

Abstract
Objective
To assess long‐term safety, tolerability, and efficacy of once‐daily oral atogepant 60 mg in adults with migraine.
Background
Atogepant is an oral, small‐molecule, calcitonin gene–related peptide receptor antagonist approved for the preventive treatment of episodic migraine.
Methods
A 52‐week, multicenter, randomized, open‐label trial of adults (18–80 years) with migraine. Lead‐in trial completers or newly enrolled participants with 4–14 migraine days/month were enrolled and randomized (5:2) to atogepant 60 mg once daily or oral standard care (SC) migraine preventive medication. The primary objective was to evaluate the safety and tolerability of atogepant; safety assessments included treatment‐emergent adverse events (TEAEs), clinical laboratory evaluations, vital signs, and Columbia‐Suicide Severity Rating Scale scores. Efficacy assessments (atogepant only) included change from baseline in mean monthly migraine days (MMDs) and the proportion of participants with reductions from baseline of ≥50%, ≥75%, and 100% in MMDs.
Results
The trial included 744 participants randomized to atogepant 60 mg (n = 546) or SC (n = 198). The atogepant safety population was 88.2% female (n = 479/543) with a mean (standard deviation) age of 42.5 (12.0) years. TEAEs occurred in 67.0% (n = 364/543) of participants treated with atogepant 60 mg. The most commonly reported TEAEs (≥5%) were upper respiratory tract infection (10.3%; 56/543), constipation (7.2%; 39/543), nausea (6.3%; 34/543), and urinary tract infection (5.2%; 28/543). Serious TEAEs were reported in 4.4% (24/543) for atogepant. Mean (standard error) change in MMDs for atogepant was −3.8 (0.1) for weeks 1–4 and −5.2 (0.2) at weeks 49–52. Similarly, the proportion of participants with ≥50%, ≥75%, and 100% reductions in MMDs increased from 60.4% (310/513), 37.2% (191/513), and 20.7% (106/513) at weeks 1–4 to 84.2% (282/335), 69.9% (234/335), and 48.4% (162/335), at weeks 49–52.
Conclusion
Daily use of oral atogepant 60 mg for preventive treatment of migraine during this 1‐year, open‐label trial was safe, well tolerated, and efficacious.
Keywords: atogepant, calcitonin gene–related peptide, gepant, migraine, migraine preventive
Abbreviations
- AE
adverse event
- ALT
alanine aminotransferase
- AST
aspartate aminotransferase
- CGRP
calcitonin gene–related peptide
- C‐SSRS
Columbia‐Suicide Severity Rating Scale
- ECG
electrocardiogram
- EM
episodic migraine
- FDA
US Food and Drug Administration
- ICH
International Conference on Harmonization
- mAb
monoclonal antibody
- MMD
monthly migraine day
- SC
standard care
- SE
standard error
- TEAE
treatment‐emergent adverse event
- ULN
upper limit of normal
INTRODUCTION
Migraine is a highly prevalent disease worldwide and is the second‐highest contributor of years lived with disability. 1 , 2 Interictal symptoms, such as anxiety and avoidance of activities, are common in people with migraine and further compromise the quality of their lives. 3 Individuals with migraine often experience high variability and frequency of migraine attacks, 4 and uncontrolled migraine is associated with decreased quality of life and ability to perform activities of daily living. 5 , 6 Despite this, less than 30% of people with migraine who experience 4 or more migraine days per month and have moderate disability due to migraine are currently taking a migraine preventive medication. 7
Migraine preventive medications are recommended in people whose migraine attacks significantly interfere with their daily routines; 4 or more headache days per month (2 days if associated with impaired quality of life); contraindication to, failure of, or overuse of acute treatments; adverse events (AEs) with acute treatment; or patient preference. 8 , 9 However, many oral preventives currently prescribed are not US Food and Drug Administration (FDA) approved for the treatment of migraine. 8 Adherence is also an issue with non–migraine‐specific oral medications. In a large cross‐sectional survey of people with episodic migraine (EM), these treatments had discontinuation rates of ~35%–50% due to side effects or lack of efficacy. 7
Intravenous infusion of calcitonin gene–related peptide (CGRP) causes migraine attacks in people with migraine, and the efficacy of medications targeting the CGRP pathway for acute and preventive treatment of migraine is well documented. 10 Currently, four monoclonal antibodies (mAbs) targeting the CGRP pathway are FDA approved for the treatment of EM. 11 , 12 , 13 , 14 Three are subcutaneous injections and one is an intravenous infusion; the three subcutaneous injections are associated with injection‐site reactions. Wearing off of efficacy before the next scheduled dose can be a concern with some mAbs. 15
Atogepant, a small‐molecule CGRP receptor antagonist approved by the FDA for the preventive treatment of EM, is the first oral medication specifically developed for the preventive treatment of migraine. 16 In a phase 2b/3 and phase 3 (ADVANCE) clinical trial, each of 12 weeks duration, atogepant significantly reduced mean monthly migraine days (MMDs) and increased the proportion of participants experiencing a 50% or greater reduction in MMDs compared to placebo. 17 , 18 Evaluation of migraine preventive medications during an extended period of time is crucial, because migraine is a chronic disease and most people require long‐term preventive treatment. 19 The objectives of this trial were to assess the long‐term safety, tolerability, and efficacy of oral once‐daily atogepant 60 mg in adults with migraine during a 52‐week period.
METHODS
This 52‐week, multicenter, randomized, open‐label trial enrolled the first participant on October 8, 2018, and the last visit was completed on May 29, 2020. The study was conducted at 111 study centers in the United States (ClinicalTrials.gov: NCT03939312). The protocol and all amendments were approved by institutional review boards or independent ethics committees for each study center; all participants provided written informed consent. All study conduct was in accordance with the Declaration of Helsinki and the International Conference on Harmonization (ICH) Guidelines for Good Clinical Practice. Data analyses were provided by the trial sponsor, Allergan (now AbbVie). All authors had full access to the study data. The trial consisted of three periods: screening/baseline (4 weeks), open label (52 weeks), and safety/follow‐up (4 weeks).
