

Abstract
Bruton tyrosine kinase (BTK) inhibitors have taken a central role in the management of patients with Waldenström macroglobulinemia and are the only agents approved by the Food and Drug Administration (FDA) to treat these patients. Although associated with high rates of durable responses, unmet needs with BTK inhibitor therapy include indefinite duration therapy, high cost, scarcity of complete responses, and lower rates and shorter duration of response in patients with CXCR4 mutations. Herein, we review the data supporting the use of covalent BTK inhibitors, selected management issues, clinical trials with covalent BTK inhibitor combination regimens, and up‐and‐coming non‐covalent BTK inhibitors.
1. INTRODUCTION
Waldenström macroglobulinemia (WM) is an indolent lymphoma characterized by the accumulation of malignant IgM‐producing lymphoplasmacytic cells in the bone marrow (BM), lymph nodes, and other organs. MYD88 and CXCR4 somatic mutations (MYD88 MUT and CXCR4 MUT , respectively) are present in 95%–97%, and 30%–40% of patients with WM, respectively, and impact the clinical presentation and outcomes of therapy with Bruton tyrosine kinase (BTK) inhibitors. 1 , 2 , 3 , 4 , 5 , 6 , 7 , 8 Many WM patients are asymptomatic at the time of the diagnosis, and treatment should be deferred in these patients, as 20% of these patients might not need therapy for over a decade, and the life expectancy of asymptomatic patients is comparable to age and sex‐matched individuals without WM. 9 , 10 However, most WM patients will eventually need therapy.
Multiple treatment options are available for patients with WM and have classically included combinations of alkylating agents, nucleoside analogues, proteasome inhibitors, and anti‐CD20 monoclonal antibodies, which have been shown to be safe and highly effective in prospective clinical trials. 11 , 12 , 13 BTK inhibitors have been added to the armamentarium against WM and are arguably the most active monotherapy agents in these patients. The United States Food and Drug Administration (FDA) granted the first approval of any drug specific for WM to ibrutinib in 2015. Other FDA approvals include ibrutinib plus rituximab in 2018 and, more recently, zanubrutinib in 2021.
In the present document, we provide a review of the emerging data on the safety and efficacy of BTK inhibitors in the treatment of patients with WM.
2. THE BTK PATHWAY IN WM
MYD88 is a member of the Toll‐like receptor (TLR) pathway that functions as an adapter protein that triggers downstream signaling in response to pathogenic activation of TLRs. In WM, mutations in MYD88 result from missense mutations in the key signaling Toll/interleukin‐1 (IL‐1) receptor TIR domain and almost exclusively involve a switch from leucine to proline at position 265 (L265P), though more rarely non‐L265P mutations have been observed. 1 , 7 Mutations in the TIR domain of MYD88 enable auto‐assembly of a complex termed the “Myddosome,” independent of TLR activation, and trigger recruitment of BTK and IRAK4/IRAK1. 8 The recruitment of both BTK and IRAK4/IRAK1 into the Myddosome triggers NF‐kB mediated pro‐survival signaling. Notably, mutated MYD88 also leads to transcriptional up‐regulation of the SRC family member hematopoietic cell kinase (HCK) through PAX‐5 mediated transcriptional regulation. 9 , 10 HCK activates BTK enabling it to join the Myddosome complex, but also triggers extracellular regulated kinases (ERK) and AKT mediated pro‐survival signaling. 9 Importantly, mutated MYD88 through HCK also triggers SYK, thereby invoking the activation of BCR signaling through TLR‐BCR crosstalk, including AKT and STAT5. 11 , 12 The activation of ERK permits inflammatory cytokine release, which modulates microenvironmental signaling, whereas STAT5 activation results in the release of IgM. 13 , 14 These cascades provide evidence of the broad reach of mutated MYD88 that enables both pro‐survival and microenvironmental signaling. The latter may be particularly of concern in propagating resistance to drug therapy. 12 A schematic representation of mutated MYD88 signaling and the central role of BTK in WM lymphomagenesis is shown in Figure 1.
FIGURE 1.
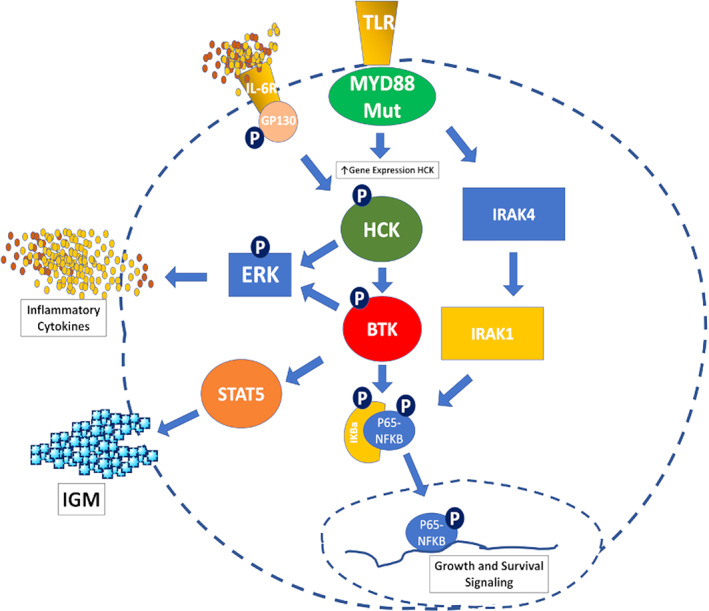
Mutated MYD88 upregulates BTK and HCK. HCK is activated by IL‐6 through IL‐6 receptor/GP‐130 signaling triggered by autocrine and paracrine IL‐6 production from the surrounding bone marrow stroma. Activated HCK triggers BTK activation that enables downstream ERK, STAT5, and NFKB activation. IRAK4 and IRAK1 are also triggered by mutated MYD88 that promote NFKB activation. [Color figure can be viewed at wileyonlinelibrary.com]
Somatic mutations in CXCR4 occur in the C‐terminal domain and are similar to those reported in the congenital WHIM syndrome. 2 , 14 In WM, over 40 different types of nonsense and frameshift mutations have been documented. 15 , 16 The somatic presence of CXCR4 mutations leads to more robust attraction of WM cells to the BM microenvironment as evidenced by the higher BM disease burden and more rare extramedullary disease involvement observed in CXCR4 mutated versus wild‐type WM. 17 Similar findings in mouse models following introduction of mutated CXCR4 WM cells have also been observed. 18 In addition, significantly less peripheral exodus of lymphocytes was observed in CXCR4 mutated versus wild‐type patients following initiation of ibrutinib, which may have also contributed to the lower activity of ibrutinib in this population in response to protective microenvironmental support. 19 Binding of SDF‐1a (CXCL12) to mutated CXCR4 also triggers exaggerated AKT and ERK signaling, which attenuate ibrutinib mediated apoptosis of CXCR4 mutated WM cells. 20 , 21 Use of CXCR4 antagonists blocks AKT and ERK signaling imparted by SDF‐1a, and sensitizes CXCR4 mutated WM cells to ibrutinib. 20
The above findings demonstrate the functional importance of BTK activation in WM pathogenesis and provide a solid scientific foundation for the development of BTK inhibitors in patients with WM.
