

Abstract
A designed N‐heterocyclic carbene (NHC) catalyst was covalently anchored on a range of mesoporous and hierarchical supports, to study the influence of pore size in the benzoin condensation of furfural. The structural and spectroscopic characteristics of the anchored catalysts were investigated, also with the help of molecular dynamics simulations, in order to rationalize the degree of stability and recyclability of the heterogenized organocatalysts. Quantitative yields (99 %) and complete recyclability were maintained after several cycles, vindicating the design rationale.
Keywords: benzoin condensation, furfural transformation, heterogeneous catalysis, N-heterocyclic carbene based catalysts, organic-inorganic hybrid catalyst
Benzimidazole exhibited divergent stability when covalently anchored on mesoporous and hierarchical supports in the benzoin condensation of furfural. The highest stability was achieved with 150 Å Davisil silica in DMF. Silica supports with smaller mesopores hindered benzimidazole stability due to van der Waals interactions with silica walls, resulting in poor recyclability of the hybrid organocatalysts.

1. Introduction
Due to the increased generation of waste and depletion of natural resources, the use of renewable resources for manufacturing fine chemicals, pharmaceuticals and fuels is gaining momentum. For this reason, biomass valorisation has emerged as an important area of research. [1] In particular, furfural (FF) is a cheap platform molecule that can be easily obtained at large scale from the dehydration of carbohydrates. [2]
In this study, we envisioned to merge the soft reaction conditions of organocatalytic chemistry to probe the recyclability of heterogeneous catalysts. [3] From the beginning of this century, organocatalysis has become a powerful tool, relying on the capability of small organic molecules to catalyse organic transformations. [4] Based on this approach, we target a furoin intermediate with potential application as chemical precursor in the synthesis of jet or diesel liquid fuels. [5]
Designing organocatalysts which can be stabilized and heterogenized on inorganic supports, can lead to extended catalyst lifetimes and enhanced recyclability for reactions encompassing FF. Herein we studied the benzoin condensation of FF promoted by a N‐heterocyclic carbene (NHC) catalyst (i. e. benzimidazole or bim, Scheme S1A, see SI), that was integrated within a range of pore apertures in solid supports, aiming to obtain a robust, reliable and recyclable bim‐supported heterogeneous catalyst. [6] We tested five different porous supports for NHC: mesoporous ordered (SBA‐15) and non‐ordered (Davisil) silicas with different pore diameters (150 Å and 30 Å), and hierarchical aluminophosphates (HP‐AlPO‐5) and silicoaluminophosphates (HP‐SAPO‐5). The acronyms used to define the five hybrids are reported in Table S1, along with the full description of the main characteristics of the solid supports used. The organic‐inorganic interface and stability of bim moieties after reaction were investigated to establish structure‐property relationships, by combining solid‐state (ss) NMR, FTIR spectroscopy and Molecular Dynamics (MD) simulations, with emphasis on the role of support pore diameters in driving the catalytic performance.
2. Results and Discussion
The established mechanism for the benzoin condensation of FF is depicted in Scheme S1B (see Supporting Information). First, the NHC catalyst is activated by deprotonation with a base (1,8‐diazabicyclo[5.4.0] undec‐7‐ene, DBU), followed by nucleophilic attack to one FF molecule. Next, proton transfer occurs, leading to the formation of a Breslow/Enders intermediate that further attacks a second FF molecule. [7] The furoin product is obtained after subsequent proton transfer and release of the NHC catalyst.
Using imidazole did not give any conversion in the benzoin condensation of FF, while a NHC catalyst based on a bim core resulted in high conversion (Table S2). The supported bim precursor was synthesized starting from bim (Scheme 1 and Supporting Information for experimental details), reacting first with 1‐bromododecane to give intermediate 1 and then with (3‐iodopropyl)trimethoxysilane to give the catalyst precursor 2. This was covalently anchored on the different supports using the trimethoxysilyl handle, to render hybrid catalyst 3. Bim anchoring was confirmed by elemental analysis, giving values in the range of 0.3–0.7 mmol/g (Table S1).
Scheme 1.
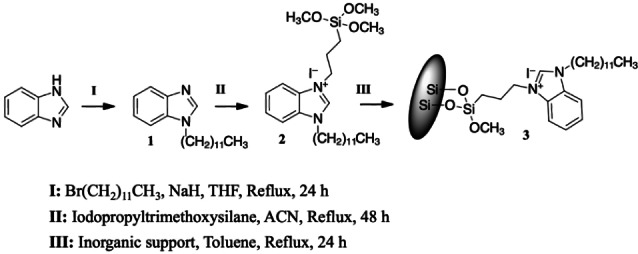
Synthesis and NHC anchoring on inorganic silica support.
