

1. WHAT IS NEW OR DIFFERENT
Addition of recently described subtypes of monogenic diabetes, including causes associated with diabetes in infancy (CNOT1, ONECUT1, YIPF5, EIF2B1, KCNMA1); and genetic causes associated with diabetes later in life (TRMT10A, DNAJC3, KCNK16, DUT).
The expanding list of genes causing monogenic diabetes further emphasizes comprehensive next‐generation sequencing (NGS) as the best approach to allow for early molecular diagnosis that can guide treatment, rather than phenotype‐based targeted testing, particularly for neonatal diabetes (NDM).
Use of increasingly available publicly accessible information about specific variants to allow for the appropriate classification of pathogenicity of gene variants according to guidelines of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology (ACMG/AMP), bolstered by the establishment of international Monogenic Diabetes Expert Panels for gene curation and variant curation with the elaboration of gene‐specific rules (https://clinicalgenome.org/affiliation/50016).
Further understanding of the neuroendocrine aspects of ATP‐sensitive potassium channel (KATP) related NDM (KATP‐NDM) is now included.
Clarification that a small fraction of NDM is likely to be autoimmune type 1 diabetes (T1D) and autoimmune etiology distinct from T1D occurring in Trisomy 21.
Among young persons with diabetes with a clinical diagnosis of type 2 diabetes (T2D), a low but significant fraction can be found to carry pathogenic MODY mutations; highlighting the importance of considering a monogenic cause even when obesity may be present.
The rate of diabetes‐related complications may be lower in HNF1A diabetes who have been treated with sulfonylureas (SU).
Liver (with or without pancreas) transplantation can improve the outcomes of individuals with Wolcott‐Rallison syndrome.
2. EXECUTIVE SUMMARY AND RECOMMENDATIONS
2.1. General aspects of monogenic diabetes
Monogenic diabetes is uncommon, but accounts for ~2.5%–6.5% of pediatric diabetes B.
NGS enables the simultaneous analysis of multiple genes at a lower cost per gene providing comprehensive testing B.
NGS is the recommended methodology for study of suspected monogenic diabetes, unless a very specific and highly suggestive clinical scenario is present, such as glucokinase (GCK) mutations, which cause a distinct phenotype of asymptomatic and stable mild fasting hyperglycemia B.
Results of genetic testing should be reported and presented to families in a clear and unambiguous manner E.
Referral to a specialist in monogenic diabetes or an interested clinical genetics unit is suggested to guide specific management considerations and/or facilitate genetic testing of other related affected or pre‐symptomatic individuals E.
2.2. Neonatal diabetes
All infants diagnosed with diabetes in the first 6 months of life are recommended to have immediate molecular genetic testing B.
Genetic testing maybe be considered in infants diagnosed between 6 and 12 months, especially in those without islet autoantibodies or who have other features suggestive of a monogenic cause C.
A molecular genetic diagnosis of NDM provides essential information regarding treatment options, associated features, and diabetes course that may have a significant clinical benefit B.
Treatment with SU, especially glibenclamide (also known as glyburide), is recommended for NDM due to KCNJ11 and ABCC8 abnormalities B.
Glibenclamide significantly improved neurological and neuro‐psychological abnormalities in individuals with neonatal onset diabetes due to KCNJ11 or ABCC8 mutations. Earlier treatment initiation was associated with greater benefits B.
2.3. Maturity onset diabetes of the young
- The diagnosis of maturity onset diabetes of the young (MODY) is recommended in the following scenarios:
- A family history of diabetes in a parent and first‐degree relatives of that affected parent in persons with diabetes who lack the characteristics of T1D and T2D B.
Testing for GCK‐MODY, which is the commonest cause of persistent, incidental hyperglycemia in the pediatric population, is recommended for mild stable fasting hyperglycemia that does not progress B.
In familial autosomal dominant symptomatic diabetes, mutations in the HNF1A gene (HNF1A‐MODY) should be considered as the first diagnostic possibility B.
Specific features can suggest subtypes of MODY, such as renal developmental disease or renal cysts (HNF1B‐MODY), macrosomia and/or neonatal hypoglycemia (HNF4A‐MODY), exocrine pancreatic dysfunction or pancreatic cysts (CEL‐MODY), or hearing impairment and maternal inheritance of diabetes (mitochondrial diabetes) C.
- Obesity alone should not preclude genetic testing in young persons, especially if: C
- family history is strongly suggestive of autosomal dominant inheritance of diabetes
- if some affected family members are NOT obese
- and/or, there are no other features of metabolic syndrome.
Some forms of MODY are sensitive to SU, such as HNF1A‐MODY and HNF4A‐MODY B
Mild fasting hyperglycemia due to GCK‐MODY is not progressive during childhood. These persons do not develop complications B and do not respond to low dose insulin or oral agents C. They should not receive treatment.
- Establishing the correct molecular diagnosis of MODY is suggested for the following reasons: C
- avoids misdiagnosis as T1D or T2D
- may offer more accurate prognosis of risk of complications
- may avoid stigma and limitation of employment opportunity (especially in the case of GCK‐MODY)
- may enable prediction of risk in relatives, including offspring
- can be cost‐effective when appropriately selected individuals are screened
3. INTRODUCTION
Monogenic diabetes results from one or more defects in a single gene or chromosomal locus. The disease may be inherited within families as a dominant, recessive, or non‐Mendelian trait or may present as a spontaneous case due to a de novo mutation.
Monogenic diabetes has been categorized as neonatal or early infancy diabetes (Table 1), MODY (Table 2), diabetes associated with extra‐pancreatic features, and monogenic insulin resistance (IR) syndromes (Table 3).
TABLE 1.
Monogenic subtypes of neonatal and infancy‐onset diabetes
| Gene | Locus | Inheritance | Other clinical features | Reference |
|---|---|---|---|---|
| Abnormal pancreatic development: | ||||
| PLAGL1/HYMAI | 6q24 | Variable (imprinting) | TNDM ± macroglossia ± umbilical hernia | 20 |
| ZFP57 | 6p22.1 | Recessive | TNDM (multiple hypomethylation syndrome) ± macroglossia ± developmental delay ± umbilical defects ± congenital heart disease | 29 |
| PDX1 | 13q12.1 | Recessive | PNDM + pancreatic agenesis (steatorrhea) | 264 |
| PTF1A | 10p12.2 | Recessive | PNDM + pancreatic agenesis (steatorrhea) + cerebellar hypoplasia/aplasia + central respiratory dysfunction | 265 |
| PTF1A enhancer | 10p12.2 | Recessive | PNDM + pancreatic agenesis without CNS features | 133 |
| HNF1B | 17q21.3 | Dominant | TNDM + pancreatic hypoplasia and renal cysts | 23 |
| RFX6 | 6q22.1 | Recessive | PNDM + intestinal atresia + gall bladder agenesis | 266, 267 |
| GATA6 | 18q11.1‐q11.2 | Dominant | PNDM + pancreatic agenesis + congenital heart defects + biliary abnormalities | 134 |
| GATA4 | 8p23.1 | Dominant | PNDM + pancreatic agenesis + congenital heart defects | 268 |
| GLIS3 | 9p24.3‐p23 | Recessive | PNDM + congenital hypothyroidism + glaucoma + hepatic fibrosis + renal cysts | 269 |
| NEUROG3 | 10q21.3 | Recessive | PNDM + enteric anendocrinosis (malabsorptive diarrhea) | 270 |
| NEUROD1 | 2q32 | Recessive | PNDM + cerebellar hypoplasia + visual impairment + deafness | 271 |
| PAX6 | 11p13 | Recessive | PNDM + microphthalmia + brain malformations | 272 |
| MNX1 | 7q36.3 | Recessive | PNDM + developmental delay + sacral agenesis + imperforate anus | 4 |
| NKX2‐2 | 20p11.22 | Recessive | PNDM + developmental delay + hypotonia + short stature + deafness + constipation | 273 |
| CNOT1 | 16q21 | Spontaneous | PNDM + pancreatic agenesis + holoprosencephaly | 274 |
| ONECUT1 | 15q21.3 | Recessive | PNDM + pancreatic hypoplasia + gall bladder hypoplasia | 275 |
| Abnormal β‐cell function: | ||||
| KCNJ11 | 11p15.1 | Spontaneous or dominant | PNDM/ TNDM ± DEND | 41 |
| ABCC8 | 11p15.1 | Spontaneous, dominant or recessive | TNDM/PNDM ± DEND | 42 |
| INS | 11p15.5 | Recessive | Isolated PNDM or TNDM | 24 |
| GCK | 7p15‐p13 | Recessive | Isolated PNDM | 107 |
| SLC2A2 (GLUT2) | 3q26.1‐q26.3 | Recessive | Fanconi‐Bickel syndrome: PNDM + hypergalactosemia, liver dysfunction | 276 |
| SLC19A2 | 1q23.3 | Recessive | Roger's syndrome: PNDM + thiamine‐responsive megaloblastic anemia, sensorineural deafness | 277 |
| KCNMA1 | 10q22.3 | Spontaneous | PNDM (not all cases) + developmental delay + intestinal malformations + cardiac malformations + bone dysplasia + dysmorphic features | 278 |
| Destruction of β cells: | ||||
| INS | 11p15.5 | Spontaneous or dominant | Isolated PNDM | 90 |
| EIF2AK3 | 2p11.2 | Recessive | Wolcott‐Rallison syndrome: PNDM + skeletal dysplasia + recurrent liver dysfunction | 98 |
| IER3IP1 | 18q21.2 | Recessive | PNDM + microcephaly + lissencephaly + epileptic encephalopathy | 279 |
| FOXP3 | Xp11.23‐p13.3 | X‐linked, recessive | IPEX syndrome (autoimmune enteropathy, eczema, autoimmune hypothyroidism, elevated IgE) | 280 |
| WFS1 | 4p16.1 | Recessive | PNDM a + optic atrophy ± diabetes insipidus ± deafness | 189 |
| WFS1 | 4p16.1 | Dominant | PNDM or infancy‐onset diabetes + congenital cataracts + deafness | 281 |
| EIF2B1 | 12q24.31 | Spontaneous | PNDM + episodic hepatic dysfunction | 282 |
| YIPF5 | 5q31.3 | Recessive | PNDM + severe microcephaly + epilepsy | 283 |
| STAT3 | 17q21.2 | Spontaneous | PNPM + enteropathy + other autoimmunity such as cytopenias | 116 |
| CTLA4 | 2q33.2 | Spontaneous | Lymphoproliferative syndrome + enteropathy + cytopenias + diabetes + thyroiditis | 127 |
| ITCH | 20q11.22 | Recessive | PNDM + facial dysmorphism + multi‐system autoimmunity | 128 |
| IL2RA | 10p15.1 | Recessive | Lymphoproliferation + multi‐system autoimmunity + diabetes | 129 |
| LRBA | 4q31.3 | Recessive | PNDM + enteropathy + hypothyroidism + autoimmune hemolytic anemia | 118 |
TABLE 2.
Most important subtypes of MODY and associated clinical features
| Gene | Locus | Clinical features | Treatment | References |
|---|---|---|---|---|
| GCK | 7p15‐p13 | Mild asymptomatic hyperglycemia | None | 284 |
| HNF1A | 12q24.2 | Renal glucosuria | Sulphonylurea | 285 |
| HNF4A | 20q12‐q13.1 | Macrosomia and neonatal hypoglycaemia, renal Fanconi syndrome (mutation specific) | Sulphonylurea | 286 |
| HNF1B | 17q12 | Renal developmental abnormalities, genital tract malformations | Insulin | 287 |
| KCNJ11 | 11p15 | Proband or relatives may have history of TNDM and/or neuropsychological difficulties | High‐dose sulphonylurea | |
| ABCC8 | 11p15 | Proband or relatives may have history of TNDM and/or neuropsychological difficulties | High‐dose sulphonylurea |
TABLE 3.
Classification of Syndromes of severe insulin resistance
| IR syndrome subtype | Gene (inheritance) | Leptin | Adiponectin | Other clinical features | |
|---|---|---|---|---|---|
| Primary insulin signaling defects | Receptor defect | INSR (AR or AD) | Decreased | Normal or elevated | No dyslipidemia or hepatic stetosis |
| Post receptor defects | AKT2, TBC1D4 (AD) | Elevated fasting triglycerides and LDL‐cholesterol, hepatic steatosis, diabetes (AKT2) | |||
| Adipose tissue abnormalities | Monogenic obesity |
MC4R (AD) LEP, LEPR, POMC (AR) Others |
Increased (low in LEP) |
Tall stature (MC4R) Hypogonadism (LEP) Hypoadrenalism (POMC) |
|
| Congenital generalized lipodystrophy |
AGPAT2, BSCL2 (AR) Others |
Decreased | Decreased |
Severe dyslipidemia (high triglycerides, low HDL‐ cholesterol) Hepatic stetosis |
|
| Partial lipodystrophy |
LMNA, PPARG, PIK3R1 (AD) Others |
Variable |
Myopathy and cardiomyopathy (LMNA) Pseudo‐acromegaly (PPARG) SHORT syndrome with partial lipodystrophy, and diabetes (PIK3R1) |
||
| Complex syndromes | Alström | ALMS1 (AR) | Cone‐rod dystrophy leading to blindness, sensorineural hearing loss, diabetes and cardiomyopathy | ||
| Bardet‐Biedl | BBS1 to BBS18 (mostly AR) | Cone‐rod dystrophy, obesity, renal dysfunction, polydactyly, learning disabilities, hypogonadism and diabetes | |||
| DNA damage repair disorders |
WRN (AR) BLM (AR) |
Scleroderma‐like skin changes, cataracts, increased cancer risk, atherosclerosis and diabetes; Sun‐sensitive, telangiectatic skin changes; increased cancer risk and diabetes |
|||
| Primordial dwarfism | PCNT (AR) | Microcephalic osteodysplastic primordial dwarfism and diabetes | |||
Abbreviations: AD, autosomal dominant; AR, autosomal recessive.
Source: modified from Parker et al. Reference 228.
4. CLINICAL RELEVANCE OF DIAGNOSING MONOGENIC DIABETES
Identification of children with monogenic diabetes usually improves their clinical care. 1
Making a specific molecular diagnosis helps predict the expected clinical course of the disease and guides the most appropriate management, including pharmacological treatment, in a particular person with diabetes.
Characterizing the specific molecular diagnosis has important implications for the family as it informs genetic counseling. It also frequently triggers extended genetic testing in other family members with diabetes or hyperglycemia who may also carry a causal mutation, thereby improving the classification of diabetes. 2 , 3
5. SELECTING CANDIDATES FOR MOLECULAR TESTING
In contrast to T1D and T2D, where there is no single definitive diagnostic test, molecular genetic testing is both sensitive and specific for diagnosing monogenic diabetes. Appropriate informed consent/assent must be prospectively obtained from the affected person and/or legal guardians and should be strongly considered in persons with a suspected monogenic cause. Genetic testing is currently available (and may be free of charge on a research basis in certain academic institutions) in many countries around the world: https://www.diabetesgenes.org; http://monogenicdiabetes.uchicago.edu; www.mody.no; http://euro-wabb.org; https://www.ospedalebambinogesu.it/test-genetici-89757/; https://robertdebre.aphp.fr/equipes‐cliniques/pole‐biologie/genetique/genetique‐moleculaire/#1461944418‐1‐40 and several commercial laboratories.
NGS enables the simultaneous analysis of multiple genes at a lower cost per gene and has mostly replaced single gene testing by Sanger sequencing or other methods. 4 , 5 , 6 , 7 , 8 Such NGS panels provide an efficient means of comprehensive testing that results in earlier genetic diagnosis, which in turn facilitates appropriate management as well as monitoring for other associated features before they become clinically apparent. It is important to note that NGS testing panels are still expensive, so it remains appropriate to use a judicious approach to selecting persons with diabetes for comprehensive molecular testing and in specific circumstances (such as living in a resource‐poor setting), Sanger sequencing of a limited number of the most treatment‐relevant genes may be the most practical approach. Moreover, some NGS panels have included genes lacking robust evidence for a causal role in monogenic diabetes and this can result in misdiagnosis and confusion for the person with diabetes and other affected family members; however, increasing international collaboration between testing laboratories has begun to limit such examples of inaccurately reported genetic testing results. Sanger sequencing remains appropriate as an efficient cost‐effective method for testing of a variant found by NGS testing of the first individual in other affected or at‐risk family members (cascade testing).
In NDM, genetic testing may be cost saving because of improved cheaper treatment; testing for MODY in appropriate populations can also be cost‐effective. 2 , 3 , 9 Targeted gene sequencing, however, may still be appropriate for some persons with diabetes; for example, a pregnant female with mild fasting hyperglycemia, in whom a rapid test to identify a GCK mutation will inform management of the pregnancy. For most people with diabetes suspected to have a monogenic cause, NGS provides an optimal approach for clinical care as it provides a genetic diagnosis that often precedes the development of additional clinical features, informs prognosis, and guides clinical management. 2 , 3 , 9
6. WHEN TO SUSPECT A DIAGNOSIS OF T1D IN CHILDREN MAY NOT BE CORRECT?
Features that suggest monogenic diabetes in children initially thought to have T1D are listed below. Except for the age of diagnosis less than 6 months, none of these are pathognomonic and should be considered together rather than in isolation:
Diabetes presenting before 6 months of age (as T1D is extremely rare in this age group), or consider NDM if the diagnosis is between 6 and 12 months and there is no evidence of autoimmunity or if the person with diabetes has other features such as congenital defects, or an unusual family history. 10 , 11
Family history of diabetes in one parent and other first‐degree relatives of that affected parent.
Absence of islet autoantibodies, especially if checked at diagnosis.
Preserved β‐cell function, with low insulin requirements and detectable C‐peptide (either in blood or urine) over an extended partial remission phase (at least 5 years after diagnosis).
7. WHEN TO SUSPECT A DIAGNOSIS OF T2D IN CHILDREN MAY NOT BE CORRECT
In young people, T2D often presents around puberty and the majority are obese. As there is no diagnostic test for T2D and because obesity has become so common in children, children and adolescents with monogenic diabetes may also be obese and can be very difficult to distinguish from T2D. 1 One recent study found that 3% of obese youth with presumed T2D in fact carried pathogenic monogenic diabetes variants. 5 Features that suggest monogenic diabetes in young people with suspected T2D are listed below:
Lack of consistent severe obesity among affected family members.
Lack of consistent acanthosis nigricans and/or other markers of metabolic syndrome (hypertension, low HDL‐cholesterol, etc.) among affected family members.
Family history of diabetes in one parent and other first‐degree relatives of that affected parent, especially if any affected family member lacks obesity and other markers of metabolic syndrome.
Unusual distribution of fat, such as central fat with thin or muscular extremities.
8. INTERPRETATION OF GENETIC FINDINGS
Despite the obvious clinical benefits derived from genetic diagnostic services:
Care needs to be exercised in the interpretation of genetic findings. The way the clinician interprets the genetic report will have a major effect on the future clinical management of the person with diabetes and his/her family.
Results should be presented in a clear and unambiguous way to ensure that both clinicians and the person with diabetes and their families receive adequate and understandable information. Specific recommendations describing the information that should be included in the molecular genetics laboratory report for MODY testing have been published. 12
This includes the method used for mutation screening, limitations of the test, classification of the variant as pathogenic/likely pathogenic or of uncertain significance (with supporting evidence included where appropriate), and information about the likelihood of the disease being inherited by the offspring.
