

Abstract

The orphan G protein-coupled receptor 35 (GPR35) is a potential target for the treatment of pain, inflammation, and metabolic diseases. Although many GPR35 agonists have been discovered, research on functional GPR35 ligands, such as fluorescent probes, is still limited. Herein, we developed a series of GPR35 fluorescent probes by conjugating a BODIPY fluorophore to DQDA, a known GPR35 agonist. All probes exhibited excellent GPR35 agonistic activity and desired spectroscopic properties, as determined by the DMR assay, bioluminescence resonance energy transfer (BRET)-based saturation, and kinetic binding experiments. Notably, compound 15 showed the highest binding potency and the weakest nonspecific BRET binding signal (Kd = 3.9 nM). A BRET-based competition binding assay with 15 was also established and used to determine the binding constants and kinetics of unlabeled GPR35 ligands.
Keywords: GPR35, fluorescent probes, bioluminescence resonance energy transfer, binding affinity
GPR35 was first identified in 1998 and is believed to be associated with many diseases including coronary artery disease, cancers, and inflammatory bowel disease.1−4 Although its endogenous activator remains controversial, a wide range of surrogate agonists are available.5−8 However, there are few studies on functionalized GPR35 ligands, which are detrimental to the study of GPR35 physiological processes. Until now, 6-bromo-8-(4-[3H]methoxybenzamido)-4-oxo-4H-chromene-2-carboxylic acid was the only molecule used in the GPR35 radioligand binding assays.9
BODIPY is considered a construction platform for fluorescent probes that are widely used in bioimaging and photodynamic therapy for their high molar absorption coefficients and fluorescence quantum yields as well as their good biocompatibility and photostability.10,11 We previously reported a series of potent GPR35 agonists with two acidic groups located at each end of a fused tricyclic aromatic scaffold.12 The structure–activity relationship (SAR) studies and docking simulations showed that the two terminal acidic groups were necessary for maintaining the high agonistic potency of the compounds, while substitutions in the middle of the scaffold were tolerated.12 Considering the high potency of the compounds and the convenience of the synthesis, we chose4,6-dihydroxy-10-methylpyrido-[3,2-g]-quinoline-2,8-dicarboxylic acid (DQDA) as a molecular moiety to target GPR35 (EC50 = 8.0 nM).
10-(p-Alkoxyphenyl)-BODIPY was selected as the fluorophore to couple with the hydroxyl of DQDA, since its absorption band between 460 and 520 nm overlaps well with the emission band of NLuc, creating a possibility for BRET between the NLuc-tagged GPR35 and the fluorescent probes.
The syntheses of the GPR35 fluorescent ligands 14–17 are depicted in Figure 1. The dimethyl 4,6-dihydroxy-10-methylpyrido- [3,2-g]-quinoline-2,8-dicarboxylate (1) was prepared as previously described.12 The iodoalkynes (2–4) reacted with compound 1 to produce intermediates 5–7. Compound 1 could be converted into intermediate 8 through the introduction of the oxa-alkyne in the Mitsunobu reaction. Using a typical copper-catalyzed azide–alkyne cycloaddition, BODIPY containing an azide group (9) was conjugated with 5–8 to yield compounds 10–13, respectively. These methyl ester products were hydrolyzed with lithium hydroxide to yield BODIPY-labeled fluorescent ligands 14–17.
Figure 1.
Syntheses of BODIPY-labeled fluorescent probes 14–17. Reagents and conditions: (a) Cs2CO3, DMF, at 50 °C overnight, 34–47% (b) 2-(prop-2-yn-1-yloxy)ethan-1-ol, PPh3, DIAD, THF, from 0 °C to room temperature overnight, 35%. (c) CuSO4·5H2O, sodium ascorbate, THF: H2O (1:1, v/v), at 50 °C overnight, 73–81%. (d) LiOH, H2O: MeOH (1:1, v/v), at room temperature for 2 h, 65–81%.