Study participants
Eligible participants had completed the phase 2b/3 trial (NCT02848326) or were newly enrolled in the current trial (Figure 1). 17 Lead‐in trial completers had a minimum gap of 6 months from the completion of the lead‐in trial to the initiation of the present trial. All eligible participants were adults (aged 18–80 years old) with 4–14 migraine days per month (recorded in an electronic diary; eDiary) for 3 months prior to screening and in the 28‐day baseline period. Participants had to have been diagnosed with migraine with or without aura according to the International Classification of Headache Disorders, 3rd edition 20 for a minimum of 1 year prior to screening, and the diagnosis was required to be made before the age of 50 years. Participants needed to be a candidate for prescription of at least one of the protocol‐defined acceptable standard care (SC) preventive treatments and were required to use a medically acceptable and effective method of birth control throughout the trial. Participants were excluded if they had difficulty distinguishing migraine from other tension‐type headaches, had a current diagnosis of chronic migraine, or had experienced a mean of 15 or more headache days per month in the previous 3 months.
FIGURE 1.
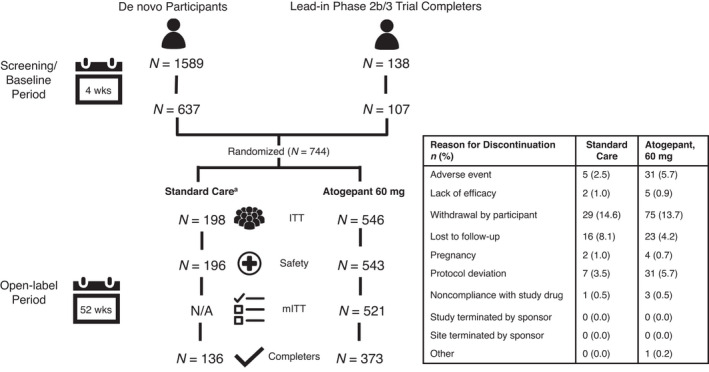
Participant disposition. aSC migraine prevention medication arm was included to contextualize hepatic laboratory data; efficacy outcomes were not collected in the SC arm; SC medication was selected by the investigator from an approved list in the protocol. ITT, intent‐to‐treat; mITT, modified intent‐to‐treat; N/A, not applicable; SC, standard care
Study design and assessments
Study investigators and staff enrolled participants into the study, and enrollment ended when the target sample size was obtained. Participants were randomized 5:2 to atogepant 60 mg once daily or oral SC migraine preventive medication; randomization was conducted using an interactive web response system to minimize bias. The sponsor provided tablets containing atogepant 60 mg but did not provide SC medications. Participants in the atogepant arm were instructed to take the tablets at approximately the same time each day. Participants randomized to the SC arm were treated with a medication recognized as safe and effective for the prevention of migraine and listed in the protocol (Table S1 in supporting information). If the initial migraine preventive medication was not tolerated or was not sufficiently effective, an investigator could prescribe an alternative preventive from the list of approved medications, prescribe an alternative preventive not listed (e.g., riboflavin, carbamazepine, gabapentin), or choose not to prescribe a migraine preventive medication; this process of updating the migraine preventive medication for the SC arm was allowed to be repeated as needed throughout the trial. Participants in the SC arm who discontinued their preventive could remain in the trial. The SC arm was used to contextualize the long‐term safety of atogepant by providing comparative data in a migraine population treated with commonly used migraine preventive medications in a manner consistent with clinical practice. Due to the flexibility to change or discontinue preventive medications as needed and the lack of efficacy assessments in the SC arm, direct comparisons of safety or efficacy between the SC and atogepant treatment arms are not appropriate. All participants were permitted to take medications for the acute treatment of migraine listed in Table S2 in supporting information.
Participants were randomized at visit 2 and study visits occurred every 4 weeks for 52 weeks. Safety assessments occurred at 4, 8, 12, 16, 20, 24, 28, 32, 36, 40, 44, 48, and 52 weeks relative to randomization. A safety follow‐up visit occurred 4 weeks after the last dose (atogepant 60 mg arm) or the last study visit (SC arm). In response to the worldwide COVID‐19 pandemic, remote visits were allowed for study visits 14, 15, and 16 (safety follow‐up), if necessary. For remote visits, safety assessments (AEs, concomitant medications, pregnancy test results review, and the Columbia‐Suicide Severity Rating Scale [C‐SSRS]), were conducted remotely, while in‐person safety assessments (clinical laboratory samples, vital signs, and electrocardiograms [ECGs]) were to be collected at the next in‐person visit. Remote visits and urine pregnancy tests were allowed for a maximum of 8 weeks (i.e., one remote visit).
Study outcomes
The primary outcome was safety and tolerability, which was assessed by monitoring AEs, clinical laboratory evaluations, vital sign measurements, ECG findings, and C‐SSRS scores. AEs of special interest included treatment‐emergent suicidal ideations with intent, with or without a plan, or any suicidal behaviors; treatment‐emergent alanine aminotransferase (ALT) or aspartate aminotransferase (AST) levels ≥3x upper limit of normal (ULN); and potential Hy's law cases. A clinical adjudication committee of independent liver experts was established for blinded adjudication of posttreatment elevations of ALT and/or AST ≥3x ULN for participants in the atogepant arm.
Efficacy outcomes included change from baseline in mean MMDs, proportion of participants with reduction from baseline of least 50%, 75%, and 100% MMDs at weeks 1–4, 9–12, 21–24, 33–36, and 49–52, and change from baseline in mean monthly acute medication use days. Definition of migraine days used in this trial was previously reported for the lead‐in trial. 17 These efficacy outcomes were collected daily via an eDiary.
Statistical analysis
The sample size of 700 randomized participants was based on the ICH‐E1 regulatory requirements of at least 300 participants exposed to atogepant 60 mg for 6 months and at least 100 exposed for at least 12 months using exposure results from prior studies. 21 , 22 Safety assessments were performed on the safety population, which included participants who received ≥1 dose of study medication, and efficacy assessments were performed on the modified intent‐to‐treat population, which included participants with an evaluable baseline period of eDiary data and ≥1 evaluable postbaseline 4‐week period of eDiary data.
