

Abstract
Efficient protocols for accessing iodo‐substituted diaryl and aryl(vinyl) sulfides have been developed using iodonium salts as reactive electrophilic arylation and vinylation reagents. The reactions take place under transition‐metal‐free conditions, employing odorless and convenient sulfur reagents. A wide variety of functional groups are tolerated in the S‐diarylation, enabling the regioselective late‐stage application of several heterocycles and drug molecules under mild reaction conditions. A novel S‐difunctionalization pathway was discovered using vinyliodonium salts, which proceeds under additive‐free reaction conditions and grants excellent stereoselectivity in the synthesis of aryl(vinyl) sulfides. A one‐pot strategy combining transition‐metal‐free diarylation and subsequent reduction provided facile access to electron‐rich thioanilines and a direct synthesis of a potential drug candidate derivative. The retained iodo group allows a wide array of further synthetic transformations. Mechanistic insights were elucidated by isolating the key intermediate, and the relevant energy profile was substantiated by DFT calculations.
Keywords: Aromatic Substitution, Difunctionalization, Hypervalent Compounds, Iodonium Salts, Sulfides
A transition‐metal‐free difunctionalization of bench‐stable sulfur salts gives facile access to iodo‐substituted sulfides using either diaryliodonium or vinyliodonium salts. The methodology was extended to one‐pot syntheses of electron‐rich thioanilines and efficient late‐stage transformations of medicinally important compounds.

Introduction
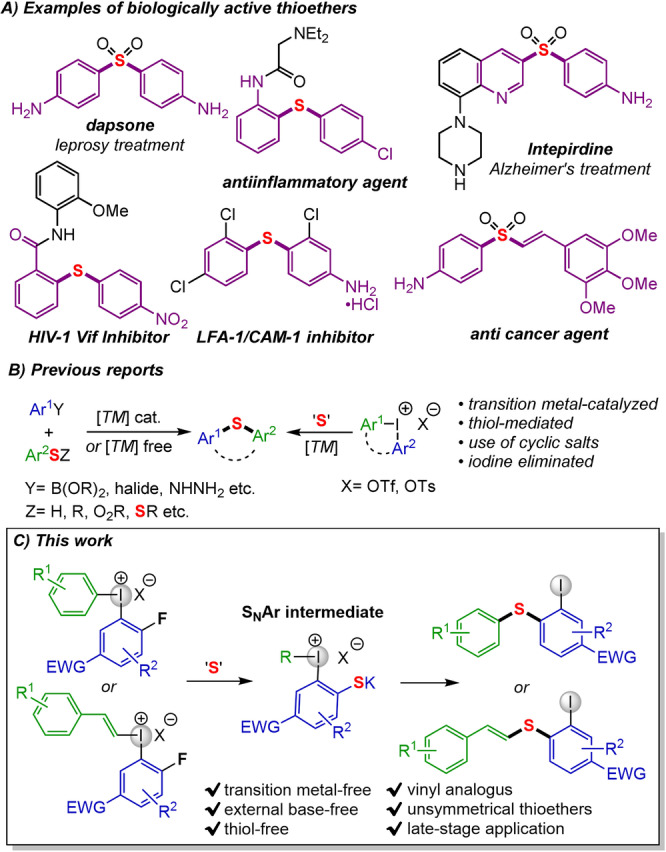
Sulfur based organic molecules have always been a prime focus for synthetic chemists, owing to their variable redox states that allow construction of several classes of organosulfur compounds. [1] Their prevalent role in biological metabolic processes makes them essential for living organisms spanning from marine to terrestrial systems. [2] Sulfide moieties make up a prominent section of building blocks and versatile intermediates, which allows for widespread applications in synthetic chemistry, material science, pharmaceutical industry and biological applications (Scheme 1A). [3] Sulfides and their derivatives can be found as one of the leading series of pharmaceutically active compounds among FDA listed 362 sulfur‐containing drugs. [4] For instance, dapsone, a well‐known antibiotic used for the treatment of leprosy, contains a sulfone scaffold. Several nitro and amino‐incorporated sulfides are reported as biologically active moieties widely used in the treatment of inflammation, Alzheimer's, HIV, breast cancer, malarial diseases and fungal related conditions.[ 4b , 5 ] Similarly, vinyl sulfones play a crucial role in several classes of drugs for cancer treatment. [6]
Scheme 1.
Difunctionalization of sulfur nucleophiles.
Synthetic routes to diaryl sulfides have been investigated thoroughly, [7] and most commonly involve a transition metal‐catalyzed cross‐coupling between thiophenols or other organosulfur reagents and aryl halides or arylboronic acids.
These methods often require ligands, a number of additives and harsh conditions (Scheme 1B). [8] The use of hazardous thiophenols as the nucleophile has several disadvantages, including unpleasant smell and oxidation sensitivity, which leads to undesired side products as well as causing irritation to skin and permanent organ damage. [9] Meanwhile, transition metal‐free methodologies often require highly functionalized substrates such as aryne precursors or disulfides, or employ harsh conditions, delivering a limited scope of sulfide products. [10]
Diaryliodonium salts (Ar2IX) are emerging electrophilic aryl sources in organic synthesis due to their high electrophilicity, ease of synthesis and air‐moisture stability. [11] These reagents enable application of user‐friendly, odorless, cheap and stable sulfur reagents in the synthesis of cyclic and acyclic sulfides. [12] Arylations with diaryliodonium salts generally suffer from poor atom economy as stoichiometric aryl iodide (ArI) is wasted. This drawback is avoided in reactions with cyclic Ar2IX, although the scope is limited with such methods.[ 12b , 13 ] Recent progress includes transition metal‐catalyzed one‐pot methods where both aryl groups of acyclic diaryliodonium salts are transferred through in situ cross‐coupling with the formed ArI. [14] Jiang and co‐workers reported such a copper‐catalyzed S‐diarylation with potassium thioacetate. [12c] Recently, the Olofsson group developed a sustainable and transition‐metal free approach for diarylation of N‐ and O‐nucleophiles using certain diaryliodonium salts under basic conditions, where both aryl groups and the iodide were retained in the product. [15] The diarylation was suggested to proceed through nucleophilic aromatic substitution (SNAr) followed by intramolecular aryl transfer to afford the product.