The results for the different hybrid catalysts, along with their recycling data, are summarized in Figure 1. Davisil and SBA‐15 are mesoporous silicas with pore apertures in the range of 80–150 Å (Table S1 in Supporting Information).[ 8 , 9 ] To study the influence of the pore size, we used a comparable mesoporous Davisil silica with a pore aperture of 30 Å. We also employed supports with hierarchical porosity, such as HP‐AlPO‐5 and HP‐SAPO‐5. Both supports were previously reported by some of us,[ 10 , 11 , 12 ] adopting a one‐pot soft‐templating strategy, where an organosilane surfactant, [13] i.e. dimethyloctadecyl[(3‐(trimethoxysilyl)propyl] ammonium chloride (DMOD), was used together with a microporous structure‐directing agent such as trimethylamine (TEA) or tetraethyl ammonium hydroxide (TEAOH) to synthesize the hierarchical structure. In this approach, the siliceous component of the surfactant incorporates within the hierarchical framework and, upon removal of the surfactant by calcination in air, Si−OH groups are generated within the inner walls of the mesoporous architecture. It is envisaged that these Si−OH groups could serve as handles for the covalent anchoring of the organocatalysts. It is worth noting that HP‐SAPO‐5 also incorporates isomorphously substituted Si(IV) Brønsted acid centres as part of the microporous framework, [13] which could lead to functionalization by the organocatalyst at the external surface of micropores. For comparison, we used HP‐AlPO‐5, which does not contain Si(IV) Brønsted acid sites, but silanol sites, owing to the synthetic strategy involving DMOD. [14]
Figure 1.

Furoin conversion obtained for the different hybrid catalysts. The furoin selectivity is 100 % in all the tests, with a carbon balance >99 %.
When bim was anchored on Davisil silica with the largest pore aperture (150 Å), the reaction yields quantitative FF conversion, and the hybrid catalyst can be recycled and reused for several runs. Using SBA‐15 with a slightly smaller pore size (80 Å), the FF conversion and recycle potential are also good, though slightly lower than the 150 Å Davisil silica support. Both supports exhibit large mesopores bearing pendant silanol groups, thus facilitating covalent bim anchoring on the inner mesopore walls. In contrast, when Davisil silica (30 Å) is used as support, it is highly likely that the steric bulk of the bim precursor facilitates functionalization to the more available external silanol groups instead of internal mesopores. The hybrid catalyst affords full FF conversion after the first run, but rapidly loses its activity after the first recycle (66 %), with no further conversion after the second recycle. This observation suggests that bim either deactivates, or is more likely removed from the support during the reaction.
The performance of HP‐SAPO‐5 mirrors that of Davisil silica (30 Å), with no FF conversion observed after the first recycle. Analogous to Davisil silica (30 Å), bim is expected to be more facilely anchored on the readily available isomorphously substituted Si−OH groups located on the external surface of micropores. Notably, when isomorphously substituted Si−OH groups are not present on the external surface of the support, as for HP‐AlPO‐5, the catalyst retains its activity after the second recycle, in stark contrast with HP‐SAPO‐5.
The loading of the benzimidazole was evaluated, both before and after the catalytic tests, for all the catalysts. We did not observe any leaching of the benzimidazole with the NHC/SiO2(150 Å), NHC/SBA‐15 and NHC/HP‐AlPO‐5 catalysts. With the NHC/SiO2(30 Å) and NHC/HP‐SAPO‐5, we observed a decrease (20 % and 38 %, respectively) in benzimidazole loading, post catalysis, which vindicates the findings in Figure 1.
In order to rationalize the divergent catalytic performance and recyclability of the five hybrid catalysts, we conducted a comprehensive characterization study combining ss NMR, FTIR spectroscopy and MD simulations. First, the thermal stability of NHC anchored on the different supports was monitored by FTIR spectroscopy using different outgassing temperatures (Figure S1). In all cases, characteristic bands ascribed to the bim moiety are visible in the range 3050–2800 cm−1, 1650–1400 cm−1 and at 1565 cm−1 that are ascribed to C−H stretching, C−C ring and C−N stretching mode of the imidazole ring in bim, respectively. [15] The intensity of all bands decreases upon outgassing at higher temperature. In particular, the latter signal decreases significantly at 250 °C and totally disappears at 400 °C, pointing out complete bim removal from the support.
