

Abstract
Rare diseases represent a highly heterogeneous group of disorders with high phenotypic and genotypic diversity within individual conditions. Due to the small numbers of people affected, there are unique challenges in understanding rare diseases and drug development for these conditions, including patient identification and recruitment, trial design, and costs. Natural history data and real‐world data (RWD) play significant roles in defining and characterizing disease progression, final patient populations, novel biomarkers, genetic relationships, and treatment effects. This review provides an introduction to rare diseases, natural history data, RWD, and real‐world evidence, the respective sources and applications of these data in several rare diseases. Considerations for data quality and limitations when using natural history and RWD are also elaborated. Opportunities are highlighted for cross‐sector collaboration, standardized and high‐quality data collection using new technologies, and more comprehensive evidence generation using quantitative approaches such as disease progression modeling, artificial intelligence, and machine learning. Advanced statistical approaches to integrate natural history data and RWD to further disease understanding and guide more efficient clinical study design and data analysis in drug development in rare diseases are also discussed.
Keywords: disease progression modeling, natural history, rare diseases, real‐world data, real‐world evidence
What Is Different About Rare Diseases and Orphan Drugs?
Rare diseases are defined by the US Food and Drug Administration (FDA) as conditions that affect fewer than 200,000 people in the United States and by the European Medicines Agency (EMA) as conditions that affect fewer than 1 in 2000 people. 1 , 2 To date, over 7000 rare diseases have been identified, and collectively they affect more than 250 million people across the world. 1 , 3 Rare diseases represent a highly heterogeneous group of disorders, and there is often high phenotypic diversity within individual conditions. 4 Due to the small numbers of people affected, opportunities to study each disease are limited, and as a result, rare diseases are usually poorly or incompletely understood. 4 Most rare diseases also lack effective treatments, and there are many challenges associated with drug development for these conditions, including patient identification and recruitment, trial design, and costs. 5 To overcome some of these challenges, the Orphan Drug Act was passed in 1983 in the United States and the Orphan Regulation was adopted in the European Union in 2000, with the aim of incentivizing pharmaceutical companies to develop orphan drugs that target rare diseases. 6
What Are Natural History Data?
Comprehensive knowledge of a disease is required to design and conduct clinical trials of adequate duration and with clinically meaningful end points. 1 For rare diseases, natural history studies play an important role in identifying appropriate patient populations and clinical outcome assessments and biomarkers, and in the design of externally controlled studies. 1 A natural history study is an observational study that is designed to track the natural course of a disease and is likely to include patients receiving the current standard of care. 1 They should be comprehensive and granular and should aim to identify demographic, genetic, environmental, and other variables that correlate with disease outcomes in the absence of treatment. 1 , 4 Beyond their role in drug development, natural history studies may also benefit patients with rare diseases by establishing communication pathways, identifying disease‐specific centers of excellence, facilitating the understanding and evaluation of current standard‐of‐care practices, evaluating signs and symptoms of a disease to improve diagnosis, and identifying ways to improve patient care. 1 , 4 Patients included in natural history studies may sometimes be used as historical controls for studies that lack an internal control, thus allowing the effectiveness of the study treatment to be determined. 4 , 7 Guidance is available from the FDA and the EMA on the use of registries for supporting regulatory decision making for drugs and biological products. 8 , 9
What Are Real‐World Data?
Real‐world data (RWD) can serve as a source of information for natural history studies. The FDA and EMA have recently developed definitions of RWD and real‐world evidence (RWE). 10 , 11 RWD is defined by the FDA as “data relating to patient health status and/or the delivery of health care routinely collected from a variety of sources” and by the EMA as “routinely collected data relating to a patient's health status or the delivery of health care from a variety of sources other than traditional clinical trials.” 10 , 11 RWE is defined by the FDA as “clinical evidence about the usage and potential benefits or risks of a medical product derived from an analysis of RWD” and by the EMA as “information derived from an analysis of RWD.” 10 , 11 Both the FDA and EMA state that evidence from traditional clinical trials will not be considered RWE, but that hybrid or pragmatic trial designs and observational studies can generate RWE. 10 , 11 RWD can be collected retrospectively as well as prospectively. There are many sources of RWD, including electronic health records; medical claims and billing data; data from product and disease registries; patient‐generated data; and data gathered from other sources, for example, mobile devices. 10 , 11
While randomized controlled trials (RCTs) continue to be considered the gold‐standard source of evidence for regulatory decision making, 12 the highly selected patient populations included in RCTs may not be representative of real‐world clinical practice and there is a growing role for RWD to bridge evidence gaps not addressed by RCTs. 12 For example, RWE can provide novel insights into the performance of medicines in everyday clinical use and is frequently used to fulfill pharmacovigilance and postmarketing commitments. 10 , 11 RWE can also be used to improve the performance of clinical trials, including generating hypotheses for testing, identifying drug development tools (eg, biomarkers), assessing trial feasibility, informing statistical models, identifying patient baseline characteristics for enrichment or stratification, and assembling geographically distributed research cohorts. 8 There may also be a role for RWE in the development of orphan drugs where performing traditional RCTs may not be feasible or ethical. 10 For example, RWD can sometimes be used as an external control arm to confirm the response rate from a single‐arm trial. 10 , 11 More detailed applications of RWD in rare diseases are presented in the subsequent sections. Recently, the FDA has established an RWE framework under the 21st Century Cures Act 13 to evaluate the use of RWE in regulatory decision making. In particular, this framework aims to evaluate the potential use of RWE to help support approval of new indications for drugs that are already approved. 11
Sources of Natural History and RWD
Natural History Data in Rare Diseases
Natural history studies have previously been employed in several rare diseases, including Duchenne muscular dystrophy (DMD), spinal muscular atrophy (SMA), and Huntington's disease (HD), and have provided data on the etiology and pathophysiology of these conditions, along with insights into the outcomes achieved with standard‐of‐care treatments. 14 , 15 , 16
An example is the Cooperative International Neuromuscular Research Group (CINRG) Duchenne Natural History Study (DNHS), which is the largest prospective natural history study performed in DMD to date. 14 Overall, 440 patients aged 2–28 years were recruited from 20 centers in 9 countries and were followed up for up to 10 years. The majority of patients (66%) were ambulatory at the initial study visit, and most (87%) received glucocorticoid therapy at some point during follow‐up.
Other examples of natural history data include Treat–Neuromuscular Diseases global DMD database, Universitair Ziekenhuis Leuven, CureDuchenne, iMDEX, and ImagingDMD. 17 , 18
Patient Registries in Rare Diseases
Patient registries are typically considered as RWD and can be used to expand knowledge on the natural history of rare diseases.
STRIDE Registry of Patients With Nonsense Mutation DMD
Strategic Targeting of Registries and International Database of Excellence (STRIDE; ClinicalTrials.gov identifier: NCT02369731) is an ongoing, multicenter, observational registry providing RWE on the use of ataluren in patients with nonsense mutations in the DMD gene in routine clinical practice. 19 It is a postapproval safety study performed to fulfill a postmarketing commitment to the Pharmacovigilance Risk Assessment Committee of the EMA. 19 , 20 STRIDE is the first drug registry to be established for patients with DMD and represents the largest real‐world registry of patients with nonsense mutations in the DMD gene to date. 20 As of July 9, 2018, 213 male patients had been enrolled in STRIDE from 11 countries (53 active study sites). 20 Corticosteroids were used by 89% of patients and mean (standard deviation) ataluren exposure was 639.0 (362.9) days. 20
RESTORE Registry of Patients With Genetically Confirmed SMA
The Registry of Patients With a Diagnosis of Spinal Muscular Atrophy (RESTORE; ClinicalTrials.gov identifier: NCT04174157) is a prospective, multicenter, observational, multinational registry with the objective of assessing long‐term outcomes for patients with genetically confirmed SMA, and providing information on the effectiveness and long‐term safety of emerging and approved treatments for SMA, including the gene therapies nusinersen and onasemnogene abeparvovec. 21 , 22 The estimated enrollment is 500 participants, and the target follow‐up duration is 15 years. 21 Recruitment began in September 2018, and as of January 3, 2020, 64 patients had been enrolled at 25 participating sites. 22
ENROLL‐HD Registry of Patients With HD
ENROLL‐HD (ClinicalTrials.gov identifier: NCT01574053) is a longitudinal, observational, multinational study that integrates 2 former HD registries (REGISTRY in Europe and COHORT in North America and Australasia), and additionally includes sites in Latin America. 23 It is the world's largest observational study for HD and has already enrolled >20,000 participants across 158 clinical sites in 22 countries. 24 The objective of this study is to develop a comprehensive repository of prospective and systematically collected clinical research data and biological specimens from individuals who manifest HD, unaffected individuals known to carry the HD mutation or at risk of carrying the HD mutation, and control research participants (eg, spouses, siblings, or offspring of HD mutation carriers known not to carry the HD mutation). 23
Other Resources on Rare Disease Registries
Additional resources on rare disease registries have been compiled by the National Organization for Rare Disorders and include EU recommendations on registration of patients with rare diseases and data collection. 25 Orphanet is another important reference source on rare diseases and includes a directory of registered patients for each of the countries in Orphanet's network. 26
Real‐World Data From Claims Databases
IBM's MarketScan Research Databases are claims databases that provide proprietary deidentified real‐world claims data for privately and publicly insured people in the United States. 27 The claims data may also be useful to understand patient characteristics, disease progression, and treatment effects.
Applications of Natural History and RWD
Natural History Studies for Drug Development
According to the FDA's guidance on conducting natural history studies, 1 drug development in rare disease can benefit from a well‐designed natural history study. Potential benefits include identification of patient population, identification or development of clinical outcome assessment and biomarkers, and design of external controls.
Patient Population to Inform Clinical Trial
Some rare diseases such as idiopathic dilated cardiomyopathy often present with substantial heterogeneity of genotypes and/or phenotypes and natural history of each subtype (eg, LMNA gene–related dilated cardiomyopathy, BLC2‐associated athanogene 3–associated dilated cardiomyopathy) is often unclear. A well‐designed natural history study can collect information on patients with disease subtypes and draw meaningful conclusions around rate, patterns of and time to progression, potential genotype versus phenotype correlations, and so on, which can be used to inform clinical trial inclusion criteria, the stage of disease to treat, duration of a trial, and other parameters.