The laboratory reporting the results should adhere to the ACMG/AMP variant classification guidelines. 13 Many genetic testing laboratories have been participating in the Monogenic Diabetes Variant Curation Expert Panel (https://clinicalgenome.org/affiliation/50016/) that has provided more definitive curation of hundreds of variants that are freely accessible and recognized by the US FDA. This resource can be utilized to check whether a variant in question has been deemed “pathogenic” or “likely pathogenic” in which case there should be confidence that this is the cause of diabetes, or if “benign” or “likely benign” that another cause should be considered. Whether or not the testing report follows ACMG/AMP guidelines, when testing reveals a variant of uncertain significance (VUS), or when predictive testing of asymptomatic individuals is requested, consultation with an expert center with experience in monogenic diabetes can often provide additional insight on the interpretation and recommendations of how to proceed.
9. SPECIFIC SUBTYPES OF MONOGENIC DIABETES AND THEIR MANAGEMENT
In children, the majority of cases of monogenic diabetes result from mutations in genes causing β‐cell loss or dysfunction, although diabetes can rarely occur from mutations resulting in very severe IR. From a clinical perspective, specific scenarios when a diagnosis of monogenic diabetes should be considered include:
Diabetes presenting before 6 months of age, which is known as NDM.
Autosomal dominant familial mild hyperglycemia or diabetes.
Diabetes associated with extra‐pancreatic features (such as, for example, congenital heart or gastrointestinal defects, brain malformations, severe diarrhea, or other autoimmune conditions in a very young child).
Monogenic IR syndromes (see below: characterized by high insulin levels or high insulin requirements; abnormal distribution of fat with a lack of subcutaneous fat, especially in extremities; dyslipidemia, especially high triglycerides; and/or significant acanthosis nigricans).
9.1. Neonatal diabetes diagnosed within the first 6–12 months of life
All infants diagnosed under 6 months should have genetic testing for a monogenic cause, regardless of islet autoantibody status.
The clinical presentation of autoimmune T1D may rarely occur before age 6 months 11 , 14 ; a recent study suggested approximately 4% of cases may be T1D (see section of autoimmune monogenic diabetes). 15
One recent study observed trisomy 21 in a much greater than expected fraction of NDM individuals, with the conclusion that trisomy 21 can cause an autoimmune form of diabetes that appears to be distinct from the more common autoimmune T1D. 16
Some cases of NDM can be diagnosed between 6–12 months 17 , 18 although the vast majority of these older infants with diabetes have T1D. Reasons to consider genetic testing in those diagnosed between 6–12 months include: negative autoantibody testing, extra‐pancreatic features such as gastrointestinal anomalies or congenital defects, unusual family history, or even the development of multiple autoimmune disorders at a young age.
Approximately half will require lifelong treatment to control hyperglycemia and are denominated as PNDM.
In the remaining cases, known as transient neonatal diabetes (TNDM), diabetes will remit within a few weeks or months although it might relapse later in life.
PNDM and TNDM present more frequently isolated or is the first feature to be noted.
Some infants with diabetes show a variety of associated extra‐pancreatic clinical features that may point to a particular gene; however, because these features often are not apparent initially, they will not always be helpful in guiding genetic testing and instead, early comprehensive testing will often allow for the genetic testing result to precede the recognition of other features (Table 1).
Many infants with NDM are born small for gestational age, which reflects a prenatal deficiency of insulin secretion as insulin exerts potent growth‐promoting effects during intrauterine development. 19
9.2. Transient neonatal diabetes from imprinting anomalies on 6q24
The genetic basis of TNDM has been mostly uncovered: approximately two‐thirds of cases are caused by abnormalities in an imprinted region on chromosome 6q24. 20 , 21
Activating mutations in either of the genes encoding the two subunits of the ATP‐sensitive potassium (KATP) channel of the β‐cell membrane (KCNJ11 or ABCC8) cause the majority of the remaining cases (KATP‐NDM). 22
A minority of cases of TNDM is caused by mutations in other genes, including HNF1B, 23 INS 24 among others.
Anomalies at the 6q24 locus, spanning two candidate genes PLAGL1 and HYMAI, are the single most common cause of NDM and always result in TNDM. 25 In normal circumstances, this region is maternally imprinted so that only the allele inherited from the father is expressed. TNDM is ultimately associated with overexpression of the imprinted genes. 26 To date, three different molecular mechanisms have been identified: (1) paternal uniparental disomy of chromosome 6 (UPD6) either complete or partial; this accounts for 50% of sporadic TNDM cases, (2) unbalanced paternal duplication of 6q24 (found in most familial cases), and (3) hypomethylation of the maternal allele (found in sporadic cases). 27 Methylation defects may result from an isolated imprinting variant affecting only the 6q24 locus or may arise in the context of a generalized hypomethylation syndrome caused by multiple imprinting alterations across the genome, that is, multi‐locus imprinting disturbance (MLIDs) along with other clinical features including congenital heart defects, brain malformations, and so forth. 28 Some cases of TNDM secondary to multiple methylation defects are caused by recessively acting mutations in ZFP57, a gene on chromosome 6p involved in the regulation of DNA methylation. 29
Neonates with diabetes caused by 6q24 abnormalities are born with severe intrauterine growth retardation (IUGR) and one‐third of them show macroglossia; more rarely, an umbilical hernia is present. They develop severe but nonketotic hyperglycemia very early, usually during the first week of life. 27 , 30
Despite the severity of the initial presentation, the insulin dose can be tapered quickly so that most infants do not require any treatment by a median age of 12–14 weeks and the rate of remission is close to 100%. 31
Because most cases exhibit some degree of endogenous β‐cell function, insulin therapy is not always necessary, and these infants may respond to oral SU or other drugs used for T2D. 31 , 32 , 33 , 34
In some, a transition to remission has been observed with no need for insulin therapy or initial SU treatment. 34
Some cases with TNDM have shown a positive response to SU. 34 , 35
Following remission, a low proportion of affected infants and children will exhibit clinically significant hypoglycemia that in some cases requires long‐term treatment. 36 , 37 During remission, transient hyperglycemia may occur during intercurrent illnesses. 38
Over time, diabetes relapses in at least 50%–60% of these young people; in one large cohort followed until 18 years of age, relapse occurred in 85%. 39 Relapse usually occurs around puberty, although recurrences have been reported as young as 4 years of age.
Therefore, parents of children with TNDM should internalize the high risk of their child's future diabetes relapse and these children may benefit from annual HbA1c testing. Relapse clinically resembles early‐onset T2D and is characterized by a loss of the first‐phase insulin secretion. 34 Long‐term metabolic and socio‐educational follow‐up has shown that these persons have decreased educational attainment, and those with diabetes have lower insulin secretion capacity. 40
The phases described above do not present uniformly in every affected child. Interestingly, some carrier relatives develop T2D or gestational diabetes in adulthood without any evidence of having had NDM, as well as in a small fraction of people with early‐onset, non‐obese, non‐autoimmune diabetes without a history of NDM. This suggests significant variability in phenotype, possibly related to other genetic or epigenetic factors that may influence the clinical expression of alterations of chromosome 6q24. 20 , 31
The role of genetic counseling depends on the underlying molecular mechanism. Uniparental disomy of chromosome 6 is generally sporadic and, therefore, the risk of recurrence in siblings and offspring is low. When paternal duplication of the 6q24 region is found, affected newborn males have a 50% chance as adults of transmitting the mutation and the disease to their children. In contrast, newborn affected females will as adults pass on the duplication, but their children will not develop the disease. In this case, TNDM may recur in the next generation as their asymptomatic sons pass on the molecular defect to their own children. Some methylation defects (i.e., ZFP57 mutations) show an autosomal recessive inheritance and hence the recurrence risk is 25% for siblings and almost negligible for the offspring of an affected individual.
9.3. Permanent neonatal diabetes due to mutations in the KATP channel genes (KATP‐NDM)
KATP‐NDM is the commonest cause of PNDM 41 , 42 , 43 , 44 , 45 and the second most common cause of TNDM. 22 The prevalence of KATP‐NDM in a specific group depends on the degree of consanguinity. In outbred populations the commonest known cause of PNDM are abnormalities in the KATP channel or INS genes. 9 , 46 If parents are related, Wolcott‐Rallison syndrome or homozygous mutations in the GCK gene are the most common etiologies. 47 The causes of up to 20% of PNDM cases remain unknown.
KATP channels are hetero‐octameric complexes formed by four pore‐forming Kir6.2 subunits and four SUR1 regulatory subunits, encoded by the genes KCNJ11 and ABCC8, respectively. 48 They regulate insulin secretion by linking the intracellular metabolic state to the β‐cell membrane electrical activity. Any increase in the intracellular metabolic activity induces a rise in the ATP/ADP ratio within the pancreatic β‐cell. The high ATP/ADP ratio closes the KATP channels and leads to cell membrane depolarization which ultimately triggers insulin secretion. 49
Activating mutations in KCNJ11 or ABCC8, prevent KATP channel closure and hence reduce insulin secretion in response to hyperglycemia, resulting in diabetes 41 , 42 , 43 , 45 (Figure 1). A loss‐of‐function nonsense mutation in ABCC8, resulting in gain‐of‐channel function, has also been reported. 50
FIGURE 1.
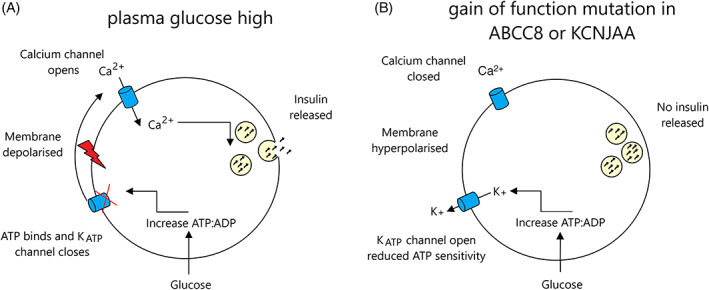
Insulin secretion from the pancreatic beta cell in (A) normal cell in a high plasma glucose environment and (B) in a cell with a K‐ATP channel mutation. Source: adapted from Reference 263 . (A) Glucose enters the cell and is metabolized, causing an increase in ATP, K‐ATP channel closure is induced via ATP binding, the membrane is depolarized, and calcium influx is triggered resulting in the release of insulin from its storage vesicles. (B) A gain of function mutation in the K‐ATP channel results in the failure of ATP to bind to the channel, causing the channel to remain open, the membrane stays hyperpolarized and no insulin is released
Approximately 90% of persons with KCNJ11 mutations have PNDM while ~10% develop TNDM, whereas ABCC8 mutations more frequently (~66%) cause TNDM. 42 , 51 There are no significant differences in the severity of IUGR or the age at diagnosis of diabetes between the two subtypes of NDM. 22 KATP channel mutations typically show milder IUGR and are diagnosed slightly later than infants with 6q24 abnormalities, indicating a less severe insulin deficiency during the last months of intrauterine development and at the time birth. In KATP‐TNDM, diabetes usually remits later and relapses earlier than in 6q24‐TNDM. 22 The low or undetectable C‐peptide levels and frequent presentation with diabetic ketoacidosis in KATP‐NDM suggest insulin dependency. 52
In addition to diabetes, about 20% of affected children with KCNJ11 mutations present with associated neurological features 41 , 53 , 54 in keeping with the expression of KATP channels in neurons and muscle cells. 49 , 55 The most severely damaging mutations are also associated with marked developmental delay and early‐onset epilepsy, known as DEND (developmental delay, epilepsy, and NDM) syndrome. An intermediate DEND syndrome characterized by NDM and less severe developmental delay without epilepsy is more common. Recent studies utilizing detailed testing have revealed that mild neurodevelopmental abnormalities occur even in those with milder mutations previously thought to cause only isolated diabetes. In some studies using sibling controls, mild but significant impairments were found in several domains, including IQ, measures of academic achievement, and executive function. Many of these children met criteria for developmental coordination disorder (particularly visual–spatial dyspraxia), attention deficit hyperactivity disorder, anxiety disorder, or autism, and/or had behavioral or sleep difficulties. 39 , 56 , 57 , 58
Approximately 90% of children with activating mutations in the KATP channel genes can be switched from insulin to off‐label SU tablets. 59 , 60 , 61 A suspension of glibenclamide has shown to be safe and effective in individuals with NDM, 62 and received authorization for use in the European Union. 63
Treatment with SU dramatically improves glycemic management, which appears to be durable long‐term with only minimal mild hypoglycemia. 64 , 65
The glibenclamide doses required when calculated based on body weight are higher compared to the dose used in adults with T2D, typically needing around 0.5 mg/kg/day, although doses as high as 2.3 mg/kg/day have been occasionally reported. 66 , 67 , 68 The dose required depends mostly on the age at which the person starts SU, as well as the specific mutation. 69 , 70
Many persons have been able to progressively reduce the dose after transition while maintaining excellent glycemic management. 71 , 72 The only side effects reported to date are transient diarrhea and staining of the teeth. 73 , 74 Recently, it has been reported that celiac disease my cause secondary SU failure not explained by lack of adherence to therapy. 75
Insulin secretion in children with diabetes treated with adequate SU doses seems to be driven mostly by food intake via non‐KATP dependent pathways. Meals composed of either all carbohydrate or only protein/fat resulted in similar insulin responses, highlighting the importance of carbohydrate intake with most meals to avoid post‐prandial hypoglycemia. 76
Some studies have shown that SU may penetrate the blood–brain barrier, but maintenance of cerebrospinal fluid levels may limit the benefit of SU on neurodevelopmental outcome, and use of other agents could be considered. 77 , 78 , 79
Although SU appears to partially improve some of the neurological symptoms, the degree of improvement likely also depends on how early treatment is started. 80 , 81 , 82 , 83
Neurological features have been reported less frequently in persons with ABCC8 mutations, who more often have TNDM. 42 , 43 However, those with PNDM due to ABCC8 mutations had a similar range of difficulties as those with PNDM due to KCNJ11 mutations. 84
The ABCC8 encoded SUR‐1 protein is crucial in retinal function and SU (glibenclamide) confers direct retinal neuroprotection through SUR‐1 mediated mechanisms. 85 , 86
A recent study utilized patient‐derived iPSCs to generate cerebral organoids and found major defects in early development of cortical neuronal network in V59M mutants compared to controls, that could partially be rescued by the SU tolbutamide. 87
Activating mutations in KCNJ11 causing NDM are always heterozygous. Since about 90% of these mutations arise de novo, there is usually no family history of NDM 88 but familial cases show an autosomal dominant pattern of inheritance. Recurrence risk for the offspring of an affected person is 50%. This is also true for most people with activating mutations in ABCC8. However, some persons are homozygous or compound heterozygous for two different mutations and NDM is recessively inherited. 43 In this case, the risk of NDM for future siblings is 25%, but almost non‐existent for the offspring of the affected person unless the other parent is also a carrier for the same mutation. Germline mosaicism (mutations present in the gonads but not detectable in blood) has been reported in several families 88 and hence unaffected parents of a child with an apparently de novo mutation should be advised that the recurrence risk in siblings is low but not negligible.
9.3.1. Neonatal diabetes due to mutations in INS gene
Mutations in the proinsulin gene (INS) are the second most common cause of PNDM after KATP channel mutations. 46 , 89 , 90 , 91 , 92 Individuals with diabetes due to INS mutations lack any extra‐pancreatic features and are insulin dependent. 89 , 91 , 93 Dominant heterozygous mutations are most common and usually result in a misfolded proinsulin molecule that is trapped and accumulates in sub‐cellular compartments, leading to endoplasmic reticulum stress and ß‐cell apoptosis. 93 , 94 , 95 Recessive biallelic (homozygous or compound heterozygous) mutations lead to loss or inactivation of proinsulin. 24 These mutations do not cause slowly progressive β‐cell destruction but result in a lack of insulin biosynthesis before and after birth, which explains much lower birth weights and earlier presentation of diabetes in affected children. Since the disease is recessively inherited, there will be a 25% recurrence risk in siblings when each parent has been confirmed to be a carrier of a causal INS variant.
The severity of IUGR in children with heterozygous INS mutations is similar to those with KATP channel mutations, but they present at somewhat later ages.
Although the diabetes is still diagnosed most often before 6 months of age, it can also occur up to a year of age or even later; therefore genetic testing should be considered in children with autoantibody negative diabetes presenting at early ages, 89 , 91 , 93 , 96 as well as in those with a MODY‐like phenotype.
Most heterozygous INS mutations are sporadic de novo mutations but about 20% of probands have a family history of autosomal dominant NDM. 91
9.3.2. Wolcott–Rallison syndrome
This rare autosomal recessive syndrome is the commonest cause of PNDM in highly inbred populations and characterized by early‐onset diabetes mellitus, spondyloepiphyseal dysplasia, and recurrent hepatic and/or renal dysfunction. 97 , 98 Wolcott–Rallison syndrome (WRS) is caused by biallelic mutations in the EIF2AK3 (eukaryotic translation initiation factor alpha 2‐kinase 3) gene, which encodes a protein involved in the regulation of the endoplasmic reticulum (ER) stress response. Pancreatic development is rather normal in the absence of the functional protein, but misfolded proteins accumulate within the endoplasmic reticulum after birth and eventually induce β‐cell apoptosis. Although diabetes usually manifests during infancy, it might not present until 3–4 years of age. Diabetes may be the first clinical manifestation of the syndrome and, therefore, this diagnosis should be considered even in children with isolated PNDM, especially if they were born to consanguineous parents or from a highly inbred population. 99 , 100 Since the disease is recessively inherited, there is a 25% recurrence risk in siblings. Fulminant hepatic failure is the main cause of death in persons with WRS and currently there is no agent to reverse this abnormality 101 ; however, recent reports indicate that liver (with or without pancreas) transplantation can be life saving and improve the outcomes of individuals with this syndrome. 101 , 102 , 103 , 104
9.3.3. Neonatal diabetes due to GCK mutations
The enzyme glucokinase is considered the glucose sensor of the β‐cells, as it catalyzes the rate‐limiting step of glucose phosphorylation and therefore enables the β‐cell to respond appropriately to the degree of glycemia. 105
Complete glucokinase deficiency secondary to mutations in both alleles, either homozygous or compound heterozygous, prevents the β‐cells from secreting insulin in response to hyperglycemia. 106 , 107
Neonates present with severe IUGR, are usually diagnosed with diabetes during the first few days of life, and require exogenous insulin therapy. Apart from diabetes, they do not show any relevant extrapancreatic features. 106 , 107 , 108 , 109 , 110 , 111 , 112 , 113
GCK is responsible for not more than 2%–3% of cases of PNDM overall, 47 but has an increased prevalence in regions with a high degree of consanguinity. 114 This type of PNDM is inherited in a recessive manner so the recurrence risk for future siblings is 25%. This diagnosis should be strongly considered in probands born to parents with asymptomatic mild hyperglycemia; therefore, measuring fasting blood glucose in the parents of any child with NDM, even when there is no known consanguinity or family history of diabetes, is often recommended.
