

Abstract
Objective
This study was undertaken to identify key disease pathways driving conventional dendritic cell (cDC) alterations in systemic sclerosis (SSc).
Methods
Transcriptomic profiling was performed on peripheral blood CD1c+ cDCs (cDC2s) isolated from 12 healthy donors and 48 patients with SSc, including all major disease subtypes. We performed differential expression analysis for the different SSc subtypes and healthy donors to uncover genes dysregulated in SSc. To identify biologically relevant pathways, we built a gene coexpression network using weighted gene correlation network analysis. We validated the role of key transcriptional regulators using chromatin immunoprecipitation (ChIP) sequencing and in vitro functional assays.
Results
We identified 17 modules of coexpressed genes in cDCs that correlated with SSc subtypes and key clinical traits, including autoantibodies, skin score, and occurrence of interstitial lung disease. A module of immunoregulatory genes was markedly down‐regulated in patients with the diffuse SSc subtype characterized by severe fibrosis. Transcriptional regulatory network analysis performed on this module predicted nuclear receptor 4A (NR4A) subfamily genes (NR4A1, NR4A2, NR4A3) as the key transcriptional regulators of inflammation. Indeed, ChIP‐sequencing analysis indicated that these NR4A members target numerous differentially expressed genes in SSc cDC2s. Inclusion of NR4A receptor agonists in culture‐based experiments provided functional proof that dysregulation of NR4As affects cytokine production by cDC2s and modulates downstream T cell activation.
Conclusion
NR4A1, NR4A2, and NR4A3 are important regulators of immunosuppressive and fibrosis‐associated pathways in SSc cDCs. Thus, the NR4A family represents novel potential targets to restore cDC homeostasis in SSc.
INTRODUCTION
Systemic sclerosis (SSc) is a complex, chronic autoimmune disease mainly characterized by vascular abnormalities, immunologic abnormalities, and fibrosis of the skin and internal organs (1). In accordance with the American College of Rheumatology (ACR) criteria and the extent of skin fibrosis in patients, patients are classified into 4 SSc subsets: early SSc (eaSSc), noncutaneous SSc (ncSSc), limited SSc (lcSSc), and diffuse SSc (dcSSc) (2, 3). Vascular injury appears to be one of the earliest events in the pathogenesis of SSc (4), which in turn leads to the recruitment and activation of immune cells secreting proinflammatory cytokines and growth factors (5). The resulting mix of inflammatory molecules induces the differentiation of resident epithelium, endothelium, monocytes, and fibroblasts into myofibroblasts that deposit excessive amounts of extracellular matrix (ECM), eventually leading to permanent tissue scarring (6, 7).
Conventional dendritic cells (cDCs) are a population of antigen‐presenting cells that play a central role in regulation of adaptive immune cell responses (8), as well as in vascular tissue and fibroblasts (9, 10). Given their placement at the crossroads of inflammation and fibrosis, cDCs have been implicated in the pathogenesis of SSc and are hypothesized to be essential for the activation of pathways that promote fibrosis (11). Indeed, in the early phases of SSc, cDCs migrate to the skin (12, 13) and display an enhanced proinflammatory and profibrotic cytokine production upon innate immune stimulation by Toll‐like receptor (TLR) ligation (14). Furthermore, cDCs are a rather heterogeneous population, with CD1c+ cDCs (cDC2s) having a high capacity for priming CD4+ T cells (15). Given the well‐established pathogenic role of T cells in SSc (16, 17, 18), cDC2s are a highly interesting subset to investigate in SSc.
Although data from previous studies support an instrumental role for cDCs in the pathogenesis of SSc, the molecular mechanisms that drive their dysregulation in the disease remain incompletely understood. To study the mechanisms behind cDC2 dysregulation in SSc, we compared the transcriptomic profile of circulating cDC2s obtained from peripheral blood (PB) of patients with SSc and healthy controls. Using coexpression network analysis and in vitro validation studies, we aimed to unravel key players of cDC2 dysregulation in SSc.
MATERIALS AND METHODS
Sample collection and demographics
The study was approved by the medical ethics committee of the University Medical Center Utrecht (METC no. 12/296 and no. 13/697). PB samples were collected from patients with SSc and from age‐ and sex‐matched healthy controls from the University Medical Center Utrecht (The Netherlands), the Maasstad Medical Center Rotterdam (The Netherlands), and the IRCCS Policlinico of Milan (Italy) (Supplementary Table 1, available on the Arthritis & Rheumatology website at https://onlinelibrary.wiley.com/doi/10.1002/art.42319). All participants signed an informed consent in accordance with the Declaration of Helsinki. Patients fulfilled the ACR/EULAR 2013 classification criteria for SSc (2). We also included ncSSc patients who fulfilled the classification criteria but did not present with skin fibrosis and eaSSc patients with Raynaud's phenomenon and positivity for SSc‐specific autoantibodies and/or typical findings on nailfold capillaroscopy (3). For functional experiments on cDC2s from healthy controls not paired with SSc patients, we used buffy coats (Sanquin).
CD1c+ cDC purification
We isolated PB mononuclear cells (PBMCs) from heparinized whole blood samples from SSc patients and from healthy controls by density‐gradient centrifugation using Ficoll‐Paque Plus solution (GE Healthcare Life Sciences). We isolated cDC2s using the MACS human CD1c (CD1c+) dendritic cell isolation kit followed by separation on the AutoMACS Pro Separator, according to the instructions of the manufacturer (Miltenyi Biotec). For the RNA‐sequencing cohort and the validation cohort, freshly isolated cDC2s were immediately lysed in RLT Plus buffer (Qiagen) supplemented with β‐mercaptoethanol and then stored at −20°C until further processing.
RNA‐sequencing process and analysis
We purified total RNA from RLT Plus lysates using a DNA/RNA/microRNA universal kit, according to the instructions of the manufacturer (Qiagen). Purified RNA was quantified with a Qubit RNA assay kit (Life Technologies) on a Qubit fluorometer (Invitrogen). RNA‐sequencing analysis was performed at the Beijing Genomics Institute (BGI). We generated cDNA libraries from total RNA using a TruSeq RNA sample preparation kit (Illumina), selecting for polyadenylated transcripts, and sequenced the libraries on the HiSeq2000 sequencing system (Illumina) using 100‐bp paired‐end reads. After quality filtering was performed according to the BGI pipeline, the reads were aligned to the GrCh38 reference genome using STAR (19). Summed exon read counts per gene were calculated using HTSeq (20). Upper quartile lane normalization was performed using EDAseq (21). To account for batch effects arising from different geographic locations (The Netherlands and Italy), we applied the Bioconductor/R package RUVseq (22) using the RUVr function for k = 2 factors of unwanted variation. We tested pairwise comparisons between SSc patients and healthy controls using the Wald's test in DESeq2 (23). Gene expression levels are given as variance‐stabilized data.
We constructed coexpression networks with WGCNA (24), using all genes with ≥1 raw count in all samples as input. We used a soft threshold power of 5 to construct an unsigned network with scale‐free topology. Modules were identified using the “cutreeDynamic” function (module size of 50). Closely related modules were merged using the “mergeCloseModules” function (height cut of 0.25).