In this paper, abundant sulfur‐based nucleophiles are employed in atom‐efficient and thiol‐free difunctionalization reactions using iodonium salts with a strong electron‐withdrawing substituent (EWG) under transition metal‐free reaction conditions (Scheme 1C). The methodology gives facile access to unsymmetrical, difunctionalized aryl sulfides with an iodide handle that is accessible for further derivatizations, and demonstrates novel reactivity of vinyliodonium salts.
Results and Discussion
Based on previous work from our group and literature precedence,[ 15 , 16 ] we commenced our initial investigation using iodonium salt 1 a, molecular sulfur (2 a), and K2CO3 as a base in DMSO at 80 °C. The desired product 3 a was not detected, but the diaryl ether 3 a′ was observed instead (Table 1, entry 1). [17] Changing the sulfur source to thiourea 2 b [18] allowed the formation of 3 a in 26 % yield (entry 2). The more nucleophilic sulfur reagents potassium thioacetate 2 c and potassium ethyl xanthogenate 2 d gave significantly increased reactivity already at room temperature. Furthermore, the formation of 3 a′ was entirely suppressed and external base was no longer necessary (entries 3–5). Other polar solvents proved to be effective for this transformation (entries 6,7), and significantly improved yields were obtained in reactions performed in the absence of light (entries 8–10). [19] Upon increased reaction scale, sulfide 3 a was isolated in excellent yield at 40 °C in DMF (entry 11), whereas surprisingly poor results were obtained in acetone (entry 12). [17]
Table 1.
Optimization of the reaction conditions.[a]
|
| ||||||
|---|---|---|---|---|---|---|
|
entry |
“S” 2 |
base |
solvent |
T [°C] |
yield 3 a [%][b] |
yield 3 a′ [%][b] |
|
1 |
2 a |
K2CO3 |
DMSO |
80 |
n.d. |
11 |
|
2 |
2 b |
K2CO3 |
DMSO |
80 |
26 |
n.d. |
|
3 |
2 c |
K2CO3 |
DMSO |
rt |
16 |
n.d. |
|
4 |
2 c |
– |
DMSO |
rt |
22 |
n.d. |
|
5 |
2 d |
– |
DMSO |
rt |
52 |
n.d. |
|
6 |
2 d |
– |
DMF |
rt |
47 |
n.d. |
|
7 |
2 d |
– |
acetone |
rt |
44 |
n.d. |
|
8[c] |
2 d |
– |
DMF |
rt |
99 |
n.d. |
|
9[c] |
2 d |
– |
acetone |
rt |
76 |
n.d. |
|
10[c] |
2 d |
– |
DMSO |
rt |
75 |
n.d. |
|
11 [c,d] |
2 d |
– |
DMF |
40 |
93[e] |
n.d. |
|
12[c,d] |
2 d |
– |
acetone |
40 |
25 |
n.d. |

[a] Reaction conditions: 1 a (0.05 mmol), 2 (0.10 mmol), base (2.0 equiv), solvent (0.1 M) for 15–18 h. [b] 1H NMR yields using 1,3,5‐trimethoxybenzene (TMB) as internal standard. [c] Reactions carried out in the absence of light. [d] Reaction conditions: 1 a (0.2 mmol), 2 (0.28 mmol), solvent (0.2 M). [e] Isolated yield.
Next, we investigated the generality of our protocol using the conditions in entry 11 (Scheme 2A). Diaryliodonium salts 1 bearing alkyl groups in the ring displayed excellent reactivity, producing diaryl sulfides 3 b–3 d in high yields. Haloarenes are primary precursors for metal‐catalyzed synthesis of sulfides, which limits the synthesis of the corresponding halide‐substituted derivatives. [20] Importantly, our route can sustain halogen substituents in the aryl ring of 1, providing moderate to excellent yields of diaryl sulfides 3 e–3 h. Electron withdrawing groups such as NO2, CF3, OCF3, CN, and COMe were endurable to the reaction conditions, furnishing the desired products 3 i–3 m in good to excellent yields. Electron‐donating substituents (EDG) like Ph, OMe, OPh, and NHAc were also tolerated (3 n–3 q). Even a free hydroxyl group could be carried through to product 3 r, which is impressive as applications of hydroxy‐substituted iodonium salts are scarce in literature. [21]
Scheme 2.

Diarylation scope. [a] Reaction conditions: 1 (0.2 mmol), 2 d (0.28 mmol), DMF (0.2 M) under argon atmosphere for 18 h in the absence of light, isolated yields are reported. [b] BF4 salt was used. [c] OTs salt was used. [d] Reaction performed at rt for 12 h and 60 °C for 6 h.