Few studies have evaluated the risk of microvascular complications in NDM, but one study showed that individuals with KATP/PNDM or abnormalities in the insulin gene (INS) do not seem prone to severe eye complications even after a median diabetes duration of 24 years. 115
10. IPEX SYNDROME AND OTHER MONOGENIC CAUSES OF AUTOIMMUNE DIABETES
Mutations in at least nine different genes are now known to cause autoimmune syndromes that can include neonatal and infancy‐onset diabetes associated with pancreatic islet autoantibodies: AIRE, CTLA4, FOXP3, IL2RA, ITCH, LRBA, STAT1, STAT3, and STAT5B.
These monogenic conditions that do cause autoimmune diabetes share basic features with pediatric T1D 15 , 116 , 117 , 118 and account for the previously mentioned rare cases of T1D in the first months of life.
Rarely, some cases of diabetes with onset during the first 6 months of life have an autoimmune basis; it is now accepted that mutations in a range of genes related to immune function (such as FOXP3, STAT3, or LRBA) are at least as likely as T1D.
Mutations in the FOXP3 gene are responsible for the immune dysregulation, polyendocrinopathy, enteropathy, X‐linked (IPEX) syndrome. 119 , 120 IPEX syndrome is clinically heterogeneous ranging from severe intrauterine forms to moderate phenotypes, as has been recently described in different cohorts. 117 , 121 , 122 Among male infants who present with diarrhea, eczema, autoimmune diabetes, immune deficiency, and/or life‐threatening infection, mutations in FOXP3 should be considered. 123 , 124 Treatment with immunosuppressive agents (sirolimus or steroids). 123 , 124 Alternatively, allogeneic hematopoietic stem cell transplantation (HSCT) with reduced‐intensity conditioning is recommended. 125 Survival is similar with both immunosuppressant treatment and HSCT, but higher rates of disease‐free survival and improved quality of life have been shown with HSCT. 126
In addition to mutations in FOXP3 “classic IPEX,” there is a group with an “IPEX‐like” phenotype with defects in other genes. Examples include individuals with heterozygous mutations in CTLA4 causing autoimmune lymphoproliferative syndrome which can include autoimmune diabetes, enteropathy, cytopenias, and thyroiditis 127 ; individuals with recessive mutations in the ubiquitin ligase gene (ITCH) present with multisystem autoimmune disease and facial dysmorphism 128 ; individuals with bi‐allelic mutations in IL2RA (interleukin 2 receptor subunit alpha) resulting in immunodeficiency 41 syndrome, with lymphoproliferation, other autoimmunity, and autoimmune diabetes, 129 , 130 as well as individuals with recessively inherited mutations in LRBA reported as a cause of immunodeficiency‐8 with autoimmune enteropathy, T1D, autoimmune hypothyroidism, and autoimmune hemolytic anemia. 118
The proteins encoded by the STAT3, STAT1, and STAT5B genes are transcription factors involved in the cellular response to cytokines and growth factors. Activating mutations in STAT3 cause multiple autoimmune disease with enteropathy, hematological autoimmune disorders, autoimmune cytopenia, and autoimmune diabetes which often presents in the neonatal period. 116 , 131 Persons with gain of function mutations in STAT1 present with chronic fungal infections including respiratory tract infections with a subset of people developing severe organ specific autoimmunity including T1D. 132 On the other hand, loss of function mutations in STAT5B are associated with disorders characterized by allergic or autoimmune manifestations.
Loss of function mutations in the AIRE gene cause polyendocrine autoimmune syndrome type 1 (APS1), characterized by chronic mucocutaneous candidiasis, hypoparathyroidism, and autoimmune adrenal insufficiency. In addition, 13% of individuals present with diabetes by 30 years of age. 128
11. OTHER CAUSES OF NEONATAL DIABETES
More than 30 genetic subtypes of NDM have been described. The clinical features seen in the more common causes of neonatal and infancy‐onset diabetes are shown in Table 1. Pancreatic scanning is unreliable in neonates and so it is best to use functional tests of exocrine pancreatic function (fecal elastase and fecal fats) when assessing if pancreatic aplasia is present. 133 , 134 Apart from KATP‐NDM and some persons with SLC19A2 mutations causing thiamine‐responsive megaloblastic anemia (TRMA) syndrome, 135 all other causes need to be treated with subcutaneous insulin. Children with pancreatic aplasia/hypoplasia will also require exocrine pancreatic supplements.
11.1. Genetic testing should be performed as soon as diabetes is diagnosed in a child aged less than 6 months
All infants diagnosed with diabetes in the first 6 months of life should have immediate molecular genetic testing to define their subtype of monogenic NDM, as T1D is extremely rare in this subgroup.
Genetic testing will allow diagnosis of a specific type of monogenic diabetes in over 80% of children whose diabetes is diagnosed before the age of 6 months. As discussed above, this will influence treatment as well as prediction of clinical features.
It is no longer necessary to wait to see if diabetes resolves or for other features to develop, as major laboratories will offer comprehensive testing of all NDM subtypes as well as very rapid testing of subtypes that alter treatment.
12. AUTOSOMAL DOMINANT FAMILIAL MILD HYPERGLYCEMIA OR DIABETES (MODY)
A familial form of mild diabetes presenting during adolescence or in early adulthood was first described many years ago. 10 , 136 Even though diabetes presented in young people, the disease clinically resembled elderly‐onset non‐insulin dependent diabetes and the newly recognized subtype of familial diabetes became known by the acronym MODY (maturity‐onset diabetes of the young). 137 As MODY persons passed on the disease to their offspring following an autosomal dominant pattern of inheritance, it was quickly suspected that it might be a monogenic disorder. 138 MODY is by far the commonest type of monogenic diabetes. All currently known subtypes of MODY are caused by dominantly acting heterozygous mutations in genes important for the development or function of β‐cells. Over the last few years, however, a number of forms of monogenic diabetes clinically and genetically different from MODY have been identified. 1 Individuals may harbor dominant mutations arising de novo; in such cases, a family history suggesting a monogenic condition is lacking. 41 , 90 , 139 These facts, along with a widespread lack of awareness, hinder clinical diagnosis so that the majority of children with genetically proven monogenic diabetes are initially misdiagnosed as having T1D 140 , 141 or T2D. 142 , 143 Although monogenic diabetes is uncommon, it accounts for 2.5%–6% of pediatric diabetes cases. 144 , 145 , 146 , 147 , 148 , 149
MODY syndromes are forms of monogenic diabetes characterized by impaired insulin secretion, with minimal or no defects in insulin action. 150
Most cause isolated diabetes and therefore may be misdiagnosed as either familial T1D or T2D. 142 , 151
Classic criteria for MODY include family history of diabetes; however, sporadic de novo mutations in several causative genes have been reported. 152
The different genetic subtypes of MODY differ in age of onset, pattern of hyperglycemia, and response to treatment.
Three genes are responsible for the majority of MODY cases (GCK, HNF1A, and HNF4A) and will be described in some detail below.
Most MODY subtypes will have a phenotype of isolated diabetes or stable mild fasting hyperglycemia, but some MODY genes have additional features such as renal cysts (see HNF1B below) or pancreatic exocrine dysfunction. 153
At least 14 different genes have been reported to cause diabetes with a MODY‐like phenotype (Table 2), and some panels will include all these genes or, possibly, also many other genes associated with exceedingly rare recessive causes. It is reasonable to consider including syndromic causes such as mitochondrial diabetes, as diabetes can often be the first presenting feature and a molecular diagnosis can thereby guide monitoring and treatment of other associated features. In the modern era of expanded testing by many different laboratories, caution must be used when interpreting test results, as often there is very little information available to support the causality of rare variants in uncommon subtypes.
13. MILD FASTING HYPERGLYCEMIA DUE TO GLUCOKINASE GENE MUTATIONS (GCK‐MODY, MODY2)
GCK‐MODY is the commonest subtype of monogenic diabetes in the pediatric diabetes clinic and its clinical phenotype is remarkably homogeneous among affected persons.
In contrast to other subtypes of monogenic diabetes, persons with GCK‐MODY regulate insulin secretion adequately but around a slightly higher set point than other people. As a result, they show nonprogressive mild hyperglycemia from birth. 154
HbA1c is mildly elevated but usually below 7.5% (59 mmol/mol). 155
Despite the mild fasting hyperglycemia, there is usually a small increment in blood glucose during an oral glucose tolerance test (OGTT) (<60 mg/dl or <3.5 mmol/L) 156 although this should not be considered an absolute criterion because of the variability of the OGTT.
Since the degree of hyperglycemia is not high enough to cause osmotic symptoms, most cases are usually diagnosed incidentally when blood glucose is measured for another reason.
The incidental finding of mild hyperglycemia (5.5–8 mmol/L or 100–145 mg/dl) in otherwise asymptomatic children and adolescents raises the possibility that they will subsequently develop T1D or T2D. In the absence of concomitant islet autoimmunity, the risk of future T1D is minimal, 157 and a significant proportion will have a heterozygous mutation in GCK. 158 In peripubertal children and adolescents with a diagnosis of T2D, the lack of obesity or other signs of IR should raise concern about the diagnosis of MODY.
Since blood glucose does not deteriorate significantly over time, this subtype of monogenic diabetes is rarely associated with chronic microvascular or macrovascular complications of diabetes. 159 , 160 and affected individuals do not generally require any treatment 161 except in the setting of pregnancy where an affected mother has an unaffected fetus and there is in utero evidence of accelerated growth. 162
When the clinical features of asymptomatic, long‐standing, stable mild fasting hyperglycemia are present, specific testing of GCK is appropriate.
Very often, the affected parent remains undiagnosed or has been misdiagnosed with early‐onset T2D. Measuring fasting glucose concentrations in apparently unaffected parents is important when considering a diagnosis of a GCK mutation. GCK‐MODY may first be diagnosed during pregnancy; it represents ~2%–6% of cases of gestational diabetes and can be differentiated from gestational diabetes based on clinical characteristics and fasting glucose concentration. 163 , 164
Of note, the presence of a GCK mutation does not protect against the concurrent development of polygenic T2D later in life, which occurs at a similar prevalence as in the general population. 165 GCK‐PNDM may manifest in GCK‐MODY families especially in the setting of consanguinity.
14. FAMILIAL DIABETES DUE TO HNF1A‐MODY (MODY3) AND HNF4A‐MODY (MODY1)
The possibility of monogenic diabetes should be considered whenever a parent of a child with diabetes also has diabetes, even if they are thought to have T1D or T2D.
Glucose intolerance associated with HNF1A‐ and HNF4A‐MODY usually becomes evident during adolescence or early adulthood. In the early stages of the disease, fasting blood glucose concentration may be normal, but there may be a large increment in blood glucose (>80 mg/dl or 5 mmol/L) after meals or at 2 h during an OGTT. 156
Over time, fasting hyperglycemia and osmotic symptoms (polyuria, polydipsia) present but they rarely develop ketosis because some residual insulin secretion persists for many years.
Chronic complications of diabetes are frequent, and their development is related to the degree of glycemic management. 166
HNF1A‐MODY is the most common form of monogenic diabetes that results in familial symptomatic diabetes, with heterozygous HNF1A mutations being about 10 times more frequent than heterozygous mutations in HNF4A. 167 Therefore, HNF1A‐MODY is the first diagnostic possibility to be considered in families with autosomal dominant symptomatic diabetes.
Persons with HNF1A‐MODY demonstrate an impaired incretin effect and inappropriate glucagon responses to OGTT. 168
Despite the association of HNF1A mutations with microvascular complications, recent data suggest that timely initiation of treatment with SUs is associated with lower rate of microvascular complications than T1D. 169 HNF1A mutations are also associated with an increased frequency of cardiovascular disease and mortality. 170
Mutations in HNF1A show a high penetrance so that 63% of mutation carriers develop diabetes before 25 years of age, 79% before age 35, and 96% before 55 years. 1 The age at diagnosis of diabetes is partly determined by the location of the mutation within the gene. 171 , 172 Persons with mutations affecting the terminal exons (8 to 10) are diagnosed, on average, 8 years later than those with mutations in exons 1 to 6. On the other hand, exposure to maternal diabetes in utero (when the mutation is maternally inherited) brings forward the age at onset of diabetes by about 12 years. 156 In the pediatric population, diabetes in HNF4A mutation carriers tends to appear at a similar age to persons with mutations in HNF1A. 146
Some differential clinical characteristics may be noted between persons with mutations in HNF4A and HNF1A; however, they do not often help in the choice of genes to be sequenced and it would be preferable to test all genes simultaneously with NGS whenever possible. 173
Persons with HNF1A mutations typically have a low renal threshold for glucose reabsorption due to impaired renal tubular transport of glucose and may present postprandial glycosuria before developing significant hyperglycemia. 174
In addition to diabetes, carriers of the p.Arg76Trp (R76W) mutation in HNF4A present with an atypical form of Fanconi syndrome including hypercalciuria and nephrocalcinosis. 175
About 50% of HNF4A mutation carriers are macrosomic at birth and 15% have diazoxide‐responsive neonatal hyperinsulinemic hypoglycemia. 176 In this case, hyperinsulinism typically remits during infancy and individuals develop diabetes from adolescence. 177 , 178 Hyperinsulinemic hypoglycemia has also been reported in HNF1A mutation carriers 179 but this is very uncommon.
Persons with both HNF1A and HNF4A‐diabetes can initially be treated with diet although they will have marked postprandial hyperglycemia with high carbohydrate food. 156
Most will need pharmacological treatment as they show progressive deterioration in glycemic management. They are extremely sensitive to SUs, 180 which usually allow better glycemic management than that achieved with insulin, especially in children and young adults. 181
The initial dose of SUs should be low (one‐quarter of the normal starting dose in adults) to avoid hypoglycemia. As long as there are no problems with hypoglycemia, they can be maintained on low‐dose SUs (e.g., 20–40 mg gliclazide daily) for decades. 182 , 183
If there is hypoglycemia despite dose titration of a once or twice daily SU preparation, a slow‐release preparation or meal time doses with a short‐acting agent such as a meglitinide may be considered. 184 A randomized controlled trial comparing a glucagon‐like peptide receptor agonist (GLP1RA) with a SU demonstrated lower fasting glucose in those treated with the GLP1RA. 168
15. DIABETES ASSOCIATED WITH EXTRA‐PANCREATIC FEATURES
A monogenic disorder should be considered in any child with diabetes associated with multi‐system extrapancreatic features, 185 or in young‐onset diabetes when consanguinity is known or suspected, even when syndromic features are not obvious. 186 These syndromes may either cause NDM (Table 1) or present later in life (see below). The Online Mendelian Inheritance in Man website (www.ncbi.nlm.nih.gov/omim or www.omim.org) can help with clinical features and to know if the gene for a particular syndrome has been defined hence molecular genetic testing is available. Genetic testing for some of these conditions is available on a research basis at www.euro-wabb.org. 187 The most common syndromes usually presenting beyond infancy are described in some detail below. A number of rare syndromes that include diabetes may also be tested through a gene panel approach (for example, see https://www.diabetesgenes.org/).
15.1. Diabetes insipidus, diabetes mellitus, optic atrophy, and deafness (DIDMOAD) syndrome (WFS)
The combination of diabetes and progressive optic atrophy below 16 years of age is diagnostic of this autosomal recessive syndrome. 188 Non‐autoimmune, insulin requiring diabetes, presenting at a mean age of 6 years, is usually the first manifestation of the disease. 189 Other reported features, including sensorineural deafness, central diabetes insipidus, urinary tract dysfunction, and neurological symptoms that develop later in a variable order even within the same family. 190 , 191 , 192 Many individuals with WFS are initially diagnosed as having T1D and subsequent loss of vision, which occurs ~4 years after diabetes diagnosis, may be misdiagnosed as diabetic retinopathy. 193 , 194 Persons with WFS die at a median age of 30 years, mainly from neurodegenerative complications. At least 90% of these people harbor biallelic mutations in the WFS1 gene. 195 This gene encodes WFS1, which is an endoplasmic reticulum (ER) transmembrane protein important for the negative regulation of ER stress and the maintenance of cellular calcium homeostasis. 196 Preclinical studies in cell and animal models suggest that therapeutic strategies targeting ER calcium homeostasis may be beneficial. However, a recent trial of using dantrolene sodium in 19 WFS subjects showed no significant improvement in β‐cell, retinal or neurological function. 197
A second variant of the syndrome (WFS2) has been described in association with mutations in CISD2 gene. 198 Persons with this rare variant do not develop diabetes insipidus but present with additional symptoms including a bleeding diathesis and peptic ulcer disease.
The current management of WFS involves symptomatic treatment of the associated features with no agents to cure or slow the disease progression.
15.2. Renal cysts and diabetes (RCAD) syndrome (HNF1B‐MODY or MODY5)
Although initially described as a rare subtype of familial diabetes, it is now clear that persons with heterozygous mutations in HNF1B rarely present with isolated diabetes. 199 In contrast, renal developmental disorders (especially renal cysts and renal dysplasia) are present in almost all persons with HNF1B mutations or gene deletions 139 and constitute the main presentation in children, even in the absence of diabetes. 200 , 201 , 202 Genital tract malformations (particularly uterine abnormalities), hyperuricemia, and gout can also occur, as well as abnormal liver function tests. 199 Diabetes develops later, typically during adolescence or early adulthood 203 , 204 although TNDM has been reported in a few cases. 23 , 203 In addition to insulin deficiency related to pancreatic hypoplasia, 205 affected persons also show some degree of hepatic IR, 206 which explains why they do not respond adequately to SU treatment and require early insulin therapy. 1 Moreover, mutation carriers have lower exocrine pancreatic function with reduced fecal elastase; this involves both ductal and acinar cells. 207 Therefore, the phenotype of RCAD is highly variable even within families sharing the same HNF1B mutation and therefore this diagnosis should be considered not only in the diabetes clinic but also in other clinics (nephrology, urology, gynecology, etc.). In people with diabetes found to have renal cysts, imaging of the pancreas is indicated, since the absence of the pancreatic body and/or tail is highly indicative of HNF1B‐MODY. 208 Fecal elastase should also be measured, as this is always abnormal in persons with HNF1B‐MODY. 207 Importantly, a family history of renal disease or diabetes is not essential to prompt genetic testing, as de novo mutations and deletions of this gene are common (one‐third to two‐thirds of cases). 139 , 200
15.3. Mitochondrial diabetes
Diabetes due to mitochondrial mutations and deletions is rarely seen (<1%) in children and adolescents 209 as most affected persons develop diabetes as young or middle‐aged adults. The most common form of mitochondrial diabetes is caused by the m.3243A>G mutation in mitochondrial DNA. Diabetes onset is usually insidious but ~20% may have an acute presentation, including diabetic ketoacidosis. 210 Although it typically presents in adulthood, some cases have been reported in adolescents with a high degree of heteroplasmy. 209 , 211 , 212 Mitochondrial diabetes should be suspected in persons presenting with diabetes and maternally inherited sensorineural hearing loss, or diabetes and progressive external ophthalmoplegia. Interestingly, the same m.3243A>G mutation also causes a much more severe clinical syndrome known as MELAS (myopathy, encephalopathy, lactic acidosis, and stroke). 213
Persons with mitochondrial diabetes may initially respond to diet or oral hypoglycemic agents but often require insulin treatment within months or years. Metformin should be avoided as it interferes with mitochondrial function and may trigger episodes of lactic acidosis. 214
The penetrance of diabetes in mutation carriers depends on age, but is estimated to be above 85% at 70 years. 210 Affected males do not transmit the disease to their offspring. In contrast, females transmit the mutation to all their children, although some may not develop the disease. 1 In addition to the m.3243A>G mutation, early‐onset diabetes (even in infancy) has been reported in other less common mitochondrial disorders such as Kearns‐Sayre syndrome 215 and Pearson syndrome. 216
15.4. Diabetes secondary to monogenic diseases of the exocrine pancreas
Heterozygous mutations in CEL, which encodes a pancreatic lipase, cause CEL‐MODY or MODY8, an autosomal dominant disorder of pancreatic exocrine insufficiency and diabetes. 153 Importantly, the exocrine component of the syndrome is evident in childhood, 10–30 years before diabetes develops, and can be revealed by reduced fecal elastase and/or pancreatic lipomatosis. 217 , 218 Diabetes typically develops in the 30–40s together with pancreatic cysts. 218 The CEL gene is highly polymorphic and extremely difficult to sequence. El Jellas et al recently described how to diagnose CEL‐MODY. 219 The disease mechanism of CEL‐MODY involves protein misfolding/aggregation, endoplasmic reticulum stress, and proteotoxicity. 220 , 221 , 222 , 223 Other autosomal dominant monogenic diseases mainly affecting the exocrine pancreas that can lead to diabetes sooner or later include cystic fibrosis (CFTR), hereditary pancreatitis (PRSS1 and SPINK1) 224 and pancreatic agenesis/hypoplasia (GATA6). 134
15.5. Syndromic diabetes due to TRMT10A and DNAJC3 deficiencies: oxidative stress, apoptosis in β‐cells
Mutations in TRMT10A, a nuclear tRNA methyltransferase, are associated with a novel syndrome of young‐onset diabetes mellitus or impaired glucose metabolism, microcephaly, intellectual disability, short stature, and delayed puberty (OMIM 616013). To date, five families are described in the literature with a total of 11 people with a mutation. Phenotypes are heterogenous with most individuals presenting with impaired glucose homeostasis, microcephaly, short stature, seizures, and intellectual disability. 225
DNAJC3 mutation, which is associated with DM and multisystemic neurodegeneration, have been described. Familial case of DNAJC3 mutation manifesting as juvenile‐onset DM, hypothyroidism, multisystemic neurodegeneration, short stature, and sensorineural hearing loss with the new finding of pancreatic fibrosis and atrophy. 226
16. MONOGENIC INSULIN RESISTANCE SYNDROMES
The cardinal features of IR syndromes include moderate to severe acanthosis nigricans in association either with markedly increased insulin concentrations (fasting insulin >150 pmoL/L) or, where there is diabetes, increased insulin requirements, usually in the absence of a corresponding degree of obesity.