CD1c+ cDC cultures
For cell cultures, cDC2s were purified from healthy control buffy coats (Sanquin) and plated in culture medium (RPMI 1640 GlutaMax [Life Technologies], supplemented with 10% heat‐inactivated fetal bovine serum [BioWest] and 1% penicillin/streptomycin [Life Technologies]) at 0.5 × 106 cells/ml (100 μl) in 96‐well round‐bottomed plates. We treated cells with one of the following stimuli: 100 ng/ml resiquimod (R‐848; InvivoGen), 800 units/ml granulocyte–macrophage colony‐stimulating factor (GM‐CSF; R&D Systems), 10 μg/ml CXCL4 (PeproTech), 100 ng/ml tumor necrosis factor (Tebu‐Bio), 1,000 units/ml interferon‐α2a (IFNα2a; Cell Sciences), 100 ng/ml lipopolysaccharide (LPS) EB Ultrapure (InvivoGen), 50 ng/ml interleukin‐6 (IL‐6; ImmunoTools), 200 ng/ml IL‐15 (ImmunoTools), or 100 ng/ml transforming growth factor β2 (TGFβ2; R&D Systems). For hypoxia experiments, we cultured cells under atmospheric or hypoxic conditions (Rasquinn Invivo2 1000 hypoxia chamber, set at 1% O2 and 5% CO2) for 24 hours. For experiments using nuclear receptor 4A (NR4A) agonists, we pretreated cells for 1 hour with DMSO (Sigma), C‐DIM5 (Tocris Bioscience), or C‐DIM12 (Tebu‐Bio). Cultures were incubated at 37°C in the presence of 5% CO2, for the time points indicated in each single experiment. Supernatants were stored at −80°C, and cDC2s were lysed in RLT Plus buffer and stored at −20°C.
PBMC cultures
Three batches (individual days) of randomly selected dcSSc and matched healthy control samples of liquid nitrogen stored PBMCs were thawed in RPMI 1640 medium (20% fetal bovine serum) and washed with phosphate buffered saline (PBS). We plated cells in culture medium at 0.75 × 106 cells/200 μl in 96‐well round‐bottomed plates. We pretreated PBMCs for 1 hour with 10 μM DMSO, C‐DIM5, or C‐DIM12 before stimulation with R‐848 (100 ng/ml) and GolgiStop (1,500 times; BD Biosciences), followed by incubation at 37°C in the presence of 5% CO2 for 4 hours.
Reverse transcription–quantitative polymerase chain reaction (RT‐qPCR)
Purified RNA was reverse transcribed using a SuperScript IV reverse transcriptase kit, according to the instructions of the manufacturer (Invitrogen). We quantified gene expression, in duplicate, using SYBR Select Master Mix (Applied Biosystems), with gene‐specific primers (Supplementary Table 2, available on the Arthritis & Rheumatology website at https://onlinelibrary.wiley.com/doi/10.1002/art.42319) on the QuantStudio 12k flex System (Applied Biosystems). Relative induction or down‐regulation of gene expression was obtained using the comparative threshold cycle (ΔΔCt) method using GUSB as the endogenous control.
Enzyme‐linked immunosorbent assay (ELISA)
We measured concentrations of IL‐6 in cell‐free supernatants using the PeliKine compact human IL‐6 sandwich ELISA kit, according to the instructions of the manufacturer (Sanquin).
Cocultures of cDC2s and CD4+ T cells
We isolated cDC2s and CD4+ T cells in parallel from PBMCs from healthy control buffy coats, cDC2 cell cultures as described above, and CD4+ T cells using the MACS human CD4+ T cell isolation kit followed by separation on the AutoMACS Pro Separator. Thereafter, cDC2s and T cells were plated, in parallel, in culture medium. We pretreated cDC2s for 1 hour with DMSO, C‐DIM5, or C‐DIM12, which were either left untreated or treated with R‐848 (100 ng/ml). After overnight incubation, we washed cDC2s twice with sterile PBS, resuspended them in culture medium, and added the suspension to T cells in a 1:5 ratio. Cells were cultured for 3 days at 37°C in the presence of 5% CO2. After 3 days, cocultures were restimulated with phorbol myristate acetate (50 ng/ml; Sigma‐Aldrich) and ionomycin (500 ng/ml; Sigma‐Aldrich) for 6 hours. During the last 3 hours, GolgiStop was added.
Flow cytometry
We washed the cells with cold PBS and incubated them with fixable viability dye eF780 (eBioscience) at room temperature for 10 minutes. We then transferred the cells to V‐bottomed plates (Greiner Bio‐One) and allowed cells to incubate for 30 minutes at 4°C in the dark with the surface‐staining antibodies (Supplementary Table 3, available on the Arthritis & Rheumatology website at https://onlinelibrary.wiley.com/doi/10.1002/art.42319). Next, we washed the cells in fluorescence‐activated cell sorting buffer (1% bovine serum albumin and 0.1% sodium azide in PBS) and then fixed/permeabilized the cells for 30 minutes at 4°C in the dark with 100 μl fixation/permeabilization concentrate and diluent (catalog nos. 00‐5123‐43 and 00‐5223‐56, eBioscience); this step was followed by intracellular staining. After 60 minutes of staining at 4°C in the dark, cells were washed and measured on the BD LSRFortessa with 4 lasers (405, 488, 561, and 635 nm) using FACSDiva software version 8.0.1. We analyzed the resulting files using FlowJo.
Chromatin immunoprecipitation (ChIP)–sequencing analysis
We allowed 1.5 × 106 freshly isolated cDC2s to culture overnight, with cDC2s either left untreated or treated with R‐848 (100 ng/ml). Cells were crosslinked using the truChIP Ultra Low chromatin shearing kit, according to the instructions of the manufacturer (Covaris). We performed chromatin shearing using the microTUBE AFA Fiber Pre‐Slit Snap‐Cap vessel (Covaris) and sonication with the Covaris S220 focused ultrasonicator (peak incident power 105, duty factor 2%, cycles per burst 200, treatment time 12 minutes). After shearing was completed, we allowed chromatin to undergo centrifugation at 10,000g for 5 minutes at 4°C to pellet insoluble material, which was then stored at −20°C. For every condition, we obtained 3 biologic replicates, for which materials from 3–4 donors were pooled to obtain enough material. We performed ChIP sequencing with 3 μg of anti‐NR4A1 (NB100‐56745, Novus Biologicals), anti‐NR4A2 (NB110‐40415, Novus Biologicals), or anti‐NR4A3 (NLS2341, Novus Biologicals), using the low cell ChIP‐Seq kit, according to the instructions of the manufacturer (Active Motif). For all conditions, 10% of input chromatin was stored. ChIP DNA was de‐crosslinked with NaCl and proteinase K (Active Motif) at 65°C overnight. DNA was extracted by phenol–chloroform–isoamyl alcohol precipitation and dissolved in low‐Tris–EDTA buffer (Active Motif). We generated the ChIP‐Seq libraries with GenomeScan and the NEBNext Ultra II DNA Library Prep kit (Illumina) and sequenced the libraries using Illumina NovaSeq6000, generating ~20 million 150‐bp paired‐end reads per sample.
We used FastQC software for quality checks. We mapped reads against the reference genome GRCh38 using Bowtie2 software (25). Peaks were identified using MACS2 (26) in BAMPE mode. We excluded peaks from ENCODE blacklist regions and X and Y chromosomes. We used ChIPseeker to annotate peaks to the nearest genes (27).
Statistical analysis
We used the Mann‐Whitney test to compare any combination of 2 groups. Comparison of multiple groups was performed using one‐ or two‐way analysis of variance. P < 0.05 was considered statistically significant. Enrichment analyses of Gene Ontology (GO) terms and KEGG pathways and genomes were performed using clusterProfiler (28). Terms with Benjamini and Hochberg–corrected P < 0.05 were considered significant. Spearman's rank correlation coefficient was calculated to assess correlations.
Processed counts and raw files for RNA‐sequencing data and ChIP sequencing were deposited in NCBI's GEO database (accession no. GSE186199).
RESULTS
Differences in the transcriptomic profiles between cDC2s from SSc patients and cDC2s from healthy controls
RNA sequencing of cDC2s from 48 SSc patients and 12 matched healthy controls was performed to assess differences in their transcriptomic profiles. We found 6,594 differentially expressed genes (DEGs) in ≥1 SSc subset compared with our findings in healthy controls (P < 0.05) (Figures 1A and 1B and Supplementary Table 4, available on the Arthritis & Rheumatology website at https://onlinelibrary.wiley.com/doi/10.1002/art.42319). Principal components analysis revealed that these genes, in general, distinguished SSc patients from healthy controls, although a few samples in the eaSSc, ncSSc, and especially lcSSc groups overlapped with healthy controls (Figure 1C). Samples from the dcSSc group showed the largest separation from healthy controls, indicating that the dcSSc transcriptome is the most distinct. GO term enrichment analysis showed that DEGs were enriched in pathways related to immune cell activation, IFN signaling, and translation (Figure 1D and Supplementary Table 5, available on the Arthritis & Rheumatology website at https://onlinelibrary.wiley.com/doi/10.1002/art.42319). These results indicate that cDC2s from SSc patients have a transcriptional profile distinct from healthy donor cDC2s. Accordingly, the identified DEGs may allude to pathways relevant for SSc pathogenesis.