AEs were collected from consent through last visit, and were reported as number and percentage of the safety population. AEs were classified by investigators by severity, potential relationship to study medication, start and stop date, and seriousness. Mean (standard error [SE]) change from baseline was calculated for MMDs and acute medication use days. The proportions of participants with a ≥50%, ≥75%, and 100% reduction in MMDs were also evaluated. Efficacy outcomes were analyzed using the mixed‐effects model for repeated measures; analyses were conducted using SAS version 9.4 or later (SAS Institute, Inc.).
The assumption of reporting mean values (SE or standard deviation) was verified using histograms, which demonstrated symmetric distribution suggesting a very close to normal distribution.
RESULTS
Participant disposition and extent of exposure
Of 1727 participants screened, 744 were randomized (Figure 1). Participant baseline demographics are listed in Table 1. A total of 509 participants completed the trial. Withdrawal by participant was the most common reason for discontinuation in both treatment arms. For atogepant participants, discontinuations due to AEs (<6%) or lack of efficacy (<1%) were not common. Of 543 atogepant participants, 428 (78.8%) and 362 (66.7%) received atogepant for ≥180 and ≥360 days, respectively. Participants had a mean of 291.6 days of exposure to atogepant and 433.6 participant‐years of treatment. The most commonly reported protocol deviation was the use of prohibited concomitant medications (15.6% [85/546] in the atogepant group and 1.5% [3/198] in the SC group). The three migraine preventive medications taken by the SC group participants for their initial choice were topiramate (35.7%), amitriptyline (21.9%), and propranolol (15.8%). The mean (SE) number of migraine preventive medications used by SC participants throughout the trial was 1.4 (0.8).
TABLE 1.
Participant baseline demographics
| Standard care (n = 196) | Atogepant 60 mg, safety population (n = 543) | Atogepant 60 mg, mITT population (n = 521) | |
|---|---|---|---|
| Age, a mean (SD), years | 41.1 (12.1) | 42.5 (12.0) | 42.5 (12.0) |
| Sex, n (%), female | 172 (87.8) | 479 (88.2) | 460 (88.3) |
| Race, n (%) | |||
| White | 145 (74.0) | 416 (76.6) | 400 (76.8) |
| Black or African American | 38 (19.4) | 100 (18.4) | 95 (18.2) |
| Asian | 5 (2.6) | 12 (2.2) | 11 (2.1) |
| American Indian or Alaska Native | 0 (0.0) | 3 (0.6) | 3 (0.6) |
| Native Hawaiian or Other Pacific Islander | 2 (1.0) | 2 (0.4) | 2 (0.4) |
| Multiple b | 5 (2.6) | 10 (1.8) | 10 (1.9) |
| Missing | 1 (0.5) | 0 (0.0) | 0 (0.0) |
| Ethnicity, n (%) | |||
| Hispanic or Latinx | 29 (14.8) | 83 (15.3) | 82 (15.7) |
| Not Hispanic or Latinx | 166 (84.7) | 460 (84.7) | 439 (84.3) |
| Missing | 1 (0.5) | 0 (0.0) | 0 (0.0) |
| BMI, mean (SD), kg/m2 | 30.6 (8.0) | 30.6 (8.0) | 30.5 (7.4) |
Abbreviations: BMI, body mass index; mITT, modified intent‐to‐treat; SC, standard care; SD, standard deviation.
Age is relative to informed consent date.
Participants who reported multiple races are only included in multiple categories.
Adverse events in the atogepant arm
Treatment‐emergent AEs (TEAEs) occurred in 364/543 (67.0%) participants treated with atogepant (Table 2). The majority of TEAEs in the atogepant group were mild (140/543, 25.8%) or moderate (187/543, 34.4%) in severity, with 37/543 (6.8%) severe TEAEs reported. All TEAEs occurring in ≥2% of atogepant‐treated participants are listed in Table 3. The most common TEAEs in atogepant‐treated participants were upper respiratory tract infection (56/543, 10.3%), constipation (39/543, 7.2%), nausea (34/543, 6.3%), and urinary tract infection (28/543, 5.2%). The most common treatment‐related TEAEs in atogepant‐treated participants were constipation (26/543, 4.8%), nausea (16/543, 2.9%), and fatigue (12/543, 2.2%). One participant (1/543, 0.2%) discontinued due to constipation, which was considered mild and related to treatment, and occurred early in the trial (day 8). Most cases of constipation were mild to moderate in severity. One participant in the atogepant group reported a TEAE of constipation that was severe in intensity (day 23). This TEAE did not result in discontinuation from the study and was downgraded to mild on day 96 and resolved by day 126.
TABLE 2.
Overall summary of treatment‐emergent adverse events (safety population)
| Atogepant 60 mg (n = 543), n (%) | |
|---|---|
| TEAE | 364 (67.0) |
| Treatment‐related TEAE | 98 (18.0) |
| Death | 2 (0.4) |
| Treatment‐emergent serious adverse event | 24 (4.4) |
| TEAE leading to study discontinuation | 31 (5.7) |
Note: Participants counted only once within each category.
Abbreviation: TEAE, treatment‐emergent adverse event.
TABLE 3.
Summary of common treatment‐emergent adverse events (≥2.0%; safety population)
| Atogepant 60 mg (n = 543) n (%) | |
|---|---|
| Upper respiratory tract infection | 56 (10.3) |
| Constipation | 39 (7.2) |
| Nausea | 34 (6.3) |
| Urinary tract infection | 28 (5.2) |
| Nasopharyngitis | 24 (4.4) |
| Influenza | 18 (3.3) |
| Dizziness | 17 (3.1) |
| Anxiety | 16 (2.9) |
| Sinusitis | 15 (2.8) |
| Fatigue | 14 (2.6) |
| Hypertension | 14 (2.6) |
| Weight decreased | 14 (2.6) |
| Aspartate aminotransferase increased | 13 (2.4) |
| Back pain | 13 (2.4) |
| Gastroenteritis | 13 (2.4) |
| Alanine aminotransferase increased | 11 (2.0) |
| Arthralgia | 11 (2.0) |
| Muscle strain | 11 (2.0) |
| Pharyngitis streptococcal | 11 (2.0) |
Note: Participants counted only once within each preferred term. Includes TEAEs occurring in ≥2.0% in the atogepant 60 mg group (after rounding).