The reactivity was not influenced by ortho‐substitution in the iodonium salt, and target products 3 t and 3 u were secured in high yields. We were also delighted by the straightforward formation of sterically congested sulfides 3 v and 3 w, even allowing the synthesis of the triisopropylphenyl derivative 3 x in high yield. The substitution pattern of the electron‐deficient ring was investigated next, and the nitro group could be replaced by SO2CF3 to give sulfide 3 y in excellent yield. While weaker EWG (CO2Me, CN) resulted in poor conversion, [17] addition of further electron‐withdrawing substituents was well tolerated (3 z, 3 aa). In the latter case, the CF3 group reinforced the SNAr reactivity along with NO2 group. The introduction of additional fluoride substituents resulted in complete regioselectivity in favor of the desired transformation, with SNAr para to the nitro group yielding 3 ab–3 ad. The reactivity was influenced by the position of the extra fluorides, with 3 ab giving excellent yield whereas 3 ac required prolonged reaction time and heating to reach full conversion. Interestingly, a symmetric, highly electron‐deficient iodonium salt could be utilized in the reaction to produce 3 ae. To our satisfaction, the introduction of an electron deficient pyridiyl ring, which could also undergo SNAr, [22] did not interfere with the reaction and the desired product 3 af was obtained in 73 % yield.
The utility of the developed diarylation methodology was demonstrated through the synthesis of more complex products using functionalized diaryliodonium salts derived from heterocycles, bioactive compounds and drug molecules. Iodonium salts 1 ag–1 am were thus synthesized with complete regioselectivity from the corresponding arenes 4 and iodoarene 5 a using literature methods (Scheme 2B). [23] They were then employed in the diarylation to yield a range of S‐functionalized products with complete regioselectivity, constituting novel late‐stage applications under transition metal‐free conditions.
Applications of heteroaryl iodonium salts are still scarce in arylation methodologies, [24] and we were pleased that pyridine‐derived sulfide 3 ag and coumarin‐derived product 3 ah could be obtained in high yields. The phthalimide‐derived sulfide 3 ai was easily obtained, and the methyl ester derivative of gemfibrozil was converted to sulfide derivative 3 aj in excellent yield. Likewise, the drugs clofibrate and isoxepac could be S‐functionalized to give the desired products 3 ak and 3 al. A derivative of another NSAID drug, flurbiprofen, was applied to the salt synthesis and S‐functionalization sequence to give the corresponding sulfide 3 am.
To examine whether the difunctionalization methodology could be expanded beyond diarylations, we envisioned the application of vinyliodonium salts[ 11a , 25 ] to reach aryl(vinyl) sulfides. Literature routes to aryl(vinyl) sulfides primarily require thiophenols as sulfur source [26] or employ transition metals to exploit the reactivity of disulfides. [27] Thiiranes have been utilized to synthesize aryl(vinyl) sulfides from diaryliodonium salts with the common atom‐economy drawback caused by elimination of ArI. [28] A primary objective for such methodologies is to ensure high E : Z selectivity, and our difunctionalization methodology would be an attractive and atom‐efficient route to highly E‐selective aryl(vinyl) sulfides under transition metal‐free conditions.
Reactions with vinyliodonium salts generally lead to the monofunctionalization of nucleophiles or fragmentation to vinyl iodides, [29] and vinylbenziodoxolones (VBX) were recently reported as a related reagent class. [30] While oxyvinylations with VBX are known, [31] it should be noted that difunctionalizations creating two C−Nu bonds from vinyliodonium salts or VBX remain unreported.[ 25a , 30b ]
When the aryl(vinyl)iodonium salt 6 a(OTf) was reacted with 2 d under the diarylation conditions, only 21 % of the desired product 7 a was observed, with 8 : 1 E : Z selectivity (Table 2, entry 1). Reactions with potassium thioacetate 2 c afforded 7 a with similar selectivity (entry 2). The reaction conditions were reoptimized using vinyliodonium salts with varying counter ions, and enhanced yields and diastereoselectivities were observed with both 6 a(BF4) and 6 a(OTs) (entries 3, 4). While reactions in acetone proceeded poorly (entries 5, 6), the transformation benefitted from lower temperature, and reactions at −15 °C gave significantly increased yield of 7 a (entries 7, 8). Further improvements were observed with 2 c upon initially lowering the temperature to −40 °C followed by reaction at rt, which gave 7 a in 92 % yield with excellent E : Z selectivity (entry 9). Increased reaction scale delivered 7 a in high isolated yield and excellent E : Z selectivity using both 6 a(BF4) and 6 a(OTs) (entries 10, 11).
Table 2.
Optimization of the reaction conditions.[a]
|
| ||||||
|---|---|---|---|---|---|---|
|
entry |
6 a(X) |
“S” |
solvent |
T [°C] |
yield of 7 a [%][b] |
E : Z |
|
1 |
OTf |
2 d |
DMF |
rt |
21 |
8 : 1 |
|
2 |
OTf |
2 c |
DMF |
rt |
32 |
9 : 1 |
|
3 |
BF4 |
2 c |
DMF |
rt |
58 |
>20 : 1 |
|
4 |
OTs |
2 c |
DMF |
rt |
72 |
17 : 1 |
|
5 |
BF4 |
2 c |
acetone |
rt |
49 |
13 : 1 |
|
6 |
OTs |
2 d |
acetone |
rt |
trace |
– |
|
7 |
BF4 |
2 c |
DMF |
−15 |
83 |
>20 : 1 |
|
8 |
BF4 |
2 d |
DMF |
−15 |
81 |
13 : 1 |
|
9[c] |
BF4 |
2 c |
DMF |
−40, rt |
92 |
>20 : 1 |
|
10[c] |
BF4 |
2 c |
DMF |
−40, rt |
80[d] |
>20 : 1 |
|
11[c] |
OTs |
2 c |
DMF |
−40, rt |
75[d] |
>20 : 1 |
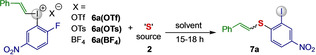
[a] Reaction conditions: 6 a (0.025 mmol), 2 (0.075 mmol), solvent (0.05 M). [b] 1H NMR yield using TMB as internal standard. [c] Reactions were done at −40 °C for 6 h and then at rt for 12 h. [d] Isolated yields using 6 a (0.2 mmol), 2 (0.4 mmol).