Figure 1.
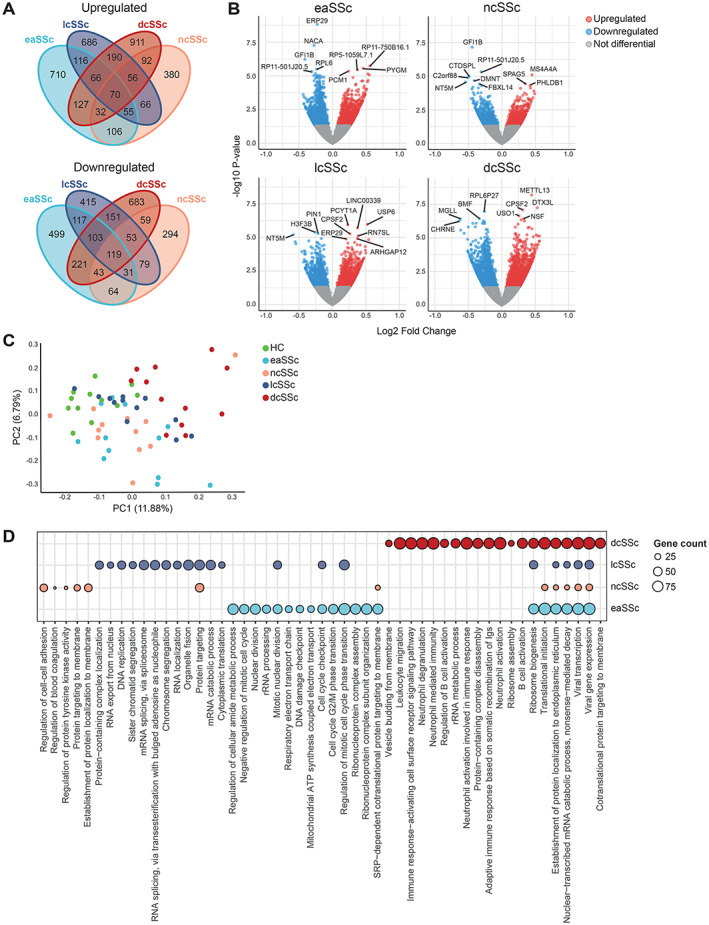
CD1c + conventional dendritic cells (cDC2s) from patients with systemic sclerosis (SSc) showing distinct transcriptomic profiles compared with cDC2s from healthy controls. A, Number of differentially expressed genes (DEGs) (P < 0.05) identified in cDC2s from different SSc subsets versus healthy controls. B, Volcano plots highlighting transcriptional differences between different SSc subsets and healthy controls (HC). Blue dots represent significantly down‐regulated genes (P < 0.05, log2 fold change <0), and red dots represent significantly up‐regulated genes (P < 0.05, log2 fold change >0). Top DEGs based on P value are indicated. C, Principal component (PC) analysis of the DEGs from all comparisons of SSc patients versus healthy controls. D, Pathway enrichment analysis of DEGs identified in SSc patients versus healthy controls. Circle size denotes DEG count associated with enriched pathways. When available, the top 10 pathways are shown (Benjamini and Hochberg–corrected P < 0.05). eaSSc = early SSc; ncSSc = noncutaneous SSc; lcSSc = limited SSc; dcSSc = diffuse SSc.
Role of NR4A as a key transcriptional regulator in the alteration of functionally relevant pathways in SSc cDC2s
To further study the molecular pathways dysregulated in SSc cDC2s, we constructed a gene coexpression network (24). We identified 42 modules of tightly coexpressed genes in cDC2s, of which 17 were clinically relevant and enriched in DEGs and displayed a significant correlation with clinical traits relevant for SSc (Figure 2A). Functional annotation of these modules showed that some were associated with molecular pathways relevant in the context of cDC biology and inflammation. These included the viral pathway and ribosome module (blue), the immune cell regulation module (dark green), and the antigen presentation and inflammation module (violet) (Figure 2B).
Figure 2.
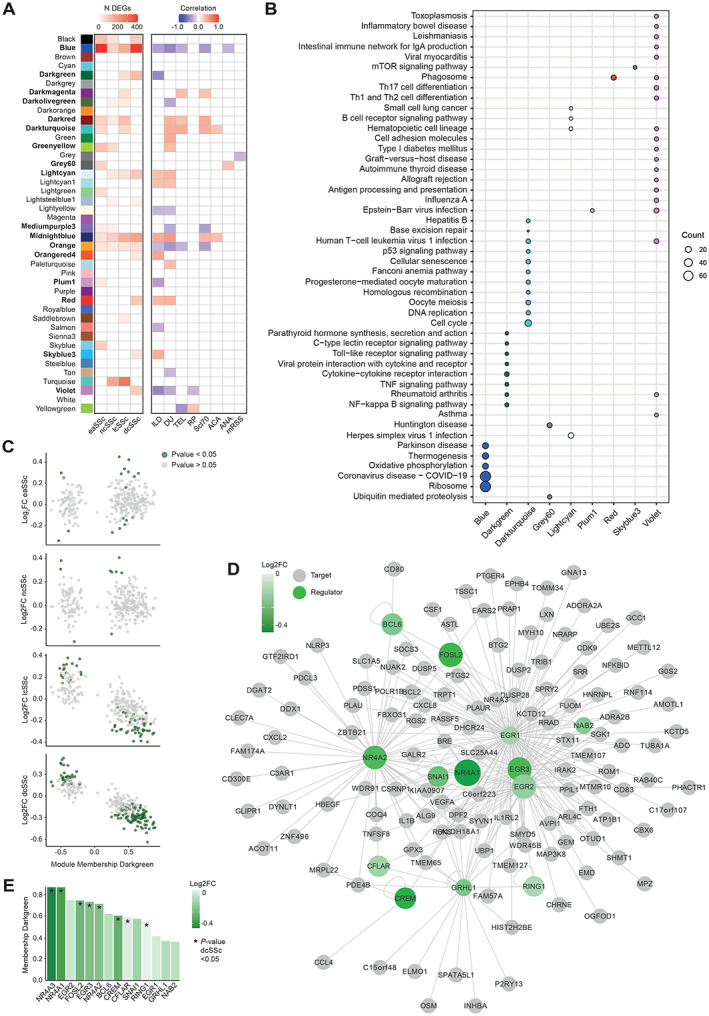
Coexpression network analysis identifying nuclear receptor 4A family members (NR4As) as regulators of immune‐regulatory pathways decreased in SSc cDC2s. A, Overall number of DEGs with coexpression modules (left) and the correlation of module eigengenes (Mes) to SSc clinical traits (right), with intensity bar highlighting significant enrichments (P < 0.05 by Fisher's exact test) and significant correlations with clinical traits (P < 0.05 by Pearson's chi‐square test). B, KEGG enrichment of relevant modules, with circle size denoting number of module genes associated with enriched pathways. When available, the top 10 pathways are shown (Benjamini and Hochberg–corrected P < 0.05). C, Module membership and log2 fold change (FC) (healthy control versus SSc subsets) for genes in the dark green module (immune cell regulation module) indicated in A. DEGs are highlighted in green (P < 0.05). D, Transcription factor network for the dark green module indicated in A. Transcriptional regulators are connected to their targets based on interactions from REGNET and TRRUST. Green shading intensity bar indicates log2 FC between dcSSc and healthy cDC2s. Circle size denotes module membership in the dark green module. E, Level of membership in the dark green module based on log2 FC between HC and dcSSc, as indicated in the intensity bar, of transcriptional regulators. ILD = interstitial lung disease; DU = digital ulcers; TEL = telangiectasia; RP = Raynaud's phenomenon; ACA = anticentromere antibodies; ANA = antinuclear antibodies; mRSS = modified Rodnan skin thickness score; mTOR = mechanistic target of rapamycin; TNF = tumor necrosis factor (see Figure 1 for other definitions).