3. THE CASE FOR IBRUTINIB
The approval of ibrutinib for WM has dramatically changed the treatment landscape of this disease and has made the vision of chemotherapy‐free treatment come to life. Data from three independent prospective clinical trials have demonstrated that ibrutinib is arguably the most effective single agent for patients with WM.
The seminal experience with ibrutinib monotherapy in 63 patients with previously treated WM reported an overall response rate (ORR) of 91% and a major response rate of 73%. 14 Importantly, the median times for minor and major responses were fast at 1 and 2 months, respectively. The median progression‐free survival (PFS) for this previously treated patient population was not reached. 15 The 5‐year PFS rate for patients with MYD88 MUT CXCR4 wildtype (CXCR4 WT ) was 70%, and 38% for patients with MYD88 MUT CXCR4 MUT . The rate of very good partial response (VGPR) increased from 16% at 18 months to 30% at 59 months of follow‐up. Similar response rates with ORR of 90% and a major response rate of 71% were reported in 31 patients with rituximab‐refractory disease in the INNOVATE Arm C study. 16 The median times for a minor and major response were 1 and 2 months, respectively. However, there was a shorter median PFS of 39 months in this heavily pretreated population. 17 The median PFS was not reached for patients with MYD88 MUT CXCR4 WT disease and was 18 months for patients with MYD88 MUT CXCR4 MUT . The single patient with MYD88 WT CXCR4 WT status progressed at 6 months. The rate of VGPR increased from 13% at 18 months to 29% at 58 months of follow‐up. A study in 30 treatment naïve patients with WM showed an ORR of 100% and a major response rate of 83%, with median times to minor and major response of 1 and 2 months, respectively. 18 The 4‐year PFS rate was 76%, 92% for patients with MYD88 MUT CXCR4 WT and 59% for patients with MYD88 MUT CXCR4 MUT disease. 19 The VGPR rate increased from 20% at 18 months to 30% at 50 months of follow‐up.
In the phase III randomized multicenter INNOVATE study, which included both treatment‐naïve and previously treated patients with WM, the combination of ibrutinib plus rituximab was associated with a higher ORR (92% and 47%) and a higher major response rate (72% and 32%) than placebo and rituximab. 20 With a median follow‐up of 50 months, the median PFS of ibrutinib plus rituximab was not reached versus 20 months for rituximab monotherapy. 21 Though patients with MYD88 MUT CXCR4 MUT had lower rates of VGPR or better than patients with MYD88 MUT CXCR4 WT disease (23% vs. 44%), the 54‐month PFS was 72% and 63% for MYD88 MUT CXCR4 WT and MYD88 MUT CXCR4 MUT patients, respectively. The results of the INNOVATE study established the superiority of ibrutinib plus rituximab of rituximab monotherapy. However, the superiority over ibrutinib monotherapy was not evaluated.
As WM is an indolent disease without curative approaches, the quality of life should not be compromised by treatment. In this respect, the low toxicity profile of a given drug will impact the acceptance by patients and treating physicians. The overwhelming utilization of ibrutinib in WM, now considered a standard treatment for this disease, clearly indicates that ibrutinib is well tolerated by most patients. In the INNOVATE study, only eight patients discontinued ibrutinib in combination with rituximab over a period of 5 years. Nevertheless, ibrutinib has side effects such as cardiac toxicity and an increased bleeding tendency in patients, although major hemorrhage occurred in only 4% of patients, similar in both treatment arms. The one fatal bleeding event occurred in the control arm. The incidence of atrial fibrillation (AF) is reported at 10% to 15%. Most of the events were mild (grade 1 or 2), and most patients continued ibrutinib with medical management of the arrhythmia.
Taken all together, ibrutinib has become a pillar in the treatment landscape for WM based on its efficacy and overall safety profile. Nevertheless, ibrutinib monotherapy has limitations, as patients with CXCR4 mutations can experience delayed, more superficial, and less durable responses. 22 , 23 In addition, ibrutinib has side effects that might limit its use in a subgroup of patients on anticoagulation or with cardiac comorbidities. Based on this, the development of second generation BTK inhibitors seemed appropriate. However, what would we expect from novel BTK inhibitors taking the place of ibrutinib? First, clinically meaningful improved efficacy in patients with WM. In this regard, it will not be sufficient to demonstrate better response rates but also a superior PFS. Substantial differences in PFS should be seen in MYD88 MUT CXCR4 MUT patients compared to ibrutinib monotherapy. Optimally, the higher efficacy should be accompanied by improved tolerability, which would mean to avoid ibrutinib‐associated side effects without adding new ones. Finally, administration should be as easy as for ibrutinib with once daily oral intake. Ibrutinib has set a very high bar, and it will not be facile to develop BTK inhibitors to outcompete ibrutinib.
4. THE CASE FOR ZANUBRUTINIB
The introduction of ibrutinib revolutionized the treatment of WM. It was an extraordinary experience as a clinical researcher to witness the dramatic fall in IgM, rise in hemoglobin, and improved quality of life of patients receiving ibrutinib. However, while serious and grade 3+ adverse events attributable to ibrutinib are uncommon, even grade 1 or 2 adverse events have a significant impact on quality of life. Recurrent or new AF occurred in 10% to 15% of patients on ibrutinib. 15 , 19 These side effects may be due to off‐target inhibition of kinases structurally related to BTK, including the epidermal growth factor receptor, tyrosine kinase expressed in hepatocellular carcinoma, SRC family kinases, and others. Zanubrutinib was designed to achieve improved therapeutic concentrations, with higher selectivity for BTK compared with other kinases and fewer off‐target effects. In clinical studies across many indolent B‐cell malignancies, including more than 250 patients with WM, this hypothesis was supported by a lower rate of toxicities seen in phase II and phase III comparisons with ibrutinib in WM and chronic lymphocytic leukemia (CLL). 24
Additional studies showed zanubrutinib's pharmacokinetics are unaffected by renal dysfunction and mild or moderate hepatic impairment, and with dose reductions, it can be administered in severe hepatic failure and with moderate or strong CYP3A inhibitors. 25 Zanubrutinib can be administered concurrently with proton pump inhibitors or acid‐reducing agents. These studies also demonstrated that zanubrutinib induces complete and sustained BTK inhibition. At a dose of 160 mg twice daily, BTK exposure levels were ~ eight‐fold higher than those observed for ibrutinib at 560 mg daily in the blood and lymph node compartments. 26 , 27 These data, along with exposure‐response analyses, were used to support the recommended dose of 320 mg once daily or 160 mg twice daily, with no difference in clinical efficacy or safety profiles.
In the monotherapy study of 77 patients, zanubrutinib was well tolerated and resulted in deep and durable responses in all molecular subtypes, including MYD88 WT . 28 ORR was 96%, and the VGPR rate was 45%, which increased over time. The estimated 3‐year progression‐free survival rate was 81%, and the overall survival rate was 85%. Discontinuation due to toxicity occurred in 13% of patients. Grade 5 AEs occurred in 6.5%, none considered treatment related. The rate of grade ≥3 AF was only 1%.
