

Summary
Globally, weedy plants are a major constraint to sustainable crop production. Much of the success of weeds rests with their ability to rapidly adapt in the face of human‐mediated management of agroecosystems. Alopecurus myosuroides (blackgrass) is a widespread and impactful weed affecting agriculture in Europe.
Here we report a chromosome‐scale genome assembly of blackgrass and use this reference genome to explore the genomic/genetic basis of non‐target site herbicide resistance (NTSR). Based on our analysis of F2 seed families derived from two distinct blackgrass populations with the same NTSR phenotype, we demonstrate that the trait is polygenic and evolves from standing genetic variation.
We present evidence that selection for NTSR has signatures of both parallel and non‐parallel evolution. There are parallel and non‐parallel changes at the transcriptional level of several stress‐ and defence‐responsive gene families. At the genomic level, however, the genetic loci underpinning NTSR are different (non‐parallel) between seed families.
We speculate that variation in the number, regulation and function of stress‐ and defence‐related gene families enable weedy species to rapidly evolve NTSR via exaptation of genes within large multi‐functional gene families. These results provide novel insights into the potential for, and nature of plant adaptation in rapidly changing environments.
Keywords: blackgrass (Alopecurus myosuroides), herbicide resistance, parallel evolution, polygenic trait, quantitative genetics, rapid plant adaptation, weed evolution, weed genomics
Introduction
Human‐mediated environmental change is driving rapid evolutionary responses in the global biota (Palumbi, 2001; Hendry et al., 2011) and it is important to understand the outcome of these changes in natural and agricultural plant populations and communities. Reference genomes offer glimpses into the adaptive potential of plants when challenged with novel stresses, while agricultural weeds have been proposed as ideal models to address fundamental questions in plant ecology and evolution (Neve et al., 2009; Vigueira et al., 2013; Kreiner et al., 2018; Baucom, 2019; Mahaut et al., 2020).
Herbicide use has become a mainstay of weed management. Unsurprisingly, heavy reliance on herbicides has resulted in the rapid and widespread evolution of resistance, making herbicide resistance a widely studied weedy trait (Heap, 2014; Gould et al., 2018). Two ‘types' of herbicide resistance are recognized (Powles & Yu, 2010; Gaines et al., 2020). Target site resistance (TSR) refers to the modification of the sequence, copy number and/or expression of the gene encoding the herbicide target enzyme. Non‐target site resistance (NTSR) encompasses a range of mechanisms that limit herbicide delivery to its site of action. Typically, NTSR is inherited in a quantitative manner, but despite some advances in identifying and/or validating causal loci (Cummins et al., 2013; Delye, 2013; Tetard‐Jones et al., 2018; Franco‐Ortega et al., 2021; Han et al., 2021), efforts to discern the genomic basis and evolutionary dynamics of this trait have been hampered by lack of access to genomic resources.
The widespread evolution of herbicide resistance is an emblematic example of repeated (or convergent) evolution of plant defence in the face of an extreme, novel selection pressure (Baucom, 2016). In general, TSR has provided an example of genetic parallelism (Martin & Orgogozo, 2013) where the convergent evolution of resistance is underpinned by parallel patterns of selection at single major loci (Powles & Yu, 2010; Gaines et al., 2020). The genetic basis of NTSR is not fully resolved, but current evidence suggests that this trait is polygenic, that the genomic architecture of NTSR may be determined by selection at parallel and non‐parallel genetic loci (Van Etten et al., 2020; Kreiner et al., 2021), via co‐option (or exaptation) of standing genetic variation in plant stress‐ and defensive‐responsive pathways (Hawkins et al., 2019). Addressing these questions through studies of the genomic basis of NTSR has power to answer fundamental questions about the importance of genetic parallelism and non‐parallelism, genomic constraint, genetic background (contingency) and standing genetic variation in rapid plant adaptation to a novel environmental stress (Allen Orr, 2005a,b; Losos, 2011; Lobkovsky & Koonin, 2012; Bolnick et al., 2018).
The diploid, allogamous grass, Alopecurus myosuroides (blackgrass) is native to the Eastern Mediterranean and West Asia (Bulcke, 1975) but is now a widespread and impactful weed in agricultural crops in much of Europe (Menchari et al., 2007; Rosenhauer et al., 2013; Hicks et al., 2021) and in China (Liu et al., 2021). Blackgrass populations are prone to the rapid and widespread evolution of herbicide resistance. In a nationwide survey in England, most blackgrass populations exhibited resistance to multiple herbicide modes of action (Hicks et al., 2018). Resistance was conferred by coexisting TSR and NTSR mechanisms, with evidence that historical herbicide‐use regimes favoured the evolution of the NTSR (Comont et al., 2020). Herbicide‐resistant blackgrass is estimated to cost UK farmers £0.4 billion per year (Varah et al., 2020) and there is no evidence for fitness costs for any of a variety of life‐history traits associated with NTSR (Comont et al., 2022).
Access to genomes and genomic resources for weed species will greatly enhance the capacity to unravel contemporary adaptation in economically and ecologically important weedy plant species (Ravet et al., 2018). Here, we present a high‐quality reference genome for blackgrass. We use these genome resources to reveal that patterns of convergent evolution of organismal‐ (whole plant assays) and gene expression‐based phenotypes for NTSR‐based resistance are conferred by non‐parallel changes at multiple genetic loci distributed widely throughout the blackgrass genome.
Materials and Methods
Plant materials for genome sequencing and annotation
Alopecurus myosuroides L. (blackgrass) seeds collected in 2017 from section 8 of the Rothamsted ‘Broadbalk’ long‐term experiment (Moss et al., 2004) were used to select an individual blackgrass plant for genome sequencing. Established in 1843, these field plots have never received herbicide application, and extensive testing of this population (Rothamsted, Hertfordshire, UK) over the last 20 yr has confirmed that it remains susceptible to all herbicides, representing a true wild‐type blackgrass genotype. In addition, two field‐collected blackgrass seed populations (Peldon and Lola91) previously characterized as being strongly NTSR to acetyl‐CoA carboxylase (ACCase) inhibiting herbicides were used to generate F2 seed families (named CC2 and CC5, respectively) for quantitative trait loci (QTL)‐seq and RNA‐seq analyses. Detailed protocols for the selection of a single herbicide‐sensitive plant for genome sequencing and for the development of CC2 and CC5 seed families are presented in Notes S1.
Genome survey
A previous study has reported that blackgrass has seven chromosomes (Johnsson, 1944). In our study, genome size was estimated through flow cytometry and k‐mer‐based analysis. Flow cytometry was conducted on four field‐collected blackgrass populations (the Rothamsted, Lola91, and Peldon populations used within this study, plus a further herbicide‐susceptible population). Genome size estimates were generated for three replicate plants from each of these populations, against a known standard of the plant Allium schoenoprasum. Using these data, the blackgrass genome size was estimated as 3312–3423 Mb. K‐mer‐based analysis from Illumina sequencing data of the Rothamsted population indicated a genome size of 3400–3550 Mb. We also estimated the heterozygosity and repeat content of the blackgrass genome with gce package (https://github.com/BioInfoTools/GCE), the results suggest the blackgrass genome exhibits high levels of heterozygosity (1.52%) and repeat content (84.2%).
Genome sequencing
A mix of single‐molecule and short read sequencing data was collected for de novo genome assembly. These data include 513 Gb (144× coverage) PacBio continuous long reads, 860 Gb (241× coverage) BioNano optical maps, 126 Gb (35× coverage) Hi‐C reads and 291 Gb (81× coverage) Illumia short reads. Detailed protocols for sequencing and assembly of the blackgrass genome are presented in Notes S1.
Genome assembly
A de novo assembly of PacBio long reads into contigs was performed with Mecat2 (Xiao et al., 2017). This produced 12 107 contigs with an N50 of 0.9 Mb and a total size of 4906 Mb. The assembled contigs were polished with PacBio long reads via Arrow (https://github.com/PacificBiosciences/SMRT‐Link) and Illumina short reads with Pilon (v.1.20) (Walker et al., 2014). Polished contigs were repeat marked using WindowMasker (Morgulis et al., 2006) and then haplotype merged using HaploMerger2 (Huang et al., 2017) to address the high heterozygosity of the blackgrass genome. BioNano data were first filtered for molecule length (> 150 kb) and then aligned to primary contigs to select mapped molecules for de novo assembly to obtain the BioNano optical maps. The primary contigs and BioNano maps were combined to produce the base hybrid scaffold assembly. The Hi‐C reads were aligned to the base assembly using the Juicer pipeline (Durand et al., 2016a). Hybrid scaffolds were then further scaffolded using the 3D‐DNA pipeline (Dudchenko et al., 2017). The results were manually examined using the Juicebox Assembly Tools, an assembly‐specific module in the Juicebox visualization system (Durand et al., 2016b). The Hi‐C scaffolding resulted in seven pseudomolecule chromosomes. Assembly gaps were identified and filled with Cobbler (v.0.6.1) (Warren, 2016). The final assembly was polished again with PacBio long reads via Arrow and Illumina short reads via Pilon (Walker et al., 2014). Detailed methods are presented in Notes S1.
Genome assembly quality assessment
The quality of the genome assembly was evaluated by the following analyses: (1) The Illumina short reads used for polishing were mapped to the genome assembly using Bwa‐Mem, and the mapping rate and genome coverage were examined. (2) The assembly was assessed for single‐copy gene ortholog content with Busco (v.4.0.1) (Simao et al., 2015) using the embryophyta_odb10 database. (3) The long terminal repeat (LTR) assembly Index (Ou et al., 2018) was calculated. (4) Correlation of the assembled chromosome length to the cytogenic chromosome length (Johnsson, 1944) was examined.
Genome annotation
A comprehensive non‐redundant repeat library for the blackgrass genome was built using EDTA, a de novo transposable element (TE) annotator that integrates structure‐ and homology‐based approaches for TE identification (Ou et al., 2019). The EDTA pipeline incorporates LTRharvest, the parallel version of Ltr_Finder, Ltr_retriever, Grf, Tir‐Learner, HelitronScanner and RepeatModeler as well as customized filtering scripts. Genome‐wide prediction of ncRNAs, such as rRNA, small nuclear RNA and miRNA, was performed using the Infernal software (Nawrocki et al., 2009) to the Rfam database. The tRNA genes were predicted using tRNAscan‐SE (Lowe & Eddy, 1997).