Abbreviation: TEAE, treatment‐emergent adverse event.
Serious TEAEs were reported in 24/543 (4.4%) atogepant‐treated participants; no participant had more than one serious TEAE. Study discontinuation due to TEAEs occurred in 31/543 (5.7%) atogepant‐treated participants. Only nausea (3/543, 0.6%), fatigue (2/543, 0.4%), dizziness (2/543, 0.4%), and rash (2/543, 0.4%) resulted in the discontinuation of more than one atogepant‐treated participant.
Description of deaths
Two deaths occurred in the trial, both in the atogepant arm. A 56‐year‐old female participant was a victim of a homicide that occurred during a car‐jacking on study day 4. A 26‐year‐old female participant died due to a Group A beta‐hemolytic streptococcal sepsis (toxic shock syndrome) on study day 43. Both deaths were considered not related to atogepant.
Hepatic laboratory values
Hepatic laboratory values are listed in Table S3 in supporting information and elevations in ALT or AST levels are summarized in Table 4. Overall mean changes in hepatic values were minimal and comparable between the atogepant and SC treatment arms. Treatment‐emergent postbaseline elevations of ALT or AST ≥3x ULN occurred in 13 (2.4%) atogepant‐treated participants and 6 (3.2%) SC participants. All cases of ALT and/or AST elevations ≥3x ULN were reviewed by the clinical adjudication committee of independent liver experts; 2 of the 13 were assessed as possibly related to atogepant. Following evaluation of the individual cases and assessment for patterns and/or trends across the cases, the committee found that no hepatic safety issues related to atogepant were identified in this trial. No cases of potential Hy's law (ALT or AST ≥3x ULN and bilirubin total ≥2x ULN and alkaline phosphatase <2x ULN during a 24‐h period) were identified.
TABLE 4.
Summary of ALT/AST values of potential clinical significance
| Parameter (unit) criteria | Standard care | Atogepant 60 mg |
|---|---|---|
| (N = 196) n/N1 | (N = 543) n/N1 | |
| ALT or AST (U/L) | ||
| ≥1 × ULN | 55/190 (28.9) | 112/531 (21.1) |
| ≥1.5 × ULN | 16/190 (8.4) | 46/531 (8.7) |
| ≥2 × ULN | 11/190 (5.8) | 26/531 (4.9) |
| ≥3 × ULN | 6/190 (3.2) | 13/531 (2.4) |
| ≥5 × ULN | 1/190 (0.5) | 7/531 (1.3) |
| ≥10 × ULN | 0/190 (0.0) | 3/531 (0.6) |
| ≥20 × ULN | 0/190 (0.0) | 0/531 (0.0) |
| Potential Hy's law a | 0/190 (0.0) | 0/531 (0.0) |
Abbreviations: ALP, alkaline phosphatase; ALT, alanine aminotransferase; AST, aspartate aminotransferase; N, number of participants in the safety population; N1, number of participants with at least one nonmissing postbaseline value; n, number of participants within a specific category; ULN, upper limit of normal.
ALT or AST ≥3 × ULN and bilirubin total ≥2 × ULN and ALP <2 × ULN.
Suicidality in the atogepant arm
Lifetime history of suicidal ideation or behavior, per C‐SSRS assessment at baseline, was reported in 37/543 (6.8%) participants in the atogepant arm. In the open‐label period, 5/543 (0.9%) atogepant‐treated participants reported suicidal ideation and 2/543 (0.4%) reported suicidal behavior with actual attempts. Two participants had suicidal ideation type 5 (with specific plan and intent; classified as a serious TEAE). Both of these participants had a medical history of depression and one participant had a previously reported suicide attempt. One additional participant had suicidal ideation type 4 (with some intent, without a specific plan; considered a TEAE). This participant had a medical history of anxiety, depression, and posttraumatic stress disorder, and reported a history of suicidal ideation and behavior.
Laboratory values
Mean changes in laboratory values from baseline throughout the trial were small and comparable between the treatment groups. Differences in laboratory analytes between the atogepant and SC arms were not clinically meaningful. Laboratory results did not result in any discontinuations and none were reported as a serious AE.
Adverse events in the standard care arm
AEs were collected for the SC participants (Table S4 in supporting information). TEAEs were experienced in 154 of 196 (78.6%) SC participants and 71 (36.2%) were considered related to treatment. Most TEAEs were mild (68/196, 34.7%) or moderate (76/196, 38.8%), while 10/196 (5.1%) were severe. Treatment‐emergent SAEs occurred in 7/196 (3.6%) SC participants; only noncardiac chest pain occurred in more than 1 participant (n = 2). Five (5/196, 2.6%) SC participants discontinued due to an AE; no AEs leading to discontinuation occurred in more than one participant. The SC arm was included as a comparator arm for liver laboratory values. Any comparison of AE rates between the SC and atogepant treatment arms should be approached with caution, as the two arms are not comparable with respect to study design (e.g., SC treatment based on investigator judgment; could stop treatment at any time; participants’ prior medication history; etc.).
Efficacy outcomes
Mean (SE) MMDs at baseline for the atogepant group was 7.3 (2.6) days. During the first month (weeks 1–4) of treatment, the atogepant group reported a −3.8 (0.1) change in MMDs with consistently greater improvement during the subsequent months of treatment (Figure 2). Mean (SE) change in MMDs reached −5.2 (0.2) at weeks 49–52.
FIGURE 2.