WM commonly occurs in older patients, who often have comorbidities; therefore, long‐term tolerability is important. The phase III ASPEN study compared the efficacy and safety between ibrutinib and zanubrutinib, in 201 patients with WM with MYD88 L265P disease. 29 Twenty‐nine (28%) patients on zanubrutinib and 19 (19%) patients on ibrutinib achieved a VGPR, a non‐statistically significant difference. Median PFS was not reached; 84% of patients on ibrutinib and 85% on zanubrutinib were progression‐free at 18 months. Therefore, the key discriminating factor in the selection of which BTK inhibitor to use for patients with WM is the tolerability of the BTK inhibitor. AF, contusion, diarrhea, peripheral edema, hemorrhage, muscle spasms, and pneumonia, as well as adverse events leading to treatment discontinuation, were more common with ibrutinib. With a median follow‐up of 19 months, there was a 15% rate of AF in patients on ibrutinib compared to 2% in patients on zanubrutinib. On an exposure‐adjusted basis, patients on ibrutinib experienced a 10‐fold higher incidence of AF or flutter and an approximately two‐fold increased frequency of hypertension. Diarrhea occurred in 32% versus 21%. The cumulative rate of Grade ≥3 hypertension was 11% versus 6%. Major bleeding events were lower with zanubrutinib (0.6 vs. 0.3 events/100 person‐months). In most quality‐of‐life assessments, zanubrutinib trended toward greater improvement.
Nonetheless, the incidence of neutropenia and febrile neutropenia was higher with zanubrutinib. Patients on zanubrutinib experienced a two‐fold incidence of any grade (25% vs. 12%) and grade ≥3 (20% vs. 8%) neutropenia vs. ibrutinib. More neutropenic patients in the zanubrutinib arm received granulocyte‐colony stimulating factor (G‐CSF; 47% vs. 31%). Nonetheless, grade ≥3 infection rates were similar in both arms. When considering infections, more ibrutinib‐treated patients received anti‐infective therapies (83% vs. 63%). Also, more patients treated with ibrutinib required dose reductions for adverse events (23% vs. 14%, respectively). Nine (9%) patients on ibrutinib vs. four (4%) on zanubrutinib discontinued study treatment for toxicity.
Another important cohort in the ASPEN study was the 26 patients with WM who were MYD88 WT , the largest cohort of such patients to be studied. Zanubrutinib monotherapy resulted in major responses of 50%, including 27% with VGPR. Median time to first major response was 3 months. The primary reason for discontinuation was disease progression (in 3 of 6 patients within the first 3 cycles).
In conclusion, zanubrutinib is less prone to pharmacokinetic modulation, leading to more consistent, sustained therapeutic exposures. Zanubrutinib's selectivity and pharmacodynamic profile have translated into durable responses and improved tolerability. The low discontinuation rate suggests that zanubrutinib has a favorable safety profile and is suitable for long‐term use as a single agent, representing an important new treatment option for patients with WM.
5. HOW DO WE CHOOSE BETWEEN IBRUTINIB AND ZANUBRUTINIB IN WM?
There are pros and cons for each of these BTK inhibitors in patients with WM. On the one hand, ibrutinib has a longer track record, with 7 years since FDA approval versus 1 year for zanubrutinib. Ibrutinib also provides a once‐daily, single‐pill therapy versus two pills twice daily or four pills once daily for zanubrutinib. Although no undue long‐term toxicity is expected with zanubrutinib, additional follow‐up is needed to confirm this as well as investigate the impact of zanubrutinib dosing on patients' compliance. In the ASPEN study, there were no statistical differences in response and PFS rates between these agents in patients with mutated MYD88. On the other hand, zanubrutinib is associated with a lower frequency of AF, hypertension, diarrhea, and skin rash, though with higher rates of neutropenia than ibrutinib. This neutropenia, however, is easily managed with the judicious use of G‐CSF to keep the neutrophil count >1.0 × 109/L.
Long‐term follow‐up data on the ASPEN study were presented at the 2022 Annual Meetings of the European Hematology Association and the American Society of Clinical Oncology. 30 , 31 With a median follow‐up of 43 months, the VGPR rate increased in both arms to 36% on zanubrutinib and 22% on ibrutinib (p = .02). Median PFS was not yet reached for both arms. The rates of AF, hypertension, diarrhea, hemorrhage, pneumonia, and adverse events leading to discontinuation or death were lower with zanubrutinib. The higher rate of neutropenia initially observed with zanubrutinib seems to have leveled off and evened out over time.
CXCR4 mutations were associated with lower rates of VGPR in patients treated with zanubrutinib (28%) and with ibrutinib (5%). Testing for CXCR4 mutations is encouraged in clinical trials, but its use in clinical care is not yet widespread despite the endorsement by the NCCN and the IWWM to perform CXCR4 mutational testing in patients being considered for BTK inhibitor therapy. An important limitation is access to next‐generation sequencing platforms, which are costly and may not be available in community settings.
The absence of MYD88 mutations has also been associated with lower rates of response in patients with WM treated with ibrutinib with an ORR of 60%, but 0% rates of PR and VGPR. 15 In a substudy of ASPEN, zanubrutinib was associated with an ORR of 81%, major response rate of 50%, and VGPR rate of 27% in 28 patients with MYD88 wildtype status. 32 In Arm C of the INNOVATE study, the combination of rituximab was associated with an ORR, major response rate, and VGPR rate of 82%, 73%, and 27%, respectively. 21 In the absence of direct comparisons, the findings of these studies suggest that zanubrutinib or the combination of ibrutinib and rituximab might be effective treatment options for this group of patients.
It is important to note, however, that there are substantial differences between the techniques used in prospective clinical trials for MYD88 and CXCR4 mutational testing, which can substantially impact the detection rate of these mutations, making the interpretation of the results across studies challenging. 33 , 34
Ibrutinib (with and without rituximab) and zanubrutinib are safe, effective, and reasonable treatment options for patients with WM. The longer experience and ease of administration with ibrutinib would need to be counterbalanced against the favorable side effect profile of zanubrutinib.
6. OTHER COVALENT BTK INHIBITORS
6.1. Acalabrutinib
Like ibrutinib and zanubrutinib, acalabrutinib is a covalent BTK inhibitor that has been found to be more selective than ibrutinib. A phase II multicenter clinical trial including 106 patients with WM, comprising of 14 treatment‐naïve and 92 previously treated patients, demonstrated an ORR of 93% in treatment naïve and previously treated patients. 35 Major response rates were 79% and 78% for treatment naïve and previously treated patients, respectively, although evaluation there were differences in response rates based on MYD88 mutational status. Of the 50 patients with mutational testing, 36 patients had MYD88 MUT and 14 had MYD88 WT status. Those with a MYD88 MUT had an ORR of 94%, compared with an ORR of 79% in the 14 patients with MYD88 WT . In addition, the major response rate was different between these two groups, with 78% of patients with MYD88 MUT achieving a major response and only 57% of those with MYD88 WT achieving a major response. No VGPRs were noted in patients with MYD88 WT . The most common adverse effects were headache, diarrhea, contusions, nausea, dizziness, fatigue, upper respiratory infections, constipation, and arthralgias. The most common grades 3–4 adverse events included neutropenia (16%), pneumonia (7%), bleeding (3%), and AF (1%). Overall, AF was noted in 5% of patients, none of whom had AF prior to starting treatment, although no patients required cessation of acalabrutinib therapy.