Three different subtypes are described, based on the underlying pathogenic mechanism: primary insulin signaling defects, IR secondary to adipose tissue abnormalities, and IR as a feature of complex syndromes. 227
Clinical and biochemical characterization of persons with severe IR may be used to guide genetic testing (Table 3).
In contrast with monogenic syndromes of β‐cell failure, hyperglycemia and diabetes tend to occur later in the genetic syndromes of severe IR and may not be a feature before the onset of puberty, 228 except for Donohue syndrome.
The phenotypes of monogenic IR syndromes tend to be more pronounced in females, who may present during adolescence with significant ovarian hyperandrogenism. The physical appearance of the partial lipodystrophies can also be less pronounced in males and so the presentation is more common in females who can present with features similar to those seen in polycystic ovarian syndrome.
16.1. Primary insulin signaling defects due to mutations in the insulin receptor (INSR) gene
INSR mutations are responsible for a number of rare IR syndromes. 229 , 230 Leptin levels are low, but adiponectin levels are paradoxically normal or elevated since insulin normally inhibits adiponectin secretion. 231 There is a spectrum of severity, depending on the effect of the mutation on the signaling function of the receptor. The most severe forms are associated with either homozygous or compound heterozygous mutations in the INSR gene responsible for Donohue and Rabson‐Mendenhall Syndromes. In Donohue Syndrome this leads to almost complete loss of insulin action at the cellular level and in Rabson‐Mendenhall Syndrome, where there is some residual insulin signaling, the phenotype can be milder. 232 Infants with Donohue Syndrome are born small for gestational age and develop diabetes in infancy with insulin concentrations over 1000 pmoL/L, often in association with cardiomyopathy and hypertrichosis. Postprandial hyperglycemia may be severe, and present early in life, but is usually accompanied by fasting hypoglycemia. There is no effective treatment, and the majority of infants sadly succumb to infection, or cardiac complications during the first year of life. Children with Rabson–Mendenhall Syndrome may not present until later in childhood, with failure to thrive, gingival hyperplasia, acanthosis nigricans, hyperandrogenism, and insulin resistant diabetes requiring very high doses of insulin developing during adolescence. 229 , 233
Type A IR Syndrome is the mildest form and results most commonly from a heterozygous mutation in the INSR gene and is inherited in an autosomal dominant manner. 229 Diabetes is rare before adolescence, but there can be significant ovarian hyperandrogenism and acanthosis nigricans during puberty.
The management of hyperglycemia in persons with INSR mutations can be challenging as insulin is largely ineffective even at high does. Insulin sensitizers such as metformin may be tried initially but most will need extraordinarily high doses of insulin, with limited effect. 229 As an alternative therapeutic method for young children, recombinant human IGF‐I has been reported to improve both fasting and postprandial glycemia although long‐term effects on survival remain unclear. 234 , 235 Recently, a trial showed benefits of long‐term treatment with metreleptin in persons with Rabson‐Mendenhall Syndrome. 236 Use of SGLT2i has also been reported to be beneficial in improving hyperglycemia. 237 , 238 For females, the hirsutism resulting from ovarian hyperandrogenism should be managed using similar strategies as for polycystic ovarian syndrome. 239
16.2. Monogenic lipodystrophies
Lipodystrophies are characterized by a partial or complete reduction in adipose tissue, which results in decreased adipokine levels and IR. 240 , 241 Mutations in either AGPAT2 or BSCL account for ~80% of cases of congenital generalized lipodystrophy (Berardinelli–Seip syndrome). 242 These are recessively inherited disorders characterized by an almost complete absence of subcutaneous and visceral fat. The clinical features are often apparent at birth. Inability to store excess dietary fat results in ectopic fat deposition in the liver, with hepatic steatosis that may progress to cirrhosis. 241 Diabetes can manifest in early infancy, but there then can be a period of remission until late childhood.
In contrast, a clinical diagnosis of familial partial lipodystrophy (FPLD) is usually made after puberty where there is failure to gain subcutaneous fat in the extremities and lower trunk during puberty, in combination with progressive accumulation of subcutaneous adipose tissue in the face and around the neck. 241 , 243 Heterozygous mutations in LMNA or PPARG account for ~50% of cases. 240 Visceral fat is greatly increased in addition to hyperinsulinemia, hypertriglyceridemia, and decreased HDL‐cholesterol levels. 244 Diabetes usually appears in late adolescence or early adulthood. More recently, there has been opportunity to make a genetic diagnosis in the offspring of persons with FPLD. In theory, this permits early intervention with lifestyle recommendations and screening for co‐morbidities in the hope that the development of co‐morbidities can be delayed but it is too early to tell whether this approach will be effective.
More rarely, lipodystrophy may occur as part of a multi‐system disorder. A mutation in POLD1, a universal DNA polymerase causes subcutaneous lipodystrophy in combination with diabetes, deafness, mandibular hypoplasia, and hypogonadism in males. 245 SHORT syndrome (short stature, hypermobility of joints, ocular depression, Rieger's anomaly, teething delay) with partial lipodystrophy, is caused by a hot spot mutation in PIK3R1 which has a central role in the insulin‐signaling pathway and growth factor resistance. 246 , 247 , 248 Mutation carriers of the dominant‐negative mutation in PIK3R1 seem to be protected from obesity and hepatic steatosis but not diabetes, 249 and the disease mechanisms is associated with unfolded protein response and reduced sensitivity to ER stress‐dependent apoptosis. 250
The mainstay of therapy for lipodystrophy is dietary intervention with a low‐fat, calorie‐neutral diet, 241 and an expert dietician as part of the multidisciplinary team is of paramount importance. In partial lipodystrophy, insulin sensitizers such as metformin and glitazones may be initially effective 251 but glitazones may exacerbate accumulation of ectopic fat in the face and neck. 228 More recently, therapy with recombinant leptin, given by daily subcutaneous injection, has been shown to be well tolerated, with sustained improvements in hypertriglyceridemia, glycemic management, and liver volume. 252 Efficacy in the partial forms of lipodystrophy is less clear, but where conventional therapy for diabetes and hypertriglyceridemia has not been successful, adjunctive therapy with metreleptin should be considered. 253
16.3. Ciliopathy‐related insulin resistance and diabetes
16.3.1. Alström syndrome
This autosomal recessive disorder shares symptoms with Bardet‐Biedl syndrome (see below), including progressive visual impairment related to cone‐rod dystrophy, sensorineural hearing loss, obesity, and diabetes mellitus. It can be distinguished from the latter syndrome by the lack of polydactyly and hypogonadism and by the absence of cognitive impairment. 254 More than 60% of individuals with ALMS develop cardiomyopathy. The syndrome is caused by mutations within the ALMS1 gene of unknown function. 255 Persons with Alström syndrome (ALMS) usually show many features of the metabolic syndrome including acanthosis nigricans, hyperlipidemia, hyperuricemia, hypertension, and slowly‐progressive insulin‐resistant diabetes. 256 Lifestyle intervention can initially ameliorate the metabolic abnormalities. 257
16.3.2. Bardet‐Biedl syndrome (BBS)
This disorder is characterized by intellectual disability, progressive visual impairment due to cone‐rod dystrophy, polydactyly, obesity, diabetes mellitus, renal dysplasia, hepatic fibrosis, and hypogonadism. Obesity is found in almost every affected individual, while diabetes affects less than 50%. 258 While the syndrome shares some similarities with Lawrence‐Moon syndrome, these two disorders can be distinguished by the presence of paraplegia and the absence of polydactyly, obesity, and diabetes mellitus in Lawrence‐Moon syndrome. Terms such as Lawrence‐Moon‐Bardet‐Biedl or Lawrence‐Moon‐Biedl syndrome should therefore be avoided. Bardet‐Biedl syndrome has been linked to 18 different genetic loci, referred to as BBS1 to BBS18. 259 , 260 The majority of cases are autosomal recessive, 261 but triallelic inheritance has been reported. 262 Genetic diagnostic laboratories and detailed clinical recommendations for persons with ALMS and BBS are present at http://www.euro-wabb.org.
17. CONCLUSIONS
Advances in molecular genetics have led to the identification of genes associated with many clinically identified subgroups of diabetes. Molecular genetic testing should now be considered an essential clinical diagnostic tool that can help define the diagnosis and determine the appropriate treatment of children with diabetes. Although the cost of NGS continues to drop, diagnostic genetic testing should be limited to those persons with diabetes who are likely to harbor a mutation based on the suggestive clinical features described above.
CONFLICT OF INTEREST
Dr Michel Polak MD, PhD has acted as scientific advisor for the development of the glibenclamide‐glyburide suspension named AMGLIDIA in the European Union. The other authors have declared no conflicts of interest.
Greeley SAW, Polak M, Njølstad PR, et al. ISPAD Clinical Practice Consensus Guidelines 2022: The diagnosis and management of monogenic diabetes in children and adolescents. Pediatr Diabetes. 2022;23(8):1188‐1211. doi: 10.1111/pedi.13426
Contributor Information
Siri Atma W. Greeley, Email: sgreeley@uchicago.edu.
Ethel Codner, Email: ecodner@med.uchile.cl.
DATA AVAILABILITY STATEMENT
Data sharing not applicable to this article as no datasets were generated or analysed during the current study.
REFERENCES
- 1. Murphy R, Ellard S, Hattersley AT. Clinical implications of a molecular genetic classification of monogenic beta‐cell diabetes. Nat Clin Pract Endocrinol Metab. 2008;4(4):200‐213. doi: 10.1038/ncpendmet0778 [DOI] [PubMed] [Google Scholar]
- 2. Greeley SA, John PM, Winn AN, et al. The cost‐effectiveness of personalized genetic medicine: the case of genetic testing in neonatal diabetes. Diabetes Care. 2011;34(3):622‐627. doi: 10.2337/dc10-1616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Naylor RN, John PM, Winn AN, et al. Cost‐effectiveness of MODY genetic testing: translating genomic advances into practical health applications. Diabetes Care. 2014;37(1):202‐209. doi: 10.2337/dc13-0410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bonnefond A, Philippe J, Durand E, et al. Highly sensitive diagnosis of 43 monogenic forms of diabetes or obesity through one‐step PCR‐based enrichment in combination with next‐generation sequencing. Diabetes Care. 2014;37(2):460‐467. doi: 10.2337/dc13-0698 [DOI] [PubMed] [Google Scholar]
- 5. Ellard S, Lango Allen H, De Franco E, et al. Improved genetic testing for monogenic diabetes using targeted next‐generation sequencing. Diabetologia. 2013;56(9):1958‐1963. doi: 10.1007/s00125-013-2962-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gao R, Liu Y, Gjesing AP, et al. Evaluation of a target region capture sequencing platform using monogenic diabetes as a study‐model. BMC Genet. 2014;15:13. doi: 10.1186/1471-2156-15-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Johansson S, Irgens H, Chudasama KK, et al. Exome sequencing and genetic testing for MODY. PLoS One. 2012;7(5):e38050. doi: 10.1371/journal.pone.0038050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Alkorta‐Aranburu G, Carmody D, Cheng YW, et al. Phenotypic heterogeneity in monogenic diabetes: the clinical and diagnostic utility of a gene panel‐based next‐generation sequencing approach. Mol Genet Metab. 2014;113(4):315‐320. doi: 10.1016/j.ymgme.2014.09.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. De Franco E, Flanagan SE, Houghton JA, et al. The effect of early, comprehensive genomic testing on clinical care in neonatal diabetes: an international cohort study. Lancet. 2015;386(9997):957‐963. doi: 10.1016/S0140-6736(15)60098-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tattersall R. Maturity‐onset diabetes of the young: a clinical history. Diabet Med. 1998;15(1):11‐14. doi: 10.1002/(SICI)1096-9136(199801)15:1<11::AID-DIA561>3.0.CO;2-0 [DOI] [PubMed] [Google Scholar]
- 11. Iafusco D, Stazi MA, Cotichini R, et al. Permanent diabetes mellitus in the first year of life. Diabetologia. 2002;45(6):798‐804. [DOI] [PubMed] [Google Scholar]
- 12. Ellard S, Bellanne‐Chantelot C, Hattersley AT. European molecular genetics quality network Mg. Best practice guidelines for the molecular genetic diagnosis of maturity‐onset diabetes of the young. Diabetologia. 2008;51(4):546‐553. doi: 10.1007/s00125-008-0942-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405‐424. doi: 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Edghill EL, Dix RJ, Flanagan SE, et al. HLA genotyping supports a nonautoimmune etiology in patients diagnosed with diabetes under the age of 6 months. Diabetes. 2006;55(6):1895‐1898. [DOI] [PubMed] [Google Scholar]
- 15. Johnson MB, Patel KA, De Franco E, et al. Type 1 diabetes can present before the age of 6 months and is characterised by autoimmunity and rapid loss of beta cells. Diabetologia. 2020;63:2605‐2615. doi: 10.1007/s00125-020-05276-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Johnson MB, De Franco E, Greeley SAW, et al. Trisomy 21 is a cause of permanent neonatal diabetes that is autoimmune but not HLA associated. Diabetes. 2019;68(7):1528‐1535. doi: 10.2337/db19-0045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rubio‐Cabezas O, Flanagan SE, Damhuis A, Hattersley AT, Ellard S. KATP channel mutations in infants with permanent diabetes diagnosed after 6 months of life. Pediatr Diabetes. 2012;13(4):322‐325. doi: 10.1111/j.1399-5448.2011.00824.x [DOI] [PubMed] [Google Scholar]
- 18. Mohamadi A, Clark LM, Lipkin PH, Mahone EM, Wodka EL, Plotnick LP. Medical and developmental impact of transition from subcutaneous insulin to oral glyburide in a 15‐yr‐old boy with neonatal diabetes mellitus and intermediate DEND syndrome: extending the age of KCNJ11 mutation testing in neonatal DM. Pediatr Diabetes. 2010;11(3):203‐207. doi: 10.1111/j.1399-5448.2009.00548.x [DOI] [PubMed] [Google Scholar]
- 19. Slingerland AS, Hattersley AT. Activating mutations in the gene encoding Kir6.2 alter fetal and postnatal growth and also cause neonatal diabetes. J Clin Endocrinol Metab. 2006;91(7):2782‐2788. doi: 10.1210/jc.2006-0201 [DOI] [PubMed] [Google Scholar]
- 20. Temple I, Gardner R, Mackay D, Barber J, Robinson D, Shield J. Transient neonatal diabetes: widening the understanding of the etiopathogenesis of diabetes. Diabetes. 2000;49(8):1359‐1366. [DOI] [PubMed] [Google Scholar]
- 21. Gardner RJ, Mackay DJ, Mungall AJ, et al. An imprinted locus associated with transient neonatal diabetes mellitus. Hum Mol Genet. 2000;9(4):589‐596. [DOI] [PubMed] [Google Scholar]
- 22. Flanagan SE, Patch AM, Mackay DJ, et al. Mutations in ATP‐sensitive K+ channel genes cause transient neonatal diabetes and permanent diabetes in childhood or adulthood. Diabetes. 2007;56(7):1930‐1937. doi: 10.2337/db07-0043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yorifuji T, Kurokawa K, Mamada M, et al. Neonatal diabetes mellitus and neonatal polycystic, dysplastic kidneys: phenotypically discordant recurrence of a mutation in the hepatocyte nuclear Factor‐1{beta} gene due to germline mosaicism. J Clin Endocrinol Metab. 2004;89(6):2905‐2908. [DOI] [PubMed] [Google Scholar]
- 24. Garin I, Edghill EL, Akerman I, et al. Recessive mutations in the INS gene result in neonatal diabetes through reduced insulin biosynthesis. Proc Natl Acad Sci U S A. 2010;107(7):3105‐3110. doi: 10.1073/pnas.0910533107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mackay D, Bens S, Perez de Nanclares G, Siebert R, Temple IK. Clinical utility gene card for: transient neonatal diabetes mellitus, 6q24‐related. Eur J Hum Genet. 2014;22(9):1153. doi: 10.1038/ejhg.2014.27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ma D, Shield JPH, Dean W, et al. Impaired glucose homeostasis in transgenic mice expressing the human transient neonatal diabetes mellitus locus, TNDM. J Clin Invest. 2004;114(3):339‐348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Temple IK, Shield JP. Transient neonatal diabetes, a disorder of imprinting. Review J Med Genet. 2002;39(12):872‐875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mackay DJ, Hahnemann JM, Boonen SE, et al. Epimutation of the TNDM locus and the Beckwith‐Wiedemann syndrome centromeric locus in individuals with transient neonatal diabetes mellitus. Hum Genet. 2006;119(1–2):179‐184. doi: 10.1007/s00439-005-0127-4 [DOI] [PubMed] [Google Scholar]