Given its association with immune cell regulation, we focused on the immune cell regulation module. A closer inspection showed that genes in the module with the highest module membership level (i.e., the best representative of overall module gene expression) were strongly down‐regulated in dcSSc patients (Figure 2C), indicating that genes down‐regulated in dcSSc cDC2s are likely driving this module. Because transcription factors (TFs) are key regulators of gene expression, we next constructed a TF‐targeted network, based on known interactions from the REGNET and TRRUST databases. We identified 13 TFs in the immune cell regulation module that had target genes present in the same module (Figure 2D). Most notable were members of the NR4A family of nuclear receptors, NR4A1 and NR4A2, which have previously been reported as important regulators of inflammation and fibrosis (29, 30, 31, 32, 33, 34). Of all TFs identified, NR4A1 and NR4A3 displayed the highest module membership (0.87) and were the most strongly down‐regulated in dcSSc patients (NR4A3: log2 fold change −0.44, P = 0.0006; NR4A1: log2 fold change −0.38, P = 0.004) (Figure 2E). These results support the importance of NR4A as a regulatory factor in cDC2s and suggested that the down‐regulated expression of NR4A family members in dcSSc patients is of clinical relevance.
In the RNA‐sequencing cohort, all NR4A family members were significantly down‐regulated in dcSSc patients (Figure 3A). To validate this, we evaluated the expression levels of NR4A members using RT‐qPCR in cDC2s from another, independent cohort of 6 dcSSc patients and 7 healthy controls (Supplementary Table 1, available at https://onlinelibrary.wiley.com/doi/10.1002/art.42319). NR4A2 (P = 0.002) and NR4A3 (P = 0.035) were significantly down‐regulated in the dcSSc patient group (Figure 3B), and NR4A1 showed a nonsignificant trend (P = 0.18) for down‐regulation (Figure 3B). Nonetheless, expression levels of all NR4A family members in the RNA‐sequencing cohort and the RT‐qPCR validation cohort were strongly correlated with each other (Figure 3C), with consistent down‐regulation of all NR4A members in the cDC2s from dcSSc patients.
Figure 3.
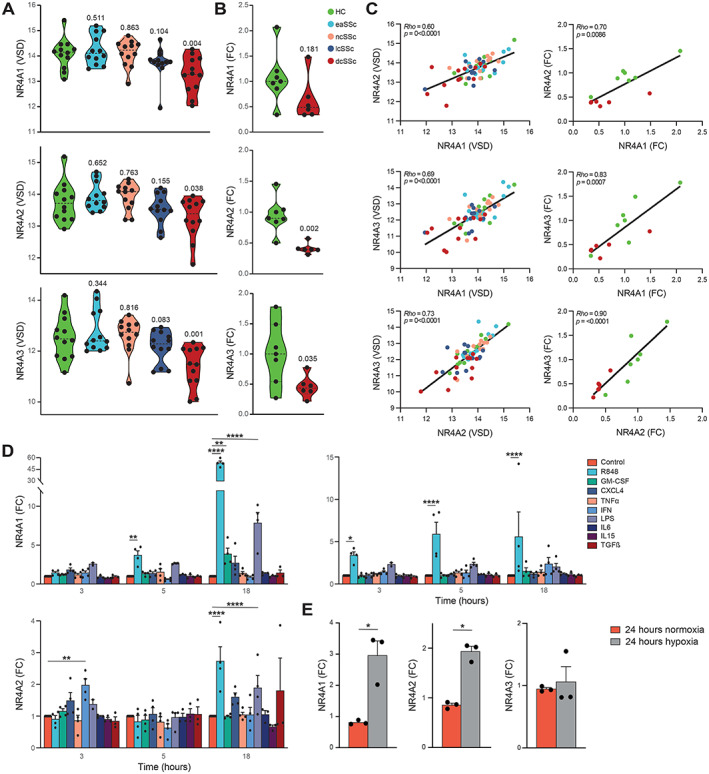
Characterization of expression of nuclear receptor 4A family members (NR4As) in cDC2s from SSc patients and healthy controls. A, Variance‐stabilized data (VSD) results comparing expression of NR4As in the SSc patient groups versus HC group from the RNA‐sequencing cohort (dashed lines indicate mean). Indicated P values comparing SSc patients with HCs were calculated using Wald's test implemented in DESeq2. B, Log2 fold change (FC) in expression of NR4As in representative samples from dcSSc patients compared to HCs in the validation cohort as measured by target‐specific reverse transcription–quantitative polymerase chain reaction. Indicated P values were calculated using Kruskal‐Wallis with Dunn's post hoc test. C, Correlations (regression lines) of expression among NR4As in the RNA‐sequencing cohort (left) and the validation cohort (right). Correlations were calculated using Spearman's rank correlation coefficient (rho). D, Fold change in expression of NR4As in cDC2s from HCs after stimulation for 3, 5, or 18 hours with Toll‐like receptor ligands and cytokines implicated in DC biology and SSc pathogenesis. E, Fold change in expression of NR4As in cDC2s from HCs cultured in normoxic or hypoxic conditions. Symbols in D and E represent individual experiments; bars show the mean ± SEM. * = P < 0.05; ** = P < 0.01; *** = P < 0.001; **** = P < 0.0001, by 2‐way analysis of variance with Dunnett's post hoc test for multiple comparisons of stimulated samples versus their own time point controls (D) or by paired‐sample t‐test (E). GM‐CSF = granulocyte–macrophage colony‐stimulating factor; IFN = interferon; LPS = lipopolysaccharide; TGFβ = transforming growth factor β (see Figure 1 for other definitions).
Association between proinflammatory stimulation of cDC2s from dcSSc patients and expression of NR4A
To investigate whether activation of cDC2s from SSc patients is associated with NR4A down‐regulation, we cultured freshly isolated cDC2s obtained from healthy control PB in the presence of various proinflammatory mediators for 3, 5, or 18 hours. NR4A expression was either induced or unaffected in all of the culture conditions studied (Figure 3D), with the strongest induction of expression observed after stimulation with the TLR‐7/TLR‐8 agonist R‐848 (resiquimod) for 18 hours (Figure 3D). To confirm that the stimuli were able to induce activation of cDC2s, we assessed IL‐6 expression on qPCR (Supplementary Figure 1A, available on the Arthritis & Rheumatology website at https://onlinelibrary.wiley.com/doi/10.1002/art.42319), which showed a strong up‐regulation of IL‐6 production in stimulated conditions.
Next, because hypoxia has been described as an important inflammatory hallmark in SSc (35), we investigated the effects of hypoxia on NR4A expression in cDC2s. Expression of NR4A1 and NR4A2 was induced under hypoxic conditions (Figure 3E).
To see whether dcSSc cDC2s had a capacity similar to healthy control cDC2s for activated NR4A expression after stimulation, we repeated the stimulation experiments on cDC2s from 5 dcSSc patients and 5 matched healthy controls. We found no significant difference in NR4A expression between R‐848–stimulated cDC2s from healthy controls and cDCs from SSc patients (Supplementary Figure 1B). These results support that an enhanced activation status of cDC2s does not underlie the decreased expression of NR4As observed in dcSSc patients but rather is a result of an intrinsic mechanism in cDC2s.