Clinical Outcome Assessments
Clinical outcome assessment (COA) is often used to assess both safety and efficacy of an investigational treatment during a clinical trial. Four types of COAs are used by the FDA including clinician‐reported outcome (eg, reasons for hospitalization), observer‐reported outcome (eg, caregiver reports), patient‐reported outcome (eg, Quality of Life, Kansas City Cardiomyopathy Questionnaire), and performance outcome (eg, test of functional capacity). A new or existing clinical outcome assessment can be used and evaluated in a natural history study to assess change or progression in a particular disease along with its performance and reproducibility for use in a clinical investigation. In addition, natural history studies can be used to assess correlation of COAs; understand minimal clinically important differences; and explore digital end points, such as gait, stride, or other digital functional assessments, as well as digital therapeutics. 28 , 29 , 30
Biomarker Development
Identification and validation of robust biomarkers are integral parts of rare disease drug development and even more important for sponsors managing early development portfolio. 31 In a natural history study for a rare disease, information collected on biomarkers can be used to diagnose the disease, serve as a prognostic factor for the disease's course, predict treatment response, or guide patient selection. Some biomarkers may serve as end points (eg, hemoglobin A1C) or surrogate end points (eg, low‐density lipoprotein as a surrogate end point for heart attack) in clinical trials if robustly validated.
External Controls
In many rare disease trials, having an adequate control group may not be feasible due to the small patient populations for recruitment or randomization to placebo being unethical for a life‐threatening disease. Data from natural history studies may provide the general medical knowledge of the disease course to contextualize the clinical trial results. They can also serve as an external control group as compared with the treatment group to demonstrate substantial evidence of effectiveness in an investigational trial or an observational registry study, as shown in the case examples of FDA approvals.
Use of Natural History Databases to Compare Treatment Outcomes in DMD
Data obtained from natural history studies can be used to compare outcomes in patients receiving different treatments. For example, the effect of glucocorticoids on clinically relevant functional outcomes can be compared in patients with DMD. These outcomes include loss of ambulation and decline in the ability to stand from supine and hand‐to‐mouth function, which are all predictors of DMD disease progression and are highly meaningful to patients, given that they are associated with delay in the onset of subsequent irreversible disease milestones and represent the degree of patients’ personal autonomy in daily life. 14
The results of the CINRG DNHS demonstrated that glucocorticoid treatment for ≥1 year was associated with a reduced risk of losing ambulation and upper limb disease progression milestones as well as a reduced risk of death compared with treatment for <1 month or no treatment. Overall, 9 clinically meaningful milestones of disease progression were established that were predictive of future trajectories of functional decline. The natural history study enabled comparison of loss of function outcomes that are applicable to clinical practice over a longer follow‐up period and in a larger rare disease cohort than would be possible in the context of an RCT. 14 The benefits of glucocorticoid therapy were also reported for patients included in the Treat–Neuromuscular Diseases global DMD database, which included clinical data from 5345 patients with DMD from 31 countries. 17
The CINRG DNHS additionally allowed for the comparison of effectiveness of individual glucocorticoids. In that study, 26% of patients received prednisone or prednisolone, and 27% received deflazacort. Decline in functional outcomes tended to be more delayed in patients treated with deflazacort compared with those treated with prednisone or prednisolone, with statistically significant 2.1–2.7‐year differences observed for age at loss of ability to stand from supine, age at loss of ambulation, and age at loss of hand‐to‐mouth function with retained hand function. 14 Similar results were obtained in an analysis of data from 4 other DMD natural history databases (Universitair Ziekenhuis Leuven, CureDuchenne, iMDEX and ImagingDMD). 18 A Cox proportional hazards model indicated that deflazacort use was significantly associated with a longer time to loss of ability to stand from supine. Observational studies have also provided insights into the safety profiles associated with corticosteroid use over a much longer period of time than can be afforded in RCTs. 14 , 32 , 33 , 34 , 35
As another example of how natural history studies can enable treatment comparison, outcomes for patients enrolled in STRIDE were compared with those reported in the CINRG DNHS to assess the effectiveness of ataluren treatment. 36 As the patients in STRIDE and the CINRG DNHS were not randomly assigned to treatment, propensity score matching was performed to identify a subset of CINRG DNHS patients who were comparable to STRIDE patients according to established predictors of disease progression. 36 This technique of using natural history data as an external control is described in more details in the Statistical Analysis Approaches section below. For the comparison of STRIDE and the CINRG DNHS, a propensity score was created using a logistic regression model that included the following covariates: age at first clinical symptoms, age at first corticosteroid use, duration of deflazacort use (<1 month, ≥1 to <12 months, and ≥12 months); and duration of other corticosteroid use (<1 month, ≥1 to <12 months, and ≥12 months). 36 Kaplan–Meier analyses of the matched STRIDE and CINRG DNHS populations demonstrated that ataluren combined with standard‐of‐care treatment significantly delayed the age at loss of ambulation and age at worsening performance in timed function tests compared with standard of care alone (P ≤ .05). 36
Real‐World Studies to Investigate Treatment Effects in DMD
Results from a retrospective real‐world study performed at the Cincinnati Children's Hospital Medical Center corroborated data from CINRG DHNS reporting differences in treatment outcomes with deflazacort versus prednisone. 37 The Cincinnati Children's Hospital Medical Center study analyzed 435 boys with DMD and demonstrated that median age at loss of ambulation was 15.6 years and 13.5 years among deflazacort and prednisone‐initiated patients, respectively. Deflazacort was also associated with better pulmonary function indicated by a higher percentage—predicted forced vital capacity, lower risk of scoliosis, higher percentage of lean body mass, shorter stature, and lower weight compared with prednisone after adjusting for age, and corticosteroid duration. 37 The benefits of switching from prednisone to deflazacort were also shown in a different real‐world study, a retrospective chart review of patients with DMD or Becker muscular dystrophy in the United States. 38 In this study, 62 patients with DMD and 30 patients with Becker muscular dystrophy switched from prednisone to deflazacort. The primary physician‐reported reasons for switching were to slow disease progression and improve tolerability. On average, the physician‐recorded clinical global impression of improvement scores were improved in patients receiving deflazacort compared with prednisone, and patients experienced fewer adverse events after switching to deflazacort compared with prednisone. 38
Real‐World Data in Understanding Disease Burden and Routine Care in HD
RWD has been used in the understanding of disease burden and routine care in HD. 39 Data from the Institute for Applied Health Research Berlin Research Database, comprising data of ≈4 million insured persons from ≈70 German statutory health insurances was analyzed. The prevalence of HD increased with advancing age, peaking at 60–69 years (16.8 per 100,000 persons; 95%CI, 13.4–21.0) and decreasing afterward. The most common medications in patients with HD were antipsychotics (66.9%) followed by antidepressants (45.1%). Physical therapy was the most often used medical aid in patients with HD (46.4%). This study provides new insights into the epidemiology and routine care of patients with HD in Germany.
RWD has also been used in longitudinal evaluation of treatment effects of commonly used medications in HD. 40 Analysis of the cross‐sectional data in the ENROLL‐HD database showed that disease progression regarding clinical, functional, and cognitive outcomes over 2 years was not affected by any of the treatment groups compared with HD control.
Model‐Based Approaches for Integrating Natural History and RWD in Rare Diseases
Model‐based data analysis plays an important role in quantitatively understanding the natural history of rare diseases, not only on the disease progression rates, but also on identification of disease onset, patient subpopulations, new end points, novel prognostic variables, optimal duration for signal detection, and the variability of the above factors. More importantly, the models will allow prediction of responses and inform efficient clinical trial design in the diseases with scarce patients. Various types of data including observational data, controlled clinical trial data, aggregated study level literature data, and RWD can be used in the modeling.
Disease Progression Modeling in HD
The longitudinal disease progression has been characterized using linear mixed effect modeling using the Unified Huntington Disease Rating Scale (UHDRS) dimension scores from 379 patients with early HD in a placebo‐controlled clinical trial European Huntington Disease Initiative study. 41 The mean linear progression rates in total motor scores (TMS) and total functional capacity (TFC) were 4.75/year and –0.44/year, respectively, and were the most significant among all the dimension scores.
Similar linear disease progression has also been reported using data from 334 patients with clinically manifesting HD in the COHORT database. 16 The mean progression rates in the TMS, Mini‐Mental State Examination, and TFC were 3.0/year (95%CI, 2.5–3.4), –0.68/year (95%CI, –0.6 to –0.8), and –0.6/year (95%CI, –0.7 to –0.5), respectively. In addition, subgroup analyses showed that more advanced disease was associated with slower progression in TFC and the chorea scores in TMS, but a more rapid progression in the Mini‐Mental State Examination. These results suggest that interventions aimed at slowing chorea progression may be better focused on individuals with less advanced disease; however, treatments seeking to demonstrate a symptomatic effect on chorea may be best studied in those with prominent chorea, who generally have more advanced disease. For interventions aimed at cognition, disease‐modifying interventions could evaluate patients with HD in the earliest symptomatic stages of the disease when deficits begin to develop. Symptomatic interventions seeking to improve, stabilize or slow down cognitive function could include those with more advanced disease, where the deficits and rates of decline are greatest.
A disease progression model using aggregated data from a total of 1821 patients with HD in 11 randomized, double‐blind, and placebo‐controlled studies also estimated similar progression rates of TMS and TFC, 6.0/year (95%CI, 5.6–6.4), and –0.60/year (95%CI, –0.72 to –0.48), respectively. 42 However, no obvious progression in chorea was estimated in this analysis, which is different from the result on chorea by Dorsey et al. 16 In addition to disease progression, Jin, et al 42 also estimated the placebo and symptomatic treatment effects by interventions.
In contrast to modeling the disease progression rates at population levels, Kuan et al 43 developed models to predict disease progression in individual patients with HD. Forty‐one of a total of 68 patients with HD exhibited a linear progression, while the remaining 17 patients showed a quadratic disease progression. Subgroup analyses also demonstrated that patients with longer CAG repeats had earlier onset and more rapid disease progression compared with patients with shorter CAG repeats. Patients sharing similar clinical profiles (age, CAG repeat length, and UHDRS) could exhibit very different patterns of disease progression. These results are consistent with the modeling results by Langbehn et al 44 using data from 2913 patients with HD from 40 centers worldwide.
A more comprehensive nonlinear mixed‐effect modeling on TMS and TFC progression was performed with a larger number of participant‐level longitudinal data from 347 participants from CARE‐HD (a controlled clinical trial), 429 from COHORT, and 815 from REGISTRY. 45 In this analysis, individual baseline disease burden (a product of age and expanded CAG repeat length) was incorporated as a continuous variable and was a significant covariate on both the progression rate constant and the baseline score in the TMS model. The model is able to predict not only population‐level but also individual‐level disease progression, and can be used to guide patient selection and enhance efficiency in clinical trials evaluating disease‐modifying therapeutics for HD by enrolling patients with more rapid disease progression.
More recently, with the advancement of data analytic tools, a disease progression model derived by machine learning (ML) has been published using pooled data from 4 observational studies (PREDICT‐HD, REGISTRY, TRACK‐HD, and ENROLL‐HD). 46 Analysis of transition times showed that the natural history of HD can be described by 9 disease states of increasing severity. “Early‐disease” states 1 and 2, which occur before motor diagnosis, lasted ≈16 years. Increasing numbers of participants had motor onset during “transition” states 3–5, which collectively lasted ≈10 years, and the “late‐disease” states 6–9 also lasted ≈10 years. The annual probability of conversion from any 1 of the 9 identified disease states to the next ranged from 5% to 27%. The insights on disease states and probabilities for progression through these states may improve trial design and participant selection.