- 29. Mackay DJ, Callaway JL, Marks SM, et al. Hypomethylation of multiple imprinted loci in individuals with transient neonatal diabetes is associated with mutations in ZFP57. Nat Genet. 2008;40(8):949‐951. doi: 10.1038/ng.187 [DOI] [PubMed] [Google Scholar]
- 30. Docherty LE, Kabwama S, Lehmann A, et al. Clinical presentation of 6q24 transient neonatal diabetes mellitus (6q24 TNDM) and genotype‐phenotype correlation in an international cohort of patients. Diabetologia. 2013;56(4):758‐762. doi: 10.1007/s00125-013-2832-1 [DOI] [PubMed] [Google Scholar]
- 31. Yorifuji T, Matsubara K, Sakakibara A, et al. Abnormalities in chromosome 6q24 as a cause of early‐onset, non‐obese, non‐autoimmune diabetes mellitus without history of neonatal diabetes. Diabet Med. 2015;32(7):963‐967. doi: 10.1111/dme.12758 [DOI] [PubMed] [Google Scholar]
- 32. Sovik O, Aagenaes O, Eide SA, et al. Familial occurrence of neonatal diabetes with duplications in chromosome 6q24: treatment with sulfonylurea and 40‐yr follow‐up. Pediatr Diabetes. 2012;13(2):155‐162. doi: 10.1111/j.1399-5448.2011.00776.x [DOI] [PubMed] [Google Scholar]
- 33. Carmody D, Beca FA, Bell CD, et al. Role of noninsulin therapies alone or in combination in chromosome 6q24‐related transient neonatal diabetes: sulfonylurea improves but does not always normalize insulin secretion. Diabetes Care. 2015;38(6):e86‐e87. doi: 10.2337/dc14-3056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bonfanti R, Iafusco D, Rabbone I, et al. Differences between transient neonatal diabetes mellitus subtypes can guide diagnosis and therapy. Eur J Endocrinol. 2021;184(4):575‐585. doi: 10.1530/EJE-20-1030 [DOI] [PubMed] [Google Scholar]
- 35. Neumann U, Buhrer C, Blankenstein O, Kuhnen P, Raile K. Primary sulphonylurea therapy in a newborn with transient neonatal diabetes attributable to a paternal uniparental disomy 6q24 (UPD6). Diabetes Obes Metab. 2018;20(2):474‐475. doi: 10.1111/dom.13085 [DOI] [PubMed] [Google Scholar]
- 36. Flanagan SE, Mackay DJ, Greeley SA, et al. Hypoglycaemia following diabetes remission in patients with 6q24 methylation defects: expanding the clinical phenotype. Diabetologia. 2013;56(1):218‐221. doi: 10.1007/s00125-012-2766-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kalaivanan P, Arya VB, Shah P, et al. Chromosome 6q24 transient neonatal diabetes mellitus and protein sensitive hyperinsulinaemic hypoglycaemia. J Pediatr Endocrinol Metab. 2014;27(11–12):1065‐1069. doi: 10.1515/jpem-2014-0031 [DOI] [PubMed] [Google Scholar]
- 38. Shield JP, Temple IK, Sabin M, et al. An assessment of pancreatic endocrine function and insulin sensitivity in patients with transient neonatal diabetes in remission. Arch Dis Child Fetal Neonatal Ed. 2004;89(4):F341‐F343. doi: 10.1136/adc.2003.030502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Busiah K, Drunat S, Vaivre‐Douret L, et al. Neuropsychological dysfunction and developmental defects associated with genetic changes in infants with neonatal diabetes mellitus: a prospective cohort study [corrected]. Lancet Diabetes Endocrinol. 2013;1(3):199‐207. doi: 10.1016/S2213-8587(13)70059-7 [DOI] [PubMed] [Google Scholar]
- 40. Le Bourgeois F, Beltrand J, Baz B, et al. Long‐term metabolic and Socioeducational outcomes of transient neonatal diabetes: a longitudinal and cross‐sectional study. Diabetes Care. 2020;43(6):1191‐1199. doi: 10.2337/dc19-0324 [DOI] [PubMed] [Google Scholar]
- 41. Gloyn AL, Pearson ER, Antcliff JF, et al. Activating mutations in the gene encoding the ATP‐sensitive Potassium‐Channel subunit Kir6.2 and permanent neonatal diabetes. N Engl J Med. 2004;350(18):1838‐1849. [DOI] [PubMed] [Google Scholar]
- 42. Babenko AP, Polak M, Cave H, et al. Activating mutations in the ABCC8 gene in neonatal diabetes mellitus. N Engl J Med. 2006;355(5):456‐466. [DOI] [PubMed] [Google Scholar]
- 43. Ellard S, Flanagan SE, Girard CA, et al. Permanent neonatal diabetes caused by dominant, recessive, or compound heterozygous SUR1 mutations with opposite functional effects. Am J Hum Genet. 2007;81(2):375‐382. doi: 10.1086/519174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Flanagan SE, Edghill EL, Gloyn AL, Ellard S, Hattersley AT. Mutations in KCNJ11, which encodes Kir6.2, are a common cause of diabetes diagnosed in the first 6 months of life, with the phenotype determined by genotype. Diabetologia. 2006;49(6):1190‐1197. [DOI] [PubMed] [Google Scholar]
- 45. Vaxillaire M, Populaire C, Busiah K, et al. Kir6.2 mutations are a common cause of permanent neonatal diabetes in a large cohort of French patients. Diabetes. 2004;53(10):2719‐2722. [DOI] [PubMed] [Google Scholar]
- 46. Russo L, Iafusco D, Brescianini S, et al. Permanent diabetes during the first year of life: multiple gene screening in 54 patients. Diabetologia. 2011;54(7):1693‐1701. doi: 10.1007/s00125-011-2094-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Rubio‐Cabezas O, Ellard S. Diabetes mellitus in neonates and infants: genetic heterogeneity, clinical approach to diagnosis, and therapeutic options. Horm Res Paediatr. 2013;80(3):137‐146. doi: 10.1159/000354219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. McTaggart JS, Clark RH, Ashcroft FM. The role of the KATP channel in glucose homeostasis in health and disease: more than meets the islet. J Physiol. 2010;588(Pt 17):3201‐3209. doi: 10.1113/jphysiol.2010.191767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ashcroft FM. ATP‐sensitive potassium channelopathies: focus on insulin secretion. J Clin Invest. 2005;115(8):2047‐2058. doi: 10.1172/JCI25495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Flanagan SE, Dung VC, Houghton JAL, et al. An ABCC8 nonsense mutation causing neonatal diabetes through altered transcript expression. J Clin Res Pediatr Endocrinol. 2017;9(3):260‐264. doi: 10.4274/jcrpe.4624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Proks P, Arnold AL, Bruining J, et al. A heterozygous activating mutation in the sulphonylurea receptor SUR1 (ABCC8) causes neonatal diabetes. Hum Mol Genet. 2006;15(11):1793‐1800. [DOI] [PubMed] [Google Scholar]
- 52. Letourneau LR, Carmody D, Wroblewski K, et al. Diabetes presentation in infancy: high risk of diabetic ketoacidosis. Diabetes Care. 2017;40:e147‐e148. doi: 10.2337/dc17-1145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Gloyn AL, Diatloff‐Zito C, Edghill EL, et al. KCNJ11 activating mutations are associated with developmental delay, epilepsy and neonatal diabetes syndrome and other neurological features. Eur J Hum Genet. 2006;14(7):824‐830. doi: 10.1038/sj.ejhg.5201629 [DOI] [PubMed] [Google Scholar]
- 54. Hattersley AT, Ashcroft FM. Activating mutations in Kir6.2 and neonatal diabetes: new clinical syndromes, new scientific insights, and new therapy. Diabetes. 2005;54(9):2503‐2513. [DOI] [PubMed] [Google Scholar]
- 55. Clark RH, McTaggart JS, Webster R, et al. Muscle dysfunction caused by a KATP channel mutation in neonatal diabetes is neuronal in origin. Science. 2010;329(5990):458‐461. doi: 10.1126/science.1186146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Carmody D, Pastore AN, Landmeier KA, et al. Patients with KCNJ11‐related diabetes frequently have neuropsychological impairments compared with sibling controls. Diabet Med. 2016;33:1380‐1386. doi: 10.1111/dme.13159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Bowman P, Broadbridge E, Knight BA, et al. Psychiatric morbidity in children with KCNJ11 neonatal diabetes. Diabet Med. 2016;33:1387‐1391. doi: 10.1111/dme.13135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Landmeier KA, Lanning M, Carmody D, Greeley SAW, Msall ME. ADHD, learning difficulties and sleep disturbances associated with KCNJ11‐related neonatal diabetes. Pediatr Diabetes. 2017;18(7):518‐523. doi: 10.1111/pedi.12428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Rafiq M, Flanagan SE, Patch AM, et al. Effective treatment with oral sulfonylureas in patients with diabetes due to sulfonylurea receptor 1 (SUR1) mutations. Diabetes Care. 2008;31(2):204‐209. doi: 10.2337/dc07-1785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Garcin L, Mericq V, Fauret‐Amsellem AL, Cave H, Polak M, Beltrand J. Neonatal diabetes due to potassium channel mutation: response to sulfonylurea according to the genotype. Pediatr Diabetes. 2020;21(6):932‐941. doi: 10.1111/pedi.13041 [DOI] [PubMed] [Google Scholar]
- 61. Ngoc CTB, Dien TM, De Franco E, et al. Molecular genetics, clinical characteristics, and treatment outcomes of KATP‐channel neonatal diabetes mellitus in Vietnam National Children's Hospital. Front Endocrinol. 2021;12:727083. doi: 10.3389/fendo.2021.727083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Beltrand J, Baptiste A, Busiah K, et al. Glibenclamide oral suspension: suitable and effective in patients with neonatal diabetes. Pediatr Diabetes. 2019;20(3):246‐254. doi: 10.1111/pedi.12823 [DOI] [PubMed] [Google Scholar]
- 63. European Medicines Agency. https://www.ema.europa.eu/en/documents/smop-initial/chmp-summary-positive-opinion-amglidia_en.pdf.
- 64. Bowman P, Sulen Å, Barbetti F, et al. Effectiveness and safety of long‐term treatment with sulfonylureas in patients with neonatal diabetes due to KCNJ11 mutations: an international cohort study. Lancet Diabetes Endocrinol. 2018;6(8):637‐646. doi: 10.1016/s2213-8587(18)30106-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Lanning MS, Carmody D, Szczerbinski L, Letourneau LR, Naylor RN, Greeley SAW. Hypoglycemia in sulfonylurea‐treated KCNJ11‐neonatal diabetes: mild‐moderate symptomatic episodes occur infrequently but none involving unconsciousness or seizures. Pediatr Diabetes. 2018;19(3):393‐397. doi: 10.1111/pedi.12599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Sagen JV, Raeder H, Hathout E, et al. Permanent neonatal diabetes due to mutations in KCNJ11 encoding Kir6.2: patient characteristics and initial response to sulfonylurea therapy. Diabetes. 2004;53(10):2713‐2718. [DOI] [PubMed] [Google Scholar]
- 67. Greeley SA, Tucker SE, Naylor RN, Bell GI, Philipson LH. Neonatal diabetes mellitus: a model for personalized medicine. Research support, N.I.H., extramural research support, non‐U.S. Gov't review. Trends Endocrinol Metab. 2010;21(8):464‐472. doi: 10.1016/j.tem.2010.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Greeley SA, Tucker SE, Worrell HI, Skowron KB, Bell GI, Philipson LH. Update in neonatal diabetes. Research support, N.I.H., extramural research support, non‐U.S. Gov't review. Curr Opin Endocrinol Diabetes Obes. 2010;17(1):13‐19. doi: 10.1097/MED.0b013e328334f158 [DOI] [PubMed] [Google Scholar]
- 69. Thurber BW, Carmody D, Tadie EC, et al. Age at the time of sulfonylurea initiation influences treatment outcomes in KCNJ11‐related neonatal diabetes. Diabetologia. 2015;58(7):1430‐1435. doi: 10.1007/s00125-015-3593-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Babiker T, Vedovato N, Patel K, et al. Successful transfer to sulfonylureas in KCNJ11 neonatal diabetes is determined by the mutation and duration of diabetes. Diabetologia. 2016;59(6):1162‐1166. doi: 10.1007/s00125-016-3921-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Klupa T, Skupien J, Mirkiewicz‐Sieradzka B, et al. Efficacy and safety of sulfonylurea use in permanent neonatal diabetes due to KCNJ11 gene mutations: 34‐month median follow‐up. Diabetes Technol Ther. 2010;12(5):387‐391. doi: 10.1089/dia.2009.0165 [DOI] [PubMed] [Google Scholar]
- 72. Pearson ER, Flechtner I, Njolstad PR, et al. Switching from insulin to oral sulfonylureas in patients with diabetes due to Kir6.2 mutations. N Engl J Med. 2006;355(5):467‐477. doi: 10.1056/NEJMoa061759 [DOI] [PubMed] [Google Scholar]
- 73. Codner E, Flanagan S, Ellard S, Garcia H, Hattersley AT. High‐dose Glibenclamide can replace insulin therapy despite transitory diarrhea in early‐onset diabetes caused by a novel R201L Kir6.2 mutation. Diabetes Care. 2005;28(3):758‐759. [DOI] [PubMed] [Google Scholar]
- 74. Kumaraguru J, Flanagan SE, Greeley SA, et al. Tooth discoloration in patients with neonatal diabetes after transfer onto glibenclamide: a previously unreported side effect. Diabetes Care. 2009;32(8):1428‐1430. doi: 10.2337/dc09-0280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Iafusco D, Zanfardino A, Piscopo A, et al. Case report: coeliac disease as a cause of secondary failure of glibenclamide therapy in a patient with permanent neonatal diabetes due to KCNJ11/R201C mutation. Diabetologia. 2021;64(7):1703‐1706. doi: 10.1007/s00125-021-05454-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Bowman P, McDonald TJ, Knight BA, et al. Patterns of postmeal insulin secretion in individuals with sulfonylurea‐treated KCNJ11 neonatal diabetes show predominance of non‐KATP‐channel pathways. BMJ Open Diabetes Res Care. 2019;7(1):e000721. doi: 10.1136/bmjdrc-2019-000721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Fendler W, Pietrzak I, Brereton MF, et al. Switching to sulphonylureas in children with iDEND syndrome caused by KCNJ11 mutations results in improved cerebellar perfusion. Diabetes Care. 2013;36(8):2311‐2316. doi: 10.2337/dc12-2166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Mlynarski W, Tarasov AI, Gach A, et al. Sulfonylurea improves CNS function in a case of intermediate DEND syndrome caused by a mutation in KCNJ11. Nat Clin Pract Neurol. 2007;3:640‐645. doi: 10.1038/ncpneuro0640 [DOI] [PubMed] [Google Scholar]
- 79. Lahmann C, Kramer HB, Ashcroft FM. Systemic Administration of Glibenclamide Fails to achieve therapeutic levels in the brain and cerebrospinal fluid of rodents. PLoS One. 2015;10(7):e0134476. doi: 10.1371/journal.pone.0134476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Battaglia D, Lin YW, Brogna C, et al. Glyburide ameliorates motor coordination and glucose homeostasis in a child with diabetes associated with the KCNJ11/S225T, del226‐232 mutation. Pediatr Diabetes. 2012;13(8):656‐660. doi: 10.1111/j.1399-5448.2012.00874.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Gurgel LC, Crispim F, Noffs MH, Belzunces E, Rahal MA, Moises RS. Sulfonylrea treatment in permanent neonatal diabetes due to G53D mutation in the KCNJ11 gene: improvement in glycemic control and neurological function. Diabetes Care. 2007;30:e108. doi: 10.2337/dc07-1196 [DOI] [PubMed] [Google Scholar]
- 82. Koster JC, Cadario F, Peruzzi C, Colombo C, Nichols CG, Barbetti F. The G53D mutation in Kir6.2 (KCNJ11) is associated with neonatal diabetes and motor dysfunction in adulthood that is improved with sulfonylurea therapy. J Clin Endocrinol Metab. 2008;93(3):1054‐1061. doi: 10.1210/jc.2007-1826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Shah RP, Spruyt K, Kragie BC, Greeley SA, Msall ME. Visuomotor performance in KCNJ11‐related neonatal diabetes is impaired in children with DEND‐associated mutations and may be improved by early treatment with sulfonylureas. Diabetes Care. 2012;35:2086‐2088. doi: 10.2337/dc11-2225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Bowman P, Mathews F, Barbetti F, et al. Long‐term follow‐up of glycemic and neurological outcomes in an international series of patients with sulfonylurea‐treated ABCC8 permanent neonatal diabetes. Diabetes Care. 2021;44(1):35‐42. doi: 10.2337/dc20-1520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Berdugo M, Delaunay K, Lebon C, et al. Long‐term Oral treatment with non‐hypoglycemic dose of Glibenclamide reduces diabetic retinopathy damage in the Goto‐KakizakiRat model. Pharmaceutics. 2021;13(7):1095. doi: 10.3390/pharmaceutics13071095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Berdugo M, Delaunay K, Naud MC, et al. The antidiabetic drug glibenclamide exerts direct retinal neuroprotection. Transl Res. 2021;229:83‐99. doi: 10.1016/j.trsl.2020.10.003 [DOI] [PubMed] [Google Scholar]
- 87. Dalgin G, Tryba AK, Cohen AP, et al. Developmental defects and impaired network excitability in a cerebral organoid model of KCNJ11 p.V59M‐related neonatal diabetes. Sci Rep. 2021;11:21590. doi: 10.1038/s41598-021-00939-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Edghill EL, Gloyn AL, Goriely A, et al. Origin of de novo KCNJ11 mutations and risk of neonatal diabetes for subsequent siblings. J Clin Endocrinol Metab. 2007;92(5):1773‐1777. doi: 10.1210/jc.2006-2817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Polak M, Dechaume A, Cave H, et al. Heterozygous missense mutations in the insulin gene are linked to permanent diabetes appearing in the neonatal period or in early infancy: a report from the French ND (neonatal diabetes) study group. Diabetes. 2008;57(4):1115‐1119. [DOI] [PubMed] [Google Scholar]
- 90. Stoy J, Edghill EL, Flanagan SE, et al. Insulin gene mutations as a cause of permanent neonatal diabetes. Proc Natl Acad Sci U S A. 2007;104(38):15040‐15044. doi: 10.1073/pnas.0707291104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Edghill EL, Flanagan SE, Patch AM, et al. Insulin mutation screening in 1,044 patients with diabetes: mutations in the INS gene are a common cause of neonatal diabetes but a rare cause of diabetes diagnosed in childhood or adulthood. Diabetes. 2008;57(4):1034‐1042. doi: 10.2337/db07-1405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Flechtner I, Vaxillaire M, Cave H, Scharfmann R, Froguel P, Polak M. Neonatal hyperglycaemia and abnormal development of the pancreas. Best Pract Res Clin Endocrinol Metab. 2008;22(1):17‐40. doi: 10.1016/j.beem.2007.08.003 [DOI] [PubMed] [Google Scholar]