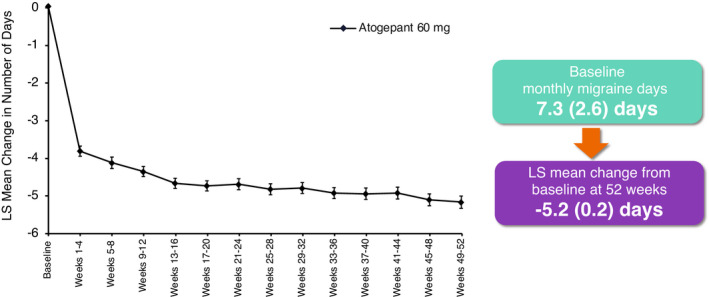
Change from baseline in number of monthly migraine days with once‐daily atogepant 60 mg (mITT population). Mixed‐effects model for repeated measures includes visit as a fixed effect, the baseline value as a covariate, and baseline‐by‐visit as an interaction term, with an unstructured covariance matrix. LS, least squares; mITT, modified intent‐to‐treat population
The proportions of participants with a ≥50%, ≥75%, and 100% reduction in MMDs were also assessed throughout the trial (Figure 3). During the first month (weeks 1–4) of treatment, a ≥50%, ≥75%, and 100% reduction in MMDs was reported by 60.4% (310/513), 37.2% (191/513), and 20.7% (106/513) of participants, respectively, in the atogepant group. At weeks 21–24, these responder rates increased to 75.3% (314/417), 55.4% (231/417), and 37.6% (157/417), respectively. Finally, at weeks 49–52, 84.2% (282/335), 69.9% (234/335), and 48.4% (162/335) of atogepant‐treated participants reported a ≥50%, ≥75%, and 100% reduction in MMDs, respectively.
FIGURE 3.
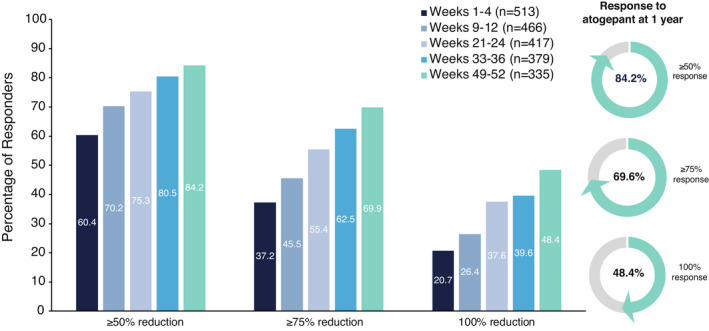
Proportion of responders with ≥50%, ≥75%, and 100% reduction in monthly migraine days (mITT population). For each individual, a percentage reduction in MMDs during each respective treatment period was calculated relative to the 28‐day baseline period. The percentage of all participants achieving a ≥50%, ≥75%, and 100% reduction in MMDs is plotted for each treatment period. LS, least squares; mITT, modified intent‐to‐treat; MMD, monthly migraine day
Consistent with the reduction observed in MMDs, a reduction in the use of acute medication was also observed throughout the trial for the atogepant group. At baseline, the mean (SE) number of acute medication use days was 6.6 (3.26) days per month in the atogepant group. During open‐label treatment with atogepant, the mean number of monthly acute medication use days was reduced by 4.0 days at weeks 1–4 and showed a gradual increase throughout treatment, with a reduction of 4.9 acute medication use days at weeks 49–52 (Figure 4). Efficacy measurements were not collected in the SC participants.
FIGURE 4.
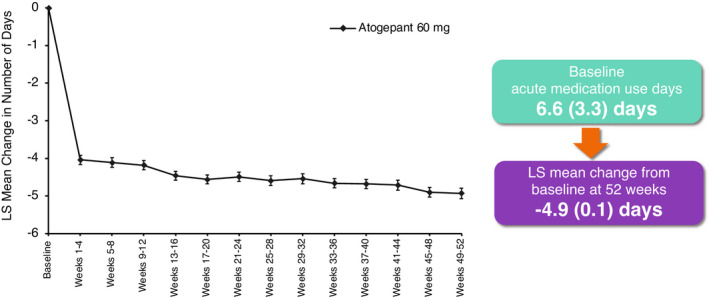
Change from baseline in number of monthly acute medication use days (mITT population). Mixed‐effects model for repeated measures includes visit as a fixed effect, the baseline value as a covariate, and baseline‐by‐visit as an interaction term, with an unstructured covariance matrix. LS, least squares; mITT, modified intent‐to‐treat
DISCUSSION
The results of this 52‐week, open‐label, randomized trial of once‐daily oral atogepant 60 mg were consistent with those previously reported in the randomized, placebo‐controlled, 12‐week phase 2b/3 trial and phase 3 (ADVANCE) trial, indicating atogepant is safe, well tolerated, and efficacious for the preventive treatment of migraine. 17 , 18
The current trial randomized 744 participants with a mean of 291.6 days of exposure to atogepant. Withdrawal by participant was the most common reason for discontinuation in both treatment arms. In a long‐term trial, withdrawal by participant can sometimes be attributed to lifestyle changes, such as moving, job changes, scheduling issues, etc. Lifestyle changes may have been more common in this trial due to the COVID‐19 pandemic. In contrast, discontinuations due to lack of efficacy or AEs in atogepant‐treated participants were rare.
The proportion of TEAEs reported in the atogepant arm was generally similar to those observed in the prior 12‐week trials. 17 , 18 Only 18% of TEAEs were considered related to treatment, which is lower than observed in the SC arm (36.2%) and on the lower end of the range reported in previous atogepant trials (15%–26%). 17 , 18 Common TEAEs considered related to treatment were constipation, nausea, and fatigue. Most treatment‐related TEAEs were mild to moderate in severity. The SC arm was used to contextualize long‐term safety of atogepant by providing comparative data in a migraine population treated with commonly used migraine preventive medications in a manner consistent with clinical practice. Due to the flexibility to change or discontinue preventive medications as needed, a direct comparison of specific TEAEs between the two treatment arms is not appropriate. No clinically meaningful changes from baseline in laboratory parameters in atogepant‐treated participants occurred throughout the study, providing additional evidence of the tolerability of atogepant. Laboratory analyte changes did not result in trial discontinuation or serious AEs in any participant. Overall, the safety results for atogepant‐treated participants in this year‐long trial were comparable to those observed in the two double‐blind, 12‐week, placebo‐controlled trials with similar rates of common TEAEs, such as nausea, upper respiratory tract infection, and constipation. 17 , 18
Changes in hepatic values were minimal and comparable between the atogepant and SC treatment arms. Of the 13 (2.4%) atogepant‐treated participants with elevations of ALT or AST ≥3, only 2 (0.4%) were determined to be “possibly” related to atogepant. The blinded clinical adjudication committee of independent liver experts evaluated the cases and found no hepatic safety issues were related to atogepant. Importantly, no cases of potential Hy's law occurred in any participants.