Next, 1H MAS NMR was used to gain insight into the nature of Si−OH groups responsible for the grafting of the organic moiety (Figure S2). All the calcined and dehydrated supports (Figure S2A) exhibit a characteristic signal ascribed to isolated Si−OH groups (1.8–2.0 ppm), while Brønsted acid sites (3.8–5.0 ppm) are clearly visible for HP‐AlPO‐5 and HP‐SAPO‐5. [16] In the latter samples, two Brønsted acid sites are clearly visible corresponding to different positions in the AFI framework, which is consistent with the FTIR spectra. [17] Upon bim grafting (Figure S2B), the signal at 1.8 ppm due to Si−OH groups vanishes, while the signals ascribed to Brønsted acid sites are still present, confirming that only Si−OH groups are used to anchor the bim precursor. Additional signals are visible in the 0–3 ppm range due to the aliphatic chain on the N‐pyrrolic ring, as well as a broad signal from 6.5 to 11 ppm that is attributed to aromatic protons. The imidazole proton, which appears at 11 ppm in the liquid phase, also appears in this range. [18]
Successful bim grafting was also confirmed by 29Si MAS NMR and 13C CPMAS NMR. The 29Si MAS NMR spectra (Figure 2A, Figure S3) display characteristic bands from −50 ppm to −70 ppm that are assigned to T‐type silicon species [T3 (RSi(OSi)3) and T2 (RSi(OSi)2OH)] having a Si−C bond. [19] Bands at around −92, −100 and −110 ppm are also apparent, which are ascribed to Q2 (Si(OH)2(OSi)2), Q3 (Si(OH)(OSi)3) and Q4 (Si(OSi)4) units. [20] In the 13C MAS NMR spectra (Figure 2C, Figure S4), the bands ascribed to 13C in aliphatic chains appear in the 20–35 ppm range, while terminal methyl and methylene carbons, as well as propyl linker carbons, resonate in the 5–15 ppm range. At around 50 ppm, the band belonging to the two N−CH2 groups is visible, while the signals of the carbon atoms of the bim moiety appear in the 110–150 ppm range. [21]
Figure 2.

29Si (A and B) and 13C (C and D) ss NMR spectra of NHC anchored on different supports before (A and C) and after reaction (B and D). Curve a: NHC/Davisil 150 Å, curve b: NHC/Davisil 30 Å, curve c: NHC/SBA‐15, curve d: NHC/HP‐SAPO‐5 and curve e: NHC/HP‐AlPO‐5. Detailed 13C assignments are given in the Supporting Information, as well as 29Si MAS NMR spectra of the pristine inorganic supports (Figure S3).
As pointed out above, bim anchored on wider silica pores, either non‐ordered (Davisil 150 A) or ordered (SBA‐15), retain a high FF conversion after two recycles, while it declines after the first run for supports with smaller pores. To elucidate the catalyst integrity after reaction, 29Si and 13C MAS NMR were used. The Si environment does not change after the reaction (Figure 2B), while 13C MAS NMR spectra (Figure 2D) reveal a new signal at ca. 165 ppm that is visible for all hybrids except for bim/Davisil 150 Å. This signal can be attributed to protonated DBU, the organic base used for in situ bim activation, [22] and can explain the better recyclability of bim/Davisil 150 Å with respect the other hybrids. The presence of protonated DBU adsorbed on the catalyst surface, in fact, can partially hinder the active sites, thus leading to lower performance.
MD simulations were used to explore the effect of the pore size and Si−OH concentration on the conformation of grafted bim moieties (Figure 3A), which can affect the catalytic activity of the hybrid catalysts. The relative stability of the hybrids was also studied by MD using the thermodynamic integration method (i. e. Bennet Acceptance Radius, BAR) as described in the Experimental Section.[ 23 , 24 , 25 ] Several models were used to describe the hybrid catalysts, anchoring a bim molecule either to a flat silica surface (Figure 3B), or within a pore width 3 or 10 nm (Figure 3C–D). All the models were evaluated at two different Si−OH surface concentrations, i. e. 3 and 7 nm−2. Then suitable MD runs (0.5 ns equilibration, 1 ns production) were performed at 298 K by monitoring the distance between the carbene atom (Figure 3A, C atom indicated by the arrow), and the closest silicon atom on the surface, to assess if bim interactions with the solid reduce the availability and hence the efficiency of the bim moiety. The simulations were conducted both in vacuo and in a box filled with dimethylformamide (DMF) molecules at the appropriate density (i. e. 3 and 7 nm−2). Representative snapshots of the MD runs (with low Si−OH concentration, 3 Si−OH.nm−2) are depicted in Figure 4. The bim‐surface average distance during the simulations is shown in the Supporting Information (Figure S5‐S6 and S7).
