

SUMMARY
The king protea (Protea cynaroides), an early‐diverging eudicot, is the most iconic species from the Megadiverse Cape Floristic Region, and the national flower of South Africa. Perhaps best known for its iconic flower head, Protea is a key genus for the South African horticulture industry and cut‐flower market. Ecologically, the genus and the family Proteaceae are important models for radiation and adaptation, particularly to soils with limited phosphorus bio‐availability. Here, we present a high‐quality chromosome‐scale assembly of the P. cynaroides genome as the first representative of the fynbos biome. We reveal an ancestral whole‐genome duplication event that occurred in the Proteaceae around the late Cretaceous that preceded the divergence of all crown groups within the family and its extant diversity in all Southern continents. The relatively stable genome structure of P. cynaroides is invaluable for comparative studies and for unveiling paleopolyploidy in other groups, such as the distantly related sister group Ranunculales. Comparative genomics in sequenced genomes of the Proteales shows loss of key arbuscular mycorrhizal symbiosis genes likely ancestral to the family, and possibly the order. The P. cynaroides genome empowers new research in plant diversification, horticulture and adaptation, particularly to nutrient‐poor soils.
Keywords: Protea cynaroides, genome assembly, genome annotation, early‐divergent eudicot, comparative genomics
Significance Statement
Our chromosome‐level Protea cynaroides genome assembly is the first endemic South African plant genome to be published, and can signify a new wave of research focusing on indigenous fynbos and endemic African plant species. We foresee this serving as increasing visibility of plant genomics research from the African continent and the Global South, and enriching global biodiversity, sustainability, evolution and conservation research.
INTRODUCTION
The order Proteales, along with the Ranunculales, Trochodendrales and Buxales, forms part of the early‐diverging Eudicots and is sister to the core Eudicots (One Thousand Plant Transcriptomes Initiative, 2019). Within this order, the Proteaceae is a classic example of a Gondwanan plant lineage, with molecular dating studies placing their origin at about 110–120 million years ago (Anderson et al., 2005), although studies on the evolutionary relationships of the genera and species within the family have indicated that intercontinental dispersal must have occurred as well (Barker et al., 2007). Proteaceae is also one of the earliest diverging lineages of the Eudicots, with approximately 1600 described species within either the so‐called ‘superrosids’ or ‘superasterids’, representing by far the most species‐diverse family outside the core Eudicots (Moore et al., 2010). Lineages within the Proteaceae have demonstrated variable diversification rates at three main periods of the Earth's history: since the origin of the family about 110 MYA; since the K‐Pg mass extinction event (66 MYA); and about the late Miocene (5–14 MYA). The most recent diversification representative of extant Proteaceae species is estimated to have occurred in parallel in South Africa and Australia due to environmental shifts towards a nutrient‐poor, fire‐dominated Mediterranean climate in the late Miocene, although there is evidence for some lineages that are older, about 20–30 MYA (Born et al., 2006; Linder, 2003, 2008).
The genus Protea currently contains 107 species, and displays a suite of floral adaptations for a diverse array of pollinators including beetles, birds and small mammals (Collins & Rebelo, 1987). It is most notably emblematic of the fynbos biome and the Cape Floristic Region (CFR). The CFR not only has an extremely high number of plant species (~9000 vascular plant species, almost exclusively flowering plants) but also close to 70% species endemism – unusual for a mainland continental range and comparable only to some islands (Born et al., 2006; Goldblatt & Manning, 2002; Linder, 2003). It is notable for having diversity that is the product of both so‐called ‘mature’ and recent rapid radiations (Linder, 2008; van Santen & Linder, 2020). The genus represents an excellent model system for understanding the genetic basis of adaptive radiation, particularly with regard to adaptation to dramatic changes in rainfall and temperature (Akman et al., 2016; Mitchell et al., 2017). Additionally, Protea contains a variety of species representing repeated loss and acquisition of both reseeding and resprouting adaptations in response to fire (Lamont et al., 2013). One of the species, the King Protea (Protea cynaroides), is an early‐diverging species in the genus (Mitchell et al., 2017). It is the National Flower of South Africa and represents a cultural icon, and also a popular member of the floriculture industry, making up a significant majority of the cut‐flower industry and export in South Africa (Coetzee & Littlejohn, 2010; Reinten et al., 2011).
Here, we present a chromosome‐scale genome for P. cynaroides (var. ‘Little Prince’), the first genome for any Protea species, and the first genome of a species endemic to the CFR and the fynbos biome. We were especially interested in determining whether large‐scale genomic events such as whole‐genome duplication (WGD) or repetitive element proliferations have occurred, and whether these may have led to genome evolutionary dynamics and potentially be linked to major radiation events in the family. Our results reveal that a WGD event in the lineage likely coincided with or preceded the K‐Pg mass extinction (57–76 MYA), prior to the second major diversification of the Proteaceae. Our analysis also revises the WGD dating reported for Macadamia integrifolia, indicating that the WGD event occurred in the common ancestor of P. cynaroides and M. integrifolia, and would therefore be shared among all members of the subfamilies Proteoideae and Grevilleoideae. A single round of WGD and conserved genome structure make Proteaceae species an irreplaceable genomic resource to further our understanding of WGDs in papaver (poppy) belonging to Ranunculales. In addition to new insights into gains and expansions in genes and gene families, analysis of Proteales genomes point to specific gene losses in the ancestor of all extant Proteaceae (and possibly Proteales) explaining their inability to form arbuscular mycorrhizal (AM) associations. The P. cynaroides genome paves the way for understanding molecular mechanisms underlying critical ecological adaptations of the Proteaceae, such as fire‐ecology, specialized root‐nutrient acquisition and the unique floral development characteristic of the Proteaceae, as well as the variation underlying the immense diversity in the unique endemism hotspot of the CFR.
RESULTS
Genome sequencing, assembly and annotation
We sequenced the genome of P. cynaroides using a combination of Illumina and Oxford Nanopore high‐throughput sequencing systems. We generated 73.4 Gb (60 ×) of Nanopore long‐reads with a read N50 length of 18.3 kb and 132 Gb (110 ×) of Illumina 151‐bp paired‐end sequence data. K‐mer (k = 21) analysis indicated P. cynaroides is a diploid with an estimated genome size of approximately 1.18 Gb (Figure S1). The Nanopore long‐reads were used for genome assembly prior to using Illumina short‐reads for further polishing. The total length of 4533 contigs assembled by Flye is about 1.22 Gb with a contig N50 size of 1.07 Mb, which is consistent with the estimated haploid genome size obtained with k‐mer analysis. The assembly was further scaffolded with 185.1 million Omni‐C paired‐end reads, this yielded a final scaffold assembly of 12 unambiguous chromosome‐level pseudomolecules with a scaffold N50 of 100.6 Mb (Figure 1a,b; Table 1) covering approximately 96.6% (1.18 Gb) of the assembled genome size.
Figure 1.
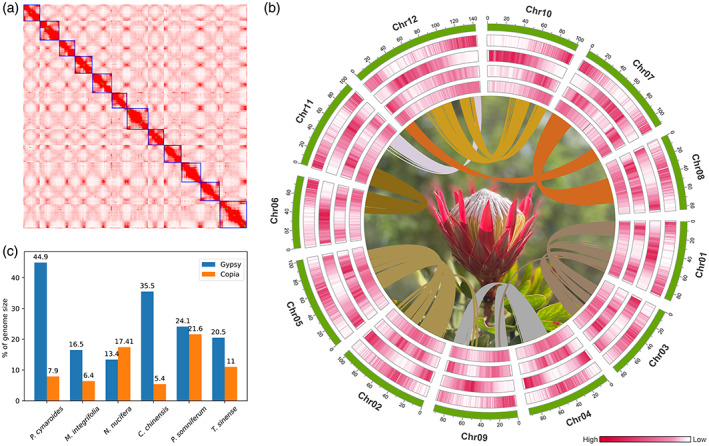
Genome assembly features of Protea.
(a) Hi‐C interaction heatmap of the assembled Protea cynaroides genome.
(b) The landscape of P. cynaroides genome assembly. Bar plots (from innermost outwards) show DNA transposable elements (TEs), Copia, Gypsy, gene density per 300 kb and chromosomes. Ribbons connect inter‐chromosomal syntenic regions.
(c) Gypsy and Copia content comparison of six early‐diverging eudicot plants.
Table 1.
Statistics of genome assembly and annotation
| Assembly features | |
|---|---|
| Estimated genome size (bp) | 1 180 302 352 |
| Number of contigs | 4533 |
| N50 contig length (bp) | 1 068 931 |
| Total size of contigs (bp) | 1 223 600 671 |
| Number of pseudo‐chromosomes | 12 |
| Total size of pseudo‐chromosomes (bp) | 1 181 608 347 |
| N50 scaffold length (bp) | 100 581 735 |
| GC content | 39.59% |
| BUSCO (complete) | 95.0% |
| LTR Assembly Index (LAI) | 14.92 |
| Gene models | |
| Number of gene models | 33 320 |
| Mean gene length (bp) | 8133 |
| Mean coding sequence length (bp) | 1176 |
| Mean number of exons per gene | 5.14 |
| Mean exon length (bp) | 229 |
| Mean intron length (bp) | 1683 |
| BUSCO (complete) | 98.0% |
| Non‐coding RNA | |
| Number of miRNA | 165 |
| Number of tRNA | 1403 |
| Number of rRNA | 245 |
| Number of snoRNA | 773 |
To evaluate the quality of the assembled P. cynaroides genome, we aligned the Illumina short‐reads to the assembled genome, resulting in a mapping rate of 99.8%. Benchmarking sets of Universal Single‐Copy Orthologs (Manni et al., 2021) analysis using the embryophyta_odb10 dataset showed that 95.0% of the BUSCO sequences were completely present in the final assembly (Figure S2; Table 1). The long terminal repeat (LTR) Assembly Index score of the final assembly was 14.92, reaching the criterion of reference quality (Ou et al., 2018). Together, these results indicate that the genome assembly of P. cynaroides is of high contiguity and sequence quality.
Repetitive elements account for 72.15% (884 Mb) of the P. cynaroides genome. LTR‐retrotransposons (LTR‐RT) are the major class of transposable elements (TEs) and account for 53.11% of the assembly, while DNA TEs comprise 10.91% of the genome (Figure S3; Table S1). Gypsy elements are the most abundant, comprising 44.88% of the assembly among the LTRs, followed by Copia elements, which occupy 7.88% of the assembly (Figure 1c). This is similar to the makeup of the Coptis chinensis genome in which a much greater abundance of Gypsy elements compared with Copia elements was observed (Liu et al., 2021); however, the Gypsy to Copia ratio is still exceptionally higher in P. cynaroides than most sequenced early‐diverging eudicot plants (Figure 1c). A total of 7630 intact LTR‐RTs in P. cynaroides were estimated to have an average insertion time of 2.74 MYA, with the burst of LTR/Gypsy insertions (Figure S4), suggesting the relatively larger P. cynaroides genome size (1200 Mb) can be explained by recent bursts of LTR/Gypsy elements, when compared with the genomes of the sister Proteaceae species M. integrifolia (~794 Mb) and Nelumbo nucifera (~813 Mb).
We used an integrated strategy including evidence‐based and ab initio gene prediction to annotate protein‐coding genes in the P. cynaroides genome. After manual curation, we annotated 33 320 gene models, 85.9% of which (28 619 genes) are supported by transcriptome data (TPM > 0) and 71.8% of which (23 909 genes) had detectable expression (TPM > 1). On average, protein‐coding genes in P. cynaroides are 8133 bp long and contain 5.14 exons (Table 1). To evaluate the quality of annotation, BUSCO (v4.1.4) was performed and the result indicated that 98% of BUSCO genes were complete, 90.6% of the BUSCO genes were single‐copy and 7.4% were found in duplicate. Only 1.2% of the BUSCO gene set (18 genes) are missing (Figure S2; Table 1). Moreover, 93.2% of these P. cynaroides genes could be assigned functions using InterProScan (Jones et al., 2014). We further annotated non‐coding RNA genes, yielding 1403 transfer RNA (tRNA), 245 ribosomal RNA (rRNA), 773 small nuclear RNA genes and 165 microRNA genes (Table 1).
