

Abstract
Aims
This study investigated the safety, tolerability, pharmacokinetics and pharmacodynamics of danuglipron (PF‐06882961), which is a novel, oral small‐molecule glucagon‐like peptide‐1 receptor agonist, in Japanese participants with type 2 diabetes mellitus (T2DM).
Materials and Methods
This phase 1, randomized, double‐blind, placebo‐controlled, parallel‐group study enrolled adult Japanese participants with T2DM inadequately controlled on diet and exercise. Participants received twice‐daily oral doses of placebo or multiple ascending doses of danuglipron titrated to 40, 80 or 120 mg twice daily over 8 weeks. The primary outcome was the safety and tolerability of danuglipron. Secondary and exploratory outcomes included plasma pharmacokinetics, glycaemic parameters and body weight.
Results
In the 37 participants randomized, the most common treatment‐emergent adverse events were nausea, vomiting, abdominal discomfort, diarrhoea and headache. Most treatment‐emergent adverse events were of mild or moderate intensity. Dose‐proportional increases in danuglipron exposure parameters were observed at steady state (Day 56). Significant reductions from baseline were observed with danuglipron on Day 56 for mean daily glucose [least squares mean (90% confidence interval) placebo‐adjusted difference of up to −67.89 (−88.98, −46.79) mg/dl] and on Day 57 for fasting plasma glucose [up to −40.87 (−53.77, −27.98) mg/dl], glycated haemoglobin [up to −1.41% (−2.01%, −0.82%)] and body weight [up to −1.87 (−3.58, −0.17) kg].
Conclusions
In Japanese adults with T2DM, danuglipron exhibited dose‐proportional increases in plasma exposure at steady state and robustly reduced glycaemic parameters and body weight after 8 weeks of dosing, with a safety profile consistent with the mechanism of action.
Keywords: danuglipron, diabetes, glucagon‐like peptide‐1 receptor agonist, phase 1 study
1. INTRODUCTION
In 2021, diabetes affected an estimated 11 million adults in Japan (age‐adjusted prevalence, 6.6%) and caused ~245 000 deaths. 1 Recent studies have found that nearly half of Japanese patients with type 2 diabetes mellitus (T2DM) do not achieve the standard glycated haemoglobin (HbA1c) target of <7.0%. 2 , 3 Japanese clinical practice guidelines 4 for the management of non‐insulin‐dependent T2DM recommend the use of an oral antihyperglycaemic agent, such as a dipeptidyl peptidase‐4 inhibitor or metformin, 5 and/or an injectable glucagon‐like peptide 1 receptor (GLP‐1R) agonist as first‐line therapy in patients where adequate glycaemic control is not achieved through diet and exercise. 4 However, a survey of injectable‐naïve Japanese patients with T2DM revealed a strong preference for drug profiles associated with oral versus injectable GLP‐1R agonists, with administration mode and frequency the most important factors contributing to this decision. 6
Danuglipron (PF‐06882961) is a novel, potent, oral small‐molecule GLP‐1R agonist being investigated as an adjunct to diet and exercise to improve glycaemic control in adults with T2DM. 7 Based on pharmacokinetic data from early phase 1 studies, 7 , 8 danuglipron is administered twice daily and can be dosed without regard to food. In non‐clinical models, 7 , 8 danuglipron stimulated glucose‐dependent insulin release and suppressed food intake with equivalent efficacy to that of injectable peptidic GLP‐1R agonists. In a recent multiple ascending‐dose phase 1 study in non‐Japanese participants with T2DM on a background of metformin, 8 danuglipron reduced glycaemic indices and body weight with favourable safety and pharmacokinetic profiles, supporting further clinical development of danuglipron for the treatment of T2DM and obesity.
This current phase 1 study investigated the safety, tolerability, pharmacokinetics and pharmacodynamics of danuglipron in Japanese participants with T2DM following oral, twice‐daily dosing for 8 weeks. This was the first clinical study of danuglipron in Japanese patients, and the first to describe the effect of danuglipron dosing beyond 28 days, in an outpatient study, and on a background of diet and exercise alone.
2. MATERIALS AND METHODS
2.1. Study oversight
The study (NCT04552470) was conducted in compliance with the Declaration of Helsinki and with International Conference on Harmonisation Good Clinical Practice guidelines. The final protocol and informed consent documentation were reviewed and approved by the Institutional Review Board at the study centre (P‐One Clinic; Keikokai Medical Corporation, Tokyo, Japan). Study recruitment took place during October‐December 2020; the study was completed in March 2021. All participants provided written informed consent before participation in the study.
2.2. Study design
This phase 1, randomized, double‐blind, placebo‐controlled, four‐arm, parallel‐group study in Japanese participants with T2DM inadequately controlled on diet and exercise alone consisted of a 4‐week screening period, an 8‐week treatment period and a 4‐week follow‐up period (Figure S1: Appendix S1). Participants were randomized 1:1:1:1 to oral placebo or danuglipron target doses of 40, 80 or 120 mg twice daily. Up to 6 weeks of the 8‐week double‐blind treatment period was used for dose escalation, utilizing a pre‐specified fixed schedule with starting doses and increments preserved across the study groups.
The study included a total of 12 visits (Figure S1: Appendix S1), two of which were inpatient visits (Visit 2 and Visit 10; consisting of 4 days/3 nights). Participants were randomized at Visit 2 to receive danuglipron or matching placebo (Day 1), with completion of dosing at Visit 10 (Day 56). Between study visits, participants continued dosing at home with blinded study intervention provided in blister packs.
Doses were administered in tablet formulation and consisted of three tablets each in the morning and evening, taken with food, approximately 10‐12 h apart and at about the same time each day. Danuglipron dosing coincided with morning and evening meals to standardize the timing of study drug administration across inpatient and outpatient site visits and with self‐administration at home on non‐visit days. However, danuglipron pharmacokinetics show similar plasma exposure values when administered in fed versus fasted states. 7
Mixed‐meal tolerance test (MMTT) assessments were performed at Visit 2 (Day −1) and Visit 10 (Day 56) (Figure S1: Appendix S1) to assess postprandial changes in pharmacodynamic parameters, as described in Section 2.6 below and in Appendix S1.