Figure 3.

Models used in MD simulations. (A) bim catalyst (the arrow indicates the carbene atom followed during MD); (B) flat silica surface; (C) 3 nm wide mesopore; (D) 10 nm wide mesopore.
Figure 4.

Snapshots extracted from the MD runs on 3 nm wide mesopore in vacuo (A1) and in DMF (A2), 10 nm wide pore in vacuo (B1) and in DMF (B2), and flat surface in vacuo (C1) and in DMF (C2), in all cases with low silanol concentration (i. e. 3 Si−OH nm−2). Bim catalyst represented as CPK, silica support as ball‐and‐stick, DMF as points.
We carried out a first series of simulations in vacuo (see snapshots in Figure 4A1–C1). Regardless of the Si−OH surface concentration, the bim moiety lies close to the surface (about or less 4 nm on average) for most of the simulation time (Figure S5 and S7 in the Supporting Information). In contrast, in DMF (snapshots in Figure 4A2–C2) the solvent passivates the surface, reducing bim interaction with the silica walls. As a result, the average bim‐surface distance in DMF rises above 6 nm on the small mesopore and above 7 nm on both the large mesopore and flat surface, with a low effect of the Si−OH surface concentration (Figure S6 and S7 in the Supporting Information). These results point out that bim interactions with the silica walls are unlikely to be the origin of its deactivation at the experimental conditions (i. e. in DMF).
We also computed the free energy of formation of the various hybrids in vacuo and in DMF (Table 1). In vacuo (entries 1–2), bim grafted inside the narrow mesopore (3 nm wide) is the most stable hybrid owing to interactions with the close walls, followed by bim grafted in the wide mesopore (10 nm wide) (+7.1 kJ/mol), and on the flat surface (+18.9 kJ/mol). The effect is larger at lower Si−OH concentration (i. e. 3 Si−OH nm−2) (Table 1, entry 2), indicating that van der Waals interactions between bim and Si−O−Si are stronger than with Si−OH groups. This trend is reversed in DMF (Table 1, entry 3), where bim grafted on the flat surface is more stable, followed by bim inside the wide (10 nm wide) (+40.1 kJ/mol) and narrow mesopore (3 nm wide) (+74.0 kJ/mol). These results confirm that bim is more stabilized by solvation than by interaction with the silica surface: owing to steric reasons, the organic moiety is better solvated on the flat surface, followed by 10 nm and 3 nm wide mesopores. Interestingly, the interactions between bim and the pore walls are overwhelmed by solvation, with no significant effect of the Si−OH concentration. Overall, these results point out that lower recyclability of the hybrid catalyst with 3 nm wide mesopores is ascribed to lower bim stability in DMF, leading to its removal during the reaction.
Table 1.
Relative free energy (kJ/mol) for the hybrid system models.
|
Entry |
System |
Pore size |
Relative free energy [kJ/mol] |
|---|---|---|---|
|
1 |
Environment=vacuum [Si−OH]=7 nm−2 |
3 nm |
0.0 |
|
10 nm |
7.1 |
||
|
flat |
18.9 |
||
|
2 |
Environment=vacuum [Si−OH]=3 nm−2 |
3 nm |
0.0 |
|
10 nm |
10.5 |
||
|
flat |
40.6 |
||
|
3 |
Environment=DMF [Si−OH]=3 nm−2 |
3 nm |
74.0 |
|
10 nm |
40.1 |
||
|
flat |
0.0 |
3. Conclusion
In conclusion, a NHC catalyst based on a benzimidazole was covalently grafted on a range of mesoporous and hierarchical supports, and the hybrid catalysts showed divergent stability in the benzoin condensation of furfural. The highest stability was achieved with 150 Å Davisil silica due to enhanced stabilization of bim moieties in DMF. The use of silica supports with smaller mesopores hinder bim stability, due to van der Waals interactions between bim and the silica walls.