The efficacy of atogepant for the preventive treatment of migraine has been demonstrated in two double‐blind, placebo‐controlled trials (a 12‐week phase 2b/3 and a 12‐week phase 3). 17 , 18 All atogepant doses reduced MMDs and acute medication use days across the 12‐week treatment period compared to placebo. Additionally, 52%–62% of all atogepant dose groups in both trials achieved a 50% or greater reduction in MMDs (3‐month average) and monthly acute medication use was decreased by approximately 4 days at the end of each 12‐week trial. The efficacy results over 52 weeks reported here are consistent with these placebo‐controlled trials. Reductions in mean MMDs and acute medication use days were observed during the first 4 weeks (weeks 1–4) and sustained over the year‐long trial. Similarly, clinically relevant responder rates were observed early and increased throughout the trial. The proportions of participants achieving a ≥50%, ≥75%, and 100% reduction in MMDs from baseline to the end of the trial were approximately 84%, 70%, and 48%, respectively. Discontinuations due to lack of efficacy occurred in less than 1% of participants in the atogepant arm, indicating that the considerable response rates at the end of the trial were not simply due to selecting for responders as the trial progressed.
Monoclonal antibodies that target the CGRP pathway are another recent innovation for migraine preventive treatment. Open‐label trials of the CGRP‐targeted mAbs erenumab, galcanezumab, and fremanezumab each reported 60%–74% of participants experiencing a ≥50% reduction in MMDs from baseline to week 52. 23 , 24 , 25 Although findings may be limited by the study's attrition rate, here we reported that 84% of participants treated with atogepant 60 mg once daily demonstrated a ≥50% reduction in MMDs from baseline to week 52. Trials of erenumab and galcanezumab reported approximately 20% of participants experienced a 100% response compared to 48% of participants treated with atogepant after 1 year of treatment (100% response results not published for fremanezumab trial). 23 , 24 Eptinezumab long‐term open‐label results have yet to be reported. Direct comparison is limited because of differences in trial design, but generally, atogepant‐treated participants had higher rates of 50%, 75%, and 100% responses compared to participants in the trials of CGRP‐targeted mAbs.
Gastrointestinal AEs, including constipation, have been reported with CGRP‐targeted mAbs. 26 Consistent with these results, constipation was reported by 7.2% of participants receiving once‐daily atogepant 60 mg in this long‐term, open‐label clinical trial. Most cases of constipation were mild to moderate in severity, with one severe case (later downgraded to mild) and one participant discontinuing due to constipation (mild intensity). Our results suggest that constipation may be managed with over‐the‐counter medications and is unlikely to interrupt treatment. If atogepant is approved for the preventive treatment of migraine, real‐world studies will be needed to fully characterize its safety and tolerability profile. Given the relatively short half‐life of atogepant (approximately 11 h) compared to that of mAbs (27–31 days), clinicians would be able to discontinue atogepant treatment and rapidly reduce pharmacologically active concentrations in response to any AE that a patient experienced. 11 , 12 , 13 , 14 The ability to immediately discontinue treatment and remove atogepant from the circulation may also be appealing for women of childbearing potential who may become pregnant and need to adjust preventive treatment accordingly.
Persons with migraine are often undertreated or are treated with nonspecific oral preventives with suboptimal risk–benefit profiles. 27 The development of atogepant, an oral, once‐daily migraine preventive medication specifically designed for the treatment of migraine, will provide clinicians and patients with a novel treatment with demonstrated safety, tolerability, and efficacy.
This study has several potential limitations. First, the open‐label design did not permit formal statistical analyses to compare the efficacy of atogepant with placebo; however, the values observed were generally similar to those in the placebo‐controlled trials. 17 , 18 Second, the attrition rate was around 35%, which may affect the generalizability of these results; however, the sample size was calculated based on data from previous long‐term studies 21 , 22 to account for approximately 60% of participants completing 6 months of atogepant treatment and 65% of those completing 12 months of atogepant treatment. Additionally, discontinuations due to AEs or lack of efficacy in participants who received atogepant treatment were not common. Third, the flexibility for investigators to switch or stop a participant's SC medication throughout the trial did not permit direct comparisons of efficacy or AE rates between the SC and atogepant arms. Because of the nature of within‐group comparisons, the effects noted could be due to factors other than treatment efficacy. Differences in trial design and differences in definitions of outcomes may limit comparisons to trials of other migraine preventive medications. Finally, this trial was not designed to assess individuals with chronic migraine; an ongoing trial is evaluating atogepant in this population (NCT03855137). The strengths of this trial include the 52‐week duration, which allowed for a robust assessment of safety and tolerability with long‐term use of atogepant.
In conclusion, daily use of oral atogepant 60 mg for the preventive treatment of migraine during this 1‐year, open‐label trial was safe and well tolerated. Atogepant use reduced MMDs, moderate/severe headache days, and acute medication use days. Efficacy was observed early, was sustained over 1 year, and suggested an increase in efficacy with the duration of treatment. Results support the potential for once‐daily oral atogepant 60 mg as a long‐term preventive treatment of migraine.
AUTHOR CONTRIBUTIONS
Study concept and design: Lawrence Severt. Acquisition of data: Lawrence Severt. Analysis and interpretation of data: Messoud Ashina, Susan Hutchinson, Stewart Tepper, Uwe Reuter. Drafting of the manuscript: Messoud Ashina, Susan Hutchinson, Stewart Tepper, Uwe Reuter. Revising it for intellectual content: Messoud Ashina, Stewart J. Tepper, Uwe Reuter, Andrew M. Blumenfeld, Susan Hutchinson, Jing Xia, Rosa Miceli, Lawrence Severt, Michelle Finnegan, Joel M. Trugman. Final approval of the completed manuscript: Messoud Ashina, Stewart J. Tepper, Uwe Reuter, Andrew M. Blumenfeld, Susan Hutchinson, Jing Xia, Rosa Miceli, Lawrence Severt, Michelle Finnegan, Joel M. Trugman.