Genome evolution and paleopolyploid ancestry of Proteaceae
We investigated gene family evolution in P. cynaroides through comparison with 18 other plant species, including representatives from monocots (outgroup), Proteales, Ranunculales, Rosids, Asterales and Caryophyllales (Table S2). Overall, 457 177 genes were clustered into 28 680 orthologous gene families and 176 genes were single‐copy in the species investigated, except for Papaver somniferum and Papaver setigerum. Phylogenomic analysis using these 176 single‐copy genes revealed that P. cynaroides (Proteoideae) is sister to M. integrifolia and Telopea speciosissima (Grevilleoideae), while their divergence was estimated at approximately 63 (60–67) MYA (Figure 2b). Within Grevilleoideae, the split between M. integrifolia and T. speciosissima was estimated to be approximately 39 (25–53) MYA (Figure 2b). N. nucifera (Nelumbonaceae) is sister to Proteaceae, including P. cynaroides, M. integrifolia, and T. speciosissima (Figure 2b).
Figure 2.
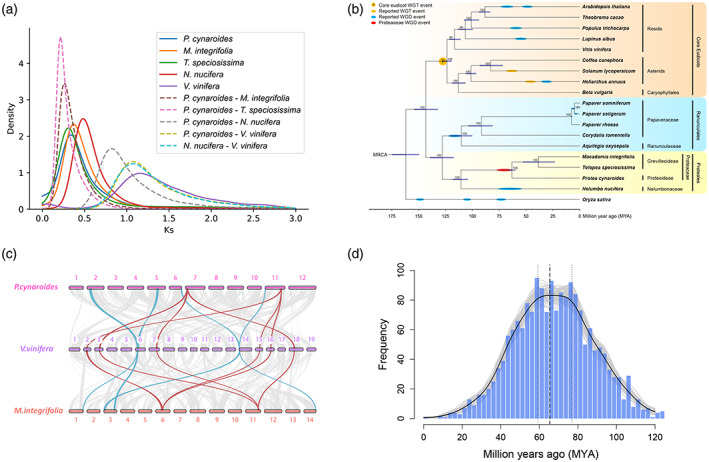
The whole‐genome duplication (WGD) event in Proteaceae.
(a) Ks distributions for anchor pairs (retained paralogs from the WGD event) within Protea cynaroides, Macadamia integrifolia and Telopea speciosissima genome, and for orthologs of P. cynaroides and related species.
(b) Inferred phylogenetic tree with 176 single‐copy genes in 17 species except for Papaver somniferum and Papaver setigerum identified by OrthoFinder. Timing of Proteaceae WGD was estimated in this study, and previously reported whole‐genome triplication (WGT)/WGD events are superimposed on the tree.
(c) Intergenomic co‐linearity among P. cynaroides, Vitis vinifera and M. integrifolia.
(d) Absolute dating of the P. cynaroides WGD event. The age distribution was obtained by phylogenomic dating of P. cynaroides paralogs. The solid black line represents the Kernel Density Estimation (KDE) of the dated paralogs, while the vertical dashed black line represents its peak at 67.9 MYA, which was used as the consensus WGD age estimate. The gray lines represent density estimates from 2500 bootstrap replicates, and the vertical black dotted lines represent the corresponding 90% confidence interval (CI) for the WGD age estimate, 59.267–76.85 MYA (see Experimental Procedures section). The histogram shows the raw distribution of dated paralogs.
We used the CAFE5 (Mendes et al., 2020) software to identify the expansion and contraction of gene families across the same 19 genomes, and considered 12 268 gene families with at least one gene both in the outgroup clade and its complement as ancestral gene families (most recent common ancestor). Of those gene families, 1345 families have undergone expansion, while 1456 families have undergone contraction in the Proteaceae. Functional annotation of the expanded genes demonstrated that they were mainly enriched in lipid metabolic processes, fatty acid (FA) biosynthetic and metabolic processes, nitrogen compound transport, sulfur compound metabolic processes, carbohydrate transmembrane transport, pollen–pistil interactions and developmental processes involved in reproduction (Figure S9a).
Age distributions of synonymous substitutions per synonymous site (KS) for paralogous genes and the syntenic depth ratio of 2:1 with Aristolochia fimbriata, which has not undergone any relatively recent WGDs (Qin et al., 2021), show clear evidence for a WGD event in P. cynaroides, M. integrifolia and T. speciosissima, with a KS peak value of 0.36, 0.34 and 0.38, respectively (Figures 2a and S5). Intragenomic syntenic analysis in P. cynaroides also identified a high level of within‐genome collinearity with 12 683 collinear genes in 874 collinear blocks (see Experimental Procedures section), representing 38.06% of the P. cynaroides gene complements (Figure 1b), which again provides strong support for the WGD event in P. cynaroides. Our results also confirm the WGD event in M. integrifolia, which has previously been reported (Lin et al., 2022). We were also interested in confirming whether species of Proteaceae shared the same WGD event. The KS peak values for orthologs between P. cynaroides, M. integrifolia and T. speciosissima are lower than those of the WGD peaks, suggesting that the WGD predated the divergence of these species (Figure 2a). In addition, the KS distribution for paralogous genes in the N. nucifera genome and intergenomic collinearity analysis again confirm a WGD peak at KS ~0.47 in N. nucifera (Figures 2a and 3), while the KS peak value of their speciation (Ks values for orthologs of P. cynaroides and N. nucifera) is considerably higher at KS ~0.82, suggesting that these two WGD events in the Proteaceae and Nelumbonaceae occurred independently after their divergence (Figure 2a). Moreover, intergenomic collinearity analysis also shows that neither Proteaceae nor Nelumbonaceae experienced γ, the hexaploidization event shared by all core eudicots (Jaillon et al., 2007; Van de Peer et al., 2017), as comparing P. cynaroides, M. integrifolia, T. speciosissima and N. nucifera with Vitis vinifera shows a 2:3 syntenic relationship (Figures 2c and S6).
Figure 3.
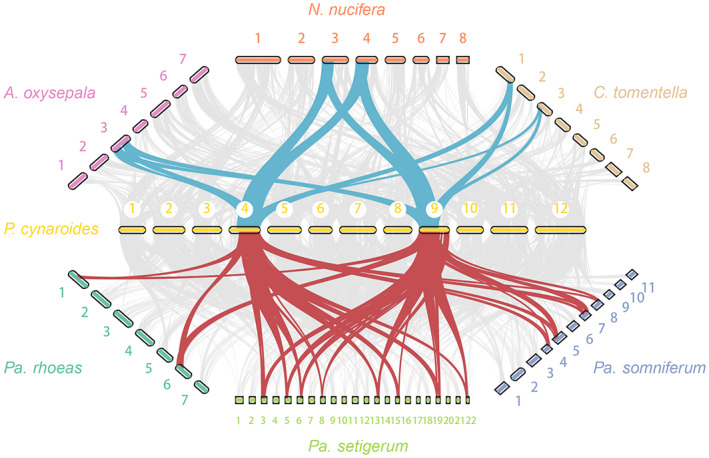
Intergenomic comparisons between Protea and early‐diverging eudicots.
Synteny between Protea cynaroides and three early‐Eudicot species (Nelumbo nucifera, Aquilegia oxysepala and Corydalis tomentella), as well as three poppies (Papaver rhoeas, Papaver somniferum and Papaver setigerum), respectively. A clear 2:2 syntenic relationship between P. cynaroides and these three early‐Eudicot species from different families can be observed. Meanwhile, a clear 2:2 synteny relationships between P. cynaroides and Pa. rhoeas, 2:4 synteny relationships between P. cynaroides and Pa. somniferum, and 2:8 synteny relationships between P. cynaroides and P. setigerum can be identified.
Absolute dating of the WGD in P. cynaroides (see Experimental Procedures section) suggests that the WGD event occurred ~68 MYA, with a 90% confidence interval (CI) giving a range of 59.27–76.85 MYA (Figure 2d). Interestingly, this date coincides with the K‐Pg mass extinction that led to the loss of about 60% of plant species (Fawcett et al., 2009). Numerous plant species seem to have undergone WGDs around the K‐Pg boundary (Cannon et al., 2015; Fawcett et al., 2009; Huang et al., 2016; Lohaus & Van de Peer, 2016; Van de Peer et al., 2017; Vanneste, Baele, et al., 2014; Vanneste, Maere, et al., 2014; Yu et al., 2017), and it has been suggested that polyploidization could have facilitated plants to survive environmental turmoil and extinction events (Linder & Barker, 2014; Van de Peer et al., 2017, 2021).
The genome structure of Proteaceae seems well conserved, which is corresponding with the lower rates of evolution (Verboom et al., 2017). Although P. cynaroides and M. integrifolia have diverged about 63 MYA (Carpenter, 2012), comparative genomic analysis of P. cynaroides and M. integrifolia shows a high level of collinearity between the two genomes, and MCScanX (Wang et al., 2012) identified 367 syntenic blocks with 16 728 P. cynaroides genes and 16 863 M. integrifolia genes representing 50% and 44% of the P. cynaroides and M. integrifolia gene compositions, respectively. As Proteaceae only underwent a single round of WGD and retained a relatively conserved genome structure, they have great value for comparative genomics. For instance, using the P. cynaroides genome, we were able to identify and reconstruct with great confidence the WGD history in Papaver (poppy), a genus of the Ranunculales sister to the Proteales. By comparing the P. cynaroides genome with that of poppies, we found a 2:2 well‐preserved intergenomic synteny relationship between P. cynaroides and Papaver rhoeas, a 2:4 clear intergenomic synteny relationship between P. cynaroides and Pa. somniferum, and a 2:8 intergenomic synteny relationship between P. cynaroides and Pa. setigerum, supporting one WGD event in Pa. rhoeas, two in Pa. somniferum and three in Pa. setigerum. Phylogenetic relationships infer that there is a shared WGD by all three poppies (i.e. the ancient WGD shared by Ranunculaceae and Papaveraceae; Xu et al., 2022), a second shared by Pa. somniferum and Pa. setigerum, while the third is specific to Pa. setigerum (Figures 2b and 3). However, only the second and the Pa. setigerum‐specific WGD have been previously reported (Guo et al., 2018; Yang et al., 2021). In addition, we also identified a clear 2:2 syntenic relationship between P. cynaroides and some other early‐diverging Eudicots, including N. nucifera (Nelumbonaceae), A. oxysepala (Ranunculaceae), and C. tomentella (Papaveraceae), thereby confirming the reported single WGD in N. nucifera, A. oxysepala and C. tomentella (Figure 3).
MADS‐box gene families in Protea cynaroides
Protea cynaroides, also known as the King Protea, has composite beautiful flower heads with a collection of flowers in the center, surrounded by large colorful bracts. MADS‐box genes encode eukaryote‐specific transcription factors controlling multiple developmental programs, and type II MADS‐box genes are well studied in terms of their expression patterns, and their functions in floral organ specification are also well characterized. We identified 79 putative MADS‐box genes in the P. cynaroides genome of which 38 are type II MADS‐box genes, as compared with the 31, 31 and 35 present in M. integrifolia, Oryza sativa and Arabidopsis thaliana, respectively (Figure 4a; Table S3), including homologous genes for the ABCE model of floral organ identities: AP1 and AGL6 (A function for sepals and petals), AP3 and PI (B function for petals and stamen), AG (C function for stamen and carpel) and SEP (E function for interacting with ABC function proteins). Collinearity analyses indicate all the genes for the ABCE model have two or three copies, derived from the Proteaceae WGD event (except AG in P. cynaroides), such as two AGL6s (AGL6a, AGL6b) and two AP3s (AP3a and AP3b; Figure 4b). There are three PIs, of which two copies are derived from P. cynaroides WGD event, followed by one tandem duplication (Figure 4b). Two E‐function SEPs are also derived from Proteaceae WGD (Figure 4b). Almost all duplicates in the ABCE model were derived from the WGD event, likely contributing to their striking inflorescence and flower morphology.