In the above disease progression modeling examples, different HD populations, different end points, and different structure models were used to address different objectives. It would be ideal that more comprehensive HD disease progression models can integrate the individual CAG repeat length data, age, and larger sample size data including all stages of patients with HD with longer observation, using consistent UHDRS item scores to assess more confidently the continuum of HD disease progression, and influential factors and to inform clinical trial design.
Disease Progression Modeling in DMD
A model‐based clinical trial simulation (CTS) platform has been developed to optimize the design of clinical trials for DMD by the Cooperative International Neuromuscular Research Group and Duchenne Regulatory Science Consortium. 47 Data from 15 clinical trials and studies, 1505 subjects, and 27,252 observations were integrated. The nonlinear mixed‐effects models captured longitudinal changes in 5 clinical measures (NorthStar Ambulatory Assessment, forced vital capacity, and the velocities of the following 3 timed functional tests: time to stand from supine, time to climb 4 stairs, and time to walk/run 10 meters), including both early disease when function improves as a result of growth and development and later disease stages when function declines. The models and the CTS platform can be used to perform trial simulations to optimize the patient inclusion/exclusion criteria, end point selection, sample size optimization, treatment enrichment, and other trial parameters, as well as dose selection by including drug‐effect models. The data sets and models have been reviewed by the FDA and EMA; have been accepted into the Fit‐for‐Purpose and Qualification for Novel Methodologies pathways, respectively; and will be submitted for potential endorsement by both agencies. The development of a DMD disease progression model and the CTS platform has been a collaborative effort from multiple sectors including academic research groups, the pharmaceutical industry, regulatory agencies, and nonprofit organizations. It is hoped that it will provide a more efficient way of rare disease drug development.
Disease progression of DMD was also quantified using magnetic resonance biomarkers of leg muscles from a prospective observational study in DMD with yearly follow‐up for up to 6 years. 48 Magnetic resonance spectroscopy fat fractions and magnetic resonance imaging quantitative T2 levels increased with DMD disease duration and provided sensitive noninvasive measures of DMD progression. The progression time constants differed markedly between individuals and across muscles. Modeling changes in these biomarkers across multiple muscles can also be used to detect and monitor the therapeutic effects of corticosteroids on disease progression and to provide prognostic information on functional outcomes.
Haber et al 49 quantified the associations between genetic mutations and loss of ambulation (LoA) among male patients diagnosed with childhood‐onset dystrophinopathy to understand the variation in DMD disease progression. The results showed that mutation type did not predict time to LoA. Controlling for corticosteroids, exons 8 (hazard ratio, 0.22; 95%CI, 0.08–0.63) and 44 (hazard ratio, 0.30; 95%CI, 0.12–0.78) were associated with delayed LoA compared with other exon deletions. These findings suggest that mutation information should be considered in clinical trials to account for the phenotypic variability.
Statistical Analysis Approaches for Using RWD in Rare Disease
RWD data can be used both prospectively and retrospectively. While the latter is common in drug development, the prospective use of RWD in drug development has been growing ever since the passage of FDA Regulatory Act 50 and the issuance of guidance documents from regulatory agencies. 8 , 9 RWD can be used in several ways to inform the study design in drug development for rare diseases. Widely accepted applications of RWD include identification of study population, validation of end point, planning inclusion and exclusion criteria, and safety monitoring. However, a more appealing use of RWD is informing the trial design prospectively (eg, for control arm) to improve precision and save sample size, especially in rare disease in which the patient recruitment for a trial is time consuming due to the nature of the disease. In this section, we provide a brief overview of strategies and statistical methodologies to leverage RWD in the design and analysis. We have covered here important frequentist and Bayesian statistical methods.
Study designs and analyses with RWD for regulatory purposes fall into 4 broad categories: (1) use of external control; (2) evidence synthesis; (3) pragmatic trials; and (4) using RWE to fulfill a postmarketing requirement for safety and/or effectiveness. 51 Detailed statistical methodologies associated with categories 1 and 2 are presented below. The example of STRIDE registry data analysis 36 mentioned in the Applications section is a good example for category 4. Pragmatic trials (3) inform a clinical or policy decision by providing evidence for adoption of the intervention into real‐world clinical practice. There is an increased interest in pragmatic RCTs recently due to the current needs for rapid, affordable, and applicable research at the policy and clinical levels. The Salford Lung Study is an important example of pragmatic trials to evaluate the effectiveness of a drug intervention. 52 Patients with chronic obstructive pulmonary disease or asthma were randomized to compare continuation of the standard therapy versus a once‐daily combination of the inhaled corticosteroid. However, the regulatory acceptance of the RWE generated by pragmatic clinical trials is still limited due to lack of rigorous collection standards, absence of blinding, and evidence quality.
Use of External Control
The use of an external control has gained significant attention in recent times due to its practical advantages, including saving sample size, effective use of resources, acceleration of drug development, ethical appeal, and increased regulatory acceptance. It has been broadly used in 2 settings. First, a control group in an RCT can be augmented with control information from external data resources. This is often referred to as a “partially randomized control” design. Second, when there is no control group in the trial, one can create a stand‐alone external control group solely from the available external data for comparison. This is referred to as a “synthetic control.” The latter is useful for rare diseases in which single‐arm trials predominate and randomizing to control is often infeasible or unethical. For either strategy, the key assumption is the similarity between external control and current trial. This needs to be thoroughly assessed along with the reliability and validity, as discussed in the previous section.
A major concern about the use of external controls is the potential mismatch of external control data and current trial data, which can introduce biases in the treatment comparison. Sources of bias include:
Selection bias: Difference in the populations between the clinical trial and the RWD.
Unmeasured confounding: Key confounding data may not be available in both data sets for appropriate adjustment.
Time bias: Significant time lag between the external control data and trial data that involves change in clinical practice as well as standard of care.
-
Operational bias: Various sources of operation bias are:
Data collection: Difference in the manner current study and RWD data are collected.
Covariate measurement error: Same variable is measured differently in the trial as compared with RWD.
Outcome measurement: Collection of outcome differs between external control sources and current trial.
The potential for each of these biases must be addressed to support the validity of any decision making from analyses involving external controls. With real‐world controls as compared with historical RCT controls, there may be less concern with time bias, as data are collected in a similar time frame. However, there is a likelihood of issues with selection bias, unmeasured confounding, missing data, and measurement errors due to quality of RWD.
A causal framework provides a general guidance for designing, conducting, analyzing, and interpreting clinical trials with external controls. However, the fundamental assumptions include ignorability of treatment assignment, assuming no unobserved difference between treatment and control, given the set of confounders, and positivity. 53 Ho et al 54 described a detailed road map for study design and analysis using RWD. A similar framework is applicable while using an external control group. There are many statistical and computational methodologies available to allow use of external control data with a tangible degree of dissimilarity. A few main approaches are described below.
Test‐Then‐Pool Methodology
The availability of the RCT controls allows in a sense for a check of the validity of using the external control data. A “test‐then‐pool” approach was proposed by Viele et al. 55 After the initial stage of finding relevant controls via propensity‐matching score methods, a statistical test of differences in the outcome variables between the external and RCT controls is performed. If the test is rejected, then no pooling takes place. If the test does not reject the null hypothesis of equal mean outcomes, then the external controls are merged with the RCT controls for the analysis. Though this approach is simple and pragmatic in nature, it completely ignores the intrinsic variability between and within different data sources. Moreover, multiplicity adjustment is required to control the overall type I error.
Propensity Score Method
Propensity score methods are commonly used for causal inference on treatment effects in observational studies. A 2‐stage propensity score study design proposed by Yue et al 56 and Li et al 57 provides a paradigm for conducting a comparative observational, nonrandomized study within the premarket regulatory setting. The key assumption for propensity score methods is that baseline covariates explain all differences between external data sources and the current trial. In the first‐stage design, a subset of the individual subjects is selected from the external data sources, applying inclusion/exclusion criteria of the current trial. In the second‐stage design, a propensity score for each of the enrolled subjects in the target trial and the external subjects from the first stage design is estimated, based on baseline information, using logistic regression or ML techniques. Finally, the propensity score is used to choose control subjects such that they are comparable to the subjects in the current trial. The last step includes methodologies for the outcome analysis such as matching, inverse probability treatment weighting, stratification, or using the propensity score as a covariate. Propensity score methods are applicable for randomized and single‐arm studies as well as observational studies. The overall goal is to adjust for imbalance to the extent explanatory factors are available in data. For example, the DMD STRIDE registry data analysis used a propensity score matching approach for analysis. 36 However, such approaches do not take into account the possibility of mismatch of outcome variable (eg, primary end point variable) between external control and internal control due to unknown confounders. Such a situation is not uncommon in rare disease development as all disease modifiers are often unknown due to lack of data. Therefore, alternative methodological considerations (eg, coarsened exact matching 58 ) can be considered to address this issue.
The second approach in this category is the propensity score–integrated composite likelihood method. 59 , 60 First, the propensity score model is used to preselect a subset of external data comparable with the current study in terms of observed covariates. Second, different strata are formed on the basis of all available trial data and selected external control data. Within each stratum, the covariates are more balanced between the external control and current study subjects. Finally, a composite likelihood is constructed for the parameter of interest in each stratum by discounting the external control data. The parameter of interest for the trial is then estimated as a weighted average maximum likelihood estimates across all strata.
Another propensity score‐based approach adapts the proposals of Stuart and Rubin. 61 The proposed algorithm matches from multiple control groups (eg, internal concurrent control arm in an RCT and external control from RWD) with adjustment for differences in observed covariates between groups. This algorithm captures the additional otherwise unobserved differences between control groups. This approach differs, as it does not assume that the outcomes (adjusted for all covariates) are the same in the control arm of RCT and external controls but allows for the outcomes to differ and adjusts for observed differences. This approach has 3 propensity‐matching steps. First, propensity matching is conducted to find a match for RCT‐treated patients with a concurrent control arm in the RCT. Second, for the remainder of the RCT‐treated patients who are not yet matched in step 1, a match is obtained from the external control group via propensity scoring. Finally, this approach computes an adjustment factor for the outcomes in the external data by matching and statistically estimating the difference in outcomes between RCT and external controls.
Bayesian Hierarchical Model
Bayesian hierarchical modeling is a methodology used to combine results from multiple studies (or data sources) in which the information is available on several different levels using Bayesian methods. The Bayesian approach provides a mathematically rigorous and principled methodology for making decisions under arbitrarily complex scenarios. Two common methods of the Bayesian hierarchical model are commensurate power and meta‐analytic approaches. These approaches are principally similar as they discount the external control information via between‐trial heterogeneity.