BODIPY derivatives with extended conjugated structures often exhibit a significant positive or negative solvatochromic effect in different polar environments.13−15 Some simple derivatives of BODIPY are characterized by a small Stokes shift and a solvatochromic effect on the polarity of the medium.16 We recorded the UV absorption and fluorescence spectra of probes 14–17 in PBS solution, a standard aqueous buffer used in in vitro pharmacological assays, and in n-octanol, an organic solvent usually used to mimic a predominantly hydrophobic environment like the receptor binding site or cell membrane (Figures S1 and S2). These fluorescent probes showed low solvatochromic behavior in the solvents, with both the absorption maxima and emission maxima showing little change (Table S1). In n-octanol, the probes displayed an absorption maximum at 502 nm and a fluorescence maximum at 512 nm with a 10 nm Stokes shift. These results were consistent with literature reports16 and were expected since the BODIPY unit of the probe was simply tethered to the target head DQDA.
Using coumarin 153 as the reference, the relative quantum yields of all probes in n-octanol were determined and are listed in Table S1. Probe 15 exhibited the highest relative quantum yield (Φ) of 0.77. However, dramatic decreases in the absorption and fluorescence intensity were observed for all probes in PBS, with the emission maxima of 14–17 decreasing by 3- to 15-fold (Figure S1). The observed results can be reasonably attributed to the formation of an intramolecular charge transfer state that was stabilized by the polar environment to give lower Φ values.17Figure S3 shows the fluorescence of in intramolecular charge transfer state that was stabilized by the polar environment to give lower Φ values.17Figure S3 shows the fluorescence spectra of probe 15 in various solvents, including PBS, H2O, DMSO, methanol, ethanol, THF, dioxane, toluene, and n-octanol, further demonstrating the significant difference in intensity between organic solvents and aqueous solutions. The results showed that the quantum yield of the probes was highly sensitive to the environment, and the high fluorescence activity was preserved in the nonaqueous environment. Therefore, the background fluorescence from unbound probes in aqueous media should not be an issue.
DMR assays were applied to profile compound activity on GPR35 endogenously expressed in the human colorectal adenocarcinoma cell line HT-29. Zaprinast was used as a full agonist and tool molecule in DMR activation and desensitization experiments.18,19 All four probes not only gave rise to the concentration-dependent DMR signals in HT-29 but also desensitized the DMR responses induced by 1 μM zaprinast after 1 h of incubation (Table 1 and Figure 2). All probes exhibited high agonistic potency, with EC50 values between 42.2 and 80.8 nM. The GPR35 inhibitor ML-145 dose-dependently blocked the DMR responses generated by these probes (Table 1). Taken together, it is suggested that these probes generated DMR responses specifically through the activation of GPR35 in the HT-29 cells. To confirm this, these four ligands were further tested on two other cell lines. The CHO-K1 cell did not endogenously express GPR35, with none of the probes inducing the signals. However, in GPR35-overexpressing CHO-K1 cells, all the probes exhibited similar agonist activity and were confirmed to be GPR35 agonists (Figure S4).
Table 1. Pharmacologic Properties of Probes 14–17.
| hGPR35 |
|||
|---|---|---|---|
| Compd | EC50a (nM) | IC50b (nM) | IC50c (nM) |
| zaprinast | 0.71 ± 0.12 | — | — |
| 14 | 80.8 ± 9.5 | 9.6 ± 0.7 | 1034.1 ± 95.6 |
| 15 | 49.5 ± 5.6 | 9.4 ± 2.4 | 542.2 ± 87.3 |
| 16 | 42.2 ± 3.3 | 5.6 ± 0.8 | 397.6 ± 65.2 |
| 17 | 58.8 ± 5.8 | 7.8 ± 1.4 | 768.7 ± 84.6 |
EC50 to trigger DMR.
IC50 to desensitize cells against repeated stimulation of 1 μM zaprinast.
IC50 of a known GPR35 antagonist ML-145 to block the agonist-induced DMR. The data represent mean ± SD from two independent measurements, each with four replicates (n = 2).
Figure 2.

DMR response in HT-29 cells induced by fluorescent probes 14–17 as a function of the concentration. The data represent mean ± SD from two independent measurements, each with four replicates (n = 2).