Figure 4.
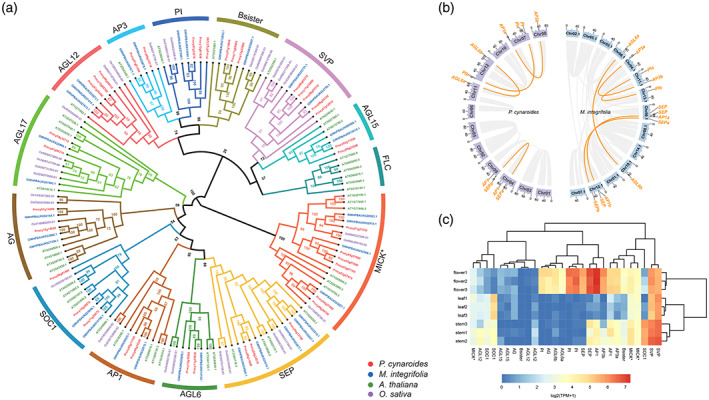
MADS‐box genes in Protea cynaroides and Macadamia integrifolia.
(a) The phylogenetic tree of type II MADS‐box genes in P. cynaroides, M. integrifolia, Arabidopsis thaliana and Oryza sativa.
(b) The syntenic relationships of MADS‐box genes in P. cynaroides and M. integrifolia. Genes in yellow are MADS‐box genes; lines link the genes in the syntenic blocks, derived from the whole‐genome duplication (WGD) event.
(c) Gene expression patterns of type II MADS‐box genes from various organs in P. cynaroides.
We further investigated the expression profiles of type II MADS‐box genes in the flower, stem and stem tissues. Notably, two P. cynaroides AGL6 homologs are mainly expressed in flower, whereas the two AP1 homologs are also expressed in stem (Figure 4c). Beyond that, three B‐function PI homologs, the C‐function AG homologs and one of E‐function SEP genes are mainly expressed in flowers, suggesting they play roles in the ABCE model, together with AGL6. In Nymphaea colorata, the B‐function AP3 homolog is mainly expressed in floral organs, whereas PI has no expression, suggesting that AP3 acts as a B‐function gene in N. colorata (Zhang et al., 2020). On the contrary, PI homologs may play a key role in B‐function for P. cynaroides as they are mainly expressed in floral organs, whereas AP3 are also expressed in stem (Figure 4c). In some cases, the evolution of new functional roles for ABCE gene duplicates may well explain morphological novelties observed in some species (Irish, 2017; Kramer et al., 2007; Sablowski, 2015), for instance, the expression of a duplicated B‐function gene imparting petalloid characteristics to the petal‐like bracts of dogwoods (Cornus florida; Irish, 2017). It would be interesting to therefore explore the roles of B‐function PI duplicates particularly in the formation of P. cynaroides floral bracts that are so iconic in the genus and are of horticultural importance. Also, three divergent Ranunculus species (Eschscholzia californica, Nigella damascena and Aquilegia coerulea) demonstrated that B gene homologs are important for the initiation and/or maintenance of the outer extent of the AG homolog expression domain (Sharma and Kramer, 2017), while expanded B‐function genes (5 versus 2–3 in A. thaliana and O. sativa) likely have implication for C‐function in P. cynaroides. Genes in the ABCE model are expressed in broader domains, but limited in floral organs (Irish, 2017; Zhang et al., 2020). Our expression profile shows (for the first time) that A‐function AP1 and B‐function AP3 are also expressed in the stem for as yet unknown reasons.
Furthermore, P. cynaroides seems to have additional copies of AGL12 (three members) and MIKC* (seven members; Figure 4a). The Arabidopsis AGL12 gene is involved in root cell differentiation (Tapia‐López et al., 2008), and both epiphytic orchids and Utricularia gibba without true roots lack AGL12 clade (Ibarra‐Laclette et al., 2013; Zhang et al., 2017). We found AGL12 expanded in P. cynaroides with low expression in flowers, leafs and stems. However, it still remains elusive whether the additional copies of AGL12 could contribute to forming cluster roots in P. cynaroides, due to the lack of the transcriptome data in the root organ. In seed plants, MIKC* genes are major regulators of male gametophytic development (Kwantes et al., 2012; Liu et al., 2013), and the increase in the number of MIKC* genes in P. cynaroides may be related to a suite of floral adaptations for a diverse array of pollinators including beetles, birds and small mammals.
Proteales have lost key AM symbiosis genes
Cluster roots represent one of three common root adaptations in plants to acquire nutrients in soils that lack phosphorous and/or nitrogen – the other two being nodule formation for which legumes (members of the Fabaceae) are most well‐known; and AM symbioses (MacLean et al., 2017; Roy et al., 2020). Although these can occur together, many species and often entire families that have evolved cluster roots are thought to have rendered AM symbioses redundant (Brundrett & Tedersoo, 2018). Interestingly, independent losses of the ability to form this symbiosis have occurred in several lineages of flowering plants, including A. thaliana (Lambers & Teste, 2013; Veiga et al., 2013). Advances in phylogenomics, and various forward‐ and reverse‐genetic approaches have revealed conserved mechanisms for signaling and regulation of AM symbiosis (Bravo et al., 2016), as well as common loss of symbiotic genes in non‐host Brassicales species via large‐scale deletion or pseudogenization (Delaux et al., 2014). The available genomes of P. cynaroides (Proteoideae), two Grevilleoids (M. integrifolia and T. speciosissima) and Sacred Lotus (N. nucifera), all non‐host species belonging to Proteales, offer a unique opportunity to investigate the deletion or pseudogenization of conserved AM ‘symbiotic toolkit’ genes in clades representing the vast majority of species in the order.
To investigate the presence of the ‘symbiotic toolkit’ in P. cynaroides and its conservation in other Proteales species, we interrogated the genomes of these Proteaceae and Nelumbonaceae species in the context of other host and non‐host plants. To this end, a previously constructed query gene list (Bravo et al., 2016; Table S4) was used as a reference to search for homologous genes in Arabidopsis (non‐host plant representative), Theobroma cacao and O. sativa (host plant representatives), and Proteaceae and Nelumbonaceae species investigated.
Previous studies have revealed that a subset of ‘symbiotic toolkit’ genes are exclusively conserved in AMS host plants, referred to as ‘AMS‐conserved’ genes (Delaux et al., 2014; Favre et al., 2014). These genes, which include a phosphate transporter (PT4; Harrison et al., 2002), two ABC transporters (STR and STR2; Zhang et al., 2010), a GRAS transcription factor (RAM1; Gobbato et al., 2012) and two lipid biosynthetic enzymes (RAM2 and FatM; Bravo et al., 2017) are all required for AMS and are present only in host plants (Bravo et al., 2016; Delaux et al., 2014). Our comparative analyses indicate that these ‘AMS‐conserved’ genes are absent in all non‐host species, including both Protea, Macadamia and the Sacred Lotus (Figure 5). In addition, we also investigated the common symbiotic pathway (CSP) genes – a well‐characterized subset of the ‘symbiotic toolkit’ involved in both AM and root nodule symbioses (MacLean et al., 2017; Roy et al., 2020) – and found that the Proteales species have consistently retained certain CSP genes (NUP85, NUP133, NENA and POLLUX/DMI1; Figure 5) and their conserved motifs (Figure S7). A previous study by Delaux et al. (2014) reported the presence of a ‘conserved’ subset of genes in Arabidopsis and other host and non‐host species, which unsurprisingly also have non‐symbiotic roles (Gomez‐Roldan et al., 2008; Liu et al., 2011). However, other CSP genes (NSP1, NSP2, SYMRK/DMI3 and CCaMK/DMI2) are inconsistently retained and/or lost in the Proteales species and Arabidopsis (Figure 5). Firstly, NSP1 and NSP2, while present in Arabidopsis, are absent in these Proteales species (Figure 5). Secondly, SYMRK (DMI2) and CCaMK (DMI3) are absent in Arabidopsis and most non‐host plants (Delaux et al., 2014), but present in Proteales species (Figure 5). In addition, CCaMK also has additional copies derived from the Proteaceae WGD event (Figure S8). Because previous studies have reported a role of NSP1 and NSP2 in strigolactone biosynthesis (Liu et al., 2011), a non‐symbiotic role for SYMRK and CCaMK in Proteales species would not be unconceivable. Our analysis proves definitively (and for the first time) that ‘AM‐conserved’ genes were lost in Proteales, and explains the inability of forming this symbiosis in Proteaceae species. Whether the loss of these key genes, which ultimately caused the loss of AM symbiosis, was followed by (or preceded by) the emergence of cluster roots (i.e. an alternative nutrient uptake strategy) remains unclear.
Figure 5.
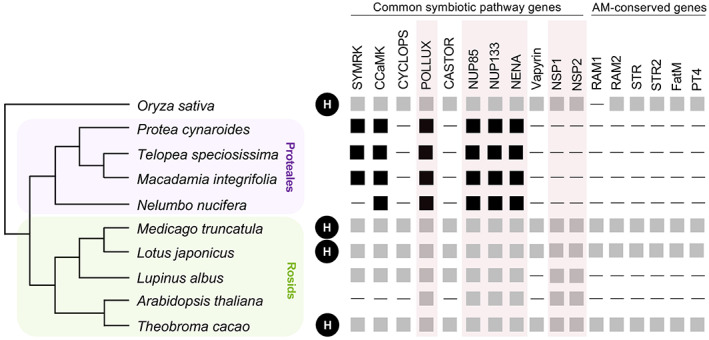
Loss of arbuscular mycorrhizal (AM)‐conserved genes in the order Proteales.
Homologs of common symbiotic pathway (CSP) genes are present in both host (H) and non‐host species. In contrast, homologs of ‘AM‐conserved’ genes were lost in known non‐hosts (Lupinus albus and Arabidopsis thaliana) as well as in the Proteales species indicated here. Boxes indicate presence, with gray indicating presence based on previous studies. Lines indicate absence. Genes indicated by a transparent red box are considered ‘conserved’ genes based on available literature (Delaux et al., 2014).
FA biosynthesis and terpene biosynthesis in Protea cynaroides
We identified gene families related to FA biosynthesis using the 30 known genes in A. thaliana as queries. The activity of acetyl‐CoA carboxylase (ACC), which catalyzes the committed and rate‐limiting step of de novo FA synthesis, includes four subunits: biotin carboxyl carrier protein, biotin carboxylase, and the α‐ and β‐subunits of carboxyltransferase. In addition to the ACC enzyme, other key catalytic components of FA biosynthesis belong to the 3‐ketoacyl‐ACP synthase (KAS) family, members of which include KASI for the elongation of FA chains from enoyl‐ACP (4:0‐ACP) to palmitoyl‐ACP (16:0‐ACP), KASII (or FAB1) for the final extension to 18:0‐ACP, and KASIII for the initial combination of acetyl‐CoA and malonyl‐ACP. KASII activity is therefore a major determinant of the ratio of C18 to C16 FAs in plant cells. Finally, there are the three sets of FA desaturases (FADs and SAD). In addition to those genes directly involved in FA biosynthesis, there are others involved in the transmembrane transport of FAs from the plastid to the ER, including acyl‐CoA binding proteins, the FATTY ACID EXPORT (FAX) gene and various long‐chain acyl‐CoA synthetases.
