

Abstract
Utreloxastat (PTC857) is a 15‐lipoxygenase inhibitor being developed to treat amyotrophic lateral sclerosis. This first‐in‐human study investigated the safety and pharmacokinetics of utreloxastat in healthy volunteers (N = 82) in a double‐blind, placebo‐controlled trial. The effects of a single ascending dose (100–1000 mg), multiple ascending doses (150–500 mg), and food (500 mg) on the pharmacokinetics and safety of utreloxastat were evaluated. Following single doses, the time to maximum plasma concentration (Cmax) was observed ≈4 hours after dosing and the terminal half‐life ranged from 20 to 25.3 hours. The Cmax and area under the concentration‐time curve (AUC) increased slightly over dose proportionally. Following multiple doses (once daily/twice daily), the apparent clearance reduced and terminal half‐life was ≥33 hours. There was no apparent difference of exposure following morning or evening doses. Varying diets increased the Cmax and AUCs of utreloxastat but did not alter time to Cmax. There were no gender‐based differences in exposure. Utreloxastat showed no marked safety signal following single doses up to 1000 mg and multiple doses over 14 days of 500 mg once daily or 250 mg twice daily. The results support further development of utreloxastat for the treatment of patients with amyotrophic lateral sclerosis at a 250‐mg twice‐daily dose administered with food.
Keywords: 15‐lipooxygenase inhibitor, ALS, amyotrophic lateral sclerosis, pharmacokinetic, utreloxastat (PTC857)
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease that affects the motor neuron system. ALS results in progressive paralysis and eventual death due to respiratory failure, 1 typically within 3–5 years of symptom onset. 2 The disease is estimated to affect ≈1–2.6 in 100,000 people per year worldwide. 3 Currently, only two approved treatments (riluzole and edaravone) for ALS are available in the United States, and in the European Union only riluzole is available. These therapies are aimed at slowing disease progression but do so with limited efficacy. 4
Oxidative stress is a central component of ALS and is hypothesized to be responsible for ferroptosis, an iron‐dependent oxidative mode of cell death, which contributes to the disease pathology. 2 Oxidative stress results in production of reactive oxygen species (ROS). ROS attacks polyunsaturated fatty acids contained in cellular membrane phospholipids, triggering lipid peroxidation, which leads to cell death through ferroptosis. 5
ROS upregulates 15‐lipoxygenase (15‐LO), an enzyme that typically uses polyunsaturated fatty acids to facilitate the removal of apoptotic cells and activate inflammatory processes. 6 , 7 This upregulation results in the production of lipid peroxide products that increase oxidative stress, activate proinflammatory glial cells, deplete reduced glutathione, accumulate iron, and ultimately cause ferroptosis. 7 , 8
These biochemical events, in particular the production of oxidized lipids, inflammation, oxidative stress, and depletion of reduced glutathione, are known contributors to the neuronal cell death that characterizes ALS. 9 ROS and the resultant oxidative damage are increased in samples of ALS patients and ALS animal models. 10 , 11 , 12 , 13 The role of 15‐LO in ferroptosis‐related cell death makes it a promising potential treatment target in ALS.
Utreloxastat (alkyl‐substituted cyclohexadienedione) (Figure 1) is an orally bioavailable small molecule being developed for the treatment of neurodegenerative diseases characterized by high levels of oxidative stress and mitochondrial pathology, such as ALS. Pharmacologically, utreloxastat can be classified as a redox‐active inhibitor of ferroptosis; it functions as an inhibitor of 15‐LO to reduce oxidative stress and block depletion of reduced glutathione. By inhibiting 15‐LO, utreloxastat is expected to slow or prevent neurodegeneration in ALS by preventing ferroptosis.
Figure 1.
Structure of utreloxastat.
The pharmacokinetics (PK) of utreloxastat has been evaluated in nonclinical studies of mice, rats, and monkeys. Utreloxastat was found to be orally bioavailable in all nonclinical studies, and following oral doses to mice, utreloxastat was well distributed into brain tissues with a favorable brain‐to‐plasma ratio of ≈10‐fold after time to maximum plasma concentration (tmax).
In vitro studies were conducted to understand utreloxastat metabolism and drug‐drug interaction potential. Multiple cytochrome P450 (CYP) enzymes, potentially but not limited to CYP1A2, 2B6, 2C8, 2C19, 3A4, and 4F2, involved in the metabolism of utreloxastat were investigated using pooled human liver microsomes or recombinant human CYP enzymes. In vitro utreloxastat was a weak inhibitor for CYP1A2 and 2B6 with half maximal inhibitory concentration > 5.3 µM and inhibition toward other CYP450 enzymes was minor (half maximal inhibitory concentration > 46 µM). In human hepatocytes, utreloxastat at 20 µM induced messenger RNA expression of CYP2B6 and 3A4 but not CYP1A2. Utreloxastat metabolism in rats was extensive. Following a single oral dose of 14C‐radiolabeled utreloxastat, unchanged utreloxastat accounted for <5% total radioactivity in plasma but was not detected in urine and bile. The safety of utreloxastat was evaluated in vitro and in rats and monkeys following repeat doses over 28 days. Overall, nonclinical data were favorable and support further development and evaluation of utreloxastat in humans.
The current study assessed the safety, tolerability, and PK of utreloxastat in a phase 1, 3‐part, single‐center, double‐blind, randomized, placebo‐controlled, single‐ascending‐dose (SAD), multiple‐ascending‐dose (MAD), and food‐effect (FE) study in healthy human subjects.
Methods
Study Design
The study was conducted in compliance with the principles of the Declaration of Helsinki and the current Dutch Medical Research Involving Human Subjects Act (Wet medisch‐wetenschappelijk onderzoek met mensen) and was approved by the appropriate institutional review boards. One clinical site (QPS Netherlands B.V, Groningen, Netherlands) participated in the study. The clinical protocol and informed consent statements were reviewed and approved by the Independent Ethics Committee of the Foundation Evaluation of Ethics in Biomedical Research (Stichting Beoordeling Ethiek Biomedisch Onderzoek; Assen, the Netherlands). Written informed consent was obtained from each subject before enrollment in the study.