2.3. Study participants
Eligible participants were Japanese adults aged 20‐70 years, inclusive, with T2DM treated by diet and exercise alone, HbA1c level ≥7% to ≤10.5% at screening, total body weight >50 kg and body mass index (BMI) 22.5‐45.4 kg/m2. Use of other medications for glycaemic control was not permitted in this study. The time frame of restriction of use of antidiabetic medications ranged from 60 days (for oral medications) to 90 days (for injectable medications and thiazolidinediones) before the screening visit and throughout the study participation until the follow‐up visit. See Appendix S1 for additional inclusion and exclusion criteria.
2.4. Safety assessments
The primary objective of the study was to evaluate the safety and tolerability of multiple oral doses of danuglipron in adult Japanese participants with T2DM. Endpoints included the incidence of treatment‐emergent adverse events (TEAEs) [including adverse events (AEs) and serious AEs (SAEs)], clinical safety laboratory measurements, vital signs and 12‐lead electrocardiograms (ECGs). Change from baseline in body weight was assessed as a pre‐specified exploratory endpoint. AEs were summarized per the Medical Dictionary for Regulatory Activities version 23.1.
2.5. Pharmacokinetic assessments
The secondary objective of the study was to characterize the plasma pharmacokinetics of danuglipron in adult Japanese participants with T2DM. Endpoints included area under the plasma concentration‐time curve (AUC) from time 0 to 24 h (AUC24), maximum plasma concentration [Cmax (0‐10 h)], time to Cmax (Tmax) and terminal half‐life (t½). See Appendix S1 for details on pharmacokinetic assays.
2.6. Pharmacodynamic assessments
Exploratory objectives of the study included characterization of the pharmacodynamic effect of danuglipron following multiple oral doses in adult Japanese participants with T2DM. Pre‐specified exploratory endpoints included change from baseline in mean daily glucose (MDG, assessed as glucose AUC24/24 h) following an MMTT, fasting plasma glucose (FPG), HbA1c, fasting plasma insulin, and homeostatic model assessment of insulin resistance (HOMA‐IR) and of β‐cell function (HOMA‐B). Additional endpoints included fasting glucagon, fasting C‐peptide and change from baseline in AUC from time 0 to 4 h (AUC0‐4) for glucose, insulin, glucagon and C‐peptide following an MMTT. After randomization, all pharmacodynamic endpoints were blinded; only glucose levels that met pre‐specified criteria for hypo‐ or hyperglycaemia were unblinded. See Appendix S1 for details on pharmacodynamic assays, including MMTT assessments.
2.7. Statistical analyses
Detailed statistical methods are provided in Appendix S1.
3. RESULTS
3.1. Participant disposition and baseline characteristics
In total, 65 participants were screened for study entry; 37 participants were randomized to receive danuglipron or placebo twice daily (Figure S2: Appendix S1). Ten participants were randomized to receive danuglipron 40 mg, nine participants to danuglipron 80 mg, nine participants to danuglipron 120 mg and nine participants to placebo. All 37 participants completed the study. Eight participants discontinued from the study drug: six participants in danuglipron dose groups discontinued treatment due to AEs (discussed below); one in the placebo group discontinued treatment because of “lack of efficacy”; and one participant in the danuglipron 40‐mg group was randomized inappropriately (participant did not meet eligibility criteria) and was discontinued from treatment.
Baseline demographic and clinical characteristics are presented in Table 1 and were generally similar across treatment groups, with some variability noted across treatment groups in duration of T2DM, body weight and BMI.
TABLE 1.
Key baseline demographic and clinical characteristics of the study participants
| Parameter | All participants (N = 37) | Placebo (N = 9) | Danuglipron dose | ||
|---|---|---|---|---|---|
| 40 mg BID (N = 10) | 80 mg BID (N = 9) | 120 mg BID (N = 9) | |||
| Age (years) | 55.8 (8.6) | 58.6 (8.8) | 55.9 (10.0) | 58.0 (6.7) | 50.7 (7.5) |
| Sex | |||||
| Male | 32 (86.5) | 8 (88.9) | 8 (80.0) | 8 (88.9) | 8 (88.9) |
| Female | 5 (13.5) | 1 (11.1) | 2 (20.0) | 1 (11.1) | 1 (11.1) |
| Asian race | 37 (100.0) | 9 (100.0) | 10 (100.0) | 9 (100.0) | 9 (100.0) |
| T2DM duration (years) | 5.7 (5.5) | 5.5 (4.1) | 2.9 (2.2) | 9.1 (5.8) | 5.7 (7.7) |
| MDG (mg/dl) | 220.8 (54.3) | 218.8 (59.8) | 223.4 (50.5) | 240.6 (52.3) | 200.0 (56.3) |
| FPG (mg/dl) | 173.9 (36.1) | 182.9 (36.3) | 173.3 (29.7) | 175.7 (32.7) | 163.7 (47.5) |
| HbA1c (%) | 8.3 (1.1) | 8.3 (1.2) | 8.2 (1.1) | 8.6 (1.0) | 8.4 (1.2) |
| Body weight (kg) | 78.6 (11.1) | 73.3 (9.9) | 79.7 (13.6) | 79.6 (10.0) | 81.5 (10.4) |
| BMI (kg/m2) | 27.8 (3.6) | 25.9 (2.7) | 28.6 (4.1) | 28.2 (3.6) | 28.6 (3.4) |
Note: data are n (%) or mean (SD). Baselines for body weight, FPG and HbA1c were defined as the measurement at Day −1, 0 h. Baseline for MDG was defined as glucose AUC24/24 h on Day −1.
