500 format tubes [3, 24, 25, 26], which required participants to spit into the tube until the contents reached the fill line per collection instructions (2 mL). Detailed DNA collection and analysis procedures have been described elsewhere [23, 25, 26]. Samples were transported from the field to the research laboratory where they were refrigerated at 4 °C until DNA extraction and analysis were completed. Standard protocol for DNA extraction and purification was conducted as indicated in the standard operating procedures guidelines using ReliaPrep kits, and the Illumina Infinium Methylation EPIC (850K) BeadChip was used for epigenome‐wide DNAm measurement. We used a quantile normalization approach in the R package “minfi” for processing EPIC data to correct for methylation signals and to generate adjusted β values for the associated analyses [27]. All individual samples passed laboratory‐based quality‐control procedures (missing rate < 10% and no sex mismatch). After merging with phenotypic data at baseline visit, 239 participants were included in the EWAS of BMI. CpG sites were excluded if they had a missing rate greater than 10% (n = 3264), overlapped with single‐nucleotide polymorphisms (n = 87,074), or were listed in the recent Illumina product quality notice. Quality‐control procedures and all analyses were uniformly performed among autosomal sites. A total of 756,544 autosomal CpG sites were included in the association analyses as previously described [23, 28]. The CpG sites measured by the EPIC BeadChip were mapped to Genome Research Consortium human build 37.
Replication study
Replication of these findings was conducted using a sample of 961 Black/AA participants from the GENOA study also using the Illumina Infinium Methylation EPIC BeadChip. In addition to all GENOA participants, replication was also conducted in women only (n = 685; Figures 1, 2, 3, 4, Table 2, Supporting Information Table S1).
FIGURE 1.
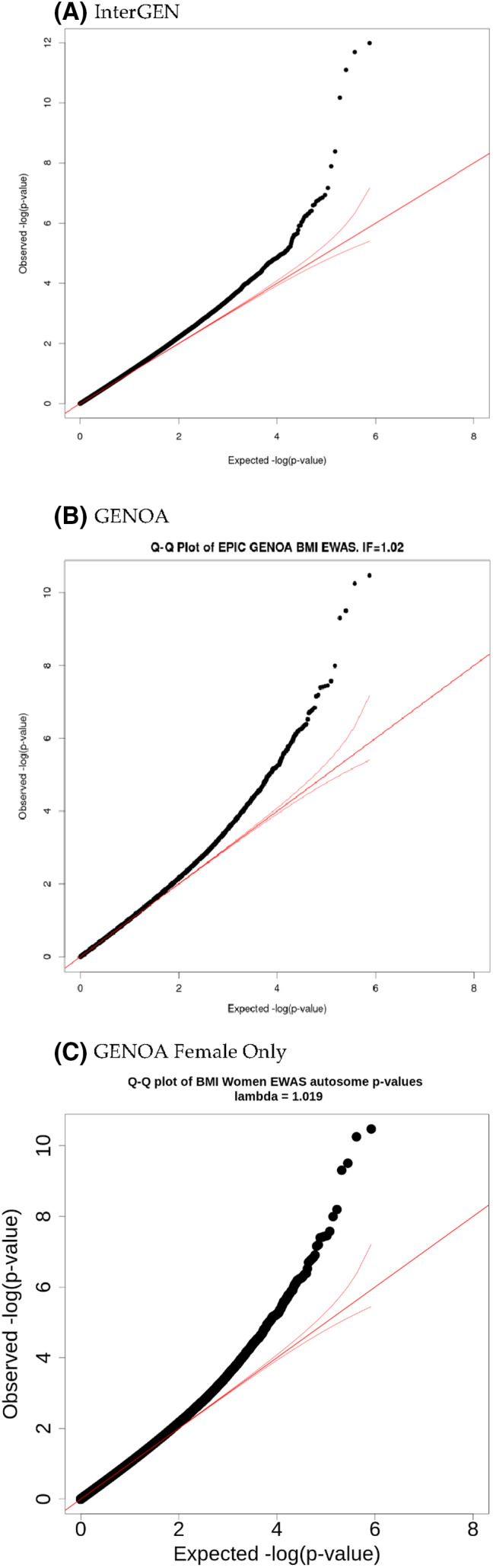
Quantile‐quantile (Q‐Q) plot of InterGEN and GENOA. Inflation factors of InterGEN, GENOA, and GENOA female‐only EWAS are 1.09, 1.01, and 1.02, respectively. EWAS, epigenome‐wide association study [Color figure can be viewed at wileyonlinelibrary.com]
FIGURE 2.
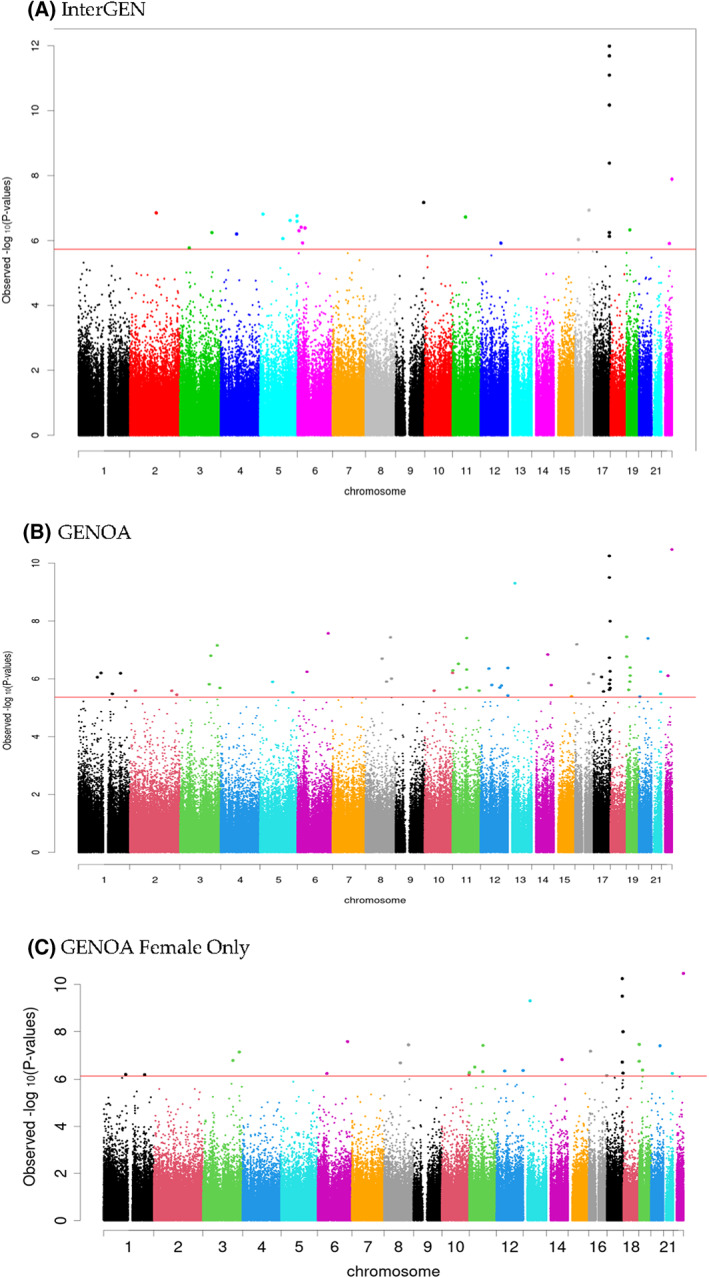
Manhattan plot of InterGEN and GENOA BMI EWAS. EWAS, epigenome‐wide association study [Color figure can be viewed at wileyonlinelibrary.com]
FIGURE 3.