Healthy subjects between 18 and 55 years of age were eligible to participate in the study. Subjects were excluded if they had a history of coagulopathy or fat malabsorption or had any prior medical or psychological history that could jeopardize their safety in the opinion of the investigator. Subjects were also excluded if they had dietary restrictions preventing them from adhering to the dietary requirements of the study, had a history of drug abuse, used nicotine‐containing products, or had a positive drug urine screen. Diet, exercise, caffeine, and smoking were restricted during the study. Known CYP3A inhibitors/inducers (eg, grapefruit) were not permitted.
This was a randomized, double‐blind, placebo‐controlled study in healthy volunteers. The study consisted of 3 parts: part 1 was a SAD study, part 2 was a MAD study; and part 3 was a FE study.
The starting dose of 100 mg was based on the most sensitive species’ no observed adverse effect level from 1‐month toxicology studies in rats and monkeys. The no observed adverse effect level found in rats (100 mg/kg) has a human equivalent dose of 16 mg/kg. The 100‐mg starting dose in humans represents one‐tenth of the human equivalent dose for an adult with a median body weight of 60 kg.
Utreloxastat was prepared in sesame oil (US Pharmacopeia/National Formulary) and packed in hard gelatin 50 mg capsules. Placebo to utreloxastat was sesame oil (US Pharmacopeia/National Formulary) packed in the same type of hard gelatin capsules.
Part 1: SAD
In part 1 (SAD), 40 subjects were randomized into 1 of 4 cohorts (10 subjects each) to receive either placebo treatment (n = 2) or single doses of utreloxastat at 100, 250, 500, or 1000 mg (n = 8). For each cohort, initial sentinel dosing was performed in 2 subjects (1 subject with utreloxastat and 1 subject with placebo) before the remaining subjects were dosed (7 subjects with utreloxastat and 1 subject with placebo).
Subjects received oral utreloxastat or placebo on day 1 after an overnight fast and after the consumption of a medium‐fat breakfast. The medium‐fat diet consisted of 500–800 total calories with 25%–50% of calories from fat. An example of a medium‐fat meal consisted of 3 slices of bread with 30 g of diet margarine; 25 g of jam, honey, or apple syrup; 1 slice (20 g) of meat (chicken filet, ham, or smoked beef); 1 slice (20 g) of cheese; 200 mL of demi milk; and 150 mL of decaffeinated or thein‐free tea. Subjects were released from the clinic after all study procedures had been completed on day 2 and returned to the clinic for sampling on day 3 (48 hours) and day 4 (72 hours). Escalation to the next dose level was based on a review of safety and tolerability data from the previous cohort by the safety review committee.
Part 2: MAD
In part 2 (MAD), 30 subjects were randomized into 1 of 3 cohorts (10 subjects each) to receive 14 days of treatment with either placebo (n = 2) or utreloxastat (n = 8) at doses of either 250 mg twice daily, 500 mg once daily, or 150 mg twice daily/once daily. Utreloxastat was administered with a medium‐fat diet as in part 1. For subjects receiving a twice‐daily dose, the evening dose was administered 12 hours after the morning dose with a medium‐fat diet.
In both parts 1 and 2, progression to the next dose level was based on a review of the safety and tolerability data from at least 7 evaluable subjects in the applicable cohort and all available data from the previous cohort(s).
Part 3: FE
Part 3 (FE) consisted of a 3 × 3 crossover design with randomized, sequential treatment periods of 12 healthy subjects. Subjects were randomized into 1 of 3 sequences and would receive 1 of the 3 treatments in a period with a single dose of utreloxastat 500 mg on day 1 of each period in either a fasted, low‐fat fed, or high‐fat fed condition. Subjects were released from the clinic on day 2 of each treatment period and returned to the clinic for sampling on day 3 (48 hours) and day 4 (72 hours). Each treatment period was separated by a 7‐day washout period, which could be shortened or prolonged depending on the PK results of part 1.
A utreloxastat dose of 500 mg QD was selected on the basis of the mean exposure in part 1 (SAD) that would be equivalent to or greater than the nonclinical model‐projected efficacious exposure, as measured by reduction of lipoxygenase‐mediated ferroptotic activity. The low‐fat and high‐fat diets were comparable to those suggested by guidance from the Food and Drug Administration. 14
Safety Assessment
The overall safety profile of utreloxastat in the SAD, MAD, and FE parts of the study was characterized by type, frequency, severity, timing, and relationship to study treatment of any adverse events (AEs), vital signs, laboratory abnormalities, physical examination abnormalities, Columbia‐Suicide Severity Rating Scale scores, or electrocardiogram (ECG) abnormalities. AEs were collected from screening of each part of the study until the end of that part of the study.
Pharmacokinetic Sample Collection and Analysis
Approximately 2.0 mL of blood for PK samples were collected before dosing and at 0.5, 1, 2, 3, 4, 6, 8, 10, 12, 24, 48, and 72 hours after dosing following utreloxastat oral administration on day 1 (SAD and FE) and before dosing and at 0.5, 1, 2, 3, 4, 6, 8, 10, 12, 14, 16, 18, and 24 hours on days 1 and 14 following the morning dose, with an additional predose sample on day 7 before the morning dose and additional postdose samples on day 14 at 48 and 72 hours (MAD).