FUNDING INFORMATION
This study was sponsored by Allergan (prior to its acquisition by AbbVie). Allergan (now AbbVie) also participated in the study design, research, analysis, data collection, interpretation of data, reviewing, and approval of the publication. All authors had access to relevant data and participated in the drafting, review, and approval of this publication. No honoraria or payments were made for authorship.
CONFLICT OF INTEREST
Messoud Ashina (MA) is a consultant, speaker, or scientific advisor for AbbVie/Allergan, Alder, Amgen, Biohaven, Eli Lilly, Lundbeck, Novartis, Pfizer and Teva, and primary investigator for AbbVie/Allergan, Alder, Amgen, Eli Lilly, Lundbeck, Novartis, and Teva trials. MA has no ownership interest and does not own stocks of any pharmaceutical company. MA serves as associate editor of Cephalalgia, associate editor of The Journal of Headache and Pain, associate editor of Brain. Stewart J. Tepper (SJT) has served as a consultant for Acorda, Alder, Alexsa, Alphasights, Amgen, ATI, Axsome Therapeutics, BioDelivery Sciences International, Biohaven, Charleston Labs, Decision Resources, DeepBench, Dr. Reddy's, electroCore, Eli Lilly, eNeura, GLG, GSK, Guidepoint Global, Impel, M3 Global Research, Magellan Rx Management, Medicxi, Navigant Consulting, Neurolief, Nordic BioTech, Novartis, Pfizer, Reckner Healthcare, Relevale, Satsuma, Scion Neurostim, Slingshot Insights, Sorrento, Sudler and Hennessey, Teva, Theranica, Thought Leader Select, Trinity Partners, XOC, and Zosano. SJT receives a salary as editor‐in‐chief of Headache Currents from the American Headache Society and receives royalties for books published by Springer. Uwe Reuter (UR) is a consultant, speaker, or scientific advisor for AbbVie, Amgen, CoLucid, Lilly, Lundbeck, Medscape, Novartis, Perfood, Pfizer, and Teva. UR serves as associate editor of Frontiers in Neurology and of The Journal of Headache and Pain. UR is board member of the European Headache Federation. Andrew M. Blumenfeld (AMB) has served on advisory boards for AbbVie, Aeon, Alder, Allergan, Amgen, Axsome, Biohaven, Impel, Lundbeck, Lilly, Novartis, Revance, Teva, Theranica, and Zoscano and has received funding for speaking from AbbVie, Allergan, Amgen, Biohaven, Lundbeck, Lilly, and Teva. AMB is a consultant for AbbVie, Alder, Allergan, Amgen, Biohaven, Lilly, Lundbeck, Novartis, Teva, and Theranica and has received grant support from Allergan and Amgen. He is a contributing author for AbbVie, Allergan, Amgen, Biohaven, Novartis, Lilly, and Teva. Susan Hutchinson (SH) has served on advisory boards for AbbVie, Alder/Lundbeck, Amgen, Biohaven, Currax, electroCore, Eli Lilly, Impel, Novartis, Teva, Theranica, and Upsher‐Smith. SH is on the speakers bureaus for AbbVie, Amgen, Biohaven, electroCore, Eli Lilly, Lundbeck, Novartis, Teva, Theranica, and Upsher‐Smith. Joel M. Trugman is an employee of AbbVie and may hold AbbVie stock. Michelle Finnegan, Jing Xia, Rosa Miceli, and Lawrence Severt were employees of AbbVie at the time of study conduct and may hold AbbVie stock.
CLINICAL TRIALS REGISTRY
ClinicalTrials.gov (Identifier: NCT03700320).
Supporting information
Appendix S1
ACKNOWLEDGMENTS
Allergan (now AbbVie) and the authors thank all the trial participants and investigators for their participation in and dedication to this trial. We also thank Cherisse Loucks, PhD, formerly of AbbVie, for medical writing assistance for the development of this publication.
Ashina M, Tepper SJ, Reuter U, et al. Once‐daily oral atogepant for the long‐term preventive treatment of migraine: Findings from a multicenter, randomized, open‐label, phase 3 trial. Headache. 2023;63:79‐88. doi: 10.1111/head.14439
DATA AVAILABILITY STATEMENT
AbbVie is committed to responsible data sharing regarding the clinical trials we sponsor. This includes access to anonymized, individual, and trial‐level data (analysis data sets), as well as other information (e.g., protocols, clinical study reports, or analysis plans), as long as the trials are not part of an ongoing or planned regulatory submission. This includes requests for clinical trial data for unlicensed products and indications. These clinical trial data can be requested by any qualified researchers who engage in rigorous, independent scientific research, and will be provided following review and approval of a research proposal, Statistical Analysis Plan (SAP), and execution of a Data Sharing Agreement (DSA). Data requests can be submitted at any time after approval in the United States and Europe and after acceptance of this manuscript for publication. The data will be accessible for 12 months, with possible extensions considered. For more information on the process or to submit a request, visit the following link: https://www.abbvie.com/our‐science/clinical‐trials/clinical‐trials‐data‐and‐information‐sharing/data‐and‐information‐sharing‐with‐qualified‐researchers.html.