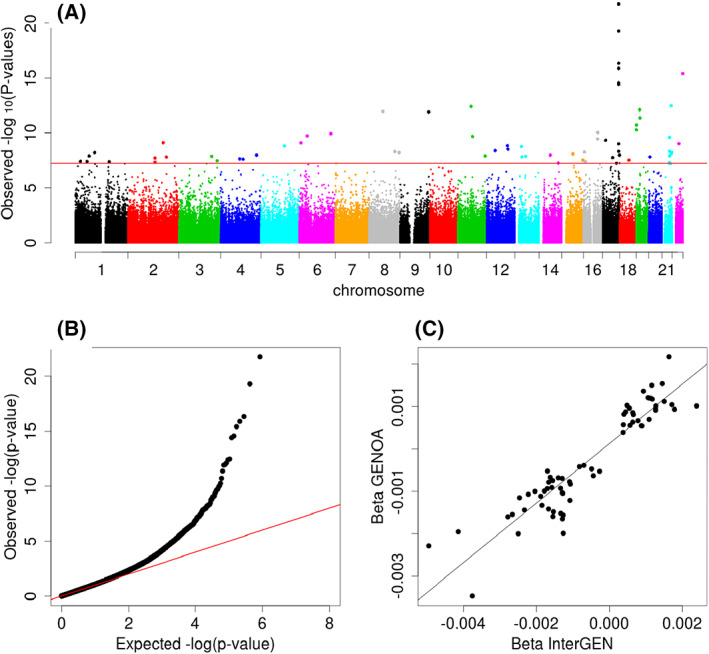
Meta‐analysis of epigenome‐wide associations with BMI from InterGEN and GENOA. (A) Manhattan plot of the meta‐analysis of BMI EWAS. Horizontal line indicates genome‐wide significance of Bonferroni‐corrected p value 0.05. (B) Quantile‐quantile plot of the meta‐analysis of BMI EWAS. Inflation factor of 1.06. (C) Scatterplot comparing the β coefficients of the top BMI associations between the InterGEN and GENOA samples. Correlation coefficient: 0.93 (p < 2.2 × 10−16); β coefficient: 0.70 (SE: 0.035, p < 2 × 10−16). EWAS, epigenome wide association study [Color figure can be viewed at wileyonlinelibrary.com]
FIGURE 4.
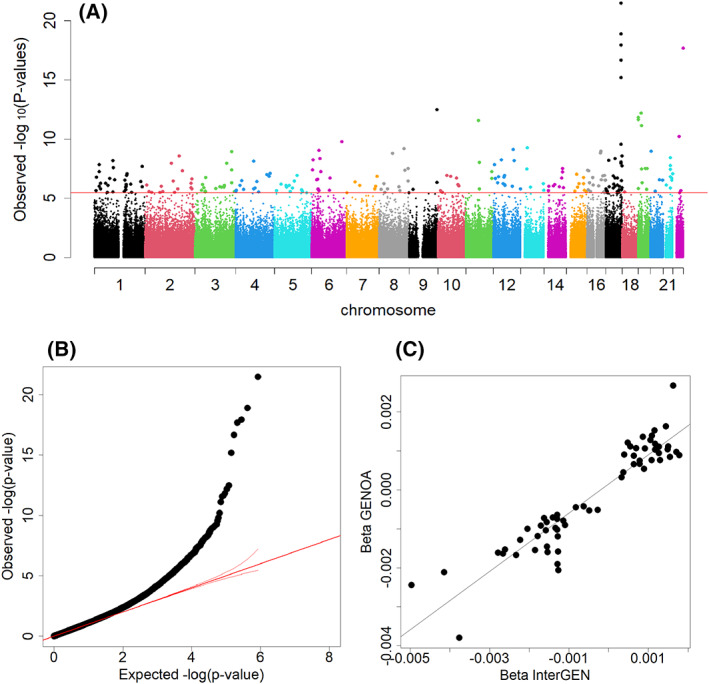
Meta‐analysis of epigenome‐wide associations with BMI from InterGEN and GENOA females only. (A) Manhattan plot of the female‐only meta‐analysis. Horizontal line indicates genome‐wide significance of Bonferroni‐corrected p value 0.05. (B) Quantile‐quantile plot: inflation factor of 1.07. (C) Scatterplot of β coefficients among 65 genome‐wide significant DNA methylation sites in the female‐only meta‐analysis. Correlation coefficient of 0.93 (p < 2.2 × 10−16); β coefficient of 0.75 (SE 0.039, p < 2 × 10−16) [Color figure can be viewed at wileyonlinelibrary.com]
TABLE 2.
Discovery of 28 epigenome‐wide significant associations in InterGEN (FDR‐q < 0.05), with replication in GENOA full sample and females only
| CpG | CHR | BP | Gene | Genomic region | Relation to UCSC CpG island | InterGEN (discovery, n = 239) | GENOA (replication, N = 961) | GENOA (replication, female only, n = 685) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| β | SE | p | β | SE | p | β | SE | p | ||||||
| cg18181703 | 17 | 76,354,621 | SOCS3 | Body | N Shore | −0.28 | 3.58 × × 10−2 | 1.02 × 10−12 | −1.60 × 10−3 | 2.91 × 10−4 | 5.13 × 10−8 | −0.0016 | 3.07 × 10−4 | 1.85 × 10−7 |
| cg11047325 | 17 | 76,354,934 | SOCS3 | Body | Island | −0.50 | 6.49 × 10−2 | 2.03 × 10−12 | −2.28 × 10−3 | 4.75 × 10−4 | 1.82 × 10−6 | −0.0024 | 5.13 × 10−4 | 2.37 × 10−6 |
| cg03067296 | 17 | 76,274,577 | LOC100996291 | TSS200 | −0.27 | 3.59 × 10−2 | 7.99 × 10−12 | −1.54 × 10−3 | 2.24 × 10−4 | 1.15 × 10−11 | −0.0016 | 2.46 × 10−4 | 5.60 × 10−11 | |
| cg13343932 | 17 | 76,355,061 | SOCS3 | Body | Island | −0.41 | 5.90 × 10−2 | 6.69 × 10−11 | −1.95 × 10−3 | 4.12 × 10−4 | 2.51 × 10−6 | −0.0021 | 4.35 × 10−4 | 1.48 × 10−6 |
| cg19748455 | 17 | 76,274,856 | LOC100996291 | TSS1500 | −0.23 | 3.73 × 10−2 | 4.11 × 10−9 | −1.44 × 10−3 | 2.49 × 10−4 | 9.89 × 10−9 | −0.0017 | 2.62 × 10−4 | 3.13 × 10−10 | |
| cg09349128 | 22 | 50,327,986 | Intergenic | N Shore | −0.22 | 3.70 × 10−2 | 1.28 × 10−8 | −1.07 × 10−3 | 1.73 × 10−4 | 9.87 × 10−10 | −0.0013 | 1.90 × 10−4 | 3.37 × 10−11 | |
| cg03770138 | 9 | 136,009,651 | RALGDS | Body | −0.13 | 2.25 × 10−2 | 6.72 × 10−8 | −1.05 × 10−3 | 2.42 × 10−4 | 1.66 × 10−5 | −0.0012 | 2.62 × 10−4 | 6.06 × 10−6 | |
| cg24382141 | 16 | 67,944,348 | PSKH1 | Body | S Shore | −0.13 | 2.33 × 10−2 | 1.16 × 10−7 | −6.91 × 10−4 | 1.77 × 10−4 | 9.69 × 10−5 | −0.0006 | 1.97 × 10−4 | 1.06 × 10−3 |
| cg02398240 | 2 | 128,256,334 | IWS1 | Body | −0.16 | 2.94 × 10−2 | 1.41 × 10−7 | −6.65 × 10−4 | 2.45 × 10−4 | 6.71 × 10−3 | −0.0007 | 2.70 × 10−4 | 7.60 × 10−3 | |
| cg11917181 | 5 | 14,108,303 | Intergenic | −0.11 | 2.05 × 10−2 | 1.53 × 10−7 | −1.66 × 10−5 | 1.21 × 10−4 | 8.91 × 10−1 | 0.0001 | 1.31 × 10−4 | 4.35 × 10−1 | ||
| cg16843099 | 5 | 178,956,830 | Intergenic | N Shore | −0.24 | 4.41 × 10−2 | 1.73 × 10−7 | −6.94 × 10−5 | 2.36 × 10−4 | 7.69 × 10−1 | −0.0002 | 2.71 × 10−4 | 4.26 × 10−1 | |
| cg19758958 | 11 | 62,319,222 | Intergenic | −0.13 | 2.37 × 10−2 | 1.88 × 10−7 | −1.03 × 10−3 | 2.12 × 10−4 | 1.42 × 10−6 | −0.0010 | 2.30 × 10−4 | 1.05 × 10−5 | ||
| cg19679801 | 5 | 145,422,431 | SH3RF2 | Body | −0.19 | 3.43 × 10−2 | 2.40 × 10−7 | 4.95 × 10−5 | 1.28 × 10−4 | 6.98 × 10−1 | 3.38 × 10−6 | 1.38 × 10−4 | 9.80 × 10−1 | |
| cg12367539 | 5 | 178,956,838 | Intergenic | N Shore | −0.25 | 4.70 × 10−2 | 2.55 × 10−7 | 1.14 × 10−4 | 2.21 × 10−4 | 6.05 × 10−1 | −4.57 × 10−5 | 2.51 × 10−4 | 8.56 × 10−1 | |
| cg19494100 | 6 | 18,387,313 | RNF144B | TSS1500 | N Shore | −0.08 | 1.45 × 10−2 | 3.88 × 10−7 | −1.23 × 10−4 | 1.48 × 10−4 | 4.05 × 10−1 | −0.0001 | 1.58 × 10−4 | 4.01 × 10−1 |
| cg15781610 | 6 | 36,992,554 | FGD2 | Body | −0.17 | 3.22 × 10−2 | 4.09 × 10−7 | −9.34 × 10−4 | 2.31 × 10−4 | 5.65 × 10−5 | −0.0009 | 2.55 × 10−4 | 3.22 × 10−4 | |
| cg00840791 | 19 | 16,453,259 | Intergenic | −0.38 | 7.14 × 10−2 | 4.70 × 10−7 | −3.47 × 10−3 | 7.12 × 10−4 | 1.31 × 10−6 | −0.0038 | 7.76 × 10−4 | 1.24 × 10−6 | ||
| cg17936938 | 6 | 7,866,213 | BMP6 | Body | 0.18 | 3.39 × 10−2 | 4.96 × 10−7 | 9.37 × 10−4 | 2.49 × 10−4 | 1.74 × 10−4 | 0.0009 | 2.75 × 10−4 | 1.23 × 10−3 | |
| cg10508317 | 17 | 76,355,146 | SOCS3 | Body | Island | −0.08 | 1.57 × 10−2 | 5.66 × 10−7 | −4.16 × 10−4 | 1.09 × 10−4 | 1.42 × 10−4 | −0.0004 | 1.11 × 10−4 | 6.24 × 10−5 |