Protein‐coding genes were predicted by a combination of de novo prediction, homology‐based and transcriptome‐based strategies. Snap (Korf, 2004), Augustus (Stanke et al., 2006) and GeneMark (Lomsadze et al., 2005) were used for ab initio gene predictions. For homology‐based prediction, protein sequences of seven species (A. thaliana, O. sativa, S. bicolor, B. distachyon, H. vulgare, Z. mays and T. aestivum) were aligned to the genome assembly using the GeMoMa program (Keilwagen et al., 2019) to provide homology‐based evidence. For transcriptome‐based prediction, RNA‐seq data were generated from a diversity of blackgrass tissues collected over developmental time (leaf, main stem, root, developing flowers, mature flowers pre‐anthesis, and mature flowers with pollen). RNA‐seq reads were processed to remove adapters and low‐quality bases and assembled both de novo and genome‐guided using Trinity (v.2.4.0) (Haas et al., 2013) followed by the Pasa program (http://pasapipeline.github.io) to improve the gene structures. All predicted gene structures were integrated into consensus gene models using the EVidenceModeler (Haas et al., 2008). Functional annotation of protein‐coding genes was carried out by comparing alignments to the SwissProt, GenBank nonredundant protein, InterProScan and EggNOG databases. Gene ontology (GO) terms for each gene were obtained from InterPro descriptions. In addition, the gene set was mapped to the Kegg pathway database using BlastKoala (https://www.kegg.jp/blastkoala/) (Kanehisa et al., 2016).
LTR retrotransposons insertion time estimation and expression analysis
As the direct repeat of an LTR‐RT is identical upon insertion, the divergence between the LTR of an individual element reflects the time of the insertion. The insertion date (T) for each LTR‐RT was computed by T = K/2μ, where K is the divergence rate and μ is the neutral mutation rate (K = −3/4 × loge(1−d × 4/3), μ = 1.3 × 10−8) (Ma & Bennetzen, 2004). Sequence identity (%) between the 5′ and 3′ direct repeats of an LTR candidate is approximated using Blastn, so the proportion of sequence differences is calculated as d = 100% − identity%. The tetranscripts package (Jin et al., 2015) was used to estimate the expression of LTR‐RTs, and differential expression between samples was analysed in R v.4.0.2 (R Core Team, 2018) using Deseq2 (Love et al., 2014).
Gene duplication and gene family expansion
To identify orthologous and paralogous gene clusters, protein‐coding genes from blackgrass and 11 other species (A. tauschii, T. urartu, H. vulgare, P. tenuiflora, B. distachyon, O. sativa, Z. mays, S. bicolor, S. italica, E. haploclada and A. thaliana) were analysed using Orthofinder2 (v.2.5.1) (Emms & Kelly, 2015). In cases where there were multiple transcript variants, the longest transcript was selected to represent the gene. A total of 476 single‐copy orthologous genes were identified. Single‐copy genes from each species were aligned using Muscle (Edgar, 2004) and the alignments were concatenated. The concatenated alignment was used to construct a maximum likelihood phylogenetic tree using RAxML (Stamatakis, 2014). The MCMCTree program (Yang & Rannala, 2006) of Paml (Yang, 2007) was used to estimate the divergence time among 12 species. Three calibration time points were used based on previous publications and the TimeTree website (http://www.timetree.org) as normal priors to restrain the age of the node, including 146–154 million years ago (Ma) between Arabidopsis and rice, 68–72 Ma between rice and sorghum, and 49–53 Ma between barley and Brachypodium. Gene family expansion and contraction was determined by comparing the gene cluster size differences between the ancestor and each species with the Café program (De Bie et al., 2006). Café uses a model of stochastic birth and death for gene family evolution and a Monte Carlo re‐sampling procedure to calculate the probability (P‐value) of a gene family with the observed family size change (expansion or contraction). The threshold for significance was set at P ≤ 0.05. To determine the possible whole genome duplication events in the blackgrass genome, we performed a self‐alignment using Last (v.963) (Kielbasa et al., 2011) and identified syntenic blocks with MCscanX (Wang et al., 2012). For each gene pair within syntenic blocks, synonymous divergence levels (K s) were calculated using the YN model in the KaKs_Calculator (Wang et al., 2010). The K s values of all gene pairs were plotted to identify putative whole genome duplication events. To calculate the genome duplication and gene family expansion events, the formula T = K s/2R was used, where R is the rate of divergence of nuclear genes in plants, which was set to 6.1 × 10−9, according to Lynch & Conery (2000).
QTL‐seq analysis (bulk segregant analysis of SNPs)
Leaf tissue was harvested from unsprayed tillers of all 25 ‘R’ and ‘S’ plants from each F2 family. In all cases, young leaf material was collected over 1 h at midday, from each plant into separate 5 ml Eppendorf tubes. Each sample was immediately flash frozen in liquid nitrogen (LN2) and stored at −80°C. Samples were homogenised in LN2 using a micro‐pestle. For bulk segregant analysis, four bulks were made by pooling DNA from all 25 selected individuals from each phenotypic group (herbicide resistant ‘R’, and susceptible ‘S’, in each of the CC2 and CC5 F2 families). Illumina paired‐end reads were processed to remove adapters and low‐quality sequences using Trimmomatic (Bolger et al., 2014). Cleaned read data were generated after removing reads with > 10% unidentified nucleotides (N), > 30% bases had Phred quality scores < 20 and < 75 bp of read length. Cleaned reads were then mapped to the blackgrass reference genome using Bwa. Variants were called using BCFtools (http://samtools.github.io/bcftools) and filtered using VCFtools (http://vcftools.sourceforge.net). Single nucleotide polymorphisms (SNPs) were subjected to quality control and removed if they met the following criteria: (1) non‐biallelic SNPs, (2) read depth for SNP > 500 or < 5, (3) mapping quality > 40, (4) genotype quality > 100, (5) missing rate < 10% and (6) SNPs within < 20 bp distance from nearby InDels. The QTL‐seq pipeline was used for calculating the SNP‐index, and the ∆SNP‐index was then calculated by subtracting the SNP‐index of one bulk from that of another bulk (Takagi et al., 2013).
RNA‐seq analysis
An RNA‐seq analysis was also conducted using the 25‐herbicide resistant ‘R’ and susceptible ‘S’ plants from each F2 family. For each phenotypic group, five replicate RNA‐bulks were created by pooling RNA from five individual plants. RNA was sequenced using standard Illumina TruSeq mRNAseq protocols. The quality of the RNA sequences derived from each sample was assessed using FastQC v.0.11.8 (Andrews, 2010) and pre‐processed as described earlier. The trimmed reads for each sample were mapped to the blackgrass genome using Hisat2 v.2.2.1 (Kim et al., 2019) with default parameters except for minimum alignment score parameters of L, 0, −0.6. Reads that mapped to coding sequences of annotated genes were counted using FeatureCounts v.1.6.4 (Liao et al., 2014) with default settings. Differential gene expression between samples was analysed in R v.4.0.2 (R Core Team, 2018) using Deseq2 (Love et al., 2014).
The expression of all technical replicates was checked before analysis. First, all counts data were transformed using the regularised log‐transform function ‘rlog()’ of the Deseq2 package. Transformed data were visualised using both a principal component analysis and hierarchical clustering of the Euclidean distance between samples. Visual inspection of these results identified one clear outlier sample (CC5 ‘S’ sample A), which was excluded from further analysis. A pre‐filtering step was used to remove genes with zero or low counts before differential expression analysis. First, counts were summed across technical replicates to leave only biological samples. Next, genes were removed if they did not have at least one read per million in at least four samples (where four is equal to the minimum number of reps per treatment level) as per Anders et al. (2013). The filtered, biological replicates were analysed using the ‘DESeq()’ function of the Deseq2 package in R, specifying four phenotypic groups: CC2 ‘S’, CC2 ‘R’, CC5 ‘S’ and CC5 ‘R’. In total, 19 937 genes and 19 biological replicate samples were included in this analysis. To generate lists of differentially expressed genes (DEGs), specific comparisons were extracted for the ‘R’ vs ‘S’ samples within each family from this fitted model. Only genes which were significant (P < 0.05) and with at least 1.5× fold difference in expression were categorised as differentially expressed. The resultant lists of DEGs for the CC2 and CC5 families were then intersected, to identify DEGs common to both.
Gene ontology information was combined from the SwissProt, EggNOG, and InterPro annotation files to create a single Gene:GO association map, containing 905 051 associations between 28 498 genes and 13 192 GO terms. Gene ontology enrichment analysis was performed for DEGs using TBtools (https://github.com/CJ‐Chen/TBtools). The Gene:GO association map was specified as a custom gene category mapping to use for analysis, and resultant P‐values were adjusted using the Benjamini and Hochberg method to control for false discovery rate. In addition, overrepresentation of DEGs on each chromosome was tested using a Fishers' exact test as per Giacomini et al. (2020). For each chromosome, the observed number of DEGs was tested against the expected number given chromosome length and number of genes encoded. Resultant P‐values were Bonferroni adjusted to account for multiple testing before ascribing significance.
Gene co‐expression network construction
Trimmed means of M‐values were calculated from mapped RNAseq data using the edge‐R package in R (Robinson et al., 2010) to construct a gene expression matrix (GEM). The GEM was log2 transformed and quantile normalized in R (R Core Team, 2018). The traditional gene co‐expression network (GCN) was created using the Knowledge Independent Network Construction tool (Kinc v.3.4.0) (Shealy et al., 2019). A gene correlation matrix was constructed using the Spearman rank correlation coefficient approach (Song et al., 2012) with the following Kinc‐specific parameters: ‐‐minsamp 15 –minexp ‐inf –mincorr 0.5 –maxcorr 0.99. A threshold for correlation was determined using the random matrix theory approach with the following parameters: ‐‐tstart 0.95 –tstep 0.001 tstop 0.5 –threads 1 –epsilon 1 e‐6 –mineigens 50 –spline true –minspace 10 –maxpace 40 –bins 60 and was determined to be 0.919. The network was extracted using the extract function and visualized in Cytoscape v.3.9.0 (Shannon et al., 2003). The condition‐specific GCN was constructed using the same GEM and Spearman‐ranked correlation coefficient approach in Kinc, but also incorporated a Gaussian mixed model to determine DEG pair clusters that represent condition‐specific sub‐graphs. Low‐powered edges were determined and filtered with the ‘corrpower’ function with an alpha of 0.001 and a power of 0.8. An annotation file was prepared in text format with samples either being annotated as ‘resistant’ or ‘susceptible’ and used to run the ‘cond‐test’. Condition‐specific sub‐graphs were extracted and visualized in Cytoscape v.3.9.0 (Shannon et al., 2003).