6.2. Tirabrutinib
The success of tirabrutinib (ONO/GS‐4059), an additional covalent BTK inhibitor, was initially evaluated in a 90‐patient, multicenter phase I trial that included three patients with WM. 36 Patients received treatment with doses of tirabrutinib that ranged from 20 to 600 mg once daily or 240 to 300 mg twice daily. Dose limiting toxicity was noted in two of the patients with WM (urticarial rash in a patient on 300 mg daily and a non‐immune drug reaction in a patient on 600 mg daily). The third patient with WM achieved a partial response and remained on treatment when the initial trial data were reported. Later, a phase I trial was initiated using tirabrutinib in 17 Japanese patients with CLL and other non‐Hodgkin lymphomas. 37 Two patients with WM were enrolled in this trial, and both patients responded to treatment, although one dose‐limiting adverse event of pneumonitis was observed. Despite the toxicities noted in the patients with WM, tirabrutinib was found to have an acceptable safety profile and robust hematologic response rates, so a subsequent trial dedicated only to patients with WM was initiated. 38 In this multicenter, phase II trial, 27 patients with WM were treated with tirabrutinib 480 mg once daily. Treatment naïve and pre‐treated patients were included in the trial, and the ORRs were 94% and 100%, respectively. Major response rate was 89% in both treatment naïve and previously treated patients. No complete responses were reported. Time to major response was 1.9 months. Response rates were higher in patients with CXCR4 WT than in those with CXCR4 MUT (91% vs. 67%). The most common adverse events, most of which were grade 1 or 2, were rash, neutropenia, leukopenia, and stomatitis. Grade ≥3 adverse events occurred in three patients and included neutropenia (11%), lymphopenia (11%), and leukopenia (7%). All bleeding events were grade 1, and no episodes of hypertension or AF were attributed to the treatment.
6.3. Orelabrutinib
Orelabrutinib is a covalent BTK inhibitor and was used in a phase II multi‐center trial that included 47 patients with previously treated WM. 39 The ORR was 87% and the major response rate was 75%, with the highest major response rates in patients with MYD88 MUT /CXCR4 WT (80%). The PFS rate at 12 months was 88%. The most common adverse events were thrombocytopenia (28%), neutropenia (15%), leukopenia (11%), upper respiratory infections (15%), weight gain (15%), flu‐like symptoms (13%), and rash (11%). Most adverse events (90%) were grades 1–2. Ongoing in vitro and in vivo investigation into the use of orelabrutinib has suggested that combination therapy may play an important role in the treatment of B‐cell lymphomas, 40 and additional clinical trials using orelabrutinib are planned in WM and other lymphomas.
7. SELECTED MANAGEMENT ISSUES
7.1. Cardiac arrhythmia
AF is a relatively common adverse event seen in 10% to 15% of patients treated with ibrutinib. The risk of AF is lower with zanubrutinib and acalabrutinib based on recently published randomized clinical trials. 29 , 41 There was a low rate of AF in single‐arm studies with tirabrutinib and orelabrutinib. 39 , 42 A preclinical study in mice showed that exposure to ibrutinib, but not acalabrutinib, induced AF, left atrial enlargement, myocardial fibrosis, and inflammation. 33 , 43 C‐terminal Src kinase (Csk) was the strongest candidate for ibrutinib‐induced AF, and cardiac‐specific Csk knockouts induced a similar phenotype to ibrutinib exposure. Clinical features such as older age, male sex, and previous history of arrhythmia, hypertension, or coronary artery disease, as well as echocardiographic findings, such as higher left atrial diameter and area have been associated with a higher risk of AF in patients treated with ibrutinib. 44 , 45 , 46 Many patients with AF report palpitations, in which case a standard 12‐lead electrocardiogram (ECG) or heart monitoring should be performed to formally make a diagnosis. Once a diagnosis is made and if no contraindications exist, anticoagulation can be promptly started if indicated based on the CHA2DS2‐VASc score for AF stroke risk. 47 Similarly, if necessary, beta‐blockers can also be started upon diagnosis, followed by a referral to a cardiologist for further management, which can include antiarrhythmic agents, cardioversion, or cardiac ablation.
7.2. Bleeding
BTK inhibitor therapy is associated with increased events of bleeding by affecting platelet adhesion and aggregation. 48 , 49 , 50 , 51 In the ASPEN study, the estimated cumulative incidence of hemorrhage at 2 years was significantly higher with ibrutinib than with zanubrutinib (65% vs. 50%, respectively), with a rate of bleeding of 7 and 4.4 events in 100 person‐months. 29 In a randomized study in CLL, acalabrutinib was associated with a lower frequency of bleeding events than ibrutinib (38% vs. 51%, respectively), though the rate of major bleeding was comparable between agents (4.5% vs. 5.3%, respectively). Preclinical studies suggest that acalabrutinib and ibrutinib might have similar effects on platelet aggregation, but ibrutinib might have a higher impact on platelet adhesion than acalabrutinib, causing dysfunctional thrombus formation. 48 A more recent study showed that acalabrutinib delayed aggregation only in ibrutinib high‐sensitive patient cells, suggesting that in patients who experience bleeding complications with ibrutinib, a switch to acalabrutinib might not reduce the risk of bleeding. 52 In most cases, the bleeding is superficial or associated with surgical procedures. The temporary discontinuation of the BTK inhibitor would predictably decrease the risk of bleeding due to surgery. Table 1 shows suggested BTK inhibitors hold durations for surgical procedures in our practice. However, the duration of the temporary hold should depend on the invasiveness of the procedure, the patient's risk of bleeding, and previous history of bleeding or withdrawal symptoms while holding BTK inhibitors.
TABLE 1.