We identified 47 genes related to FA chains elongation, desaturation and intermembrane transport in P. cynaroides (Table S5). Based on copy number, there indeed appear to be more genes associated with FA biosynthesis in P. cynaroides than in A. thaliana, especially KAS genes. There were eight members (four KASI, two KASII and two KASIII) in P. cynaroides, and only three members in Arabidopsis (Figure S9). All of these expanded KAS genes appeared to reside within larger syntenic blocks, suggesting that these copies likely originated in the WGD (Figure S9). Furthermore, FAD (two FAD2 and two FAD3 members) and SAD genes (four members) are also expanded in P. cynaroides (Figure S10). The duplicates of FAD2 and FAD3 also reside within the syntenic blocks, whereas the three more copies of SAD are the tandem duplications.
Previous studies have demonstrated terpene (volatile isoprenoid) concentrations are assumed to influence plant flammability (De Lillis et al., 2009; Dewhirst et al., 2020; Myburg et al., 2014; Ormeño et al., 2009). Fire‐prone species likely have higher terpene abundance and are intrinsically the most flammable. A total of 39 TPS genes, assigned to six gene families, were identified in P. cynaroides (Table S6). Notably, the TPS‐b gene family, which are either monoterpene synthases or isoprene synthases, significantly expanded in P. cynaroides (18 members) compared with Arabidopsis (six members; Figure 4c; Table S6). Beyond that, P. cynaroides has five TPS‐c genes (one TPS‐c gene in A. thaliana and three in O. sativa), which catalyzes the conversion of geranylgeranyl pyrophosphate to copalyl pyrophosphate of gibberellin biosynthesis and five TPS‐g genes (Figure S10).
DISCUSSION
The CFR and the fynbos biome represent ideal environments in which to study multiple dimensions relating to adaptation, evolution, diversification, threat and rates of change, fundamental biology such as cluster root formation, as well as possibly convergent adaptations, such as sclerophylly, reseeding/resprouting life history strategies and pollination syndromes. A similar Mediterranean climate occurs in South West Australia, which hosts numerous species and genera of the Proteaceae, thus enabling inter‐continental comparisons. The fynbos Proteaceae play ecologically important functional roles – being the dominant overstory component of fynbos vegetation, and comprising more than 300 species with close to 90% endemicity. Additionally, they play a symbolic role as keystone species, due to their horticultural value as well as iconic status in society for conservation of this biome (Schurr et al., 2012). Genome sequences from Proteales representatives to date have been extremely limited and included only the Sacred Lotus N. nucifera (Gui et al., 2018; Ming et al., 2013), as well as a handful of Proteaceae members native to Australia (Macadamia spp., T. speciosissima) and New Zealand (Knightia excelsa). For these reasons, and to begin to understand the unique adaptation and adaptability of the Proteaceae to their representative climates and specific biomes, sequencing genomes in this lineage particularly from the CFR can inform not only fundamental questions for plant genome evolution and adaptation, but also for understanding implications for both plant ecology and biotechnology in the context of climate change. Apart from their striking inflorescence and flower morphology, the Proteaceae are perhaps best known for possessing cluster or ‘Proteoid’ roots for phosphorus acquisition – an adaptation for growth in nutrient‐poor soils. Our comparison of the P. cynaroides genome and other previously sequenced Proteales genomes demonstrates a genetic basis for the inability of all extant Proteaceae to form AM symbioses genes. The genome also empowers research into understanding the origin and emergence of cluster roots as an alternative nutrient uptake strategy in the family, and a basis for comparative evo‐devo work with parallel evolution of phosphorus‐scavenging roots in other lineages.
Finally, with the Proteaceae being a keystone family and one of the major components of the fynbos biome (along with Ericoid and Restoid components), as well as the many species within the family being of major horticultural significance, it is our hope that the P. cynaroides genome (and that of future fynbos species) enables the next wave of molecular research that builds on the foundational wealth of adaptation, speciation, ecology and eco‐physiology research, to inform both conservation and future biotechnology and synthetic biology research on the African continent.
EXPERIMENTAL PROCEDURES
DNA extraction
We collected three whole young flower buds of approximately 6 cm each from three P. cynaroides horticultural variety ‘Little Prince’ for DNA extraction and genome sequencing. The QIAGEN Genomic‐tip 100/G column (QIAGEN, Hilden, Germany) was used for DNA extraction. The tissue sample was grounded into a fine powder in liquid nitrogen. A total of 2.5 g of ground tissue powder was resuspended in a 50‐ml conical centrifuge tube containing 25 ml lysis buffer (10 mm Tris–HCl pH 7.9; 20 mm EDTA; 1% Triton X‐100; 500 mm Guanidince‐HCl; 200 mm NaCl), to which 20 mg of cellulase (Sigma‐Aldrich) and 100 mg of lysing enzymes from Trichoderma harzianum (Sigma‐Aldrich, St. Louis, MO, USA) was added. The tube was mixed by vortexing at maximum speed for 20 sec, followed by an incubation step at 42°C for 1.5 h. After the first incubation step, 5 μl of RNase A (7000 units ml−1; QIAGEN) was added and the tube was incubated at 37°C for 30 min, followed by an additional incubation step at 50°C for an hour. Proteinase K (Sigma‐Aldrich) was added to the digestion mixture (0.8 mg ml−1) and the tube was incubated at 50°C for 2 h. All incubation steps were done with constant gentle agitation. After the enzymatic digestion steps, the tube was centrifuged for 20 min at 7500 g at room temperature. The supernatant was collected and loaded onto a QIAGEN Genomic‐tip 100/G column. The column was washed and DNA eluted according to the manufacturer's protocol. The quality of the extracted DNA was assessed using agarose gel electrophoresis and concentration quantified using the Qubit™ dsDNA BR assay kit (ThermoFisher Scientific, Waltham, MA, USA).
Illumina and nanopore sequencing
We performed both short‐read Illumina sequencing and long‐read Nanopore sequencing to assemble the genome. Illumina sequencing was done by Macrogen (Seoul, Korea) on the HiSeq X platform. A library with a 550‐bp median insert size was prepared using the TruSeq DNA PCR Free kit (Illumina, San Diego, CA, USA) and ran on the sequencer to obtain 151‐bp paired‐end reads. Nanopore sequencing was carried out on the MinION Mk1B sequencer (Oxford Nanopore, Oxford, UK). We used the Genomic DNA by Ligation (SQK‐LSK109) kit to generate sequencing libraries following the manufacturer's protocol and performed nanopore sequencing on the R9.4.1 flow cells. We obtained raw data from all MinION runs and performed base calling with Guppy (v2.3.7 or v4.0.14) after the runs have been completed.
Hi‐C library construction and sequencing
We used the restriction enzyme‐free Dovetail® Omni‐C® Kit (Cantata Bio, Scotts Valley, CA, USA) to construct a Hi‐C library. Young leaves (1–2 weeks old) were collected and used as starting material for the construction of the Hi‐C library following the Omni‐C™ proximity ligation protocol for non‐mammalian samples version 1.0. Briefly, 300 mg of flash‐frozen leaf tissue was ground into a fine powder in liquid nitrogen. Next, the chromatin was crosslinked in formaldehyde. The crosslinking solution containing nuclei was washed and filtered through a Steriflip® filter 20‐μm unit (Millipore, Burlington, MA, USA) to remove cell debris. In situ nuclease digestion was carried out according to the protocol. The digestion profile was assessed by means of fragment analysis on the TapeStation (Agilent, Santa Clara, CA, USA), and the digested sample with optimal digestion profile was taken into proximity ligation and library construction according to the instructions from the protocol. The constructed library was sequenced on the Illumina NovaSeq 600 platform to obtain around 45 × coverage of the expected haploid genome size (about 185 million paired‐end, 151‐bp reads).
Genome assembly and assessment
We trimmed the Illumina data using Trimmomatic (Bolger et al., 2014) (v0.38) and Nanopore data using Porechop (https://github.com/rrwick/Porechop) before using them for further analysis. We used GenomeScope 2.0 (Ranallo‐Benavidez et al., 2020) to estimate genome size, ploidy and heterozygosity with Illumina paired‐end data. We assembled the genome using nanopore data with Flye (Kolmogorov et al., 2019; v2.8.1). The assembled scaffolds obtained from Flye were polished three rounds with Racon (Vaser et al., 2017; v1.3.1), followed by one with Medaka (v1.0.3, https://github.com/nanoporetech/medaka), both making use of the trimmed nanopore reads mapped to the genome with Minimap2 (Li, 2018). Next, we used Purge Haplotigs (v1.0.2; Roach et al., 2018) to identify and remove redundant scaffolds representing different haplotypes. The curated assembly obtained with Purge Haplotig was polished twice with Pilon (Walker et al., 2014; v1.23) and then once with Racon (v1.3.1), both making use of paired‐end Illumina data mapped to the assembled scaffolds with BWA (Li & Durbin, 2009; v0.7.17). Finally, the assembly was filtered to remove any contigs that had less than 10% median Illumina data coverage as these could represent artifacts due to the noisy nanopore data. We used BUSCO (Manni et al., 2021; v4.0.6) to evaluate the genome completeness of the intermediate results and also that of the final assembly.
Reads obtained from Hi‐C sequencing were preprocessed with adapters and low‐quality reads removed. Hi‐C reads were mapped to the contig level assembly using BWA (v0.7.17) and the BAM alignment was used as input for Juicer (v1.9.8). The contigs were clustered, ordered and oriented using 3D‐DNA (Dudchenko et al., 2017; v180922). Juicebox (Durand et al., 2016; v1.11.08) was used to visualize the contact matrix, misassembled scaffolds were manually corrected based on contact frequency.
Transcriptome sequencing
For Illumina RNA sequencing, flash frozen tissue from flower buds, leaves and stems from three individual P. cynaroides variety Little Prince were ground and sent to Novogene (Beijing, China) for RNA extraction and sequencing. Paired‐end complementary cDNA libraries with insert size of 150 bp were constructed and sequenced using Illumina HiSeq 2500 instrument.
Repeat annotation
A repeats library of P. cynaroides genome was ab initio constructed using RepeatModeler (Flynn et al., 2020; v2.0.1; http://www.repeatmasker.org/RepeatModeler). The consensus TE sequences generated by RepeatModeler software were used as a repeats library in RepeatMasker (v4.1.1; http://www.repeatmasker.org) for repetitive element identification in the P. cynaroides genome. TEs not classified by RepeatModeler were analyzed using DeepTE (Yan et al., 2020). A preliminary list of candidate LTR‐RT was generated using LTR_FINDER_parallel (Ou & Jiang, 2019; v1.1) and LTR_harvest (Ellinghaus et al., 2008; v1.5.9) with default parameters. The identification of high‐quality intact LTR‐RTs and the calculation of insertion age for intact LTR‐RTs were carried out using LTR_retriever (Ou & Jiang, 2018; v2.8). We calculate synonymous substitutions per site per year (r) in P. cynaroides through Equation (1):
| (1) |
using the absolute dating result of WGD in P. cynaroides and the peak value of its Ks distribution, as the average level of nucleotide substitution in intergenic regions (‐u) was ~twofold higher than that of synonymous substitution in coding regions of genes (Ma & Bennetzen, 2004), thus we set ‐u 5.30 e‐9 parameter for LTR_retriever to infer the insertion time of LTR.