Results
Genome assembly and annotation
Genome analysis indicated that blackgrass (A. myosuroides) has a large genome (3.31–3.55 Gb) and exhibits heterozygosity of 1.52% and repeat content of 84.2% (Tables S1, S2). The high repeat content likely accounts for the large genome size. We adopted a hierarchical sequencing approach that includes complementary single‐molecule sequencing/mapping technologies coupled with deep coverage short read sequences to generate a pseudo‐chromosome reference genome assembly for blackgrass (Fig. S1). The total primary contig length is 3475 Mb, which is consistent with our genome size estimations based on flow cytometry and k‐mer analysis (3312–3423 and 3400–3550 Mb, respectively). The final polished blackgrass genome assembly size was 3572 Mb, including 3400 (95.2%) Mb ordered as seven pseudo‐chromosomes with only 172 Mb of unanchored sequences (Fig. 1; Tables 1, S3).
Fig. 1.
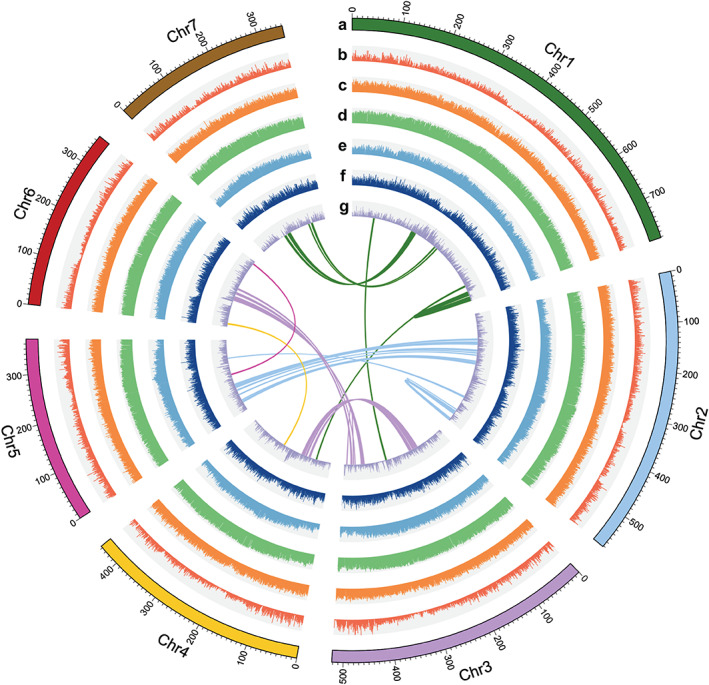
Overview of the A. myosuroides genome. A circos graph shows the assembled seven chromosomes (a), distribution of protein‐coding genes (b), distribution of GC content across the genome (c), distribution of transposable elements (d), distribution of Gypsy family of long terminal repeats retrotransposons (LTR RTs) (e), distribution of Copia family of LTR RTs (f), distribution of SNP/Indel (g). All the histograms (from ‘a’ to ‘g’) were featured in a 1‐Mb sliding window. Connecting line in the centre of the diagram represents a genomic syntenic region covering at least 10 paralogues.
Table 1.
Assembly statistics of the blackgrass genome.
| Characteristics | Values |
|---|---|
| Assembly size (bp) | 3572 044 634 |
| Number of scaffolds | 2512 |
| N50 scaffold length (bp) | 2255 730 |
| The largest scaffold (bp) | 17 744 454 |
| Number of contigs | 7866 |
| N50 contig length (bp) | 1189 615 |
| The largest contig (bp) | 9284 242 |
| GC content (%) | 44.66 |
| Total size of pseudomolecules (bp) | 3400 051 202 |
| Total size of unanchored sequences | 171 993 432 |
| Ns in the assembly | 80 915 468 |
| Total size of retrotransposons (bp) | 2302 477 515 |
| Total size of DNA transposons (bp) | 507 120 408 |
| Total size of repeat sequences (bp) | 2851 385 969 |
| Number of genes | 45 263 |
| Average length of genes (bp) | 2864 |
| Average number of exons per gene | 4.3 |
| Total size of genes (bp) | 129 639 341 |
| Number of annotated genes | 35 999 |
Both the euchromatic and heterochromatic components of the blackgrass genome are highly complete as supported by Busco scores (96.9% from the Embryophyta lineage) (Simao et al., 2015) and a LTR assembly index (Ou et al., 2018) (LAI: 9.6–35.2, a mean value of 21.9, Table S4; Fig. S2). In addition, the Illumina short reads (81×) returned a 99.6% mapping rate and covered 99.9% of the assembled genome. We identified 8026 403 polymorphisms as SNPs or InDels (Fig. 1g), which corroborates the predicted heterozygosity level of the blackgrass genome. We also observed a high correlation (r = 0.98, Table S5) between the assembled chromosome and cytogenic chromosome lengths based on published data (Johnsson, 1944).
We annotated 45 263 protein‐coding genes (mean gene length of 2864 bp) based on de novo, homology‐based predictions and transcriptome data from multiple tissues (Fig. S3). Genes were unevenly distributed across the chromosomes with increased gene density towards the distal ends of chromosomes that recedes to low densities in the centre (Fig. 1b). Among these protein‐coding genes, 2385 were annotated as transcription factors. In addition, 4258 non‐coding RNAs were identified, including 1369 transfer RNAs, 941 ribosomal RNAs, 513 micro RNAs and 1425 small nuclear RNAs (Fig. 1 for genome overview).
Genome dynamics and NTSR
We annotated 2851 Mb (81.7%) of sequence in the assembled genome as TEs (Table S6). The dominant type of TE was LTR retrotransposons (RTs), representing c. 80.3% (2290 Mb) of annotated TEs and amounting for 65.6% of the blackgrass genome size (Fig. 1d). Gypsy, Copia and unclassified RT elements contributed to 39.2%, 8.6% and 17.9% of the genome size, respectively (Fig. 1e,f). DNA transposons contributed to 14.5% of the genome (Fig. 2a). LTR‐RTs are highly unstable and have played an important role in the evolution of plant genomes (Fedoroff, 2000). We observed a single peak of insertion time, occurring c. 0.1 Ma, for Gypsy, Copia and unclassified RTs in blackgrass, which suggests a recent burst of LTR RTs in the genome (Fig. 2b). In addition, we observed a burst of RTs in blackgrass that occurred more recently than those in barley (Hordeum vulgare) and goatgrass (Aegilops tauschii) but occurred at a similar time in rice (Oryza sativa) (Fig. 2c). We observed that LTR‐RTs exhibited different expression profiles between susceptible and resistant plants. In all, 33 and 19 LTR‐RTs exhibited different expression levels between susceptible and resistant plants in CC2 and CC5 families, respectively (Fig. 2d,e). However, common differentially expressed LTR‐RTs were not observed between CC2 and CC5 (Fig. 2f). These results indicate that different LTR‐RTs were historically activated and potentially contribute to NTSR in our different blackgrass populations.
Fig. 2.
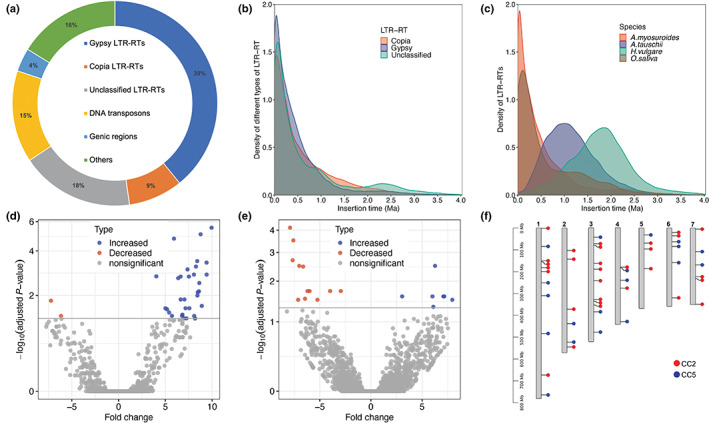
The burst and expression of long terminal repeat retrotransposons (LTR‐RTs) in the A. myosuroides genome. (a) Proportions of the major elements in the blackgrass genome, including Gypsy LTR‐RTs, Copia LTR‐RTs, unclassified LTR‐RTs, DNA transposons, coding DNA and unannotated sequences. (b) The insertion time distribution of different types of LTR‐RT in the blackgrass genome. (c) The insertion time distribution of intact LTR‐RTs in the blackgrass genome compared with those in goatgrass (progenitor of the wheat D genome), barley and rice. (d, e) Volcano plots show differentially expressed LTR‐RTs in CC2 and CC5 family, respectively. Resultant P‐values were Bonferroni‐adjusted to account for multiple testing before ascribing significance. (f) The distribution of differentially expressed LTR‐RTs on seven blackgrass chromosomes.
Genomic duplications, including gene family expansions, can be a result of polyploidization events and signatures of stress adaptations. In blackgrass, we observed two distinct peaks at K s values of 0.1 and 0.8 (Fig. 3a). The peak at c. 0.8 was shared in all grass species investigated, suggesting blackgrass underwent the same ancient whole genome duplication in the ancestor of Poaceae species c. 65.6 Ma (Paterson et al., 2004). The peak at 0.1 is not apparent in these other species, suggesting that this duplication event is unique to blackgrass. We further examined paralogous gene content within the duplication events and found that the peak at 0.1 (corresponding to 8.2 Ma) was evidenced by a high density of ‘co‐located’ paralogous genes on chromosomes 1, 2 and 3 (Fig. 1), accounting for 10% of total paralogous genes. These results suggest the blackgrass genome underwent small‐scale local duplication events after the occurrence of whole genome duplication. To proximate gene family evolution between blackgrass and other grasses, we constructed a phylogenetic tree based on the concatenated sequence alignment of the 476 single‐copy orthologous genes shared by blackgrass and 11 other species (Fig. 3b). We next examined gene family evolution through expansion and contraction events. A total of 33 757 orthologous gene families composed of 382 550 genes were identified from 12 species, of which 6470 gene families were shared by all the species (Fig. S4). In blackgrass, a total of 559 and 352 gene families were identified with significant expansion and contraction, respectively (P <0.05). Gene ontology enrichment analysis of the expanded genes revealed that they were mainly related to multiple enzymatic functions, including glutathione S‐transferase (GST), UDP‐glycosyltransferase (UGT) and monooxygenases, all of which have been reported (Gaines et al., 2020) to be associated with non‐target site herbicide resistance (Fig. 3c). Here, we define ‘NTSR‐related gene families’ as GST, UGT, cytochrome P450 (P450), ATP‐binding cassette (ABC) transporters, and aldo‐keto reductase (AKR). Using that definition, out of 5440 genes from 559 expanded families, 408 (7.5%) were identified as NTSR‐related genes. The K s values for these NTSR‐related gene family expansion events were plotted for all paralog pairs within expanded gene set (Fig. 3d). Peaks at 0.09 (corresponding to 7.3 Ma) and 0.26 (corresponding to 21.7 Ma) were observed for NTSR‐related and non‐NTSR gene families, respectively. Therefore, the expansion of the NTSR‐related gene families greatly predated the use of herbicide, suggesting the possibility that standing genetic variation may have facilitated the rapid evolution of herbicide resistance contributing to weediness in this species.