| cg01671681 | 3 | 155,421,735 | PLCH1 | 5' UTR | −0.14 | 2.69 × 10−2 | 5.69 × 10−7 | −6.78 × 10−4 | 2.21 × 10−4 | 2.27 × 10−3 | −0.0007 | 2.35 × 10−4 | 2.59 × 10−3 | |
| cg03578005 | 4 | 77,170,672 | FAM47E | Body | N Shore | −0.06 | 1.07 × 10−2 | 6.25 × 10−7 | −2.76 × 10−5 | 3.29 × 10−5 | 4.03 × 10−1 | −2.34 × 10−5 | 3.65 × 10−5 | 5.22 × 10−1 |
| cg04610187 | 17 | 76,360,794 | LOC101928674 | Body | S Shelf | −0.14 | 2.77 × 10−2 | 7.43 × 10−7 | −4.97 × 10−4 | 3.03 × 10−4 | 1.02 × 10−1 | −0.0003 | 3.20 × 10−4 | 3.14 × 10−1 |
| cg20710777 | 5 | 110,411,740 | TSLP | Body | S Shelf | −0.17 | 3.31 × 10−2 | 8.70 × 10−7 | −5.19 × 10−4 | 1.12 × 10−4 | 3.96 × 10−6 | −0.0005 | 1.25 × 10−4 | 2.19 × 10−4 |
| cg04730825 | 16 | 16,116,191 | ABCC1 | Body | −0.07 | 1.29 × 10−2 | 9.31 × 10−7 | 8.72 × 10−5 | 1.55 × 10−4 | 5.74 × 10−1 | 7.56 × 10−5 | 1.72 × 10−4 | 6.60 × 10−1 | |
| cg13258453 | 6 | 25,180,502 | Intergenic | −0.19 | 3.73 × 10−2 | 1.18 × 10−6 | −3.76 × 10−4 | 2.45 × 10−4 | 1.26 × 10−1 | −0.0003 | 2.73 × 10−4 | 2.12 × 10−1 | ||
| cg20803896 | 12 | 99,006,941 | Intergenic | 0.11 | 2.15 × 10−2 | 1.20 × 10−6 | 6.98 × 10−4 | 1.95 × 10−4 | 3.72 × 10−4 | 0.0008 | 2.08 × 10−4 | 2.58 × 10−4 | ||
| cg15227014 | 22 | 38,037,179 | SH3BP1 | Body | S Shore | 0.12 | 2.43 × 10−2 | 1.23 × 10−6 | 2.50 × 10−4 | 2.19 × 10−4 | 2.55 × 10−1 | 0.0003 | 2.37 × 10−4 | 2.72 × 10−1 |
| cg20647087 | 3 | 45,709,608 | LIMD1 | Body | −0.11 | 2.19 × 10−2 | 1.69 × 10−6 | −1.16 × 10−4 | 1.67 × 10−4 | 4.86 × 10−1 | −5.19 × 10−5 | 1.73 × 10−4 | 7.64 × 10−1 | |
Note: The CpG sites were mapped to Genome Research Consortium human build 37 (GRCh37). The genes mapped to each CpG site were identified according to Illumina's annotation file of the EPIC BeadChip.
Abbreviations: BP, base pair location; body, gene body; CHR, chromosome; FDR−q, false discovery rate; GENOA, Genetic Epidemiology Network of Arteriopathy; InterGEN, Intergenerational Impact of Genetic and Psychological Factors on Blood Pressure; TSS, transcription start site; 5' UTR, 5′ untranslated region; UCSC, UCSC Genome Browser.
GENOA sample
GENOA is a multiphase, community‐based, prospective study of sibships with two or more siblings diagnosed with primary hypertension before the age of 60. Participants self‐identified as Black/AA, all siblings with a sibship were invited to participate regardless of hypertension status, and recruitment took place in Jackson, Mississippi. A total of N = 1854 Black/AA participants were recruited in Phase I (1995‐2000) from 683 sibships, and n = 1482 returned in Phase II (2000‐2005). Most participants (71%) in GENOA were women. In each phase, demographics, medical history, clinical characteristics, lifestyle factors, and fasting blood samples were collected [24, 29]. Sample characteristics for both InterGEN and GENOA can be found in Table 1. In Phase I, DNAm was measured from peripheral blood samples, and BMI and smoking status were also assessed. Smoking status was assessed through questions regarding whether the individual smokes and the frequency and type of cigarettes used (i.e., filtered, unfiltered, e‐cigarettes, other). IRB approval was received for the GENOA study protocol through the University of Mississippi Medical Center and the University of Michigan IRB.
Genomic DNA was extracted from stored peripheral blood leukocytes collected at Phase I (n = 1106) or Phase II (n = 304) using AutoGen FlexStar (AutoGen). Bisulfite conversion was performed with the EZ DNA Methylation Kit (Zymo Research), and methylation was assessed using the Illumina EPIC BeadChip. After obtaining the raw intensity data, the shinyMethyl R package [30] was used to generate the density plot to identify sex mismatch or sample outliers [24]. Sample identity was further checked using 59 single‐nucleotide polymorphism probes implemented in the EPIC chip, and mismatched samples were removed. Samples with incomplete bisulfite conversion identified using the QCinfo() function in the ENmix R package [31] were also removed. The minfi R package was used to perform background correction and normalization [24, 32]. The regression on correlated probes method [33] was used to adjust probe‐type bias. Principal variance component analysis [34] was used to quantify the corresponding effect of each batch factor (sample plate, chip, and row), and ComBat [33, 35] was used to adjust for batch effects. The effects of GENOA phase, age, and gender were preserved when adjusting for batch effects. Sample plate, chip, and row were adjusted for sequentially, until the weighted proportion of variation explained by any batch factor in the first principal components (containing 60% of the variation) in the methylation was below 5%. Detection p value for each sample at each probe was obtained, and individual probes with detection p < 10−16 were considered successfully detected. Samples and probes with detection rate < 10% were removed. After merging with phenotypic data, 857,121 CpG sites for 961 Black/AA participants were used for this analysis. Cell counts were estimated and adjusted using the Houseman method [36, 37].