Meta‐analytic approaches are the methods of choice for evidence synthesis from clinical trials and have also been applied to derive external control information. The random‐effects meta‐analysis (MA) allows the parameter of interest in the different external control and current trial to be different. Therefore, taking account of between‐trial heterogeneity among the external control data source trials as well as heterogeneity between the current trials. The main assumption of meta‐analytic approaches is “exchangeability” or “similarity” between external control data and current trial data. 62 , 63 , 64 , 65 The method is flexible enough to adapt different types of source data, including individual subject data and aggregate data from publications. At the planning stage, a random‐effects MA is used to construct a “pseudo‐control” using the available relevant external control data. Once the trial data are observed, statistical inference on the difference between test and control is straightforward, using all available data (from trial and relevant external control) and Bayesian inference. For example, in case of continuous end point, the posterior distribution of difference between test and control follows a normal distribution if the prior for the control parameter is derived from RWD using a meta‐analytic approach and approximated by a normal distribution. 62 Therefore, all the inference related to the difference can be done easily using a closed‐form formula. For more complex settings, Markov chain Monte Carlo sampling is required. 63
The degree of borrowing can be approximated by the “effective sample size.” 66 , 67 , 68 Depending on the type of relevant source data, more complex meta‐analytic approaches may be needed, including meta‐regression, which use baseline covariate information to explain part of the between‐trial heterogeneity. Furthermore, the methodology is further extended to handle possible conflict between external control and concurrent control of the current trial using mixture with a weakly informative prior. 63 This additional component acknowledges the possibility of mismatch in outcome between external data and the current trial, despite a careful selection of the historical trials and provide robust statistical inference compared with a nonmixture prior.
In the meta‐analytic approach, the degree of borrowing from external data depends on the “between‐trial data source heterogeneity parameter.” The heterogeneity parameter requires special attention while using this model in practice. 62 , 69 When the number of external data sources is small, it is not feasible to estimate the between‐trial data source heterogeneity. Therefore, prior judgments are required.
Power Prior and Propensity Score–Integrated Power Prior
The power prior is a useful class of informative priors for external control data. The power prior discounts the likelihood of the external control data directly using a power parameter. 70 , 71 This parameter controls the amount of borrowing from the external control while taking into account both the current study and the external data source. The key assumption of power prior is “equality” of parameter of interest between external control data and the current trial. When this “power parameter” is set to 0, no external data are used in the analysis, whereas when the power parameter is set to 1, no down‐weighting occurs. In practice, the power parameter is often thought of as the “proportion of the external data” being used by the procedure. The power parameter can be fixed or random. Adding a prior on the power parameter will allow for uncertainty in the analysis when the similarity between external control and current trial is unknown. However, several authors caution against the use of the prior for power prior, as it violates the likelihood principle. 72 Therefore, normalization of the posterior distribution is required for proper inference. 73 , 74
However, rather than directly constructing the power prior based on external data, one can use an integrated propensity score–based method along with prior power for practical application. Similarly to the composite likelihood approach, trial data and external control data are divided into homogeneous strata using a propensity score. The power prior method is then implemented in each stratum to obtain stratum‐specific posterior distributions, which are then combined to complete the Bayesian inference for the parameters of interest. 75
Other notable methods to borrow external control include the use of advanced ML methods like random forests, neural networks, and cluster analysis. These advanced analytical tools have enhanced many aspects of drug development by identifying clinically meaningful patterns from large, unstructured, and heterogeneous data sources. These novel techniques are powerful in exploring both linear and nonlinear relationships between outcomes and large number of covariates. Also, the use of these advanced techniques allows better identification of the homogeneous strata for composite likelihood and power prior application, as described previously. For example, the EMA has recently released a draft qualification opinion on their 3‐step Prognostic Covariate Adjustment procedure for phase 2 and 3 trials. 76 This procedure uses novel “digital twins” technology to construct an external control. 77 Currently, application of these methods is restricted to exploratory analysis and requires separate statistical analysis.
Evidence Synthesis
Evidence synthesis refers to a broader framework that allows the use of RWD data of investigational treatment and competitors for informed decision making. MA and network MA (NMA) are commonly used methodologies to combine different sources of external data, especially in rare diseases for which clinical trials are usually small. MA and NMA are widely applied in drug development. These analyses are usually planned prospectively. Flexible statistical models are used to combine heterogeneous types of data sources with summary‐level and individual‐subject‐level data. Both fixed‐effects and random‐effects models are used for MA. In contrast to RCT, RWD presents some unique challenges for MA due to the difference in the data collection process among different sources (eg, nonrandomized studies). Use of data sources other than RCT generates a biased treatment effect, which poses further challenges to the interpretation of the MA estimates. One possible solution is the use of generalized evidence synthesis techniques. 78 This is an extension of the standard random‐effects MA model incorporating a third level in the hierarchy to account for heterogeneity between study design types explicitly, as well as heterogeneity between individual studies with the same design type. In addition, several methods are available to assess the impact of bias in MA. These include the informal assessment of a funnel plot, statistical tests, and adjustment models and procedures. Additional sensitivity analyses are also necessary to ensure robustness of the result.
Indirect comparisons and NMA, an extension of conventional MA, in which multiple treatments are being compared using both direct comparisons of interventions within RCTs and indirect comparisons across trials based on a common comparator. It is useful when multiple potential treatments are available in a disease area. NMAs are often used to synthesize evidence from RCTs. Including RWD sources in NMA and decision making is growing. Like standard MA, inclusion of RWD in NMA needs careful consideration. Without appropriate handling of the confounders, the treatment effect will be biased and requires cautious interpretation. This is particularly useful for rare disease drug development, as the evidence is often generated from nonrandomized studies, which can introduce systematic bias. Generalized evidence synthesis methods 79 , 80 can be useful to address this issue.
Data Quality Considerations in Using Natural History and RWD
The intention to use RWD and more specifically natural history data for the benefit of rare disease drug development is certainly growing, along with an increased scrutiny about the quality of RWD. The question of whether the data are good enough for decision making is often raised. 81 It is important to understand that quality must be evaluated in the context of its intended use and cannot be assessed by looking at data in isolation. Any evaluation of RWD quality underpinning RWE must consider whether any individual or combined data source has the information to answer a given research question (ie, its information value). Specific data quality issues that come to bear when attempting to curate and integrate various rare disease data sources include missing data, a lack of core common data elements, no or incomplete data dictionaries (eg, missing definition of scoring values, data derivation formulas, or units), a lack of longitudinality, and a lack of validated questionnaires. 82 As various stakeholders conduct individual landscape analyses on available RWD sources, it would be important in the rare disease effort to share these assessments particularly in the context of data quality in the context of intended use. Most of these issues are not unique to the rare diseases data ecosystem but rather widespread issues common to all patient‐level data. Issues in quality, completeness, and relevance of source data inevitably lead to uncertainty in downstream findings. When these data are used in applications for regulatory decision making, uncertainties that cannot be addressed through available evidence will translate into gaps in knowledge that should be addressed in the design and conduct of clinical trials for the intended indication.
Necessarily, rare outcomes raise issues in sampling populations, particularly in elucidating factors that influence the onset of diseases. At the same time, characterizing disease progression after onset for rare conditions requires identification of affected patients, often from multiple data sources. Patient identification also raises the issues of missing values and duplicate data and the potential solution of using global unique identifiers (GUIDs) to address the duplication issue. A GUID is a 128‐bit text string that represents an identification. In general, organizations generate GUIDs when a unique reference number is needed to identify information on a computer or network. A GUID can be used to identify hardware, software, accounts, documents, and other items. A GUID in the context of rare disease research is a computer‐generated alphanumeric code that is unique to each research participant. A GUID protects personally identifiable information and enables deidentified data from 1 person to be integrated and tracked over time across multiple projects, databases, and biobanks. 83 , 84 A GUID model is essential for the linking of study data to other data sets, particularly registry data. As the majority of registry data sources come from surveys, there is also the issue of data integration with more structured data sources (eg, clinical trial data).
Recently, some have suggested the establishment of a framework for evaluating data appropriateness, often referred to as “fitness for purpose” or the degree to which the chosen data source aligns with the ability to address the research question posed accurately and reliably. 85 As part of the Sentinel Initiative, 86 the FDA and other organizations are developing detailed recommendations on standards‐based approaches to describing data and presenting data quality metrics. There are some efforts to address the limitations of natural history data more rigorously, but there are still no universally accepted standards, and the variation in data quality among natural history data sources continues to make the integration of such sources difficult. 82 As such, integration is often rate limiting and a critical initial step in the creation of drug development tools to facilitate rare disease research and development. This is an important area to continue to support, as it will no doubt improve quality considerations for data that are included in drug product registration and fidelity in models and tools constructed from them.
Currently, pharmaceutical sponsors seeking to use these data to support registration do so at their own risk, but also with the intention to expose the risk‐benefit for using such data given that the lack of data among rare diseases is still the most pressing issue. Good recommendations for use given the limitations and risks are still valuable and often justify the use given that the benefit outweighs the risk. 86 , 87 For their part, regulators remain open‐minded and new regulatory guidance by the FDA and the EMA on using RWD suggests that they are receptive to receiving well‐characterized assessments and proposals. 88 , 89 , 90
Even if all the metrics for evaluating RWD quality were established and agreed upon, such criteria are unlikely to be enough for every scientific research purpose. Most RWD sources will be a good fit for some research questions but not others. Data appropriateness needs to be gauged by reviewing the strengths and weaknesses of any data set under consideration in the context of the research initiative, study design, budget, and time available to assemble relevant information. Data quality is not an absolute metric that can predict utility in isolation. A quality assessment can be evaluated only with full knowledge of the research question and immediacy of the need of information. Only through the optimal pairing of data source and research question parameters can you have confidence in delivering a reliable conclusion.
Considerations and Examples in Using Natural History and RWD for Regulatory Decision Making
The 21st Century Cures Act 13 was signed into law by the US Congress on December 13, 2016. The Cures Act is designed to bring new innovations and accelerate medical product development more efficiently, thus benefiting the patient. It added section 505F to the Federal Food, Drug, and Cosmetic Act, which mandates the FDA to develop guidance for use of RWD/RWE for regulatory decisions. As a result, the FDA issued a framework for an RWE program in 2018 to support the approval of a new indication for a drug and support or stratify postapproval study requirement. 11
It is well recognized that there has been rapid advancement in RWD collection and curation efforts. For example, for a patient with a genetic cardiomyopathy, health care data may be captured in a rare disease–specific registry in addition to medical records, claims data, laboratory data, genetic data, and other RWD sources. While these data sources are often siloed, harmonizing them can provide researchers a holistic, longitudinal view into the disease's natural history, the patient's health status, their interactions with health care systems, and insights into a drug's long‐term safety and effectiveness (Figure 1).
Figure 1.