Abbreviations: BID, twice daily; BMI, body mass index; FPG, fasting plasma glucose; HbA1c, glycated haemoglobin; MDG, mean daily glucose; SD, standard deviation; T2DM, type 2 diabetes mellitus.
3.2. Safety and tolerability
Seventy all‐causality TEAEs were reported by 28 participants (75.7%); of these, 54 AEs (54 of 70; 77.1%) reported by 24 participants (64.9%) were considered treatment‐related (Table S1: Appendix S1). The number of TEAEs (all‐causality and treatment‐related) was higher in the danuglipron groups compared with the placebo group but did not appear to increase with increasing dose. There were no deaths or SAEs during the study. Six participants (16.2%) permanently discontinued from the study drug due to TEAEs. All treatment discontinuations occurred in danuglipron groups and were due to treatment‐related AEs of nausea (two participants in the 40‐mg group), vomiting (two participants in the 80‐mg group) and abdominal discomfort (one participant each in the 80‐ and 120‐mg groups). Two participants had temporary discontinuations from the study drug because of TEAEs: one participant (80‐mg group) experienced treatment‐related nausea; the other participant (40‐mg group) experienced two AEs of increased alanine or aspartate aminotransferase levels that were not considered treatment‐related. These AEs resolved and permitted continuation with study medication.
All but one of the TEAEs reported during the study were mild (n = 36) or moderate (n = 33) in severity. One participant (40‐mg group) experienced a severe TEAE of increased alanine aminotransferase level, which resolved on Day 36 and was not considered treatment‐related; the study drug was temporarily discontinued because of this TEAE, as noted above.
The most frequently reported all‐causality TEAEs were related to gastrointestinal disorders (reported by 67.6% of participants), most commonly nausea (48.6%), vomiting (43.2%), abdominal discomfort (35.1%) and diarrhoea (10.8%), and to nervous system disorders (reported by 13.5% of participants), most commonly headache (10.8%) (Table 2). The incidence of TEAEs related to gastrointestinal disorders was higher in the danuglipron groups compared with the placebo group but did not appear to increase in a dose‐dependent manner. Time courses of the incidence of nausea, vomiting and diarrhoea TEAEs over the duration of the study are shown in Figure S3: Appendix S1. The incidence of these TEAEs varied over time according to the target dose of danuglipron and the associated dose titration scheme. With the exception of nausea in the danuglipron 80‐mg group, the incidence of gastrointestinal TEAEs tended to decrease or stabilize after the first ~3‐4 weeks of dosing, and incidence rates varied markedly between danuglipron dose groups. All TEAEs of nausea, vomiting, abdominal discomfort, diarrhoea and decreased appetite in the danuglipron dose groups were considered treatment‐related, whereas TEAEs of headache and dental caries were considered not treatment‐related. One participant in the danuglipron 40‐mg group experienced a probable event of symptomatic hypoglycaemia (blood glucose was not measured) of mild severity, which was considered treatment‐related; symptoms resolved on the day of onset following treatment with oral carbohydrates. No other hypoglycaemic events were reported in the study.
TABLE 2.
All‐causality treatment‐emergent adverse events by preferred term following twice‐daily oral dosing of danuglipron a
| Preferred term | All participants (N = 37) | Placebo (N = 9) | Danuglipron dose | ||
|---|---|---|---|---|---|
| 40 mg BID (N = 10) | 80 mg BID (N = 9) | 120 mg BID (N = 9) | |||
| Nausea | 18 (48.6) | 0 | 6 (60.0) | 8 (88.9) | 4 (44.4) |
| Vomiting | 16 (43.2) | 0 | 5 (50.0) | 6 (66.7) | 5 (55.6) |
| Abdominal discomfort | 13 (35.1) | 0 | 4 (40.0) | 4 (44.4) | 5 (55.6) |
| Diarrhoea | 4 (10.8) | 0 | 1 (10.0) | 2 (22.2) | 1 (11.1) |
| Headache | 4 (10.8) | 1 (11.1) | 2 (20.0) | 1 (11.1) | 0 |
| Decreased appetite | 2 (5.4) | 0 | 0 | 1 (11.1) | 1 (11.1) |
| Dental caries | 2 (5.4) | 1 (11.1) | 0 | 0 | 1 (11.1) |
Note: data are n (%). Participants were counted only once per treatment per event.
Abbreviation: BID, twice daily.
Treatment‐emergent adverse events with ≥2 occurrences.
There were no apparent dose‐related increases in the frequency of laboratory, vital sign or ECG abnormalities by categorical analysis. A trend for increases in mean time‐matched double differences in systolic blood pressure (SBP) or diastolic blood pressure (DBP) over the course of the day was observed on Day 56 in the danuglipron 80‐ and 120‐mg groups, relative to the placebo and danuglipron 40‐mg groups; no apparent increases were observed in the danuglipron 40‐mg group relative to placebo (Figure S4A,B: Appendix S1). Mean SBP and DBP values for all dose groups were in the normal range. A trend for increases in mean time‐matched double differences in pulse rate and heart rate by ECG over the course of the day was observed on Day 56 in all danuglipron dose groups compared with placebo, with a greater observed increase in the danuglipron 120‐mg group compared with other groups (Figure S4C,D: Appendix S1). Mean pulse rate and heart rate values for all dose groups were generally in the normal range. No TEAEs related to vital sign or ECG abnormalities were reported, and there were no occurrences of heart rates >120 bpm. Compared with placebo, there were no dose‐related trends in mean time‐matched double differences from baseline in the ECG parameters of PR, QRS, QT or QTcF intervals over 8 weeks, and no ECG findings were reported as clinically meaningful by the investigator.