Structural and functional annotation
We adapted a combination of three strategies that include homology‐based predictions, ab initio predictions and transcriptome‐assisted predictions to annotate the protein‐coding genes in our genome assembly. For the homology‐based predictions, the protein sequences of A. thaliana, Populus trichocarpa, N. nucifera and O. sativa were used as query sequences to search the reference genome using TBLASTN (Camacho et al., 2009; v2.6.0) with a cutoff E‐value of 1e‐5, and regions mapped by these query sequences were subjected to Exonerate (Slater & Birney, 2005; v2.4.0) to predict gene models. For ab initio predictions, BRAKER2 (Brůna et al., 2021; v2.1.2) was used on the soft‐masked genome assembly with the incorporation of RNA‐Seq data for gene model training. To achieve transcriptome‐assisted predictions, the RNA‐Seq data were assembled using Trinity (Grabherr et al., 2011) with (−‐genome_guided_max_intron 500000 ‐‐genome_guided_bam ‐‐min_kmer_cov 4) reference guidance. The assembled transcripts were subject to the PASA (Haas, 2003) pipeline (v2.4.1) to improve the gene structures. To finalize the gene set, EvidenceModeler (Haas et al., 2008; v1.1.1) was employed to combine all the predictions to produce the non‐redundant gene set, and the gene annotation results were evaluated by BUSCO. Non‐coding RNAs were identified using Rfam (Kalvari et al., 2021; v14.7) and Infernal (Nawrocki and Eddy, 2013; v1.1.2). Putative gene function was identified using InterProScan with different databases, including PFAM, Gene3D, PANTHER, CDD, SUPERFAMILY, ProSite and GO.
WGD identification and dating
Ks‐based age distributions were constructed as previously described by Vanneste et al. (2013). In brief, an all‐against‐all protein sequence similarity search was performed using BLASTP with an E‐value cutoff of 1 × 10−10 to construct the paranome (the entire collection of duplicated genes in a genome), after which gene families were built with the mclblastline pipeline (Enright et al., 2002; v10‐201; http://micans.org/mcl). Each gene family was aligned using MUSCLE (Edgar, 2004; v3.8.31), and Ks estimates for all pairwise comparisons within a gene family were obtained through maximum likelihood estimation using the CODEML program (Goldman & Yang, 1994) of the PAML package (Yang, 2007; v4.4c). Gene families were then subdivided into subfamilies for which Ks estimates between members did not exceed a value of 5.
To correct for the redundancy of Ks values [a gene family of n members produces n(n − 1)/2 pairwise Ks estimates for n − 1 retained duplication events], a phylogenetic tree was constructed for each subfamily using PhyML (Guindon et al., 2010) under default settings. For each duplication node in the resulting phylogenetic tree, all m Ks estimates between the two child clades were added to the Ks distribution with a weight of 1/m (in which m is the number of Ks estimates for a duplication event), so that the weights of all Ks estimates for a single duplication event summed to 1.
Phylogenetic dating of this WGD event was performed as previously described (Cai et al., 2015; Vanneste, Baele, et al., 2014). In brief, paralogous gene pairs located in duplicated segments (so‐called anchor genes, anchor pairs or anchors) and duplicated pairs lying under the WGD peak (so‐called peak‐based duplicates) were collected for phylogenetic dating. Anchors, assumed to be corresponding to the most recent WGD, were detected using i‐ADHoRe (Fostier et al., 2011; Proost et al., 2012; v3.0). We selected anchor pairs and peak‐based duplicates present under the P. cynaroides WGD peak and with Ks values between 0.2 and 0.7 for absolute dating. For each supposed WGD paralogous pair, an orthogroup was created that included the two paralogs plus several orthologs from other plant species as identified by InParanoid (Ostlund et al., 2010; v4.1) using a broad taxonomic sampling: one representative ortholog from the order Cucurbitales, two from the Rosales, two from the Fabales, two from the Malpighiales, two from the Brassicales, one from the Malvales, one from the Solanales and two from the Poales, and one from either Musa acuminata (D'Hont et al., 2012; Zingiberales) or Phoenix dactylifera (Al‐Dous et al., 2011; Arecales). In total, 1000 orthogroups based on anchors and 993 orthogroups based on peak‐based duplicates could be collected. The node joining the two P. cynaroides WGD paralogs was then dated using the BEAST (Drummond et al., 2012; v1.7) package under an uncorrelated relaxed clock model and a LG + G (four rate categories) evolutionary model.
A starting tree with branch lengths satisfying all fossil prior constraints was created according to the consensus APG IV phylogeny (Chase et al., 2016). Fossil calibrations were implemented using log‐normal calibration priors on the following nodes: the node uniting the Malvidae based on the fossil Dressiantha bicarpellata (Gandolfo et al., 1998) with prior offset = 82.8, mean = 3.8528 and SD = 0.5 (Beilstein et al., 2010); the node uniting the Fabidae based on the fossil Paleoclusia chevalieri (Crepet & Nixon, 1998) with prior offset = 82.8, mean = 3.9314 and SD = 0.5 (Xi et al., 2012); the node uniting the monocots based on fossil Liliacidites (Ramírez et al., 2007) with prior offset = 93.0, mean = 3.5458 and SD = 0.5 (Janssen & Bremer, 2004); and the root with prior offset = 124, mean = 4.0786 and SD = 0.5 (Smith et al., 2010). The offsets of these calibrations represent hard minimum boundaries, and their means represent locations for their respective peak mass probabilities in accordance with some recent and most taxonomically complete dating studies available for these specific clades (Clarke et al., 2011).
A run without data was performed to ensure proper placement of the marginal calibration prior distributions (Heled & Drummond, 2012). The MCMC for each orthogroup was run for 10 million generations, sampling every 1000 generations resulting in a sample size of 10 000. The resulting trace files of all orthogroups were evaluated manually using Tracer (Drummond et al., 2012; v1.5) with a burn‐in of 1000 samples to ensure proper convergence (minimum effective sample size for all statistics at least 200). In total, 1859 orthogroups were accepted and absolute age estimates of the node uniting the WGD paralogous pairs based on both anchor pairs and peak‐based duplicates were grouped into one absolute age distribution, for which kernel density estimation and a bootstrapping procedure were used to find the peak consensus WGD age estimate and its 90% CI boundaries, respectively. More detailed information is available in Vanneste, Baele, et al. (2014).
Phylogenetic tree construction and estimation of divergence times
To retrieve the evolutionary history of Proteaceae, we chose two representative species in Grevilleoideae that were available [M. integrifolia (Lin et al., 2022) and T. speciosissima (Chen et al., 2022)] and our P. cynaroides (Proteoideae), as well as 16 other species representing extended diversity within Eudicot (Table S2). OrthoFinder (Emms and Kelly, 2019; v2.3) was used to identify orthologous groups. All‐versus‐all BLASTP with an E‐value cutoff of 1e‐05 was performed and orthologous genes were clustered using OrthoFinder. Single‐copy orthologous genes were extracted from the clustering results. MAFFT (Katoh and Standley, 2013) with default parameters was used to perform multiple sequence alignment of protein sequences for each set of single‐copy orthologous genes, and to transform the protein sequence alignments into codon alignments. Poorly aligned or divergent regions were removed from the multiple sequence alignment results using trimAl (Capella‐Gutiérrez et al., 2009). The resulting codon alignments from all single‐copy orthologs were then concatenated into one supergene for species phylogenetic analysis. A maximum‐likelihood phylogenetic tree of single‐copy protein alignments and codon alignments was constructed using IQ‐TREE (Nguyen et al., 2015) with the GTR + G model and 1000 bootstrap replicates. Divergence times between the 19 plant species were estimated using MCMCtree from the PAML package (Yang, 2007) under the GTR + G with reference speciation times of 110 MYA for the divergence between Papaveraceae and Ranunculaceae (Guo et al., 2018), 116–126 MYA for the divergence time between asterids and rosids (Li et al., 2019), 95–106 MYA for the divergence time between Coffea canephora and Helianthus annuus from Timetree (Kumar et al., 2017), 82–109 MYA for the divergence time between Fumarioideae and Papaveroideae (Xu et al., 2022), and 7.7 MYA for the divergence time between Pa. somniferum and Pa. rhoeas (Yang et al., 2021). We used MCMCTree to obtain 10 000 trees from the posterior sampling every 150 iterations after a burn‐in of 500 000 iterations. We compared two independent runs with each other to verify convergence and with a run of the MCMC algorithm under the prior alone to compare the posterior distribution for the node ages to the effective prior implied by the fossil calibrations.
Genome and gene family evolution
Syntenic analysis of genomes was performed using MCScanX with parameters ‘‐s 10’ and the Circos figures were drawn using Tbtools (Chen et al., 2020). CAFE5 (Mendes et al., 2020) was used to identify the expansion and contraction of gene families following divergence predicted by the phylogenetic tree above. TBtools was also used to determine the enrichment of GO terms in expanded and contracted families.
For MADS‐box genes, hidden Markov Model (HMM) was employed to identify the MADS‐box genes in P. cynaroides and M. integrifolia genomes. The HMM profile of the SRF‐TF domain (PF00319) was obtained from the Pfam (Mistry et al., 2021) database. To acquire the P. cynaroides and M. integrifolia MADS‐box genes, the HMM profile was used to search against the local protein database by HMMER software with E‐value < 1e‐5. MADS‐box protein sequences of A. thaliana and O. sativa were obtained by the same method above. Subsequently, all candidate proteins in these four species were aligned using MAFFT and concatenated for phylogenetic analysis.
Genes linked to AM symbiosis were used as queries for homologous gene searches in Arabidopsis (non‐host plant representative), Th. cacao and O. sativa (host plant representatives) and Proteales species investigated, with an E‐value of 1e‐5. MAFFTMultiple sequence alignment of gene families was conducted using MAFFT (v7.453) with default parameters, and a maximum‐likelihood phylogenetic tree was constructed using Figtree (v1.7) with JTT + R model and 1000 bootstrap replicates (Figure S11), then we used MEME suite (https://meme‐suite.org/meme/) to identify the conserved motifs for each gene.
AUTHOR CONTRIBUTIONS
EM, NB and YVdP designed and supervised the research. TAD conducted all genomic DNA preparation, and generated Nanopore, illumina and Omni‐C data. DR conducted all RNA preparation and generated RNA‐Seq data. TAD and JC performed genome assembly and evaluation. JC conducted genome annotation, synteny analysis and phylogenomic analysis. JC and XM interpreted the WGD events and performed the analysis of gene families. CS and XM performed the investigation of AMS genes. JC, TAD, CS and XM prepared figures and tables. JC, TAD, CS, XM, NB, ZL, YVdP and EM wrote the paper. All authors read and approved the final version of this manuscript.
CONFLICT OF INTEREST
The authors declare that they have no conflicts of interest associated with this work.
Supporting information
Figure S1. Genome profiling with GenomeScope2 using Illumina short‐read data. P. cynaroides is estimated to have a haploid genome size of 1.18 Gb with a 1.07% genome‐wide heterozygosity level.
Figure S2. BUSCO scores were obtained from runs with the embryophyta_odb10 dataset (n = 1614) on the intermediate stages and the final assembly.
Figure S3. Repeat content of the assembly showing the relative proportions of the DNA element, long terminal (LTR), long interspersed (LINE), and other and unclassified repeats.
Figure S4. Insertion time of Gypsy and Copia in P. cynaroides, M. integrifolia and N. nucifera.
Figure S5. Dot plot comparing P. cynaroides, M. integrifolia and T. speciosissima with Aristolochia fimbriata.
Figure S6. Syntenic relationships comparing P. cynaroides, M. integrifolia and T. speciosissima with V. vinifera.
Figure S7. The conserved motifs of NUP85, NUP133, POLLUX/DMI1 and NENA using MEME suite.
Figure S8. The duplications of CCaMK (DMI3) in Proteaceae species derived from the whole‐genome duplication event.