With the optimized conditions in hand (entry 10), the generality of the protocol was evaluated using aryl(vinyl)iodonium salts 6 bearing electronically varied styryl handles (Scheme 3). A set of 4‐substituted vinyl salts were investigated first, and the desired products 7 b–7 c were obtained in high yields and excellent E : Z selectivities. Extension of the conjugation in the styryl moiety gave 7 d in high isolated yield but diminished E : Z selectivity, due to isomerization in the purification process. Similar selectivities were observed when electron rich and electron deficient substituents were utilized, as exemplified by 4‐OMe substituted 7 e and NO2‐substituted product 7 f. On the contrary, the addition of a fluoride substituent did not affect the reactivity and desired product 7 g was isolated in 89 % yield with 20 : 1 E : Z selectivity. When the aryl group was decorated with an additional fluoride, the SNAr reaction still took place para to the nitro group to yield the desired product 7 h with excellent E‐selectivity.
Scheme 3.
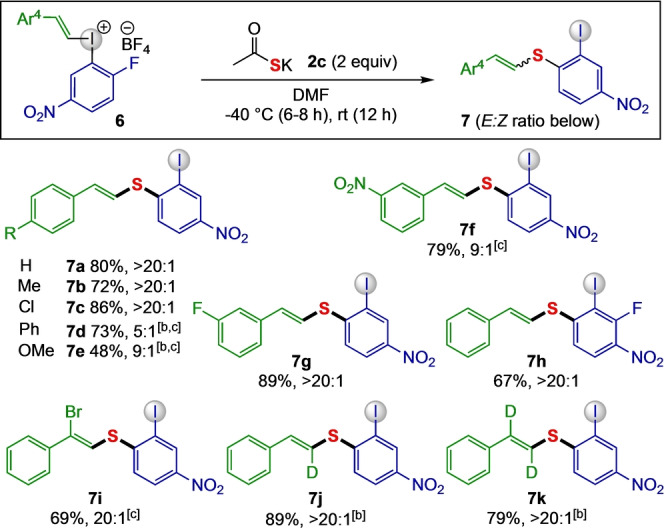
Scope of aryl(vinyl) sulfides 7. [a] Reaction conditions: 6 (0.20 mmol), 2 c (0.40 mmol), DMF (0.2 M) under argon atmosphere for 18 h, isolated yields are reported. [b] OTs salt was used. [c] The diastereoselectivity was >20 : 1 in crude 1H NMR.
To broaden the scope of vinyl sulfides, several trisubstituted aryl(vinyl)iodonium salts were synthesized from the corresponding vinylboronic acids. [32] To our satisfaction, the 2‐bromo substituted vinyliodonium salt 6 i reacted swiftly to give trisubstituted vinyl sulfide 7 i. This bromo‐substituted sulfide enables orthogonal reactivity due to the presence of the vinyl bromide. Reactions with deuterium‐labelled vinyliodonium salts proceeded smoothly to generate the corresponding sulfides 7 j and 7 k in excellent yields with exclusive E selectivity.
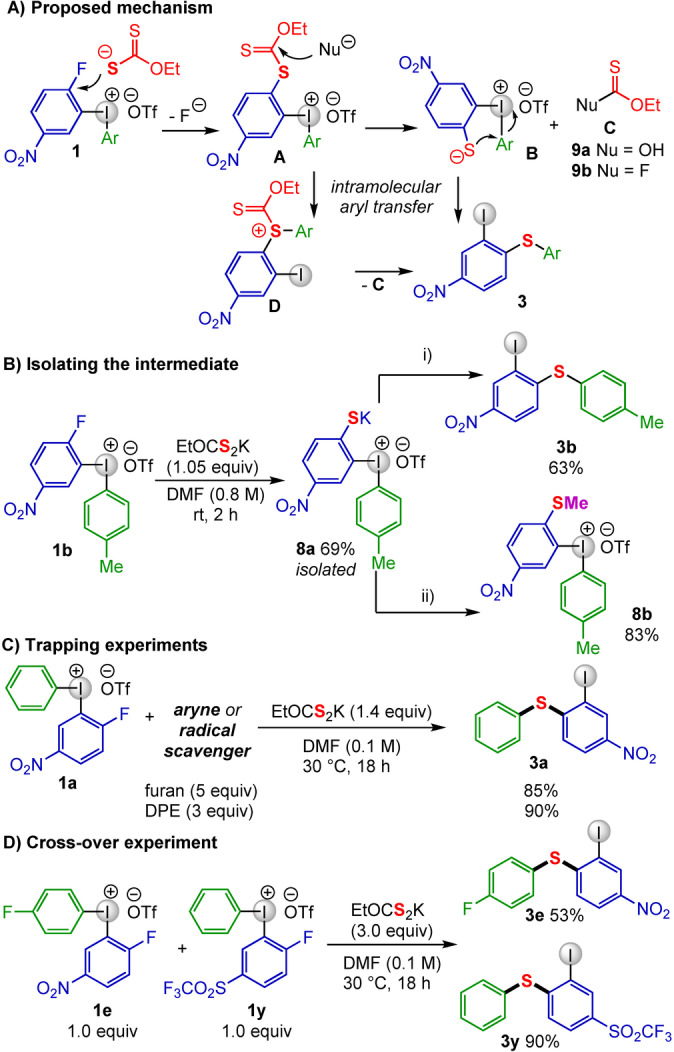
The diarylation of N‐ and O‐nucleophiles was proposed to proceed via initial SNAr to form a Meisenheimer complex followed by intramolecular aryl transfer. [15] A related mechanism can be proposed for the S‐diarylation, where reaction of 1 with the S‐nucleophile forms the SNAr product A, followed by nucleophilic cleavage of the xanthogenate fragment to B and carbonothioate derivative C, then intramolecular aryl transfer would yield diarylated product 3 (Scheme 4A). Alternatively, the aryl transfer could proceed before hydrolysis via intermediate D. The SNAr step could proceed via a stepwise or concerted mechanism, as previously reported in S‐arylation. [33] We propose that fluoride or trace water cleaves the xanthogenate intermediate, as a Chugaev elimination is unlikely due to the low reaction temperature. [22]
Scheme 4.