3.3. Pharmacodynamics
3.3.1. Mean daily glucose
Statistically significant reductions from baseline in MDG were observed on Day 56 in all danuglipron groups compared with placebo (Table 3; Figure 1A). Modelled least squares (LS)‐mean [90% confidence interval (CI)] placebo‐adjusted changes from baseline to Day 56 in MDG were similar across the danuglipron 40‐, 80‐ and 120‐mg groups: −67.89 (−88.98, −46.79), −64.82 (−87.66, −41.98) and −67.29 (−88.22, −46.36) mg/dl, respectively.
TABLE 3.
Summary of key pharmacodynamic biomarkers at baseline and at 8 weeks a following twice‐daily oral dosing of danuglipron
| Biomarker | Placebo (N = 9) | Danuglipron dose | ||
|---|---|---|---|---|
| 40 mg BID (N = 10) | 80 mg BID (N = 9) | 120 mg BID (N = 9) | ||
| MDG (mg/dl) | ||||
| Absolute mean (SD) at baseline | 218.8 (59.8) | 223.4 (50.5) | 240.6 (52.3) | 200.0 (56.3) |
| Absolute mean (SD) at Day 56 | 195.5 (42.0) | 138.8 (19.2) | 152.7 (49.7) | 120.4 (24.0) |
| Absolute mean (SD) CFB | −8.34 (10.34) | −87.60 (44.27) | −95.55 (28.45) | −67.64 (48.83) |
| LS‐mean (90% CI) CFB b | −13.98 (−28.29, 0.32) | −81.87 (−97.15, −66.59) | −78.80 (−96.05, −61.55) | −81.27 (−97.07, −65.48) |
| LS‐mean (90% CI) placebo‐adjusted CFB b | – | −67.89 (−88.98, −46.79) | −64.82 (−87.66, −41.98) | −67.29 (−88.22, −46.36) |
| FPG (mg/dl) | ||||
| Absolute mean (SD) at baseline | 182.9 (36.3) | 173.3 (29.7) | 175.7 (32.7) | 163.7 (47.5) |
| Absolute mean (SD) at Day 57 | 163.6 (29.0) | 137.3 (18.2) | 130.5 (22.3) | 112.7 (18.7) |
| Absolute mean (SD) CFB | −11.88 (6.81) | −39.71 (14.74) | −46.17 (24.63) | −41.86 (36.22) |
| LS‐mean (90% CI) CFB c | −9.26 (−18.16, −0.37) | −36.51 (−45.60, −27.42) | −46.04 (−55.47, −36.62) | −50.14 (−59.35, −40.92) |
| LS‐mean (90% CI) placebo‐adjusted CFB c | – | −27.25 (−39.95, −14.55) | −36.78 (−49.72, −23.85) | −40.87 (−53.77, −27.98) |
| HbA1c (%) | ||||
| Absolute mean (SD) at baseline | 8.3 (1.2) | 8.2 (1.1) | 8.6 (1.0) | 8.4 (1.2) |
| Absolute mean (SD) at Day 57 | 7.8 (0.9) | 6.8 (0.7) | 7.3 (1.5) | 6.6 (0.4) |
| Absolute mean (SD) CFB | −0.18 (0.21) | −1.29 (0.58) | −1.40 (0.89) | −1.60 (1.15) |
| LS‐mean (90% CI) CFB c | −0.14 (−0.56, 0.28) | −1.34 (−1.76, −0.92) | −1.00 (−1.42, −0.57) | −1.55 (−1.97, −1.13) |
| LS‐mean (90% CI) placebo‐adjusted CFB c | – | −1.20 (−1.80, −0.61) | −0.86 (−1.46, −0.26) | −1.41 (−2.01, −0.82) |
| Body weight (kg) | ||||
| Absolute mean (SD) at baseline | 73.3 (9.9) | 79.7 (13.6) | 79.6 (10.0) | 81.5 (10.4) |
| Absolute mean (SD) at Day 57 | 74.1 (8.8) | 74.4 (12.1) | 78.4 (8.6) | 77.0 (9.3) |
| Absolute mean (SD) CFB | −0.93 (0.94) | −2.47 (1.37) | −3.68 (2.26) | −3.26 (2.95) |
| LS‐mean (90% CI) CFB c | −1.21 (−2.36, −0.05) | −2.61 (−3.77, −1.45) | −3.08 (−4.30, −1.86) | −2.87 (−3.98, −1.75) |
| LS‐mean (90% CI) placebo‐adjusted CFB c | – | −1.40 (−3.03, 0.23) | −1.87 (−3.58, −0.17) | −1.66 (−3.28, −0.04) |
Note: data are absolute mean (SD) or LS‐mean (90% CI) as indicated. Baseline for MDG was defined as glucose AUC24/24 h on Day −1. Baselines for FPG, HbA1c and body weight were defined as the measurement at Day −1, 0 h. For calculation of MDG, for glucose at 24 h on Day –1 and 24 h on Day 56, glucose at 0 h on Day –1 and 0 h on Day 57 were used from the same participant, respectively.
Abbreviations: ANCOVA, analysis of covariance; BID, twice daily; CFB, change from baseline; CI, confidence interval; FPG, fasting plasma glucose; HbA1c, glycated haemoglobin; LS, least squares; MDG, mean daily glucose; MMRM, mixed model repeated measures; SD, standard deviation.
Day 56 for MDG; Day 57 for FPG, HbA1c and body weight.
From an ANCOVA model that included the change from baseline in MDG on Day 56 as the dependent variable, treatment as a fixed effect and baseline MDG as a covariate.
From an MMRM analysis that included treatment, baseline value, day, the baseline‐by‐day interaction and the day‐by‐treatment interaction, with day fitted as a repeated effect and participant as a random effect.
FIGURE 1.