Experimental Section
Preparation of N‐heterocyclic carbene (NHC) precursor and derived hybrids. [26]
I‐ Alkylation of benzimidazole (compound 1 in Scheme 1): in a 50 mL round bottom flask kept under inert atmosphere, benzimidazole (8.5 mmol) was slowly added to a suspension of NaH (1 equiv.) in tetrahydrofuran (THF) (6 mL) at 0–5 °C under stirring; the solution was further stirred for 30 min at room temperature. Subsequently, dodecyl bromide (8.5 mmol) was added dropwise, and the solution was left to reflux for 24 h. The solution was filtered and washed with dichloromethane over a pad of celite. The solvent was evaporated, and the product was purified by column chromatography, giving compound 1 with 97 % yield. 1H NMR (CDCl3, 500 MHz, ppm): 0.90 (t, −CH2−CH3), 1.25‐ 1.35 (m, N−CH2−CH2−(CH2)9−CH3), 1.89 (q, N−CH2−CH2−(CH2)9−CH3), 4.17 (t, N−CH2−CH2− (CH2)9−CH3), 7.83 (m, Ar), 7.90 (s, N−CH−N). 13C NMR (CDCl3, 125 MHz, ppm): 14 (−N−(CH2)11−CH3), 22 (−N− (CH2)10−CH2−CH3), 26.8, 29.1, 29.3, 29.4, 29.58, 29.6, 29.85 (aliphatic chain), 31.9 (N−CH2−CH2−(CH2)9), 45.12 (N−CH2−CH2−(CH2)9), 109.65, 120.38, 121.98, 122.75, 133.84, 143.90 (aromatic except N−CH−N), 142.93 (N−CH−N).
II‐ Silylation (compound 2 in Scheme 1): in a round bottom flask kept under inert atmosphere and under stirring, 1‐alkylated benzimidazole (1, 8.2 mmol) was dissolved in acetonitrile (ACN) (12 mL). Iodopropyltrimethoxysilane (12.3 mmol) was then slowly added. After 30 min stirring at room temperature, the solution was left to reflux for 48 h under inert atmosphere, then, the product was recovered and washed with anhydrous pentane, giving compound 2 with 95 % yield. 1H NMR (CDCl3, 500 MHz, ppm): 0.80 (t, −CH2−CH3), 0.699 (O−Si−CH2−CH2), 1.11‐1.41 (m, N−CH2−CH2−(CH2)9−CH3), 2 (q, N+−CH2−CH2−CH2−Si), 2.11 (q, N−CH2−CH2−(CH2)9−CH3), 3.55 (O−CH3), 4.53 (t, N−CH2−CH2−(CH2)9−CH3), 4.58 (t, N+−CH2−CH2−CH2‐Si), 7.58−7.69 (m, Ar), 11.1 (s, N−CH−N). 13C NMR (CDCl3, 125 MHz, ppm): 14 (−N−(CH2)11−CH3), 22.3, 22.7, 23.3, 26.6, 29, 29.31, 29.37, 29.49, 29.5, 29.57, 31.9, 34.1 (aliphatic chain), 50.8 (CH2−Si−O−(CH3)3), 113, 113.24, 127.21, 127.24 (aromatic except N−CH−N), 141.94 (N−CH−N).
III‐ Anchoring on solid supports: in a round bottom flask kept under stirring and inert atmosphere, the silylated NHC precursor (compound 3, 0.5 mmol) was dissolved in anhydrous toluene, then the inorganic support (Davisil silicas, SBA‐15, HP‐SAPO‐5 and HP‐AlPO‐5, 0.5 g) was added, and the mixture was refluxed and stirred for 24 h. The reaction mixture was filtered and washed with toluene, giving a yellow solid, which was cured in air at 80 °C overnight.
Characterization methods: The FTIR spectra of self‐supporting pellets were collected at variable temperature under vacuum conditions (residual pressure <10‐4mbar) using a Bruker Equinox 55 spectrometer equipped with a pyroelectric detector (DTGS type) with a resolution of 4 cm−1. FTIR spectra were normalized with respect the pellet weight.