The concentrations of utreloxastat in plasma were determined using validated liquid chromatography with tandem mass spectrometry (LC‐MS/MS) assays. Utreloxastat concentrations were calculated by interpolation from a calibration curve. The calibration range for quantitation of utreloxastat in human plasma was 0.5–500 ng/mL. Utreloxastat in dipotassium ethylenediaminetetraacetic acid human plasma (100 µL) and the internal standard (IS), utreloxastat‐d6, were extracted by a liquid‐liquid extraction procedure using tertbutyl methyl ether. The mixture was mixed and centrifuged, and the organic layer was transferred to a clean glass tube. After evaporation of the organic layer under nitrogen at 40 °C, the residue was reconstituted into a small amount of reconstitution solution (0.5% formic acid in 80:20, v/v acetonitrile/water). A 5‐µL sample of the reconstituted extract was injected on a high‐performance liquid chromatography system and quantified using tandem mass spectrometers. The chromatographic separation was performed using Acquity UPLC CSH Phenyl‐hexyl column (1.7 µm, 2.1 × 50 mm, Waters Corp., Milford, Massachusetts), maintained at 0.8 mL/min flow rate and using mobile phase as water and acetonitrile (60:40, v/v). The multiple reaction monitoring transition ions monitored were m/z 277.2→165.2 and m/z 283.3→168.2 for utreloxastat and the IS (utreloxastat d6), respectively. Analytes to IS peak area ratios for the standards were used to obtain a linear calibration curve using 1/x2 weighted least‐squares regression analysis. Owing to the photolabile nature of utreloxastat, the method was validated in yellow light conditions (in the absence of white light). Due to high concentrations expected in escalating‐dose cohorts in the first‐in‐human study, an additional LC‐MS/MS method with a concentration range of 400–40000 ng/mL was fully validated with methodology like the low‐range method.
The method for quantitation of utreloxastat in human urine that had been pretreated with 2% human serum albumin was validated in the range of 0.1–100 ng/mL using LC‐MS/MS. In addition to maintaining a yellow light environment, extraction was performed in glass tubes to avoid nonspecific adsorption of utreloxastat to urine collection containers. Similar chromatographic and mass spectrometric conditions were used for quantification of urine samples as for plasma specimen.
Plasma specimens were stored deep frozen at −80 °C in polypropylene containers, whereas urine specimen glass containers were stored at −20 °C until analysis instead of −80 °C to avoid the potential breaking of glass containers at ultra‐low temperature. Quality control samples were analyzed throughout the study, and their measured concentrations were used to determine between‐run and overall precision and accuracy of the assays. The lower limit of quantitation of plasma and urine assays was 0.5 and 0.1 ng/mL, respectively. The assay performance during the study was demonstrated by specificity, precision, and accuracy of plasma quality control samples, and reproducibility was demonstrated by successful performance of incurred sample reanalysis that met acceptance criteria. PK parameters were calculated using a noncompartmental model with Phoenix WinNonlin (Certara, Princeton, New Jersey). Terminal half‐life (t1/2) was calculated with at least 3 data points after tmax in the regression and reported only when the adjusted R 2 was ≥0.80 and the ratio of the area under the concentration‐time curve (AUC) from time 0 to the last measured (or measurable) concentration (AUC0‐t) to the AUC from time 0 to infinity (AUC0‐inf) was ≥0.80.
Dose proportionality was examined across dose groups in part 1 (SAD) using a power model 15 with ln‐transformed maximum plasma concentration (Cmax), AUC0‐t, and AUC0‐inf with the following equation:
where α was the intercept and β was the slope. Dose proportionality was accepted if the 90%CI constructed on the estimate of slope parameter β included 1.
Results
Subject Demographics
A total of 40 subjects (19 men, 21 women) participated in part 1 (SAD), 30 subjects (19 men, 11 women) participated in part 2 (MAD), and 12 subjects (6 men, 6 women) participated in part 3 (FE) of the study (Table 1). All subjects completed the study aside from 1 subject in part 2 (MAD) who withdrew for family reasons. The median age of participants was between 29.5 and 35.2 years for subjects in each part of the study. The majority of subjects were White (79%) and non‐Hispanic or Latino (98%).
Table 1.
Summary of Subject Demographics
| Variable/Status |
Part 1 (SAD) All Subjects |
Part 2 (MAD) All Subjects |
Part 3 (FE) All Subjects |
|---|---|---|---|
| N | 40 | 30 | 12 |
| Age, y | |||
| Mean (SD) | 29.5 (9.54) | 35.0 (8.82) | 35.2 (8.04) |
| Median (min‐max) | 27.0 (18–55) | 32.0 (21–53) | 36.0 (23–46) |
| Sex, n (%) | |||
| Female | 21 (52.50) | 11 (36.67) | 6 (50) |
| Male | 19 (47.50) | 19 (63.33) | 6 (50) |
| Ethnicity, n (%) | |||
| Hispanic or Latino | 1 (2.50) | 1 (3.33) | 0 |
| Not Hispanic or Latino | 39 (97.50) | 29 (96.67) | 12 (100) |
| Race, n (%) | |||
| Asian | 1 (2.50) | 4 (13.33) | 0 |
| Black or African American | 1 (2.50) | 6 (20) | 1 (8.33) |
| White | 36 (90) | 19 (63.33) | 10 (83.33) |
| American Indian or Alaska Native | 0 | 1 (3.33) | 0 |
| Other | 2 (5) | 0 | 1 (8.33) |
| Body weight, kg | |||
| Mean (SD) | 73.7 (11.2) | 76.1 (10.9) | 76.9 (11.8) |
| Median (min‐max) | 73.7 (52.3–101.6) | 76.7 (58.4–96.5) | 77.8 (57.1–95.5) |
Abbreviations: FE, food‐effect; MAD, multiple ascending dose; max, maximum; min, minimum; SAD, single ascending dose; SD, standard deviation.