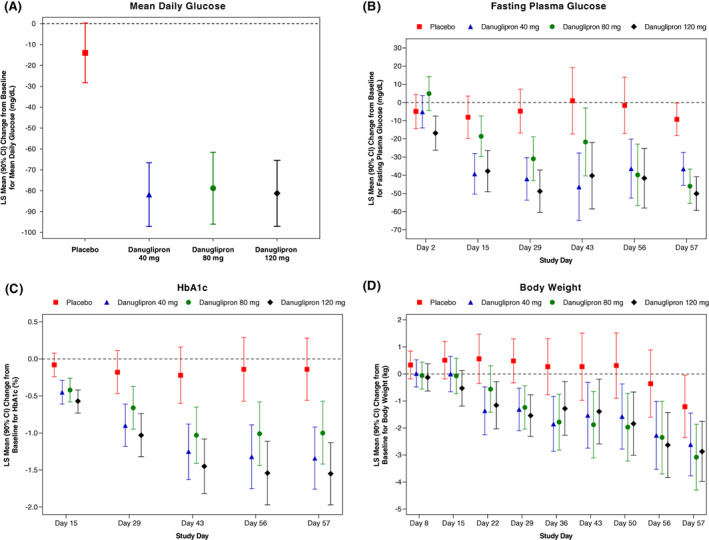
LS‐mean change from baseline in A, MDG at Day 56, and B, FPG; C, HbA1c; and D, body weight over time, following twice‐daily oral dosing of danuglipron. Dose escalation, as shown in Figure S1 (Appendix S1), was utilized to achieve danuglipron target doses of 40, 80 and 120 mg twice daily. A, MDG was defined as glucose AUC24/24 h. For glucose at 24 h on Day −1 and 24 h on Day 56, glucose at 0 h on Day −1 and 0 h on Day 57 were used from the same participant, respectively. Values are LS‐mean (90% CI). From an ANCOVA model that included the change from baseline in MDG on Day 56 as the dependent variable, treatment as a fixed effect, and baseline MDG as a covariate. B‐D, Values are LS‐mean (90% CI). From an MMRM analysis that included treatment, baseline value, day, the baseline‐by‐day interaction, and the day‐by‐treatment interaction, with day fitted as a repeated effect and participant as a random effect. AUC24, area under the plasma concentration‐time curve from time 0 to 24 h; ANCOVA, analysis of covariance; CI, confidence interval; FPG, fasting plasma glucose; HbA1c, glycated haemoglobin; LS, least squares; MDG, mean daily glucose; MMRM, mixed model repeated measures
3.3.2. Fasting plasma glucose
Statistically significant reductions from baseline in FPG were observed on Day 57 in all danuglipron groups compared with placebo (Table 3; Figure 1B). Modelled LS‐mean (90% CI) placebo‐adjusted changes from baseline to Day 57 in FPG found dose‐proportional reductions of −27.25 (−39.95, −14.55), −36.78 (−49.72, −23.85) and −40.87 (−53.77, −27.98) mg/dl for the danuglipron 40‐, 80‐ and 120‐mg groups, respectively. Reductions in FPG levels were observed across all danuglipron groups relative to placebo from Day 15 through to Day 57. There were no occurrences of FPG ≤70 mg/dl in any of the randomized participants during the study.
3.3.3. Glycated haemoglobin
Statistically significant reductions from baseline on Day 57 were also observed for HbA1c across all danuglipron groups compared with placebo (Table 3; Figure 1C). The modelled LS‐mean (90% CI) placebo‐adjusted reductions from baseline to Day 57 in HbA1c were −1.20% (−1.80%, −0.61%), −0.86% (−1.46%, −0.26%) and −1.41% (−2.01%, −0.82%) across the danuglipron 40‐, 80‐ and 120‐mg groups, respectively. Reductions in HbA1c were observed in all danuglipron dose groups relative to placebo over the course of the study, with reductions generally increasing in magnitude over time up to Day 57.
3.3.4. Body weight
Greater mean reductions from baseline in body weight were observed on Day 57 in all danuglipron groups compared with placebo (Table 3; Figure 1D). Modelled LS‐mean (90% CI) placebo‐adjusted reductions in body weight from baseline to Day 57 were similar across all danuglipron groups and were statistically significant versus placebo for the 80‐ and 120‐mg groups: −1.40 (−3.03, 0.23), −1.87 (−3.58, −0.17) and −1.66 (−3.28, −0.04) kg for the danuglipron 40‐, 80‐ and 120‐mg groups, respectively. Greater mean reductions from baseline in body weight were evident in all danuglipron dose groups compared with placebo from Day 22, with reductions generally increasing in magnitude over time. A higher proportion of participants in the danuglipron groups achieved a weight loss of ≥3% or ≥5% from baseline at Day 57 compared with the placebo group (Table S2: Appendix S1).
3.3.5. Other pharmacodynamic parameters
Results for fasting plasma insulin, fasting glucagon, fasting C‐peptide, HOMA‐IR and HOMA‐B, and for postprandial changes in glucose, insulin, glucagon and C‐peptide, are provided in Appendix S1 (Supplementary Results, Table S3, Figures S5 and S6).
3.4. Pharmacokinetics
Following multiple dosing, danuglipron plasma exposures on Day 56 (steady state) increased in an approximately dose‐proportional manner (Figures S7 and S8: Appendix S1), with dose‐proportional increases in geometric mean AUC24 and Cmax values, and similar dose‐normalized (dn) exposures (AUC24(dn); Cmax(dn)), observed across the dose groups (Table 4). Lower median danuglipron concentrations were observed for the 120‐mg group compared with the 80‐mg group at 24, 36 and 48 h post‐dose on Day 56 (Figure S7: Appendix S1); high interindividual variability at these time points and the small number of participants per cohort most likely contributed to this observation.
TABLE 4.