- 93. Colombo C, Porzio O, Liu M, et al. Seven mutations in the human insulin gene linked to permanent neonatal/infancy‐onset diabetes mellitus. J Clin Invest. 2008;118(6):2148‐2156. doi: 10.1172/JCI33777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Liu M, Sun J, Cui J, et al. INS‐gene mutations: from genetics and beta cell biology to clinical disease. Mol Asp Med. 2015;42:3‐18. doi: 10.1016/j.mam.2014.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Wang H, Saint‐Martin C, Xu J, et al. Biological behaviors of mutant proinsulin contribute to the phenotypic spectrum of diabetes associated with insulin gene mutations. Mol Cell Endocrinol. 2020;518:111025. doi: 10.1016/j.mce.2020.111025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Molven A, Ringdal M, Nordbo AM, et al. Mutations in the insulin gene can cause MODY and autoantibody‐negative type 1 diabetes. Diabetes. 2008;57(4):1131‐1135. doi: 10.2337/db07-1467 [DOI] [PubMed] [Google Scholar]
- 97. Senee V, Vattem KM, Delepine M, et al. Wolcott‐Rallison syndrome: clinical, genetic, and functional study of EIF2AK3 mutations and suggestion of genetic heterogeneity. Diabetes. 2004;53(7):1876‐1883. [DOI] [PubMed] [Google Scholar]
- 98. Delepine M, Nicolino M, Barrett T, Golamaully M, Lathrop GM, Julier C. EIF2AK3, encoding translation initiation factor 2‐alpha kinase 3, is mutated in patients with Wolcott‐Rallison syndrome. Nat Genet. 2000;25(4):406‐409. doi: 10.1038/78085 [DOI] [PubMed] [Google Scholar]
- 99. Rubio‐Cabezas O, Patch AM, Minton JA, et al. Wolcott‐Rallison syndrome is the most common genetic cause of permanent neonatal diabetes in consanguineous families. J Clin Endocrinol Metab. 2009;94(11):4162‐4170. doi: 10.1210/jc.2009-1137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Habeb AM, Flanagan SE, Deeb A, et al. Permanent neonatal diabetes: different aetiology in Arabs compared to Europeans. Arch Dis Child. 2012;97(8):721‐723. doi: 10.1136/archdischild-2012-301744 [DOI] [PubMed] [Google Scholar]
- 101. Habeb AM, Deeb A, Johnson M, et al. Liver disease and other comorbidities in Wolcott‐Rallison syndrome: different phenotype and variable associations in a large cohort. Horm Res Paediatr. 2015;83(3):190‐197. doi: 10.1159/000369804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Tzakis AG, Nunnelley MJ, Tekin A, et al. Liver, pancreas and kidney transplantation for the treatment of Wolcott‐Rallison syndrome. Am J Transplant. 2015;15(2):565‐567. doi: 10.1111/ajt.13005 [DOI] [PubMed] [Google Scholar]
- 103. Nordstrom J, Lundgren M, Jorns C, et al. First European case of simultaneous liver and pancreas transplantation as treatment of Wolcott‐Rallison syndrome in a small child. Transplantation. 2020;104(3):522‐525. doi: 10.1097/TP.0000000000002869 [DOI] [PubMed] [Google Scholar]
- 104. Elsabbagh AM, Hawksworth J, Khan KM, Yazigi N, Matsumoto CS, Fishbein TM. World's smallest combined en bloc liver‐pancreas transplantation. Pediatr Transplant. 2018;22:13082. doi: 10.1111/petr.13082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Matschinsky FM. Glucokinase, glucose homeostasis, and diabetes mellitus. Curr Diab Rep. 2005;5(3):171‐176. doi: 10.1007/s11892-005-0005-4 [DOI] [PubMed] [Google Scholar]
- 106. Njølstad PR, Sagen JV, Bjorkhaug L, et al. Permanent neonatal diabetes caused by glucokinase deficiency: inborn error of the glucose‐insulin signaling pathway. Diabetes. 2003;52(11):2854‐2860. doi: 10.2337/diabetes.52.11.2854 [DOI] [PubMed] [Google Scholar]
- 107. Njolstad PR, Sovik O, Cuesta‐Munoz A, et al. Neonatal diabetes mellitus due to complete glucokinase deficiency. N Engl J Med. 2001;344(21):1588‐1592. [DOI] [PubMed] [Google Scholar]
- 108. Raimondo A, Chakera AJ, Thomsen SK, et al. Phenotypic severity of homozygous GCK mutations causing neonatal or childhood‐onset diabetes is primarily mediated through effects on protein stability. Hum Mol Genet. 2014;23(24):6432‐6440. doi: 10.1093/hmg/ddu360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Esquiaveto‐Aun AM, De Mello MP, Paulino MF, Minicucci WJ, Guerra‐Junior G, De Lemos‐Marini SH. A new compound heterozygosis for inactivating mutations in the glucokinase gene as cause of permanent neonatal diabetes mellitus (PNDM) in double‐first cousins. Diabetol Metab Syndr. 2015;7:101. doi: 10.1186/s13098-015-0101-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Lin DC, Huang CY, Ting WH, et al. Mutations in glucokinase and other genes detected in neonatal and type 1B diabetes patient using whole exome sequencing may lead to disease‐causing changes in protein activity. Biochim Biophys Acta Mol basis Dis. 2019;1865:428‐433. doi: 10.1016/j.bbadis.2018.11.013 [DOI] [PubMed] [Google Scholar]
- 111. Bolu S, Eroz R, Dogan M, Arslanoglu I, Uzun H, Timur F. A family with novel homozygous deletion mutation (c.1255delT; p.Phe419Serfs*12) in the glucokinase gene, which is a rare cause of permanent neonatal diabetes mellitus. Turk Pediatri Ars. 2020;55(4):434‐437. doi: 10.14744/TurkPediatriArs.2019.05882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Shepherd M, Knight BA, Laskey K, McDonald TJ. Parental experiences of a diagnosis of neonatal diabetes and perceptions of newborn screening for glucose: a qualitative study. BMJ Open. 2020;10(11):e037312. doi: 10.1136/bmjopen-2020-037312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Oza CM, Karguppikar MB, Khadilkar V, Khadilkar A. Variable presentations of GCK gene mutation in a family. BMJ Case Rep. 2022;15(2):e246699. doi: 10.1136/bcr-2021-246699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Al Senani A, Hamza N, Al Azkawi H, et al. Genetic mutations associated with neonatal diabetes mellitus in Omani patients. J Pediatr Endocrinol Metab. 2018;31(2):195‐204. doi: 10.1515/jpem-2017-0284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Iafusco D, Salardi S, Chiari G, et al. No sign of proliferative retinopathy in 15 patients with permanent neonatal diabetes with a median diabetes duration of 24 years. Diabetes Care. 2014;37(8):e181‐e182. doi: 10.2337/dc14-0471 [DOI] [PubMed] [Google Scholar]
- 116. Flanagan SE, Haapaniemi E, Russell MA, et al. Activating germline mutations in STAT3 cause early‐onset multi‐organ autoimmune disease. Nat Genet. 2014;46(8):812‐814. doi: 10.1038/ng.3040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Rubio‐Cabezas O, Minton JA, Caswell R, et al. Clinical heterogeneity in patients with FOXP3 mutations presenting with permanent neonatal diabetes. Diabetes Care. 2009;32(1):111‐116. doi: 10.2337/dc08-1188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Johnson MB, De Franco E, Lango Allen H, et al. Recessively inherited LRBA mutations cause autoimmunity presenting as neonatal diabetes. Diabetes. 2017;66(8):2316‐2322. doi: 10.2337/db17-0040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Bennett CL, Christie J, Ramsdell F, et al. The immune dysregulation, polyendocrinopathy, enteropathy, X‐linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet. 2001;27(1):20‐21. [DOI] [PubMed] [Google Scholar]
- 120. Verbsky JW, Chatila TA. Immune dysregulation, polyendocrinopathy, enteropathy, X‐linked (IPEX) and IPEX‐related disorders: an evolving web of heritable autoimmune diseases. Curr Opin Pediatr. 2013;25(6):708‐714. doi: 10.1097/MOP.0000000000000029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Duclaux‐Loras R, Charbit‐Henrion F, Neven B, et al. Clinical heterogeneity of immune dysregulation, polyendocrinopathy, enteropathy, X‐linked syndrome: a French multicenter retrospective study. Clin Transl Gastroenterol. 2018;9(10):201. doi: 10.1038/s41424-018-0064-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Gambineri E, Ciullini Mannurita S, Hagin D, et al. Clinical, immunological, and molecular heterogeneity of 173 patients with the phenotype of immune dysregulation, polyendocrinopathy, enteropathy, X‐linked (IPEX) syndrome. Front Immunol. 2018;9:2411. doi: 10.3389/fimmu.2018.02411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Yong PL, Russo P, Sullivan KE. Use of sirolimus in IPEX and IPEX‐like children. J Clin Immunol. 2008;28(5):581‐587. doi: 10.1007/s10875-008-9196-1 [DOI] [PubMed] [Google Scholar]
- 124. Bindl L, Torgerson T, Perroni L, et al. Successful use of the new immune‐suppressor sirolimus in IPEX (immune dysregulation, polyendocrinopathy, enteropathy, X‐linked syndrome). J Pediatr. 2005;147(2):256‐259. doi: 10.1016/j.jpeds.2005.04.017 [DOI] [PubMed] [Google Scholar]
- 125. Rao A, Kamani N, Filipovich A, et al. Successful bone marrow transplantation for IPEX syndrome after reduced‐intensity conditioning. Blood. 2007;109(1):383‐385. doi: 10.1182/blood-2006-05-025072 [DOI] [PubMed] [Google Scholar]
- 126. Barzaghi F, Amaya Hernandez LC, Neven B, et al. Long‐term follow‐up of IPEX syndrome patients after different therapeutic strategies: an international multicenter retrospective study. J Allergy Clin Immunol. 2018;141:1036‐1049.e5. doi: 10.1016/j.jaci.2017.10.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Schubert D, Bode C, Kenefeck R, et al. Autosomal dominant immune dysregulation syndrome in humans with CTLA4 mutations. Nat Med. 2014;20(12):1410‐1416. doi: 10.1038/nm.3746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Johnson MB, Hattersley AT, Flanagan SE. Monogenic autoimmune diseases of the endocrine system. Lancet Diabetes Endocrinol. 2016;4(10):862‐872. doi: 10.1016/S2213-8587(16)30095-X [DOI] [PubMed] [Google Scholar]
- 129. Goudy K, Aydin D, Barzaghi F, et al. Human IL2RA null mutation mediates immunodeficiency with lymphoproliferation and autoimmunity. Clin Immunol. 2013;146(3):248‐261. doi: 10.1016/j.clim.2013.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Roth TL, Puig‐Saus C, Yu R, et al. Reprogramming human T cell function and specificity with non‐viral genome targeting. Nature. 2018;559(7714):405‐409. doi: 10.1038/s41586-018-0326-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Velayos T, Martinez R, Alonso M, et al. An activating mutation in STAT3 results in neonatal diabetes through reduced insulin synthesis. Diabetes. 2017;66(4):1022‐1029. doi: 10.2337/db16-0867 [DOI] [PubMed] [Google Scholar]
- 132. Toubiana J, Okada S, Hiller J, et al. Heterozygous STAT1 gain‐of‐function mutations underlie an unexpectedly broad clinical phenotype. Blood. 2016;127:3154‐3164. doi: 10.1182/blood-2015-11-679902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Weedon MN, Cebola I, Patch AM, et al. Recessive mutations in a distal PTF1A enhancer cause isolated pancreatic agenesis. Nat Genet. 2014;46(1):61‐64. doi: 10.1038/ng.2826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Allen HL, Flanagan SE, Shaw‐Smith C, et al. GATA6 haploinsufficiency causes pancreatic agenesis in humans. Research support, non‐U.S. Gov't. Nat Genet. 2012;44(1):20‐22. doi: 10.1038/ng.1035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Habeb AM, Flanagan SE, Zulali MA, et al. Pharmacogenomics in diabetes: outcomes of thiamine therapy in TRMA syndrome. Diabetologia. 2018;61(5):1027‐1036. doi: 10.1007/s00125-018-4554-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Fajans SS, Bell GI. MODY: history, genetics, pathophysiology, and clinical decision making. Diabetes Care. 2011;34(8):1878‐1884. doi: 10.2337/dc11-0035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Tattersall RB, Fajans SS. A difference between the inheritance of classical juvenile‐onset and maturity‐onset type diabetes of young people. Diabetes. 1975;24(1):44‐53. doi: 10.2337/diab.24.1.44 [DOI] [PubMed] [Google Scholar]
- 138. Tattersall RB. Mild familial diabetes with dominant inheritance. Q J Med. 1974;43(170):339‐357. [PubMed] [Google Scholar]
- 139. Bellanne‐Chantelot C, Clauin S, Chauveau D, et al. Large genomic rearrangements in the hepatocyte nuclear factor‐1beta (TCF2) gene are the most frequent cause of maturity‐onset diabetes of the young type 5. Diabetes. 2005;54(11):3126‐3132. doi: 10.2337/diabetes.54.11.3126 [DOI] [PubMed] [Google Scholar]
- 140. Moller AM, Dalgaard LT, Pociot F, Nerup J, Hansen T, Pedersen O. Mutations in the hepatocyte nuclear factor‐1alpha gene in Caucasian families originally classified as having type I diabetes. Diabetologia. 1998;41(12):1528‐1531. doi: 10.1007/s001250051101 [DOI] [PubMed] [Google Scholar]
- 141. Lambert AP, Ellard S, Allen LI, et al. Identifying hepatic nuclear factor 1alpha mutations in children and young adults with a clinical diagnosis of type 1 diabetes. Diabetes Care. 2003;26(2):333‐337. [DOI] [PubMed] [Google Scholar]
- 142. Awa WL, Schober E, Wiegand S, et al. Reclassification of diabetes type in pediatric patients initially classified as type 2 diabetes mellitus: 15 years follow‐up using routine data from the German/Austrian DPV database. Diabetes Res Clin Pract. 2011;94(3):463‐467. doi: 10.1016/j.diabres.2011.09.011 [DOI] [PubMed] [Google Scholar]
- 143. Kleinberger JW, Copeland KC, Gandica RG, et al. Monogenic diabetes in overweight and obese youth diagnosed with type 2 diabetes: the TODAY clinical trial. Genet Med. 2018;20(6):583‐590. doi: 10.1038/gim.2017.150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Fendler W, Borowiec M, Baranowska‐Jazwiecka A, et al. Prevalence of monogenic diabetes amongst polish children after a nationwide genetic screening campaign. Diabetologia. 2012;55(10):2631‐2635. doi: 10.1007/s00125-012-2621-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Irgens HU, Molnes J, Johansson BB, et al. Prevalence of monogenic diabetes in the population‐based Norwegian childhood diabetes registry. Diabetologia. 2013;56(7):1512‐1519. doi: 10.1007/s00125-013-2916-y [DOI] [PubMed] [Google Scholar]
- 146. Pihoker C, Gilliam LK, Ellard S, et al. Prevalence, characteristics and clinical diagnosis of maturity onset diabetes of the young due to mutations in HNF1A, HNF4A, and Glucokinase: results from the SEARCH for diabetes in youth. J Clin Endocrinol Metab. 2013;98:4055‐4062. doi: 10.1210/jc.2013-1279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Johansson BB, Irgens HU, Molnes J, et al. Targeted next‐generation sequencing reveals MODY in up to 6.5% of antibody‐negative diabetes cases listed in the Norwegian childhood diabetes registry. Diabetologia. 2017;60(4):625‐635. doi: 10.1007/s00125-016-4167-1 [DOI] [PubMed] [Google Scholar]
- 148. Delvecchio M, Mozzillo E, Salzano G, et al. Monogenic diabetes accounts for 6.3% of cases referred to 15 Italian pediatric diabetes centers during 2007 to 2012. J Clin Endocrinol Metab. 2017;102(6):1826‐1834. doi: 10.1210/jc.2016-2490 [DOI] [PubMed] [Google Scholar]
- 149. Shepherd M, Shields B, Hammersley S, et al. Systematic population screening, using biomarkers and genetic testing, identifies 2.5% of the U.K. pediatric diabetes population with monogenic diabetes. Diabetes Care. 2016;39(11):1879‐1888. doi: 10.2337/dc16-0645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. American Diabetes Association Professional Practice C . 2. Classification and diagnosis of diabetes: standards of medical Care in Diabetes‐2022. Diabetes Care. 2022;45:S17‐S38. doi: 10.2337/dc22-S002 [DOI] [PubMed] [Google Scholar]
- 151. Shields BM, Hicks S, Shepherd MH, Colclough K, Hattersley AT, Ellard S. Maturity‐onset diabetes of the young (MODY): how many cases are we missing? Diabetologia. 2010;53(12):2504‐2508. doi: 10.1007/s00125-010-1799-4 [DOI] [PubMed] [Google Scholar]
- 152. Stanik J, Dusatkova P, Cinek O, et al. De novo mutations of GCK, HNF1A and HNF4A may be more frequent in MODY than previously assumed. Diabetologia. 2014;57(3):480‐484. doi: 10.1007/s00125-013-3119-2 [DOI] [PubMed] [Google Scholar]
- 153. Raeder H, Johansson S, Holm PI, et al. Mutations in the CEL VNTR cause a syndrome of diabetes and pancreatic exocrine dysfunction. Nat Genet. 2006;38(1):54‐62. doi: 10.1038/ng1708 [DOI] [PubMed] [Google Scholar]
- 154. Prisco F, Iafusco D, Franzese A, Sulli N, Barbetti F. MODY 2 presenting as neonatal hyperglycaemia: a need to reshape the definition of "neonatal diabetes"? Diabetologia. 2000;43(10):1331‐1332. doi: 10.1007/s001250051531 [DOI] [PubMed] [Google Scholar]
- 155. Steele AM, Wensley KJ, Ellard S, et al. Use of HbA1c in the identification of patients with hyperglycaemia caused by a glucokinase mutation: observational case control studies. PLoS One. 2013;8(6):e65326. doi: 10.1371/journal.pone.0065326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Stride A, Vaxillaire M, Tuomi T, et al. The genetic abnormality in the beta cell determines the response to an oral glucose load. Diabetologia. 2002;45(3):427‐435. [DOI] [PubMed] [Google Scholar]
- 157. Lorini R, Alibrandi A, Vitali L, et al. Risk of type 1 diabetes development in children with incidental hyperglycemia: a multicenter Italian study. Diabetes Care. 2001;24(7):1210‐1216. [DOI] [PubMed] [Google Scholar]