Statistical analysis
We conducted an EWAS of BMI among women enrolled in the InterGEN study. DNAm β‐value was modeled as the dependent variable using linear mixed‐effects model. Age and smoking status were controlled for as potential confounders. To control for potential heterogeneity in cell proportions from saliva, we implemented a reference‐free deconvolution method to estimate putative cell type proportions and included them as covariates [24, 36, 38]. We also adjusted for batch effects as random intercepts in the analysis. False discovery rate (FDR) was used to correct for multiple comparisons. We applied the Benjamini and Hochberg method to calculate the FDR‐corrected p value [39]. CpG sites with FDR‐corrected p < 0.05 were considered as statistically significant in the discovery EWAS to pursue replication. In GENOA, linear mixed‐effects models were implemented to assess the association between DNAm and BMI adjusting for age, sex, smoking status, cell type proportions, and the top 10 genetic principal components. Family ID was included as a random effect to account for relatedness. We performed inverse‐variance weighted fixed‐effects meta‐analysis among DNAm sites available from both InterGEN and GENOA studies using the computationally efficient software METAL [40]. We reported the epigenome‐wide significant (Bonferroni‐corrected p value < 0.05, nominal p value < 6.50 × 10−8) DNAm sites considering a total of 749,801 tests and their heterogeneity between two Black/AA EWASs. All statistical analyses were performed in the R statistical environment version 3.4.1 (http://www.r-project.org/). We conducted pathway enrichment analysis based on annotated genes from the top DNAm sites from meta‐analysis results. The analysis was completed using the Analysis35R package missMethyl, which corrects for the distribution of the DNAm sites on the array [41].
RESULTS
InterGEN EWAS
The descriptive statistics can be found in Table 1. In the discovery EWAS using the InterGEN sample, 28 CpG sites were significantly associated with BMI after adjusting for age and smoking status (FDR‐corrected p < 0.05; Table 2, Figures 1 and 2). These CpG sites were mapped to the SOCS3, LOC100996291, RALGDS, PSKH1, IWS1, SH3RF2, RNF144B, FGD2, BMP6, PLCH1, FAM47E, LOC101928674, TSLP, ABCC1, SH3BP1, and LIMD1 genes. Twenty‐five CpG sites were found to be hypomethylated with higher BMI, and three were found hypermethylated (Table 2). Among the 28 significant CpG sites, we identified gap signals of cg19679801 (3.64% and 96.36% in groups 1 and 2) using GapHunter (R package minfi) after excluding outlier signals with <1% of study samples. The association of cg19679801 might be affected by an underlying genetic effect with low minor allele frequency. GapHunter was run on the GENOA sample and there was no signal gap.
GENOA replication
A total of 17 out of 28 CpG sites in the InterGEN EWAS were replicated in the GENOA full sample EWAS with consistent directionality and statistical significance after Bonferroni correction (p < 0.0018 = 0.05/28; Table 2). Twelve CpG sites were novel compared with the previous reports [1, 16, 17, 19, 24, 42, 43], of which eleven were new to the Illumina arrays (i.e., they were not available on earlier arrays, including the 450 K). Genes mapped by these newly identified EPIC 850K array‐specific CpG sites included SOCS3, RALGDS, PSKH1, FGD2, BMP6, and TSLP (Table 2).
Meta‐analysis
To maximize the statistical power, we also conducted a fixed‐effects meta‐analysis combining EWAS results from the InterGEN and GENOA cohort studies (Figure 3). Using a stringent significance threshold of Bonferroni‐corrected p < 0.05 to reduce the false‐positive rate, 65 CpG sites were significantly associated with BMI (Supporting Information Table S2). Among those, 48 were not significant in the discovery analysis, mapping to genes including VASN, CPT1A, MAFG, and SNTB1 (Supporting Information Table S2). The β coefficients among 65 genome‐wide significant DNAm sites between InterGEN and GENOA were highly correlated, with a correlation coefficient of 0.93 (p < 2.2 × 10−16; Figure 3C). We also conducted a fixed‐effects meta‐analysis combining female‐only EWAS results from the two studies (n = 239 for InterGEN and n = 685 for GENOA; Figure 4). A total of 61 CpG sites were significantly associated with BMI (Bonferroni‐corrected meta‐analysis p < 0.05, Supporting Information Tables S1 and S2). Similarly, the β coefficients among 61 genome‐wide significant DNAm sites between InterGEN and GENOA were highly correlated, with a correlation coefficient of 0.93 (p < 2.2 × 10−16; Figure 4C). Of those CpG sites in the meta‐analysis, most BMI‐associated DNAm sites were consistent between two meta‐analyses. However, 12 DNAm sites were genome‐wide significant (p < 6.50 × 10−8) only in the female‐only meta‐analysis but were not genome‐wide significant in the meta‐analysis including both men and women (Supporting Information Table S2). Because some DNAm sites are correlated, the Bonferroni correction for multiple testing can increase the false‐negative rate. Therefore, we also reported BMI‐associated DNAm sites from the sex‐combined meta‐analysis (658 sites; Supporting Information Table S3) and the female‐only meta‐analysis (626 sites; Supporting Information Table S4). Volcano plots of the meta‐analysis of BMI EWAS offer additional supporting information (Supporting Information Figures S1A and B).
Pathway enrichment analysis
We tested pathway enrichment using two sets of top associations, 65 Bonferroni significant CpG sites (42 unique genes) and 154 CpG sites with nominal p < 10−6 (124 unique genes). After multiple testing correction using FDR 0.05, no pathway was significantly enriched among BMI‐associated CpG sites.
DISCUSSION
We completed a cross‐sectional epigenome‐wide analysis of BMI and DNAm from two cohorts of Black/AA individuals in the United States. Of the 28 CpG sites from our discovery cohort of all Black/AA women, we identified 12 novel CpG sites not previously associated with BMI in previous BMI analyses and replicated among a second all Black/AA cohort [1, 6, 16, 17, 19, 24, 42, 43]. Although other EWASs have been conducted, they were primarily White cohorts and they did not conduct any stratified analyses [1, 10, 21]. Our findings are notable in that we were able to identify differences in DNAm associated with BMI in saliva samples and to replicate our findings in a separate cohort using peripheral blood samples. Most previous work has conducted EWASs using peripheral blood samples, and our study supports that less invasive saliva samples can be used to evaluate or replicate previously identified differences in DNAm associated with BMI. The New England Family Study used adipose tissue, rather than blood, to examine sex‐specific methylation associated with obesity [44]. A majority of CpG sites that were associated with BMI were hypomethylated and the direction of change was consistent across both cohorts using different tissue types. Results from our meta‐analysis identified 17 CpG sites previously reported in BMI EWAS. We identified DNAm differences within 19 genes, of which previous BMI EWASs observed significant DNAm differences at different CpG sites.
Of the 12 novel CpG sites from our discovery and replication cohorts, 2 CpG sites (cg110473253, cg13343932) are located in the SOCS3 gene. Two additional CpG sites (cg18181703, cg09349128) were also significant in SOCS3 in our meta‐analysis, and differential methylation at these sites has been associated with BMI in previous EWASs [1, 17, 40, 45, 46, 47]. Hypomethylation of the SOCS3 mapped CpG sites was consistently found for all studies. In addition to evaluating the relationship between BMI and DNAm, Li and colleagues [46] evaluated the change in DNA methylation over time related to BMI. When they evaluated the change in BMI and DNAm across identical twin pairs, the association was significant only when BMI was used as the predictor and DNAm as the outcome variable. Furthermore, their results suggest that hypomethylation at the SOCS3 site (cg18181703) is the result of increased BMI. In murine models, expression of SOCS3 was shown to improve leptin and insulin sensitivity [48]. Ali and colleagues also observed hypomethylation at SOCS3 cg18181703 was associated with increased BMI, waist to height ratio, triglycerides, and metabolic syndrome [47, 49]. Interestingly, in multivariate analysis, Chambers et al. noted that DNA methylation differences at the SOCS3 site were not significantly associated with type 2 diabetes after adjusting for BMI, and they suggest that DNAm at these sites may serve as a biomarker for insulin resistance and risk for metabolic disease [21]. Therefore, loss of methylation across SOCS3 could contribute to the development of obesity‐induced metabolic syndrome and type 2 diabetes if associated with decreased gene expression.