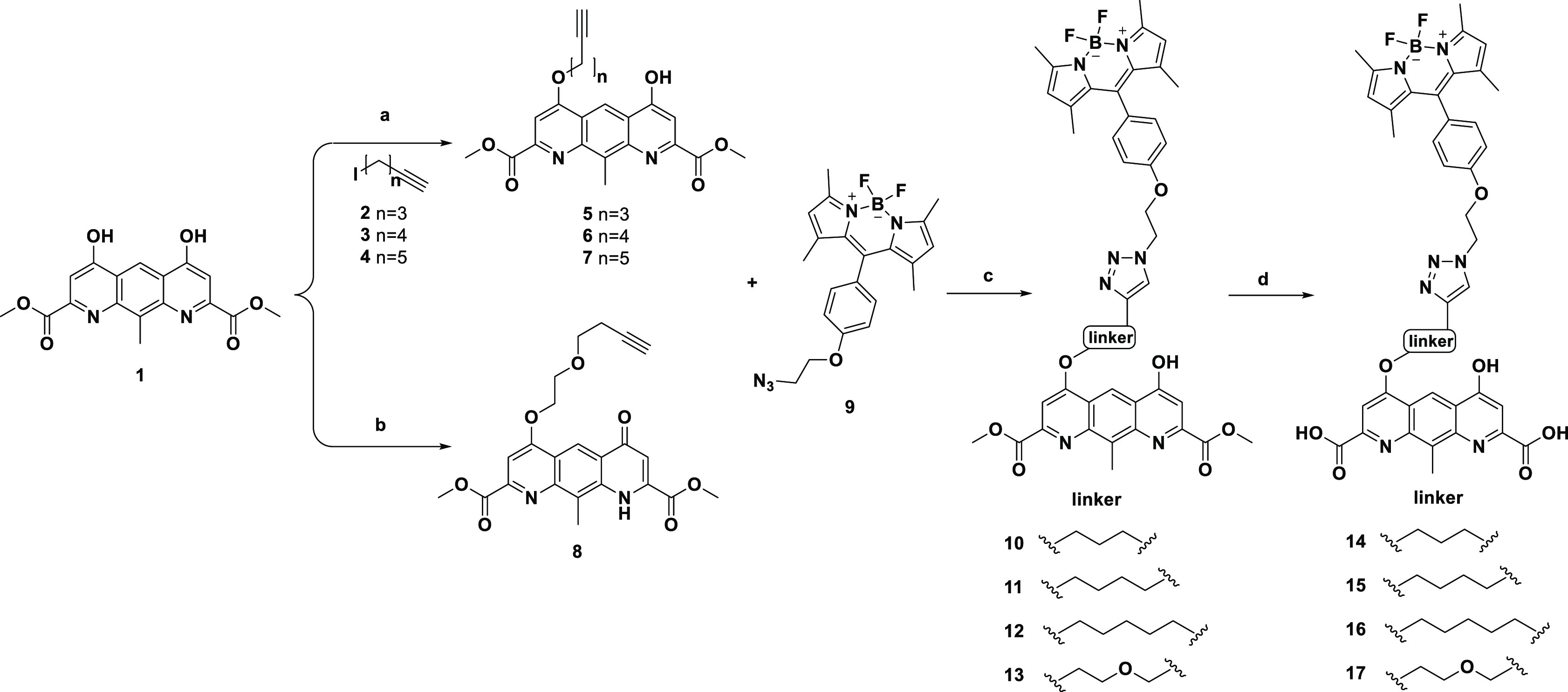
A BRET-based GPR35 binding assay was then developed by using these fluorescent probes. It was performed as a homogeneous “mix and measure” assay well suited for kinetics and high-throughput equilibrium binding experiments.20 CHO-K1 cells were transfected with hGPR35, which was N-terminally tagged with a nano-luciferase (NLUC).21 NLUC has an emission maximum between 410 and 560 nm, displaying a partial overlap with the excitation spectra of probes, such as 15 (Figure 3). When the probe binds to NLUC-GPR35, energy transfer at 485 nm will occur between the NLUC and the BODIPY moiety, resulting in BODIPY emission at 528 nm. As BRET methods strictly depend on the distance between the bioluminescent and fluorescent partners, this approach would eliminate the need to remove the unbound probes from the assay medium.
Figure 3.
Emission spectrum of NLUC (dark green) and excitation (light red solid) and emission (light red dot) spectra of 15 were measured under the binding assay conditions.