Pharmacokinetic Results
Single Dose
Following the first dose of utreloxastat between 100 and 1000 mg, the first plasma sample with measurable utreloxastat concentration ranged from the first sampling time 0.5 hour to 4 hours after dosing (Table 2). Utreloxastat plasma tmax was ≈4 hours, with a large range from 1 to 6 hours following a single dose. The elimination of utreloxastat followed a biphasic model, with a rapid distribution t1/2 of ≈1.8 hours and a relative slower t1/2 ≈23 hours after dosing. A second concentration peak (shoulder peak) of utreloxastat was observed around 10 hours for some subjects. The likelihood of the appearance of the second peak was higher with higher utreloxastat dose (Figure 2). The elimination rate was consistent in the dose range from 100 to 1000 mg.
Table 2.
Summary of Plasma Pharmacokinetic Parameters of Utreloxastat Following a Single Oral Dose of Utreloxastat in Healthy Volunteers After a Medium‐Fat Breakfast on Day 1 (Part 1 SAD)
| Parameters a |
100 mg Mean (SD) (N = 8) b |
250 mg Mean (SD) (N = 8) |
500 mg Mean (SD) (N = 8) b |
1000 mg Mean (SD) (N = 8) |
|---|---|---|---|---|
| Cmax, ng/mL | 264 (111) | 670 (3.11) | 2310 (922) | 3930 (1680) |
| tmax, h | 4.0 (1.0, 4.0) | 4.0 (2.0, 6.0) | 3.5 (2.0, 4.0) | 4.0 (3.0, 6.0) |
| AUC0‐t, ng ⋅ h/mL | 1160 (350) | 3640 (748) | 9960 (2380) | 19 500 (5020) |
| AUC0‐inf, ng ⋅ h/mL | 1310 (322) | 3810 (748) | 10 700 (2380) | 20 600 (4960) |
| t1/2, h | 22.6 (4.9) | 20.6 (3.7) | 25.3 (8.8) | 24.8 (10.1) |
| CL/F, L/h | 81.9 (26.5) | 67.5 (11.5) | 49.4 (13.9) | 51.0 (11.7) |
| Cmax/D, ng/mL/mg | 2.64 (1.11) | 2.68 (1.24) | 4.62 (1.84) | 3.93 (1.68) |
| AUC0‐t/D, ng ⋅ h/mL/mg | 11.6 (3.50) | 14.6 (2.99) | 19.9 (4.76) | 19.5 (5.02) |
Abbreviations: AUC0‐inf, area under the concentration‐time curve from time 0 to infinity; AUC0‐t, area under the concentration‐time curve from time 0 to time t, where t is the time of the last measured (or measurable) concentration; AUC0‐t/D, dose normalized AUC0‐t; CL/F, total body clearance; Cmax, the maximum observed plasma concentration; Cmax/D, dose normalized Cmax; t1/2, apparent terminal half‐life.
Data presented are mean (SD) for all parameters except for tmax, which are median (minimum‐maximum).
One subject who received 100 mg and 1 subject who received 500 mg had an adjusted R 2 value or AUC0‐t/AUC0‐inf ratio <0.80. Their CL/F, AUC0‐inf, and t1/2 were not reportable and were not included in the summary statistics (therefore, N = 7 for these statistics).
Figure 2.
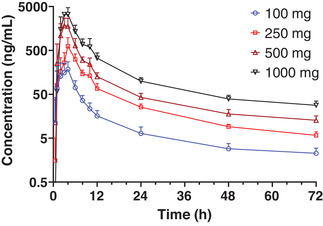
Mean (+SD) utreloxastat plasma concentration versus time following a single oral administration of utreloxastat with a medium‐fat diet in healthy subjects (part 1 SAD). SAD, single ascending dose; SD, standard deviation.
The utreloxastat exposures (Cmax, AUC0‐t, and AUC0‐inf) increased slightly over dose proportionally in the dose range from 100 to 1000 mg following a single dose of utreloxastat (Figure 3). The slope β was ≈1.225, 1.257, and 1.223 for Cmax, AUC0‐t, and AUC0‐inf, respectively, following the power model, with 90%CIs of the slope parameter β lower bound just above the value of 1.
Figure 3.
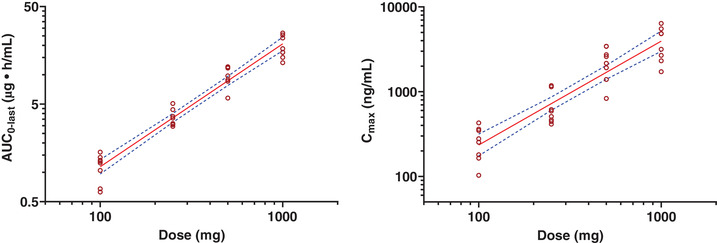
Dose linearity of utreloxastat plasma exposure following a single oral dose of utreloxastat (part 1 SAD). For AUC, β = 1.26 (1.15, 1.38), R 2= 0.9431; for Cmax, β = 1.23 (1.03, 1.42), R 2 = 0.8403. The dashed line represents the 90%CI. AUC, area under the concentration‐time curve; AUC0‐last, area under the concentration‐time curve from time 0 to the last observable time point; Cmax, maximum observed plasma concentration; SAD, single ascending dose.
Multiple Dose
Subjects in the MAD 250‐mg twice‐daily cohort received only the morning dose on day 14 by mistake. Subjects in the 150‐mg twice‐daily/once‐daily cohort received utreloxastat 150 mg twice daily from day 1 to day 6 and 150 mg once daily from day 7 to day 14. Following multiple doses of utreloxastat at doses of 250 mg twice daily, 150 mg twice daily/once daily, and 500 mg once daily for 14 days (MAD), the absorption rate and mean tmax were similar to the first doses administered (between 2.5 and 3.5 hours on day 1 following the first dose, and between 2.0 and 4.0 hours on day 14) (Figure 4, Table 3). The t1/2 was longer on day 14 with a mean value between 33.2 and 38.5 hours for evaluable subjects, who comprised less than half of the total number of subjects in the MAD study. The mean t1/2 of the remaining subjects was ≈70 hours; this statistic was not reported formally because the AUC0‐t/AUC0‐inf ratio was greater than 0.80 for those subjects.