A previous study [50] of lean, young Norwegian women and Norwegian women with obesity (N = 120, mean age = 27.2), identified 10 CpG sites across eight genes associated with obesity (Supporting Information Table S5) [1, 4, 6, 8, 9, 10, 15, 16, 17, 18, 43, 47, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59, 60]. We identified significantly different methylation at CpG sites in three of the same genes in our discovery cohort (SOCS3, SBNO2, and FGD2) and three CpG sites located in the gene bodies of SOCS3 (cg18181703) and SBNO2 (cg12170787, cg18608055). Similar to Kvaløy and colleagues, we observed that more differentially methylated sites were associated with hypomethylation versus hypermethylation [29, 47, 48, 49, 50]. Many of the CpG sites identified in the Kvaløy et al. study were also associated with serum C‐reactive protein levels [61]. Among sites identified in our meta‐analysis, five of the CpG sites were consistent with differential DNAm among Black/AA C‐reactive protein‐EWAS results with consistent directionality (hypomethylation: SNOB2 [cg18608055], SOCS3 [cg18181703], PIK3IP1 [cg08548559], GPRIN3 [cg02734358], and hypermethylation: MYO5C [cg06192883]) [61]. Some of the sites in the Ligthart et al. study (SNOB2 [cg18608055], SOCS3 [cg18181703], MYO5C [cg06192883]) were also associated with other cardiometabolic traits, including total cholesterol, incidence of coronary heart disease, triglycerides, and fasting glucose and insulin levels. Although this study was not designed to assess these long‐term outcomes, replication of these sites suggests that differential DNAm associated with BMI can be detected via saliva samples of women prior to the development of other cardiometabolic complications. Further work in this area could illuminate whether differential DNAm at these sites could serve as a biomarker for increased risk of developing obesity‐induced cardiometabolic disorders.
A large proportion of the GENOA sample had hypertension, which may influence methylation [29]. None of the CpG sites identified in the InterGEN discovery cohort were significantly associated with systolic or diastolic BP in the largest EWAS of BP to date [9]. However, one CpG site that was identified only in the InterGEN/GENOA meta‐analysis, cg00574958, was also identified in the BP EWAS [9, 29]. Differential DNAm has consistently been reported in cg00574958 carnitine palmitoyltransferase 1A (CPT1A) in EWASs for BMI and other cardiometabolic disorders [1, 17, 29, 48, 50, 61, 62, 63]. Although we did not observe significant differences in cg00574958 of CPT1A in our discovery cohort, we did observe significant hypomethylation at this site in our meta‐analysis and in the GENOA cohort. The site in the InterGEN cohort was directionally congruent but it did not each statistical significance after multiple corrections. Differences in CPT1A methylation are likely to influence biosynthesis of cholesterol and triglyceride metabolism [17] and may be moderated by dietary intake of fats and carbohydrates [62]. Lai and colleagues determined that CPTA1 methylation was induced by carbohydrates and reduced by fats [61, 63]. Although the mechanism is unknown, they hypothesize that methylation at CPTA1 could mediate the influence of fat intake on BMI and increase risk of developing cardiometabolic disease.
To better characterize the function of the CpGs identified in the InterGEN study and the meta‐analysis (Table 2; Supporting Information Tables S1 and S2), we looked them up in databases that report associations between CpGs and transcripts of nearby genes (i.e., identify methylation sites associated with transcript expression, or cis‐eQTMs). We considered a CpG and a transcript to be associated using the significance criteria reported in the original publication for each database [22, 64, 65]. In monocytes from the Multi‐Ethnic Sample of Atherosclerosis (n = 1264), the following pairs of CpGs and transcripts were associated (FDR < 0.001): cg12367539 with HNRNPH1 (p = 4.85 × 10−07), cg21650866 with PTP4A3 (p = 8.84 × 10−08), and cg00926657 with BAIAP2 (p = 2.72 × 10−06). In whole blood from the Framingham Heart Study (N = 4170), the following pairs of CpGs and transcripts were associated (FDR < 0.05): cg12367539 with RUFY1 (p = 1.38 × 10−14), cg00574958 with CPT1A (p = 5.89 × 10−26), cg08548559 with PISD (2.51 × 10−21), cg17901584 with DHCR24 (3.98 × 10−18), and cg21650866 with PTP4A3 (p = 6.76 × 10−09) [22]. In peripheral blood mononuclear cells from the iMETHYL database, the following pairs of CpGs and transcripts were associated (FDR < 0.05): cg17901584 with DHCR24 (p = 1.027 × 10−13) and RP11‐67 L3 (p = 0.0058), cg07458272 with KIAA0355 (p = 0.0054), and cg09349128 with ALG12 (p = 0.0064) [65]. Methylation at these CpGs decreased gene expression for all associations noted, with the exception of cg07458272 with KIAA0355 and cg09349128 with ALG12 [22, 64, 65]. The following significant CpG sites and associated genes from the Multi‐Ethnic Sample of Atherosclerosis or Framingham Heart Study noted previously were also replicated in the InterGEN and GENOA analyses for this study (cg12367539 with HNRNPH1, cg12367539 with RUFY1, and cg09349128 with ALG12), and they can be found in Table 2 with additional data in Supporting Information Tables S1 and S2.
The potential for mediation and/or pleiotropic effects of BMI‐associated DNAm differences with other BMI related traits has been reported across the spectrum. For example, many of the previous works cited here have found differential DNAm associations with other traits, including SOCS3 with type 2 diabetes [58, 66], CPT1A with type 2 diabetes [21, 67] and lipid profiles [48], and ABCG1 with BP [42]. However, the fact that many of these CpG sites have been replicated specifically for BMI across multiple studies with different cardiometabolic phenotypes, populations, and sample characteristics such as sex suggests a consistent association signal with BMI. Additional evidence is provided by the difference between the InterGEN and GENOA cohort tissue types (saliva vs. blood), age range, sex, and geographic locations with replication remaining significant for many CpG sites.
We note that this study was unable to determine whether the DNAm changes associated with BMI are causal or a consequence of BMI. Previous studies have suggested that, for the most part, associated methylation differences tend to follow BMI changes, and thus they may be biomarkers of adiposity [1]. Previous studies that included Mendelian randomization or mediation analysis suggest that many of the observed epigenetic changes associated with BMI may also be precursors to the development of cardiometabolic traits that are associated with differences at these loci [1, 16, 17, 23, 42, 46, 67, 68]. Future work investigating mechanistic underpinnings of DNAm conversion at these sites could lead to novel targets for preventive measures and interventions to reduce and/or prevent the development of BMI‐associated cardiometabolic disease. Research increasingly illustrates the potential influence of sex on the association of SES and obesity, whereas education was a significant (negative) factor only among women. Ultimately, findings have demonstrated that epigenetic mediation pathways linking SES to obesity are stronger for women [44, 45].
In conclusion, we were able to identify differential DNAm at several loci, some within genes that have previously been associated with obesity and cardiometabolic disorders. Some of the sites identified may be unique to Black/AA populations, specifically women. Given our results, it is notable that considering sex differences and diverse racial/ethnic groups can improve EWAS discovery. Further replication studies among larger cohorts of Black/AA populations and women (as the results were significantly replicated in women only, but not men and women in GENOA) will help identify the generalizability of these results and whether they represent a consistent epigenetic signature associated with BMI.