- 158. Lorini R, Klersy C, d'Annunzio G, et al. Maturity‐onset diabetes of the young in children with incidental hyperglycemia: a multicenter Italian study of 172 families. Diabetes Care. 2009;32(10):1864‐1866. doi: 10.2337/dc08-2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Steele AM, Shields BM, Wensley KJ, Colclough K, Ellard S, Hattersley AT. Prevalence of vascular complications among patients with glucokinase mutations and prolonged, mild hyperglycemia. JAMA. 2014;311(3):279‐286. doi: 10.1001/jama.2013.283980 [DOI] [PubMed] [Google Scholar]
- 160. Velho G, Blanche H, Vaxillaire M, et al. Identification of 14 new glucokinase mutations and description of the clinical profile of 42 MODY‐2 families. Diabetologia. 1997;40(2):217‐224. [DOI] [PubMed] [Google Scholar]
- 161. Stride A, Shields B, Gill‐Carey O, et al. Cross‐sectional and longitudinal studies suggest pharmacological treatment used in patients with glucokinase mutations does not alter glycaemia. Diabetologia. 2014;57(1):54‐56. doi: 10.1007/s00125-013-3075-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Chakera AJ, Steele AM, Gloyn AL, et al. Recognition and Management of Individuals with Hyperglycemia because of a heterozygous Glucokinase mutation. Diabetes Care. 2015;38(7):1383‐1392. doi: 10.2337/dc14-2769 [DOI] [PubMed] [Google Scholar]
- 163. Chakera AJ, Spyer G, Vincent N, Ellard S, Hattersley AT, Dunne FP. The 0.1% of the population with glucokinase monogenic diabetes can be recognized by clinical characteristics in pregnancy: the Atlantic diabetes in pregnancy cohort. Diabetes Care. 2014;37(5):1230‐1236. doi: 10.2337/dc13-2248 [DOI] [PubMed] [Google Scholar]
- 164. Rudland VL, Hinchcliffe M, Pinner J, et al. Identifying Glucokinase monogenic diabetes in a multiethnic gestational diabetes mellitus cohort: new pregnancy screening criteria and utility of HbA1c. Diabetes Care. 2016;39(1):50‐52. doi: 10.2337/dc15-1001 [DOI] [PubMed] [Google Scholar]
- 165. Fendler W, Malachowska B, Baranowska‐Jazwiecka A, et al. Population‐based estimates for double diabetes amongst people with glucokinase monogenic diabetes, GCK‐MODY. Diabet Med. 2014;31(7):881‐883. doi: 10.1111/dme.12449 [DOI] [PubMed] [Google Scholar]
- 166. Isomaa B, Henricsson M, Lehto M, et al. Chronic diabetic complications in patients with MODY3 diabetes. Diabetologia. 1998;41(4):467‐473. doi: 10.1007/s001250050931 [DOI] [PubMed] [Google Scholar]
- 167. Pearson ER, Pruhova S, Tack CJ, et al. Molecular genetics and phenotypic characteristics of MODY caused by hepatocyte nuclear factor 4alpha mutations in a large European collection. Diabetologia. 2005;48(5):878‐885. doi: 10.1007/s00125-005-1738-y [DOI] [PubMed] [Google Scholar]
- 168. Ostoft SH, Bagger JI, Hansen T, et al. Glucose‐lowering effects and low risk of hypoglycemia in patients with maturity‐onset diabetes of the young when treated with a GLP‐1 receptor agonist: a double‐blind, randomized, crossover trial. Diabetes Care. 2014;37(7):1797‐1805. doi: 10.2337/dc13-3007 [DOI] [PubMed] [Google Scholar]
- 169. Bacon S, Kyithar MP, Rizvi SR, et al. Successful maintenance on sulphonylurea therapy and low diabetes complication rates in a HNF1A‐MODY cohort. Diabet Med. 2016;33(7):976‐984. doi: 10.1111/dme.12992 [DOI] [PubMed] [Google Scholar]
- 170. Steele AM, Shields BM, Shepherd M, Ellard S, Hattersley AT, Pearson ER. Increased all‐cause and cardiovascular mortality in monogenic diabetes as a result of mutations in the HNF1A gene. Diabet Med. 2010;27(2):157‐161. doi: 10.1111/j.1464-5491.2009.02913.x [DOI] [PubMed] [Google Scholar]
- 171. Bellanne‐Chantelot C, Carette C, Riveline JP, et al. The type and the position of HNF1A mutation modulate age at diagnosis of diabetes in patients with maturity‐onset diabetes of the young (MODY)‐3. Diabetes. 2008;57(2):503‐508. doi: 10.2337/db07-0859 [DOI] [PubMed] [Google Scholar]
- 172. Harries LW, Ellard S, Stride A, Morgan NG, Hattersley AT. Isomers of the TCF1 gene encoding hepatocyte nuclear factor‐1 alpha show differential expression in the pancreas and define the relationship between mutation position and clinical phenotype in monogenic diabetes. Hum Mol Genet. 2006;15(14):2216‐2224. doi: 10.1093/hmg/ddl147 [DOI] [PubMed] [Google Scholar]
- 173. Donath X, Saint‐Martin C, Dubois‐Laforgue D, et al. Next‐generation sequencing identifies monogenic diabetes in 16% of patients with late adolescence/adult‐onset diabetes selected on a clinical basis: a cross‐sectional analysis. BMC Med. 2019;17(1):132. doi: 10.1186/s12916-019-1363-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Stride A, Ellard S, Clark P, et al. Beta‐cell dysfunction, insulin sensitivity, and glycosuria precede diabetes in hepatocyte nuclear factor‐1alpha mutation carriers. Diabetes Care. 2005;28(7):1751‐1756. doi: 10.2337/diacare.28.7.1751 [DOI] [PubMed] [Google Scholar]
- 175. Hamilton AJ, Bingham C, McDonald TJ, et al. The HNF4A R76W mutation causes atypical dominant Fanconi syndrome in addition to a beta cell phenotype. J Med Genet. 2014;51(3):165‐169. doi: 10.1136/jmedgenet-2013-102066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Pearson ER, Boj SF, Steele AM, et al. Macrosomia and hyperinsulinaemic hypoglycaemia in patients with heterozygous mutations in the HNF4A gene. PLoS Med. 2007;4(4):e118. doi: 10.1371/journal.pmed.0040118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Flanagan SE, Kapoor RR, Mali G, et al. Diazoxide‐responsive hyperinsulinemic hypoglycemia caused by HNF4A gene mutations. Eur J Endocrinol. 2010;162(5):987‐992. doi: 10.1530/EJE-09-0861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Kapoor RR, Locke J, Colclough K, et al. Persistent hyperinsulinemic hypoglycemia and maturity‐onset diabetes of the young due to heterozygous HNF4A mutations. Diabetes. 2008;57(6):1659‐1663. doi: 10.2337/db07-1657 [DOI] [PubMed] [Google Scholar]
- 179. Stanescu DE, Hughes N, Kaplan B, Stanley CA, De Leon DD. Novel presentations of congenital hyperinsulinism due to mutations in the MODY genes: HNF1A and HNF4A. Research support, N.I.H., extramural. J Clin Endocrinol Metab. 2012;97(10):E2026‐E2030. doi: 10.1210/jc.2012-1356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Pearson ER, Starkey BJ, Powell RJ, Gribble FM, Clark PM, Hattersley AT. Genetic cause of hyperglycaemia and response to treatment in diabetes. Lancet. 2003;362:1275‐1281. [DOI] [PubMed] [Google Scholar]
- 181. Byrne MM, Sturis J, Menzel S, et al. Altered insulin secretory responses to glucose in diabetic and nondiabetic subjects with mutations in the diabetes susceptibility gene MODY3 on chromosome 12. Diabetes. 1996;45(11):1503‐1510. doi: 10.2337/diab.45.11.1503 [DOI] [PubMed] [Google Scholar]
- 182. Fajans SS, Brown MB. Administration of sulfonylureas can increase glucose‐induced insulin secretion for decades in patients with maturity‐onset diabetes of the young. Diabetes Care. 1993;16(9):1254‐1261. doi: 10.2337/diacare.16.9.1254 [DOI] [PubMed] [Google Scholar]
- 183. Shepherd M, Shields B, Ellard S, Rubio‐Cabezas O, Hattersley AT. A genetic diagnosis of HNF1A diabetes alters treatment and improves glycaemic control in the majority of insulin‐treated patients. Diabet Med. 2009;26(4):437‐441. doi: 10.1111/j.1464-5491.2009.02690.x [DOI] [PubMed] [Google Scholar]
- 184. Raile K, Schober E, Konrad K, et al. Treatment of young patients with HNF1A mutations (HNF1A‐MODY). Diabet Med. 2015;32(4):526‐530. doi: 10.1111/dme.12662 [DOI] [PubMed] [Google Scholar]
- 185. Schmidt F, Kapellen TM, Wiegand S, et al. Diabetes mellitus in children and adolescents with genetic syndromes. Exp Clin Endocrinol Diabetes. 2012;120(10):579‐585. doi: 10.1055/s-0032-1306330 [DOI] [PubMed] [Google Scholar]
- 186. Patel KA, Ozbek MN, Yildiz M, et al. Systematic genetic testing for recessively inherited monogenic diabetes: a cross‐sectional study in paediatric diabetes clinics. Diabetologia. 2022;65(2):336‐342. doi: 10.1007/s00125-021-05597-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Farmer A, Ayme S, de Heredia ML, et al. EURO‐WABB: an EU rare diseases registry for Wolfram syndrome, Alstrom syndrome and Bardet‐Biedl syndrome. BMC Pediatr. 2013;13:130. doi: 10.1186/1471-2431-13-130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Inoue H, Tanizawa Y, Wasson J, et al. A gene encoding a transmembrane protein is mutated in patients with diabetes mellitus and optic atrophy (Wolfram syndrome). Nat Genet. 1998;20(2):143‐148. doi: 10.1038/2441 [DOI] [PubMed] [Google Scholar]
- 189. Barrett TG, Bundey SE, Macleod AF. Neurodegeneration and diabetes: UK nationwide study of Wolfram (DIDMOAD) syndrome. Lancet. 1995;346(8988):1458‐1463. doi: 10.1016/s0140-6736(95)92473-6 [DOI] [PubMed] [Google Scholar]
- 190. Marshall BA, Permutt MA, Paciorkowski AR, et al. Phenotypic characteristics of early Wolfram syndrome. Orphanet J Rare Dis. 2013;8:64. doi: 10.1186/1750-1172-8-64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Karzon R, Narayanan A, Chen L, Lieu JEC, Hershey T. Longitudinal hearing loss in Wolfram syndrome. Orphanet J Rare Dis. 2018;13(1):102. doi: 10.1186/s13023-018-0852-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Bueno GE, Ruiz‐Castañeda D, Martínez JR, Muñoz MR, Alascio PC. Natural history and clinical characteristics of 50 patients with Wolfram syndrome. Endocrine. 2018;61(3):440‐446. doi: 10.1007/s12020-018-1608-2 [DOI] [PubMed] [Google Scholar]
- 193. de Heredia ML, Cleries R, Nunes V. Genotypic classification of patients with Wolfram syndrome: insights into the natural history of the disease and correlation with phenotype. Genet Med. 2013;15(7):497‐506. doi: 10.1038/gim.2012.180 [DOI] [PubMed] [Google Scholar]
- 194. Zmyslowska A, Borowiec M, Fichna P, et al. Delayed recognition of Wolfram syndrome frequently misdiagnosed as type 1 diabetes with early chronic complications. Exp Clin Endocrinol Diabetes. 2014;122(1):35‐38. doi: 10.1055/s-0033-1357160 [DOI] [PubMed] [Google Scholar]
- 195. Khanim F, Kirk J, Latif F, Barrett TG. WFS1/wolframin mutations, Wolfram syndrome, and associated diseases. Hum Mutat. 2001;17(5):357‐367. doi: 10.1002/humu.1110 [DOI] [PubMed] [Google Scholar]
- 196. Fonseca SG, Ishigaki S, Oslowski CM, et al. Wolfram syndrome 1 gene negatively regulates ER stress signaling in rodent and human cells. J Clin Invest. 2010;120(3):744‐755. doi: 10.1172/JCI39678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Abreu D, Stone SI, Pearson TS, et al. A phase Ib/IIa clinical trial of dantrolene sodium in patients with Wolfram syndrome. JCI Insight. 2021;6(15):145188. doi: 10.1172/jci.insight.145188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Amr S, Heisey C, Zhang M, et al. A homozygous mutation in a novel zinc‐finger protein, ERIS, is responsible for Wolfram syndrome 2. Am J Hum Genet. 2007;81(4):673‐683. doi: 10.1086/520961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Bingham C, Hattersley AT. Renal cysts and diabetes syndrome resulting from mutations in hepatocyte nuclear factor‐1beta. Nephrol Dial Transplant. 2004;19(11):2703‐2708. doi: 10.1093/ndt/gfh348 [DOI] [PubMed] [Google Scholar]
- 200. Ulinski T, Lescure S, Beaufils S, et al. Renal phenotypes related to hepatocyte nuclear factor‐1beta (TCF2) mutations in a pediatric cohort. J Am Soc Nephrol. 2006;17(2):497‐503. doi: 10.1681/ASN.2005101040 [DOI] [PubMed] [Google Scholar]
- 201. Madariaga L, Garcia‐Castano A, Ariceta G, et al. Variable phenotype in HNF1B mutations: extrarenal manifestations distinguish affected individuals from the population with congenital anomalies of the kidney and urinary tract. Clin Kidney J. 2019;12(3):373‐379. doi: 10.1093/ckj/sfy102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Dubois‐Laforgue D, Cornu E, Saint‐Martin C, et al. Diabetes, associated clinical Spectrum, Long‐term prognosis, and genotype/phenotype correlations in 201 adult patients with hepatocyte nuclear factor 1B (HNF1B) molecular defects. Diabetes Care. 2017;40(11):1436‐1443. doi: 10.2337/dc16-2462 [DOI] [PubMed] [Google Scholar]
- 203. Edghill EL, Bingham C, Ellard S, Hattersley AT. Mutations in hepatocyte nuclear factor‐1beta and their related phenotypes. J Med Genet. 2006;43(1):84‐90. doi: 10.1136/jmg.2005.032854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Raile K, Klopocki E, Holder M, et al. Expanded clinical spectrum in hepatocyte nuclear factor 1b‐maturity‐onset diabetes of the young. J Clin Endocrinol Metab. 2009;94(7):2658‐2664. doi: 10.1210/jc.2008-2189 [DOI] [PubMed] [Google Scholar]
- 205. Bellanne‐Chantelot C, Chauveau D, Gautier J‐F, et al. Clinical Spectrum associated with hepatocyte nuclear Factor‐1{beta} mutations. Ann Intern Med. 2004;140(7):510‐517. [DOI] [PubMed] [Google Scholar]
- 206. Pearson ER, Badman MK, Lockwood CR, et al. Contrasting diabetes phenotypes associated with hepatocyte nuclear factor‐1alpha and ‐1beta mutations. Diabetes Care. 2004;27(5):1102‐1107. doi: 10.2337/diacare.27.5.1102 [DOI] [PubMed] [Google Scholar]
- 207. Tjora E, Wathle G, Erchinger F, et al. Exocrine pancreatic function in hepatocyte nuclear factor 1beta‐maturity‐onset diabetes of the young (HNF1B‐MODY) is only moderately reduced: compensatory hypersecretion from a hypoplastic pancreas. Diabet Med. 2013;30(8):946‐955. doi: 10.1111/dme.12190 [DOI] [PubMed] [Google Scholar]
- 208. Haldorsen IS, Vesterhus M, Raeder H, et al. Lack of pancreatic body and tail in HNF1B mutation carriers. Diabet Med. 2008;25(7):782‐787. doi: 10.1111/j.1464-5491.2008.02460.x [DOI] [PubMed] [Google Scholar]
- 209. Reinauer C, Meissner T, Roden M, et al. Low prevalence of patients with mitochondrial disease in the German/Austrian DPV diabetes registry. Eur J Pediatr. 2016;175(5):613‐622. doi: 10.1007/s00431-015-2675-5 [DOI] [PubMed] [Google Scholar]
- 210. Maassen JA, 't Hart LM, van Essen E, et al. Mitochondrial diabetes: molecular mechanisms and clinical presentation. Diabetes. 2004;53(90001):S103‐S109. doi: 10.2337/diabetes.53.2007.S103 [DOI] [PubMed] [Google Scholar]
- 211. Guillausseau PJ, Dubois‐Laforgue D, Massin P, et al. Heterogeneity of diabetes phenotype in patients with 3243 bp mutation of mitochondrial DNA (maternally inherited diabetes and deafness or MIDD). Diabetes Metab. 2004;30(2):181‐186. doi: 10.1016/S1262-3636(07)70105-2 [DOI] [PubMed] [Google Scholar]
- 212. Laloi‐Michelin M, Meas T, Ambonville C, et al. The clinical variability of maternally inherited diabetes and deafness is associated with the degree of heteroplasmy in blood leukocytes. J Clin Endocrinol Metab. 2009;94(8):3025‐3030. doi: 10.1210/jc.2008-2680 [DOI] [PubMed] [Google Scholar]
- 213. Goto Y, Nonaka I, Horai S. A mutation in the tRNA(Leu)(UUR) gene associated with the MELAS subgroup of mitochondrial encephalomyopathies. Nature. 1990;348(6302):651‐653. doi: 10.1038/348651a0 [DOI] [PubMed] [Google Scholar]
- 214. Lalau JD. Lactic acidosis induced by metformin: incidence, management and prevention. Drug Saf. 2010;33(9):727‐740. doi: 10.2165/11536790-000000000-00000 [DOI] [PubMed] [Google Scholar]
- 215. Laloi‐Michelin M, Virally M, Jardel C, et al. Kearns Sayre syndrome: an unusual form of mitochondrial diabetes. Diabetes Metab. 2006;32(2):182‐186. doi: 10.1016/s1262-3636(07)70267-7 [DOI] [PubMed] [Google Scholar]
- 216. Superti‐Furga A, Schoenle E, Tuchschmid P, et al. Pearson bone marrow‐pancreas syndrome with insulin‐dependent diabetes, progressive renal tubulopathy, organic aciduria and elevated fetal haemoglobin caused by deletion and duplication of mitochondrial DNA. Eur J Pediatr. 1993;152(1):44‐50. doi: 10.1007/BF02072515 [DOI] [PubMed] [Google Scholar]
- 217. Raeder H, Haldorsen IS, Ersland L, et al. Pancreatic lipomatosis is a structural marker in nondiabetic children with mutations in carboxyl‐ester lipase. Diabetes. 2007;56(2):444‐449. [DOI] [PubMed] [Google Scholar]
- 218. Raeder H, McAllister FE, Tjora E, et al. Carboxyl‐ester lipase maturity‐onset diabetes of the young is associated with development of pancreatic cysts and upregulated MAPK signaling in secretin‐stimulated duodenal fluid. Diabetes. 2014;63(1):259‐269. doi: 10.2337/db13-1012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219. El Jellas K, Dusatkova P, Haldorsen IS, et al. Two new mutations in the CEL gene causing diabetes and hereditary pancreatitis: how to correctly identify MODY8 cases. J Clin Endocrinol Metab. 2022;107(4):e1455‐e1466. doi: 10.1210/clinem/dgab864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220. Johansson BB, Fjeld K, El Jellas K, et al. The role of the carboxyl ester lipase (CEL) gene in pancreatic disease. Pancreatology. 2018;18(1):12‐19. doi: 10.1016/j.pan.2017.12.001 [DOI] [PubMed] [Google Scholar]
- 221. Gravdal A, Xiao X, Cnop M, et al. The position of single‐base deletions in the VNTR sequence of the carboxyl ester lipase (CEL) gene determines proteotoxicity. J Biol Chem. 2021;296:100661. doi: 10.1016/j.jbc.2021.100661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222. Xiao X, Jones G, Sevilla WA, et al. A carboxyl ester lipase (CEL) mutant causes chronic pancreatitis by forming intracellular aggregates that activate apoptosis. J Biol Chem. 2017;292(19):7744. doi: 10.1074/jbc.A116.734384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223. Dalva M, Lavik IK, El Jellas K, et al. Pathogenic carboxyl ester lipase (CEL) variants interact with the normal CEL protein in pancreatic cells. Cell. 2020;9:244. doi: 10.3390/cells9010244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. Rebours V, Boutron‐Ruault MC, Schnee M, et al. The natural history of hereditary pancreatitis: a national series. Gut. 2009;58(1):97‐103. doi: 10.1136/gut.2008.149179 [DOI] [PubMed] [Google Scholar]