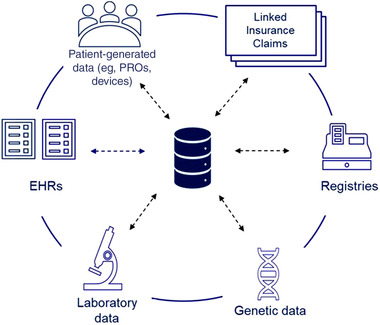
Harmonization of real‐world and natural history source data sets. EHRs, electronic health records; PROs, patient reported outcomes
The FDA recently issued a guidance on RWD regarding the use of registries to support regulatory decision making. 8 The guidance highlighted that use of RWD including data collected directly from the registry and additional data linked from other RWD sources such as insurance claims, electronic health records (EHRs), laboratory data sources, blood bank, and/or medical device outputs can be used to support the development of disease natural history studies. However, data relevance and reliability need to be considered when assessing data sources. This guidance coupled with guidance on EHR and medical claims data can serve as a foundation for using RWD to build natural history studies for rare disease. 91
Examples of FDA Approvals in Rare Diseases Based on RWE in Supporting Effectiveness
As of February 2, 2019, research showed that there were ≈26 orphan drugs that were approved with at least 1 source of RWD. 92 All these approvals have RWD/RWE described in Section 14 of the drug label. For most labels, the RWE was descriptive and presented in tables or figures and the statistical analysis was typically simple. With rare diseases, the use of natural history and/or RWD as the control in single‐arm clinical studies is the most common historical use of non‐RCT data when it comes to regulatory approvals. The intent of a natural history study is to follow the progression of a patient's disease in the absence of treatment for the entire course of the disease from onset until its outcome, such as death, disability, or recovery. In this type of single‐arm clinical trial, all subjects receive the same treatment. Subsequently, the results of this treatment are then compared against the natural history progression of the disease. 93 There were several notable observations. Patients in the treatment arm of the clinical trials were matched to historical control patients. Statistical models were used to adjust for differences in baseline covariates between the historical control cohort and the treated clinical trial arm, that is, Cox regression.
The acceptance of using natural history data is especially critical when there is no approved treatment for diseases in which the patient's condition continues to worsen due to lack of medical intervention. In such situation, it is logical to use natural history data due to the challenge in recruiting an adequate number of patients to conduct an RCT and the ethical issues related to the administration of placebos that do not provide any benefits to the patients. 94 Following are 6 examples of FDA approvals for rare disease indications that used natural history and/or RWD. They are listed in reverse chronological order by approval year, starting with the most recent approval. Two of the examples, Brineura and Defitelio, provide details on the basis for the statistical analysis that was used in the submissions and the labels. 95 The other examples consist of shorter excerpts highlighting the type of natural history or RWD data that was used.
Case 1: Besremi
In 2021, the FDA approved Besremi (ropeginterferon alfa‐2b‐njft) for treatment of adults with polycythemia vera (PV) using clinical trial data and natural history data. Given the statistical issues in ropeginterferon alfa‐2b‐njft clinical studies, understanding of the natural history of PV and that spontaneous remissions do not occur randomly in this disease, the high unmet medical need, and the rarity of the disease, the review team evaluated the effectiveness of ropeginterferon alfa‐2b from the Safety Study of Pegylated Interferon Alpha 2b to Treat Polycythemia Vera (PEGINVERA; a phase 1/2) study and the published studies on the natural history of the disease. In addition, the review team evaluated the objective change from baseline for the hematology laboratory parameters in the active treatment group from the PROUD‐PV (a randomized phase III study comparing hydroxyurea to ropeginterferon alfa‐2b) as mechanistic and confirmatory evidence to establish substantial evidence of effectiveness for ropeginterferon alfa‐2b in the treatment of patients with PV without symptomatic splenomegaly. 96
Case 2: Brineura
Brineura (cerliponase alfa) was approved by the FDA on April 27, 2017. The indication is for a form of Batten disease, neuronal ceroid lipofuscinosis type 2 (CLN2). The target population is pediatric patients aged ≥3 years. The incidence of CLN2 is between ≈0.56 and 4 patients per 100,000 live births in the United States and Europe, and thus CLN2 is considered an orphan rare disease. The efficacy claim was based on the comparison of data from a single‐arm clinical study with historical controls. 94 , 97 , 98
The sponsor initially proposed a primary analysis that uses a 1‐sample t‐test aimed to compare patient data to their baseline value instead of a control arm. However, the FDA recommended using historical control data obtained from the DEM‐CHILD (a treatment‐oriented research project of neuronal ceroid lipofuscinosis disorders as a major cause of dementia in childhood) database, which consists of patients with CLN2. This DEM‐CHILD database was created to provide a tool for experimental therapy studies. 99 The FDA statistics review discussed the fact that the CLN2 rating scales used in the DEM‐CHILD database and the current study were different, that is, slightly different assessment scales, different assessment frequency/times, and different methodologies for data collection. Following quantitative evaluation of the comparability of the rating scales, it was advised to change the score and the definition of a responder in the single‐arm trial. 100
For the natural history cohort, a subset of 69 patients with similar eligibility criteria to the clinical study was initially selected from the DEM‐CHILD database. A further 27 patients were then excluded due to insufficient assessments and overlap with the participants in the Brineura clinical study.
The Brineura single‐arm study recruited 24 patients, and the natural history cohort consisted of 42 subjects. The FDA requested analysis based on matched pairs. Matched pairs are defined as exact matching on baseline motor score, age ±3 months, and genotype (0, 1, or 2 key mutations). Six patients in the single‐arm study could not be matched to the natural history cohort on the basis of this criterion. 101 If there was >1 match, further matching variables were considered in the following order: detailed genotype, sex, and age of first symptom. The result of the effectiveness analyses that are reported in the label was based on this matched subset of 17 pairs.
Case 3: Defitelio
Defitelio (defibrotide sodium) was approved by the FDA on March 30, 2016, for the treatment of adult and pediatric patients with hepatic veno‐occlusive disease (VOD), also known as sinusoidal obstruction syndrome. These patients have renal or pulmonary dysfunction following hematopoietic stem‐cell transplantation (HSCT). 102 Defitelio is the first FDA‐approved therapy for this rare and life‐threatening liver disease. Per the US label, the efficacy assessment was approved on the basis of 1 pivotal single‐arm study, a pediatric clinical study, and an expanded access study.
The pivotal study was conducted as an open‐label, historically controlled, multicenter study. 103 There were 2 cohorts: a defibrotide cohort with 102 subjects and a historical control cohort with 32 subjects. Subjects in the historical control cohort consisted of 6867 subjects undergoing HSCT from 35 medical centers. These subjects received the best supportive care. The primary end point was the percentage of patients who were still alive 100 days after HSCT.
As described in the Medical Review, 104 the historical cohort was selected by a medical review committee. Two independent hematologists monitored the process by reviewing the subjects’ medical charts. Confounding effects from the potential prognostic factors were adjusted by the following method. A propensity score was performed for the primary and selected supportive and sensitivity efficacy analyses. For everyone, a propensity score for cohort membership (Defitelio cohort vs historical control cohort) was calculated using a logistic regression, with “cohort membership” as the outcome and 4 baseline prognostic factors of survival as the independent variables. Patients were then categorized into quintiles or quartiles based on this propensity score. The strata created by quintiles/quartiles were used to perform stratified treatment difference analysis. Although the selection was rigorous, the FDA reviewers still had concerns that the historical control cohort versus the clinical trial patients may not consist of the same underlying patient population. 105
Furthermore, the time frame was different for the 2 cohorts. For the defibrotide cohort, the overall time frame from the earliest date of HSCT to the latest date of HSCT was 2 years, while the overall time frame spanned 12 years for the historical control cohort. Additionally, many of the potential prognostic factors that might confound the evaluation of temporal effect were not collected. To address the concerns, the medical review committee subsequently reviewed the source data and reduced the historical control patients from 86 to 32. The resulting smaller sample size led the FDA statistical reviewer to question the sponsor's interpretation of the primary efficacy results, as the behavior of propensity score methodology is not well established for small sample sizes.
Therefore, in the current US label for this product, there is no detail about the source of this historical control cohort, and no statistical comparison of the pivotal study with this historical control is presented. The 100‐day survival for the historical cohort is presented as a range. To accommodate, an expanded access program was also included in the label. 103 This was for pediatric patients with hepatic VOD. In this study, the 100‐day survival after HSCT was evaluated to be 45% for 351 patients who had received an HSCT and developed hepatic VOD with renal or pulmonary dysfunction.
Case 4: Vimizin
In 2014, the FDA used RWD generated from studies of related diseases to approve Vimizin (elosulfase alfa), an enzyme replacement therapy (ERT), as the first approved treatment for Morquio syndrome. Morquio syndrome is one of a class of mucopolysaccharidoses (MPSs), a group of rare inherited disorders caused by the lack or inability of lysosomal enzymes to break down certain carbohydrates. Vimizin was the fourth ERT to receive approval for the treatment of an MPS disorder, although the prior treatments were each approved for different MPS disorders. The FDA relied on regulatory histories of the other MPS ERTs, as well as historical data from other lysosomal disorders, along with a single‐arm trial of a small premarket population, 106 with comparison to historical controls, which included data from the Morquio A Clinical Assessment Program (MorCAP) natural history study. 107
Case 5: Carbaglu
In 2010, the FDA relied solely on historical studies to approve Carbaglu (carglumic acid) for treating hyperammonia caused by N‐acetylglutamate synthase deficiency, a rare genetic disorder that can be fatal or cause permanent central nervous system damage in infants soon after birth if not detected and treated quickly. The FDA relied on a retrospective review of 23 N‐acetylglutamate synthase–deficient patients who received Carbaglu for a median of ≈8 years. The clinical observations in the 23‐patient case series were unblinded and uncontrolled and preclude any meaningful formal statistical analyses of the data. However, short‐term efficacy was evaluated using mean and median change in plasma ammonia levels from baseline to days 1–3. Persistence of efficacy was evaluated using long‐term mean and median change in plasma ammonia level. Carbaglu reduced blood ammonia levels within 24 hours and brought them to normal levels within 3 days. 108 , 109
Case 6: Myozyme
In 2006, the FDA approved Myozyme (alglucosidase alfa), an ERT, for the treatment of Pompe disease, a rare inherited lysosomal storage disorder. The approval was based on a clinical study that used a historical cohort of untreated individuals as a benchmark. The sponsor demonstrated efficacy with respect to the infantile‐onset form of the disease using a single‐arm open‐label clinical study of 18 patients conducted between 2003 and 2005. The efficacy was assessed by comparing the treated group over a period ranging from 52 to 106 weeks to a historical cohort of 61 untreated patients born between 1982 and 2002 who were identified through a retrospective medical chart review (only 1 of whom was still alive) using improved ventilator‐free survival as the primary efficacy end point. The patients with untreated infantile‐onset Pompe with similar age and disease severity were diagnosed by age 6 months. 110
All 6 cases led to regulatory approvals by using natural history from various databases as the external study control (cases 1–4 and 6), and/or using RWD (case 5). The external control patients were carefully selected to closely match the demographic and disease status to the patients in the treated arm. Among these cases, extensive statistical analyses were conducted to support the approvals. With increasing experience and expanded databases, it is anticipated that the use of natural history data and RWD will become more widely used and continue to support the approval of rare disease indications.
