

Abstract
Pancreatic ductal adenocarcinoma (PDAC) is an extremely aggressive malignancy with minimal treatment options and a global rise in prevalence. PDAC is characterized by frequent driver mutations including KRAS and TP53 (p53), and a dense, acidic tumor microenvironment (TME). The relation between genotype and TME in PDAC development is unknown. Strikingly, when wild type (WT) Panc02 PDAC cells were adapted to growth in an acidic TME and returned to normal pH to mimic invasive cells escaping acidic regions, they displayed a strong increase of aggressive traits such as increased growth in 3‐dimensional (3D) culture, adhesion‐independent colony formation and invasive outgrowth. This pattern of acidosis‐induced aggressiveness was observed in 3D spheroid culture as well as upon organotypic growth in matrigel, collagen‐I and combination thereof, mimicking early and later stages of PDAC development. Acid‐adaptation‐induced gain of cancerous traits was further increased by p53 knockout (KO), but only in specific extracellular matrix (ECM) compositions. Akt‐ and Transforming growth factor‐β (TGFβ) signaling, as well as expression of the Na+/H+ exchanger NHE1, were increased by acid adaptation. Whereas Akt inhibition decreased spheroid growth regardless of treatment and genotype, stimulation with TGFβI increased growth of WT control spheroids, and inhibition of TGFβ signaling tended to limit growth under acidic conditions only. Our results indicate that a complex crosstalk between tumor acidosis, ECM composition and genotype contributes to PDAC development. The findings may guide future strategies for acidosis‐targeted therapies.
Keywords: extracellular matrix, invasion, organotypic culture, spheroids, tumor microenvironment
What's new?
Pancreatic ductal adenocarcinoma (PDAC) is characterized by specific driver mutations and by the presence of extracellular acidosis within a dense, fibrotic, extracellular matrix (ECM). While these features are independently linked to increased PDAC invasiveness, whether genotype impacts the pancreatic tumor microenvironment remains unclear. Our study shows that three‐dimensional and anchorage‐independent PDAC cell growth and PDAC progression are enhanced by acid adaptation, specifically when normal ECM pH is reestablished, as occurs during vascularization and metastasis. The findings suggest that interplay between acidosis, ECM composition and driver mutations strongly influences PDAC aggressiveness, with possible relevance for the development of novel therapeutic strategies.
Abbreviations
- ECM
extracellular matrix
- ERK1/2
extracellular signal regulated kinase‐1/2
- KO
knockout
- NBCn1
electroneutral Na+,HCO3 − cotransporter‐1
- NHE1
Na+/H+ exchanger‐1
- PDAC
pancreatic ductal adenocarcinoma
- pHe
extracellular pH
- pHi
intracellular pH
- TGFβ
transforming growth factor‐β
- TME
tumor microenvironment
- TXNIP
thioredoxin‐interacting protein
- WT
wild type
1. INTRODUCTION
Pancreatic ductal adenocarcinoma (PDAC) is a devastating disease with a 5‐year survival of less than 7%. 1 Patients are rarely diagnosed until late‐stage disease, leaving chemotherapy as the only treatment option, further limited by extensive chemotherapy resistance. 1 Underscoring the urgent need for better therapeutic options, PDAC is projected to become the second leading cause of global cancer mortality by 2030. 2
Activating KRAS mutations are the most ubiquitous PDAC driver mutation, found in over 90% of PDAC tumors. The tumor suppressor p53, the TP53 gene product 3 is inactivated in about 75% of PDAC cases 4 and this is associated with increased invasiveness, dedifferentiated malignant disease, and worse prognosis. 5 , 6 Two additional characteristics of PDAC tumors are extensive desmoplasia—the accumulation of a dense, fibrotic, extracellular matrix (ECM) by tumor stromal cells 7 —and profound extracellular acidosis. Desmoplasia is linked to increased proliferation, migration/invasion, metabolism and chemotherapy resistance in experimental PDAC models 8 and associated with aggressiveness and shorter survival in PDAC patients. 9 , 10 Altered ECM architecture has also been linked to upregulation of a vasculogenic mimicry transcriptional program predicting patient survival in multiple cancers. 11 Extracellular acidosis is a hallmark of the tumor microenvironment (TME) across cancers 12 , 13 , 14 , 15 including PDAC. 16 , 17 The acidosis reflects extensive metabolic acid production and limited perfusion in solid tumors and contributes to numerous aspects of cancer development, including genetic instability, metabolic rewiring, invasiveness and chemotherapy resistance. 12 , 18 In vivo, acidic preconditioning of cancer cells increases local invasion 19 and metastasis. 20 , 21 On the other hand, low extracellular pH (pHe) challenges intracellular pH (pHi), which per se is a limitation for cell proliferation and growth. 22 , 23 We therefore previously proposed that extracellular acidosis may initially contribute to cancer dormancy, and that additional events, such as oncogenic mutations or selection for upregulated acid extrusion and other reprogramming events overcoming this limitation are required for acidosis to become protumorigenic. 24 However, it is unknown how interplay between tumor acidosis, ECM properties and driver mutations impact PDAC progression.
Here, we used a combination of 3D spheroid culture and organotypic growth in matrigel, collagen, or combinations thereof, to test the hypothesis that prolonged extracellular acidosis synergizes with p53 inactivation and ECM composition to drive a transcriptional reprogramming contributing to PDAC aggressiveness. We show that adaptation to acidosis drives a marked, p53‐dependent potentiation of cancer cell growth, adhesion‐independent colony formation and invasiveness, which is only seen upon return to normal tissue pH to mimic invasive growth. Our results demonstrate for the first time a crosstalk between acidosis, ECM properties and genotype, which leads to increased growth and invasiveness of PDAC cells.
2. MATERIALS AND METHODS
2.1. Cell lines and acid adaptation
All experiments were performed with mycoplasma‐free cells. Panc02 mouse pancreatic cancer cells (RRID: CVCL_D627), a gift from A. Trauzold, University of Kiel, Germany, were cultured in DMEM (Gibco Life Technologies) with 10% FBS (Sigma, F9665‐500ML) and 1% Penicillin/Streptomycin (Sigma, P0781‐100ML) at 5% CO2/37°C. Panc02 cells were adapted to growth at pH 6.7 by decreasing the pH of the culture medium by ~0.2 pH units weekly. Panc‐1 cells (RRID CVCL_0480) and AsPC‐1 cells (RRID CVCL_0152) were obtained from ATCC (Manassas, VA, USA) and were cultured in AsPC‐1 and Panc‐1 cells were cultured in DMEM (10569010 and 41966029, respectively, Gibco Life Technologies) with 10% FBS (Sigma, F9665‐500ML) and 1% Penicillin/Streptomycin (Sigma, P0781‐100ML) at 5% CO2/37°C and acid‐adapted by transfer to pH 6.7 medium followed by a month of growth under these conditions. Culture media were pH‐adjusted by titration with hydrochloric acid and measured at 5% CO2. Once pH 6.7 was reached, cells were cultured for maximally 8 weeks. HUVECs were cultured in Endothelial Cell Growth Medium (C‐22210, PromoCell) with Growth Medium Supplement Mix (C‐39215, PromoCell) and 1% Penicillin/Streptomycin (Sigma, P0781‐100ML) at 5% CO2/37°C.
2.2. CRISPR/Cas9 p53 knock‐out
The Panc02‐p53 knockout (KO) cell line was established as in Reference 25 and analyzed as in Reference 26. Briefly, guide RNA (gRNA) sequence 5’‐CACCGTGCACATAACAGACT‐3′ targeting exon 4 of Trp53 was designed in CRISPR Finder software (https://wge.stemcell.sanger.ac.uk/find_crisprs) and cloned into the pSpCas9(BB)‐2A‐GFP (PX458) plasmid (Addgene #48138) and transfected into Panc02 WT cells using Lipofectamine 3000 (Invitrogen) according to the manufacturer's protocol. After 3 days, cells were sorted for GFP positive cells using BD FACSAriaTM III (BD Biosciences‐US). Transfection and FACS sorting were repeated and followed by single cell clone isolation and expansion, and Trp53 KO was verified by western blotting.