Solid‐state NMR spectra were acquired with a wide bore 11.75 Tesla magnet on a Bruker Avance III 500 spectrometer at operational frequencies for 1H, 29Si and 13C of 500.13, 99.35 and 125.77 MHz, respectively. A 4 mm triple resonance probe (in double resonance mode) with magic angle spinning (MAS) was employed in all the experiments, and the samples were packed in a Zirconia rotor and closed with a Kel‐F cap and spun at a MAS rate between 10 and 15 kHz. The magnitudes of radio frequency (RF) field for 90‐degree pulse were 100 and 42 kHz for 1H and 29Si, respectively. The relaxation delay between accumulations was 5 and 60 s for 1H and 29Si, respectively. For 13C and 29Si cross polarization (CP) MAS experiments, RF fields of 55 and 28 kHz were used for initial proton excitation and decoupling, respectively. During the CP period, the 1H RF field was ramped using 100 increments, whereas the 13C/29Si RF fields were maintained at a constant level. A moderate ramped RF field of 62 kHz was used for spin locking, while the carbon/silicon RF field was matched to obtain optimal signal, and a CP contact time of 2 ms was used. During the acquisition, the protons were decoupled from the carbons/silicons by using a two‐pulse phase‐modulated (TPPM) decoupling scheme. All chemical shifts were reported using the chemicals shift (δ) scale and were externally referenced to TMS for 1H, 13C and 29Si.
Theoretical methods: The MD calculations were carried out with GROMACS2020 package. [27] After initial energy minimization to remove spurious close contacts, the MD simulations were performed with 2×105 steps of 0.5 fs for equilibration, and 2×106 steps of 1 fs for production runs. A 3 nm cut‐off was used for Van der Waals interactions, while electrostatic interactions were computed with a 2 nm cut‐off using the PME method for longer distances. In all the simulations, Si atoms not belonging to Si−OH groups were kept frozen. The furfural parameters were taken from GROMOS 54A7 set as provided by the Automated Topology Builder (ATB) website. [28]
The free energy of formation of the hybrid systems was computed by decoupling bim from the silica support [29] using the Bennet Acceptance Ratio (BAR) method [30] implemented in GROMACS (gmx BAR procedure).
The coupling parameter λ (varying from 1 to 0 as the system shifts from real to decoupled) was defined and used to gradually switch off intermolecular interactions between the target molecule and the rest of the system: first, Coulomb interactions were removed in 20 steps, while vdW interactions remained unaltered. Next, vdW terms were also eliminated in 20 further steps, until the target unit was completely decoupled. For each λ value, a MD run was performed comprising 0.5 ns equilibration (timestep 0.5 fs) and 1 ns production (timestep 1 fs).
Catalytic tests: In a glass reactor were added in sequence the heterogeneous catalyst (10 mol%), anhydrous DMF (0.5 ml), furfural (1 equiv, 0.2 mmol) and DBU (10 mol%). To account for the difference in NHC loadings on the various supports (Table S1), the catalyst weight was normalized, to maintain the same substrate‐to‐catalyst mole ratio (as above). Argon was bubbled for 20 min, and the reaction was carried at room temperature for 20 h. The crude mixture was treated with concentrated HCl to regenerate the stable catalyst precursor. A key step in the protocol of catalyst recyclability is acidification at the end of the reaction. As the carbene is a reactive species that can dimerize, the catalyst must be kept as benzoimidazolinium salt. The crude was then filtered and washed with MeOH, dried under vacuum and reused in the next cycle. The furfural conversion was monitored by 1H NMR.
Conflict of interest
The authors declare no conflict of interest.
4.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
The project leading to these results has received funding from the European Union's Horizon 2020 research and innovation program under grant agreement N. 720783‐MULTI2HYCAT.
Miletto I., Meazza M., Paul G., Cossi M., Gianotti E., Marchese L., Rios R., Pera-Titus M., Raja R., Chem. Eur. J. 2022, 28, e202202771.
Contributor Information
Prof. Marc Pera‐Titus, Email: peratitusm@cardiff.ac.uk.
Prof. Robert Raja, Email: r.raja@soton.ac.uk.
Data Availability Statement
The data that support the findings of this study are openly available in HAL at https://orca.cardiff.ac.uk, reference number 1234.
References
- 1.