Mechanistic studies. Reaction conditions i) DMF, 40 °C, 12 h, ii) MeI, DMF, rt, 2 h. NMR yields are reported using TMB as internal standard.
The mechanism was investigated by isolation of key intermediates through shortening the reaction time and adding diethyl ether to precipitate out key intermediates. To our satisfaction, SNAr product 8 a could be isolated as a light‐yellow solid in 69 % yield (Scheme 4B). As 8 a was unstable in the NMR solvent and gradually transformed to product 3 b when subjected to the standard conditions, it was transformed to the methylated sulfide 8 b to confirm the structure of 8 a.
Kinetic experiments suggest fast formation of the intermediate and gradual transformation to the product. [17] At the end of the reaction, trace amounts of carbonothioate derivative 9 a were detected in crude 1H NMR and HRMS. Furthermore, a fluorine‐containing intermediate was observed in 19F NMR, suggesting that fluoride acts as the primary nucleophile in the xanthogenate cleavage to give 9 b, which can be hydrolyzed to 9 a in the presence of moisture (Scheme 4A).
Standard control experiments in the presence of aryne or radical scavengers (furan or 1,1‐diphenylethylene (DPE)), did not influence the reaction outcome, indicating that such pathways are unlikely (Scheme 4C). When a crossover experiment was performed with diaryliodonium salts 1 e and 1 y, no traces of cross‐over product was detected (Scheme 4D). All the experimental findings support the proposed mechanism with initial SNAr followed by nucleophilic cleavage of the xanthogenate and intramolecular aryl transfer.
Density functional theory (DFT) calculations were performed at the M06‐2X level of theory to substantiate the experimental observations and detail the mechanistic pathway further with fluoride as the nucleophile. [17] The key intermediates and transition states are depicted in Figure 1. Initial coordination of xanthate 2 d to the iodine gives intermediate I (−2.5 kcal mol−1), followed by a concerted SNAr via transition state TS1 (21.8 kcal mol−1) to deliver iodine(III) intermediate II. From intermediate II, the mechanism could proceed via an ene‐type elimination assisted by the fluoride or via nucleophilic attack of the fluoride onto the xanthate carbon.
Figure 1.

Calculated free energy profile for diarylation of sulfur nucleophiles. [M06‐2X(SMD)/def2‐TZVP//M06‐2X(SMD)/def2‐SVP level in implicit DMF solvent].
The ene‐type reaction resulted in a very high calculated energy profile, [17] whereas the nucleophilic attack via TS2 (10.7 kcal mol−1) led to intermediate III. Then, intermediate IV and fluorocarbonothioate derivative 9 b form via TS3 (10.2 kcal mol−1). 9 b is easily hydrolyzed in presence of trace amount of water to yield 9 a, which was experimentally observed. Next, the sulfur anion attacks the ipso‐carbon of the iodine in a Smiles‐type rearrangement [34] to yield the desired product 3 a via TS4 (21.7 kcal mol−1). This is the rate‐determining step of the reaction, in agreement with the kinetic experiments. [17] The energy profile was also calculated without the potassium cation, [17] which resulted in a considerably higher energy profile. This indicates that the potassium cation plays a key role in the mechanism by stabilizing the ionic intermediate and the fluoride. Such influence has been previously reported in reactions involving hypervalent iodine reagents. [35]
Post‐synthetic modifications were achieved by transforming the diaryl sulfide 3 a using literature reactions (Scheme 5). Interestingly, the nitro group can easily be reduced to the corresponding amino group using transition metal‐catalyzed or transition metal‐free pathways, enabling a straightforward route to electron‐rich amino sulfide 10 a using either iron and ammonium chloride, or B2(OH)4 and 2,2′‐bipyridine. Transition metal‐free oxidation of the sulfide using meta‐chloroperbenzoic acid (mCPBA) gave facile access to sulfone derivative 10 b, which makes the difunctionalization route interesting in the synthesis of biologically interesting sulfones (see Scheme 1A). Also, diaryl sulfides can be smoothly oxidized to sulfoxides 10 c using urea peroxide and phthalic anhydride. The iodide substituent present in all sulfides 3 and 7 allows several post‐synthetic cross‐coupling reactions. Indeed, a Pd‐catalyzed Sonogashira coupling resulted in alkynylated sulfide 10 d, and a Suzuki coupling delivered arylated sulfide 10 e.
Scheme 5.
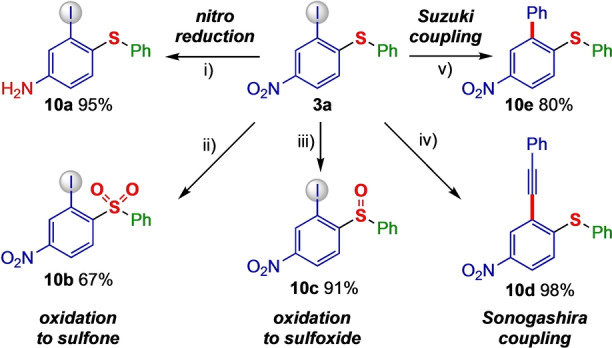
Post synthetic applications of sulfide 3 a. Reaction conditions: i) B2(OH)4, 2,2′‐bipyridine, DMF, 40 °C, 4 h or Fe, NH4Cl, MeOH/THF/H2O, 70 °C, 5 h; ii) mCPBA, acetone, 16 h, 40 °C.; iii) phthalic anhydride, urea‐H2O2, EtOAc, 16 h, rt iv) PhCCH, PdCl2(PPh3)2, CuI, Et3N, 14 h, rt; iv) PhB(OH)2, Pd(PPh3)4, K2CO3, DMF, 15 h, 100 °C.