2.3. Spheroid culture, imaging and pharmacological treatments
Cells were seeded at 1000 cells/well in round‐bottomed ultra‐low attachment (ULA) 96‐well‐plates (VWR, #4441020) in 200 μL of the appropriate medium and grown in a CO2‐incubator at 37°C. Medium was changed every 2‐3 days. Images were acquired on the indicated days with an Olympus IX83 microscope, using a SP 10× objective and CellSens Dimension software. Spheroid area was determined in ImageJ software using freehand selection to mark spheroid outline, followed by area quantification in ImageJ. Necrotic core quantification (in three independent biological replicates) was performed using ImageJ software. Bright field images were analyzed using a macros function where images were converted to 8‐bit format, adjusted to Threshold (0.50) to define the necrotic core in each spheroid. Area in pixels2 was measured using “Analyze Particles” function with size of particles bigger than 2000 pixels2, and converted to mm2. In each experiment 4‐6 biological replicates were used. Where indicated, media were supplemented with SB 431542 (#1614, Tocris Bioscience), human TGFβ1 (#100‐21‐2UG, PeproTech), Capivasertib (AZD5363, #S8019, Selleckchem), or MK‐2206 (#S1078, Selleckchem).
2.4. 3D organotypic culture
Single Panc02 cell suspensions were seeded in 50 μL domes of matrigel (Corning, # 356237; 3000 cells/dome), collagen‐I (3 mg/mL, Collagen G, Sigma L7213‐100ML, 5000 cells/dome) or 1:1 matrigel:collagen‐I mix (2000 cells/dome) in 24‐well‐plates (BioLite Multidish, Thermo Fisher Scientific) and cultured at 5% CO2/37°C. Solidified 3D domes were subsequently overlaid with 0.5 mL of appropriate culture media. Media were replaced every third day. Brightfield images were acquired after 5 (matrigel), 7 (collagen‐I) or 3 days (mix) with an Olympus IX83 microscope, using 4× and 10× objectives and CellSens software.
2.5. Flow cytometry
One thousand cells/well were seeded in 96‐well‐plates. On day nine, 20 spheroids per condition were collected, washed once with PBS, centrifuged at 50 RCF for 3 minutes and dissociated with Accutase solution (Sigma, A6964) for 20 minutes. The resulting cell suspension was spun down, washed in ice‐cold PBS with 2% FBS (flow buffer) (Gibco, #10106‐177), followed by resuspension in flow buffer containing 5 μg/mL PI (Invitrogen, #P3566). The samples were incubated at 37°C for 30 minutes, followed by a wash and resuspension in flow buffer. PI negative sample was included to control for autofluorescence. Samples were subsequently transferred to polystyrene tubes (BD Falcon) and mixed with 50 μL CountBright Absolute Counting Beads (Invitrogen Cat. # C36950), followed by analysis on a BD FACS Calibur cytometer and CellQuest software (BD Biosciences). Cell concentrations were determined according to manufacturer's instructions. Briefly, samples were analyzed in an FSC vs SSC plot and gates were drawn around the population of beads and the cell population of interest. The calculation of cell concentrations was made by the following equation: A/B * C/D = concentration of sample as cells/μL, where A = number of cell events in gate, B = number of bead events in gate, C = assigned bead count of the lot (beads/50 μL) and D = volume of sample (μL). The measurement of dead cells was made by analyzing the samples in a SSC vs FL2 (PI signal) plot, and obtaining percentages for the negative and positive populations (gate discriminating neg/pos population was based on the unstained [PI negative] sample). The resulting quadrant statistics where subsequently plotted in prism.
2.6. Total viability assays
Viability was assessed using the CellTiter GLO kit (Promega, #G9683). To evaluate cell viability in spheroid cultures, colonies were recovered from matrices after 9 days, and ATP levels were measured, following the manufacturers protocol. For organotypic cultures, culture medium was removed from the well, the 3D dome was disrupted by pipetting ice‐cold medium up and down, and the well content was transferred to a Corning 96‐well black polystyrene microplate (VWR, #4441020). One hundred microliters of Cell Titer Glo 3D Reagent (Promega, #G9683) was added to the well followed by shaking for 5 minutes and incubation at room temperature for 25 minutes. Luminescence was measured using a FluoStar Optima platereader and Optima software.
2.7. Immunofluorescence analysis
Organotypic cultures were seeded as above, except in 8‐well chamber slides (Lab‐TekTM, Thermo Fisher Scientific) and stained as in Reference 27. Briefly, cultures were fixed with 4% PFA for 15 minutes and permeabilized in 0.5% Triton X‐100 in PBS for 20 minutes. After 1 hour of blocking in 1% BSA solution, cells were incubated overnight at 4°C with primary antibody (Table S1) against α‐tubulin (1:1000, T5168, Sigma‐Aldrich). Next day, specimens were incubated f1 hour with Alexa‐488 donkey‐anti‐mouse secondary antibody (1:400, Invitrogen) and 10 minutes with DAPI (1 μg/mL). Samples were fixed with Fluoromount‐G antifade (Invitrogen, 00‐4988‐02) and analyzed employing an Olympus IX83 microscope with a Yokogawa scanning unit, a PLANAPO 60×/1.4 NA oil immersion objective and CellSens Dimension software.
2.8. Live/dead staining of organotypic cultures
Single Panc02 cell suspensions (in three independent biological replicates) were seeded in 50 μL domes of matrigel (3000 cells/dome), collagen‐I (5000 cells/dome) in 24‐well‐plates, as described above, and cultured at 5% CO2/37°C. Solidified 3D domes were subsequently overlaid with 0.5 mL of appropriate culture media. 3D cultures of Panc02 cells were cultured for 5 (Matrigel) and 7 days (Collagen‐I). Media were replaced every third day. On the last day of culture, 3D domes were washed with sterile 1× PBS twice and stained with 3 μM Calcein‐AM (C1430, ThermoFisher Scientific) and 2.5 μM Propidium Iodide (PI) (P3566, ThermoFisher Scientific) for 30 minutes and then imaged using an Olympus IX83 microscope. Calcein‐AM and PI absorbances were detected at 485 nm/535 nm and 530 nm/620 nm respective emission wavelengths.
2.9. PAS staining
18 × 18 mm glass coverslips (ROTH, 0657) were ethanol‐washed, air dried, placed in 6‐well plates and coated with 50 μL of matrigel‐collagen I Mix (50:50%). Coverslips were air‐dried at room temperature for 1 hour prior to seeding of cells. Cells were trypsinized, counted and resuspended in 200 μL of appropriate culture medium. The coverslips were then incubated for 1.5 hours in a CO2 incubator to allow cells to adhere to the matrix. Three milliliters of culture medium was carefully added to the coverslips and changed every 2 days. PAS staining was performed as described in. 28
2.10. Boyden chamber invasion assay
Cells were starved for 24 hours in DMEM medium containing 2% FBS. Fifty thousand cells were seeded in this medium in the inner compartment of Boyden chamber inserts (8 μm pores, Falcon #353097) containing a polycarbonate membrane precoated with matrigel (300 mg/mL in 2% FBS DMEM). The lower chamber contained DMEM with 10% FBS. After 24 hours, cells in the upper chamber were removed using a cotton swab, and nuclei of migrated cells were stained with DAPI (1 μg/mL). Images of six random fields per membrane were taken and nuclei counted using the Cell Counter ImageJ plugin.