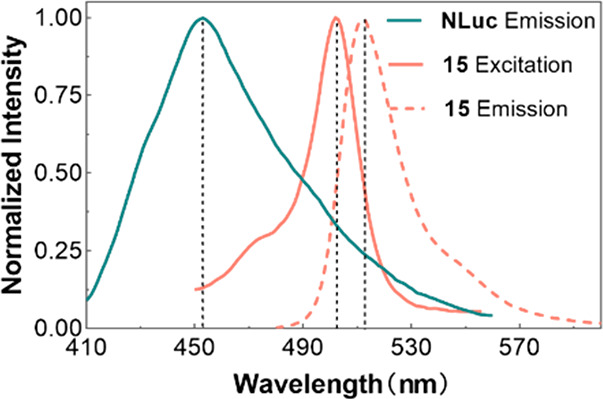
Saturation binding experiments were performed by adding fluorescent probes at varying concentrations to the CHO-K1-hGPR35-NLUC cells. The results suggested that probes 14–17 could effectively respond to NLUC-tagged GPR35 via BRET (Figure 4). The nonspecific BRET signal was determined by coincubation with excess zaprinast (10 μM). Probes 14 and 16 displayed a considerable nonspecific BRET signal (Figure 4A and C), probe 15 showed a minor nonspecific BRET signal even at concentrations of up to 100 nM (Figure 4B), and probe 17 was in between (Figure 4D). The obvious difference was unexpected, as these probe linkers only differed slightly and may be related to the change of their fluorescence intensity in different polar environments. As shown in Figure S2, the fluorescence intensity of probe 15 in PBS decreased by a factor of 15 times that in n-octanol, changing more drastically than the other three probes. This difference might further reduce the signal of the nonspecific BRET signal shown in Figure 4B. The subtraction of the nonspecific BRET signal from the total binding signals generated monophasic binding curves with Kd = 7.4 ± 0.5 nM for 14, Kd = 3.9 ± 2.5 nM for 15, Kd = 8.3 ± 0.8 nM for 16, and Kd = 17.9 ± 5.1 nM for 17. Among these potent candidates, probe 15 was selected for subsequent studies because of its high ratio of specific to nonspecific signals.
Figure 4.
Saturation binding experiments of probes (A) 14, (B) 15, (C) 16, and (D) 17 in CHO-K1-hGPR35-NLUC cells. Nonspecific bindings were measured by pretreatment with 10 μM zaprinast. The data represent mean ± SD from three independent measurements, each with three replicates (n = 3).
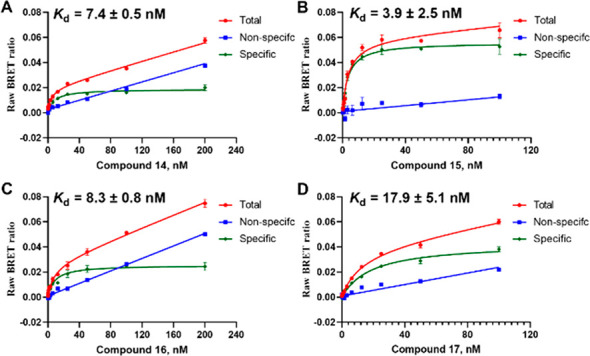
In the BRET kinetic binding experiments, the BRET emission ratio of 15 decreased drastically when GPR35 antagonist ML-145 was added, indicating a successful BRET between GPR35-NLUC and the probe molecule as a completely reversible binding process (Figure 6).
Figure 6.
Kinetic binding experiment of probe 15; 10 min after the addition of probe 15 (5 nM), ML-145 (10 μM) was added to dissociate the probe from GPR35.
Next, we further investigated whether this BRET-based strategy using compound 15 as the probe could be applied to assess the affinity of GPR35 ligands, the structures are shown in Figure 5. The prerequisites for this application were that the binding of all of the active ligands was reversible and competed with probe 15 at the same GPR35 site. Competitive binding assays were conducted in CHO-K1 transferred with GPR35-NLUC by using 15 to assess the affinity of a range of previously reported GPR35 ligands (Figure S6). Some known GPR35 ligands were selected, such as agonists 18–20 and antagonists 21 and 22 (Figure 5). The results indicated that the selected molecules exhibited competitive binding against 15, which allowed estimation of the Ki binding affinity constants. The Ki values measured by our BRET method were in agreement with those measured by the radioligand method reported in the literature (Table 2). These results indicated that this BRET-based binding assay should be a reliable strategy to measure the Ki values of the GPR35 ligands.