Suggested duration of temporary BTK inhibitor hold for surgical procedures
| Procedure | No. of days to hold pre‐procedure | Day of procedure | No. of days to hold post‐procedure |
|---|---|---|---|
| Bronchoscopy | 2 days | Hold | 2 days |
| Colonoscopy | 2 days | Hold | 2 days |
| Fine needle aspiration | 1 day | Hold | 1 day |
| Core needle biopsy | 1 day | Hold | 1 day |
| Paraspinal injections (e.g., for nerve injections) | 1 day | Hold | 1 day |
| Lumbar puncture | 3 days | Hold | 1 day |
| Tooth extraction | No hold | No hold | No hold |
| Multiple dental extractions, more invasive dental work (e.g., implants, root canal) | 2 days | Hold | 2 days |
| Joint replacement | 3 days | Hold | 3 days |
| Joint injection | 1 day | Hold | 1 day |
| Skin punch biopsy | No hold | No hold | No hold |
| Mohs procedure | 2 days | Hold | 2 days |
| Carpal tunnel release | 3 days | Hold | 3 days |
| Renal biopsy | 3 days | Hold | 3 days |
| Laminectomy | 3 days | Hold | 3 days |
| Neurosurgery | 7 days | Hold | 7 days |
| Cardiac ablation, pacemaker placement, or other arterial access required | 1 day | Hold | 1 day |
7.3. Iron supplementation
The initial response to any BTK inhibitor is so universal that should any patient not have a corresponding rapid rise in their hemoglobin within weeks of commencing therapy, iron studies should be re‐checked to ensure sufficient stores to accommodate the sudden increase in hematopoietic demand. Given the hepcidin‐driven suppression of iron utilization, we aim for a ferritin >200 ug/L and transferrin saturation > 20%.
7.4. Secondary malignancies
In the ASPEN study, the rate of secondary malignancies was similar between ibrutinib and zanubrutinib, mainly driven by the development of non‐melanomatous skin cancers . 27 These rates were consistent with the experience reported in a phase II study with zanubrutinib, in which 12% of patients had emergent basal cell skin carcinoma . 28 This high rate can be explained by the predominance of patients from Australia and New Zealand, countries with a high prevalence of skin cancers.
7.5. Withdrawal symptoms
BTK inhibitors are temporarily held for surgical procedures or to manage adverse events. In a retrospective study of 189 patients with WM on ibrutinib, approximately 20% of patients with WM who temporarily held ibrutinib experienced withdrawal symptoms characterized by fever, body ache, night sweats, arthralgia, headache, and fatigue. 53 Patients who experienced withdrawal symptoms were more likely to be in a VGPR than patients who were asymptomatic during the temporary hold (32% vs. 14%; p = .01), and in 65% of the patients who experienced withdrawal symptoms, there was not a concurrent increase in serum IgM associated with the symptoms. Withdrawal symptoms are promptly managed with prednisone 10–20 mg (or equivalent) orally twice daily. It is useful to enquire about past withdrawal symptoms when BTK inhibitors are paused during Nirmatrelvir and Ritonavir (Paxlovid) administration for COVID‐19 infection management. In that setting, dose reduction may be a more prudent approach to mitigate the increased plasma concentration of the BTK inhibitor caused by suppression of the CYP3A enzyme.
7.6. Dose reductions
One approach to managing BTK inhibitor‐associated toxicity would be dose reduction. In a retrospective study of 385 patients with WM treated with ibrutinib, a dose reduction was pursued in 25% of patients. 54 The most common reasons for dose reduction were musculoskeletal (e.g., myalgia, arthralgia), cardiac (e.g., arrhythmia, hypertension), and cytopenia. Patients with dose reductions were older (71 vs. 66 years; p < .001) and were more likely to be women (47% vs. 34%; p = .02) than patients who did not necessitate a dose reduction. Two thirds of the patients had resolution or improvement of the symptoms prompting the dose reduction, and 94% of patients maintained or improved their response despite the dose reduction.
7.7. Switch to other covalent BTK inhibitors
There are no data on the outcomes of patients with WM switching from one BTK inhibitor to another. In CLL, an initial experience evaluated 33 patients who enrolled in a phase I/II study with acalabrutinib and were labeled as intolerant to ibrutinib by the investigator prior to enrollment. 55 The most common adverse events that prompted the switch were rash, arthralgia, diarrhea, fatigue, and hemorrhage. Two thirds of the patients did not have a recurrence of the symptoms that prompted the switch, and the ORR to acalabrutinib was 76%. A second, dedicated phase II study included 60 CLL patients who were ibrutinib‐intolerant and switched therapy to acalabrutinib. 56 Ibrutinib intolerance was defined as having experienced a grade 3 or 4 adverse event or a grade 2 adverse event that persisted for at least 2 weeks or recurred at least twice on ibrutinib. The most common adverse events to ibrutinib were AF, diarrhea, arthralgia, and rash. Of the 74 ibrutinib‐intolerance events, 42 (57%) did not recur on acalabrutinib but 27 (36%) did. Of the latter, 18 events (67%) were of lower grade on acalabrutinib than on prior ibrutinib therapy. An ongoing study is evaluating zanubrutinib in patients with B‐cell malignancies, including 10 patients with WM, who were intolerant to ibrutinib or acalabrutinib. 57 Overall, 73% of patients did not experience a recurrence of the events on ibrutinib or acalabrutinib, and 79% of the recurrent events were of lower severity, while 84% of patients maintained or improved the response attained with the previous BTK inhibitor.
8. NON‐COVALENT BTK INHIBITORS
Mutations in BTK at C481, the covalent binding site of covalent BTK inhibitors, lead to ERK re‐activation in the presence of ibrutinib. ERK re‐activation in turn triggers IL‐6 and IL‐10 signaling, which renders neighboring BTK wild‐type cells resistant to ibrutinib therapy, demonstrating a paracrine function for MYD88 directed signaling. 58
8.1. Pirtobrutinib
Patients treated with covalent BTK inhibitors may acquire BTK mutations that lead to the development of resistance to binding at the C481 location on BTK. Novel non‐covalent BTK inhibitors, such as pirtobrutinib (LOXO‐305), have recently been developed and can overcome resistance related to C481S mutations. An initial phase I/II clinical trial with pirtobrutinib enrolled 323 patients with multiple types of non‐Hodgkin lymphoma, including 26 patients with WM. 59 During this trial, the recommended phase II dose was established at 200 mg once daily. Of the patients with WM, 67% had progressive disease prior to enrollment, and 33% had toxicity to prior therapy. Nineteen patients with WM were evaluable for response, and the ORR was 60%, including nine partial responses and four minor responses. Six of the eight patients in this study who were progressing on a covalent BTK inhibitor responded to pirtobrutinib. The adverse events that occurred in >10% of patients included fatigue (20%), diarrhea (17%), and contusion (13%). Neutropenia was the only grade ≥3 adverse event that occurred in more than 10% of patients. No new cases of AF were detected in the study. Additional clinical trials with pirtobrutinib are planned for patients with WM.
8.2. Nemtabrutinib (MK‐1026, formerly ARQ‐531)
Like pirtobrutinib, nemtabrutinib is a non‐covalent, reversible inhibitor that binds BTK independent of the mutational status of C481. This inhibitor has been studied in a phase I/II dose escalation and dose expansion trial, in which 65 mg once daily was established as the recommended phase II dosing. 60 In the phase II portion of this trial, 118 patients with a variety of non‐Hodgkin lymphomas, including WM, were enrolled. Data presented to date from this trial report an ORR of 58% in CLL, with the most common drug‐related adverse events including dysgeusia (15%), nausea (11%), fatigue (11%), and neutropenia (10%). Further results describing the outcomes in WM and other lymphomas are pending.