2.11. Spheroid outgrowth invasion experiments
Panc02 spheroids (in three independent biological replicates) were formed in ULA 96‐well‐plates, as above, and cultured for 3 days. On the third day of spheroid formation, spheroids (3‐5 spheroids/dome) were transferred to 50 μL Matrigel domes and treated or not with SB 431542 (2, 10 μM) or TGFβ1 (10, 20 ng/mL) in 24‐well‐plates and cultured at 5% CO2/37°C. Solidified 3D domes were subsequently overlaid with 0.5 mL of appropriate culture medium. Media were replaced every third day. Light microscopy images were taken right after transferring the spheroids to Matrigel (day 3) and 6 days later (day 9). Brightfield images were acquired with an Olympus IX83 microscope, using 4× and 10× objectives and CellSens software. Images were analyzed using ImageJ software. Bright field pictures were analyzed using macros function where images were converted to 8‐bit format, adjusted to Threshold (0, 138) to define the edges of initial spheroid area (day 3). Area in pixels2 was measured using the “Analyze Particles” function with size of particles bigger than 500 pixels2. The initial area of each individual spheroid was displayed as an overlay mask on the processed image and saved for a reference for day 9. Bright field pictures from day 9 were analyzed using a freehand selection to measure area in pixels2 and later converted to μm2. A reference image of initial spheroid area with overlay mask from day 3 was used to follow the invasion of each individual spheroid and correctly quantify a relative invasion spheroid area on day 9. The area from day 9 was divided by initial area from day 3 to obtain the relative spheroid invasion area.
2.12. Anchorage‐independent growth
In a 6‐well plate, 500 000 cells/well were suspended in 3 mL of 0.5% low gelling temperature agarose (Sigma, A9414‐25G) prepared in 10% FBS DMEM at appropriate pH and overlaid on 3 mL solidified 1% agarose. The top layer was covered with 3 mL of 10% DMEM. Media was refreshed every 2 or 3 days. After 4 weeks of culture, colonies were counted and measured using brightfield imaging and ImageJ software.
2.13. Western blotting
Western blotting was performed essentially as in Reference 29. A full description is provided in the online Supplementary Material.
2.14. Immunohistochemistry analysis of spheroids
Immunohistochemistry (IHC) analysis of spheroids was performed essentially as in Reference 30. A full description of the methodological procedures is provided in the online Supplementary Material. Quantification of intensity profiles of labeling across the spheroids, mean pixel intensity profiles through the spheroids were calculated using ImageJ software, based on rectangular Regions of Interest (ROIs) of 200 μm width, going from the spheroid periphery to core (2‐3 independent biological replicates per transporter/condition was measured). For each intensity profile, the x‐axis was set to start at zero and x‐values were normalized to the largest x‐value resulting in an x‐axis ranging from 0‐1 (“0” indicates region in a necrotic core, and “1” indicates the region in periphery). The intensity values (y‐axis) were likewise normalized to the largest intensity, generating y‐axis ranging from 0‐1. The resulting intensity profiles were plotted in a scatter plot and LOWESS curves with 20 points in the smoothing window were drawn using GraphPad PRISM 9 in order to better visualize trends of the data.
2.15. RNA isolation, RT and qPCR analysis
Total RNA from monolayer cells was isolated using the RNeasy Mini Kit (Qiagen, 74 104) according to the manufacturer's protocol. Reverse transcription and qPCR analysis were carried out as in Reference 31. A full description is provided in the online Supplementary Material.
2.16. Statistical analysis
R (4.0.2) was used to compute all statistics. Two‐sample one‐sided t‐test, pairing the data by passaging line, was used whenever relevant. Unpaired test was used when comparing data from two different genotypes, or when data points were missing. Using Benjamini‐Hochberg correction, we adjusted P values separately for each subfigure and for each type of comparisons, that is, [same genotype—different conditions] and [same condition—different genotypes]. In Figure 3E, two‐sided Kolmogorov‐Smirnov (KS) test and two‐sided Mann‐Whitney U test were used for the comparisons mentioned above.
FIGURE 3.
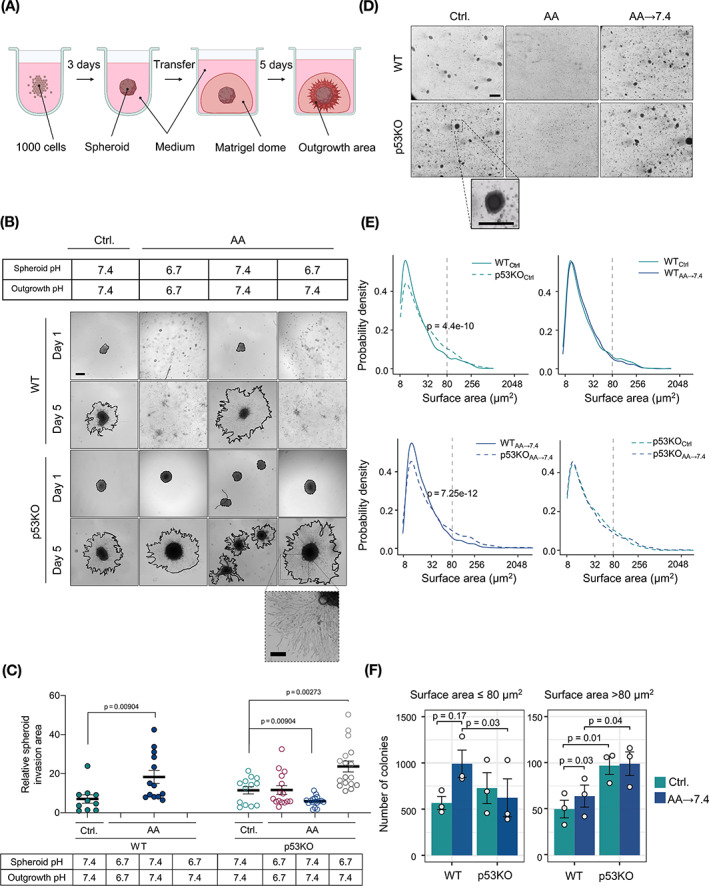
Acid adaptation promotes 3D invasion and anchorage‐independent growth. (A) Panc02 spheroids were formed in ultra‐low attachment plates and after 3 days embedded in 3D matrigel domes to assess their invasive potential. (B) Representative images of spheroids transferred to the matrigel domes. Comparison of initial spheroid area (day 1) and invasive area (day 5, red line). Scale bar: 200 μm. (C) Pixel‐based quantification of relative spheroid invasive area. Total spheroid area on day 5 was divided by initial spheroid area on day 1. Each dot represents one spheroid. n = 3, SEM error bars. (D) Representative light microscopic images of Panc02 cells grown in anchorage‐independent conditions in 0.6% soft agarose for 30 days. Scalebar: 200 μm. (E) Surface area distribution plots. The y‐axis shows the probability density, and the x‐axis shows log‐scaled surface area. Colonies with surface area ≤80 μm2 were defined as “small colonies,” and colonies with surface area >80 μm2 as “large colonies.” Comparison of WT Ctrl. and p53KO Ctrl.: two‐sided KS test: P = 4.4 e−10, two‐sided Mann‐Whitney U test: P = 2.12 e−11; Comparison of WT AA‐7.4 and p53KO AA‐7.4: two‐sided KS test: P = 7.25 e−12, two‐sided Mann‐Whitney U test: P = 1.01 e−8. (F) Number of colonies developed in soft agarose, divided by size as above. Bars show mean counts, error bars denote SEM. P values denote paired t test results
3. RESULTS
3.1. Acid adaptation does not increase viability in 2D cultures
To test our hypothesis that adaptation to an acidic TME synergizes with cancer driver mutations in promoting an aggressive cancer cell phenotype, we used CRISPR/Cas9 to knock‐out (KO) Trp53 in Panc02 mouse pancreatic cancer cells, starting from a Trp53 wild type (WT) (Figure 1A,B). 32 To simulate the gradual acidification during tumor growth we next subjected WT and Trp53 KO (denoted p53KO) cell lines to gradual acidification over 4 weeks from pHe 7.4 to final pHe 6.7, corresponding to that commonly found in solid tumors 12 (Figure 1A). The acid adapted cell lines are hereafter denoted AA cells. To mimic the reexposure of cancer cells to physiological pH during metastatic spreading or altered vascularization, we next acutely returned the cells to pHe 7.4 medium (denoted AA → 7.4) (Figure 1A).