Role of NR4As in the direct regulation of crucial pathways dysregulated in cDC2s from SSc patients
To scrutinize the regulatory function of NR4As in cDC2s in steady state and after stimulation, we performed ChIP sequencing for NR4A1, NR4A2, and NR4A3 in resting and stimulated cDC2s. We identified numerous NR4A‐binding sites, of which some were specific to resting conditions (medium) and others specific to inflammatory conditions (R‐848) (Supplementary Figure 2 and Supplementary Table 6, available on the Arthritis & Rheumatology website at https://onlinelibrary.wiley.com/doi/10.1002/art.42319). In line with the known role of NR4A in neuronal development (36) and cardiac tissue development (37), we found a significant enrichment of NR4A binding at genes related to these processes (Figure 4A). Moreover, we identified NR4A binding in the promoters of genes related to cell morphogenesis, ECM organization, and chemotaxis (Figure 4A and Supplementary Table 7, available on the Arthritis & Rheumatology website at https://onlinelibrary.wiley.com/doi/10.1002/art.42319). These results demonstrate that NR4As are involved in the direct transcriptional regulation of various key SSc pathology–related processes in cDC2s.
Figure 4.
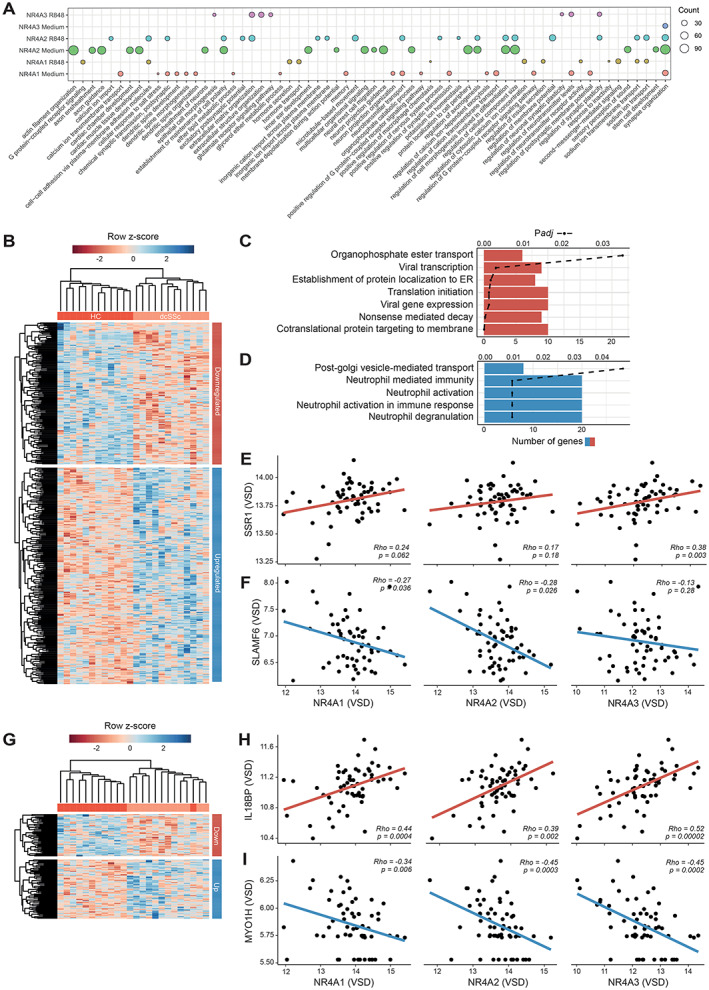
Chromatin immunoprecipitation (ChIP) sequencing of transcriptional regulation of cDC2s by nuclear receptor 4A family members (NR4As). A, Gene Ontology–term enrichment of genes that show NR4A binding within their promoter region. Circle size denotes number of genes associated with enriched biologic processes. Top 20 genes are shown (Benjamini and Hochberg–corrected P < 0.05). B, Heatmap of DEGs in dcSSc that display binding of NR4As at their promoters in resting cDC2s. C and D, Pathway enrichment of genes down‐regulated (C) and up‐regulated (D) in dcSSc with NR4A binding at their gene promoters in resting cDC2s. Bars depict the number of genes identified within the enriched pathway, and dashed lines indicate Benjamini and Hochberg–corrected P value. E and F, Scatterplots showing correlations between expression in NR4As and SSR1 (E) or SLAMF6 (F), calculated using Spearman's rank correlation coefficient. G, Heatmap of DEGs in dcSSc that display binding of NR4As at their promoters in stimulated cDC2s. H and I, Scatterplots showing correlations between expression of NR4As and IL18BP (H) or MYOH1 (I). In scatterplots (E, F, H, I), red regression lines (or “line of best fit”) indicate positive correlations and blue regression lines indicate negative correlations. ER = endoplasmic reticulum; VSD = variance‐stabilized data (see Figure 1 for other definitions).
Many genes directly bound by NR4As in resting cDC2s were also differentially expressed in cDC2s from dcSSc patients in the RNA‐sequencing cohort (Figure 4B). Moreover, in the RNA‐sequencing cohort, NR4A expression was correlated with genes associated with peptide translocation into the endoplasmic reticulum, including SSR1 (38) (Figure 4E), and inversely correlated with genes related to immune activation, including SLAMF6 (39) (Figure 4F). We also observed a strikingly lower overlap of DEGs with NR4A‐binding sites obtained from R‐848–stimulated cDC2s compared with resting cDC2s (Figure 4G). This observation reflects a potential loss of NR4A binding during cDC2 activation in the SSc group. Among the genes directly bound by NR4A in stimulated cDC2s, we identified anti‐inflammatory IL18BP (40), which was down‐regulated in dcSSc cDC2s and displayed a direct correlation with NR4A in the RNA‐sequencing cohort (Figure 4H). Among the up‐regulated genes, we found MYO1H, a molecule involved in cytokinesis, cell shape maintenance, and cell motility (41), which was inversely correlated with NR4A expression (Figure 4I). Taken together, these results demonstrate that NR4As are directly involved in transcriptional programs limiting cDC2 activation in vitro, at least in the culture‐based experimental conditions that we tested. Accordingly, the loss of NR4A expression in SSc cDC2s leads to induction of transcriptional programs associated with increased cellular activation.
Role of NR4A activation in limiting proinflammatory cytokine production by cDC2s from healthy controls and dcSSc patients
In our ChIP‐sequencing analysis, we found many inflammation‐related genes up‐regulated in dcSSc patients to be under the direct transcriptional control of NR4A TFs. We therefore hypothesized that down‐regulation of NR4As contributes to increased cytokine production previously observed in cDC2s from dcSSc patients (14). To test this hypothesis, we sought to identify whether activation of NR4A signaling could attenuate proinflammatory cytokine production by cDC2s from healthy controls and dcSSc patients. We treated freshly isolated cDC2s with increasing concentrations of C‐DIM5 (NR4A1 agonist) or C‐DIM12 (NR4A2 agonist) and measured the expression of GUSB to evaluate the effect of NR4A activation on viability. For C‐DIM5 and C‐DIM12, concentrations up to 10 μM and 25 μM were well tolerated (Figure 5A). Flow cytometry analysis also did not show a decrease in viability of cDC2s after treatment with 10 μM CDIM‐5 or 10 μM CDIM‐12 (Supplementary Figure 3A, available on the Arthritis & Rheumatology website at https://onlinelibrary.wiley.com/doi/10.1002/art.42319). Preincubation of cDC2s with 10 μM C‐DIM5 or 10 μM C‐DIM12 before stimulation with R‐848 led to a significant decrease in IL‐6 production, as shown in both messenger RNA and protein levels (Figure 5B). These data confirm the suppressive role of NR4As in proinflammatory cytokine production in cDC2s.
Figure 5.