This study's strengths include data from two well established and well characterized Black/AA study cohorts. The InterGEN study included healthy Black/AA women and children and collected weight and height for BMI calculation, percent body fat, and percent body water (mother only) at four time points. Findings from the study were significantly replicated with the GENOA data set. Study samples were deidentified and replication was conducted to prevent bias. Deidentification ensures the anonymity between those who provided the samples and those conducting the data analyses. Therefore, assumptions or biases among participants will not impact the analysis process. The InterGEN and GENOA study protocols established methods that ensured rigor in recruitment, data collection, and statistical analyses. Rigor for the studies was enhanced through ongoing training and supervision of research staff and use of robust and reproducible methods that have been published. For instance, all research staff received intensive initial and ongoing training in data collection and in distress protocols. We enhanced reproducibility in several ways: (1) published two methods papers (cited previously [25, 27]); (2) made study data available on Database of Genotypes and Phenotypes (dbGaP); (3) and disseminated findings through more than 30 publications and publicly via our study website (study website link: https://intergen.yale.edu/; recent publications are found here: nursing.columbia.edu/crpc).
The focus of this paper was on BMI and not on other potential comorbidities. The snapshot that our results gives us provides valuable information for methodologies that could be applied to other at‐risk populations. The analysis methods outlined here may have utility for future studies evaluating epigenetic risks related to cardiometabolic outcomes. Another limitation of the study is that we did not evaluate any downstream protein products that may result from DNAm sites identified as significant. Future studies we have planned may include other methods such as metabolomics and nutritional data to obtain a more complete picture of the participants' environment and allow us to determine how DNAm may influence physiology. Despite higher incidence of obesity and cardiometabolic disorders, Black/AA women have been underrepresented in research studies focused on EWAS analysis of BMI. Including minority populations in epigenomic studies is important and this can be used to develop individual health care plans and identify needs for the individual and community. The InterGEN and GENOA cohort studies were preexisting data sets that did not include data on lifestyle choices, dietary intake, nutritional habits, and physical activity. Therefore, this information could not be assessed in the analysis [7].
AUTHOR CONTRIBUTIONS
Conceptualization, Jacquelyn Y. Taylor, Jennifer A. Smith, Yan V. Sun; data analysis, Yunfeng Huang, Wei Zhao, Zeyuan Wang, Qin Hui, Sharon L.R. Kardia, Jennifer A. Smith, Yan V. Sun, Stephanie Potts‐Thompson, Laura Prescott; writing—original draft preparation, review, and editing, Jacquelyn Y. Taylor, Yunfeng Huang, Yutong Yao, Wei Zhao, Michelle L. Wright, Zeyuan Wang, Qin Hui, Stephanie Potts‐Thompson, Veronica Barcelona, Laura Prescott, Cindy Crusto, Sharon L.R. Kardia, Jennifer A. Smith, Yan V. Sun; supervision, Jacquelyn Y. Taylor; funding acquisition, Jacquelyn Y. Taylor, Cindy Crusto, Sharon L.R. Kardia, Jennifer A. Smith, Yan V. Sun. All authors have read and agreed to the published version of the manuscript.
FUNDING INFORMATION
The Intergenerational Impact of Genetic and Psychological Factors on Blood Pressure (InterGEN) research study was funded by the National Institute for Nursing Research under grant R01NR013520 and the National Heart, Lung and Blood Institute under grants U01HL054457, RC1HL100185, R01HL087660, R01HL119443, R01HL133221, and R01HL141292. The Genetic Epidemiology Network of Arteriopathy (GENOA) research study was funded by the National Institute of Diabetes and Digestive and Kidney Diseases under grant R01DK125187 and the National Institute for Nursing Research under grant K01NR017010 and K01NR017903. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
CONFLICT OF INTEREST
The authors declared no conflict of interest.
Supporting information
Supporting Information Figure S1A Volcano plot of the meta‐analysis of BMI EWAS. BMI, body mass index; EWAS, epigenome‐wide association study.
Supporting Information Figure S1B Volcano plot of the female‐only meta‐analysis of BMI EWAS. BMI, body mass index; EWAS, epigenome‐wide association study.
Supporting Information Table S1 Epigenome‐wide associations of 61 significant (Bonferroni corrected p < 0.05) CpG sites with BMI from the meta‐analysis of InterGEN and GENOA. Female only. BMI, body mass index; CpG, XXXX; GENOA, Genetic Epidemiology Network of Arteriopathy; InterGEN, Intergenerational Impact of Genetic and Psychological Factors on Blood Pressure.
Supporting Information Table S2 Epigenome‐wide associations of 65 significant (Bonferroni corrected p < 0.05) CpG sites with BMI from the meta‐analysis of InterGEN and GENOA. BMI, body mass index; CpG, XXXX; GENOA, Genetic Epidemiology Network of Arteriopathy; InterGEN, Intergenerational Impact of Genetic and Psychological Factors on Blood Pressure.
Supporting Information Table S3 Epigenome‐wide associations of 658 significant (FDR 0.05 threshold) CpG sites with BMI from the meta‐analysis of InterGEN and GENOA. BMI, body mass index; FDR, false discovery rate; GENOA, Genetic Epidemiology Network of Arteriopathy; InterGEN, Intergenerational Impact of Genetic and Psychological Factors on Blood Pressure.
Supporting Information Table S4 Epigenome‐wide associations of 628 significant (FDR 0.05 threshold) CpG sites with BMI from the meta‐analysis of InterGEN and GENOA. Female only. BMI, body mass index; CpG, XXXX; FDR, false discovery rate; GENOA, Genetic Epidemiology Network of Arteriopathy; InterGEN, Intergenerational Impact of Genetic and Psychological Factors on Blood Pressure.
Supporting Information Table S5 Summary of literature review on EWAS of BMI with discovery set cohort characteristics and study design if available. All studies consist of mixed gender. None of the studies include participants with HIV. All samples came from peripheral blood except noted otherwise. BMI, body mass index; EWAS, epigenome‐wide association study.
Taylor JY, Huang Y, Zhao W, et al. Epigenome‐wide association study of BMI in Black populations from InterGEN and GENOA. Obesity (Silver Spring). 2023;31(1):243‐255. doi: 10.1002/oby.23589
Funding information National Heart, Lung, and Blood Institute, Grant/Award Numbers: R01HL087660, R01HL119443, R01HL133221, R01HL141292, RC1HL100185, U01HL054457; National Institute of Diabetes and Digestive and Kidney Diseases, Grant/Award Number: R01DK125187; National Institute of Nursing Research, Grant/Award Numbers: K01NR017010, K01NR017903, R01NR013520
DATA AVAILABILITY STATEMENT
InterGEN genotype and phenotype data are available through the Database of Genotypes and Phenotypes (dbGaP accession number phs001792.v1.p1). DNA methylation data are available with a Data Use Agreement upon reasonable request to the InterGEN study investigators Jacquelyn Y. Taylor (jyt2116@cumc.columbia.edu) and Yan V. Sun (yan.v.sun@emory.edu). GENOA genotype and phenotype data are available through the Database of Genotypes and Phenotypes (dbGaP accession number phs001238.v2.p1). Methylation data are from the Gene Expression Omnibus (GEO): GSE157131. Owing to Institutional Review Board restriction, mapping of the sample IDs between genotype data (dbGaP) and DNA methylation data (GEO) cannot be provided publicly but is available upon written request to the study investigators Jennifer A. Smith (smjenn@umich.edu) and Sharon L.R. Kardia (skardia@umich.edu).