FIGURE 1.
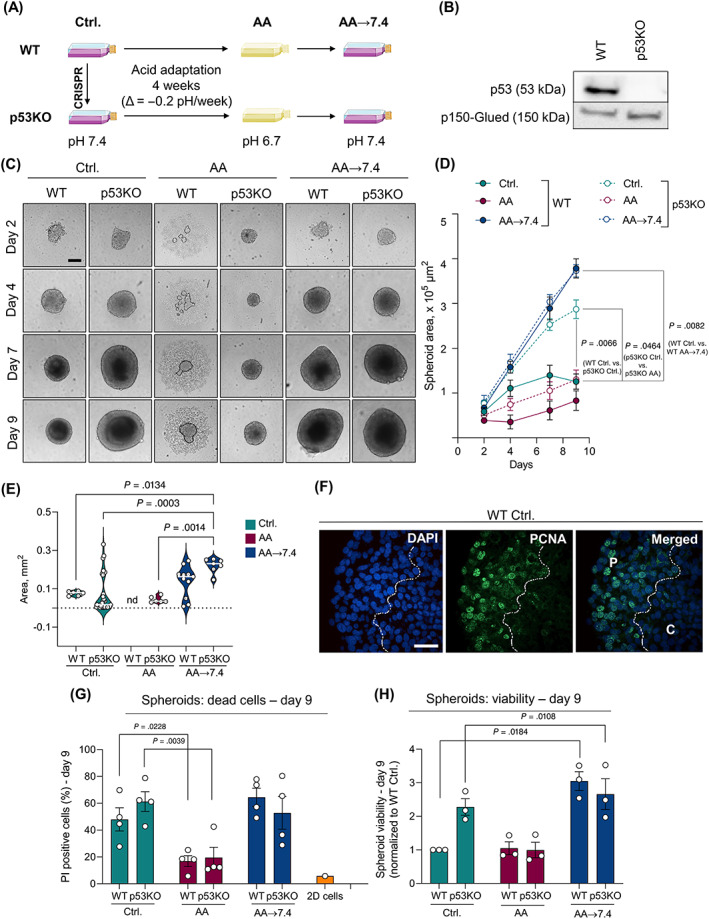
Acid adaptation promotes 3D growth of Panc02 cells. (A) Panc02 WT and p53KO cells were cultured for 4 weeks in gradually decreasing pH (from 7.4 to 6.7) to develop their acid‐adapted (AA) counterparts. Acid adapted cells were then returned to pH 7.4 (AA → 7.4). (B) Western blot demonstrating the successful knock‐out (KO) of the Trp53 gene. p150‐Glued was used as a loading control. (C) AA → 7.4 cells form significantly larger spheroids than AA and control cells. Representative light microscopic images of Panc02 spheroids on day 2, 4, 7 and 9. Scalebar: 200 μm. The dotted line shows the border of the small spheroid formed under acidic conditions, surrounded by dispersed cells. (D) Quantification of average spheroid area shown in C. n = 3, means with SEM error bars. (E) IHC analysis of WT Ctrl. spheroids. PCNA and DAPI staining was mainly found in nuclei in peripheral regions (“P”) of the spheroid and absent from the core, indicated by the dotted line and “C.” Scalebar: 40 μm. (F) Quantification of necrotic core area, determined as described in Materials and Methods. n = 3, means with SEM error bars. Statistics: ANOVA followed by Tukey's post‐test. (G) Percent apoptotic cells. Panc02 spheroids were dissociated, PI stained, and analyzed by flow cytometry. n = 4, SEM error bars. (H) Total viability of Panc02 spheroids after 9 days of culture. n = 3, means with SEM error bars
In standard 2D monolayer culture, viability assays showed that AA cells exhibited lower viability than their pH 7.4 control counterparts (25% for WT, ~50% for p53KO), largely returning to a viability similar to that of control cells when returned to pH 7.4 (Figure S1A). The same pattern was observed when the confluence of control‐, AA‐ and AA → 7.4 cells was monitored using time‐lapse microscopy (Figure S1B‐E). Thus, in 2D culture, acid adaptation and return to control conditions does not significantly increase Panc02 cell growth.
3.2. Acid adaptation and p53 loss increase spheroid growth
To mimic the architecture and microenvironment conditions of tumor tissue, 33 , 34 we cultured Panc02 cells as 3D spheroids and monitored spheroid growth over 9 days (Figure 1C). Under control conditions, p53KO spheroids grew to more than twice the area and were more viable compared to WT cells at day 9 (Figure 1C‐E). At pH 6.7, WTAA cells grew much slower in 3D than their non‐acid adapted counterparts and almost failed to form tight spheroid structures (illustrated by the dotted line), whereas p53KOAA spheroids were small (Figure 1C,D) but remained coherent (Figure 1C). AA cells of both genotypes were largely viable, with fewer dead cells than under control conditions and near‐absence of a necrotic spheroid core. This was also illustrated by quantification of necrotic core area, which generally correlated with spheroid size (Figure 1E). Strikingly, upon return to pH 7.4, WT spheroids (WTAA→7.4) grew nearly 4‐fold larger, and p53KOAA→7.4 spheroids about 30% larger compared to their nonacid‐adapted controls, with an obvious necrotic core (Figure 1C‐E). Accordingly, proliferating cells (nuclear PCNA staining) were localized only in the rim of the spheroids, and nuclei in the spheroid core were pyknotic (Figure 1F, necrotic core marked by a dotted line). Similarly, large spheroids in the control and return groups contained a greater fraction of propidium iodide (PI)‐positive (dead) cells than pH 6.7 spheroids (Figure 1G), confirmed by measurements of total spheroid viability at day 9 (Figure 1H).
Collectively, these results show that Panc02 cells adapted to growth at pH 6.7 exhibit markedly increased 3D growth upon return to pH 7.4. This suggests that acid adaptation rewires Panc02 into a new state that strongly favors cell growth at physiological pH. Notably, the growth‐enhancing effects of p53KO and acid adaptation were only partially additive.
3.3. Acid adaptation, p53KO and matrix composition interact to regulate cell growth and morphology
In PDAC, the stroma is dominated by collagen‐I, whereas the basement membrane is better mimicked by the basement membrane composition of matrigel. 7 To address how ECM composition impacts the effects of acid adaptation and p53 KO on invasive growth, we grew control‐ and AA Panc02 cells in organotypic culture, using conditions mimicking early stages (matrigel); advanced stages (collagen‐I); and an intermediate stage (1:1 matrigel‐collagen‐I mix).
In matrigel domes, the pattern of differences in growth between the different cell lines was reminiscent to that observed for spheroid growth, however, quantitative differences were less pronounced and growth morphology reflected that the cells were now embedded in ECMs (Figure 2A‐C). Live/dead staining confirmed that except in the center of large colonies, cells were mostly viable in all conditions (Figure 2B). Viability was increased by ~50% by p53KO, was reduced in p53KOAA cells but not in WTAA cells compared to their control counterparts, and increased after return to pH 7.4, albeit less so for p53KO than for WT (~doubled compared to Ctrl.) cells (Figure 2B). Both WTAA and p53KOAA cells developed long protrusions into the matrigel (Figure 2A, arrow), which immunofluorescence analysis revealed to represent rows of migrating cells (Figure S2A).
FIGURE 2.