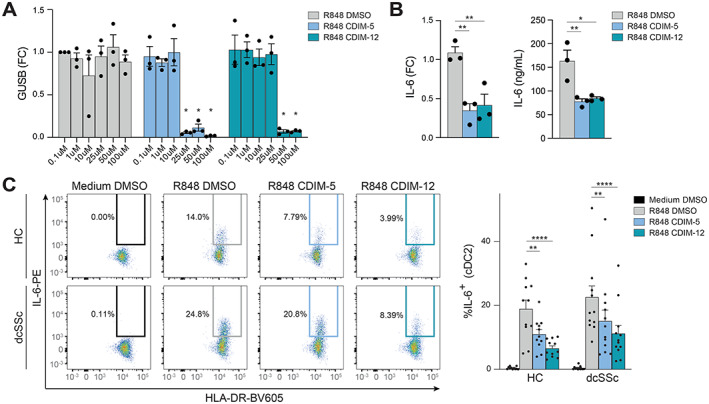
Nuclear receptor 4A (NR4A) activation leads to a decrease in the production of interleukin‐6 (IL‐6) in healthy control cDC2s and dcSSc cDC2s. A, Reverse transcription–quantitative polymerase chain reaction of GUSB mRNA expression by freshly isolated cDC2s after preincubation with increasing concentrations of DMSO (negative control) or NR4A agonists C‐DIM5 and C‐DIM12, followed by overnight stimulation with R‐848. Results are shown as fold change (FC) compared with 0.1 μM DMSO. * = P < 0.05, by 2‐way analysis of variance (ANOVA) with Dunnett's post hoc test comparing treated samples versus DMSO control with matching concentrations. B, IL‐6 mRNA (left) and protein expression (right) in cDC2s pretreated with 10 μM DMSO, C‐DIM5, or C‐DIM12, followed by overnight stimulation with R‐848. Relative mRNA expression levels (FC) are normalized to GUSB housekeeping levels. * = P < 0.05; ** = P < 0.01, by 1‐way ANOVA followed by Friedman's test for multiple comparisons. C, Left, Representative flow cytometry plots of percentage of IL‐6–positive cells within the cDC2 fraction in peripheral blood mononuclear cell cultures pretreated with 10 μM DMSO, C‐DIM5, or C‐DIM12, followed by overnight stimulation with R‐848. Right, Quantification of flow cytometry data. Symbols represent individual experiments; bars show the mean ± SEM. ** = P < 0.01; **** = P < 0.001, by 2‐way ANOVA with Dunnett's post hoc test. See Figure 1 for definitions.
To substantiate these results and evaluate the effects of NR4A agonists on dcSSc cDC2s, we repeated this experiment using PBMC samples obtained from 12 dcSSc patients and 11 matched healthy controls. Intracellular flow cytometry (Supplementary Figure 3B) again showed that NR4A activation led to a significant decrease of IL‐6 production by cDC2s (Figure 5C). Notably, NR4A activation also led to a significant decrease of IL‐6 production in dcSSc cDC2s, demonstrating that NR4A activation can effectively attenuate proinflammatory cytokine production in these patients.
Role of NR4A activation in decreasing the CD4+ T cell stimulatory capacity of cDC2s
Considering that NR4As are regulators of IL‐6 production by cDC2s (Figure 5) and IL‐6 is a critical contributor to the priming of CD4+ T cells by cDC2s, we next investigated the role of NR4As in priming cDC2s for autologous CD4+ T cell activation (Figure 6A and Supplementary Figure 4A, available on the Arthritis & Rheumatology website at https://onlinelibrary.wiley.com/doi/10.1002/art.42319). When pretreated with the NR4A agonists C‐DIM5 and C‐DIM12, cDC2s were less capable of inducing IFNγ production by CD4+ T cells (Figure 6B). These results held true for cDC2s stimulated with R‐848, showing that NR4A activation also attenuates CD4+ T cell induction by cDC2s under proinflammatory conditions (Figure 6C). We also detected intracellular IFNγ in the cDC2 cocultures. No significant difference was shown after treatment with different agonists, although we observed a trend for lower production in agonist‐treated cDC2s (Supplementary Figure 4B). These data demonstrate that, beside controlling the expression of proinflammatory cytokines, NR4As also have the capacity to control T cell activation by cDC2s.
Figure 6.
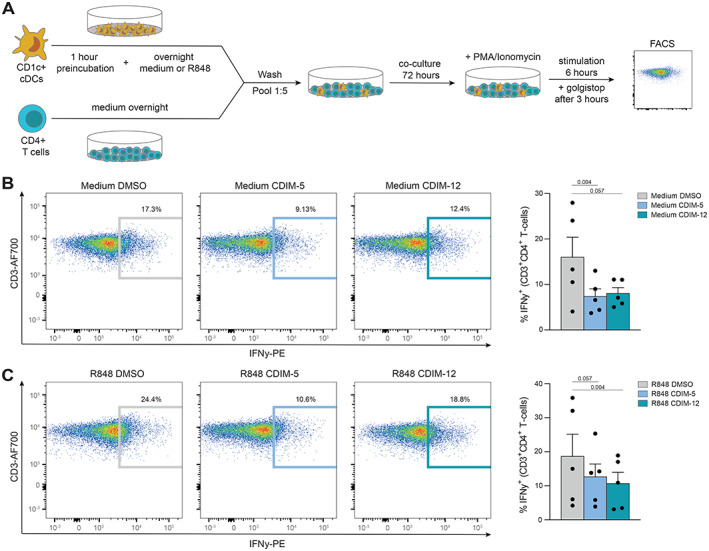
Nuclear receptor 4A (NR4A) activation leads to decreased CD4+ T cell activation by cDC2s. A, Schematic overview of the coculture setup of freshly isolated autologous CD4+ T cells and cDC2s. B and C, Representative flow cytometry plots of the percentage of interferon‐γ (IFNγ)–positive T cells within the CD4+ T cell fraction after 3 days of coculture with cDC2s pretreated with 10 μM DMSO, C‐DIM5, or C‐DIM12, followed by overnight culture in medium (B) or overnight stimulation with R‐848 (C). Symbols in B and C represent individual experiments; bars show the mean ± SEM. Indicated P values, comparing C‐DIM5 or C‐DIM12 versus DMSO treatment, were calculated by one‐way analysis of variance followed by Friedman's test for multiple comparisons. PMA = phorbol myristate acetate; FACS = fluorescence‐activated cell sorting; PE = phycoerythrin (see Figure 1 for other definitions).
DISCUSSION
Previous studies have shown that cDCs are important immune cells involved in SSc pathogenesis (11, 12, 13, 14). However, the key molecular mechanisms underlying their dysregulation are so far unknown. Here, we provide detailed transcriptomic profiles of cDC2s from SSc patients and from healthy controls, which allowed us to characterize their transcriptomic landscape. We identified various clinically relevant modules of coexpressed genes, enriched in pathways highly relevant for SSc and cDC biology, including immune cell regulation, antigen presentation, antiviral mechanisms, and translation. In particular, a module of immunoregulatory genes was down‐regulated in cDC2s from dcSSc patients. TF network analysis supported that the NR4A family of orphan nuclear receptors may be important regulators of this module. Our results implicated NR4As as key regulators of inflammatory responses by cDC2s in SSc. In addition to NR4As, our results showed that other TFs may also be involved in this process. These include the early growth response (EGR) family TFs, which are known to repress proinflammatory responses in macrophages (42). Future studies could investigate the possible implication of these TFs in SSc pathogenesis.
NR4As regulate gene expression in a ligand‐independent manner, and their activity is largely dependent on expression levels and posttranslational modifications (43, 44). NR4As play critical roles in the regulation of immune cell activation (45, 46, 47), and NR4A1 in particular has been established as a regulator of proinflammatory responses in DCs (32). Our ChIP‐sequencing analysis provided detailed insights into the direct binding of NR4As at their target gene promoters in human cDC2s. Because studies on ChIP sequencing of these cells are rare and mostly limited to model systems like monocyte‐derived DCs, our data can provide a valuable resource for future studies on NR4As in general, as well as on cDC2s and SSc. We showed that NR4As are strongly involved in transcriptional programs underlying DC dysregulation in SSc. In addition to inflammation, these programs include the regulation of morphology and ECM production under stimulated conditions. Given the reduced expression of NR4As in circulating cDC2s from dcSSc patients, our results suggest that dcSSc cDC2s might show an enhanced expression of ECM‐related genes once they get activated, for example, upon migration to the skin. Interestingly, inflammatory DCs have been implicated in fibrosis in SSc via the increased secretion of ECM molecules such as fibronectin and α‐smooth muscle actin, which promotes myofibroblast differentiation (48). Although more detailed ChIP‐sequencing analyses are needed to quantify NR4A binding in dcSSc cDC2s and healthy donor cDC2s, as well as to evaluate modulation of target genes at the protein level, so that these results can be validated, our analysis points toward NR4As as major transcriptional regulators of pathways implicated in cDC2 dysregulation in SSc.