Figure S9. Reconstruction of metabolic pathways involved in fatty acid biosynthesis and terpene biosynthesis in P. cynaroides. (a) GO enrichment of 1345 expanded families in P. cynaroides (P < 0.05). (b) Fatty acid synthesis pathway in P. cynaroides. Number in red means the gene number in P. cynaroides, number in black means the gene number in Arabidopsis. (c) Terpenoid biosynthesis pathway in P. cynaroides. Number in red means the gene number in P. cynaroides, number in black means the gene number in Arabidopsis. Synteny means the expanded genes in the syntenic blocks from the whole‐genome duplication. Tandem duplication means the expanded genes are tandem duplication.
Figure S10. The phylogenetic tree of TPS genes in P. cynaroides, M. integrifolia, A. thaliana and O. sativa.
Figure S11. The phylogenetic tree of common symbiotic pathway (CSP) genes involved in arbuscular mycorrhizal symbiosis. Different genes are indicated by different colors.
Figure S12. GO enrichment of retained genes after WGD.
Table S1. Statistics of repeat predict.
Table S2. Species used in this study.
Table S3. Type II MADS‐box genes.
Table S4. AMS genes investigated in this study.
Table S5. FAS genes in the P. cynaroides.
Table S6. TPS genes in the P. cynaroides
ACKNOWLEDGEMENTS
EM acknowledges funding from the Technology Innovation Agency of South Africa, the Genomics Research Institute, University of Pretoria, and from South Africa's National Research Foundation (Grant UID 116239). YVdP acknowledges funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program (No. 833522) and from Ghent University (Methusalem funding, BOF.MET.2021.0005.01). TAD acknowledges infrastructure support from Prof. Brenda D. Wingfield, made possible by the DSI‐NRF SARChI Chair in Fungal Genomics (Grant UID 98353). NB acknowledges funding from the National Research Foundation of South Africa (Grant UID 113296) and the Genomics Research Institute, University of Pretoria. The authors acknowledge that the King Protea is a cultural icon of South Africa, and that it is “an emblem of the beauty of our land and the flowering of our potential as a nation in pursuit of the African Renaissance. It also symbolizes the holistic integration of forces that grow from the earth, nurtured from above” (Government Communication and Information System, n.d.). We dedicate the completion of this genome to all past, present and future researchers working on – as well as peoples benefiting from – South African Biodiversity.
DATA AVAILABILITY STATEMENT
Protea cynaroides genome assembly and associated annotation files have been deposited at DDBJ/ENA/GenBank under the accession JAMYWD000000000. The version described in this paper is version JAMYWD010000000. Raw sequence reads (whole genome, transcriptome and Hi‐C data) generated in this study have been deposited in the Sequence Read Archive (SRA) under the BioProject PRJNA847781.
REFERENCES
- Akman, M. , Carlson, J.E. , Holsinger, K.E. & Latimer, A.M. (2016) Transcriptome sequencing reveals population differentiation in gene expression linked to functional traits and environmental gradients in the South African shrub Protea repens . The New Phytologist, 210, 295–309. [DOI] [PubMed] [Google Scholar]
- Al-Dous, E.K. , George, B. , Al-Mahmoud, M.E. , AI-Jaber, M.Y. , Wang, H. , Salemeh, Y.M. et al. (2011) De novo genome sequencing and comparative genomics of date palm (Phoenix dactylifera). Nature Biotechnology, 29, 521–527. [DOI] [PubMed] [Google Scholar]
- Anderson, C.L. , Bremer, K. & Friis, E.M. (2005) Dating phylogenetically basal eudicots using rbcL sequences and multiple fossil reference points. American Journal of Botany, 92, 1737–1748. [DOI] [PubMed] [Google Scholar]
- Barker, N.P. , Weston, P.H. , Rutschmann, F. & Sauquet, H. (2007) Molecular dating of the “Gondwanan” plant family Proteaceae is only partially congruent with the timing of the break‐up of Gondwana. Journal of Biogeography, 34, 2012–2027. [Google Scholar]
- Beilstein, M.A. , Nagalingum, N.S. , Clements, M.D. , Manchester, S.R. & Mathews, S. (2010) Dated molecular phylogenies indicate a Miocene origin for Arabidopsis thaliana . Proceedings of the National Academy of Sciences of the United States of America, 107, 18724–18728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger, A.M. , Lohse, M. & Usadel, B. (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics, 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Born, J. , Linder, H.P. & Desmet, P. (2006) Original article: the greater Cape Floristic Region. Journal of Biogeography, 34, 147–162. [Google Scholar]
- Bravo, A. , Brands, M. , Wewer, V. , Dörmann, P. & Harrison, M.J. (2017) Arbuscular mycorrhiza‐specific enzymes FatM and RAM2 fine‐tune lipid biosynthesis to promote development of arbuscular mycorrhiza. The New Phytologist, 214, 1631–1645. [DOI] [PubMed] [Google Scholar]
- Bravo, A. , York, T. , Pumplin, N. , Mueller, L.A. & Harrison, M.J. (2016) Genes conserved for arbuscular mycorrhizal symbiosis identified through phylogenomics. Nature Plants, 2, 15208. [DOI] [PubMed] [Google Scholar]
- Brůna, T. , Hoff, K.J. , Lomsadze, A. , Stanke, M. & Borodovsky, M. (2021) BRAKER2: automatic eukaryotic genome annotation with GeneMark‐EP+ and AUGUSTUS supported by a protein database. NAR: Genomics and Bioinformatics, 3, lqaa108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brundrett, M.C. & Tedersoo, L. (2018) Evolutionary history of mycorrhizal symbioses and global host plant diversity. The New Phytologist, 220, 1108–1115. [DOI] [PubMed] [Google Scholar]
- Cai, J. , Liu, X. , Vanneste, K. , Proost, S. , Tsai, W.C. , Liu, K.W. et al. (2015) The genome sequence of the orchid Phalaenopsis equestris . Nature Genetics, 47, 65–72. [DOI] [PubMed] [Google Scholar]
- Camacho, C. , Coulouris, G. , Avagyan, V. , Ma, N. , Papadopoulos, J. , Bealer, K. et al. (2009) BLAST+: architecture and applications. BMC Bioinformatics, 10, 421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon, S.B. , McKain, M.R. , Harkess, A. , Nelson, M.N. , Dash, S. , Deyholos, M.K. et al. (2015) Multiple polyploidy events in the early radiation of nodulating and nonnodulating legumes. Molecular Biology and Evolution, 32, 193–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capella‐Gutiérrez, S. , Silla‐Martínez, J.M. & Gabaldón, T. (2009) trimAl: a tool for automated alignment trimming in large‐scale phylogenetic analyses. Bioinformatics, 25, 1972–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter, R.J. (2012) Proteaceae leaf fossils: phylogeny, diversity, ecology and austral distributions. The Botanical Review, 78, 261–287. [Google Scholar]
- Chase, M.W. , Christenhusz, M.J.M. , Fay, M.F. , Byng, J.W. , Judd, W.S. , Soltis, D.E. et al. (2016) An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG IV. Botanical Journal of the Linnean Society, 181, 1–20. [Google Scholar]
- Chen, C. , Chen, H. , Zhang, Y. , Thomas, H.R. , Frank, M.H. , He, Y. et al. (2020) TBtools: an integrative toolkit developed for interactive analyses of big biological data. Molecular Plant, 13, 1194–1202. [DOI] [PubMed] [Google Scholar]
- Chen, S.H. , Rossetto, M. , van der Merwe, M. , Lu‐lrving, P. , Yap, J.S. , Sauquet, H. et al. (2022) Chromosome‐level de novo genome assembly of Telopea speciosissima (New South Wales waratah) using long‐reads, linked‐reads and Hi‐C. Molecular Ecology Resources, 22, 1836–1854. [DOI] [PubMed] [Google Scholar]
- Clarke, J.T. , Warnock, R.C.M. & Donoghue, P.C.J. (2011) Establishing a time‐scale for plant evolution. The New Phytologist, 192, 266–301. [DOI] [PubMed] [Google Scholar]
- Coetzee, J.H. & Littlejohn, G.M. (2010) Protea: a floricultural crop from the Cape Floristic Kingdom. In: Janick, J. (Ed.) Horticultural reviews. Oxford, UK: John Wiley & Sons, Inc., pp. 1–48. [Google Scholar]
- Collins, B.G. & Rebelo, T. (1987) Pollination biology of the Proteaceae in Australia and southern Africa. Austral Ecology, 12, 387–421. [Google Scholar]
- Crepet, W. & Nixon, K. (1998) Fossil Clusiaceae from the late Cretaceous (Turonian) of New Jersey and implications regarding the history of bee pollination. American Journal of Botany, 85, 1122–1133. [PubMed] [Google Scholar]
- De Lillis, M. , Bianco, P.M. & Loreto, F. (2009) The influence of leaf water content and isoprenoids on flammability of some Mediterranean woody species. International Journal of Wildland Fire, 18, 203–212. [Google Scholar]
- Delaux, P.‐M. , Varala, K. , Edger, P.P. , Coruzzi, G.M. , Pires, J.C. & Ané, J.‐M. (2014) Comparative phylogenomics uncovers the impact of symbiotic associations on host genome evolution. PLoS Genetics, 10, e1004487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewhirst, R.A. , Smirnoff, N. & Belcher, C.M. (2020) Pine species that support crown fire regimes have lower leaf‐level terpene contents than those native to surface fire regimes. Fire, 3, 17. [Google Scholar]
- D'Hont, A. , Denoeud, F. , Aury, J.M. , Baurens, F.C. , Carreel, F. , Garsmeur, O. et al. (2012) The banana (Musa acuminata) genome and the evolution of monocotyledonous plants. Nature, 488, 213–217. [DOI] [PubMed] [Google Scholar]
- Drummond, A.J. , Suchard, M.A. , Xie, D. & Rambaut, A. (2012) Bayesian phylogenetics with BEAUti and the BEAST 1.7. Molecular Biology and Evolution, 29, 1969–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudchenko, O. , Batra, S.S. , Omer, A.D. , Nyquist, S.K. , Hoeger, M. , Durand N.C. et al. (2017) De novo assembly of the Aedes aegypti genome using Hi‐C yields chromosome‐length scaffolds. Science, 356, 92–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand, N.C. , Robinson, J.T. , Shamim, M.S. , Machol, I. , Mesirov, J.P. , Lander, E.S. et al. (2016) Juicebox provides a visualization system for Hi‐C contact maps with unlimited zoom. Cell Systems, 3, 99–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar, R.C. (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Research, 32, 1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellinghaus, D. , Kurtz, S. & Willhoeft, U. (2008) LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. BMC Bioinformatics, 9, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emms, D.M. & Kelly, S. (2019) OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biology, 20, 238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enright, A.J. , Van Dongen, S. & Ouzounis, C.A. (2002) An efficient algorithm for large‐scale detection of protein families. Nucleic Acids Research, 30, 1575–1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favre, P. , Bapaume, L. , Bossolini, E. , Delorenzi, M. , Falquet, L. & Reinhardt, D. (2014) A novel bioinformatics pipeline to discover genes related to arbuscular mycorrhizal symbiosis based on their evolutionary conservation pattern among higher plants. BMC Plant Biology, 14, 333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fawcett, J.A. , Maere, S. & Van de Peer, Y. (2009) Plants with double genomes might have had a better chance to survive the Cretaceous‐Tertiary extinction event. Proceedings of the National Academy of Sciences of the United States of America, 106, 5737–5742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynn, J.M. , Hubley, R. , Goubert, C. , Rosen, J. , Clark, A.G. , Feschotte, C. et al. (2020) RepeatModeler2 for automated genomic discovery of transposable element families. Proceedings of the National Academy of Sciences of the United States of America, 117, 9451–9457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fostier, J. , Proost, S. , Dhoedt, B. , Saeys, Y. , Demeester, P. , Van de Peer, Y. et al. (2011) A greedy, graph‐based algorithm for the alignment of multiple homologous gene lists. Bioinformatics, 27, 749–756. [DOI] [PubMed] [Google Scholar]
- Gandolfo, M. , Nixon, K. & Crepet, W. (1998) A new fossil flower from the Turonian of New Jersey: Dressiantha bicarpellata gen. et sp. nov. (Capparales). American Journal of Botany, 85, 964–974. [PubMed] [Google Scholar]
- Gobbato, E. , Marsh, J.F. , Vernié, T ., Wang, E. , Maillet, F. , Kim, J. et al. (2012) A GRAS‐type transcription factor with a specific function in mycorrhizal signaling. Current Biology, 22, 2236–2241. [DOI] [PubMed] [Google Scholar]
- Goldblatt, P. & Manning, J.C. (2002) Plant diversity of the Cape region of Southern Africa. Annals of the Missouri Botanical Garden, 89, 281–302. [Google Scholar]
- Goldman, N. & Yang, Z. (1994) A codon‐based model of nucleotide substitution for protein‐coding DNA sequences. Molecular Biology and Evolution, 11, 725–736. [DOI] [PubMed] [Google Scholar]
- Gomez-Roldan, V. , Fermas, S. , Brewer, P.B. , Puech-Pagè, V. , Dun, E.A. , Pillot, J.P. et al. (2008) Strigolactone inhibition of shoot branching. Nature, 455, 189–194. [DOI] [PubMed] [Google Scholar]
- Government Communication and Information System . (n.d.) Corporate identity and branding guidelines. Government Communication and Information System. Available at: https://www.gcis.gov.za/sites/default/files/docs/resourcecentre/guidelines/corpid/3_2.pdf [Accessed 20th October 2022].