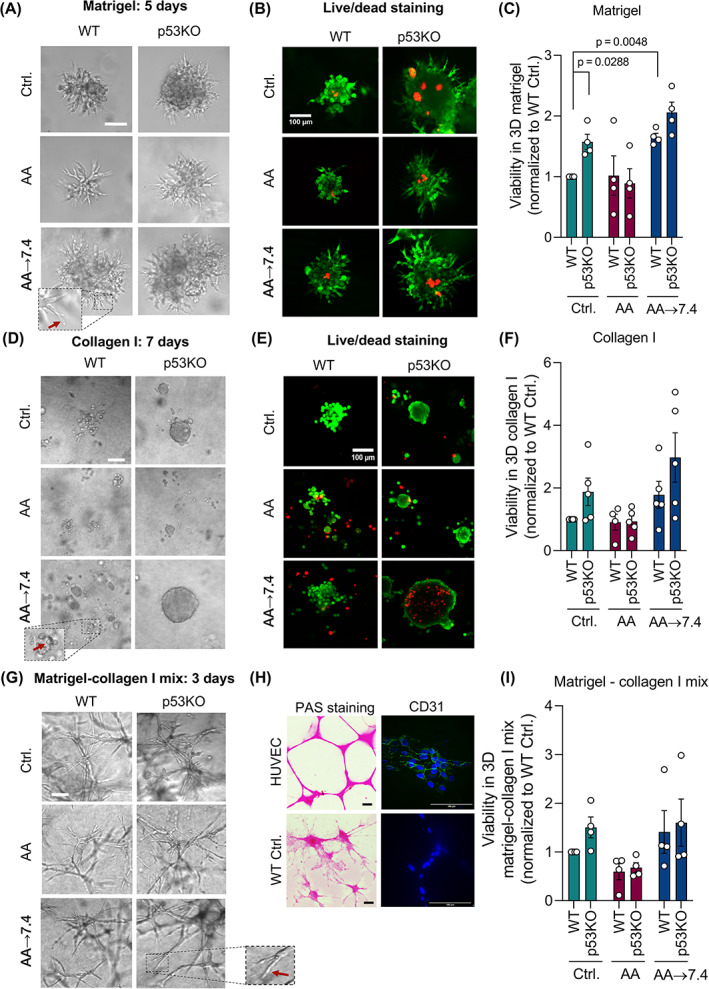
Acid adaptation promotes Panc02 growth and viability in different 3D matrices. (A) AA → 7.4 cells develop protrusions (demonstrated in higher magnification, red arrow) and form substantially larger colonies in 3D matrigel than their control counterparts and AA cells at pH 6.7. Representative light microscopic images of Panc02 cells grown in 3D matrigel on day 5. Scalebars: 200 μm. (B) Live/dead staining of cultures as in (A). Live cells label green, dead cells red. Represents 3 independent experiments. (C) Total viability of Panc02 cells cultured in 3D matrigel for 5 days. n = 4, mean with SEM error bars. (D) Representative light microscopic images of Panc02 cells grown in 3D collagen I on day 7 showing solid colonies with no protrusions. Panc02 AA → 7.4 cells form significantly bigger colonies in 3D collagen I than their control counterparts and than AA cells at pH 6.7. Scalebars: 100 μm. (E) Live/dead staining of cultures as in (D). Live cells label green, dead cells red. Representative of 3 independent experiments. (F) Viability of Panc02 cells cultured in 3D collagen I for 7 days. n = 4. (G) Representative light microscopic images of Panc02 cells grown in 1:1 matrigel:collagen‐I on day 5 showing elongated vascular‐like structures. Scalebar: 200 μm. (H) Representative images of Periodic Acid‐Schiff (PAS) staining of human umbilical vein endothelial cells (HUVEC, upper left) and Panc02 WT Ctrl. cells (bottom left). Scalebar: 200 μm. Immunofluorescence analysis, CD31 staining of HUVEC (upper right) and Panc02 WT Ctrl. cells (bottom right). Scalebar: 100 μm. (I) Viability of Panc02 cells cultured in 1:1 matrigel:collagen‐I for 3 days. n = 4. Unless P‐values are shown, data were not statistically different
In marked contrast, embedding cells in collagen‐I elicited growth as compact colonies completely lacking protrusive outgrowths. Under control conditions, p53KO cells grew as tight colonies with a total viability that was about twice that of WT cells (Figures 2D‐F and S2B). At acidic pH, the viability of both genotypes was similar to that of WT control cells. Upon return to pH 7.4, total viability of WTAA cells was about doubled and that of p53KOAA cells increased by 50% compared to control p53KO cells, and to 3‐fold that of AA p53KO cells, although with substantial variation (Figure 2F). Notably, live/dead analysis revealed that this reflected a combination of greatly increased growth and increased death at the substantial necrotic core formed under these conditions (Figure 2E).
Remarkably, when seeded in 1:1 matrigel:collagen‐I (Figure 2G), Panc02 cells developed a morphology highly reminiscent of vasculogenic mimicry, that is, the differentiation of cancer stem cells to endothelial‐like cells which can provide partial neo‐vascularization. In accordance with this phenotype, 35 the vascular‐like Panc02 cell networks consisted of long strands of cells (Figure S2C). The cells stained positive for Periodic acid‐Schiff (PAS), a marker for vascular mimicry, 28 but were negative for CD31, a marker for normal endothelial cells, which strongly stained HUVECs as positive control (Figure 2H). Furthermore, growth was markedly accelerated in this ECM, and accordingly the cultures in Figure 2G, represent 33% fewer cells seeded than Figure 2A, were cultured for only 3 days. Despite this striking phenotype, the relative viability between treatments and genotypes was similar to that of matrigel or collagen‐I alone, although differences were less pronounced and did not reach statistical significance (Figure 2I).
To explore in more depth the relationship between the growth‐promoting effects of acid adaptation and p53 KO, we repeated the acid adaptation and organotypic growth experiments in two human pancreatic cancer cell lines with aberrant p53 expression: AsPC1 cells, which are p53 negative, and Panc1 cells, which exhibit the common p53 R273H mutation 36 that renders p53‐driven transcription pHi sensitive. 37 When grown as organotypic cultures in either matrigel or collagen, AsPC1 cells exhibited a growth pattern similar to that of Panc02 cells, with a clear tendency of decreased growth at acidic conditions and increased growth after return of acid‐adapted cells to physiological pH (Figure S3A‐C). In contrast, Panc1 cells similarly adapted to acidic growth failed to increase their growth rate after return to physiological pH, in both matrigel and collagen‐I culture (Figure S3D‐F).
These results show that growth of Panc02 and AsPC1 cells in matrigel and collagen‐I is increased by acid adaptation upon return to pH 7.4. Growth is also potentiated by p53KO, in a manner not fully additive to that of acid adaptation. Finally, Panc02 cell growth in a 1:1 matrigel:collagen‐I mix elicits a phenotype resembling vascular mimicry, independent of genotype and treatment.
3.4. Acid adaptation increases 3D invasiveness
To assess how acid adaptation impacts cell motility, Panc02 cells were first subjected to 2D wound healing assays (Figure S4A,B). Wound closure rate was similar for control and AA cells and both genotypes at pH 6.7, and returning cells to pH 7.4 slowed 2D migration by 20‐30%. Similarly, acid adaptation did not promote invasion of 2D‐cultured cells through matrigel (Figure S4C).
We next asked how invasiveness was altered by acid adaptation when cells were cultured in 3D to simulate tumor conditions. Spheroids were embedded in 3D matrigel domes, and outgrowth was imaged and quantified at day 5 (Figure 3A‐C). Under control conditions, both WT and p53KO spheroids exhibited outgrowth into the matrigel, with a tendency for more outgrowth from p53KO spheroids. Since WT cells do not form compact spheroids at pH 6.7 (see Figure 1), we seeded WTAA spheroids at pH 7.4 and transferred them to matrigel at pH 7.4. Under these conditions, their outgrowth was more than doubled compared to that of control spheroids, whereas, surprisingly, outgrowth of p53KO spheroids was decreased by ~50%. The largest invasive area—twice that of p53KO control spheroids—was formed by p53KOAA spheroids formed at pH 6.7 and embedded in matrigel at pH 7.4 (Figure 3A‐C).