We showed that activation of NR4As in cDC2s by selective agonists attenuates the release of the proinflammatory cytokine IL‐6 and the downstream activation of CD4+ T cells. Importantly, although cDC2s from dcSSc patients in our study demonstrated down‐regulated expression of NR4A1, NR4A2, and NR4A3, we observed that activation of NR4A signaling by the agonists C‐DIM5 and C‐DIM12 inhibited IL‐6 production in dcSSc cDC2s. Interestingly, IL‐6 is an important cytokine linked to SSc pathogenesis, and levels of this cytokine are linked to worsening of clinical outcomes and increased fibrosis (49). Additionally, the NR4A1 agonist cytosporone B has been previously shown to ameliorate collagen deposition and myofibroblast differentiation in mouse models of bleomycin‐induced fibrosis (33), highlighting a role for NR4As at the crossroads of fibrosis and inflammation. Although it remains to be investigated to what extent these antifibrotic effects are mediated through modulation of cDC2s, small‐molecule agonists that overcome the reduced expression of NR4A in cDC2s may represent potential targets for immunotherapy in patients with SSc. Because SSc patients with early diffuse phenotypes display signs of enhanced DC activation with increased IL‐6 production (14), targeting NR4As early in SSc pathogenesis might prevent DC activation at prefibrotic stages and limit disease progression. With cDC2s having an indispensable role in CD4+ T cell activation, the early targeting of NR4As also has the potential to modulate the downstream adaptive immune response in SSc patients toward a more tolerant state.
Although the factors that underlie NR4A down‐regulation in dcSSc cDC2s remain to be resolved, our experiments do provide new insights. Consistent with the roles of NR4As as immediate early response genes (43), stimulation of cDC2s with TLR ligands R‐848 and LPS, as well as hypoxia (which has been linked to SSc pathogenesis [35]), did overall induce the expression of NR4A1, NR4A2, and NR4A3. Also, the cytokines known to be increased in the PB of SSc patients or related to SSc pathogenesis, including CXCL4, IFNα, TGFβ2, GM‐CSF, IL‐6, and IL‐15, did not reduce NR4A expression, at least at the concentrations and time points that we included in our study. Moreover, stimulation of freshly isolated cDC2s from dcSSc patients with the TLR‐7/TLR‐8 ligand R‐848 also led to an induction of NR4A expression, comparable to levels in cDC2s from healthy donors, suggesting that the upstream transcriptional regulation of NR4As is not defective.
Given the well‐described heterogeneity of the cDC2 subset (50, 51), one might propose that the down‐regulation of NR4A expression that we observed in bulk cDC2s from dcSSc patients may reflect an imbalance among distinct populations within the cDC2 subset. Indeed, expression of NR4A2 and NR4A3 is low in CD1c+Tbet− cDCs (also known as cDC2B), an inflammatory DC population within the cDC2 compartment (52). However, in a recent analysis from Dutertre et al of the composition of cDC2 subsets in the blood of SSc patients, the proportions of distinct cDC2 subpopulations in SSc were not different from those in healthy samples (51). Thus, the down‐regulation of NR4As that we observed in dcSSc cDC2s may not be attributed to heterogeneity within the DC compartment. The exact molecular mechanisms causing NR4A down‐regulation in dcSSc cDC2s remain to be investigated. These mechanisms include alterations in the chromatin landscape or regulation at the posttranscriptional level.
In conclusion, we show that the NR4A TF family members NR4A1, NR4A2, and NR4A3 are important regulators underlying cDC2 dysregulation in SSc. We propose that the pharmacologic activation of NR4As is an attractive therapeutic candidate to attenuate proinflammatory and profibrotic responses in SSc patients, in an untargeted manner or through the use of DC‐directed approaches.
AUTHOR CONTRIBUTIONS
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Dr. Servaas had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design
Servaas, Rossato, Radstake, Kuiper, Boes, Pandit.
Acquisition of data
Servaas, Chouri, Wichers, Affandi, Ottria, Bekker, Cossu, Silva‐Cardoso, van der Kroef, Hinrichs, Carvalheiro, Vazirpanah, Beretta, Rossato, Bonte‐Mineur, Radstake, Kuiper, Boes, Pandit.
Analysis and interpretation of data
Servaas, Hiddingh, Kuiper, Boes, Pandit.
Supporting information
Disclosure Form
Figure S1
Figure S2
Figure S3
Figure S4
Table S1
Table S4
Table S5
Table S6
Table S7
ACKNOWLEDGMENTS
We thank A. Pinheiro Lopes for her experimental advice on functional experiments with cDCs. We also thank E. Ton, J. M. van Laar, and J. Spierings from the Department of Rheumatology & Clinical Immunology at the University Medical Centre Utrecht for their help with patient inclusion. We are grateful to the patients and healthy control donors who participated in this study.
Dr. Pandit's work was supported by The Netherlands Organisation for Scientific Research (grant 016.Veni.178.027).
Drs. Kuiper, Boes, and Pandit contributed equally to this work.
Author disclosures are available at https://onlinelibrary.wiley.com/action/downloadSupplement?doi=10.1002%2Fart.42319&file=art42319‐sup‐0001‐Disclosureform.pdf.
Contributor Information
Marianne Boes, Email: m.l.boes@umcutrecht.nl.
Aridaman Pandit, Email: a.pandit@umcutrecht.nl.