REFERENCES
- 1. Ashina M. Migraine. N Engl J Med. 2020;383:1866‐1876. [DOI] [PubMed] [Google Scholar]
- 2. Ashina M, Katsarava Z, Do TP, et al. Migraine: epidemiology and systems of care. Lancet. 2021;397:1485‐1495. [DOI] [PubMed] [Google Scholar]
- 3. Lampl C, Thomas H, Stovner LJ, et al. Interictal burden attributable to episodic headache: findings from the Eurolight project. J Headache Pain. 2016;17:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Serrano D, Lipton RB, Scher AI, et al. Fluctuations in episodic and chronic migraine status over the course of 1 year: implications for diagnosis, treatment and clinical trial design. J Headache Pain. 2017;18:101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Schulman E. Refractory migraine—a review. Headache. 2013;53:599‐613. [DOI] [PubMed] [Google Scholar]
- 6. Dahlof C, Loder E, Diamond M, Rupnow M, Papadopoulos G, Mao L. The impact of migraine prevention on daily activities: a longitudinal and responder analysis from three topiramate placebo‐controlled clinical trials. Health Qual Life Outcomes. 2007;5:56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Blumenfeld AM, Bloudek LM, Becker WJ, et al. Patterns of use and reasons for discontinuation of prophylactic medications for episodic migraine and chronic migraine: results from the second international burden of migraine study (IBMS‐II). Headache. 2013;53:644‐655. [DOI] [PubMed] [Google Scholar]
- 8. American Headache Society . The American Headache Society position statement on integrating new migraine treatments into clinical practice. Headache. 2019;59:1‐18. [DOI] [PubMed] [Google Scholar]
- 9. Ashina M, Buse DC, Ashina H, et al. Migraine: integrated approaches to clinical management and emerging treatments. Lancet. 2021;397:1505‐1518. [DOI] [PubMed] [Google Scholar]
- 10. Ashina M, Terwindt GM, Al‐Karagholi MA, et al. Migraine: disease characterisation, biomarkers, and precision medicine. Lancet. 2021;397:1496‐1504. [DOI] [PubMed] [Google Scholar]
- 11. EMGALITY (Galcanezumab‐gnim) injection [package insert]. Eli Lilly and Company; 2019. [Google Scholar]
- 12. AIMOVIG (Erenumab‐aooe) [package insert]. Amgen Inc.; 2020. [Google Scholar]
- 13. AJOVY (Fremanezumab‐vfrm) injection [package insert]. Teva Pharmaceuticals USA, Inc.; 2020. [Google Scholar]
- 14. VYEPTI (Eptinezumab‐jjmr) injection [package insert]. Lundbeck Seattle BioPharmaceuticals, Inc.; 2020. [Google Scholar]
- 15. Robblee J, Devick KL, Mendez N, Potter J, Slonaker J, Starling AJ. Real‐world patient experience with erenumab for the preventive treatment of migraine. Headache. 2020;60:2024‐2025. [DOI] [PubMed] [Google Scholar]
- 16. FDA approves QULIPTA (Atogepant), the first and only oral CGRP receptor antagonist specifically developed for the preventive treatment of migraine [press release]. AbbVie Inc.; 2021. [Google Scholar]
- 17. Goadsby PJ, Dodick DW, Ailani J, Trugman JM, Finnegan M, Lu K. Safety, tolerability, and efficacy of orally administered atogepant for the prevention of episodic migraine in adults: a double‐blind, randomised phase 2b/3 trial. Lancet Neurol. 2020;19:727‐737. [DOI] [PubMed] [Google Scholar]
- 18. Ailani J, Lipton RB, Goadsby PJ, et al. Atogepant significantly reduces mean monthly migraine days in the phase 3 trial (ADVANCE) for the prevention of migraine [abstract MTV20‐OR‐018]. Cephalalgia. 2020;40(1 suppl):15‐16. [Google Scholar]
- 19. Bhoi SK, Kalita J, Misra UK. Is 6 months of migraine prophylaxis adequate? Neurol Res. 2013;35:1009‐1014. [DOI] [PubMed] [Google Scholar]
- 20. Headache Classification Committee of the International Headache Society (IHS) . The International Classification of Headache Disorders, 3rd edition. Cephalalgia. 2018;38:1‐211. [DOI] [PubMed] [Google Scholar]
- 21. Aurora SK, Winner P, Freeman MC, et al. OnabotulinumtoxinA for treatment of chronic migraine: pooled analyses of the 56‐week PREEMPT clinical program. Headache. 2011;51:1358‐1373. [DOI] [PubMed] [Google Scholar]
- 22. Rapoport A, Mauskop A, Diener HC, Schwalen S, Pfeil J. Long‐term migraine prevention with topiramate: open‐label extension of pivotal trials. Headache. 2006;46:1151‐1160. [DOI] [PubMed] [Google Scholar]
- 23. Ashina M, Goadsby PJ, Reuter U, et al. Long‐term efficacy and safety of erenumab in migraine prevention: results from a 5‐year, open‐label treatment phase of a randomized clinical trial. Eur J Neurol. 2021;28:1716‐1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Camporeale A, Kudrow D, Sides R, et al. A phase 3, long‐term, open‐label safety study of galcanezumab in patients with migraine. BMC Neurol. 2018;18:188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Goadsby PJ, Silberstein SD, Yeung PP, et al. Long‐term safety, tolerability, and efficacy of fremanezumab in migraine: a randomized study. Neurology. 2020;95:e2487‐e2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Goadsby PJ, Reuter U, Hallstrom Y, et al. A controlled trial of erenumab for episodic migraine. N Engl J Med. 2017;377:2123‐2132. [DOI] [PubMed] [Google Scholar]
- 27. Olesen J, Ashina M. Emerging migraine treatments and drug targets. Trends Pharmacol Sci. 2011;32:352‐359. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1
Data Availability Statement
AbbVie is committed to responsible data sharing regarding the clinical trials we sponsor. This includes access to anonymized, individual, and trial‐level data (analysis data sets), as well as other information (e.g., protocols, clinical study reports, or analysis plans), as long as the trials are not part of an ongoing or planned regulatory submission. This includes requests for clinical trial data for unlicensed products and indications. These clinical trial data can be requested by any qualified researchers who engage in rigorous, independent scientific research, and will be provided following review and approval of a research proposal, Statistical Analysis Plan (SAP), and execution of a Data Sharing Agreement (DSA). Data requests can be submitted at any time after approval in the United States and Europe and after acceptance of this manuscript for publication. The data will be accessible for 12 months, with possible extensions considered. For more information on the process or to submit a request, visit the following link: https://www.abbvie.com/our‐science/clinical‐trials/clinical‐trials‐data‐and‐information‐sharing/data‐and‐information‐sharing‐with‐qualified‐researchers.html.