Figure 5.

Structures of selected GPR35 ligands.
Table 2. Potencies and Binding Affinities of the Selected GPR35 Ligands.
| Compd | BRET assay Ki (μM)a | Reference Ki (μM)b | DMR assay (μM)c | β-arrestin assay (μM)d |
|---|---|---|---|---|
| 18 | 2.97 ± 0.87 | 2.340 ± 0.040 | (EC50) 0.5222 | (EC50) 722 |
| 19 | 0.612 ± 0.233 | 0.401 ± 0.015 | (EC50) 0.1623 | (EC50) 4.223 |
| 20 | 0.023 ± 0.006 | 0.012 ± 0.001 | (EC50) 0.00324 | (EC50) 1.20 ± 0.1325 |
| 21 | 0.004 ± 0.001 | 0.009 ± 0.001 | / | (IC50) 0.02725 |
| 22 | 0.236 ± 0.049 | 0.042 ± 0.003 | (IC50) 10.423 | (IC50) 0.2025 |
| 23 | 0.332 ± 0.063 | / | (EC50) 0.071 ± 0.01012 | / |
| 24 | 0.051 ± 0.015 | / | (EC50) 0.006 ± 0.00126 | (EC50) 0.197 ± 0.03823 |
| 25 | 0.010 ± 0.002 | / | (EC50) 0.059 ± 0.00728 | / |
| 26 | 0.352 ± 0.107 | / | (EC50) 0.150 ± 0.02026 | (EC50) 3.63 ± 0.9523 |
| 27 | 0.028 ± 0.006 | / | (EC50) 0.083 ± 0.00612 | / |
| 28 (DQDA) | 0.006 ± 0.002 | / | (EC50) 0.008 ± 0.00112 | / |
Affinities were determined through displacement BRET binding assays using 25 nM 15. The data represent mean ± SD from three independent measurements, each with three replicates (n = 3).
Ki values were acquired from radio–ligand binding assays from the literature.9 /: not measured or reported.
EC50 values were tested by DMR assays in HT-29 cells endogenously expressing GPR35.
Previously published. /: not measured or reported.
We further applied the BRET method to more GPR35 active ligands. Compounds 23–28 are the GPR35 agonists that we have previously discovered,12,26,27 but their Ki values were not determined. Using the BRET method with compound 15 as a probe, these Ki values were measured for the first time and are listed in Table 2. Overall, a good correlation between agonist potency and binding Ki values was observed. Compound 28 the second most potent agonist in the functional assays displayed the highest binding affinity (Table 2). Likewise, 25–27 showed Ki values similar to those of their functional potencies. Unexpectedly, compound 24 was the most potent agonist in the DMR functional assays (EC50 = 0.006 μM) but showed a lower affinity in the BRET binding assay (Ki = 0.051 μM). Such a discrepancy between affinity and potency also existed in other similar studies and can be attributed to the different intrinsic efficacies of various compounds in the activation of GPR35.27
In addition, we evaluated whether the probe concentrations would affect the Ki values. Figure 7 shows the application of probe 15 in a competitive binding experiment with pamoic acid 20. Higher concentrations of 15 required increasing concentrations of 20 to compete for the receptor binding site. However, the resulting Ki values of 20 varied in a narrow range (adding 15 at concentrations of 100, 50, 40, and 20 nM respectively provided Ki values of 9.6, 10.3, 8.9, and 14.9 nM) comparable to the Ki value of 12 nM reported previously (Table 2). It is worth noting that the BRET signals might be too low to be detected with the addition of probe 15 at 5 or 10 nM.
Figure 7.

Application of 15 in competition binding experiments of pamoic acid 20. Data represent mean ± SD from three independent measurements, each with three replicates (n = 3).