To showcase the synthetic flexibility of the difunctionalization methodology, it was combined with a one‐pot transition metal‐free reduction to the corresponding anilines (Scheme 6). In this fashion, the synthesis of thioaniline 10 a could be performed in a sequential one‐pot setup without the need for solvent change or workup between the steps. Furthermore, the functionalized iodonium salt 1 an could be transformed into heterocyclic arylamine 10 f through diarylation followed by reduction in the presence of iron and ammonium chloride. Product 10 f exhibits specific binding ability towards Cytochrome P450 in Mycobacterium tuberculosis, [36] hence it provides a new derivative of a potential drug candidate (drug bank reg. no DB07971). [37] The iodine handle available for further derivatizations of the synthesized drug derivative is an extra feature of this methodology.
Scheme 6.
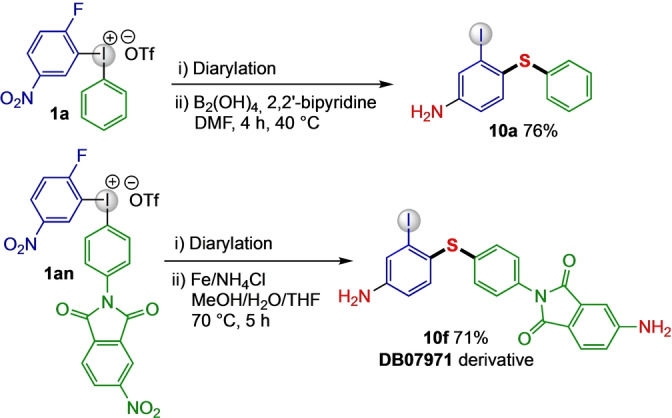
One pot synthesis of amino sulfides.
Conclusion
In conclusion, we have developed a strategy for the synthesis of iodo‐containing diaryl sulfides from iodonium salts in the absence of transition metals, external bases and thiols. Aryl groups containing acid‐ or base‐sensitive substituents, as well as sterically congested substituents could be transferred to S‐nucleophiles under mild reaction conditions. The applicability of the diarylation was demonstrated using heteroaryl and drug‐derived iodonium salts, outlining a route to the corresponding sulfides in excellent yields. A novel type of difunctionalization strategy was discovered using aryl(vinyl)iodonium salts, whereby aryl(vinyl) sulfides could be obtained in excellent stereoselectivity through a sequential arylation and vinylation. The developed difunctionalization was amenable to one‐pot transition metal free reduction to reach thioanilines and a potential drug candidate derivative. A mechanistic proposal is given, substantiated by DFT studies and isolation of the key intermediate. Overall, a general synthetic methodology harnessing the reactivity of iodonium salts has been presented that enables further applications.
Conflict of interest
The authors declare no conflict of interest.
1.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgments
Financial support for this study was provided through Olle Engkvist Foundation (207‐0615) and the Swedish Research Council (2019‐04232). The computations were enabled by resources provided by the Swedish National Infrastructure for Computing (SNIC), partially funded by the Swedish Research Council through grant agreement no. 2018‐05973.
Mondal S., Di Tommaso E. M., Olofsson B., Angew. Chem. Int. Ed. 2023, 62, e202216296; Angew. Chem. 2023, 135, e202216296.
A previous version of this manuscript has been deposited on a preprint server (https://doi.org/10.26434/chemrxiv‐2022‐c1bt4).
Data Availability Statement
The data that support the findings of this study are available in the supplementary material of this article.
References
- 1. Cremlyn R. J., Cremlyn R. J. W., An introduction to organosulfur chemistry, John Wiley & Sons, New York, 1996. [Google Scholar]
- 2.
- 2a. Hell R., Khan M. S., Wirtz M., in Cell Biology of Metals and Nutrients (Eds.: Hell R., Mendel R.-R.), Springer, Berlin, Heidelberg, 2010, pp. 43–279; [Google Scholar]
- 2b. Wang N., Saidhareddy P., Jiang X., Nat. Prod. Rep. 2020, 37, 246–275. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Diaz-Ruiz A., Zavala C., Montes S., Ortiz-Plata A., Salgado-Ceballos H., Orozco-Suarez S., Nava-Ruiz C., Pérez-Neri I., Perez-Severiano F., Ríos C., J. Neurosci. Res. 2008, 86, 3410–3419; [DOI] [PubMed] [Google Scholar]
- 3b. De Martino G., Edler M. C., La Regina G., Coluccia A., Barbera M. C., Barrow D., Nicholson R. I., Chiosis G., Brancale A., Hamel E., Artico M., Silvestri R., J. Med. Chem. 2006, 49, 947–954; [DOI] [PubMed] [Google Scholar]
- 3c. Dénès F., Schiesser C. H., Renaud P., Chem. Soc. Rev. 2013, 42, 7900–7942. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Feng M., Tang B., Liang S. H., Jiang X., Curr. Top. Med. Chem. 2016, 16, 1200–1216; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4b. Scott K. A., Njardarson J. T., Top. Curr. Chem. 2018, 376, 5. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Wang Y., Chackalamannil S., Hu Z., Clader J. W., Greenlee W., Billard W., Binch H., Crosby G., Ruperto V., Duffy R. A., McQuade R., Lachowicz J. E., Bioorg. Med. Chem. 2000, 10, 2247–2250; [DOI] [PubMed] [Google Scholar]
- 5b. Liu G., Huth J. R., Olejniczak E. T., Mendoza R., DeVries P., Leitza S., Reilly E. B., Okasinski G. F., Fesik S. W., von Geldern T. W., J. Med. Chem. 2001, 44, 1202–1210; [DOI] [PubMed] [Google Scholar]
- 5c. Mustafa M., Winum J.-Y., Expert Opin. Drug Discovery 2022, 17, 501–512; [DOI] [PubMed] [Google Scholar]
- 5d. Liu Y., Lam L. Y., Ye J., Blanchard N., Ma C., Adv. Synth. Catal. 2020, 362, 2326–2331. [Google Scholar]
- 6.