Figure 4.
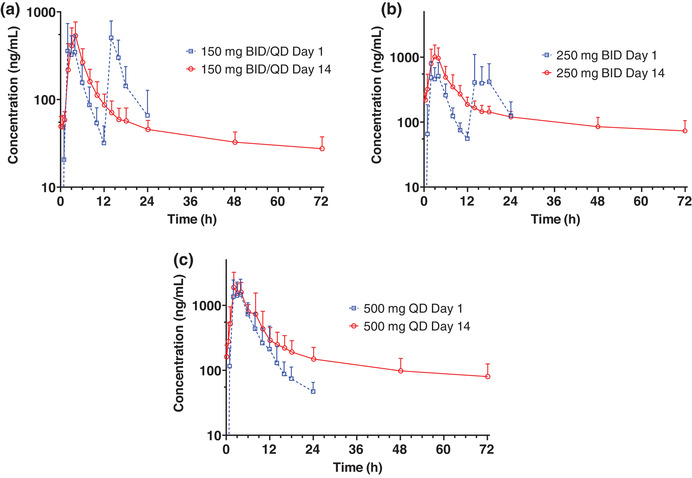
Mean (+SD) utreloxastat plasma concentration versus time following oral administration of utreloxastat at doses of (a) 150 mg, (b) 250 mg, or (c) 500 mg with a medium‐fat diet in healthy subjects on days 1 and 14 (part 2 MAD). For subjects who received the 150‐mg dose, days 1–6 were dosed twice daily (BID) and days 7–14 were dosed once daily (QD), with only the morning dose administered on day 14. For subjects who received the 250‐mg dose BID, only the morning dose was administered on day 14. MAD, multiple ascending doses; SD, standard deviation.
Table 3.
Summary of Plasma Pharmacokinetic Parameters of Utreloxastat Following Multiple Oral Doses of Utreloxastat (Part 2 MAD) After a Medium‐Fat Breakfast (Days 1 and 14)
| 250 mg Twice Daily Mean (SD) a | 500 mg Once Daily Mean (SD) a | 150 mg Twice Daily/Once Daily b Mean (SD) a | ||||||
|---|---|---|---|---|---|---|---|---|
| Parameters |
Day 1 First Dose (N = 8) |
Day 1 Second Dose (N = 8) |
Day 14 (N = 8) | Day 1 First Dose (N = 8) | Day 14 (N = 7) | Day 1 First Dose (N = 8) |
Day 1 Second Dose (N = 8) |
Day 14 (N = 8) |
| Cmax, ng/mL |
788 (213) |
876 (551) |
1300 (299) |
2270 (754) |
2630 (773) |
569 (249) |
588 (173) |
603 (262) |
| tmax, h a |
2.5 (2.0–4.0) |
4.0 (2.0–6.0) |
3.0 (1.0, 6.0) |
2.50 (1.98–6.00) |
2.0 (2.0–8.0) |
3.5 (2.0–4.0) |
2.0 (2.0–4.0) |
4.0 (2.0–6.0) |
| Ctrough, ng/mL | NA | 282 (73.9) | NA | 162 (82.1) | NA | 49.4 (14.3) | ||
| AUC0‐tau, ng ⋅ h/mL |
2650 (579) |
3400 (1400) |
5960 (1190) |
9010 (1840) |
12800 (3270) |
1780 (544) |
2290 (838) |
3320 (969) |
| AUC0‐12, ng ⋅ h/mL |
2650 (579) |
5960 (1190) |
NC | NC |
1780 (544) |
2610 (753) | ||
| AUC0‐24, ng ⋅ h/mL |
6070 (1860) |
NC |
9010 (1840) |
12,800 (3270) |
4110 (1330) |
3320 (969) | ||
| t1/2, h | NA | 35.5 (1.77) c | NA | 38.5 (3.73) d | NA | 33.2 (NA) e | ||
| Racc f | NA |
2.32 (0.639) |
NA | 1.45 (0.289) | NA | 1.50 (0.335) g | ||
| tEHL, h | NA | 14.7 (5.46) | NA | 14.1 (5.29) | NA | 16.4 (5.07) | ||
Abbreviations: AUC0‐12, area under the concentration‐time curve from time 0 to 12 hours; AUC0‐24, area under the concentration‐time curve from time 0 to 24 hours; AUC0‐inf, area under the concentration‐time curve from time 0 to infinity; AUC0‐t, area under the concentration‐time curve from time 0 to time t, where t is the time of the last measured (or measurable) concentration; AUC0‐tau, area under the concentration‐time curve within the dosing interval, calculated by linear up/log down trapezoidal method; Cmax, maximum observed plasma concentration; Ctrough, plasma concentration on day 14 before the morning dose; MAD, multiple ascending doses; NA, not applicable; NC, not calculated; Racc, accumulation ratio; R 2, coefficient of determination; SD, standard deviation; t1/2, apparent terminal half‐life; tEHL, effective half‐life; tmax, time to reach Cmax.
Data presented are mean (SD) for all parameters except for tmax, which are medians (minimum‐maximum).
From day 1 to day 6, subjects were dosed twice daily, and from day 7 to day 14, subjects were dosed once daily.
N = 3: There were 5 subjects whose adjusted R 2 value or AUC0‐t/AUC0‐inf ratio was <0.80; therefore, their t1/2 values were not reportable and were not included in the summary statistics.
N = 3: There were 4 subjects whose adjusted R 2 value or AUC0‐t/AUC0‐inf ratio was <0.80; therefore, their t1/2 values were not reportable and were not included in the summary statistics.