8.3. Future directions
Despite the growing armamentarium of safe and effective drugs to treat WM, there are still substantial unmet needs, which include a scarcity of complete responses, the lower efficacy of BTK inhibitors in WM patients with nonsense CXCR4 mutations, and the indefinite duration of currently FDA‐approved treatments. Table 2 shows the response and survival outcomes of WM patients with CXCR4 mutations to BTK inhibitors. Based on the low toxicity profile, high anti‐lymphoma activity in WM, and oral administration, BTK inhibitors are ideal candidates for developing highly effective, chemotherapy‐free combination regimens aiming at safely increasing the depth and durability of the response. The combination of ibrutinib and ulocuplumab showed encouraging results in a phase I proof‐of‐principle study, 61 and the combination of ibrutinib and mavorixafor, an oral CXCR4 inhibitor, has shown early efficacy as well. 62 Prospective clinical trials are currently evaluating BTK inhibitor‐based doublets, such as ibrutinib and daratumumab (NCT03679624), ibrutinib and carfilzomib (NCT04263480), ibrutinib and ixazomib (NCT03506373), ibrutinib and venetoclax (NCT04273139), acalabrutinib and rituximab (NCT05065554), and zanubrutinib and ixazomib (NCT04463953), as well as triple regimens, such as ibrutinib, venetoclax and rituximab (NCT04840602), ibrutinib, bortezomib and rituximab (NCT03620903), and acalabrutinib, bendamustine and rituximab (NCT04624906) to cite a few examples. Of great interest is that several studies will investigate fixed‐duration BTK inhibitor‐based therapies with the goal of minimizing drug exposure, expense, and long‐term toxicity.
TABLE 2.
Response and progression‐free survival outcomes to Bruton tyrosine kinase inhibitors in patients with Waldenström macroglobulinemia according to CXCR4 mutational status
| Study (Setting) | Regimen | CXCR4 mutational status | Time to major response | PR or better | VGPR or better | PFS |
|---|---|---|---|---|---|---|
| Treon et al. 15 (RR) | Ibrutinib | Wild type | 4.7 months | 97% | 47% | 70% at 5 years |
| Mutated | 1.8 months | 68% | 9% | 38% at 5 years | ||
| Trotman et al. 17 (RR) | Ibrutinib | Wild type | NA | 88% | 41% | NR at 5 years |
| Mutated | NA | 71% | 14% | Median: 18 months | ||
| Castillo et al. 19 (TN) | Ibrutinib | Wild type | 7.3 months | 94% | 44% | 90% at 4 years |
| Mutated | 1.8 months | 78% | 14% | 59% at 4 years | ||
| Buske et al. 21 (TN, RR) | Ibrutinib plus rituximab | Wild type | 3 months | 81% | 44% | 72% at 4.5 years |
| Mutated | 2 months | 77% | 23% | 63% at 4.5 years | ||
| Tam et al. 29 (TN, RR) | Ibrutinib | Wild type | NA | 82% | 24% | NA |
| Mutated | NA | 65% | 10% | NA | ||
| Zanubrutinib | Wild type | NA | 82% | 34% | NA | |
| Mutated | NA | 70% | 18% | NA |
Abbreviations: N/A, not available; NR, not reached; PFS, progression‐free survival; PR, partial response; RR, relapsed or refractory; TN, treatment naïve; VGPR, very good partial response.
AUTHORS CONTRIBUTIONS
All the authors designed the review, wrote and reviewed the manuscript, and approved the final version.
FUNDING INFORMATION
There was no special funding for this manuscript.
CONFLICT OF INTEREST
JJC received research funds and/or consulting fees from AbbVie, AstraZeneca, BeiGene, Casma Therapeutics, Cellectar, Janssen, Pharmacyclics, Roche/Genentech, and TG Therapeutics. CB received research funds and/or consulting fees from AbbVie, Amgen, BeiGene, Celltrion, Gilead Sciences, Janssen, Incyte, Morphosys, MSD, Novartis, Pfizer, Regeneron, Roche/Genentech. JT received research funds from BeiGene, BMS, Janssen, Pharmacyclics, Roche/Genentech, Takeda, Cellectar. SS received research funds and/or consulting fees from Acrotech Biopharma, ADC Therapeutics, and BeiGene. SPT received research funds and/or consulting fees from AbbVie, BeiGene, BMS, Janssen, Lilly, Pharmacyclics, X4 Pharmaceuticals.
Castillo JJ, Buske C, Trotman J, Sarosiek S, Treon SP. Bruton tyrosine kinase inhibitors in the management of Waldenström macroglobulinemia. Am J Hematol. 2023;98(2):338‐347. doi: 10.1002/ajh.26788
DATA AVAILABILITY STATEMENT
No data are associated with this manuscript.
REFERENCES
- 1. Jimenez C, Sebastian E, Chillon MC, et al. MYD88 L265P is a marker highly characteristic of, but not restricted to Waldenstrom's macroglobulinemia. Leukemia. 2013;27(8):1722‐1728. [DOI] [PubMed] [Google Scholar]
- 2. Poulain S, Roumier C, Decambron A, et al. MYD88 L265P mutation in Waldenstrom macroglobulinemia. Blood. 2013;121(22):4504‐4511. [DOI] [PubMed] [Google Scholar]
- 3. Schmidt J, Federmann B, Schindler N, et al. MYD88 L265P and CXCR4 mutations in lymphoplasmacytic lymphoma identify cases with high disease activity. Br J Haematol. 2015;169(6):795‐803. [DOI] [PubMed] [Google Scholar]
- 4. Treon SP, Xu L, Yang G, et al. MYD88 L265P somatic mutation in Waldenstrom's macroglobulinemia. N Engl J Med. 2012;367(9):826‐833. [DOI] [PubMed] [Google Scholar]
- 5. Castillo JJ, Moreno DF, Arbelaez MI, Hunter ZR, Treon SP. CXCR4 mutations affect presentation and outcomes in patients with Waldenstrom macroglobulinemia: a systematic review. Expert Rev Hematol. 2019;12(10):873‐881. [DOI] [PubMed] [Google Scholar]
- 6. Hunter ZR, Xu L, Yang G, et al. The genomic landscape of Waldenstrom macroglobulinemia is characterized by highly recurring MYD88 and WHIM‐like CXCR4 mutations, and small somatic deletions associated with B‐cell lymphomagenesis. Blood. 2014;123(11):1637‐1646. [DOI] [PubMed] [Google Scholar]
- 7. Roccaro AM, Sacco A, Jimenez C, et al. C1013G/CXCR4 acts as a driver mutation of tumor progression and modulator of drug resistance in lymphoplasmacytic lymphoma. Blood. 2014;123(26):4120‐4131. [DOI] [PubMed] [Google Scholar]
- 8. Treon SP, Cao Y, Xu L, Yang G, Liu X, Hunter ZR. Somatic mutations in MYD88 and CXCR4 are determinants of clinical presentation and overall survival in Waldenstrom macroglobulinemia. Blood. 2014;123(18):2791‐2796. [DOI] [PubMed] [Google Scholar]
- 9. Bustoros M, Sklavenitis‐Pistofidis R, Kapoor P, et al. Progression risk stratification of asymptomatic Waldenstrom Macroglobulinemia. J Clin Oncol. 2019;37(16):1403‐1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zanwar S, Abeykoon JP, Ansell SM, et al. Disease outcomes and biomarkers of progression in smouldering Waldenström macroglobulinaemia. Br J Haematol. 2021;195(2):210‐216. [DOI] [PubMed] [Google Scholar]