- 6a. Ahmadi R., Emami S., Eur. J. Med. Chem. 2022, 234, 114255; [DOI] [PubMed] [Google Scholar]
- 6b. Reeves J. T., Camara K., Han Z. S., Xu Y., Lee H., Busacca C. A., Senanayake C. H., Org. Lett. 2014, 16, 1196–1199. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Shen C., Zhang P., Sun Q., Bai S., Hor T. S. A., Liu X., Chem. Soc. Rev. 2015, 44, 291–314; [DOI] [PubMed] [Google Scholar]
- 7b. Abedinifar F., Bahadorikhalili S., Larijani B., Mahdavi M., Verpoort F., Appl. Organomet. Chem. 2022, 36, e6482. [Google Scholar]
- 8.
- 8a. Chen C.-W., Chen Y.-L., Reddy D. M., Du K., Li C.-E., Shih B.-H., Xue Y.-J., Lee C.-F., Chem. Eur. J. 2017, 23, 10087–10091; [DOI] [PubMed] [Google Scholar]
- 8b. Isshiki R., Kurosawa M. B., Muto K., Yamaguchi J., J. Am. Chem. Soc. 2021, 143, 10333–10340; [DOI] [PubMed] [Google Scholar]
- 8c. Ichiishi N., Malapit C. A., Woźniak Ł., Sanford M. S., Org. Lett. 2018, 20, 44–47; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8d. Kanemoto K., Sugimura Y., Shimizu S., Yoshida S., Hosoya T., Chem. Commun. 2017, 53, 10640–10643. [DOI] [PubMed] [Google Scholar]
- 9. Jang H.-Y., Org. Biomol. Chem. 2021, 19, 8656–8686. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. Wang X., Chen J.-Q., Yang X.-X., Hao E.-J., Dong Z.-B., Eur. J. Org. Chem. 2022, e202200015; [Google Scholar]
- 10b. Li F., Wang D., Chen H., He Z., Zhou L., Zeng Q., Chem. Commun. 2020, 56, 13029–13032; [DOI] [PubMed] [Google Scholar]
- 10c. Zou L.-H., Zhao C., Li P.-G., Wang Y., Li J., J. Org. Chem. 2017, 82, 12892–12898; [DOI] [PubMed] [Google Scholar]
- 10d. Prasad C. D., Balkrishna S. J., Kumar A., Bhakuni B. S., Shrimali K., Biswas S., Kumar S., J. Org. Chem. 2013, 78, 1434–1443; [DOI] [PubMed] [Google Scholar]
- 10e. Fernández-Salas J. A., Pulis A. P., Procter D. J., Chem. Commun. 2016, 52, 12364–12367. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Yoshimura A., Zhdankin V. V., Chem. Rev. 2016, 116, 3328–3435; [DOI] [PubMed] [Google Scholar]
- 11b. Villo P., Olofsson B., in PATAI′S Chemistry of Functional Groups, Wiley, New York, 2019, pp. 461–522; [Google Scholar]
- 11c. Merritt E. A., Olofsson B., Angew. Chem. Int. Ed. 2009, 48, 9052–9070; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 9214–9234. [Google Scholar]
- 12.
- 12a. Wagner A. M., Sanford M. S., J. Org. Chem. 2014, 79, 2263–2267; [DOI] [PubMed] [Google Scholar]
- 12b. Wang M., Fan Q., Jiang X., Org. Lett. 2016, 18, 5756–5759; [DOI] [PubMed] [Google Scholar]
- 12c. Wang M., Wei J., Fan Q., Jiang X., Chem. Commun. 2017, 53, 2918–2921; [DOI] [PubMed] [Google Scholar]
- 12d. Kikushima K., Elboray E. E., Jiménez-Halla J. O. C., Solorio-Alvarado C. R., Dohi T., Org. Biomol. Chem. 2022, 20, 3231–3248. [DOI] [PubMed] [Google Scholar]
- 13.
- 13a. Wang M., Chen S., Jiang X., Chem. Asian J. 2018, 13, 2195–2207; [DOI] [PubMed] [Google Scholar]
- 13b. Boelke A., Finkbeiner P., Nachtsheim B. J., Beilstein J. Org. Chem. 2018, 14, 1263–1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.For diarylation examples see
- 14a. Modha S. G., Greaney M. F., J. Am. Chem. Soc. 2015, 137, 1416–1419; [DOI] [PubMed] [Google Scholar]
- 14b. Teskey C. J., Sohel S. M. A., Bunting D. L., Modha S. G., Greaney M. F., Angew. Chem. Int. Ed. 2017, 56, 5263–5266; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 5347–5350. [Google Scholar]
- 15. Linde E., Bulfield D., Kervefors G., Purkait N., Olofsson B., Chem 2022, 8, 850–865. [Google Scholar]
- 16. Nguyen T. B., Adv. Synth. Catal. 2017, 359, 1066–1130. [Google Scholar]
- 17.See the Supporting Information for details.