- Grabherr, M.G. , Haas, B.J. , Yassour, M. , Levin, J.Z. , Thompsom, D.A. , Amit, I. et al. (2011) Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nature Biotechnology, 29, 644–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gui, S. , Peng, J. , Wang, X. , Wu, Z. , Cao, R. , Salse, J. et al. (2018) Improving Nelumbo nucifera genome assemblies using high-resolution genetic maps and BioNano genome mapping reveals ancient chromosome rearrangements. The Plant Journal, 94, 721–734. [DOI] [PubMed] [Google Scholar]
- Guindon, S. , Dufayard, J.F. , Lefort, V. , Anisimova, M. , Hordijk, W. & Gascuel, O. (2010) New algorithms and methods to estimate maximum‐likelihood phylogenies: assessing the performance of PhyML 3.0. Systematic Biology, 59, 307–321. [DOI] [PubMed] [Google Scholar]
- Guo, L. , Winzer, T. , Yang, X. , Li, Y., Ning, Z. , He, Z. et al. (2018) The opium poppy genome and morphinan production. Science, 362, 343–347. [DOI] [PubMed] [Google Scholar]
- Haas, B.J. (2003) Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Research, 31, 5654–5666. Available from: 10.1093/nar/gkg770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas, B.J. , Salzberg, S.L. , Zhu, W. , Pertea, M. , Allen, J.E. , Orvis, J. et al. (2008) Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biology, 9, R7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison, M.J. , Dewbre, G.R. & Liu, J. (2002) A phosphate transporter from Medicago truncatula involved in the acquisition of phosphate released by arbuscular mycorrhizal fungi. Plant Cell, 14, 2413–2429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heled, J. & Drummond, A.J. (2012) Calibrated tree priors for relaxed phylogenetics and divergence time estimation. Systematic Biology, 61, 138–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, C.H. , Zhang, C. , Liu, M. , Hu, Y. , Gao, T. , Qi, J. et al. (2016) Multiple polyploidization events across Asteraceae with two nested events in the early history revealed by nuclear phylogenomics. Molecular Biology and Evolution, 33, 2820–2835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibarra-Laclette, E. , Lyons, E. , Hernández-Guzmán, G. , Pérez-Torres, C.A. , Carretero-Paulet, L. , Tien-Hao, C. et al. (2013) Architecture and evolution of a minute plant genome. Nature, 498, 94–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irish, V. (2017) The ABC model of floral development. Current Biology, 27, R887–R890. [DOI] [PubMed] [Google Scholar]
- Jaillon, O. , Aury, J.-M. , Noel, B. , Policriti, A. , Clepet, C. , Casagrande, A. et al. (2007) The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature, 449, 463–467. [DOI] [PubMed] [Google Scholar]
- Janssen, T. & Bremer, K. (2004) The age of major monocot groups inferred from 800+rbcL sequences. Botanical Journal of the Linnean Society, 146, 385–398. [Google Scholar]
- Jones, P. , Binns, D. , Chang, H.Y. , Fraser, M. , Li, W. , McAnulla, C. et al. (2014) InterProScan 5: genome‐scale protein function classification. Bioinformatics, 30, 1236–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalvari, I. , Nawrocki, E.P. , Ontiveros-Palacios, N. , Argasinska, J. , Lamkiewicz, K. , Marz, M. et al. (2021) Rfam 14: expanded coverage of metagenomic, viral and microRNA families. Nucleic Acids Research, 49, D192–D200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh, K. & Standley, D.M. (2013) MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Molecular Biology and Evolution, 30, 772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolmogorov, M. , Yuan, J. , Lin, Y. & Pevzner, P.A. (2019) Assembly of long, error‐prone reads using repeat graphs. Nature Biotechnology, 37, 540–546. [DOI] [PubMed] [Google Scholar]
- Kramer, E.M. , Holappa, L. , Gould, B. , Jaramillo, M.A. , Setnikov, D. & Santiago, P.M. (2007) Elaboration of B gene function to include the identity of novel floral organs in the lower eudicot Aquilegia . Plant Cell, 19, 750–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar, S. , Stecher, G. , Suleski, M. & Hedges, S.B. (2017) TimeTree: a resource for timelines, timetrees, and divergence times. Molecular Biology and Evolution, 34, 1812–1819. [DOI] [PubMed] [Google Scholar]
- Kwantes, M. , Liebsch, D. & Verelst, W. (2012) How MIKC* MADS‐box genes originated and evidence for their conserved function throughout the evolution of vascular plant gametophytes. Molecular Biology and Evolution, 29, 293–302. [DOI] [PubMed] [Google Scholar]
- Lambers, H. & Teste, F.P. (2013) Interactions between arbuscular mycorrhizal and non‐mycorrhizal plants: do non‐mycorrhizal species at both extremes of nutrient availability play the same game? Plant, Cell & Environment, 36, 1911–1915. [DOI] [PubMed] [Google Scholar]
- Lamont, B.B. , He, T. & Downes, K.S. (2013) Adaptive responses to directional trait selection in the Miocene enabled Cape proteas to colonize the savanna grasslands. Evolutionary Ecology, 27, 1099–1115. [Google Scholar]
- Li, H. (2018) Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics, 34, 3094–3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. & Durbin, R. (2009) Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinformatics, 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H.T. , Yi, T.S. , Gao, L.M. , Ma, P.F. , Zhang, T. , Yang, J.B. et al. (2019) Origin of angiosperms and the puzzle of the Jurassic gap. Nature Plants, 5, 461–470. [DOI] [PubMed] [Google Scholar]
- Lin, J. , Zhang, W. , Zhang, X. , Ma, X. , Zhang, S. , Chen, S. et al. (2022) Signatures of selection in recently domesticated macadamia. Nature Communications, 13, 242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linder, H.P. (2003) The radiation of the Cape flora, southern Africa. Biological Reviews of the Cambridge Philosophical Society, 78, 597–638. [DOI] [PubMed] [Google Scholar]
- Linder, H.P. (2008) Plant species radiations: where, when, why? Philosophical Transactions of the Royal Society B: Biological Sciences, 363, 3097–3105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linder, H.P. & Barker, N.P. (2014) Does polyploidy facilitate long‐distance dispersal? Annals of Botany, 113, 1175–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, W. , Kohlen, W. , Lillo, A. , Op den Camp, R. , Ivanov, S. , Hartog, M. et al. (2011) Strigolactone biosynthesis in Medicago truncatula and rice requires the symbiotic GRAS‐type transcription factors NSP1 and NSP2. Plant Cell, 23, 3853–3865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, Y. , Cui, S. , Wu, F. , Yan, S. , Lin, X. , Du, X. et al. (2013) Functional conservation of MIKC*‐Type MADS box genes in Arabidopsis and rice pollen maturation. Plant Cell, 25, 1288–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, Y. , Wang, B. , Shu, S. , Li, Z. , Song, C. , Liu, D. et al. (2021) Analysis of the Coptis chinensis genome reveals the diversification of protoberberine‐type alkaloids. Nature Communications, 12, 3276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohaus, R. & Van de Peer, Y. (2016) Of dups and dinos: evolution at the K/Pg boundary. Current Opinion in Plant Biology, 30, 62–69. [DOI] [PubMed] [Google Scholar]
- Ma, J. & Bennetzen, J.L. (2004) Rapid recent growth and divergence of rice nuclear genomes. Proceedings of the National Academy of Sciences of the United States of America, 101, 12404–12410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLean, A.M. , Bravo, A. & Harrison, M.J. (2017) Plant signaling and metabolic pathways enabling arbuscular mycorrhizal symbiosis. Plant Cell, 29, 2319–2335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manni, M. , Berkeley, M.R. , Seppey, M. , Simão, F.A. & Zdobnov, E.M. (2021) BUSCO update: novel and streamlined workflows along with broader and deeper phylogenetic coverage for scoring of eukaryotic, prokaryotic, and viral genomes. Molecular Biology and Evolution, 38, 4647–4654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendes, F.K. , Vanderpool, D. , Fulton, B. & Hahn, M.W. (2020) CAFE 5 models variation in evolutionary rates among gene families. Bioinformatics, 36, 5516–5518. Available from: 10.1093/bioinformatics/btaa1022 [DOI] [PubMed] [Google Scholar]
- Ming, R. , VanBuren, R. , Liu, Y. , Yang, M. , Han, Y. , Li, L.T. et al. (2013) Genome of the long‐living sacred lotus (Nelumbo nucifera Gaertn.). Genome Biology, 14, R41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mistry, J. , Chuguransky, S. , Williams, L. , Qureshi, M. , Salazar, G.A. , Sonnhammer, E.L.L. et al. (2021) Pfam: the protein families database in 2021. Nucleic Acids Research, 49, D412–D419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell, N. , Lewis, P.O. , Lemmon, E.M. , Lemmon, A.R. & Holsinger, K.E. (2017) Anchored phylogenomics improves the resolution of evolutionary relationships in the rapid radiation of Protea L. American Journal of Botany, 104, 102–115. [DOI] [PubMed] [Google Scholar]
- Moore, M.J. , Soltis, P.S. , Bell, C.D. , Burleigh, J.G. & Soltis, D.E. (2010) Phylogenetic analysis of 83 plastid genes further resolves the early diversification of eudicots. Proceedings of the National Academy of Sciences of the United States of America, 107, 4623–4628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myburg, A.A. , Grattapaglia, D. , Tuskan, G.A. , Hellsten, U. , Hayes, R.D. , Grimwood, J. et al. (2014) The genome of Eucalyptus grandis . Nature, 510, 356–362. [DOI] [PubMed] [Google Scholar]
- Nawrocki, E.P. & Eddy, S.R. (2013) Infernal 1.1: 100‐fold faster RNA homology searches. Bioinformatics, 29, 2933–2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen, L.T. , Schmidt, H.A. , von Haeseler, A. & Minh, B.Q. (2015) IQ‐TREE: a fast and effective stochastic algorithm for estimating maximum‐likelihood phylogenies. Molecular Biology and Evolution, 32, 268–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- One Thousand Plant Transcriptomes Initiative . (2019) One thousand plant transcriptomes and the phylogenomics of green plants. Nature, 574, 679–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ormeño, E. , Céspedes, B. , Sánchez, I.A. , Velasco‐García, A. , Moreno, J.M. , Fernandez, C. et al. (2009) The relationship between terpenes and flammability of leaf litter. Forest Ecology and Management, 257, 471–482. [Google Scholar]