Fig. 3.
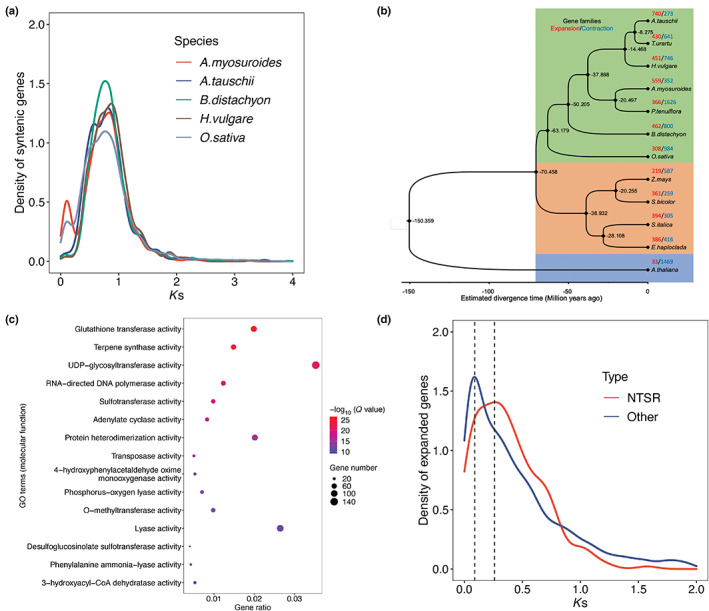
Whole genome duplication and gene expansion in the A. myosuroides genome. (a) The frequency distribution of synonymous substitution rates (K s) of paralogous genes within each plant genome. A shared whole genome duplication event for grasses was assigned to the peak. (b) Phylogenetic tree of 12 plant species and gene family expansion and contraction. Inferred divergence time is denoted at each node in black. Numerical values beside each node show the estimated divergence time of each node. The red and blue numbers above the species name indicate the total number of expanded and contracted gene families, respectively. (c) Gene ontology (GO) enrichment analysis of expanded gene families in the blackgrass genome (molecular function category). The significant levels were Bonferroni‐adjusted to account for multiple testing. (d) The frequency distribution of synonymous substitution rates (K s) of expanded genes. Expanded genes include those from either NTSR‐related or non‐NTSR (other) gene families.
QTL‐seq bulk segregant analysis for NTSR
To identify the genomic regions controlling herbicide resistance, we performed bulk segregant analysis in the CC2 and CC5 families to identify ΔSNP values with trait significance (Takagi et al., 2013; Kumar et al., 2020). We obtained 3402 057 and 3205 888 reliable SNPs for each of the CC2 and CC5 families, respectively (Fig. S5). We identified seven significant QTLs in the CC2 family distributed among chromosomes 2, 3, 5 and 6 (Table S7). In the CC5 family, we identified eight QTLs distributed mainly on chromosome 3, with one region on chromosome 2 (Table S7). A total of 371 genes were encoded within the 15 identified QTLs, with each QTL containing between 10 and 58 genes. Interestingly, there was no overlap between QTL regions identified in the two seed families (Fig. 4). Among the 15 identified QTL regions, seven contain genes that are differentially expressed between susceptible and resistant plants of either family; six of them contain transcription factors. The most significant QTL was identified on chromosome 2 in the CC2 family, which covered 2.5 Mb and contains 33 candidate genes. An NADPH‐dependent AKR gene was present in this region which is upregulated in the resistant plants of both CC2 and CC5 families. Members of this gene family have been reported to be associated with herbicide detoxification in other weed species (Pan et al., 2019). These results suggest that although single large effect genes (like this AKR gene) could be conferring resistance, it is more likely that NTSR in blackgrass involves multiple independent loci (polygenic trait), and our data provide strong evidence that NTSR mutations are population specific.
Fig. 4.

Location across the A. myosuroides genome of the differentially expressed genes (DEGs) associated with the NTSR trait. Green and purple circles show the position of DEGs identified in the CC2 and CC5 seed families, respectively. Circle sizes are relative to the adjusted P‐value, whereby larger circles denote stronger significance. DEGs consistent across both families are marked with black labels, while orange labels show the position of previously reported NTSR candidate genes. Lower sections show the change in ΔSNP‐index across these chromosomes for the CC2 (top) and CC5 (bottom) families. Shaded regions represent the 95% and 99% confidence bounds for each SNP. Vertical green and purple bars show the quantitative trait loci (QTL) regions for the CC2 and CC5 families, identified from their ΔSNP‐index. P‐values were computed based on the Wald test in the Deseq2 package and then adjusted using the Benjamini and Hochberg method for multiple test correction.
RNA‐seq analysis of NTSR
To identify DEGs between susceptible and resistant plants, we performed RNA‐seq analysis in the most and least resistant fractions of the two seed families (CC2 and CC5). Principal component analysis of gene expression data (19 937 genes across 19 biological samples) indicates that both seed families and resistance phenotypes contain significant sources of variation between samples (Fig. 5a). Seed family (CC2 vs CC5) was the stronger source of variance accounting for c. 58% of the total variance on the first Principal Component (PC1). Within each seed family, the herbicide‐resistant ‘R’ samples form separate clusters from their susceptible ‘S’ counterparts on PC2, with this axis representing 12% of the total variance in gene expression. In each seed family, the ‘direction’ of separation of ‘R’ samples from ‘S’ on PC2 is the same. Principal component analysis of each seed family independently (Fig. S6) revealed that the ‘R’ and ‘S’ samples in each family formed separate clusters on the first principal component (PC1). Within the CC2 and CC5 families, respectively, this PC accounted for 35% and 39% of the total variance in gene expression. These results demonstrate that the presence of the NTSR trait has a considerable impact on constitutive gene expression, even in the absence of herbicide treatment.
Fig. 5.
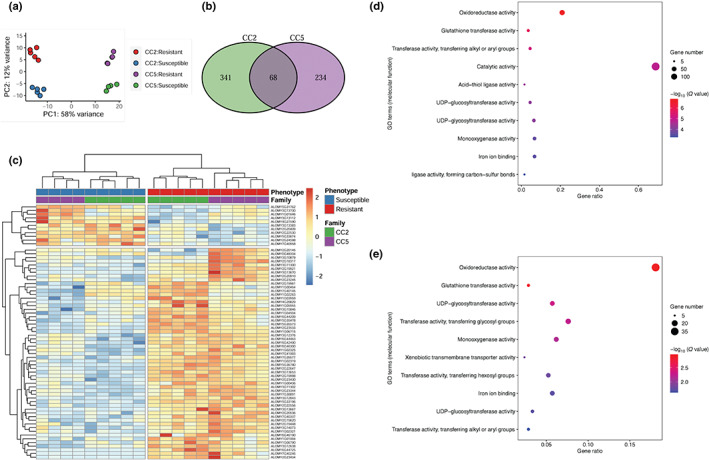
Differential gene expression analysis of the A. myosuroides seed families CC2 and CC5, segregating for the NTSR herbicide‐resistant trait. (a) Principal component analysis using all gene expression data. (b) Numbers of differentially expressed genes (DEGs) comparing the ‘R’ (green) and ‘S’ (purple) groups within each family. (c) Heatmap and hierarchical clustering of the 68 DEGs consistently associated with NTSR across both seed families. (d, e) Gene ontology terms, significantly overrepresented in the CC2 and CC5 families, respectively. The significant levels were Bonferroni‐adjusted to account for multiple testing.
Differential expression analysis between ‘R’ and ‘S’ samples across the two seed families identified 643 DEGs (Fig. 5b). A subset of 68 genes were found to be differentially expressed in both seed families. Hierarchical clustering of these 68 genes confirmed that resistance phenotype was a greater source of variability than seed family, and 81% (55) of these 68 genes were upregulated in ‘R’ samples relative to ‘S’ for both families (Fig. 5c). Within the 68 consistent DEGs, we found three of eight previously recorded blackgrass NTSR candidate genes: AmGSTF1, AmGSTU2 and AmOPR1 (Cummins et al., 2013; Tetard‐Jones et al., 2018). In each case, the expression of these three candidate genes was significantly higher in the ‘R’ phenotype than the ‘S’ (Fig. S7), agreeing with previously reported findings. In addition, three additional significant genes, ALOMY2G19998 (paralog of AKR4‐1 in Echinochloa colona), ALOMY1G02321 (paralog of CYP81A10v7 in Lolium rigidum) and ALOMY6G42490 (paralog of ABCC8 in Echinochloa colona), were all upregulated in both CC2 and CC5 families (Fig. S7). The orthologs of these three NTSR‐related genes have been validated to endow NTSR to various herbicides in other weed species (Pan et al., 2019, 2021; Han et al., 2021). These findings further reinforce the potential importance of these six genes, having now been implicated in the regulation of the herbicide metabolism phenotype across multiple populations, multiple independent studies and even multiple species.
Nevertheless, although 68 DEGs overlapped between the two families, the majority (341 for the CC2 family and 234 for the CC5 family) were unique to one family or the other (Fig. 5b). Differential expression associated with herbicide resistance was observed for another 12 P450s and five GSTs within the CC2 family, while five P450 genes displayed differential expression unique to the CC5 family. Two ABC transporters were identified as significant; one differentially expressed in the CC2 family, the other within the CC5 family. Comparably, several genes within the glycosyltransferase, drug/metabolite transporter and disease‐resistant NB‐LRR families were shown to have differential expression unique to one family or the other. Separate gene set enrichment analysis of DEGs for each family identified both shared and unique GO terms. Most of the shared overrepresented GO terms have been reported to be associated with NTSR, including GST, UGT and some cytochrome P450 superfamilies. ‘Xenobiotic transmembrane transporter’ activity was only overrepresented in CC5 (Fig. 5d,e), indicating the possible family‐specific mechanism of resistance for CC5. These results add to the growing evidence supporting a role for these gene families in herbicide detoxification, while the extent of DEGs specific to each family implies that different NTSR blackgrass populations may acquire an individual ‘profile’ of DEGs from these gene families.