N = 1: There were 7 subjects whose adjusted R 2 value or AUC0‐t/AUC0‐inf ratio was <0.80; therefore, their t1/2 values were not reportable and were not included in the summary statistics.
Racc was based on AUC0‐12 on day 14/AUC0‐12 on day 1 for MAD cohort 2:1 (250 mg twice daily) and cohort 2:3 (150 mg twice daily/once daily). Racc was based on AUC0‐tau on day 14/AUC0‐tau on day 1 for MAD cohort 2:2 (500 mg once daily). AUC0‐tau = AUC0‐24 for cohort 2:2 on days 1 and 14.
N = 7: There was 1 subject whose tEHL could not be estimated.
On day 1, utreloxastat exposures were assessed following administration of 250 or 150 mg utreloxastat with a medium‐fat diet after both the morning dose (intensive PK sampling) and evening dose (reduced PK sampling due to overnight schedule).
Overall, no significant difference of exposure after the morning and evening doses was observed. Following the morning and evening dose of 250 mg of utreloxastat, the mean Cmax was 788 and 876 ng/mL, respectively, and the mean AUC within the dosing interval (AUC0‐tau) was 2650 and 3400 hour × ng/mL (Table 3). After the morning and evening dose of 150 mg of utreloxastat, the mean Cmax was 569 and 588 ng/mL, respectively, and mean AUC0‐tau was 1780 and 2290 hour × ng/mL, respectively. The slightly greater AUC0‐tau following evening dose could be due to reduced sampling and inadequate capture of the concentration‐time profiles.
The predose concentrations increased only slightly from day 7 to day 14 in the 250‐mg twice‐daily and 500‐mg once‐daily dose groups, indicating that concentrations approached a steady state following 7 days of consecutive dosing. The plasma concentration before the morning dose on day 14 increased following dose escalation from 150 mg once daily to 500 mg once daily and 250 mg daily. The accumulation ratio of AUC0‐tau or the AUC from time 0 to 12 hours after dosing on day 14 to day 1 (Racc) was ≈1.5 following 150 mg twice daily/once daily and 500 mg once daily and 2.32 following 250 mg twice daily. The effective half‐life based on Racc was ≈15 hours in the dose range following either twice‐daily or once‐daily dosing.
The clearance was reduced following multiple doses of utreloxastat over 14 days (Figure 4) for both the alpha phase of distribution and the terminal elimination phase. This was also reflected in the much longer t1/2 on day 14.
The amounts of unchanged utreloxastat excreted in urine on day 1 and day 14 following multiple doses were limited (Fe <0.23%), suggesting that the majority of absorbed utreloxastat was either eliminated by metabolism or biliary excretion.
Food Effect
The effect of food on utreloxastat was assessed in a crossover study with 12 subjects following a single dose of 500 mg of utreloxastat with a low‐ and high‐fat diet (FE) (Figure 5). The effect of a medium‐fat diet on utreloxastat from 8 subjects in the SAD study who received a single dose of 500 mg was also included in the analysis. Administration of utreloxastat with food increased the rate of absorption (Cmax) and extent of absorption (AUC0‐t and AUC0‐inf) (Table 4).
Figure 5.
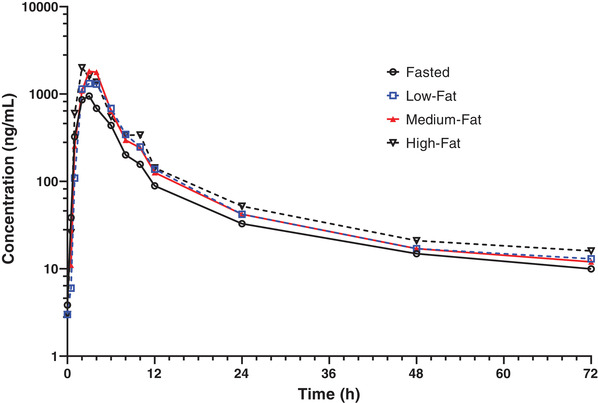
Mean utreloxastat plasma concentration versus time following a single oral administration of 500 mg of utreloxastat under various food conditions in healthy subjects (part 3, FE). FE, food effect.
Table 4.
Evaluation of the Effect of Food on the Pharmacokinetics of Utreloxastat Following a Single Oral Dose of 500 mg of Utreloxastat in the Fasted Condition (Part 3 FE) Versus Fed Condition (Part 3 FE and Part 1 SAD) in Healthy Subjects
| Fasted Condition | Fed Condition | Fed Condition (Test)/Fasted Condition (reference) | ||
|---|---|---|---|---|
| PK Parameter a | GM | GM | GMR | 90%CI |
| Low‐fat | ||||
| Cmax, ng/mL | 1143.03 | 2060.29 | 180.25 | 147.09–220.88 |
| AUC0‐t, ng ⋅ h/mL | 5891.03 | 8757.77 | 148.66 | 138.76–159.27 |
| AUC0‐inf, ng ⋅ h/mL | 6329.74 | 9234.79 | 145.90 | 135.18–157.46 |
| Medium‐fat | ||||
| Cmax, ng/mL | 1244.40 | 2111.91 b | 169.71 | 130.37–220.93 |
| AUC0‐t, ng ⋅ h/mL | 6011.19 | 9678.75 b | 161.01 | 133.98–193.50 |
| AUC0‐inf, ng ⋅ h/mL | 6418.90 | 10,414.90 c | 162.25 | 134.10–196.32 |
| High‐fat | ||||
| Cmax, ng/mL | 1143.03 | 2758.10 | 241.30 | 188.12–309.51 |
| AUC0‐t, ng ⋅ h/mL | 5891.03 | 10,748.37 | 182.45 | 167.68–198.52 |
| AUC0‐inf, ng ⋅ h/mL | 6329.74 | 11,334.88 | 179.07 | 163.10–196.61 |
Abbreviations: AUC0‐inf, area under the concentration‐time curve from time 0 to infinity; AUC0‐t, area under the concentration‐time curve from time 0 to time t, where t is the time of the last measured (or measurable) concentration; Cmax, maximum plasma concentration; FE, food‐effect; GM, geometric mean; GMR, geometric least squares mean ratio; PK, pharmacokinetic; R 2, coefficient of determination; SAD, single ascending dose
Various diets in the fed condition included a medium‐fat diet (part 1 SAD), low‐fat diet (part 3 FE), and high‐fat diet (part 3 FE).