- Ostlund, G. , Schmitt, T. , Forslund, K. , Köstler, T. , Messina, D.N. , Roopra, S. et al. (2010) InParanoid 7: new algorithms and tools for eukaryotic orthology analysis. Nucleic Acids Research, 38, D196–D203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou, S. , Chen, J. & Jiang, N. (2018) Assessing genome assembly quality using the LTR Assembly Index (LAI). Nucleic Acids Research, 46, e126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou, S. & Jiang, N. (2018) LTR_retriever: a highly accurate and sensitive program for identification of long terminal repeat retrotransposons. Plant Physiology, 176, 1410–1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou, S. & Jiang, N. (2019) LTR_FINDER_parallel: parallelization of LTR_FINDER enabling rapid identification of long terminal repeat retrotransposons. Mobile DNA, 10, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proost, S. , Fostier, J. , De Witte, D. , Dhoedt, B. , Demeester, P. , Van de Peer, Y. et al. (2012) i‐ADHoRe 3.0‐‐fast and sensitive detection of genomic homology in extremely large data sets. Nucleic Acids Research, 40, e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin, L. , Hu, Y. , Wang, J. , Wang, X. , Zhao, R. , Shan, H. et al. (2021) Insights into angiosperm evolution, floral development and chemical biosynthesis from the Aristolochia fimbriata genome. Nature Plants, 7, 1239–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez, S.R. , Gravendeel, B. , Singer, R.B. , Marshall, C.R. & Pierce, N.E. (2007) Dating the origin of the Orchidaceae from a fossil orchid with its pollinator. Nature, 448, 1042–1045. [DOI] [PubMed] [Google Scholar]
- Ranallo‐Benavidez, T.R. , Jaron, K.S. & Schatz, M.C. (2020) GenomeScope 2.0 and Smudgeplot for reference‐free profiling of polyploid genomes. Nature Communication, 11, 1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinten, E.Y. , Coetzee, J.H. & van Wyk, B.E. (2011) The potential of South African indigenous plants for the international cut flower trade. South African Journal of Botany, 77, 934–946. [Google Scholar]
- Roach, M.J. , Schmidt, S.A. & Borneman, A.R. (2018) Purge Haplotigs: allelic contig reassignment for third‐gen diploid genome assemblies. BMC Bioinformatics, 19, 460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy, S. , Liu, W. , Nandety, R.S. , Crook, A. , Mysore, K.S. , Pislariu, C.I. et al. (2020) Celebrating 20 years of genetic discoveries in legume nodulation and symbiotic nitrogen fixation. Plant Cell, 32, 15–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sablowski, R. (2015) Control of patterning, growth, and differentiation by floral organ identity genes. Journal of Experimental Botany, 66, 1065–1073. [DOI] [PubMed] [Google Scholar]
- Schurr, F.M. , Esler, K.J. , Slingsby, J.A. & Allsopp, N. (2012) Fynbos Proteaceae as model organisms for biodiversity research and conservation. South African Journal of Science, 108, Art. #1446 Available from: http://archive.sajs.co.za/index.php/SAJS/article/view/1446 [Google Scholar]
- Sharma, B. & Kramer, E.M. (2017) Aquilegia B gene homologs promote petaloidy of the sepals and maintenance of the C domain boundary. EvoDevo, 8, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slater, G.S.C. & Birney, E. (2005) Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics, 6, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, S.A. , Beaulieu, J.M. & Donoghue, M.J. (2010) An uncorrelated relaxed‐clock analysis suggests an earlier origin for flowering plants. Proceedings of the National Academy of Sciences of the United States of America, 107, 5897–5902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapia‐López, R. , García‐Ponce, B. , Dubrovsky, J.G. , Garay‐Arroyo, A. , Pérez‐Ruíz, R.V. , Kim, S.H. et al. (2008) An AGAMOUS‐related MADS‐box gene, XAL1 (AGL12), regulates root meristem cell proliferation and flowering transition in Arabidopsis. Plant Physiology, 146, 1182–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van de Peer, Y. , Ashman, T.L. , Soltis, P.S. & Soltis, D.E. (2021) Polyploidy: an evolutionary and ecological force in stressful times. Plant Cell, 33, 11–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van de Peer, Y. , Mizrachi, E. & Marchal, K. (2017) The evolutionary significance of polyploidy. Nature Reviews. Genetics, 18, 411–424. [DOI] [PubMed] [Google Scholar]
- van Santen, M. & Linder, H.P. (2020) The assembly of the Cape flora is consistent with an edaphic rather than climatic filter. Molecular Phylogenetics and Evolution, 142, 106645. [DOI] [PubMed] [Google Scholar]
- Vanneste, K. , Baele, G. , Maere, S. & Van de Peer, Y. (2014) Analysis of 41 plant genomes supports a wave of successful genome duplications in association with the Cretaceous‐Paleogene boundary. Genome Research, 24, 1334–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanneste, K. , Maere, S. & Van de Peer, Y. (2014) Tangled up in two: a burst of genome duplications at the end of the Cretaceous and the consequences for plant evolution. Philosophical Transactions of the Royal Society of London. Series B, Biological Sciences, 369, 20130353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanneste, K. , Van de Peer, Y. & Maere, S. (2013) Inference of genome duplications from age distributions revisited. Molecular Biology and Evolution, 30, 177–190. [DOI] [PubMed] [Google Scholar]
- Vaser, R. , Sović, I. , Nagarajan, N. & Šikić, M. (2017) Fast and accurate de novo genome assembly from long uncorrected reads. Genome Research, 27, 737–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veiga, R.S.L. , Faccio, A. , Genre, A. , Pieterse, C.M.J. , Bonfante, P. & van der Heijden, M.G.A. (2013) Arbuscular mycorrhizal fungi reduce growth and infect roots of the non‐host plant Arabidopsis thaliana . Plant, Cell & Environment, 36, 1926–1937. [DOI] [PubMed] [Google Scholar]
- Verboom, G.A. , Stock, W.D. & Cramer, M.D. (2017) Specialization to extremely low‐nutrient soils limits the nutritional adaptability of plant lineages. The American Naturalist, 189, 684–699. [DOI] [PubMed] [Google Scholar]
- Walker, B.J. , Abeel, T. , Shea, T. , Priest, M. , Abouelliel, A. , Sakthikumar, S. et al. (2014) Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One, 9, e112963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Y. , Tang, H. , Debarry, J.D. , Tan, X. , Li, J. , Wang, X. et al. (2012) MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Research, 40, e49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi, Z. , Ruhfel, B.R. , Schaefer, H. , Amorim, A.M. , Sugumaran, M. , Wurdack, K.J. et al. (2012) Phylogenomics and a posteriori data partitioning resolve the Cretaceous angiosperm radiation Malpighiales. Proceedings of the National Academy of Sciences of the United States of America, 109, 17519–17524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, Z. , Li, Z. , Ren, F. , Gao, R. , Wang, Z. , Zhang, J. et al. (2022) The genome of Corydalis reveals the evolution of benzylisoquinoline alkaloid biosynthesis in Ranunculales. The Plant Journal, 111, 217–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan, H. , Bombarely, A. & Li, S. (2020) DeepTE: a computational method for de novo classification of transposons with convolutional neural network. Bioinformatics, 36, 4269–4275. [DOI] [PubMed] [Google Scholar]
- Yang, X. , Gao, S. , Guo, L. , Wang, B. , Jia, Y. , Zhou, J. et al. (2021) Three chromosome‐scale Papaver genomes reveal punctuated patchwork evolution of the morphinan and noscapine biosynthesis pathway. Nature Communications, 12, 6030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Z. (2007) PAML 4: phylogenetic analysis by maximum likelihood. Molecular Biology and Evolution, 24, 1586–1591. [DOI] [PubMed] [Google Scholar]
- Yu, Y. , Xiang, Q. , Manos, P.S. , Soltis, D.E. , Soltis, P.S. , Song, B.‐H. et al. (2017) Whole‐genome duplication and molecular evolution in Cornus L. (Cornaceae) ‐ insights from transcriptome sequences. PLoS One, 12, e0171361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, G.Q. , Liu, K.W. , Li, Z. , Lohaus, R. , Hsiao, Y.Y. , Niu, S.C. et al. (2017) The Apostasia genome and the evolution of orchids. Nature, 549, 379–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, L. , Chen, F. , Zhang, X. , Li, Z. , Zhao, Y. , Lohaus, R. et al. (2020) The water lily genome and the early evolution of flowering plants. Nature, 577, 79–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Q. , Blaylock, L.A. & Harrison, M.J. (2010) Two Medicago truncatula half‐ABC transporters are essential for arbuscule development in arbuscular mycorrhizal symbiosis. Plant Cell, 22, 1483–1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Genome profiling with GenomeScope2 using Illumina short‐read data. P. cynaroides is estimated to have a haploid genome size of 1.18 Gb with a 1.07% genome‐wide heterozygosity level.
Figure S2. BUSCO scores were obtained from runs with the embryophyta_odb10 dataset (n = 1614) on the intermediate stages and the final assembly.
Figure S3. Repeat content of the assembly showing the relative proportions of the DNA element, long terminal (LTR), long interspersed (LINE), and other and unclassified repeats.
Figure S4. Insertion time of Gypsy and Copia in P. cynaroides, M. integrifolia and N. nucifera.
Figure S5. Dot plot comparing P. cynaroides, M. integrifolia and T. speciosissima with Aristolochia fimbriata.
Figure S6. Syntenic relationships comparing P. cynaroides, M. integrifolia and T. speciosissima with V. vinifera.
Figure S7. The conserved motifs of NUP85, NUP133, POLLUX/DMI1 and NENA using MEME suite.
Figure S8. The duplications of CCaMK (DMI3) in Proteaceae species derived from the whole‐genome duplication event.
Figure S9. Reconstruction of metabolic pathways involved in fatty acid biosynthesis and terpene biosynthesis in P. cynaroides. (a) GO enrichment of 1345 expanded families in P. cynaroides (P < 0.05). (b) Fatty acid synthesis pathway in P. cynaroides. Number in red means the gene number in P. cynaroides, number in black means the gene number in Arabidopsis. (c) Terpenoid biosynthesis pathway in P. cynaroides. Number in red means the gene number in P. cynaroides, number in black means the gene number in Arabidopsis. Synteny means the expanded genes in the syntenic blocks from the whole‐genome duplication. Tandem duplication means the expanded genes are tandem duplication.
Figure S10. The phylogenetic tree of TPS genes in P. cynaroides, M. integrifolia, A. thaliana and O. sativa.
Figure S11. The phylogenetic tree of common symbiotic pathway (CSP) genes involved in arbuscular mycorrhizal symbiosis. Different genes are indicated by different colors.
Figure S12. GO enrichment of retained genes after WGD.
Table S1. Statistics of repeat predict.
Table S2. Species used in this study.
Table S3. Type II MADS‐box genes.
Table S4. AMS genes investigated in this study.
Table S5. FAS genes in the P. cynaroides.
Table S6. TPS genes in the P. cynaroides
Data Availability Statement
Protea cynaroides genome assembly and associated annotation files have been deposited at DDBJ/ENA/GenBank under the accession JAMYWD000000000. The version described in this paper is version JAMYWD010000000. Raw sequence reads (whole genome, transcriptome and Hi‐C data) generated in this study have been deposited in the Sequence Read Archive (SRA) under the BioProject PRJNA847781.