Descriptive summary of key plasma pharmacokinetic parameters on Day 56 (steady state) following twice‐daily oral dosing of danuglipron
| Parameter | Danuglipron dose | ||
|---|---|---|---|
| 40 mg BID (N = 10) | 80 mg BID (N = 9) | 120 mg BID (N = 9) | |
| N2, N3 | 7, 6 | 6, 3 | 7, 7 |
| AUC24 (ng·h/ml) | 2424 (45) | 4691 (75) | 6953 (148) |
| AUC24(dn) (ng·h/ml/mg) | 30.29 (45) | 29.35 (75) | 28.94 (148) |
| Cmax (ng/ml) | 136.0 (51) | 245.3 (67) | 415.8 (153) |
| Cmax(dn) (ng/ml/mg) | 3.401 (51) | 3.068 (67) | 3.465 (152) |
| Tmax (h) | 6.00 (2.00‐6.00) | 6.00 (2.00‐8.00) | 6.00 (0.000‐10.1) |
| t1/2 (h) | 6.373 ± 1.7404 | 5.543 ± 0.30827 | 5.300 ± 0.80594 |
Note: data are geometric mean (% CV) for all except arithmetic mean ± standard deviation for t1/2 and median (range) for Tmax. N is the total number of participants in the indicated treatment group. N2 is the number of participants contributing to the summary statistics. N3 is the number of participants contributing to the summary statistics for t1/2. If a participant received a dose that was not assigned based on the randomized titration scheme, the data from that day were not included in summary statistics.
Abbreviations: AUC, area under the plasma concentration‐time curve; AUC24, AUC from time 0 to 24 h; BID, twice daily; Cmax, maximum plasma concentration during the dosing interval τ1 = 0‐10 h; CV, coefficient of variation; dn, dose normalized to a 1‐mg dose; t½, terminal half‐life; Tmax, time to Cmax.
Following the morning dose on Day 56, median Cmax was observed within a median Tmax of 6 h post‐dose across all danuglipron doses; mean t½ values on Day 56 ranged from 5.300 to 6.373 h (Table 4). Observed accumulation ratios for AUC24 (Rac) and Cmax (Rac,Cmax) on Day 56 of multiple dosing ranged between 1.202‐1.349 and 1.118‐1.392, respectively, across dose groups (Table S4: Appendix S1), indicating that accumulation was minimal following this dosing regimen.
4. DISCUSSION
This phase 1 study of the novel GLP‐1R agonist danuglipron showed that administration of ascending, multiple oral doses of danuglipron over 56 days was generally safe in adult Japanese participants with T2DM inadequately controlled on diet and exercise, with a tolerability profile consistent with the mechanism of action. The most frequently reported all‐causality TEAEs were nausea, vomiting, abdominal discomfort, diarrhoea and headache, and most AEs were of mild or moderate intensity. Although a trend for increases in SBP, DBP and pulse rate was observed on Day 56 with higher doses of danuglipron relative to placebo, mean SBP, DBP and pulse rate values were generally in the normal range. Pharmacodynamic analyses revealed significant reductions from baseline in glycaemic indices and reductions from baseline in body weight across all danuglipron groups compared with placebo. Analysis of the danuglipron pharmacokinetic profile showed dose‐proportional increases in plasma exposure at steady state (Day 56) following titration to 40‐, 80‐ or 120‐mg doses.
The safety profile of danuglipron observed in this phase 1 study of Japanese participants with T2DM is consistent with previous phase 1 studies of danuglipron conducted in Western participants with or without T2DM. 7 , 8 Previously, in healthy Western participants who received single doses of danuglipron ranging from 3 to 300 mg, 7 or in Western participants with T2DM who received multiple doses of danuglipron ranging from 10 to 200 mg administered twice daily or once daily over 28 days, 8 the most commonly reported TEAEs were related to gastrointestinal disorders (nausea, vomiting, dyspepsia, diarrhoea, constipation and abdominal discomfort or distension), in line with the current study in Japanese participants with T2DM. Gastrointestinal TEAEs were anticipated following multiple dosing of danuglipron because of the mechanism of action of this oral GLP‐1R agonist. Although dose escalation was incorporated into the protocols of both multiple‐dose studies to maximize danuglipron tolerability, the relatively short treatment durations of the studies (28 days in Western participants 8 ; 56 days in Japanese participants) dictated a rapid dose‐escalation schedule to achieve the higher doses. More gradual dose‐escalation schemes over longer treatment durations are anticipated to result in lower incidence rates of gastrointestinal AEs. 9 However, both the overall and gastrointestinal safety profiles of danuglipron are generally consistent with those described for peptidic GLP‐1R agonists, 10 , 11 including the recent oral formulation of semaglutide. 12 , 13 , 14 , 15
In this study, increases in heart rate were observed in danuglipron dose groups after 8 weeks of dosing, which was observed in the previous study in Western participants 8 and in other studies with peptidic GLP‐1R agonists. 16 The magnitude of heart‐rate increases observed in this study is in line with short‐term clinical data published with peptidic GLP‐1R agonists. 17 , 18 A trend toward mild increases in blood pressure was noted on Day 56 with the 80‐ and 120‐mg doses of danuglipron relative to the 40‐mg dose and placebo, with mean blood pressure in the normal range. This pattern was not observed in the previous study in Western participants, 8 where there was a trend for reductions in SBP with higher doses of danuglipron. However, mild increases in blood pressure have been observed in short‐term studies with peptidic GLP‐1R agonists, 18 which have subsequently showed benefits in cardiovascular outcomes. 19
Substantial reductions from baseline in glycaemic biomarkers and body weight with danuglipron versus placebo have been observed in Western participants with T2DM following 28 days of twice daily or once daily dosing. 8 This current study shows that danuglipron has similar robust effects on glycaemic parameters in Japanese participants over 56 days. This study was also the first to show sustained glycaemic efficacy with danuglipron following periods of home self‐administration in between study site visits. Relative to placebo, significant declines in MDG of up to −67.9 mg/dl from baseline were observed in all danuglipron groups on Day 56, consistent with the placebo‐adjusted reductions in MDG of up to −65.5 mg/dl with the danuglipron 120‐mg twice daily dose observed on Day 28 in Western participants with T2DM. 8 Substantial reductions in FPG, observed from Day 15, were maintained through to study end (up to −40.9 mg/dl with danuglipron 120 mg twice daily on Day 57) and were in line with the reductions in FPG seen in Western participants (up to −44.3 mg/dl with the danuglipron 120‐mg twice daily dose on Day 28). 8 Consistent with the mechanism of action, there were no occurrences of FPG levels ≤70 mg/dl during the study, and only one hypoglycaemic AE of mild severity (danuglipron 40‐mg group) was reported as probable symptomatic hypoglycaemia.