PK parameters include back‐transformed least squares mean and confidence intervals from an analysis of variance model performed on log‐transformed values. Subjects who received the medium‐fat diet (part 1 SAD) were compared to subjects under the fasted condition (part 3 FE).
Subjects included those from the SAD study (N = 8).
N = 7. There was one subject whose adjusted R 2 value or AUC0‐t/AUC0‐inf ratio was <0.80, and therefore the AUC0‐inf was not reportable and was not included in the summary statistics.
Food did not alter the tmax or clearance of utreloxastat. The mean tmax was within the range of 2.5–3.5 hours under fasted and low‐, medium‐, and high‐fat conditions. The geometric least square mean ratios (GMRs) in the low‐fat diet condition versus the fasted condition for Cmax, AUC0‐t, and AUC0‐inf were 180.25%, 148.66%, and 145.90%, respectively. With a high‐fat diet, the GMRs for Cmax, AUC0‐t, and AUC0‐inf were 241.30%, 182.45%, and 179.07%, respectively.
In addition, 8 subjects in the SAD study were administered a single dose of 500 mg of utreloxastat with a medium‐fat diet. The GMRs for Cmax, AUC0‐t, and AUC0‐inf versus the 12 subjects in FE study under fasted condition were 169.71%, 161.01%, and 162.25%, respectively.
Sex Effect
Six men and 6 women were enrolled in the FE study. The median (min‐max) body weight of male and female subjects was 86.1 (73.4–95.5) kg and 67.8 (57.5–79.6) kg, respectively. The median (min‐max) age for male and female subjects was 40.0 (28.0–46.0) and 29.5 (23.0–42.0) years, respectively.
Overall, no apparent difference of exposure was seen between male and female subjects following a single oral dose of utreloxastat. The AUC0‐t and AUC0‐inf were comparable between male and female subjects under fasted, low‐fat, and high‐fat conditions (Figure 6). The Cmax for male and female subjects was comparable in the fasted and high‐fat conditions and lower for female subjects in the low‐fat condition.
Figure 6.
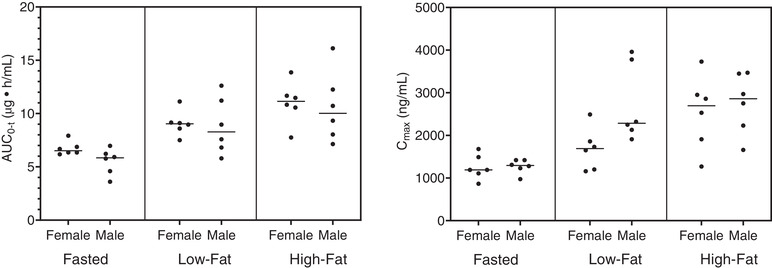
The effect of sex on utreloxastat exposure (AUC0‐t) following a single oral dose of 500 mg of utreloxastat. N = 6 each for female and male subjects. The bar represents the median value. AUC0‐t, area under the concentration‐time curve from time 0 to time t, where t is the time of the last measured (or measurable) concentration; Cmax, maximum plasma concentration.
Safety Assessment
Utreloxastat showed no marked safety signal in healthy subjects following a single dose up to 1000 mg and multiple doses for 14 days at 500 mg once daily or 250 mg twice daily. All AEs reported were mild (grade 1) and had resolved by the end of the study. No deaths or treatment‐emergent serious AEs or treatment‐emergent AEs leading to discontinuation of utreloxastat occurred over the duration of the studies. ECGs were monitored in a supine position at screening, check‐in, before dosing (before morning dose in twice daily), and 3, 6, and 8 hours after dosing (after morning dose in twice daily) on day 1 (SAD, MAD, and FE), and days 7 and 14 (MAD). No Fridericia corrected QT interval increases of ≥60 milliseconds were observed. An exploratory exposure‐response analysis indicated that there was no apparent effect of utreloxastat concentration on corrected QT interval elongation. Overall, there were no clinically abnormal ECG results.
During the MAD study, it was observed that high‐density lipoprotein (HDL) and total cholesterol decreased following multiple doses of utreloxastat. A greater reduction in HDL was observed with increasing utreloxastat dose. This decrease in HDL was fully reversible once utreloxastat administration was stopped. There were no AEs associated with these laboratory findings, which were not considered clinically significant.
Discussion and Conclusion
The current phase 1 study of utreloxastat in healthy volunteers indicated a predictable dose‐PK relationship. Utreloxastat was readily absorbed and reached Cmax ≈4 hours after dosing. The PK of utreloxastat followed a biphasic decay. A shoulder peak during the elimination phase around 10 hours after dosing was observed in some subjects during the SAD, MAD, and FE studies. The probability of this shoulder peak increased with high dose or high‐fat diet. It was hypothesized that this could be attributed to enterohepatic circulations. In doses ranging from 100 to 1000 mg, exposures to utreloxastat Cmax, AUC0‐t, and AUC0‐inf increased slightly over dose proportionally. No apparent utreloxastat exposure difference was observed between male and female subjects. The majority of absorbed utreloxastat was eliminated either by metabolism or biliary excretion. The amount of unchanged utreloxastat detected in urine was limited (Fe <0.23%).