Limitations in Using Natural History and RWD
Data Access and Availability
Many sources are accessible with a fee (insurance claims, EHRs, hospital charge master, etc). However, some RWD sources are not accessible to researchers such as registry and laboratory data. As shown in the above case examples of FDA approvals in rare diseases, the published natural history data and results allowed decision making by sponsors and regulators. Building public‐private consortia and federated data networks may further improve data accessibility. Successful examples include the Rare Disease Cures Accelerator–Data and Analytics Platform from the Critical Path Institute 111 and European Health Data and Evidence Network of the Innovative Health Initiative. 112 Some RWD are not routinely collected outside of clinical trials. For example, as a measure of functional motor abilities in ambulant children with DMD, the North Star Ambulatory Assessment is commonly used in DMD trials as a clinical end point to assess treatment efficacy. However, such measure is not routinely collected in clinical practices, making it difficult to assess real‐world disease progression after innovative treatment (eg, gene therapy, exon skipping, and readthrough therapies). Missing data are a common occurrence in RWD. For example, genetic and laboratory data may not be available in EHRs and claims without linking to specific genetic and lab sources.
Data Linkage Across Databases
In the United States, individuals, particularly patients with rare diseases, seek care with multiple providers and laboratories, and may change insurance plans often. To construct a complete, longitudinal patient journey without data gaps using RWD (see Figure 1) for a natural history study, it is necessary to link a variety of data sources generated through a patient's interactions with the health care system. However, data linkage is often not available due to privacy concerns in using personally identifiable information to link data sources. New technology such as tokenization and GUID as described in the Data Quality section, long used by the financial industry, may be used to connect siloed data sources in a privacy‐preserving way.
Structured and Unstructured RWD
The most commonly used RWD are from structured fields of EHR or billing systems. However, using structured data (eg, diagnosis, procedure, and drug codes) to construct key study variables such as exposure, end points, and covariates are often challenging. Validity of these variables from RWD must be scrutinized for use in regulatory decision making. For example, the FDA's Sentinel Initiative has led a series of validation assessments of health outcomes of interest. 86 Some variables can be reliably identified using structured data with high positive predictive values, 113 while others may not. 114 , 115 , 116 Unstructured data often contain clinical information such as disease severity, tumor stages, health behaviors, pathology report, and imaging, which may be important prognostic factors that need to be considered in analysis. However, unstructured data are usually not available or very difficult to extract from medical records, leading to risk of confounding and bias of analysis if not addressed. Artificial intelligence may be leveraged to process unstructured data and advances in ML and natural language processing could be used to extract unstructured data elements, build algorithms for outcomes identification, and assess image and laboratory results.
Other Limitations
As the RWD and natural history data are from a practical clinical setting, the patient pool may include a more heterogeneous population than what would be included in an RCT, which could affect the data collection and interpretation. Because of the difficulties associated with patient finding in rare diseases, the sample sizes of these types of studies may be small, leading to a limited ability to draw firm conclusions from the data gathered. Pooling data from different studies and more advanced analytical techniques may be necessary to better understand the rare diseases and evaluate treatment effects.
Conclusions and Future Directions
The scarcity of patients with rare diseases can pose several challenges toward their understanding as well as the development of drugs for their treatment. Natural history data and RWD provide significant value toward the understanding of disease progression, patient populations, novel biomarkers, genetic relationships, and treatment effects. However, the utility of these data in rare diseases can be limited by their heterogenicity, quality, and accessibility. A few initial regulatory guidelines have been published to provide directions to overcome these limitations. Collaboration across regulatory authorities, researchers, patients/caregivers, and drug developers on the application of natural history data and RWD is critical to expedite understanding of a rare disease and foster development of new drugs for it. Standardized data collection and data quality can further facilitate data sharing/pooling. Finally, the use of quantitative model‐based approaches such as disease progression modeling, artificial intelligence and ML, and advanced statistical approaches in natural history data and RWD will enhance disease understanding and guide more efficient clinical study design in drug development, which is particularly valuable in rare diseases.
Conflicts of Interest
The authors declare no conflicts of interest related to this work. J.L., S.R., and Y.C. are employees and shareholders of Pfizer Inc. S.R. is also a shareholder of Novartis AG. J.S.B. is an employee of the Critical Path Institute. E.T.L., L.L., and P.T. are employees and shareholders of PTC Therapeutics Inc.
Funding
Medical writing assistance was funded by PTC Therapeutics, Inc.
Acknowledgments
Medical writing assistance was provided by Tim Ellison, PhD, of PharmaGenesis London, UK, and was funded by PTC Therapeutics, Inc.
Data Availability Statement
Study protocol will be shared upon reasonable request.
References
- 1. US Food and Drug Administration. Rare Diseases: Natural History Studies for Drug Development: Draft Guidance for Industry. 2019. Accessed March 4, 2022. https://www.fda.gov/media/122425/download
- 2. European Medicines Agency. Procedural advice for orphan medicinal product designation: Guidance for sponsors. 2021. Accessed March 4, 2022. https://www.ema.europa.eu/en/documents/regulatory‐procedural‐guideline/procedural‐advice‐orphan‐medicinal‐product‐designation‐guidance‐sponsors_en.pdf
- 3. Nguengang Wakap S, Lambert DM, Olry A, et al. Estimating cumulative point prevalence of rare diseases: analysis of the Orphanet database. Eur J Hum Genet. 2020;28(2):165‐173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. US Food and Drug Administration. Importance of natural history studies in rare diseases. 2019. Accessed March 4, 2022. https://events‐support.com/Documents/Pariser.pdf
- 5. Burton A, Castaño A, Bruno M, et al. Drug discovery and development in rare diseases: taking a closer look at the tafamidis story. Drug Des Devel Ther. 2021;15:1225‐1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Abbas A, Vella Szijj J, Azzopardi LM, Serracino Inglott A. Orphan drug policies in different countries. J Pharm Health Serv Res. 2019;10(3):295‐302. [Google Scholar]
- 7. Jahanshahi M, Gregg K, Davis G, et al. The use of external controls in FDA regulatory decision making. Ther Innov Regul Sci. 2021;55(5):1019‐1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. US Food and Drug Administration. Real‐World Data: Assessing Registries to Support Regulatory Decision‐Making for Drug and Biological Products, Draft Guidance for Industry. 2021. Accessed March 18, 2022. https://www.fda.gov/media/154449/download
- 9. European Medicines Agency. Guideline on registry‐based studies. 2021. Accessed March 18, 2022. https://www.ema.europa.eu/en/human‐regulatory/post‐authorisation/patient‐registries#guideline‐on‐registry‐based‐studies‐section
- 10. Cave A, Kurz X, Arlett P. Real‐world data for regulatory decision making: challenges and possible solutions for Europe. Clin Pharmacol Ther. 2019;106(1):36‐39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. US Food and Drug Administration. Framework for FDA's real‐world evidence program 2018. Accessed March 4, 2022. https://www.fda.gov/media/120060/download
- 12. Roberts MH, Ferguson GT. Real‐world evidence: bridging gaps in evidence to guide payer decisions. Pharmacoecon Open. 2021;5(1):3‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. 21st Century Cures Act (Congress.gov) 2016. Accessed April 19, 2022. https://www.congress.gov/114/plaws/publ255/PLAW‐114publ255.pdf
- 14. McDonald CM, Henricson EK, Abresch RT, et al. Long‐term effects of glucocorticoids on function, quality of life, and survival in patients with Duchenne muscular dystrophy: a prospective cohort study. Lancet. 2018;391(10119):451‐461. [DOI] [PubMed] [Google Scholar]
- 15. Mercuri E, Lucibello S, Perulli M, et al. Longitudinal natural history of type I spinal muscular atrophy: a critical review. Orphanet J Rare Dis. 2020;15(1):84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dorsey ER, Beck CA, Darwin K, et al. Natural history of Huntington disease. JAMA Neurol. 2013;70(12):1520‐1530. [DOI] [PubMed] [Google Scholar]
- 17. Koeks Z, Bladen CL, Salgado D, et al. Clinical outcomes in Duchenne muscular dystrophy: a study of 5345 patients from the TREAT‐NMD DMD global database. J Neuromuscul Dis. 2017;4(4):293‐306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Goemans N, McDonald CM, Signorovitch J, et al. Prognostic factors for loss of ability to rise from supine in Duchenne muscular dystrophy (DMD). Neuromusc Disord. 2020;30(Suppl 1):S63‐S64. [Google Scholar]
- 19. ClinicalTrials.gov. NCT02369731: Registry of Translarna (Ataluren) in Nonsense Mutation Duchenne Muscular Dystrophy (nmDMD). 2022. Accessed March 4, 2022. https://clinicaltrials.gov/ct2/show/NCT02369731
- 20. Muntoni F, Desguerre I, Guglieri M, et al. Ataluren use in patients with nonsense mutation Duchenne muscular dystrophy: patient demographics and characteristics from the STRIDE Registry. J Comp Eff Res. 2019;8(14):1187‐1200. [DOI] [PubMed] [Google Scholar]
- 21. ClinicalTrials.gov. NCT04174157: Registry of Patients With a Diagnosis of Spinal Muscular Atrophy (SMA). 2022. Accessed March 4, 2022. https://clinicaltrials.gov/ct2/show/NCT04174157
- 22. Finkel RS, Day JW, De Vivo DC, et al. RESTORE: A prospective multinational registry of patients with genetically confirmed spinal muscular atrophy ‐ rationale and study design. J Neuromuscul Dis. 2020;7(2):145‐152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. ClinicalTrials.gov. NCT01574053: Enroll‐HD: A Prospective Registry Study in a Global Huntington's Disease Cohort. 2021. Accessed March 4, 2022. https://clinicaltrials.gov/ct2/show/NCT01574053
- 24. Enroll‐HD. 2022. Accessed March 4, 2022. https://enroll‐hd.org/
- 25. NORD. Additional Resources. 2021. Accessed March 18, 2022. https://rarediseases.org/iamrare‐registry‐program/
- 26. The portal for rare diseases and orphan drugs. Accessed June 18, 2022. https://www.orpha.net/consor/cgi‐bin/Education_AboutOrphanet.php?lng=EN
- 27. IBM MarketScan Research Databases. Accessed June 18, 2022. https://www.ibm.com/products/marketscan‐research‐databases/databases
- 28. Sverdlov O, van Dam J, Hannesdottir K, Thornton‐Wells T. Digital therapeutics: an integral component of digital innovation in drug development. Clin Pharmacol Ther. 2018;104(1):72‐80. [DOI] [PubMed] [Google Scholar]