- 18. Lu G.-p., Cai C., Adv. Synth. Catal. 2013, 355, 1271–1276. [Google Scholar]
- 19.The xanthogenate reagent 2 d is known to be light sensitive.
- 20. Uyeda C., Tan Y., Fu G. C., Peters J. C., J. Am. Chem. Soc. 2013, 135, 9548–9552. [DOI] [PubMed] [Google Scholar]
- 21. Yoshimura A., Shea M. T., Guselnikova O., Postnikov P. S., Rohde G. T., Saito A., Yusubov M. S., Nemykin V. N., Zhdankin V. V., Chem. Commun. 2018, 54, 10363–10366. [DOI] [PubMed] [Google Scholar]
- 22. He R., Liu Y., Feng Y., Chen L., Huang Y., Xie F., Li Y., Org. Lett. 2022, 24, 3167–3172. [DOI] [PubMed] [Google Scholar]
- 23.Methods used in the synthesis of iodonium salts:
- 23a. Bielawski M., Zhu M., Olofsson B., Adv. Synth. Catal. 2007, 349, 2610–2618; [Google Scholar]
- 23b. Bielawski M., Olofsson B., Chem. Commun. 2007, 2521–2523; [DOI] [PubMed] [Google Scholar]
- 23c. Dohi T., Ito M., Morimoto K., Minamitsuji Y., Takenaga N., Kita Y., Chem. Commun. 2007, 4152–4154; [DOI] [PubMed] [Google Scholar]
- 23d. Carroll M. A., Pike V. W., Widdowson D. A., Tetrahedron Lett. 2000, 41, 5393–5396; [Google Scholar]
- 23e. Chen Y., Gu Y., Meng H., Shao Q., Xu Z., Bao W., Gu Y., Xue X.-S., Zhao Y., Angew. Chem. Int. Ed. 2022, 61, e202201240; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2022, 134, e202201240. [DOI] [PubMed] [Google Scholar]
- 24. Bielawski M., Malmgren J., Pardo L. M., Wikmark Y., Olofsson B., ChemistryOpen 2014, 3, 19–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.
- 25a. Hari D. P., Nicolai S., Waser J., in Patai's Chemistry of Functional Groups: The Chemistry of Hypervalent Halogen Compounds (Eds.: Olofsson B., Marek I., Rappoport Z.), Wiley, Hoboken, 2019, pp. 523–580; [Google Scholar]
- 25b. Okuyama T., Acc. Chem. Res. 2002, 35, 12–18; [DOI] [PubMed] [Google Scholar]
- 25c. Okuyama T., Fujita M., Acc. Chem. Res. 2005, 38, 679–686. [DOI] [PubMed] [Google Scholar]
- 26.
- 26a. Castoldi L., Di Tommaso E. M., Reitti M., Gräfen B., Olofsson B., Angew. Chem. Int. Ed. 2020, 59, 15512–15516; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 15642–15646; [Google Scholar]
- 26b. Ogawa A., Ikeda T., Kimura K., Hirao T., J. Am. Chem. Soc. 1999, 121, 5108–5114. [Google Scholar]
- 27.
- 27a. Tu H.-Y., Hu B.-L., Deng C.-L., Zhang X.-G., Chem. Commun. 2015, 51, 15558–15561; [DOI] [PubMed] [Google Scholar]
- 27b. Ranu B. C., Chattopadhyay K., Banerjee S., J. Org. Chem. 2006, 71, 423–425. [DOI] [PubMed] [Google Scholar]
- 28. Dong J., Xu J., Synthesis 2018, 50, 2407–2415. [Google Scholar]
- 29.
- 29a. Skucas E., MacMillan D. W., J. Am. Chem. Soc. 2012, 134, 9090–9093; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29b. Mondal K., Pan S. C., Eur. J. Org. Chem. 2015, 2129–2132; [Google Scholar]
- 29c. Kepski K., Rice C. R., Moran W. J., Org. Lett. 2019, 21, 6936–6939. [DOI] [PubMed] [Google Scholar]
- 30.
- 30a. Stridfeldt E., Seemann A., Bouma M. J., Dey C., Ertan A., Olofsson B., Chem. Eur. J. 2016, 22, 16066–16070; [DOI] [PubMed] [Google Scholar]
- 30b. Declas N., Pisella G., Waser J., Helv. Chim. Acta 2020, 103, e2000191. [Google Scholar]
- 31.
- 31a. Borrel J., Pisella G., Waser J., Org. Lett. 2020, 22, 422–427; [DOI] [PubMed] [Google Scholar]
- 31b. Declas N., Waser J., Angew. Chem. Int. Ed. 2020, 59, 18256–18260; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 18413–18417. [Google Scholar]
- 32.
- 32a. Ochiai M., Toyonari M., Nagaoka T., Chen D.-W., Kida M., Tetrahedron Lett. 1997, 38, 6709–6712; [Google Scholar]
- 32b. Altomonte S., Telu S., Lu S., Pike V. W., J. Org. Chem. 2017, 82, 11925–11932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Singh H., Struct. Chem. 2020, 31, 2205–2213. [Google Scholar]
- 34. Holden C. M., Greaney M. F., Chemistry 2017, 23, 8992–9008. [DOI] [PubMed] [Google Scholar]
- 35. Brea O., Szabo K. J., Himo F., J. Org. Chem. 2020, 85, 15577–15585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Podust L. M., Ouellet H., Von Kries J. P., Ortiz de Montellano P. R., J. Biol. Chem. 2009, 284, 25211–25219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Flude B. M., Nannetti G., Mitchell P., Compton N., Richards C., Heurich M., Brancale A., Ferla S., Bassetto M., Viruses 2021, 13, 312. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Data Availability Statement
The data that support the findings of this study are available in the supplementary material of this article.