REFERENCES
- 1. Wahl S, Drong A, Lehne B, et al. Epigenome‐wide association study of body mass index, and the adverse outcomes of adiposity. Nature. 2017;541:81‐86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Afshin A, Forouzanfar MH, Reitsma MB, et al. Health effects of overweight and obesity in 195 countries over 25 years. N Engl J Med. 2017;377:13‐27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hales CM, Carroll MD, Fryar CD, Ogden CL. Prevalence of obesity and severe obesity among adults: United States, 2017–2018. NCHS Data Brief, no. 360. National Center for Health Statistics; 2020. [PubMed] [Google Scholar]
- 4. Sayols‐Baixeras S, Subirana I, Fernández‐Sanlés A, et al. DNA methylation and obesity traits: an epigenome‐wide association study. The REGICOR study. Epigenetics. 2017;12:909‐916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Centers for Disease Control and Prevention . About adult BMI. Updated June 3, 2022. https://www.cdc.gov/healthyweight/assessing/bmi/adult_bmi/index.html
- 6. Aslibekyan S, Demerath EW, Mendelson M, et al. Epigenome‐wide study identifies novel methylation loci associated with body mass index and waist circumference. Obesity (Silver Spring). 2015;23:1493‐1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Centers for Disease Control and Prevention . Causes of obesity. Updated March 21, 2022. Accessed January 15, 2021. https://www.cdc.gov/obesity/adult/causes.html
- 8. Meeks K, Henneman P, Venema A, et al. An epigenome‐wide association study in whole blood of measures of adiposity among Ghanaians: the RODAM study. Clin Epigenetics. 2017;9:103. doi: 10.1186/s13148-017-0403-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Li C, Wang Z, Hardy T, et al. Association of obesity with DNA methylation age acceleration in African American mothers from the InterGEN study. Int J Mol Sci. 2019;20:4273. doi: 10.3390/ijms20174273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dick KJ, Nelson CP, Tsaprouni L, et al. DNA methylation and body‐mass index: a genome‐wide analysis. Lancet. 2014;383:1990‐1998. [DOI] [PubMed] [Google Scholar]
- 11. Levine M, Crimmins E. Evidence of accelerated aging among African Americans and its implications for mortality. Soc Sci Med. 2014;118:27‐32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Thorpe RJ Jr, Fesahazion RG, Parker L, et al. Accelerated health declines among African Americans in the USA. J Urban Health. 2016;93:808‐819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Williams DR, Lawrence JA, Davis BA. Racism and health: evidence and needed research. Annu Rev Public Health. 2019;40:105‐125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Barcelona de Mendoza V, Huang Y, Crusto CA, Sun YV, Taylor JY. Perceived racial discrimination and DNA methylation among African American women in the InterGEN study. Biol Res Nurs. 2018;20:145‐152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dhana K, Braun K, Nano J, et al. An epigenome‐wide association study of obesity‐related traits. Am J Epidemiol. 2018;187:1662‐1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Demerath EW, Guan W, Grove ML, et al. Epigenome‐wide association study (EWAS) of BMI, BMI change and waist circumference in African American adults identifies multiple replicated loci. Hum Mol Genet. 2015;24:4464‐4479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Geurts YM, Dugué PA, Joo JE, et al. Novel associations between blood DNA methylation and body mass index in middle‐aged and older adults. Int J Obes (Lond). 2018;42:887‐896. [DOI] [PubMed] [Google Scholar]
- 18. Sun D, Zhang T, Su S, et al. Body mass index drives changes in DNA methylation: a longitudinal study. Circ Res. 2019;125:824‐833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Coassin S, Hermann‐Kleiter N, Haun M, et al. A genome‐wide analysis of DNA methylation identifies a novel association signal for Lp(a) concentrations in the LPA promoter. PLoS One. 2020;15:e0232073. doi: 10.1371/journal.pone.0232073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Heitkamp M, Siegrist M, Molnos S, et al. Obesity genes and weight loss during lifestyle intervention in children with obesity. JAMA Pediatr. 2021;175:e205142. doi: 10.1001/jamapediatrics.2020.5142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chambers JC, Loh M, Lehne B, et al. Epigenome‐wide association of DNA methylation markers in peripheral blood from Indian Asians and Europeans with incident type 2 diabetes: a nested case‐control study. Lancet Diabetes Endocrinol. 2015;3:526‐534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yao C, Joehanes R, Wilson R, et al. Epigenome‐wide association study of whole blood gene expression in Framingham Heart Study participants provides molecular insight into the potential role of CHRNA5 in cigarette smoking‐related lung diseases. Clin Epigenetics. 2021;13:60. doi: 10.1186/s13148-021-01041-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gagnon F, Aïssi D, Carrié A, Morange PE, Trégouët DA. Robust validation of methylation levels association at CPT1A locus with lipid plasma levels. J Lipid Res. 2014;55:1189‐1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Barcelona V, Huang Y, Brown K, et al. Novel DNA methylation sites associated with cigarette smoking among African Americans. Epigenetics. 2019;14:383‐391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Crusto CA, Barcelona de Mendoza V, Connell CM, Sun YV, Taylor JY. The Intergenerational Impact of Genetic and Psychological Factors on Blood Pressure Study (InterGEN): design and methods for recruitment and psychological measures. Nurs Res. 2016;65:331‐338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bahlo M, Stankovich J, Danoy P, et al. Saliva‐derived DNA performs well in large‐scale, high‐density single‐nucleotide polymorphism microarray studies. Cancer Epidemiol Biomarkers Prev. 2010;19:794‐798. [DOI] [PubMed] [Google Scholar]
- 27. Taylor JY, Wright ML, Crusto CA, Sun YV. The Intergenerational Impact of Genetic and Psychological Factors on Blood Pressure (InterGEN) study: design and methods for complex DNA analysis. Biol Res Nurs. 2016;18:521‐530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Fortin JP, Triche TJ Jr, Hansen KD. Preprocessing, normalization and integration of the Illumina HumanMethylationEPIC array with minfi. Bioinformatics. 2017;33:558‐560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Daniels PR, Kardia SL, Hanis CL, et al. Familial aggregation of hypertension treatment and control in the Genetic Epidemiology Network of Arteriopathy (GENOA) study. Am J Med. 2004;116:676‐681. [DOI] [PubMed] [Google Scholar]
- 30. Fortin JP, Fertig E, Hansen K. shinyMethyl: interactive quality control of illumina 450k DNA methylation arrays in R. F1000Res. 2014;3:175. doi: 10.12688/f1000research.4680.2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Xu Z, Niu L, Li L, Taylor JA. ENmix: a novel background correction method for Illumina HumanMethylation450 BeadChip. Nucleic Acids Res. 2016;44:e20. doi: 10.1093/nar/gkv907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Aryee MJ, Jaffe AE, Corrada‐Bravo H, et al. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics. 2014;30:1363‐1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Niu L, Xu Z, Taylor JA. RCP: a novel probe design bias correction method for Illumina Methylation BeadChip. Bioinformatics. 2016;32:2659‐2663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chen C, Grennan K, Badner J, et al. Removing batch effects in analysis of expression microarray data: an evaluation of six batch adjustment methods. PLoS One. 2011;6:e17238. doi: 10.1371/journal.pone.0017238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Johnson WE, Li C, Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. 2007;8:118‐127. [DOI] [PubMed] [Google Scholar]
- 36. Houseman EA, Kile ML, Christiani DC, Ince TA, Kelsey KT, Marsit CJ. Reference‐free deconvolution of DNA methylation data and mediation by cell composition effects. BMC Bioinformatics. 2016;17:259. doi: 10.1186/s12859-016-1140-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Houseman EA, Molitor J, Marsit CJ. Reference‐free cell mixture adjustments in analysis of DNA methylation data. Bioinformatics. 2014;30:1431‐1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Klebaner D, Huang Y, Hui Q, et al. X chromosome‐wide analysis identifies DNA methylation sites influenced by cigarette smoking. Clin Epigenetics. 2016;8:20. doi: 10.1186/s13148-016-0189-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series B Stat Methodol. 1995;57:289‐300. [Google Scholar]
- 40. Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta‐analysis of genomewide association scans. Bioinformatics. 2010;26:2190‐2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Phipson B, Maksimovic J, Oshlack A. missMethyl: an R package for analyzing data from Illumina's HumanMethylation450 platform. Bioinformatics. 2016;32:286‐288. [DOI] [PubMed] [Google Scholar]