- 225. Yew TW, McCreight L, Colclough K, Ellard S, Pearson ER. tRNA methyltransferase homologue gene TRMT10A mutation in young adult‐onset diabetes with intellectual disability, microcephaly and epilepsy. Diabet Med. 2016;33(9):e21‐e25. doi: 10.1111/dme.13024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Alwatban S, Alfaraidi H, Alosaimi A, et al. Case report: homozygous DNAJC3 mutation causes monogenic diabetes mellitus associated with pancreatic atrophy. Front Endocrinol. 2021;12:742278. doi: 10.3389/fendo.2021.742278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227. Semple RK, Savage DB, Cochran EK, Gorden P, O'Rahilly S. Genetic syndromes of severe insulin resistance. Research support, non‐U.S. Gov't review. Endocr Rev. 2011;32(4):498‐514. doi: 10.1210/er.2010-0020 [DOI] [PubMed] [Google Scholar]
- 228. Parker VE, Semple RK. Genetics in endocrinology: genetic forms of severe insulin resistance: what endocrinologists should know. Eur J Endocrinol. 2013;169(4):R71‐R80. doi: 10.1530/EJE-13-0327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229. Musso C, Cochran E, Moran SA, et al. Clinical course of genetic diseases of the insulin receptor (type A and Rabson‐Mendenhall syndromes): a 30‐year prospective. Medicine. 2004;83(4):209‐222. [DOI] [PubMed] [Google Scholar]
- 230. Taylor SI, Cama A, Accili D, et al. Mutations in the insulin receptor gene. Endocr Rev. 1992;13(3):566‐595. doi: 10.1210/edrv-13-3-566 [DOI] [PubMed] [Google Scholar]
- 231. Groeneveld MP, Huang‐Doran I, Semple RK. Adiponectin and leptin in human severe insulin resistance ‐ diagnostic utility and biological insights. Biochimie. 2012;94(10):2172‐2179. doi: 10.1016/j.biochi.2012.01.021 [DOI] [PubMed] [Google Scholar]
- 232. Maassen JA, Tobias ES, Kayserilli H, et al. Identification and functional assessment of novel and known insulin receptor mutations in five patients with syndromes of severe insulin resistance. J Clin Endocrinol Metab. 2003;88(9):4251‐4257. doi: 10.1210/jc.2003-030034 [DOI] [PubMed] [Google Scholar]
- 233. Melvin A, O'Rahilly S, Savage DB. Genetic syndromes of severe insulin resistance. Curr Opin Genet Dev. 2018;50:60‐67. doi: 10.1016/j.gde.2018.02.002 [DOI] [PubMed] [Google Scholar]
- 234. Regan FM, Williams RM, McDonald A, et al. Treatment with recombinant human insulin‐like growth factor (rhIGF)‐I/rhIGF binding protein‐3 complex improves metabolic control in subjects with severe insulin resistance. J Clin Endocrinol Metab. 2010;95(5):2113‐2122. doi: 10.1210/jc.2009-2088 [DOI] [PubMed] [Google Scholar]
- 235. Carmody D, Ladsaria SS, Buikema RK, Semple RK, Greeley SA. Successful rhIGF1 treatment for over 5 years in a patient with severe insulin resistance due to homozygous insulin receptor mutation. Diabet Med. 2016;33(3):e8‐e12. doi: 10.1111/dme.12884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236. Okawa MC, Cochran E, Lightbourne M, Brown RJ. Long‐term effects of Metreleptin in Rabson‐Mendenhall syndrome on Glycemia, growth, and kidney function. J Clin Endocrinol Metab. 2022;107(3):e1032‐e1046. doi: 10.1210/clinem/dgab782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237. Galderisi A, Tamborlane W, Taylor SI, Attia N, Moretti C, Barbetti F. SGLT2i improves glycemic control in patients with congenital severe insulin resistance. Pediatrics. 2022;150(1):e2021055671. doi: 10.1542/peds.2021-055671 [DOI] [PubMed] [Google Scholar]
- 238. Hosokawa Y, Ogawa W. SGLT2 inhibitors for genetic and acquired insulin resistance: considerations for clinical use. J Diabetes Investig. 2020;11(6):1431‐1433. doi: 10.1111/jdi.13309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239. Legro RS, Arslanian SA, Ehrmann DA, et al. Diagnosis and treatment of polycystic ovary syndrome: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2013;98(12):4565‐4592. doi: 10.1210/jc.2013-2350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240. Garg A. Acquired and inherited lipodystrophies. N Engl J Med. 2004;350(12):1220‐1234. doi: 10.1056/NEJMra025261 [DOI] [PubMed] [Google Scholar]
- 241. Brown RJ, Araujo‐Vilar D, Cheung PT, et al. The diagnosis and Management of Lipodystrophy Syndromes: a multi‐society practice guideline. J Clin Endocrinol Metab. 2016;101(12):4500‐4511. doi: 10.1210/jc.2016-2466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242. Agarwal AK, Simha V, Oral EA, et al. Phenotypic and genetic heterogeneity in congenital generalized lipodystrophy. J Clin Endocrinol Metab. 2003;88(10):4840‐4847. doi: 10.1210/jc.2003-030855 [DOI] [PubMed] [Google Scholar]
- 243. Patni N, Li X, Adams‐Huet B, Vasandani C, Gomez‐Diaz RA, Garg A. Regional body fat changes and metabolic complications in children with Dunnigan lipodystrophy‐causing LMNA variants. J Clin Endocrinol Metab. 2019;104(4):1099‐1108. doi: 10.1210/jc.2018-01922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244. Huang‐Doran I, Sleigh A, Rochford JJ, O'Rahilly S, Savage DB. Lipodystrophy: metabolic insights from a rare disorder. J Endocrinol. 2010;207(3):245‐255. doi: 10.1677/JOE-10-0272 [DOI] [PubMed] [Google Scholar]
- 245. Weedon MN, Ellard S, Prindle MJ, et al. An in‐frame deletion at the polymerase active site of POLD1 causes a multisystem disorder with lipodystrophy. Nat Genet. 2013;45(8):947‐950. doi: 10.1038/ng.2670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246. Chudasama KK, Winnay J, Johansson S, et al. SHORT syndrome with partial lipodystrophy due to impaired phosphatidylinositol 3 kinase signaling. Am J Hum Genet. 2013;93(1):150‐157. doi: 10.1016/j.ajhg.2013.05.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247. Winnay JN, Solheim MH, Dirice E, et al. PI3‐kinase mutation linked to insulin and growth factor resistance in vivo. J Clin Invest. 2016;126(4):1401‐1412. doi: 10.1172/JCI84005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248. Solheim MH, Clermont AC, Winnay JN, et al. Iris malformation and anterior segment dysgenesis in mice and humans with a mutation in PI 3‐kinase. Invest Ophthalmol Vis Sci. 2017;58(7):3100‐3106. doi: 10.1167/iovs.16-21347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249. Solheim MH, Winnay JN, Batista TM, Molven A, Njolstad PR, Kahn CR. Mice carrying a dominant‐negative human PI3K mutation are protected from obesity and hepatic steatosis but not diabetes. Diabetes. 2018;67(7):1297‐1309. doi: 10.2337/db17-1509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250. Winnay JN, Solheim MH, Sakaguchi M, Njolstad PR, Kahn CR. Inhibition of the PI 3‐kinase pathway disrupts the unfolded protein response and reduces sensitivity to ER stress‐dependent apoptosis. FASEB J. 2020;34(9):12521‐12532. doi: 10.1096/fj.202000892R [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251. Owen KR, Donohoe M, Ellard S, Hattersley AT. Response to treatment with rosiglitazone in familial partial lipodystrophy due to a mutation in the LMNA gene. Diabet Med. 2003;20(10):823‐827. doi: 10.1046/j.1464-5491.2003.01034.x [DOI] [PubMed] [Google Scholar]
- 252. Brown RJ, Oral EA, Cochran E, et al. Long‐term effectiveness and safety of metreleptin in the treatment of patients with generalized lipodystrophy. Endocrine. 2018;60(3):479‐489. doi: 10.1007/s12020-018-1589-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253. Simha V, Subramanyam L, Szczepaniak L, et al. Comparison of efficacy and safety of leptin replacement therapy in moderately and severely hypoleptinemic patients with familial partial lipodystrophy of the Dunnigan variety. J Clin Endocrinol Metab. 2012;97(3):785‐792. doi: 10.1210/jc.2011-2229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254. Alstrom CH, Hallgren B, Nilsson LB, Asander H. Retinal degeneration combined with obesity, diabetes mellitus and neurogenous deafness: a specific syndrome (not hitherto described) distinct from the Laurence‐moon‐Bardet‐Biedl syndrome: a clinical, endocrinological and genetic examination based on a large pedigree. Acta Psychiatr Neurol Scand Suppl. 1959;129:1‐35. [PubMed] [Google Scholar]
- 255. Hearn T, Renforth GL, Spalluto C, et al. Mutation of ALMS1, a large gene with a tandem repeat encoding 47 amino acids, causes Alstrom syndrome. Nat Genet. 2002;31(1):79‐83. doi: 10.1038/ng874 [DOI] [PubMed] [Google Scholar]
- 256. Mokashi A, Cummings EA. Presentation and course of diabetes in children and adolescents with Alstrom syndrome. Pediatr Diabetes. 2011;12(3 Pt 2):270‐275. doi: 10.1111/j.1399-5448.2010.00698.x [DOI] [PubMed] [Google Scholar]
- 257. Paisey RB, Geberhiwot T, Waterson M, et al. Modification of severe insulin resistant diabetes in response to lifestyle changes in Alstrom syndrome. Eur J Med Genet. 2014;57(2–3):71‐75. doi: 10.1016/j.ejmg.2013.12.008 [DOI] [PubMed] [Google Scholar]
- 258. Tobin JL, Beales PL. Bardet‐Biedl syndrome: beyond the cilium. Pediatr Nephrol. 2007;22(7):926‐936. doi: 10.1007/s00467-007-0435-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259. Scheidecker S, Etard C, Pierce NW, et al. Exome sequencing of Bardet‐Biedl syndrome patient identifies a null mutation in the BBSome subunit BBIP1 (BBS18). J Med Genet. 2014;51(2):132‐136. doi: 10.1136/jmedgenet-2013-101785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260. Guo DF, Rahmouni K. Molecular basis of the obesity associated with Bardet‐Biedl syndrome. Trends Endocrinol Metab. 2011;22(7):286‐293. doi: 10.1016/j.tem.2011.02.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261. Abu‐Safieh L, Al‐Anazi S, Al‐Abdi L, et al. In search of triallelism in Bardet‐Biedl syndrome. Eur J Hum Genet. 2012;20(4):420‐427. doi: 10.1038/ejhg.2011.205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262. Katsanis N, Ansley SJ, Badano JL, et al. Triallelic inheritance in Bardet‐Biedl syndrome, a Mendelian recessive disorder. Science. 2001;293(5538):2256‐2259. doi: 10.1126/science.1063525 [DOI] [PubMed] [Google Scholar]
- 263. Edghill EL, Flanagan SE, Ellard S. Permanent neonatal diabetes due to activating mutations in ABCC8 and KCNJ11. Research support, non‐U.S. Gov't review. Rev Endocr Metab Disord. 2010;11(3):193‐198. doi: 10.1007/s11154-010-9149-x [DOI] [PubMed] [Google Scholar]
- 264. Stoffers DA, Zinkin NT, Stanojevic V, Clarke WL, Habener JF. Pancreatic agenesis attributable to a single nucleotide deletion in the human IPF1 gene coding sequence. Nat Genet. 1997;15(1):106‐110. [DOI] [PubMed] [Google Scholar]
- 265. Sellick GS, Barker KT, Stolte‐Dijkstra I, et al. Mutations in PTF1A cause pancreatic and cerebellar agenesis. Nat Genet. 2004;36(12):1301‐1305. doi: 10.1038/ng1475 [DOI] [PubMed] [Google Scholar]
- 266. Smith SB, Qu HQ, Taleb N, et al. Rfx6 directs islet formation and insulin production in mice and humans. Nature. 2010;463(7282):775‐780. doi: 10.1038/nature08748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267. Passone CGB, Vermillac G, Staels W, et al. Mitchell‐Riley syndrome: improving clinical outcomes and searching for functional impact of RFX‐6 mutations. Front Endocrinol. 2022;13:802351. doi: 10.3389/fendo.2022.802351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268. D'Amato E, Giacopelli F, Giannattasio A, et al. Genetic investigation in an Italian child with an unusual association of atrial septal defect, attributable to a new familial GATA4 gene mutation, and neonatal diabetes due to pancreatic agenesis. Diabet Med. 2010;27(10):1195‐1200. doi: 10.1111/j.1464-5491.2010.03046.x [DOI] [PubMed] [Google Scholar]
- 269. Senee V, Chelala C, Duchatelet S, et al. Mutations in GLIS3 are responsible for a rare syndrome with neonatal diabetes mellitus and congenital hypothyroidism. Nat Genet. 2006;38(6):682‐687. doi: 10.1038/ng1802 [DOI] [PubMed] [Google Scholar]
- 270. Rubio‐Cabezas O, Jensen JN, Hodgson MI, et al. Permanent neonatal diabetes and enteric Anendocrinosis associated with Biallelic mutations in NEUROG3. Research support, non‐U.S. Gov't. Diabetes. 2011;60(4):1349‐1353. doi: 10.2337/db10-1008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271. Rubio‐Cabezas O, Minton JAL, Kantor I, Williams D, Ellard S, Hattersley AT. Homozygous mutations in NEUROD1 are responsible for a novel syndrome of permanent neonatal diabetes and neurological abnormalities. Diabetes. 2010;59(9):2326‐2331. doi: 10.2337/db10-0011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272. Solomon BD, Pineda‐Alvarez DE, Balog JZ, et al. Compound heterozygosity for mutations in PAX6 in a patient with complex brain anomaly, neonatal diabetes mellitus, and microophthalmia. Am J Med Genet A. 2009;149A(11):2543‐2546. doi: 10.1002/ajmg.a.33081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273. Flanagan SE, De Franco E, Lango Allen H, et al. Analysis of transcription factors key for mouse pancreatic development establishes NKX2‐2 and MNX1 mutations as causes of neonatal diabetes in man. Cell Metab. 2014;19(1):146‐154. doi: 10.1016/j.cmet.2013.11.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274. De Franco E, Watson RA, Weninger WJ, et al. A specific CNOT1 mutation results in a novel syndrome of pancreatic agenesis and holoprosencephaly through impaired pancreatic and neurological development. Am J Hum Genet. 2019;104(5):985‐989. doi: 10.1016/j.ajhg.2019.03.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275. Philippi A, Heller S, Costa IG, et al. Mutations and variants of ONECUT1 in diabetes. Nat Med. 2021;27(11):1928‐1940. doi: 10.1038/s41591-021-01502-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276. Sansbury FH, Flanagan SE, Houghton JA, et al. SLC2A2 mutations can cause neonatal diabetes, suggesting GLUT2 may have a role in human insulin secretion. Diabetologia. 2012;55(9):2381‐2385. doi: 10.1007/s00125-012-2595-0 [DOI] [PubMed] [Google Scholar]
- 277. Shaw‐Smith C, Flanagan SE, Patch AM, et al. Recessive SLC19A2 mutations are a cause of neonatal diabetes mellitus in thiamine‐responsive megaloblastic anaemia. Research support, non‐U.S. Gov't. Pediatr Diabetes. 2012;13(4):314‐321. doi: 10.1111/j.1399-5448.2012.00855.x [DOI] [PubMed] [Google Scholar]
- 278. Mameli C, Cazzola R, Spaccini L, et al. Neonatal diabetes in patients affected by Liang‐Wang syndrome carrying KCNMA1 variant p.(Gly375Arg) suggest a potential role of Ca(2+) and voltage‐activated K(+) channel activity in human insulin secretion. Curr Issues Mol Biol. 2021;43(2):1036‐1042. doi: 10.3390/cimb43020073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279. Abdel‐Salam GM, Schaffer AE, Zaki MS, et al. A homozygous IER3IP1 mutation causes microcephaly with simplified gyral pattern, epilepsy, and permanent neonatal diabetes syndrome (MEDS). Am J Med Genet A. 2012;158A(11):2788‐2796. doi: 10.1002/ajmg.a.35583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280. Petrie JR, Chaturvedi N, Ford I, et al. Cardiovascular and metabolic effects of metformin in patients with type 1 diabetes (REMOVAL): a double‐blind, randomised, placebo‐controlled trial. Lancet Diabetes Endocrinol. 2017;5(8):597‐609. doi: 10.1016/S2213-8587(17)30194-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281. De Franco E, Flanagan SE, Yagi T, et al. Dominant ER stress‐inducing WFS1 mutations underlie a genetic syndrome of neonatal/infancy‐onset diabetes, congenital sensorineural deafness, and congenital cataracts. Diabetes. 2017;66(7):2044‐2053. doi: 10.2337/db16-1296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282. De Franco E, Caswell R, Johnson MB, et al. De novo mutations in EIF2B1 affecting eIF2 signaling cause neonatal/early‐onset diabetes and transient hepatic dysfunction. Diabetes. 2020;69(3):477‐483. doi: 10.2337/db19-1029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283. De Franco E, Lytrivi M, Ibrahim H, et al. YIPF5 mutations cause neonatal diabetes and microcephaly through endoplasmic reticulum stress. J Clin Invest. 2020;130(12):6338‐6353. doi: 10.1172/JCI141455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284. Vionnet N, Stoffel M, Takeda J, et al. Nonsense mutation in the glucokinase gene causes early‐onset non‐insulin‐dependent diabetes mellitus. Nature. 1992;356(6371):721‐722. doi: 10.1038/356721a0 [DOI] [PubMed] [Google Scholar]
- 285. Yamagata K, Oda N, Kaisaki PJ, et al. Mutations in the hepatocyte nuclear factor‐1alpha gene in maturity‐onset diabetes of the young (MODY3). Nature. 1996;384(6608):455‐458. doi: 10.1038/384455a0 [DOI] [PubMed] [Google Scholar]
- 286. Yamagata K, Furuta H, Oda N, et al. Mutations in the hepatocyte nuclear factor‐4alpha gene in maturity‐onset diabetes of the young (MODY1). Nature. 1996;384(6608):458‐460. doi: 10.1038/384458a0 [DOI] [PubMed] [Google Scholar]
- 287. Horikawa Y, Iwasaki N, Hara M, et al. Mutation in hepatocyte nuclear factor‐1 beta gene (TCF2) associated with MODY. Nat Genet. 1997;17(4):384‐385. doi: 10.1038/ng1297-384 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing not applicable to this article as no datasets were generated or analysed during the current study.