- 1a. Huber G. W., Iborra S., Corma A., Chem. Rev. 2006, 106, 4044–4098; [DOI] [PubMed] [Google Scholar]
- 1b. Koutinas A., Du C., Wang R. H., Webb C., Introduction to Chemicals from Biomass, Wiley, Hoboken, 2008, pp. 77; [Google Scholar]
- 1c. Alonso D. M., Bond J. Q., Dumesic J. A., Green Chem. 2010, 12, 1493–1513; [Google Scholar]
- 1d. Dusselier M., Mascal M., Sels B. F., Top. Curr. Chem. 2014, 353, 1–40. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. van Putten R. J., van der Waal J. C., de Jong E., Rasrendra C. B., Heeres H. J., de Vries J. G., Chem. Rev. 2013, 113, 1499–1597; [DOI] [PubMed] [Google Scholar]
- 2b. Mariscal R., Maireles-Torres P., Ojeda M., Sádaba I., López Granados M., Energy Environ. Sci. 2016, 9, 1144–1189. [Google Scholar]
- 3.
- 3a. Nicolaou K. C., Edmonds D. J., Bulger P. G., Angew. Chem. Int. Ed. 2006, 45, 7134–7186; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2006, 118, 7292–7344; [Google Scholar]
- 3b. Pellissier H., Chem. Rev. 2013, 113, 442–524; [DOI] [PubMed] [Google Scholar]
- 3c. Tietze L. F., Chem. Rev. 1996, 96, 115–136. [DOI] [PubMed] [Google Scholar]
- 4. Rios R., Stereoselective Organocatalysis: bond formation methodologies and activation modes, Hoboken, US (Ed: Rios R.), Wiley, 2013. [Google Scholar]
- 5.
- 5a. Kabro A., Escudero-Adan E. C., Grushin V. V., van Leeuwen P. W. N. M., Org. Lett. 2012, 12, 4014–4017; [DOI] [PubMed] [Google Scholar]
- 5b. Liu D., Chen E. Y.-X., ChemSusChem 2013, 6, 2236–2239; [DOI] [PubMed] [Google Scholar]
- 5c. Wilson J., Chen E. Y.-X., ACS Sustainable Chem. Eng. 2016, 4, 4927–4936. [Google Scholar]
- 6.
- 6a. Enders D., Niemeier O., Henseler A., Chem. Rev. 2007, 107 (12), 5606–5655; [DOI] [PubMed] [Google Scholar]
- 6b. Menon R. S., Biju A. T., Nair V., Beilstein J. Org. Chem. 2016, 12, 444–461; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6c. Smith C. A., Narouz M. R., Lummis P. A., Singh I., Nazemi A., Li C.-H., Crudden C. M., Chem. Rev. 2019, 119, 4986–5056; [DOI] [PubMed] [Google Scholar]
- 6d. Wang L., Chen E. Y.-X., ACS Catal. 2015, 5, 6907–6917; [Google Scholar]
- 6e.E. Y.-X. Chen, L. Wang, Y. Eguchi, US 20160346774 A1, 2016.
- 7. Breslow R., J. Am. Chem. Soc. 1958, 80, 3719–3726. [Google Scholar]
- 8. Thomas J. M., Raja R., Acc. Chem. Res. 2008, 41 (6), 708–720. [DOI] [PubMed] [Google Scholar]
- 9. Verma P., Kuwahara Y., Mori K., Raja R., Yamashita H., Nanoscale 2020, 12, 11333–11363. [DOI] [PubMed] [Google Scholar]
- 10. Potter M. E., Light M. E., Irving D. J. M., Oakley A. E., Chapman S., Chater P., Cutts G., Watts A., Wharmby M., Vandegehuchte B. D., Schreiber M. W., Raja R., Phys. Chem. Chem. Phys. 2020, 22, 18860–18867. [DOI] [PubMed] [Google Scholar]
- 11. Newland S. H., Sinkler W., Mezza T., Bare S. R., Raja R., Proc. R. Soc. A 2016, 472, 20160095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chapman S., Raja R., Adv. Sci. Lett. 2017, 23, 5995–5997. [Google Scholar]
- 13. Newland S. H., Sinkler W., Mezza T., Bare S. R., Carravetta M., Haies I. M., Levy A., Keenan S., Raja R., ACS Catal. 2015, 5 (11), 6587–6593. [Google Scholar]
- 14. Chapman S., O'Malley A. J., Miletto I., Carravetta M., Cox P., Gianotti E., Marchese L., Parker S. F., Raja R., Chem. Eur. J. 2019, 25, 9938–9947. [DOI] [PubMed] [Google Scholar]
- 15.