- 29. Rochester L, Mazzà C, Mueller A, et al. A roadmap to inform development, validation and approval of digital mobility outcomes: the Mobilise‐D approach. Digit Biomark. 2020;4(Suppl 1):13‐27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mueller A, Paterson E, McIntosh A, et al. Digital endpoints for self‐administered home‐based functional assessment in pediatric Friedreich's ataxia. Ann Clin Transl Neurol. 2021;8(9):1845‐1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Janicki Hsieh S, Alexopoulou Z, Mehrotra N, Struyk A, Stoch SA. Neurodegenerative diseases: the value of early predictive end points. Clin Pharmacol Ther. 2022;111(4):835‐839. [DOI] [PubMed] [Google Scholar]
- 32. Joseph S, Wang C, Bushby K, et al. Fractures and linear growth in a nationwide cohort of boys with Duchenne muscular dystrophy with and without glucocorticoid treatment: results from the UK NorthStar database. JAMA Neurol. 2019;76(6):701‐709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bodor M, McDonald CM. Why short stature is beneficial in Duchenne muscular dystrophy. Muscle Nerve. 2013;48(3):336‐342. [DOI] [PubMed] [Google Scholar]
- 34. Lamb MM, West NA, Ouyang L, et al. Corticosteroid treatment and growth patterns in ambulatory males with Duchenne muscular dystrophy. J Pediatr. 2016;173:207‐213.e203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Labove L, Jaworski M, Nguyen C‐T. Duchenne muscular dystrophy: do boys with a shorter stature maintain ambulation longer? J Muscular Dis. 2018;5(Suppl 1):S142‐S143. [Google Scholar]
- 36. Mercuri E, Muntoni F, Osorio AN, et al. Safety and effectiveness of ataluren: comparison of results from the STRIDE Registry and CINRG DMD Natural History Study. J Comp Eff Res. 2020;9(5):341‐360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Marden JR, Freimark J, Yao Z, Signorovitch J, Tian C, Wong BL. Real‐world outcomes of long‐term prednisone and deflazacort use in patients with Duchenne muscular dystrophy: experience at a single, large care center. J Comp Eff Res. 2020;9(3):177‐189. [DOI] [PubMed] [Google Scholar]
- 38. Marden JR, Santos C, Pfister B, et al. Steroid switching in dystrophinopathy treatment: a US chart review of patient characteristics and clinical outcomes. J Comp Eff Res. 2021;10(14):1065‐1078. [DOI] [PubMed] [Google Scholar]
- 39. Ohlmeier C, Saum KU, Galetzka W, Beier D, Gothe H. Epidemiology and health care utilization of patients suffering from Huntington's disease in Germany: real world evidence based on German claims data. BMC Neurol. 2019;19:318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Achenbach J, Saft C, Faissner S. Longitudinal evaluation of the effect of tricyclic antidepressants and neuroleptics on the course of Huntington's disease‐data from a real world cohort. Brain Sci. 2021;11(4):413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Meyer C, Landwehrmeyer B, Schwenke C, et al. Rate of change in early Huntington's disease: a clinicometric analysis. Mov Disord. 2012;27(1):118‐124. [DOI] [PubMed] [Google Scholar]
- 42. Jin Y, Ahadieh S, Pickering E, Evans R, Liu J. Model‐based meta‐analysis of total motor score, chorea score, and total functional capacity for patients with Huntington's disease. Clin Pharmacol Ther. 2012;91(Suppl 1):S37‐S37. [Google Scholar]
- 43. Kuan WL, Kasis A, Yuan Y, et al. Modelling the natural history of Huntington's disease progression. J Neurol Neurosurg Psychiatry. 2015;86(10):1143‐1149. [DOI] [PubMed] [Google Scholar]
- 44. Langbehn DR, Brinkman RR, Falush D, et al. A new model for prediction of the age of onset and penetrance for Huntington's disease based on CAG length. Clin Genet. 2004;65:267‐277. [DOI] [PubMed] [Google Scholar]
- 45. Sun W, Zhou D, Warner J, Langbehn D, Hochhaus G, Wang Y. Huntington's disease progression: a population modeling approach to characterization using clinical rating scales. J Clin Pharmacol. 2020;60(8):1051‐1060. [DOI] [PubMed] [Google Scholar]
- 46. Mohan A, Sun Z, Ghosh S, et al. A machine‐learning derived Huntington's disease progression model: insights for clinical trial design. Mov Disord. 2022;37(3):553‐562. [DOI] [PubMed] [Google Scholar]
- 47. Lingineni K, Aggarwal V, Morales JF, et al. Development of a model‐based clinical trial simulation platform to optimize the design of clinical trials for Duchenne muscular dystrophy. CPT Pharmacometrics Syst Pharmacol. 2022;11(3):318‐332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Rooney WD, Berlow YA, Triplett WT, et al. Modeling disease trajectory in Duchenne muscular dystrophy. Neurology. 2020;94(15):e1622‐e1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Haber G, Conway K, Paramsothy P, et al. Association of genetic mutations and loss of ambulation in childhood‐onset dystrophinopathy. Muscle Nerve. 2021;63(2):181‐191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. US Food and Drug Administration. The FDA Reauthorization Act of 2017 (FDARA). 2017. Accessed April 19, 2022. https://www.fda.gov/regulatory‐information/selected‐amendments‐fdc‐act/fda‐reauthorization‐act‐2017‐fdara
- 51. Levenson M, He W, Chen J, et al. Biostatistical considerations when using RWD and RWE in clinical studies for regulatory purposes: a landscape assessment. Stat Biopharm Res. 2021;2:1‐20. [Google Scholar]
- 52. Albertson TE, Murin S, Sutter ME, Chenoweth JA. The Salford Lung Study: a pioneering comparative effectiveness approach to COPD and asthma in clinical trials. Pragmat Obs Res. 2017;8:175‐181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Rubin, DB . Inference and missing data. Biometrika. 1976;63(3):581‐592. [Google Scholar]
- 54. Ho M, van der Laan M, Lee H, et al. The current landscape in biostatistics of real‐world data and evidence: causal inference frameworks for study design and analysis. Stat Biopharm Res. 2021;12:1‐14. [Google Scholar]
- 55. Viele K, Berry S, Neuenschwander B, et al. Use of historical control data for assessing treatment effects in clinical trials. Pharmaceut Statist. 2014;13(1):41‐54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Yue LQ, Lu N, Xu Y. Designing premarket observational comparative studies using existing data as controls: challenges and opportunities. J Biopharm Stat. 2014;24(5):994‐1010. [DOI] [PubMed] [Google Scholar]
- 57. Li H, Mukhi V, Lu N, Xu YL, Yue LQ. A note on good practice of objective propensity score design for premarket nonrandomized medical device studies with an example. Stat Biopharm Res. 2016;8(3):282‐286. [Google Scholar]
- 58. King G, Nielsen R. Why propensity scores should not be used for matching. Political Anal. 2019;27(4):435‐454. [Google Scholar]
- 59. Chen WC, Wang C, Li H, et al. Propensity score‐integrated composite likelihood approach for augmenting the control arm of a randomized controlled trial by incorporating real‐world data. J Biopharm Stat. 2020;30(3):508‐520. [DOI] [PubMed] [Google Scholar]
- 60. Wang C, Lu N, Chen WC, et al. Propensity score‐integrated composite likelihood approach for incorporating real‐world evidence in single‐arm clinical studies. J Biopharm Stat. 2020;30(3):495‐507. [DOI] [PubMed] [Google Scholar]
- 61. Stuart EA, Rubin DB. Best practices in quasi‐experimental designs: matching methods for causal inference. In: Osborne J, ed. Best Practices in Quantitative Methods. Sage Publications; 2007: 155‐176. [Google Scholar]
- 62. Neuenschwander B, Capkun‐Niggli G, Branson M, Spiegelhalter DJ. Summarizing historical information on controls in clinical trials. Clin Trials. 2010;7(1):5‐18. [DOI] [PubMed] [Google Scholar]
- 63. Schmidli H, Gsteiger S, Roychoudhury S, O'Hagan A, Spiegelhalter D, Neuenschwander B. Robust meta‐analytic‐predictive priors in clinical trials with historical control information. Biometrics. 2014;70(4):1023‐1032. [DOI] [PubMed] [Google Scholar]
- 64. Neuenschwander B, Roychoudhury S, Schmidli H. On the use of co‐data in clinical trials. Stat Biopharm Res. 2016;8(3):345‐354. [Google Scholar]
- 65. Roychoudhury S, Neuenschwander B. Bayesian leveraging of historical control data for a clinical trial with time‐to‐event endpoint. Stat Med. 2020;39(7):984‐995. [DOI] [PubMed] [Google Scholar]
- 66. Neuenschwander B, Weber S, Schmidli H, O'Hagan A. Predictively consistent prior effective sample sizes. Biometrics. 2020;76:578‐587. [DOI] [PubMed] [Google Scholar]
- 67. Morita S, Thall PF, Müller P. Determining the effective sample size of a parametric prior. Biometrics. 2008;64(2):595‐602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Pennello G, Thompson L. Experience with reviewing Bayesian medical device trials. J Biopharm Stat. 2008;18(1):81‐115. [DOI] [PubMed] [Google Scholar]
- 69. Gelman A. Prior distributions for variance parameters in hierarchical models (comment on article by Browne and Draper). Bayesian Anal. 2006;1(3):515‐534. [Google Scholar]
- 70. Ibrahim JG, Chen MH. Power prior distributions for regression models. Statist Sci. 2020;15(1):46‐60. [Google Scholar]
- 71. Chen MH, Ibrahim JG. The relationship between the power prior and hierarchical models. Bayesian Anal. 2006;1(3):551‐574. [Google Scholar]
- 72. Neuenschwander B, Branson M, Spiegelhalter DJ. A note on the power prior. Stat Med. 2009;28(28):3562‐3566. [DOI] [PubMed] [Google Scholar]
- 73. Duan Y, Ye K, Smith EP. Evaluating water quality using power priors to incorporate historical information. Environmetrics. 2006;17:95‐106. [Google Scholar]
- 74. Ibrahim JG, Chen MH, Gwon Y, Chen F. The power prior: theory and applications. Stat Med. 2015;34(28):3724‐3749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Wang C, Li H, Chen WC, et al. Propensity score‐integrated power prior approach for incorporating real‐world evidence in single‐arm clinical studies. J Biopharm Stat. 2019;29(5):731‐748. [DOI] [PubMed] [Google Scholar]
- 76. European Medicines Agency. DRAFT Qualification opinion for Prognostic Covariate Adjustment (PROCOVA™). Accessed June 18, 2022. https://www.ema.europa.eu/en/documents/other/draft‐qualification‐opinion‐prognostic‐covariate‐adjustment‐procovatm_en.pdf
- 77. Foster JC, Taylor JM, Ruberg SJ. Subgroup identification from randomized clinical trial data. Statist Med. 2011;30(24):2867‐2880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Welton N, Sutton A, Cooper N, Abrams K, Ades AE. Evidence synthesis for decision making in healthcare. In: Welton/Evidence Synthesis for Decision Making in Healthcare. John Wiley & Sons; 2012. [Google Scholar]
- 79. Lin L, Chu H, Hodges JS. Sensitivity to excluding treatments in network meta‐analysis. Epidemiology. 2016;27(4):562‐569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Jenkins DA, Hussein H, Martina R, Dequen‐O'Byrne P, Abrams KR, Bujkiewicz S. Methods for the inclusion of real‐world evidence in network meta‐analysis. BMC Med Res Methodol. 2021;21:207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Reynolds MW, Bourke A, Dreyer NA. Considerations when evaluating real‐world data quality in the context of fitness for purpose. Pharmacoepidemiol Drug Saf. 2020;29:1316‐1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Bétourné A, Walls RL, Bateman‐House A, et al. Rare Diseases Cures Accelerator Data and Analytics Platform (RDCA‐DAP) best practices and recommendations for FAIR data, toward alignment with International Regulatory agencies. Accessed March 1, 2022. https://c‐path.org/wp‐content/uploads/2022/01/RDCA‐DAP‐Best‐Practices‐Recommendations.pdf
- 83. Posada MR, Rubinstein Y, McAuliffe M, Taruscio D, Groft SC. The GRDR‐GUID: a model for global sharing of patients de‐identified data. figshare. Poster. 2014.