In conclusion, we describe the synthesis and evaluation of GPR35 fluorescent probes by tethering the BODIPY fluorophore to a known GPR35 agonist DQDA. The resulting probes 14–17 maintained excellent GPR35 agonistic activity with EC50 values between 42.2 and 80.8 nM. Probe 15 exhibits desirable spectroscopic properties, including good photostability, a Stokes shift of 10 nm, low fluorescence activity in aqueous solution, and a high quantum yield in nonpolar environments. In the NLuc construct built into the N-terminal domain of the GPR35 receptor and BRET-based binding experiments, probes 14–17 exhibited Kd values of 7.4, 3.9, 8.3, and 17.9 nM, respectively. Notably, probe 15 exhibited a favorable specific to nonspecific BRET signal at concentrations of up to 100 nM. The BRET-based binding assay with 15 was further applied to determine the kinetic binding parameters of unlabeled GPR35 ligands, resulting in Ki values comparable to those obtained previously using a radiolabeled ligand. This is the first report about the GPR35 fluorescent probe, demonstrating its utility for further study of the GPR35 receptor and ligand characterization.
Acknowledgments
This work was supported by the innovation program of science and research from DICP, CAS (DICP ZZBS201803).
Glossary
Abbreviations
- GPCR
G protein-coupled receptor
- hGPR35
human G protein-coupled receptor 35
- BRET
bioluminescent resonance energy transfer
- NLUC
nano-luciferase
- DMR
dynamic mass redistribution
- BODIPY
4,4-difluoro-4-bora-3a,4a-diaza-s-in-dacene
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.2c00461.
1H NMR, 13C NMR spectral data and HPLC-MS analysis of compounds 5–8 and 10–17; molecular formula strings (PDF)
Author Contributions
† Lai Wei and Kaijing Xiang contributed equally to this work. Author contributions are the following: conceptualization, Yaopeng Zhao, Xinmiao Liang; chemistry: Lai Wei, Yonglin Ge; biology, Lai Wei, Kaijing Xiang, Hongjian Kang, Hongjie Fan, Tao Hou, Yang Chen; writings, Yancheng Yu, Han Zhou, Jixia Wang, Zhimou Guo. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- O’Dowd B. F.; Nguyen T.; Marchese A.; Cheng R.; Lynch K. R.; Heng H. H. Q.; Kolakowski L. F.; George S. R. Discovery of three novel G-protein-coupled receptor genes. Genomics 1998, 47, 310–313. 10.1006/geno.1998.5095. [DOI] [PubMed] [Google Scholar]
- Baumgartner R.; Casagrande F. B.; Mikkelsen R. B.; Berg M.; Polyzos K. A.; Forteza M. J.; Arora A.; Schwartz T. W.; Hjorth S. A.; Ketelhuth D. F. J. Disruption of GPR35 Signaling in Bone Marrow-Derived Cells Does Not Influence Vascular Inflammation and Atherosclerosis in Hyperlipidemic Mice. Metabolites 2021, 11, 411. 10.3390/metabo11070411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boleij A.; Fathi P.; Dalton W.; Park B.; Wu X.; Huso D.; Allen J.; Besharati S.; Anders R. A.; Housseau F. G-protein coupled receptor 35 (GPR35) regulates the colonic epithelial cell response to enterotoxigenic Bacteroides fragilis. Commun. Biol. 2021, 4, 585. 10.1038/s42003-021-02014-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quon T.; Lin L.-C.; Ganguly A.; Tobin A. B.; Milligan G. Therapeutic Opportunities and Challenges in Targeting the Orphan G Protein-Coupled Receptor GPR35. ACS Pharmacol Transl 2020, 3, 801–812. 10.1021/acsptsci.0c00079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maravillas-Montero J. L.; Burkhardt A. M.; Hevezi P. A.; Carnevale C. D.; Smit M. J.; Zlotnik A. Cutting Edge: GPR35/CXCR8 Is the Receptor of the Mucosal Chemokine CXCL17. J. Immunol 2015, 194, 29–33. 