These results show that 3D—but not 2D—invasiveness of Panc02 cells is increased by acid adaptation, and further accelerated by p53 KO.
3.5. Acid adaptation increases anchorage‐independent growth
To assess their potential for anchorage‐independent growth, a well‐established readout of aggressiveness, Panc02 cells were cultured in soft agarose for 30 days (Figure 3D‐F). Both WT and p53KO cells formed colonies at pH 7.4. AA cells failed to form colonies at pH 6.7 but regained this ability upon return to pH 7.4 (Figure 3D). Analysis of colony sizes revealed several interesting differences in formation of “Small” (surface area ≤80 μm2) and “large” (surface area >80 μm2) colonies (Figure 3E,F). Under control conditions, the number of small colonies was similar for WT and p53KO cells, but the number of large colonies was higher for p53KO than WT cells. When returned to pH 7.4, WTAA cells developed more colonies than WT control cells in both categories, whereas the development of p53KOAA colonies was unaffected by acid adaptation (Figure 3E,F).
These results show that acid adaptation and return to pH 7.4 favors WT Panc02 cell anchorage‐independent growth, whereas p53 KO increases colony size independent of acid adaptation.
3.6. Akt activity, and NHE1 and TXNIP expression, are increased in acid adapted Panc02 cells in a p53‐dependent manner
We next sought to gain insight into the mechanisms through which acid adaptation and p53 interact to regulate growth. Protein expression of both p53 and the p53 effector p21 was increased in AA cells, presumably reflecting stress induced by growth at acidic pH, while, as expected, p53KO cells completely lacked p21 (Figure 4A, densitometric quantification in Figure S5). Acidic pH has been suggested to increase DNA damage 12 and p53 signaling plays a key role in DNA damage protection. 3 Despite this, the cellular level of phosphorylated histone 2AX (γH2AX)—a marker of DNA damage—was unaffected by the treatments and only marginally elevated in p53KO cells (Figures 4A and S5).
FIGURE 4.
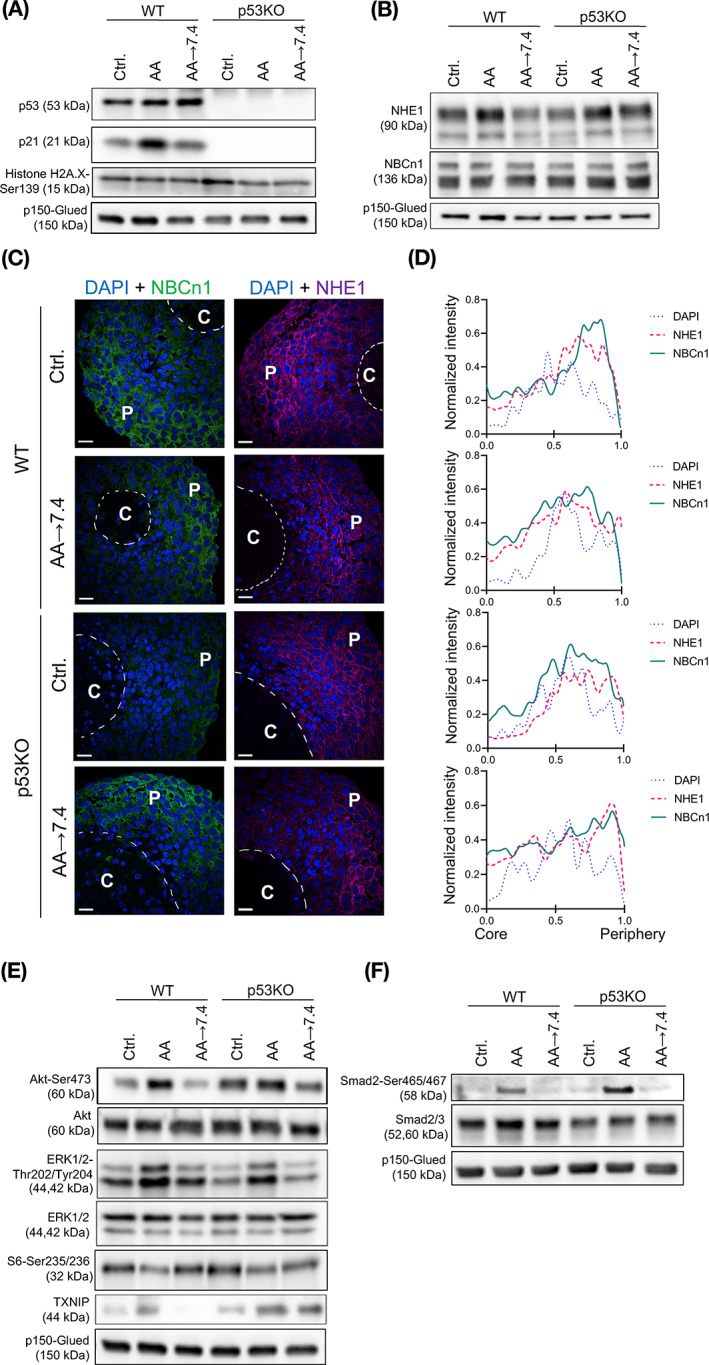
Akt activity and NHE1 and TXNIP expression are increased in acid adapted Panc02 cells. (A) Western blot analysis of protein levels of p53, its downstream effector p21, and the DNA damage marker γH2A.X. (B) Western blot analysis of the key net acid extruders Na+/H+ exchanger NHE1 and Na+,HCO3 − cotransporter NBCn1. (C) IHC analysis of NHE1 and NBCn1 localization in paraffin sections of Panc02 WT and p53KO spheroids. DAPI staining visualizes nuclei. NHE1 and NBCn1 are expressed in the cells in the peripheral regions (“P”) of the spheroids and less visible in the core region (“C”). The dotted line illustrates the border of the necrotic core, identified based on pyknotic nuclei. Scale bar: 20 μm. Individual images and controls are shown in Figure S6A‐C. (D) Quantitative analysis of NHE1 and NBCn1 distribution in spheroids, based on images as in (C), confirms the predominantly peripheral localization of the transporters. Representative of 2‐3 independent experiments per condition (E) Western blot analysis of Akt and ERK1/2 protein kinases, S6 ribosomal protein and thioredoxin interacting protein (TXNIP). p150‐Glued is used as a loading control. All results are representative of at least 3 independent experiments. (F) Western blot analysis shows that the TGFβ signaling effector Smad2 is activated in acid‐adapted cells. p150‐Glued is used as a loading control. Representative of at least 3 independent experiments. Densitometry analysis of all Western blots is found in Figure S5
We and others have shown that increased expression of net acid extruding transporters such as the Na+/H+ exchanger NHE1 and Na+,HCO3 − cotransporter NBCn1 supports cancer cell growth in acidic microenvironments. 12 Protein levels of NHE1, especially the immature precursor version, tended to be upregulated in AA cells, while NBCn1 levels were unaffected (Figures 4B and S5). Consistent with our previous work in human breast cancer spheroids, 30 NHE1 localized evenly through the viable cell population in the spheroids, whereas NBCn1 expression was higher toward the spheroid periphery, with no detectable differences between conditions (Figure 4C, controls and individual images in Figure S6A‐C). This pattern was confirmed by quantification of the fluorescence staining from spheroid core to periphery, showing clearly the lack of transporter expression in the necrotic core, and increasing expression toward the spheroid periphery (Figure 4D).