- 84. Maaroufi M, Landais P, Messiaen C, Jaulent MC, Choquet R. Federating patient's identities: the case of rare diseases. Orphanet J Rare Dis. 2018;13:199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Girman CJ, Ritchey ME, Zhou W, Dreyer NA. Considerations in characterizing real‐world data relevance and quality for regulatory purposes: a commentary. Pharmacoepidemiol Drug Saf. 2019;28(4):439‐442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Sentinel. Standardization and querying of data quality metrics and characteristics for electronic health data. 2018. Accessed April 19, 2019. https://www.sentinelinitiative.org/sentinel/methods/standardization‐and‐querying‐data‐quality‐metrics‐and‐characteristics‐electronic
- 87. Richesson RL, Krischer J. Data standards in clinical research: gaps, overlaps, challenges and future directions. J Am Med Inform Assoc. 2007;14(6):687‐696. Erratum in: J Am Med Inform Assoc. 2008;15(2):265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. US Food and Drug Administration. Considerations for the Use of Real‐World Data and Real‐World Evidence to Support Regulatory Decision‐Making for Drug and Biological Products, Draft Guidance for Industry. 2021. Accessed April 19, 2022. https://www.fda.gov/media/154714/download.
- 89. Arlett P, Kjaer J, Broich K, Cooke E. Real‐world evidence in EU medicines regulation: enabling use and establishing value. Clin Pharmacol Ther. 2022;111(1):21‐23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Burns L, Le Roux N, Kalesnik‐Orszulak R, et al. Real‐world evidence for regulatory decision‐making: guidance from around the world. Clin Ther. 2022;44(3):420‐437. [DOI] [PubMed] [Google Scholar]
- 91. US Food and Drug Administration . Real‐World Data: Assessing Electronic Health Records and Medical Claims Data to Support Regulatory Decision‐Making for Drug and Biological Products, Draft Guidance for Industry. 2021. Accessed April 19, 2022. https://www.fda.gov/media/152503/download [DOI] [PMC free article] [PubMed]
- 92. US Food and Drug Administration . Search Orphan Drug Designations and Approvals. Accessed April 19, 2022. https://www.accessdata.fda.gov/scripts/opdlisting/oopd/
- 93. Schmidli H, Häring DA, Thomas M, Cassidy A, Weber S, Bretz F. Beyond randomized clinical trials: use of external controls. Clin Pharmacol Ther. 2020;107(4):806‐816. [DOI] [PubMed] [Google Scholar]
- 94. Belson NA. Food and Drug Law Institute. FDA's Historical Use of “Real World Evidence”. Accessed December 9, 2019. https://www.fdli.org/2018/08/update‐fdas‐historical‐use‐of‐real‐world‐evidence/
- 95. Seifu Y, Gamalo‐Siebers M, Barthel FM, et al. Real‑world evidence utilization in clinical development reflected by US product labeling: Statistical Review. Ther Innov Regul Sci. 2020;54(6):1436‐1443. [DOI] [PubMed] [Google Scholar]
- 96. US Food and Drug Administration Centre for Drug Evaluation and Research. Besremi Medical Reviews. Application Number: 761166Orig1s000. 2018. Accessed April 19, 2022. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2021/761166Orig1s000MedR.pdf
- 97. Hayes E, Sutter S. BioMarin's Brineura Approval Shows FDA's Open Door for Orphan Drugs. Pink Sheet. Subscription required. Published May 02, 2017. Accessed December 5, 2019. https://pink.pharmaintelligence.informa.com/PS120539/BioMarins‐Brineura‐Approval‐Shows‐FDAs‐Open‐Door‐For‐Orphan‐Drugs
- 98. Silverman B. A Baker's dozen of US FDA efficacy approvals using real world evidence. Pink sheet. Published August 07, 2018. Accessed April 19, 2022. https://pink.pharmaintelligence.informa.com/PS123648/A‐Bakers‐Dozen‐Of‐US‐FDA‐Efficacy‐Approvals‐Using‐Real‐World‐Evidence
- 99. Schulz A, Simonati A, Laine M, Williams R, Kohlschütter A, Nickel M. OP48 – 2893: The DEM‐CHILD NCL Patient Database: a tool for the evaluation of therapies in neuronal ceroid lipofuscinoses (NCL). Eur J Paediatr Neurol. 2015;19(Suppl 1):S16. [Google Scholar]
- 100. US Food and Drug Administration Centre for Drug Evaluation and Research. Brineura Statistical Reviews. Application Number: 761052Orig1s000. Accessed April 19, 2022. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2017/761052Orig1s000StatR.pdf
- 101. Schulz A, Ajayi T, Specchio N, et al. Study of intraventricular cerliponase alfa for CLN2 disease. New Engl J Med. 2018;378(20):1898‐1907. [DOI] [PubMed] [Google Scholar]
- 102. US Food and Drug Administration. FDA approves first treatment for rare disease in patients who receive stem cell transplant from blood or bone marrow. Published March 30, 2016. Accessed November 28, 2019. https://www.fda.gov/news‐events/press‐announcements/fda‐approves‐first‐treatment‐rare‐disease‐patients‐who‐receive‐stem‐cell‐transplant‐blood‐or‐bone#:~:text=The%20U.S.%20Food%20and%20Drug,marrow%20called%20hematopoietic%20stem%20cell
- 103. Defitelio Prescribing Information. Initial U.S. Approval: 2016. Accessed December 9, 2019. https://www.accessdata.fda.gov/drugsatfda_docs/label/2016/208114lbl.pdf
- 104. US Food and Drug Administration Centre for Drug Evaluation and Research. Defitelio Medical Reviews. Application Number: 208114Orig1s000. Accessed 28 Nov 2019. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2016/208114Orig1s000MedR.pdf
- 105. US Food and Drug Administration Centre for Drug Evaluation and Research. Defitelio Statistical Reviews. Application Number: 208114Orig1s000 Accessed January 6, 2020. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2016/208114Orig1s000StatR.pdf
- 106. Hendriksz CJ, Burton B, Fleming TR, et al. Efficacy and safety of enzyme replacement therapy with BMN 110 (elosulfase alfa) for Morquio A syndrome (mucopolysaccharidosis IVA): a phase 3 randomised placebo‐controlled study. J Inherit Metab Dis. 2014;37(6):979‐990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Hendriksz CJ, Parini R, AlSayed MD, et al. Long‐term endurance and safety of elosulfase alfa enzyme replacement therapy in patients with Morquio A syndrome. Mol Genet Metab. 2016;119(1‐2):131‐143. [DOI] [PubMed] [Google Scholar]
- 108. Dearment A. FDA approves Carbaglu. Drug Store News. March 18, 2010. Accessed April 19, 2022. https://www.drugstorenews.com/news/fda‐approves‐carbaglu/
- 109. Carbaglu Prescribing Information. Initial U.S. Approval: 2010. Accessed April 19, 2022. https://www.accessdata.fda.gov/drugsatfda_docs/label/2010/022562lbl.pdf
- 110. Text of the labeling of the drug: Myozyme® (alglucosidase alfa). 2006. Accessed April 19, 2022. https://www.accessdata.fda.gov/drugsatfda_docs/label/2006/125141LBLini.pdf
- 111. Critical Path Institute. Rare Disease Cures Accelerator‐Data and Analytics Platform. Accessed June 18, 2022. https://c‐path.org/programs/rdca‐dap/
- 112. Innovative Health Initiative European Health Data and Evidence Network. Accessed June 18, 2022. https://www.ihi.europa.eu/projects‐results/project‐factsheets/ehden
- 113. Saczynski J, Andrade S, Harrold L, et al. Mini‐Sentinel systematic evaluation of health outcome of interest definitions for studies using administrative data: congestive heart failure report. December 19, 2010. Accessed April 19, 2022. https://www.sentinelinitiative.org/sites/default/files/surveillance‐tools/validations‐literature/MS_HOI_CHFReport_0.pdf
- 114. Fuller CC, Zinderman C, Spencer‐Smith C, et al. Sentinel protocol based assessment report: transfusion related acute lung injury after red blood cell, plasma, and platelet administration 2013–2015. September 10, 2019. Accessed April 19, 2022. https://www.sentinelinitiative.org/sites/default/files/Methods/TRALI_After_Red_Blood_Cell_Plasma_and_Platelet_Administration_2013_2015_ReportFinal.pdf
- 115. Lo Re V, Haynes K, Goldberg D, et al. Validity of diagnostic codes to identify cases of severe acute liver injury in the Mini‐Sentinel Distributed Database. December 6, 2012. Accessed April 19, 2022. https://www.sentinelinitiative.org/sites/default/files/surveillance‐tools/validations‐literature/Mini‐Sentinel_Validation‐of‐Severe‐Acute‐Liver‐Injury‐Cases.pdf [DOI] [PMC free article] [PubMed]
- 116. Walsh KE, Cutrona SL, Pawloski P, et al. Validity of administrative and claims data for the identification of cases of anaphylaxis in the Mini‐Sentinel Distributed Database. February 15, 2013. Accessed April 19, 2022. https://www.sentinelinitiative.org/sites/default/files/surveillance‐tools/validations‐literature/Mini‐Sentinel_Validation‐of‐Anaphylaxis‐Cases.pdf
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Study protocol will be shared upon reasonable request.