- 11. Castillo JJ, Advani RH, Branagan AR, et al. Consensus treatment recommendations from the tenth international workshop for Waldenstrom Macroglobulinaemia. Lancet Haematol. 2020;7(11):e827‐e837. [DOI] [PubMed] [Google Scholar]
- 12. Dimopoulos MA, Kastritis E, Owen RG, et al. Treatment recommendations for patients with Waldenstrom macroglobulinemia (WM) and related disorders: IWWM‐7 consensus. Blood. 2014;124(9):1404‐1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Leblond V, Kastritis E, Advani R, et al. Treatment recommendations from the eighth international workshop on Waldenstrom's Macroglobulinemia. Blood. 2016;128(10):1321‐1328. [DOI] [PubMed] [Google Scholar]
- 14. Treon SP, Tripsas CK, Meid K, et al. Ibrutinib in previously treated Waldenstrom's macroglobulinemia. N Engl J Med. 2015;372(15):1430‐1440. [DOI] [PubMed] [Google Scholar]
- 15. Treon SP, Meid K, Gustine J, et al. Long‐term follow‐up of Ibrutinib monotherapy in symptomatic, previously treated patients with Waldenstrom Macroglobulinemia. J Clin Oncol. 2021;39(6):565‐575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dimopoulos MA, Trotman J, Tedeschi A, et al. Ibrutinib for patients with rituximab‐refractory Waldenstrom's macroglobulinaemia (iNNOVATE): an open‐label substudy of an international, multicentre, phase 3 trial. Lancet Oncol. 2017;18(2):241‐250. [DOI] [PubMed] [Google Scholar]
- 17. Trotman J, Buske C, Tedeschi A, et al. Single‐agent Ibrutinib for rituximab‐refractory Waldenstrom Macroglobulinemia: final analysis of the substudy of the phase III innovate(TM) trial. Clin Cancer Res. 2021;27(21):5793‐5800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Treon SP, Gustine J, Meid K, et al. Ibrutinib monotherapy in symptomatic, treatment‐naive patients with Waldenstrom Macroglobulinemia. J Clin Oncol. 2018;36(27):2755‐2761. [DOI] [PubMed] [Google Scholar]
- 19. Castillo JJ, Meid K, Gustine JN, et al. Long‐term follow‐up of ibrutinib monotherapy in treatment‐naive patients with Waldenstrom macroglobulinemia. Leukemia. 2022;36(2):532‐539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dimopoulos MA, Tedeschi A, Trotman J, et al. Phase 3 trial of Ibrutinib plus rituximab in Waldenstrom's macroglobulinemia. N Engl J Med. 2018;378(25):2399‐2410. [DOI] [PubMed] [Google Scholar]
- 21. Buske C, Tedeschi A, Trotman J, et al. Ibrutinib plus rituximab versus placebo plus rituximab for Waldenstrom's Macroglobulinemia: final analysis from the randomized phase III iNNOVATE study. J Clin Oncol. 2022;40(1):52‐62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Castillo JJ, Xu L, Gustine JN, et al. CXCR4 mutation subtypes impact response and survival outcomes in patients with Waldenstrom macroglobulinaemia treated with ibrutinib. Br J Haematol. 2019;187(3):356‐363. [DOI] [PubMed] [Google Scholar]
- 23. Castillo JJ, Sarosiek SR, Gustine JN, et al. Response and survival predictors in a cohort of 319 patients with Waldenstrom macroglobulinemia treated with ibrutinib monotherapy. Blood Adv. 2022;6(3):1015‐1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tam CS, Dimopoulos MA, Garcia‐Sanz R, et al. Pooled safety analysis of zanubrutinib monotherapy in patients with B‐cell malignancies. Blood Adv. 2021;6:1296‐1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tam CS, Ou YC, Trotman J, Opat S. Clinical pharmacology and PK/PD translation of the second‐generation Bruton's tyrosine kinase inhibitor, zanubrutinib. Expert Rev Clin Pharmacol. 2021;14(11):1329‐1344. [DOI] [PubMed] [Google Scholar]
- 26. Advani RH, Buggy JJ, Sharman JP, et al. Bruton tyrosine kinase inhibitor ibrutinib (PCI‐32765) has significant activity in patients with relapsed/refractory B‐cell malignancies. J Clin Oncol. 2013;31(1):88‐94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tam CS, Trotman J, Opat S, et al. Phase 1 study of selective BTK inhibitor zanubrutinib in B‐cell malignancies and safety and efficacy evaluation in CLL. Blood. 2019;134(11):851‐859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Trotman J, Opat S, Gottlieb D, et al. Zanubrutinib for the treatment of patients with Waldenstrom macroglobulinemia: 3 years of follow‐up. Blood. 2020;136(18):2027‐2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tam CS, Opat S, D'Sa S, et al. A randomized phase 3 trial of zanubrutinib vs ibrutinib in symptomatic Waldenstrom macroglobulinemia: the ASPEN study. Blood. 2020;136(18):2038‐2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tam CSL, Garcia‐Sanz R, Opat S, et al. ASPEN: long‐term follow‐up results of a phase 3 randomized trial of zanubrutinib (ZANU) versus ibrutinib (IBR) in patients with Waldenström macroglobulinemia (WM). J Clin Oncol. 2022;40(16 Suppl):7521. [Google Scholar]
- 31. Dimopoulos M, Opat S, D'Sa S, et al. P1161: ASPEN: long‐term follow‐up results of a phase 3 randomized trial of ZANUBRUTINIB (ZANU) vs IBRUTINIB (IBR) IN patients (pts) with WALDENSTRÖM MACROGLOBULINEMIA (WM). HemaSphere. 2022;6:1048‐1049. [Google Scholar]
- 32. Dimopoulos M, Sanz RG, Lee HP, et al. Zanubrutinib for the treatment of MYD88 wild‐type Waldenstrom macroglobulinemia: a substudy of the phase 3 ASPEN trial. Blood Adv. 2020;4(23):6009‐6018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kofides A, Hunter ZR, Xu L, et al. Diagnostic next‐generation sequencing frequently fails to detect MYD88(L265P) in Waldenstrom Macroglobulinemia. Hema. 2021;5(8):e624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gustine JN, Xu L, Yang G, et al. Bone marrow involvement and subclonal diversity impairs detection of mutated CXCR4 by diagnostic next‐generation sequencing in Waldenstrom macroglobulinaemia. Br J Haematol. 2021;194(4):730‐733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Owen RG, McCarthy H, Rule S, et al. Acalabrutinib monotherapy in patients with Waldenstrom macroglobulinemia: a single‐arm, multicentre, phase 2 study. Lancet Haematol. 2020;7(2):e112‐e121. [DOI] [PubMed] [Google Scholar]