The Ser/Thr kinases Akt and extracellular signal regulated kinase‐1/2 (ERK1/2) are essential regulators of growth and upregulated in most cancers. 38 Total Akt and ERK1/2 levels were similar in all conditions tested (Figures 4E and S5). Akt activity was increased in AA cells, and tended to be exacerbated by p53KO. ERK1/2 activity tended to be increased in both WT and p53KO AA cells, and phosphorylation of the ERK1/2 downstream effector S6 was, interestingly, increased after return of WT AA cells to pH 7.4 (Figures 4E and S5). Thioredoxin‐interacting protein (TXNIP), an inhibitor of the antioxidant protein thioredoxin and a global regulator of cell metabolism, has been shown to be upregulated upon acidic stress in other cell types. 39 Strikingly, TXNIP was upregulated in AA cells, especially in AA cells with p53KO. Upon return to pH 7.4, TXNIP levels returned to control level in WT cells, but interestingly remained elevated in p53KO cells (Figures 4E and S5).
In summary, acid adaptation increases ERK1/2 activity in a p53‐independent manner, and NHE1 and TXNIP expression and Akt activity in a p53‐dependent manner.
3.7. Akt signaling favors Panc02 spheroid growth irrespective of acid adaptation and genotype
Given the increased Akt activity in AA cells, we assessed the role of Akt in Panc02 spheroid growth, using the allosteric Akt inhibitor MK‐2206 and the ATP‐competitive Akt inhibitor Capivasertib. Treatment of spheroids with MK‐2206 (confirmed by western blotting to efficiently inhibit Akt, Figure S7A) resulted in smaller and less viable spheroids in all conditions and both genotypes. Capivasertib had a similar, although less potent, effect (Figure 5A,B). For unknown reasons, very low concentrations of both inhibitors caused WTAA cells, which under control conditions form only loose aggregates, to form small, well‐defined and compact spheroids (Figure 5A) and increased rather than decreased WT control spheroid size and viability (Figure 5B). Thus, although Akt signaling was increased in acid adapted‐ and p53KO cells, Akt appeared to be equally important for spheroid growth under all conditions tested.
FIGURE 5.
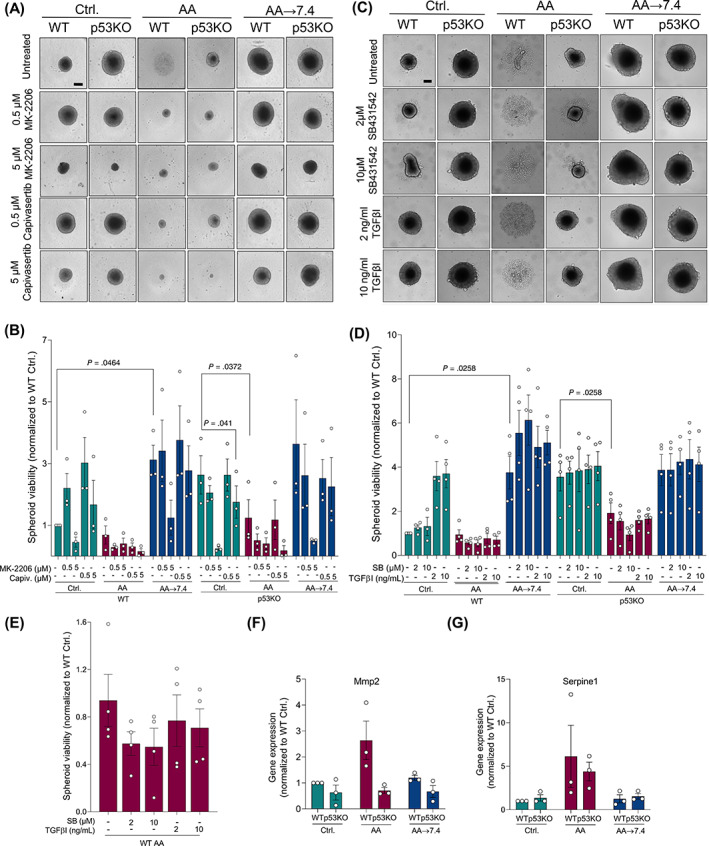
TGFβ signaling is upregulated in acid adapted Panc02 cells and regulates WT Ctrl. spheroid growth and Serpine1 and Mmp2 expression. (A) Spheroids were treated with 0.5 or 5 μM of the allosteric Akt inhibitor MK‐2206 and the ATP‐competitive Akt inhibitor Capivasertib, as shown. Scale bar: 200 μm. (B) Total viability of MK‐2206‐ and Capivasertib‐treated Panc02 spheroids for 9 days. n = 4, mean with SEM error bars. (C) Spheroids were treated with a selective inhibitor of TGFβRI (SB431542, SB) or 2 or 10 ng/mL of TGFβI for 9 days, as indicated. Scale bar: 200 μm. (D) Viability of Ctrl., AA (zoom in (E)) and AA‐7.4 spheroids treated with SB431542 or TGFβI. n = 4, mean with SEM error bars. (E) Zoom of Panc02 WT AA bars shown in D. (F and G) qPCR analysis of TGFβ target genes Serpine1 (F) and Mmp2 (G). Relative expression was normalized to mean of Actb and Gapdh expression. n = 3. Error bars denote SEM
3.8. TGFβ signaling regulates Panc02 spheroid growth in WT control cells only
Adaptation to acidosis was recently shown to promote autocrine TGFβ2 signaling in cervical cancer cells. 20 Indeed, phosphorylation of TGFβ effectors SMAD2/3 was strongly increased in AA cells, in a manner potentiated by p53KO (Figure 4F). We therefore asked whether spheroid growth and viability were affected by TGFβ signaling stimulation by the TGFβ ligand (TGFβI) or inhibition by the selective TGFβRI inhibitor SB‐431542. Western blot analysis confirmed the efficacy of both agents (Figure S7B). Panc02 cells harbor an inactivating SMAD4 mutation, 40 and consistent with the growth‐stimulatory effect of TGFβ in absence of SMAD4 [55], stimulation with TGFβI increased WT control spheroid viability ~4‐fold, while TGFβRI inhibition had no effect under these conditions (Figure 5C,D, left green bars). Remarkably, in WTAA cells, TGFβI had no detectable effect and treatment with SB431542 tended to reduce the already low viability (Figure 5D, left red bars, magnified in Figure 5E). Interestingly, both treatments slightly increased WTAA spheroid viability upon return to pH 7.4 (left blue bars), possibly indicating a shift in the balance of pro‐ and antiproliferative effects of TGFβ under these conditions. p53 is required for TGFβ‐induced regulation of some promoters, including that of Serpine1. 41 Consistent with a p53‐dependent role of TGFβ, p53KO spheroid growth was unaffected by TGFβ stimulation regardless of treatment (Figure 5D, right bars), despite the potentiation of SMAD2/3 phosphorylation by p53KO (Figure 4F).
These results prompted us to ask whether elements of TGFβ signaling were altered in WTAA cells. Indeed, TGFβ target genes, Serpine1 (encoding plasminogen activator inhibitor‐1, PAI‐1) and matrix metalloprotease‐2 (Mmp2) 41 were upregulated in AA WT cells and returned to normal when AA cells were returned to pH 7.4 (Figure 5F,G). Consistent with the p53‐dependence of TGFβ‐induced regulation of this promoter, 41 p53KO cells lacked AA‐induced upregulation of Serpine1 and a similar tendency was seen for Mmp2 (Figure 5F,G). In contrast, mRNA levels of Tgfb1 and Tgfb2 (the latter previously found to be regulated by acid adaptation, 33 ), and TGFβ receptor 3‐like (Tgfb3l), upregulated in acid adapted human cancer cells 42 were not significantly altered by any of the treatments (Figure S7C‐E).
These results show that acid adaptation increases TGFβ signaling in a manner potentiated by p53KO. Stimulation of TGFβ signaling enhances 3D growth of WT control spheroids, and its inhibition tends to inhibit growth of AA cells under acidic conditions.
4. DISCUSSION
The key finding of this work is that adaptation to growth at pHe 6.7 to mimic acidic tumor regions strongly increases pancreatic cancer cell 3D growth, anchorage‐independent growth and invasiveness after return to normal pHe, in a manner dependent on p53 and ECM.