The observed reductions in glucose levels with danuglipron treatment were reflected in changes from baseline in HbA1c, where statistically significant reductions versus placebo were observed from Day 15 through to Day 57 across all danuglipron groups. Placebo‐adjusted reductions in HbA1c of up to −1.4% (danuglipron 120‐mg group) were observed on Day 57. Administration of stable danuglipron doses over a longer treatment period may achieve even greater reductions in HbA1c, providing patients with a clinically meaningful glycaemic benefit over the longer term.
Compared with the large declines in body weight observed following danuglipron treatment in Western participants with T2DM, where placebo‐adjusted reductions of up to −5.5 kg were observed with the danuglipron 120‐mg dose on Day 28, 8 the reductions in body weight in Japanese participants with T2DM observed in this study were lower by comparison (up to −1.9 kg). Differences in the magnitude of weight reductions between Western and Japanese study populations may be due to differences in baseline body weight and BMI between the two groups (92.4 kg and 32.9 kg/m2, respectively, in Western participants 8 ; 78.6 kg and 27.8 kg/m2, respectively, in Japanese participants). Higher baseline BMI has been associated with greater degrees of weight loss in trials with peptidic GLP‐1R agonists. 20 , 21 , 22
In previous phase 1 studies in Western participants, 7 , 8 danuglipron plasma exposure increased in a dose‐proportional manner, with median Tmax values of 2‐6 h following a single dose or multiple twice‐daily dosing; the mean t½ values were 4.3‐6.1 h (single dose) 7 and 4.7‐8.1 h (multiple dosing). 8 This is in line with the current study of multiple twice‐daily dosing in Japanese participants, where danuglipron plasma exposure at steady state increased in an approximately dose‐proportional manner, with a median Tmax following the morning dose of 6 h, and mean t½ of 5.3‐6.4 h. Steady‐state AUC24(dn) and Cmax values were also broadly comparable following multiple dosing in Western 8 and Japanese participants. Administration of danuglipron occurred in the fed state in this study; however, a recent assessment of the effect of food on danuglipron pharmacokinetics revealed similar plasma exposure and t½ values when danuglipron was administered in the fed versus fasted state, 7 indicating that danuglipron can be dosed without regard to food. In contrast, the oral formulation of the peptidic GLP‐1R agonist semaglutide has fasting requirements because minimal to no measurable systemic exposure is observed when semaglutide is administered in the fed state. 23
Limitations of this phase 1 study include assessment of safety and pharmacodynamic endpoints in a relatively small number of participants, and evaluation of glucose lowering and weight loss over a short treatment duration, which required rapid dose escalation. In addition, no placebo run‐in period was incorporated because of the two inpatient stays and frequent study visits involved in the study, which may explain the mild declines in pharmacodynamic biomarkers observed in the placebo group.
5. CONCLUSIONS
In Japanese participants with T2DM, multiple doses of the novel, oral small‐molecule GLP‐1R agonist danuglipron showed a favourable safety profile that was consistent with phase 1 studies of danuglipron in Western participants 7 , 8 and with the peptidic GLP‐1R agonist class overall. 10 , 11 , 15 Approximately dose‐proportional increases in danuglipron plasma exposure were observed at steady state. Danuglipron robustly reduced plasma glucose levels, HbA1c and body weight after 8 weeks of dosing in Japanese adults with T2DM. The overall profile of danuglipron in this cohort supports further clinical development in Japanese patients with T2DM.
AUTHOR CONTRIBUTIONS
All authors contributed to the design of the study and the analysis/interpretation of the data. KF also contributed to the conduct of the study and the collection of study data. All authors contributed to the writing of the manuscript and approved the final version of the manuscript for submission.
CONFLICT OF INTEREST
KF served as a Principal Investigator for this Pfizer‐sponsored study, with funding support directed to his institution. All other authors are employees of Pfizer and may own shares/stock options in Pfizer.
PRIOR PRESENTATION
These data were presented, in part, at the American Diabetes Association 82nd Scientific Sessions, 3‐7 June 2022, New Orleans, LA, USA (oral presentation 339‐OR).
Supporting information
Appendix S1. Supporting information.
ACKNOWLEDGMENTS
Medical writing support was provided by Shirley Smith, PhD, of Engage Scientific Solutions (Horsham, UK) and was funded by Pfizer.
Ono R, Furihata K, Ichikawa Y, et al. A phase 1 study to evaluate the safety, tolerability, pharmacokinetics and pharmacodynamics of danuglipron (PF‐06882961), an oral small‐molecule glucagon‐like peptide‐1 receptor agonist, in Japanese adults with type 2 diabetes mellitus. Diabetes Obes Metab. 2023;25(3):805‐814. doi: 10.1111/dom.14928
Funding information Pfizer
DATA AVAILABILITY STATEMENT
Upon request, and subject to review, Pfizer will provide the data that support the findings of this study. Subject to certain criteria, conditions and exceptions, Pfizer may also provide access to the related individual de‐identified participant data. See https://www.pfizer.com/science/clinical-trials/trial-data-and-results for more information.
REFERENCES
- 1. International Diabetes Federation . IDF Diabetes Atlas 10th Edition 2021. https://diabetesatlas.org/data/en/. Accessed July 19, 2022.