- 42. Huang Y, Ollikainen M, Muniandy M, et al. Identification, heritability, and relation with gene expression of novel DNA methylation loci for blood pressure. Hypertension. 2020;76:195‐205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wang X, Pan Y, Zhu H, et al. An epigenome‐wide study of obesity in African American youth and young adults: novel findings, replication in neutrophils, and relationship with gene expression. Clin Epigenetics. 2018;10:3. doi: 10.1186/s13148-017-0435-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Loucks EB, Huang YT, Agha G, et al. Epigenetic mediators between childhood socioeconomic disadvantage and mid‐life body mass index: the New England Family Study. Psychosom Med. 2016;78:1053‐1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chu SH, Loucks EB, Kelsey KT, et al. Sex‐specific epigenetic mediators between early life social disadvantage and adulthood BMI. Epigenomics. 2018;10:707‐722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Li S, Wong EM, Bui M, et al. Inference about causation between body mass index and DNA methylation in blood from a twin family study. Int J Obes (Lond). 2019;43:243‐252. [DOI] [PubMed] [Google Scholar]
- 47. Ali O, Cerjak D, Kent JW Jr, et al. Methylation of SOCS3 is inversely associated with metabolic syndrome in an epigenome‐wide association study of obesity. Epigenetics. 2016;11:699‐707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kievit P, Howard JK, Badman MK, et al. Enhanced leptin sensitivity and improved glucose homeostasis in mice lacking suppressor of cytokine signaling‐3 in POMC‐expressing cells. Cell Metab. 2006;4:123‐132. [DOI] [PubMed] [Google Scholar]
- 49. Huang D, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44‐57. [DOI] [PubMed] [Google Scholar]
- 50. Kvaløy K, Page CM, Holmen TL. Epigenome‐wide methylation differences in a group of lean and obese women ‐ a HUNT study. Sci Rep. 2018;8:16330. doi: 10.1038/s41598-018-34003-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sharp GC, Alfano R, Ghantous A, et al. Paternal body mass index and offspring DNA methylation: findings from the PACE consortium. Int J Epidemiol. 2021;50:1297‐1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sharma NK, Comeau ME, Montoya D, et al. Integrative analysis of Glucometabolic traits, adipose tissue DNA methylation, and gene expression identifies epigenetic regulatory mechanisms of insulin resistance and obesity in African Americans. Diabetes. 2020;69:2779‐2793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Crocker KC, Domingo‐Relloso A, Haack K, et al. DNA methylation and adiposity phenotypes: an epigenome‐wide association study among adults in the Strong Heart Study. Int J Obes (Lond). 2020;44:2313‐2322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. He F, Berg A, Imamura Kawasawa Y, et al. Association between DNA methylation in obesity‐related genes and body mass index percentile in adolescents. Sci Rep. 2019;9:2079. doi: 10.1038/s41598-019-38587-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Campanella G, Gunter MJ, Polidoro S, et al. Epigenome‐wide association study of adiposity and future risk of obesity‐related diseases. Int J Obes (Lond). 2018;42:2022‐2035. [DOI] [PubMed] [Google Scholar]
- 56. Mendelson MM, Marioni RE, Joehanes R, et al. Association of body mass index with DNA methylation and gene expression in blood cells and relations to cardiometabolic disease: a Mendelian randomization approach. PLoS Med. 2017;14:e1002215. doi: 10.1371/journal.pmed.1002215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Crujeiras AB, Diaz‐Lagares A, Sandoval J, et al. DNA methylation map in circulating leukocytes mirrors subcutaneous adipose tissue methylation pattern: a genome‐wide analysis from non‐obese and obese patients. Sci Rep. 2017;7:41903. doi: 10.1038/srep41903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. al Muftah WA, Al‐Shafai M, Zaghlool SB, et al. Epigenetic associations of type 2 diabetes and BMI in an Arab population. Clin Epigenetics. 2016;8:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Wang S, Song J, Yang Y, Zhang Y, Wang H, Ma J. HIF3A DNA methylation is associated with childhood obesity and ALT. PLoS One. 2015;10:e0145944. doi: 10.1371/journal.pone.0145944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Karlsson IK, Ericsson M, Wang Y, et al. Replicating associations between DNA methylation and body mass index in a longitudinal sample of older twins. Int J Obes (Lond). 2020;44:1397‐1405. [DOI] [PubMed] [Google Scholar]
- 61. Ligthart S, Marzi C, Aslibekyan S, et al. DNA methylation signatures of chronic low‐grade inflammation are associated with complex diseases. Genome Biol. 2016;17:255. doi: 10.1186/s13059-016-1119-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Meeks K, Henneman P, Venema A, et al. Epigenome‐wide association study in whole blood on type 2 diabetes among sub‐Saharan African individuals: findings from the RODAM study. Int J Epidemiol. 2019;48:58‐70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Lai CQ, Parnell LD, Smith CE, et al. Carbohydrate and fat intake associated with risk of metabolic diseases through epigenetics of CPT1A. Am J Clin Nutr. 2020;112:1200‐1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Liu Y, Ding J, Reynolds LM, et al. Methylomics of gene expression in human monocytes. Hum Mol Genet. 2013;22:5065‐5074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Komaki S, Shiwa Y, Furukawa R, et al. iMETHYL: an integrative database of human DNA methylation, gene expression, and genomic variation. Hum Genome Var. 2018;5:18008. doi: 10.1038/hgv.2018.8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Mathur R, Hui Q, Huang Y, et al. DNA methylation markers of type 2 diabetes mellitus among male veterans with or without human immunodeficiency virus infection. J Infect Dis. 2019;219:1959‐1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Frazier‐Wood AC, Aslibekyan S, Absher DM, et al. Methylation at CPT1A locus is associated with lipoprotein subfraction profiles. J Lipid Res. 2014;55:1324‐1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Richard MA, Huan T, Ligthart S, et al. DNA methylation analysis identifies loci for blood pressure regulation. Am J Hum Genet. 2017;101:888‐902. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information Figure S1A Volcano plot of the meta‐analysis of BMI EWAS. BMI, body mass index; EWAS, epigenome‐wide association study.
Supporting Information Figure S1B Volcano plot of the female‐only meta‐analysis of BMI EWAS. BMI, body mass index; EWAS, epigenome‐wide association study.
Supporting Information Table S1 Epigenome‐wide associations of 61 significant (Bonferroni corrected p < 0.05) CpG sites with BMI from the meta‐analysis of InterGEN and GENOA. Female only. BMI, body mass index; CpG, XXXX; GENOA, Genetic Epidemiology Network of Arteriopathy; InterGEN, Intergenerational Impact of Genetic and Psychological Factors on Blood Pressure.
Supporting Information Table S2 Epigenome‐wide associations of 65 significant (Bonferroni corrected p < 0.05) CpG sites with BMI from the meta‐analysis of InterGEN and GENOA. BMI, body mass index; CpG, XXXX; GENOA, Genetic Epidemiology Network of Arteriopathy; InterGEN, Intergenerational Impact of Genetic and Psychological Factors on Blood Pressure.
Supporting Information Table S3 Epigenome‐wide associations of 658 significant (FDR 0.05 threshold) CpG sites with BMI from the meta‐analysis of InterGEN and GENOA. BMI, body mass index; FDR, false discovery rate; GENOA, Genetic Epidemiology Network of Arteriopathy; InterGEN, Intergenerational Impact of Genetic and Psychological Factors on Blood Pressure.
Supporting Information Table S4 Epigenome‐wide associations of 628 significant (FDR 0.05 threshold) CpG sites with BMI from the meta‐analysis of InterGEN and GENOA. Female only. BMI, body mass index; CpG, XXXX; FDR, false discovery rate; GENOA, Genetic Epidemiology Network of Arteriopathy; InterGEN, Intergenerational Impact of Genetic and Psychological Factors on Blood Pressure.
Supporting Information Table S5 Summary of literature review on EWAS of BMI with discovery set cohort characteristics and study design if available. All studies consist of mixed gender. None of the studies include participants with HIV. All samples came from peripheral blood except noted otherwise. BMI, body mass index; EWAS, epigenome‐wide association study.
Data Availability Statement
InterGEN genotype and phenotype data are available through the Database of Genotypes and Phenotypes (dbGaP accession number phs001792.v1.p1). DNA methylation data are available with a Data Use Agreement upon reasonable request to the InterGEN study investigators Jacquelyn Y. Taylor (jyt2116@cumc.columbia.edu) and Yan V. Sun (yan.v.sun@emory.edu). GENOA genotype and phenotype data are available through the Database of Genotypes and Phenotypes (dbGaP accession number phs001238.v2.p1). Methylation data are from the Gene Expression Omnibus (GEO): GSE157131. Owing to Institutional Review Board restriction, mapping of the sample IDs between genotype data (dbGaP) and DNA methylation data (GEO) cannot be provided publicly but is available upon written request to the study investigators Jennifer A. Smith (smjenn@umich.edu) and Sharon L.R. Kardia (skardia@umich.edu).