In addition to previously reported genes and gene families, we found two transcription factors (ALOMY1G01646 and ALOMY2G19620), which were differentially expressed in both families, and the corresponding GO term (GO:0042221, ‘response to chemical’) was significantly enriched in both CC2 (P = 0.016) and CC5 (P = 0.006). A further nine and seven transcription factors with altered expression were identified in the CC2 and CC5 family, respectively. Although not a causal link, this may represent some involvement of these genes in regulation of NTSR gene expression. Interestingly, two Acetyl‐CoA synthetase‐like ATP‐dependent AMP‐binding enzymes were consistently upregulated in resistant plants: ALOMY2G20910 and ALOMY6G44209. Their corresponding GO term (GO:0032787, ‘monocarboxylic acid metabolic process’) was also significantly enriched in both CC2 (P = 0.006) and CC5 (P = 0.031). AMP‐forming acetyl‐CoA synthetases (ACS) catalyse the formation of acetyl‐CoA, substrate for the ACCase enzyme which is the target for herbicidal inhibition. Altered expression of these two genes could signify some remodelling of this pathway upstream of the point of herbicidal inhibition.
Co‐localisation of NTSR‐related features (QTLs and DEGs)
In addition to the population‐specific profile of DEGs, no overlap was observed between QTL regions identified in the two seed families (Table S7). However, 12 of the 15 total QTL regions were located on chromosomes 2 and 3 (Fig. 4). These two chromosomes also showed the greatest density of DEGs identified in the RNA‐seq analysis, with almost half (33) of the 68 consistent DEGs located on these two chromosomes, along with half of the previously reported NTSR candidate loci for this species (Fig. 4). In total, chromosomes 2 and 3 combined accounted for 45% and 55% of the total DEGs observed in the CC2 and CC5 families, respectively. Results of a Fishers' exact test for overrepresentation of DEGs per chromosome confirmed that chromosome 2 (CC2 family) and chromosomes 2 and 3 (CC5 family) were significantly enriched in resistance‐associated DEGs (Table S8). These results suggest that chromosomes 2 and 3 are ‘hot‐spots’ for NTSR evolution in this species.
To examine this further, we performed a comprehensive genome‐wide analysis of the five principal NTSR‐related gene families (GST, UGT, P450, ABC and AKR) (Fig. 6). A total of 506, 93, 146, 278 and 45 genes were identified in the blackgrass genome for P450, GST, ABC, UGT and AKR, respectively. Overall, blackgrass has a larger proportion of NTSR‐related genes (1069, 2.48% of total gene number) in the genome compared with those in Arabidopsis (2.04%) and rice (2.18%) genomes (Table S9; Fig. 6a). Among the five NTSR‐related gene families, blackgrass only has a smaller proportion for ABC family compared with Arabidopsis and rice. This observation is in line with the gene family expansion analysis, where P450, UGT and GST families each contain a high ratio of expanded genes. For example, 180 (35.6%), 61 (65.6%) and 129 (46.4%) genes were expanded in P450, GST and UGT gene families, respectively. However, only 50 and 28 NTSR‐related genes were differentially expressed between susceptible and resistant plants in CC2 and CC5, respectively. A large proportion (50%) of these differentially expressed NTSR‐related genes were located across chromosomes 2 and 3 combined in both seed families (Table S10; Fig. 6b). Nevertheless, tests for overrepresentation were non‐significant, in part due to the number of total NTSR‐related genes also being high across these two chromosomes (416 of 1068 NTSR‐related genes). Most of the differentially expressed NTSR‐related genes were upregulated in resistant plants and 23 of them were shared between families, including 6, 6, 3, 7 and 1 shared genes for P450, GST, ABC, UGT and AKR, respectively (Fig. 6c). Predominantly, these results highlight that while genomic features associated with NTSR (QTLs and DEGs) are largely population specific, their significant co‐localisation on chromosomes 2 and 3 reflects the importance of these two chromosomes during NTSR evolution.
Fig. 6.
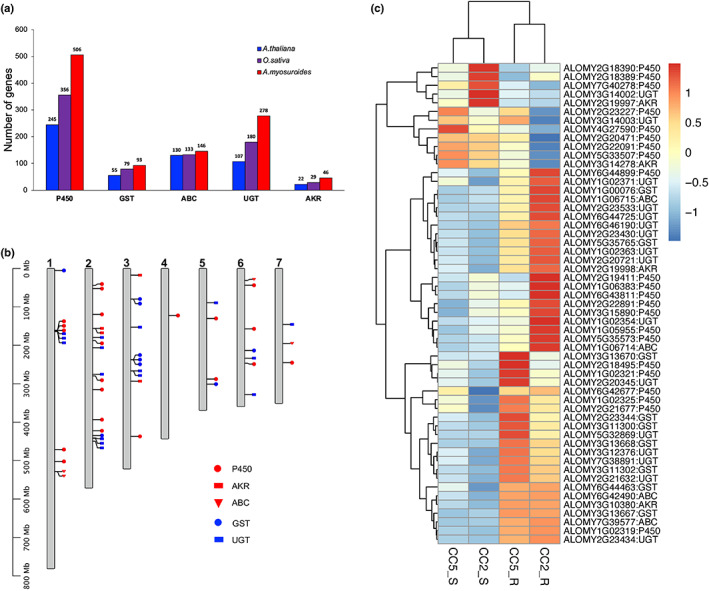
The gene number, distribution and expression for five non‐target site resistance (NTSR)‐related gene families in the A. myosuroides genome. (a) The number of genes identified for five NTSR‐related gene families including cytochrome P450 (P450), glutathione S‐transferase (GST), ATP‐binding cassette transporters (ABC), UDP‐glycosyltransferases (UGT) and aldo‐keto reductase (AKR). (b) The distribution of differentially expressed genes (DEGs) from five NTSR‐related gene families on seven blackgrass chromosomes. (c) Heatmap of the DEGs from five NTSR‐related gene families.
Genetic coordination of NTSR via GCN analysis
Gene co‐expression networks were constructed using traditional Spearman‐ranked and condition‐specific approaches that enable alternate strategies to examine the genetic coordination of NTSR mechanisms (Fig. 7a,b, respectively). The traditional Spearman‐ranked coefficient approaches resulted in a total of 16 601 nodes connected by 16 130 edges (Fig. 7a). Hub gene sub‐graphs display significant co‐expressed gene interaction pairs that include candidate genes from the bulk segregant and RNA‐seq studies. We identified a total of 13 CC2 specific sub‐graphs and 20 for CC5 (Fig. S8a–d). In CC2, we found the NTSR‐related gene families identified in the QTL‐seq analysis, such as GST, AKR and beta‐keto acyl synthase co‐expressed with various transcription factors and other genes that could be involved in their regulation (Fig. S8a,b). An HMG transcriptional regulator is also positively correlated with two genes involved in metabolism: tubulin/FtsZ family gene and a ubiquitin carboxyl‐terminal hydrolase, and negatively correlated with an α‐N‐acetylglucosaminidase (Fig. S8e). In the CC5 family sub‐graphs, we identified alternate active genetic machinery that are co‐expressed with genes identified in the QTL regions, such as cytochrome p450s, thioesterase, glycosyl hydrolase, pectinesterase, exostensin gene family and others connected with various classes of transporters and transcription factors/regulators (Fig. S8c,d). The condition‐specific network also partitioned clusters of co‐expressed gene interactions pairs in both a family specific and overlapping manner (Fig. 7b). For example, this approach also identified an AKR and protein tyrosine/serine/threonine kinase unique to CC2. Oxioreductase, peroxidase and vacuolar sorting were among CC5‐specific clusters (Fig. S8f). This approach also identified a largely connected subgraph of connected genes discovered in both CC2 and CC5 bulk‐segregant and RNA‐seq analysis (Fig. S8g). These network analyses further highlight that NTSR in blackgrass is likely to be due to a combination of core and population‐specific loci that act in concert with one another.
Fig. 7.
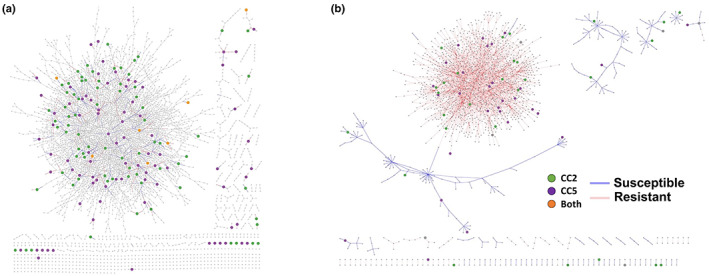
Genetic coordination of non‐target site resistance (NTSR) in CC2 and CC5 families of A. myosuroides. (a) Traditional Spearman‐ranked gene co‐expression network derived from RNA‐seq expression that depicts common and unique genetic architecture underpinning NTSR in both the CC2 and CC5 families. Green nodes are unique to CC2, purple nodes are unique to CC5 and orange are common between both families. The graph was filtered for nodes with at least two connections. (b) Condition‐specific gene co‐expression network derived from the RNA‐seq data taking into consideration plant phenotype (herbicide susceptible/resistant).
Discussion
Despite the global impacts of weedy plants, few genomic resources have been developed for weed species (see Sharma et al., 2021). Here, we present a reference‐grade genome assembly for Europe's most devastating agricultural weed, A. myosuroides (Hicks et al., 2018; Varah et al., 2020), and demonstrate that (1) NTSR in blackgrass is a complex, polygenic trait that evolves from standing genetic variation within stress‐ and defence‐related gene families and (2) that the convergent evolution of the NTSR phenotype among two field‐evolved populations has a notably different genetic basis among populations.