- 15a. Zhou Z., Meng Q., Seifert A., Wagener A., Sun Y., Ernst S., Thiel W., Microporous Mesoporous Mater. 2009, 121, 145–151; [Google Scholar]
- 15b. Colthup N. B., in An introduction to infrared and Raman spectroscopy, (ed. L. H. Daly and S. E. Wiberley) Academic Press, Boston, USA, 1990, Chapter 8, p. 261–288; [Google Scholar]
- 15c. Arjunan V., Raj A., Ravindran P., Mohan S., Spectrochim. Acta Part A 2014, 118, 951–965. [DOI] [PubMed] [Google Scholar]
- 16.
- 16a. Jiang Y., Huang J., Dai W., Hunger M., Solid State Nucl. Magn. Reson. 2011, 39, 116–141; [DOI] [PubMed] [Google Scholar]
- 16b. Hunger M., Ernst S., Steuernagel S., Weitkamp J., Microporous Mater. 1996, 6, 349–353; [Google Scholar]
- 16c. Hensen E. J. M., Poduval D. G., Degirmenci V., Ligthart D. A. J. M., Chen W., Mauge F., Rigutto M. S., van Veen J. A. R., J. Phys. Chem. C 2012, 116, 21416–21429. [Google Scholar]
- 17.
- 17a. Zibrowius B., Loffler E., Hunger M., Zeolites 1992, 12, 167–174; [Google Scholar]
- 17b. Chen J., Wright P. A., Natarajan S., Thomas J. M, Stud. Surf. Sci. Catal. 1994, 843, 1731–1738; [Google Scholar]
- 17c. Lee S. I., Chon H. J., J. Chem. Soc. Faraday Trans. 1997, 93, 1855–1860. [Google Scholar]
- 18. Gianotti E., Miletto I., Ivaldi C., Paul G., Marchese L., Meazza M., Rios R., Raja R., RSC Adv. 2019, 9, 35336–35344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.
- 19a. Derouet D., Forgeard S., Brosse J.-C., Emery J., Buzare J.-Y., J. Polym. Sci. Part A 1998, 36, 437–453; [Google Scholar]
- 19b. Paul G., Bisio C., Braschi I., Cossi M., Gatti G., Gianotti E., Marchese L., Chem. Soc. Rev. 2018, 47, 5684–5739; [DOI] [PubMed] [Google Scholar]
- 19c. Jitianu A., Cadars S., Zhang F., Rodriguez G., Picard Q., Aparicio M., Mosa J., Klein L. C., Dalton Trans. 2017, 46, 3729–3741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Luhmer M., d′Espinose J. B., Hommel H., Legrand A. P., Magn. Reson. Imaging 1996, 14, 911–913. [DOI] [PubMed] [Google Scholar]
- 21. Friebolin H. in Basic One- and Two-Dimensional NMR Spectroscopy (3rd Ed.) Wiley, Weinheim, Germany, 1998, p. 60–68 and 147–158. [Google Scholar]
- 22.
- 22a. Coady D. J., Fukushima K., Horn H. W., Rice J. E., Hedrick J. L., Chem. Commun. 2011, 47, 3105–3107; [DOI] [PubMed] [Google Scholar]
- 22b. Khokarale S. G., Mikkola J.-P., RSC Adv. 2018, 8, 18531–18541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Abraham M. J., Murtola T., Schulz R., Páll S., Smith J. C., Hess B., Lindahl E., SoftwareX 2015, 1, 19–25. [Google Scholar]
- 24. Bennet C. H., J. Comput. Phys. 1976, 22, 245–268. [Google Scholar]
- 25. Khanna V., Monroe J. I., Doherty M. F., Peters B., J. Comput. Aided Molec. Des. 2020, 34, 614–646. [DOI] [PubMed] [Google Scholar]
- 26. Gianotti E., Miletto I., Ivaldi C., Paul G., Marchese L., Meazza M., Rios R., Raja R., RSC Adv. 2019, 9, 35336–35344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Abraham M. J., Murtola T., Schulz R., Páll S., Smith J. C., Hess B., Lindahl E., SoftwareX 2015, 1, 19–25. [Google Scholar]
- 28.https://atb.uq.edu.au 2021.
- 29. Khanna V., Monroe J. I., Doherty M. F., Peters B., J. Comput. Aided Molec. Des. 2020, 34, 614–646. [DOI] [PubMed] [Google Scholar]
- 30. Bennet C. H., J. Comput. Phys. 1976, 22, 245–268. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Data Availability Statement
The data that support the findings of this study are openly available in HAL at https://orca.cardiff.ac.uk, reference number 1234.