REFERENCES
- 1. Gabrielli A, Avvedimento EV, Krieg T. Scleroderma. N Engl J Med 2009;360:1989–2003. [DOI] [PubMed] [Google Scholar]
- 2. Van den Hoogen F, Khanna D, Fransen J, et al. 2013 classification criteria for systemic sclerosis: an American College of Rheumatology/European League against Rheumatism collaborative initiative. Arthritis Rheum 2013;65:2737–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. LeRoy EC, Medsger TA. Criteria for the classification of early systemic sclerosis. J Rheumatol 2001;28:1573–6. [PubMed] [Google Scholar]
- 4. Guiducci S, Giacomelli R, Cerinic MM. Vascular complications of scleroderma. Autoimmun Rev 2007;6:520–3. [DOI] [PubMed] [Google Scholar]
- 5. Cutolo M, Soldano S, Smith V. Pathophysiology of systemic sclerosis: current understanding and new insights. Expert Rev Clin Immunol 2019;15:753–64. [DOI] [PubMed] [Google Scholar]
- 6. Rajkumar VS, Howell K, Csiszar K, et al. Shared expression of phenotypic markers in systemic sclerosis indicates a convergence of pericytes and fibroblasts to a myofibroblast lineage in fibrosis. Arthritis Res Ther 2005;7:R1113–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Binai N, O'Reilly S, Griffiths B, et al. Differentiation potential of CD14 + monocytes into myofibroblasts in patients with systemic sclerosis. PLoS One 2012;7:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Guermonprez P, Valladeau J, Zitvogel L, et al. Antigen presentation and T cell stimulation by dendritic cells. Annu Rev Immunol 2002;20:621–67. [DOI] [PubMed] [Google Scholar]
- 9. Tzeng TC, Chyou S, Tian S, et al. CD11c(hi) dendritic cells regulate the reestablishment of vascular quiescence and stabilization after immune stimulation of lymph nodes. J Immunol 2010;184:4247–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ding L, Liu T, Wu Z, et al. Bone marrow CD11c+ cell–derived amphiregulin promotes pulmonary fibrosis. J Immunol 2016;197:303–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Carvalheiro T, Zimmermann M, Radstake TR, et al. Novel insights into dendritic cells in the pathogenesis of systemic sclerosis. Clin Exp Immunol 2020;201:25–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mathes AL, Christmann RB, Stifano G, et al. Global chemokine expression in systemic sclerosis (SSc): CCL19 expression correlates with vascular inflammation in SSc skin. Ann Rheum Dis 2014;73:1864–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mokuda S, Miyazaki T, Ubara Y, et al. CD1a+ survivin+ dendritic cell infiltration in dermal lesions of systemic sclerosis. Arthritis Res Ther 2015;17:275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Van Bon L, Popa C, Vonk M, et al. Distinct evolution of TLR‐mediated dendritic cell cytokine secretion in patients with limited and diffuse cutaneous systemic sclerosis. Ann Rheum Dis 2009;69:1539–47. [DOI] [PubMed] [Google Scholar]
- 15. Sittig SP, Bakdash G, Weiden J, et al. A comparative study of the T cell stimulatory and polarizing capacity of human primary blood dendritic cell subsets. Mediators Inflamm 2016:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kalogerou A, Gelou E, Mountantonakis S, et al. Early T cell activation in the skin from patients with systemic sclerosis. Ann Rheum Dis 2005;64:1233–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. O'Reilly S, Hugle T, Van Laar JM. T cells in systemic sclerosis: a reappraisal. Rheumatology (Oxford) 2012;51:1540–9. [DOI] [PubMed] [Google Scholar]
- 18. Servaas NH, Zaaraoui‐Boutahar F, Wicher CG, et al. Longitudinal analysis of T‐cell receptor repertoires reveals persistence of antigen‐driven CD4+ and CD8+ T‐cell clusters in systemic sclerosis. J Autoimmun 2021;117:102574. [DOI] [PubMed] [Google Scholar]
- 19. Dobin A, Davis CA, Schlesinger F, et al. STAR: ultrafast universal RNA‐seq aligner. Bioinformatics 2013;29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Anders S, Pyl PT, Huber W. HTSeq—a Python framework to work with high‐throughput sequencing data. Bioinformatics 2015;31:166–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Risso D, Schwartz K, Sherlock G, et al. GC‐content normalization for RNASeq data. BMC Bioinformatics 2011;12:480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Risso D, Ngai J, Speed TP, et al. Normalization of RNA‐seq data using factor analysis of control genes or samples. Nat Biotechnol 2014;32:896–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biol 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 2008;9:559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Langmead B, Salzberg SL. Fast gapped‐read alignment with Bowtie 2. Nat Methods 2012:9:357–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gaspar JM. Improved peak‐calling with MACS2. bioRxiv. 2018. [Google Scholar]
- 27. Yu G, Wang LG, He QY. ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 2015;31:2382–3. [DOI] [PubMed] [Google Scholar]
- 28. Yu G, Wang LG, Han Y, et al. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 2012;16:284–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Haub J, Roehrig N, Uhrin P, et al. Intervention of inflammatory monocyte activity limits dermal fibrosis. J Invest Dermatol 2019;139:2144–53. [DOI] [PubMed] [Google Scholar]
- 30. Zeng X, Yue Z, Gao Y, et al. NR4A1 is involved in fibrogenesis in ovarian endometriosis. Cell Physiol Biochem 2018;46:1078–90. [DOI] [PubMed] [Google Scholar]
- 31. Mahajan S, Saini A, Chandra V, et al. Nuclear receptor Nr4a2 promotes alternative polarization of macrophages and confers protection in sepsis. J Biol Chem 2015;290:18304–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tel‐Karthaus N, Kers‐Rebel ED, Looman MW, et al. Nuclear receptor Nur77 deficiency alters dendritic cell function. Front Immunol 2018;9:1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Palumbo‐Zerr K, Zerr A, Distler P, et al. Orphan nuclear receptor NR4A1 regulates transforming growth factorβ signaling and fibrosis. Nat Med 2015;21:150–8. [DOI] [PubMed] [Google Scholar]
- 34. Ma C, Wu L, Song L, et al. The pro‐inflammatory effect of NR4A3 in osteoarthritis. J Cell Mol Med 2020;24:930–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Van Hal TW, van Bon L, Radstake TR. A system out of breath: how hypoxia possibly contributes to the pathogenesis of systemic sclerosis. Int J Rheumatol 2011;2011:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Eells JB, Wilcots J, Sisk S. NR4A gene expression is dynamically regulated in the ventral tegmental area dopamine neurons and is related to expression of dopamine neurotransmission genes. J Mol Neurosci 2012;46:545–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Medzikovic L, de Vries CJ, de Waard V. NR4A nuclear receptors in cardiac remodeling and neurohormonal regulation. Trends Cardiovasc Med 2019;29:429–37. [DOI] [PubMed] [Google Scholar]
- 38. Vogel F, Hartmann E, Gorlich D, et al. Segregation of the signal sequence receptor protein in the rough endoplasmic reticulum membrane. Eur J Cell Biol 1990;53:197–202. [PubMed] [Google Scholar]
- 39. Chatterjee M, Rauen T, Kis‐Toth K, et al. Increased expression of SLAM receptors SLAMF3 and SLAMF6 in systemic lupus erythematosus T lymphocytes promotes Th17 differentiation. J Immunol 2012;188:1206–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Dinarello CA, Novick D, Kim S, et al. Interleukin‐18 and IL‐18 binding protein. Front Immunol 2013;4:289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Maravillas‐Montero JL, Santos‐Argumedo L. The myosin family: unconventional roles of actin‐dependent molecular motors in immune cells. J Leukoc Biol 2012;91:35–46. [DOI] [PubMed] [Google Scholar]
- 42. Trizzino M, Zucco A, Deliard S, et al. EGR1 is a gatekeeper of inflammatory enhancers in human macrophages. Sci Adv 2021;7:eaaz8836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Maxwell MA, Muscat GE. The NR4A subgroup: immediate early response genes with pleiotropic physiological roles. Nucl Recept Signal 2006;4:nrs.04002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Fahrner TJ, Carroll SL, Milbrandt J. The NGFI‐B protein, an inducible member of the thyroid/steroid receptor family, is rapidly modified posttranslationally. Mol Cell Biol 1990;10:6454–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Koenis DS, Medzikovic L, van Loenen PB, et al. Nuclear receptor Nur77 limits the macrophage inflammatory response through transcriptional reprogramming of mitochondrial metabolism. Cell Rep 2018;24:2127–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Chen J, López‐Moyado IF, Seo H, et al. NR4A transcription factors limit CAR T cell function in solid tumours. Nature 2019;567:530–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. McEvoy C, de Gaetano M, Giffney HE, et al. NR4A receptors differentially regulate NFκB signaling in myeloid cells. Front Immunol 2017;8:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Silva‐Cardoso SC, Tao W, Angiolilli C, et al. CXCL4 links inflammation and fibrosis by reprogramming monocyte‐derived dendritic cells in vitro. Front Immunol 2020;11:2149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Khan K, Xu S, Nihtyanova S, et al. Clinical and pathological significance of interleukin‐6 overexpression in systemic sclerosis. Ann Rheum Dis 2012;71:1235–42. [DOI] [PubMed] [Google Scholar]
- 50. Villani AC, Satija R, Reynolds G, et al. Single‐cell RNA‐seq reveals new types of human blood dendritic cells, monocytes, and progenitors. Science 2017;356:eaah4573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Dutertre CA, Becht E, Irac SE, et al. Single‐cell analysis of human mononuclear phagocytes reveals subset‐defining markers and identifies circulating inflammatory dendritic cells. Immunity 2019;51:573–89. [DOI] [PubMed] [Google Scholar]
- 52. Brown CC, Gudjonson H, Pritykin Y, et al. Transcriptional basis of mouse and human dendritic cell heterogeneity. Cell 2019;179:846–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Disclosure Form
Figure S1
Figure S2
Figure S3
Figure S4
Table S1
Table S4
Table S5
Table S6
Table S7