F2 seed families were produced for two field‐evolved blackgrass populations with very similar phenotypic resistance to the ACCase inhibiting herbicides and global, constitutive transcript profiles were compared for bulked resistant and susceptible individuals derived from the two seed families. At the transcriptional level, 11% of the resistance‐associated DEGs were common to both seed families. These 68 common DEGs include several genes and gene families previously implicated in NTSR in blackgrass (Cummins et al., 2013; Tetard‐Jones et al., 2018) and in a range of other weed species with evolved NTSR (Yang et al., 2018; Fang et al., 2019; Davies et al., 2020; Wang et al., 2020; Suzukawa et al., 2021; Torra et al., 2021). There were also commonalities in the localization of genomic signatures for NTSR across the two populations. For example, we found that chromosomes 2 and 3 were significantly enriched in both DEGs and QTL regions associated with resistance in both tested families. These results concur with those of Giacomini et al. (2020), who found physical clustering of DEGs in Amaranthus tuberculatus. Together, these results highlight that there are commonalities observed in the metabolic basis and broad genomic localisation of features associated with independently evolved NTSR.
Our results are consistent with a growing body of evidence from studies that explore the transcriptomic basis of NTSR. These studies confirm that NTSR is conferred by the upregulation of individual genes that are members of large stress‐ and defence‐related gene families; the cytochrome P450 monooxygenases, GSTs, ABC transporters, AKRs, glucosyl transferases and others (e.g. Cummins et al., 2013; Pan et al., 2019, 2021; Dimaano & Iwakami, 2021; Huang et al., 2021). Mounting evidence shows that these gene families are regularly implicated, and common genes were upregulated in both NTSR phenotypes studied here, and in NTSR populations of other species (Pan et al., 2019, 2021; Han et al., 2021). Together, these studies highlight a notable degree of parallelism in the metabolic NTSR phenotype. This would appear to indicate some constraint in the possible pathways via which NTSR can evolve. It is important to note, however, that the majority of DEGs identified in our study were not common among the two seed families; 89% were specific to one family or the other. This suggests that though a core of commonly over‐expressed genes is key for the metabolic expression of NTSR among populations, a significant number of population‐specific genes also contribute, indicating that both parallel and non‐parallel changes occur at the level of plant metabolism associated with the independent evolution of NTSR. Further studies that explore associated functional alleles in a greater number of evolved populations are warranted, and will be required to more completely understand the relative importance of parallelism and non‐parallelism in the evolution of the NTSR transcriptome.
Although the metabolic basis of NTSR among populations provides evidence for parallelism, our results unequivocally indicate that there is a discrete, non‐parallel basis to NTSR at the genomic level. Of the 15 significant QTLs identified (8 and 7 in the two seed families, respectively), there were no overlapping QTL regions, though significant QTLs were over‐represented on chromosomes 2 and 3 in each population. These observations suggest that while selection for NTSR may be localized to certain regions of the genome, the genetic basis of these traits is quite different among blackgrass populations. These results are consistent with the conclusion in Van Etten et al. (2020) and Gupta et al. (2021), that NTSR in Ipomoea purpurea is conferred by multiple loci. Similarly, separate studies of HPPD resistance in different Amaranthus tuberculatus populations have highlighted the distal regions of the genome showing signatures of selection for resistance (Kohlhase et al., 2018; Murphy et al., 2021). Our findings, combined with our co‐expression network analysis, provide strong evidence that NTSR among blackgrass populations is divergent at the genomic level.
Assessment of repetitive genome structure and duplication arrays suggests that these elements themselves might serve as an underlying mechanism facilitating rapid adaptation in blackgrass. For instance, high heterozygosity, expanded gene families (Fig. 1), a relatively recent (and unique) genome duplication event (Fig. 2), and clusters of TEs and LTR‐RTs are signatures of a dynamic genome. It is notable that the paralogous genes associated with genome duplication in this species are located on chromosomes 1, 2 and 3, among the densest regions of significant QTLs and DEGs (Fig. 4). We speculate that high levels of variation in the number, regulation and function of these defence‐related gene families enable weedy species such as blackgrass to rapidly evolve NTSR via exaptation of genes within these large multi‐functional gene families. Variation within these gene families distributed among discrete genetic backgrounds of blackgrass likely underpin the potential for non‐parallel evolution of NTSR.
Conclusion
We have established that NTSR in blackgrass is a polygenic trait, and that the genetic basis of NTSR can be markedly different between independently evolved populations; albeit underpinned by the upregulation of some common metabolic pathways. Notably, we find evidence for multiple QTL associated with the NTSR phenotype, but no evidence that these QTLs are the same among independently evolved populations. On this basis, we conclude that the landscape‐scale evolution of NTSR results from both parallel and non‐parallel patterns of evolution across the genome, as reported by Van Etten et al. (2020) and Kreiner et al. (2021). These findings have wide significance for understanding the potential for rapid plant adaptation under novel and changing environments. They suggest that large and plastic plant genomes harbour sufficient standing genetic variation to enable rapid adaptation to novel stresses. The associated duplication and redundancy in plant genomes means that adaptation may not be mutation limited and that the repeated evolution of resistance and/or tolerance relies on neither rare mutational events nor hard selective sweeps. They also hint that complex adaptations to abiotic and biotic stresses are not constrained by genetic variation and architecture and that convergent phenotypes are shaped by population‐specific genome structure and plasticity.
Competing interests
None declared.
Author contributions
PN, CS and RB conceived the study and assembled project funding. CL, DC and PN provided characterised plant material for sequencing. LC and CS assembled and annotated the blackgrass genome. LC, DC and CS analysed genomics and transcriptomics datasets and PN, DM and RB contributed to discussion and interpretation of data. LC, DC, CS and PN wrote the first draft of the paper and all authors contributed to subsequent editing and improvement. LC and DC contributed equally to this work. RB, PN and CS senior authors contributed equally to this work.
Supporting information
Fig. S1 Pipeline of genome assembly for the blackgrass.
Fig. S2 LAI values across seven assembled blackgrass chromosomes.
Fig. S3 Pipeline of gene annotation for the blackgrass.
Fig. S4 The distribution of different types of gene family.
Fig. S5 SNP marker density for CC2 and CC5 population, respectively.
Fig. S6 Principal component analysis of all gene expression data.
Fig. S7 Differential expression of previously reported NTSR candidate genes.
Fig. S8 Parallel and overlapping sub‐graphs in CC2 and CC5.
Notes S1 The development of research materials and genome sequencing and assembling.
Table S1 Genome size estimation based on flow cytometry.
Table S2 Genome size estimation based on k‐mer analysis.
Table S3 Statistics of the assembled seven chromosomes of A. myosuroides.
Table S4 Busco analysis of genome completeness.
Table S5 Summary of cytogenic and assembly length of each blackgrass chromosome.
Table S6 Statistics of the annotated transposon elements.
Table S7 Summary of identified QTLs from CC2 and CC5 populations.
Table S8 Statistical assessment of over‐representation of differentially expressed genes.
Table S9 Summary of gene number for NTSR‐related gene families.
Table S10 Summary of differentially expressed NTSR‐related gene number.
Please note: Wiley is not responsible for the content or functionality of any Supporting Information supplied by the authors. Any queries (other than missing material) should be directed to the New Phytologist Central Office.
Acknowledgements
DC, DM and PN were supported by the Smart Crop Protection Industrial Strategy Challenge Fund (grant no. BBS/OS/CP/000001) and Rothamsted Research as part of the Lawes Agricultural Trust. LC was supported by the Clemson University Research Fellows program. Rothamsted Research, Clemson University and Bayer Crop Science were equal contributors to costs associated with genomic and transcriptomic sequencing. The authors wish to thank Richard Hull and Laura Crook (Rothamsted Research, RR) for the growth and maintenance of plant material throughout this study, and David Hughes (RR) for bioinformatics assistance and advice. This research was also made possible, in part, with support from the Clemson University Genomics and Bioinformatics Facility, which receives support from an Institutional Development Award (IDeA) from the National Institute of General Medical Sciences of the National Institutes of Health under grant no. P20GM109094.
Data availability
This Whole Genome Shotgun project has been deposited at DDBJ/ENA/GenBank under the accession no. JAPCYS000000000. The version described in this paper is version JAPCYS010000000. Raw genome and transcriptome sequencing reads have been deposited in the National Center for Biotechnology Information BioProject database under the accession no. PRJNA889547. Code availability: the code for RNA‐seq and QTL‐seq analyses is freely available at GitHub (https://github.com/Lichuncai/Blackgrass‐Genome‐Project).
References
- Anders S, McCarthy DJ, Chen YS, Okoniewski M, Smyth GK, Huber W, Robinson MD. 2013. Count‐based differential expression analysis of RNA sequencing data using R and bioconductor. Nature Protocols 8: 1765–1786. [DOI] [PubMed] [Google Scholar]
- Andrews S. 2010. FastQC: a quality control tool for high throughput sequence data. [WWW document] URL http://www.bioinformatics.babraham.ac.uk/projects/fastqc [accessed 2 January 2020]. [Google Scholar]
- Baucom RS. 2016. The remarkable repeated evolution of herbicide resistance. American Journal of Botany 103: 181–183. [DOI] [PubMed] [Google Scholar]
- Baucom RS. 2019. Evolutionary and ecological insights from herbicide‐resistant weeds: what have we learned about plant adaptation, and what is left to uncover? New Phytologist 223: 68–82. [DOI] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30: 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolnick DI, Barrett RD, Oke KB, Rennison DJ, Stuart YE. 2018. (Non) parallel evolution. Annual Review of Ecology, Evolution, and Systematics 49: 303–330. [Google Scholar]
- Bulcke MHR. 1975. The distribution, spread and importance of Alopecurus myosuroides Huds. in Europe. In: Symposium on status, biology and control of grassweeds in Europe, organised by E.W.R.S. and COLUMA. Paris, France, 23–54.