10.4049/jimmunol.1401704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S.-J.; Lee S.-J.; Nam S.-Y.; Im D.-S. GPR35 mediates lodoxamide-induced migration inhibitory response but not CXCL17-induced migration stimulatory response in THP-1 cells; is GPR35 a receptor for CXCL17?. Br. J. Pharmacol. 2018, 175, 154–161. 10.1111/bph.14082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shore D. M.; Reggio P. H.. The therapeutic potential of orphan GPCRs, GPR35 and GPR55. Front Pharmacol 2015, 6, 10.3389/fphar.2015.00069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binti Mohd Amir N. A. S.; Mackenzie A. E.; Jenkins L.; Boustani K.; Hillier M. C.; Tsuchiya T.; Milligan G.; Pease J. E. Evidence for the Existence of a CXCL17 Receptor Distinct from GPR35. J. Immunol 2018, 201, 714–724. 10.4049/jimmunol.1700884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thimm D.; Funke M.; Meyer A.; Mueller C. E. 6-Bromo-8-(4- H-3 methoxybenzamido)-4-oxo-4H-chromene-2-carboxylic Acid: A Powerful Tool for Studying Orphan G Protein-Coupled Receptor GPR35. J. Med. Chem. 2013, 56, 7084–7099. 10.1021/jm4009373. [DOI] [PubMed] [Google Scholar]
- Loudet A.; Burgess K. BODIPY dyes and their derivatives: Syntheses and spectroscopic properties. Chem. Rev. 2007, 107, 4891–4932. 10.1021/cr078381n. [DOI] [PubMed] [Google Scholar]
- Norager N. G.; Jensen C. B.; Rathje M.; Andersen J.; Madsen K. L.; Kristensen A. S.; Stromgaard K. Development of Potent Fluorescent Polyamine Toxins and Application in Labeling of lonotropic Glutamate Receptors in Hippocampal Neurons. ACS Chem. Biol. 2013, 8, 2033–2041. 10.1021/cb400272m. [DOI] [PubMed] [Google Scholar]
- Wei L.; Hou T.; Li J.; Zhang X.; Zhou H.; Wang Z.; Cheng J.; Xiang K.; Wang J.; Zhao Y.; Liang X. Structure-Activity Relationship Studies of Coumarin-like Diacid Derivatives as Human G Protein-Coupled Receptor-35 (hGPR35) Agonists and a Consequent New Design Principle. J. Med. Chem. 2021, 64, 2634–2647. 10.1021/acs.jmedchem.0c01624. [DOI] [PubMed] [Google Scholar]
- Telegin F. Y.; Marfin Y. S. Polarity and Structure of BODIPYs: A Semiempirical and Chemoinformation Analysis. Russ. J. Inorg. Chem. 2022, 67, 362–374. 10.1134/S0036023622030135. [DOI] [Google Scholar]
- Nano A.; Ziessel R.; Stachelek P.; Harriman A. Charge-Recombination Fluorescence from Push-Pull Electronic Systems Constructed around Amino-Substituted Styryl-BODIPY Dyes. Chem.—Eur. J. 2013, 19, 13528–13537. 10.1002/chem.201301045. [DOI] [PubMed] [Google Scholar]
- Petrushenko K. B.; Petrushenko I. K.; Petrova O. V.; Sobenina L. N.; Ushakov I. A.; Trofimov B. A. Environment-Responsive 8-CF3-BODIPY Dyes with Aniline Groups at the 3 Position: Synthesis, Optical Properties and RI-CC2 Calculations. Asia J. Org. Chem. 2017, 6, 852–861. 10.1002/ajoc.201700117. [DOI] [Google Scholar]
- Conroy S.; Kindon N. D.; Glenn J.; Stoddart L. A.; Lewis R. J.; Hill S. J.; Kellam B.; Stocks M. J. Synthesis and Evaluation of the First Fluorescent Antagonists of the Human P2Y(2) Receptor Based on AR-C118925. J. Med. Chem. 2018, 61, 3089–3113. 10.1021/acs.jmedchem.8b00139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakare S. S.; Chakraborty G.; Kothavale S.; Mula S.; Ray A. K.; Sekar N. Proton Induced Modulation of ICT and PET Processes in an Imidazo-phenanthroline Based BODIPY Fluorophores. J. Fluoresc 2017, 27, 2313–2322. 10.1007/s10895-017-2173-4. [DOI] [PubMed] [Google Scholar]