- 36. Walter HS, Rule SA, Dyer MJ, et al. A phase 1 clinical trial of the selective BTK inhibitor ONO/GS‐4059 in relapsed and refractory mature B‐cell malignancies. Blood. 2016;127(4):411‐419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Munakata W, Ando K, Hatake K, et al. Phase I study of tirabrutinib (ONO‐4059/GS‐4059) in patients with relapsed or refractory B‐cell malignancies in Japan. Cancer Sci. 2019;110(5):1686‐1694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sekiguchi N, Rai S, Munakata W, et al. A multicenter, open‐label, phase II study of tirabrutinib (ONO/GS‐4059) in patients with Waldenstrom's macroglobulinemia. Cancer Sci. 2020;111(9):3327‐3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhou D, Jin J, Fu Z‐z, et al. Efficacy and safety of Orelabrutinib in relapsed/refractory Waldenstrom's Macroglobulinemia patients. Blood. 2021;138(Suppl 1):46. [Google Scholar]
- 40. Yu H, Wang X, Li J, et al. Addition of BTK inhibitor orelabrutinib to rituximab improved anti‐tumor effects in B cell lymphoma. Mol Ther Oncolytics. 2021;21:158‐170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Byrd JC, Hillmen P, Ghia P, et al. Acalabrutinib versus Ibrutinib in previously treated chronic lymphocytic leukemia: results of the first randomized phase III trial. J Clin Oncol. 2021;39(31):3441‐3452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Munakata W, Sekiguchi N, Shinya R, et al. Phase 2 study of Tirabrutinib (ONO/GS‐4059), a second‐generation Bruton's tyrosine kinase inhibitor, monotherapy in patients with treatment‐Naïve or relapsed/refractory Waldenström Macroglobulinemia. Blood. 2019;134(Suppl 1):345. [Google Scholar]
- 43. Xiao L, Salem JE, Clauss S, et al. Ibrutinib‐mediated atrial fibrillation attributable to inhibition of C‐terminal Src kinase. Circulation. 2020;142(25):2443‐2455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Reda G, Fattizzo B, Cassin R, et al. Predictors of atrial fibrillation in ibrutinib‐treated CLL patients: a prospective study. J Hematol Oncol. 2018;11(1):79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lentz R, Feinglass J, Ma S, Akhter N. Risk factors for the development of atrial fibrillation on ibrutinib treatment. Leuk Lymphoma. 2019;60(6):1447‐1453. [DOI] [PubMed] [Google Scholar]
- 46. Onitilo AA, Piwuna TO, Islam N, Furuya‐Kanamori L, Kumar S, SAR D. Determinants of atrial fibrillation development among patients undergoing ibrutinib therapy. Clin Med Res. 2022;20(1):16‐22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lip GY, Nieuwlaat R, Pisters R, Lane DA, Crijns HJ. Refining clinical risk stratification for predicting stroke and thromboembolism in atrial fibrillation using a novel risk factor‐based approach: the euro heart survey on atrial fibrillation. Chest. 2010;137(2):263‐272. [DOI] [PubMed] [Google Scholar]
- 48. Bye AP, Unsworth AJ, Desborough MJ, et al. Severe platelet dysfunction in NHL patients receiving ibrutinib is absent in patients receiving acalabrutinib. Blood Adv. 2017;1(26):2610‐2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kamel S, Horton L, Ysebaert L, et al. Ibrutinib inhibits collagen‐mediated but not ADP‐mediated platelet aggregation. Leukemia. 2015;29(4):783‐787. [DOI] [PubMed] [Google Scholar]
- 50. Kazianka L, Drucker C, Skrabs C, et al. Ristocetin‐induced platelet aggregation for monitoring of bleeding tendency in CLL treated with ibrutinib. Leukemia. 2017;31(5):1117‐1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Levade M, David E, Garcia C, et al. Ibrutinib treatment affects collagen and von Willebrand factor‐dependent platelet functions. Blood. 2014;124(26):3991‐3995. [DOI] [PubMed] [Google Scholar]
- 52. Series J, Garcia C, Levade M, et al. Differences and similarities in the effects of ibrutinib and acalabrutinib on platelet functions. Haematologica. 2019;104(11):2292‐2299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Castillo JJ, Gustine JN, Meid K, Dubeau T, Severns P, Treon SP. Ibrutinib withdrawal symptoms in patients with Waldenstrom macroglobulinemia. Haematologica. 2018;103(7):e307‐e310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sarosiek S, Gustine J, Flynn CA, et al. Dose reductions related to adverse effects in patients with Waldenström Macroglobulinemia treated with the BTK‐inhibitor Ibrutinib. Blood. 2021;138(Suppl 1):3529. [Google Scholar]
- 55. Awan FT, Schuh A, Brown JR, et al. Acalabrutinib monotherapy in patients with chronic lymphocytic leukemia who are intolerant to ibrutinib. Blood Adv. 2019;3(9):1553‐1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Rogers KA, Thompson PA, Allan JN, et al. Phase II study of acalabrutinib in ibrutinib‐intolerant patients with relapsed/refractory chronic lymphocytic leukemia. Haematologica. 2021;106(9):2364‐2373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Shadman M, Flinn IW, Levy MY, et al. Phase 2 study of Zanubrutinib in BTK inhibitor‐intolerant patients (pts) with relapsed/refractory B‐cell malignancies. Blood. 2021;138(Suppl 1):1410. [Google Scholar]
- 58. Chen JG, Liu X, Munshi M, et al. BTK(Cys481Ser) drives ibrutinib resistance via ERK1/2 and protects BTK(wild‐type) MYD88‐mutated cells by a paracrine mechanism. Blood. 2018;131(18):2047‐2059. [DOI] [PubMed] [Google Scholar]
- 59. Mato AR, Shah NN, Jurczak W, et al. Pirtobrutinib in relapsed or refractory B‐cell malignancies (BRUIN): a phase 1/2 study. Lancet. 2021;397(10277):892‐901. [DOI] [PubMed] [Google Scholar]
- 60. Woyach JA, Flinn IW, Awan FT, et al. Preliminary efficacy and safety of MK‐1026, a non‐covalent inhibitor of wild‐type and C481S mutated Bruton tyrosine kinase, in B‐cell malignancies: a phase 2 dose expansion study. Blood. 2021;392:392. [Google Scholar]
- 61. Treon SP, Meid K, Hunter ZR, et al. Phase 1 study of ibrutinib and the CXCR4 antagonist ulocuplumab in CXCR4‐mutated Waldenstrom macroglobulinemia. Blood. 2021;138(17):1535‐1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Treon SP, Buske C, Thomas SK, et al. Preliminary clinical response data from a phase 1b study of Mavorixafor in combination with Ibrutinib in patients with Waldenström's macroglobulinemia with MYD88 and CXCR4 mutations. Blood. 2021;138(Suppl 1):1362. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
No data are associated with this manuscript.