- Comont D, Lowe C, Hull R, Crook L, Hicks HL, Onkokesung N, Beffa R, Childs DZ, Edwards R, Freckleton RP et al. 2020. Evolution of generalist resistance to herbicide mixtures reveals a trade‐off in resistance management. Nature Communications 11: 3086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comont D, MacGregor DR, Crook L, Hull R, Nguyen L, Freckleton RP, Childs DZ, Neve P. 2022. Dissecting weed adaptation: fitness and trait correlations in herbicide‐resistant Alopecurus myosuroides . Pest Management Science 78: 3039–3050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummins I, Wortley DJ, Sabbadin F, He ZS, Coxon CR, Straker HE, Sellars JD, Knight K, Edwards L, Hughes D et al. 2013. Key role for a glutathione transferase in multiple‐herbicide resistance in grass weeds. Proceedings of the National Academy of Sciences, USA 110: 5812–5817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies LR, Onkokesung N, Brazier‐Hicks M, Edwards R, Moss S. 2020. Detection and characterization of resistance to acetolactate synthase inhibiting herbicides in Anisantha and Bromus species in the United Kingdom. Pest Management Science 76: 2473–2482. [DOI] [PubMed] [Google Scholar]
- De Bie T, Cristianini N, Demuth JP, Hahn MW. 2006. Cafe: a computational tool for the study of gene family evolution. Bioinformatics 22: 1269–1271. [DOI] [PubMed] [Google Scholar]
- Delye C. 2013. Unravelling the genetic bases of non‐target‐site‐based resistance (NTSR) to herbicides: a major challenge for weed science in the forthcoming decade. Pest Management Science 69: 176–187. [DOI] [PubMed] [Google Scholar]
- Dimaano NG, Iwakami S. 2021. Cytochrome P450-mediated herbicide metabolism in plants: current understanding and prospects. Pest Management Science 77: 22–32. [DOI] [PubMed] [Google Scholar]
- Dudchenko O, Batra SS, Omer AD, Nyquist SK, Hoeger M, Durand NC, Shamim MS, Machol I, Lander ES, Aiden AP et al. 2017. De novo assembly of the Aedes aegypti genome using Hi‐C yields chromosome‐length scaffolds. Science 356: 92–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand NC, Robinson JT, Shamim MS, Machol I, Mesirov JP, Lander ES, Aiden EL. 2016b. Juicebox provides a visualization system for Hi‐C contact maps with unlimited zoom. Cell Systems 3: 99–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand NC, Shamim MS, Machol I, Rao SSP, Huntley MH, Lander ES, Aiden EL. 2016a. Juicer provides a one‐click system for analyzing loop‐resolution Hi‐C experiments. Cell Systems 3: 95–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC. 2004. Muscle: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Research 32: 1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emms DM, Kelly S. 2015. OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biology 16: 157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang JP, Zhang YH, Liu TT, Yan BJ, Li J, Dong LY. 2019. Target‐site and metabolic resistance mechanisms to Penoxsulam in Barnyardgrass (Echinochloa crus‐galli (L.) P. Beauv). Journal of Agricultural and Food Chemistry 67: 8085–8095. [DOI] [PubMed] [Google Scholar]
- Fedoroff N. 2000. Transposons and genome evolution in plants. Proceedings of the National Academy of Sciences, USA 97: 7002–7007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco‐Ortega S, Goldberg‐Cavalleri A, Walker A, Brazier‐Hicks M, Onkokesung N, Edwards R. 2021. Non‐target site herbicide resistance is conferred by two distinct mechanisms in black‐grass (Alopecurus myosuroides). Frontiers in Plant Science 12. doi: 10.3389/fpls.2021.636652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaines TA, Duke SO, Morran S, Rigon CAG, Tranel PJ, Kupper A, Dayan FE. 2020. Mechanisms of evolved herbicide resistance. Journal of Biological Chemistry 295: 10307–10330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giacomini DA, Patterson EL, Kupper A, Beffa R, Gaines TA, Tranel PJ. 2020. Coexpression clusters and allele‐specific expression in metabolism‐based herbicide resistance. Genome Biology and Evolution 12: 2267–2278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould F, Brown ZS, Kuzma J. 2018. Wicked evolution: can we address the sociobiological dilemma of pesticide resistance? Science 360: 728–732. [DOI] [PubMed] [Google Scholar]
- Gupta S, Harkess A, Soble A, Van Etten M, Leebens‐Mark J, Baucom RS. 2021. Inter‐chromosomal linkage disequilibrium and linked fitness cost loci associated with selection for herbicide resistance. bioRxiv. doi: 10.1101/2021.04.04.438381. [DOI] [PubMed]
- Haas BJ, Papanicolaou A, Yassour M, Grabherr M, Blood PD, Bowden J, Couger MB, Eccles D, Li B, Lieber M et al. 2013. De novo transcript sequence reconstruction from RNA‐seq using the Trinity platform for reference generation and analysis. Nature Protocols 8: 1494–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas BJ, Salzberg SL, Zhu W, Pertea M, Allen JE, Orvis J, White O, Buell CR, Wortman JR. 2008. Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biology 9: R7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han H, Yu Q, Beffa R, González S, Maiwald F, Wang J, Powles S. 2021. Cytochrome P450 CYP81A10v7 in Lolium rigidum confers metabolic resistance to herbicides across at least five modes of action. The Plant Journal 105: 79–92. [DOI] [PubMed] [Google Scholar]
- Hawkins NJ, Bass C, Dixon A, Neve P. 2019. The evolutionary origins of pesticide resistance. Biological Reviews 94: 135–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heap I. 2014. Global perspective of herbicide‐resistant weeds. Pest Management Science 70: 1306–1315. [DOI] [PubMed] [Google Scholar]
- Hendry AP, Kinnison MT, Heino M, Day T, Smith TB, Fitt G, Bergstrom CT, Oakeshott J, Jorgensen PS, Zalucki MP et al. 2011. Evolutionary principles and their practical application. Evolutionary Applications 4: 159–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hicks H, Lambert J, Pywell R, Hulmes L, Hulmes S, Walker K, Childs DZ, Freckleton RP. 2021. Characterizing the environmental drivers of the abundance and distribution of Alopecurus myosuroides on a national scale. Pest Management Science 77: 2726–2736. [DOI] [PubMed] [Google Scholar]
- Hicks HL, Comont D, Coutts SR, Crook L, Hull R, Norris K, Neve P, Childs DZ, Freckleton RP. 2018. The factors driving evolved herbicide resistance at a national scale. Nature Ecology & Evolution 2: 529–536. [DOI] [PubMed] [Google Scholar]
- Huang SF, Kang MJ, Xu AL. 2017. HaploMerger2: rebuilding both haploid sub‐assemblies from high‐heterozygosity diploid genome assembly. Bioinformatics 33: 2577–2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang XX, Zhao SM, Zhang YY, Li YJ, Shen HN, Li X, Hou BK. 2021. A novel UDP-glycosyltransferase 91C1 confers specific herbicide resistance through detoxification reaction in Arabidopsis. Plant Physiology and Biochemistry 159: 226–233. [DOI] [PubMed] [Google Scholar]
- Jin Y, Tam OH, Paniagua E, Hammell M. 2015. tetranscripts: a package for including transposable elements in differential expression analysis of RNA‐seq datasets. Bioinformatics 31: 3593–3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnsson H. 1944. Meiotic aberrations and sterility in Alopecurus myosuroides Huds. Hereditas 30: 469–566. [Google Scholar]
- Kanehisa M, Sato Y, Morishima K. 2016. BlastKOALA and GhostKOALA: Kegg tools for functional characterization of genome and metagenome sequences. Journal of Molecular Biology 428: 726–731. [DOI] [PubMed] [Google Scholar]
- Keilwagen J, Hartung F, Grau J. 2019. GeMoMa: homology‐based gene prediction utilizing intron position conservation and RNA‐seq data. Gene Prediction: Methods and Protocols 1962: 161–177. [DOI] [PubMed] [Google Scholar]
- Kielbasa SM, Wan R, Sato K, Horton P, Frith MC. 2011. Adaptive seeds tame genomic sequence comparison. Genome Research 21: 487–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Paggi JM, Park C, Bennett C, Salzberg SL. 2019. Graph‐based genome alignment and genotyping with Hisat2 and Hisat‐genotype. Nature Biotechnology 37: 907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohlhase DR, Edwards JW, Owen MDK. 2018. Inheritance of 4‐hydroxyphenylpyruvate dioxygenase inhibitor herbicide resistance in an Amaranthus tuberculatus population from Iowa, USA. Plant Science 274: 360–368. [DOI] [PubMed] [Google Scholar]
- Korf I. 2004. Gene finding in novel genomes. BMC Bioinformatics 5: 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreiner JM, Stinchcombe JR, Wright SI. 2018. Population genomics of herbicide resistance: adaptation via evolutionary rescue. Annual Review of Plant Biology 69: 611–635. [DOI] [PubMed] [Google Scholar]
- Kreiner JM, Tranel PJ, Weigel D, Stinchcombe JR, Wright SI. 2021. The genetic architecture and population genomic signatures of glyphosate resistance in Amaranthus tuberculatus . Molecular Ecology 30: 5373–5389. [DOI] [PubMed] [Google Scholar]
- Kumar R, Janila P, Vishwakarma MK, Khan AW, Manohar SS, Gangurde SS, Variath MT, Shasidhar Y, Pandey MK, Varshney RK. 2020. Whole‐genome resequencing‐based QTL‐seq identified candidate genes and molecular markers for fresh seed dormancy in groundnut. Plant Biotechnology Journal 18: 992–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, Shi W. 2014. FeatureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30: 923–930. [DOI] [PubMed] [Google Scholar]
- Liu XY, Merchant A, Xiang SH, Zong T, Zhou XG, Bai LY. 2021. Managing herbicide resistance in China. Weed Science 69: 4–17. [Google Scholar]
- Lobkovsky AE, Koonin EV. 2012. Replaying the tape of life: quantification of the predictability of evolution. Frontiers in Genetics 3: 246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomsadze A, Ter‐Hovhannisyan V, Chernoff YO, Borodovsky M. 2005. Gene identification in novel eukaryotic genomes by self‐training algorithm. Nucleic Acids Research 33: 6494–6506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losos JB. 2011. Convergence, adaptation, and constraint. Evolution 65: 1827–1840. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S. 2014. Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biology 15: 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe TM, Eddy SR. 1997. tRNAscan‐SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Research 25: 955–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M, Conery JS. 2000. The evolutionary fate and consequences of duplicate genes. Science 290: 1151–1155. [DOI] [PubMed] [Google Scholar]
- Ma J, Bennetzen JL. 2004. Rapid recent growth and divergence of rice nuclear genomes. Proceedings of the National Academy of Sciences, USA 101: 12404–12410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahaut L, Cheptou PO, Fried G, Munoz F, Storkey J, Vasseur F, Violle C, Bretagnolle F. 2020. Weeds: against the rules? Trends in Plant Science 25: 1107–1116. [DOI] [PubMed] [Google Scholar]
- Martin A, Orgogozo V. 2013. The loci of repeated evolution: a catalog of genetic hotspots of phenotypic variation. Evolution 67: 1235–1250. [DOI] [PubMed] [Google Scholar]