- 2. Yokoyama H, Oishi M, Takamura H, et al. Large‐scale survey of rates of achieving targets for blood glucose, blood pressure, and lipids and prevalence of complications in type 2 diabetes (JDDM 40). BMJ Open Diabetes Res Care. 2016;4:e000294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Takahara M, Mita T, Katakami N, et al. Three‐year glycaemic control and management in patients with type 2 diabetes initiating second‐line treatment in Japan: a prospective observational study, J‐DISCOVER. Diabetes Ther. 2022;13:251‐264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Araki E, Goto A, Kondo T, et al. Japanese clinical practice guideline for diabetes 2019. Diabetol Int. 2020;11:165‐223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Morita Y, Murayama H, Odawara M, Bauer M. Treatment patterns of drug‐naive patients with type 2 diabetes mellitus: a retrospective cohort study using a Japanese hospital database. Diabetol Metab Syndr. 2019;11:90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Igarashi A, Bekker Hansen B, Langer J, et al. Preference for oral and injectable GLP‐1 RA therapy profiles in Japanese patients with type 2 diabetes: a discrete choice experiment. Adv Ther. 2021;38:721‐738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Griffith DA, Edmonds DJ, Fortin J‐P, et al. A small‐molecule oral agonist of the human glucagon‐like peptide‐1 receptor. J Med Chem. 2022;65:8208‐8226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Saxena AR, Gorman DN, Esquejo RM, et al. Danuglipron (PF‐06882961) in type 2 diabetes: a randomized, placebo‐controlled, multiple ascending‐dose phase 1 trial. Nat Med. 2021;27:1079‐1087. [DOI] [PubMed] [Google Scholar]
- 9. Drucker DJ, Habener JF, Holst JJ. Discovery, characterization, and clinical development of the glucagon‐like peptides. J Clin Invest. 2017;127:4217‐4227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bain EK, Bain SC. Recent developments in GLP‐1RA therapy: a review of the latest evidence of efficacy and safety and differences within the class. Diabetes Obes Metab. 2021;23:30‐39. [DOI] [PubMed] [Google Scholar]
- 11. De Block CEM, Dirinck E, Verhaegen A, Van Gaal LF. Efficacy and safety of high‐dose glucagon‐like peptide‐1, glucagon‐like peptide‐1/glucose‐dependent insulinotropic peptide, and glucagon‐like peptide‐1/glucagon receptor agonists in type 2 diabetes. Diabetes Obes Metab. 2022;24:788‐805. [DOI] [PubMed] [Google Scholar]
- 12. Yabe D, Nakamura J, Kaneto H, et al. Safety and efficacy of oral semaglutide versus dulaglutide in Japanese patients with type 2 diabetes (PIONEER 10): an open‐label, randomised, active‐controlled, phase 3a trial. Lancet Diabetes Endocrinol. 2020;8:392‐406. [DOI] [PubMed] [Google Scholar]
- 13. Yamada Y, Katagiri H, Hamamoto Y, et al. Dose‐response, efficacy, and safety of oral semaglutide monotherapy in Japanese patients with type 2 diabetes (PIONEER 9): a 52‐week, phase 2/3a, randomised, controlled trial. Lancet Diabetes Endocrinol. 2020;8:377‐391. [DOI] [PubMed] [Google Scholar]
- 14. Araki E, Terauchi Y, Watada H, et al. Efficacy and safety of oral semaglutide in Japanese patients with type 2 diabetes: a post hoc subgroup analysis of the PIONEER 1, 3, 4 and 8 trials. Diabetes Obes Metab. 2021;23:2785‐2794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Niman S, Hardy J, Goldfaden RF, et al. A review on the efficacy and safety of oral semaglutide. Drugs R D. 2021;21:133‐148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Drucker DJ. The cardiovascular biology of glucagon‐like peptide‐1. Cell Metab. 2016;24:15‐30. [DOI] [PubMed] [Google Scholar]
- 17. Coskun T, Sloop KW, Loghin C, et al. LY3298176, a novel dual GIP and GLP‐1 receptor agonist for the treatment of type 2 diabetes mellitus: from discovery to clinical proof of concept. Mol Metab. 2018;18:3‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lovshin JA, Barnie A, DeAlmeida A, Logan A, Zinman B, Drucker DJ. Liraglutide promotes natriuresis but does not increase circulating levels of atrial natriuretic peptide in hypertensive subjects with type 2 diabetes. Diabetes Care. 2015;38:132‐139. [DOI] [PubMed] [Google Scholar]
- 19. Marso SP, Daniels GH, Brown‐Frandsen K, et al. Liraglutide and cardiovascular outcomes in type 2 diabetes. N Engl J Med. 2016;375:311‐322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Niswender K, Pi‐Sunyer X, Buse J, et al. Weight change with liraglutide and comparator therapies: an analysis of seven phase 3 trials from the liraglutide diabetes development programme. Diabetes Obes Metab. 2013;15:42‐54. [DOI] [PubMed] [Google Scholar]
- 21. Ahren B, Atkin SL, Charpentier G, et al. Semaglutide induces weight loss in subjects with type 2 diabetes regardless of baseline BMI or gastrointestinal adverse events in the SUSTAIN 1 to 5 trials. Diabetes Obes Metab. 2018;20:2210‐2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pratley RE, Aroda VR, Catarig AM, et al. Impact of patient characteristics on efficacy and safety of once‐weekly semaglutide versus dulaglutide: SUSTAIN 7 post hoc analyses. BMJ Open. 2020;10:e037883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Buckley ST, Baekdal TA, Vegge A, et al. Transcellular stomach absorption of a derivatized glucagon‐like peptide‐1 receptor agonist. Sci Transl Med. 2018;10:eaar7047. doi: 10.1126/scitranslmed.aar7047. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1. Supporting information.
Data Availability Statement
Upon request, and subject to review, Pfizer will provide the data that support the findings of this study. Subject to certain criteria, conditions and exceptions, Pfizer may also provide access to the related individual de‐identified participant data. See https://www.pfizer.com/science/clinical-trials/trial-data-and-results for more information.