- Deng H.; Hu H.; Ling S.; Ferrie A. M.; Fang Y. Discovery of Natural Phenols as G Protein-Coupled Receptor-35 (GPR35) Agonists. ACS Med. Chem. Lett. 2012, 3, 165–169. 10.1021/ml2003058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng H.; Hu H.; Fang Y. Multiple tyrosine metabolites are GPR35 agonists. Sci. Rep. 2012, 2, 373. 10.1038/srep00373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall M. P.; Unch J.; Binkowski B. F.; Valley M. P.; Butler B. L.; Wood M. G.; Otto P.; Zimmerman K.; Vidugiris G.; Machleidt T.; Robers M. B.; Benink H. A.; Eggers C. T.; Slater M. R.; Meisenheimer P. L.; Klaubert D. H.; Fan F.; Encell L. P.; Wood K. V. Engineered luciferase reporter from a deep sea shrimp utilizing a novel imidazopyrazinone substrate. ACS Chem. Biol. 2012, 7, 1848–1857. 10.1021/cb3002478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoddart L. A.; Johnstone E. K. M.; Wheal A. J.; Goulding J.; Robers M. B.; Machleidt T.; Wood K. V.; Hill S. J.; Pfleger K. D. G. Application of BRET to monitor ligand binding to GPCRs. Nat. Methods 2015, 12, 661–663. 10.1038/nmeth.3398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y.; Lu J. Y.-L.; Wu X.; Summer S.; Whoriskey J.; Saris C.; Reagan J. D. G-Protein-Coupled Receptor 35 Is a Target of the Asthma Drugs Cromolyn Disodium and Nedocromil Sodium. Pharmacology 2010, 86, 1–5. 10.1159/000314164. [DOI] [PubMed] [Google Scholar]
- Deng H.; Hu H.; He M.; Hu J.; Niu W.; Ferrie A. M.; Fang Y. Discovery of 2-(4-Methylfuran-2(5H)-ylidene)malononitrile and Thieno 3,2-b thiophene-2-carboxylic Acid Derivatives as G Protein-Coupled Receptor 35 (GPR35) Agonists. J. Med. Chem. 2011, 54, 7385–7396. 10.1021/jm200999f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu H.; Deng H.; Fang Y. Label-Free Phenotypic Profiling Identified D-Luciferin as a GPR35 Agonist. PLoS One 2012, 7, e34934. 10.1371/journal.pone.0034934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao P.; Sharir H.; Kapur A.; Cowan A.; Geller E. B.; Adler M. W.; Seltzman H. H.; Reggio P. H.; Heynen-Genel S.; Sauer M.; Chung T. D. Y.; Bai Y.; Chen W.; Caron M. G.; Barak L. S.; Abood M. E. Targeting of the Orphan Receptor GPR35 by Pamoic Acid: A Potent Activator of Extracellular Signal-Regulated Kinase and beta-Arrestin2 with Antinociceptive Activity. Mol. Pharmacol. 2010, 78, 560–568. 10.1124/mol.110.066746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei L.; Wang J.; Zhang X.; Wang P.; Zhao Y.; Li J.; Hou T.; Qu L.; Shi L.; Liang X.; Fang Y. Discovery of 2H-Chromen-2-one Derivatives as G Protein-Coupled Receptor-35 Agonists. J. Med. Chem. 2017, 60, 362–372. 10.1021/acs.jmedchem.6b01431. [DOI] [PubMed] [Google Scholar]
- Wei L.; Hou T.; Lu C.; Wang J.; Zhang X.; Fang Y.; Zhao Y.; Feng J.; Li J.; Qu L.; Piao H. L.; Liang X. SAR Studies of N-[2-(1H-Tetrazol-5-yl)phenyl]benzamide Derivatives as Potent G Protein-Coupled Receptor-35 Agonists. ACS Med. Chem. Lett. 2018, 9, 422–427. 10.1021/acsmedchemlett.7b00510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christiansen E.; Hudson B. D.; Hansen A. H.; Milligan G.; Ulven T. Development and Characterization of a Potent Free Fatty Acid Receptor 1 (FFA1) Fluorescent Tracer. J. Med. Chem. 2016, 59, 4849–58. 10.1021/acs.jmedchem.6b00202. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.