Administration of utreloxastat with food increased both the Cmax and AUCs of utreloxastat, and maximum increase was observed with a high‐fat diet. However, the difference between various diets (low‐, medium‐, and high‐fat) was considered nonsignificant (GMRs between 170% and 241% for Cmax, between 149% and 182% for AUC0‐t, and between 146% and 179% for AUC0‐inf) considering the intersubject variability (≈40% for Cmax and 23% for AUC0‐t and AUC0‐inf).
Following repeated dosing for 14 days, accumulation of utreloxastat was observed (Racc of 1.5 for once‐daily and 2.32 for twice‐daily dosing). The effective half‐life was 15 hours following either once‐daily or twice‐daily dosing, and the apparent clearance was reduced. Both the alpha phase and the terminal phase elimination rates were reduced, and the t1/2 was ≥33 hours on day 14 compared to around 23 hours following the first dose.
The current study also indicated a favorable safety profile of utreloxastat in healthy subjects following a single dose up to 1000 mg and multiple doses for 14 days at 500 mg once daily or 250 mg twice daily. All AEs reported during the study were mild and were resolved by the end of the study.
These PK and safety data indicate that utreloxastat is an appropriate candidate for further development for the treatment of ALS. Utreloxastat inhibits 15‐LO to reduce oxidative stress and prevent the depletion of glutathione. Specifically, it inhibits ferroptosis through the reduction of the nonheme iron in 15‐LO from its active Fe3+ state to its inactive Fe2+ state. While ALS has several proposed mechanisms, oxidative stress is widely accepted as a factor in disease progression.
Preclinical studies indicate that utreloxastat is more potent in inhibiting ferroptosis than either edaravone or riluzole. These studies found that utreloxastat inhibited ferroptosis with half maximal effective concentration values ≤98 nM with little effect on cell viability and was 70 times more potent than edaravone at preventing ferroptotic death of human spinal astrocytes. In these experiments and >50 times more potent than riluzole.
ALS represents a high unmet medical need considering the limited treatment options currently available to patients and the severe symptoms and high fatality associated with the disease. Based on the safety profile and PK of utreloxastat, the dose of 250 mg twice daily to be administered with food was selected for further investigation for treatment of patients with ALS in a randomized, placebo‐controlled phase 2 study to evaluate safety and efficacy.
Conflicts of Interest
This study is funded by PTC Therapeutics, Inc. All authors are employees and stock owners of PTC Therapeutics, Inc.
Supporting information
Acknowledgements
The authors would like to thank Susie Bryan and Suzanne Keating for their editorial support.
References
- 1. Kiernan MC, Vucic S, Cheah BC, et al. Amyotrophic lateral sclerosis. Lancet (London, England). 2011;377(9769):942‐955. [DOI] [PubMed] [Google Scholar]
- 2. Park H, Yang E. Oxidative stress as a therapeutic target in amyotrophic lateral sclerosis: opportunities and limitations. Diagnostics. 2021;11(1546). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Talbott EO, Malek AM, Lacomis D. The epidemiology of amyotrophic lateral sclerosis. In: Handbook of Clinical Neurology. 3rd ed.: Elsevier; 2016:225‐238. [DOI] [PubMed] [Google Scholar]
- 4. Petrov D, Mansfield C, Moussy A, Hermine O. ALS clinical trials review: 20 years of failure. are we any closer to registering a new treatment? Front Aging Neurosci. 2017;9:68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lewerenz J, Ates G, Methner A, Conrad M, Maher P. Oxytosis/Ferroptosis—(Re‐) emerging roles for oxidative stress‐dependent non‐apoptotic cell death in diseases of the central nervous system. Front Neurosci. 2018;12:214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Snodgrass RG, Brüne B. Regulation and Functions of 15‐Lipoxygenases in Human Macrophages. Front Pharmacol. 2019;10(719). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wenzel SE, Tyurina YY, Zhao J, et al. PEBP1 wardens ferroptosis by enabling lipoxygenase generation of lipid death signals. Cell. 2017;171(3):628‐641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ye L, Stockwell B. Transforming lipoxygenases: PE‐specific enzymes in disguise. Cell. 2017;171:501‐502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Masaldan S, Bush AI, Devos D, Rolland AS, Moreau C. Striking while the iron is hot: iron metabolism and ferroptosis in neurodegeneration. Free Radical Biol Med. 2019;133:221‐233. [DOI] [PubMed] [Google Scholar]
- 10. Bogdanov M, Brown RH, Matson W, et al. Increased oxidative damage to DNA in ALS patients. Free Radical Biol Med. 2000;29(7):652‐658. [DOI] [PubMed] [Google Scholar]
- 11. Ihara Y, Nobukuni K, Takata H, Hayabara T. Oxidative stress and metal content in blood and cerebrospinal fluid of amyotrophic lateral sclerosis patients with and without a Cu, Zn‐superoxide dismutase mutation. Neurol Res. 2005;27(1):105‐108. [DOI] [PubMed] [Google Scholar]
- 12. Perluigi M, Fai Poon H, Hensley K, et al. Proteomic analysis of 4‐hydroxy‐2‐nonenal‐modified proteins in G93A‐SOD1 transgenic mice–a model of familial amyotrophic lateral sclerosis. Free Radical Biol Med. 2005;38(7):960‐968. [DOI] [PubMed] [Google Scholar]
- 13. Mitsumoto H, Santella RM, Liu X, et al. Oxidative stress biomarkers in sporadic ALS. Amyotrophic Lateral Sclerosis. 2008;9(3):177‐183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. FDA . Guidance for Industry: Assessing the Effects of Food on Drugs in INDs and NDAs — Clinical Pharmacology Considerations. In: 2022.
- 15. Hummel J, McKendrick S, Brindley C, French R. Exploratory assessment of dose proportionality: review of current approaches and proposal for a practical criterion. Pharm Stat. 2009;8(1):38‐49. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.