- Menchari Y, Delye C, Le Corre V. 2007. Genetic variation and population structure in black‐grass (Alopecurus myosuroides Huds.), a successful, herbicide‐resistant, annual grass weed of winter cereal fields. Molecular Ecology 16: 3161–3172. [DOI] [PubMed] [Google Scholar]
- Morgulis A, Gertz EM, Schaffer AA, Agarwala R. 2006. WindowMasker: window‐based masker for sequenced genomes. Bioinformatics 22: 134–141. [DOI] [PubMed] [Google Scholar]
- Moss SR, Storkey J, Cussans JW, Perryman SAM, Hewitt MV. 2004. The Broadbalk long‐term experiment at Rothamsted: what has it told us about weeds? Weed Science 52: 864–873. [Google Scholar]
- Murphy BP, Beffa R, Tranel PJ. 2021. Genetic architecture underlying HPPD‐inhibitor resistance in a Nebraska Amaranthus tuberculatus population. Pest Management Science 77: 4884–4891. [DOI] [PubMed] [Google Scholar]
- Nawrocki EP, Kolbe DL, Eddy SR. 2009. Infernal 1.0: inference of RNA alignments. Bioinformatics 25: 1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neve P, Vila‐Aiub M, Roux F. 2009. Evolutionary‐thinking in agricultural weed management. New Phytologist 184: 783–793. [DOI] [PubMed] [Google Scholar]
- Orr HA. 2005a. The genetic theory of adaptation: a brief history. Nature Reviews Genetics 6: 119–127. [DOI] [PubMed] [Google Scholar]
- Orr HA. 2005b. The probability of parallel evolution. Evolution 59: 216–220. [PubMed] [Google Scholar]
- Ou SJ, Chen JF, Jiang N. 2018. Assessing genome assembly quality using the LTR Assembly Index (LAI). Nucleic Acids Research 46: e126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou SJ, Su WJ, Liao Y, Chougule K, Agda JRA, Hellinga AJ, Lugo CSB, Elliott TA, Ware D, Peterson T et al. 2019. Benchmarking transposable element annotation methods for creation of a streamlined, comprehensive pipeline. Genome Biology 20: 275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palumbi SR. 2001. Evolution – humans as the world's greatest evolutionary force. Science 293: 1786–1790. [DOI] [PubMed] [Google Scholar]
- Pan L, Yu Q, Han H, Mao L, Nyporko A, Fan L, Bai L, Powles S. 2019. Aldo‐keto reductase metabolizes glyphosate and confers glyphosate resistance in Echinochloa colona . Plant Physiology 181: 1519–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan L, Yu Q, Wang J, Han H, Mao L, Nyporko A, Maguza A, Fan L, Bai L, Powles S. 2021. An ABCC‐type transporter endowing glyphosate resistance in plants. Proceedings of the National Academy of Sciences, USA 118: e2100136118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paterson AH, Bowers JE, Chapman BA. 2004. Ancient polyploidization predating divergence of the cereals, and its consequences for comparative genomics. Proceedings of the National Academy of Sciences, USA 101: 9903–9908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powles SB, Yu Q. 2010. Evolution in action: plants resistant to herbicides. Annual Review of Plant Biology 61: 317–347. [DOI] [PubMed] [Google Scholar]
- R Core Team . 2018. R: a language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing. [Google Scholar]
- Ravet K, Patterson EL, Krahmer H, Hamouzova K, Fan LJ, Jasieniuk M, Lawton‐Rauh A, Malone JM, McElroy JS, Merotto A et al. 2018. The power and potential of genomics in weed biology and management. Pest Management Science 74: 2216–2225. [DOI] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK. 2010. edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26: 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenhauer M, Jaser B, Felsenstein FG, Petersen J. 2013. Development of target‐site resistance (TSR) in Alopecurus myosuroides in Germany between 2004 and 2012. Journal of Plant Diseases and Protection 120: 179–187. [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. 2003. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Research 13: 2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma G, Barney JN, Westwood JH, Haak DC. 2021. Into the weeds: new insights in plant stress. Trends in Plant Science 26: 1050–1060. [DOI] [PubMed] [Google Scholar]
- Shealy BT, Burns JJR, Smith MC, Feltus FA, Ficklin SP. 2019. GPU implementation of pairwise Gaussian mixture models for multi‐modal gene co‐expression networks. IEEE Access 7: 160845–160857. [Google Scholar]
- Simao FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM. 2015. Busco: assessing genome assembly and annotation completeness with single‐copy orthologs. Bioinformatics 31: 3210–3212. [DOI] [PubMed] [Google Scholar]
- Song L, Langfelder P, Horvath S. 2012. Comparison of co‐expression measures: mutual information, correlation, and model based indices. BMC Bioinformatics 13: 328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis A. 2014. RAxML v.8: a tool for phylogenetic analysis and post‐analysis of large phylogenies. Bioinformatics 30: 1312–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanke M, Keller O, Gunduz I, Hayes A, Waack S, Morgenstern B. 2006. Augustus: ab initio prediction of alternative transcripts. Nucleic Acids Research 34: W435–W439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzukawa AK, Bobadilla LK, Mallory‐Smith C, Brunharo CACG. 2021. Non‐target‐site resistance in Lolium spp. globally: a review. Frontiers in Plant Science 11. doi: 10.3389/fpls.2020.609209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takagi H, Abe A, Yoshida K, Kosugi S, Natsume S, Mitsuoka C, Uemura A, Utsushi H, Tamiru M, Takuno S et al. 2013. QTL‐seq: rapid mapping of quantitative trait loci in rice by whole genome resequencing of DNA from two bulked populations. The Plant Journal 74: 174–183. [DOI] [PubMed] [Google Scholar]
- Tetard‐Jones C, Sabbadin F, Moss S, Hull R, Neve P, Edwards R. 2018. Changes in the proteome of the problem weed blackgrass correlating with multiple‐herbicide resistance. The Plant Journal 94: 709–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torra J, Montull JM, Taberner A, Onkokesung N, Boonham N, Edwards R. 2021. Target‐site and non‐target‐site resistance mechanisms confer multiple and cross‐ resistance to ALS and ACCase inhibiting herbicides in Lolium rigidum from Spain. Frontiers in Plant Science 12: 57–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Etten M, Lee KM, Chang SM, Baucom RS. 2020. Parallel and nonparallel genomic responses contribute to herbicide resistance in Ipomoea purpurea, a common agricultural weed. PLoS Genetics 16: 1008593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varah A, Ahodo K, Coutts SR, Hicks HL, Comont D, Crook L, Hull R, Neve P, Childs DZ, Freckleton RP et al. 2020. The costs of human‐induced evolution in an agricultural system. Nature Sustainability 3: 63–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigueira CC, Olsen KM, Caicedo AL. 2013. The red queen in the corn: agricultural weeds as models of rapid adaptive evolution. Heredity 110: 303–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker BJ, Abeel T, Shea T, Priest M, Abouelliel A, Sakthikumar S, Cuomo CA, Zeng Q, Wortman J, Young SK et al. 2014. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 9: 0112963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Zhang Y, Zhang Z, Zhu J, Yu J. 2010. KaKs_calculator 2.0: a toolkit incorporating gamma‐series methods and sliding window strategies. Genomics, Proteomics & Bioinformatics 8: 77–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Chen J, Li X, Li D, Li Z, Cui H. 2020. Pro‐197‐Ser mutation in ALS and high‐level GST activities: multiple resistance to ALS and ACCase inhibitors in Beckmannia syzigachne. Frontiers in Plant Science 11: 572610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YP, Tang HB, DeBarry JD, Tan X, Li JP, Wang XY, Lee TH, Jin HZ, Marler B, Guo H et al. 2012. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Research 40: e49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren RL. 2016. RAILS and Cobbler: scaffolding and automated finishing of draft genomes using long DNA sequences. Journal of Open Source Software 1: 116. [Google Scholar]
- Xiao CL, Chen Y, Xie SQ, Chen KN, Wang Y, Han Y, Luo F, Xie Z. 2017. Mecat: fast mapping, error correction, and de novo assembly for single‐molecule sequencing reads. Nature Methods 14: 1072–1074. [DOI] [PubMed] [Google Scholar]
- Yang Q, Li J, Shen J, Xu Y, Liu H, Deng W, Li X, Zheng M. 2018. Metabolic resistance to acetolactate synthase inhibiting herbicide tribenuron‐methyl in Descurainia sophia L. mediated by cytochrome P450 enzymes. Journal of Agricultural and Food Chemistry 66: 4319–4327. [DOI] [PubMed] [Google Scholar]
- Yang ZH. 2007. Paml 4: phylogenetic analysis by maximum likelihood. Molecular Biology and Evolution 24: 1586–1591. [DOI] [PubMed] [Google Scholar]
- Yang ZH, Rannala B. 2006. Bayesian estimation of species divergence times under a molecular clock using multiple fossil calibrations with soft bounds. Molecular Biology and Evolution 23: 212–226. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 Pipeline of genome assembly for the blackgrass.
Fig. S2 LAI values across seven assembled blackgrass chromosomes.
Fig. S3 Pipeline of gene annotation for the blackgrass.
Fig. S4 The distribution of different types of gene family.
Fig. S5 SNP marker density for CC2 and CC5 population, respectively.
Fig. S6 Principal component analysis of all gene expression data.
Fig. S7 Differential expression of previously reported NTSR candidate genes.
Fig. S8 Parallel and overlapping sub‐graphs in CC2 and CC5.
Notes S1 The development of research materials and genome sequencing and assembling.
Table S1 Genome size estimation based on flow cytometry.
Table S2 Genome size estimation based on k‐mer analysis.
Table S3 Statistics of the assembled seven chromosomes of A. myosuroides.
Table S4 Busco analysis of genome completeness.
Table S5 Summary of cytogenic and assembly length of each blackgrass chromosome.
Table S6 Statistics of the annotated transposon elements.
Table S7 Summary of identified QTLs from CC2 and CC5 populations.
Table S8 Statistical assessment of over‐representation of differentially expressed genes.
Table S9 Summary of gene number for NTSR‐related gene families.
Table S10 Summary of differentially expressed NTSR‐related gene number.
Please note: Wiley is not responsible for the content or functionality of any Supporting Information supplied by the authors. Any queries (other than missing material) should be directed to the New Phytologist Central Office.
Data Availability Statement
This Whole Genome Shotgun project has been deposited at DDBJ/ENA/GenBank under the accession no. JAPCYS000000000. The version described in this paper is version JAPCYS010000000. Raw genome and transcriptome sequencing reads have been deposited in the National Center for Biotechnology Information BioProject database under the accession no. PRJNA889547. Code availability: the code for RNA‐seq and QTL‐seq analyses is freely available at GitHub (https://github.com/Lichuncai/Blackgrass‐Genome‐Project).