While heterogeneous extracellular acidosis is an important feature of solid tumors from a wide range of cancers, 12 , 40 pancreas physiology makes acid‐base dysregulation particularly interesting in this organ. Postprandially, massive secretion of HCO3 − from the epithelial cells to the ducts is accompanied by a corresponding acid flux to the bloodstream to sustain epithelial pHi. Pancreatic ductal epithelial cells are thus exposed to recurrent pHe variations, alkaline on the luminal and acidic on the basolateral side, and we previously hypothesized that this may prime cells for survival in the increasingly acidic microenvironment as the tumor grows. 24 Here we show that indeed, preadaptation to growth under acidic conditions supports PDAC cell aggressiveness. Importantly, potentiated aggressive behavior was observed not under acidic conditions per se, but when cells were returned to pH 7.4 to mimic reexposure to normal or less acidic pHe conditions upon increased perfusion or invasive outgrowth. We show that this transition favors spheroid‐ and organotypic 3D growth, anchorage‐independent colony formation, and invasiveness—key traits of aggressive cancers. This strongly suggest that cells acquire additional cancerous potential during acid adaptation, yet this potential is only fully realized at normal pH when cells are relieved of the stress of growth in acid conditions.
4.1. Microenvironmental drivers of acid potentiation
Studies using simple 2D culture have found cell growth to be decreased or unaltered by acid adaptation. 20 , 43 , 44 This is extended by our study of 3D cultures and consistent with our suggestion that tissue acidosis contributes to tumor dormancy in early PDAC stages. 24 Importantly, we show a potent, growth‐enhancing effect of acid adaptation which is revealed only when cells are grown in 3D as spheroids or organotypic cultures embedded in ECM, and returned to pH 7.4. The extent and morphology of growth were dependent on ECM composition: Growth was promoted by acid adaptation under all ECM conditions tested, but was quantitatively most profound for nonadherent 3D spheroids and least significant in a matrigel:collagen‐I mix, perhaps because the latter condition in itself potently accelerated growth. Also anchorage‐independent colony formation—a reliable measure of the ability to form tumors in vivo 45 —was increased by acid adaptation. This effect, to our knowledge reported for the first time, underscores the aggressiveness driven by acid adaptation.
Consistent with the key role of the dense ECM in PDAC, 8 , 9 , 10 cell morphology was profoundly ECM‐dependent. Growth in matrigel:collagen‐I appeared to support vasculogenic mimicry, independent of genotype and acid treatment. Notably, this ECM also markedly increased growth, suggesting that the link between vascular mimicry and aggressiveness 35 may not be limited to a role in supporting tumor perfusion.
Our work also demonstrated that acid adapted cells exhibited a marked increase in 3D invasiveness. Whereas these are to our knowledge the first such 3D analyses, our data is well in line with earlier reports showing increased in vitro invasiveness and in vivo metastasis of acid adapted cells 12 , 19 , 20 and substantiates the notion that increased invasiveness is an important property driven by acid adaptation.
4.2. Genetic drivers of acid potentiation
A key question motivating this work was the interplay between microenvironment acidosis and driver mutations. We focused on p53‐inactivating mutations, found in ~75% of PDAC tumors 4 and associated with highly aggressive disease. 5 , 6 As expected, p53KO by itself increased growth of control cells in all conditions tested. This effect was partially additive to that of acid adaptation in matrigel or collagen‐I, but not when cells were grown as nonadherent spheroids. Interestingly, p53 negative human pancreatic cancer cells (AsPC1) showed an organotypic growth pattern very reminiscent of that of p53KO Panc02 cells, whereas Panc1 cells, which harbor the p53 R273H mutation, grew poorly in matrigel and collagen at acidic pH and failed to increase their growth rate upon return to physiological extracellular pH. Because this mutation introduces a histidine in the DNA‐binding site, it renders p53‐driven transcription higher at more acidic, and lower at more alkaline pHi values. 37 While these differences should not be overinterpreted because these cell lines are not syngeneic, it is interesting to consider that the acidic pHi of the cells during acid adaptation may activate a potentiated p53R273H‐driven transcriptional program of growth inhibition that persists after return to normal pH, preventing growth recovery.
Importantly, p53KO completely alleviated the loss of cell‐cell adhesion that largely prevented spheroid growth of WT cells at pH 6.7, suggesting that p53KO can support 3D growth in an acidic TME. The interaction between p53 status and acid adaptation was underscored by the effect of acid adaptation on adhesion‐independent colony formation: Colonies formed by p53KO cells were larger than those formed by control cells, but their size was unaffected by acid adaptation, whereas WT colony size was increased. In contrast, the acid adaptation‐induced increase in 3D invasion was potentiated by p53KO, suggesting that p53 loss and acidic TME may synergize to drive PDAC metastasis. A similar synergy was reported between p53 KO and hypoxia, 46 substantiating the notion that interaction between TME properties and p53KO enhances cancer aggressiveness.
4.3. A model for acid adaptation and potentiation
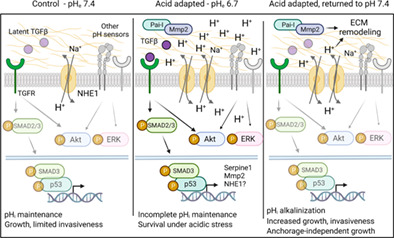
Acquisition of cancerous potential involves (i) genetic selection, where acidic environment selects for cells with cancerous potential, and/or (ii) transcriptional/epigenetic adaptive reprogramming. While we have not focused on genetic selection, our study strongly indicates that transcriptional/epigenetic reprogramming is important, as we identified upstream signals for changes on transcriptional and protein levels, leading to a hypothesized initial model (Figure 6). First, consistent with recent work in other cancers, 20 acid adaptation was associated with changes in TGFβ signaling. Similar to many PDAC patient tumors, Panc02 cells harbor an inactivating SMAD4 mutation, 32 causing TGFβ signaling to be redirected from canonical antiproliferative to noncanonical, growth‐stimulatory pathways. 4 SMAD2/3 phosphorylation was increased by acid adaptation, yet mRNA levels of main TGFβ ligands were not increased. This suggests that acid adaptation may increase TGFβ bioavailability, a notion consistent with recent findings 20 and in congruence with the release of latent TGFβ by acidic pH. 47 Akt and ERK1/2 phosphorylation was also increased in acid adapted cells, consistent with, albeit not unique to, the activation of both pathways by noncanonical TGFβ signaling 48 (Figure 6). The marked upregulation of Na+/H+ exchanger NHE1 in acid adapted cells is likely a prerequisite for survival under the strong inward driving force for H+ during growth at pH 6.7. Upon return to pH 7.4, this likely renders the cells equipped with excess acid extrusion capacity, contributing to both invasiveness and growth driven by acid adaptation, as previously demonstrated by us and others 29 , 49 , 50 , 51 (Figure 6). However, as acid‐base transporters are highly posttranslationally regulated, activity is not a direct function of expression, and future work should assess the specific contributions of NHE1 and other net acid extruding transporters to the acid extrusion capacity. The NHE1 upregulation could be at least in part TGFβ‐driven as we recently showed in Panc‐1 human pancreatic cancer cells. 31 Spheroid viability was increased by TGFβI in control cells only, and tended to be reduced by TGFβ receptor inhibition in acid adapted cells. While this shift, in conjunction with the increase in TGFβ signaling (Smad2/3 phosphorylation, target upregulation) suggests that upregulated TGFβ signaling helps support 3D growth at acidic pH, it cannot account for the potent increase in growth and invasiveness upon return to pH 7.4 where Smad2/3 phosphorylation and TGFβ target expression were not increased. Future work should therefore map the roles of other acid adaptation‐regulated signaling pathways and cell‐ECM interactions in this process (Figure 6).
FIGURE 6.
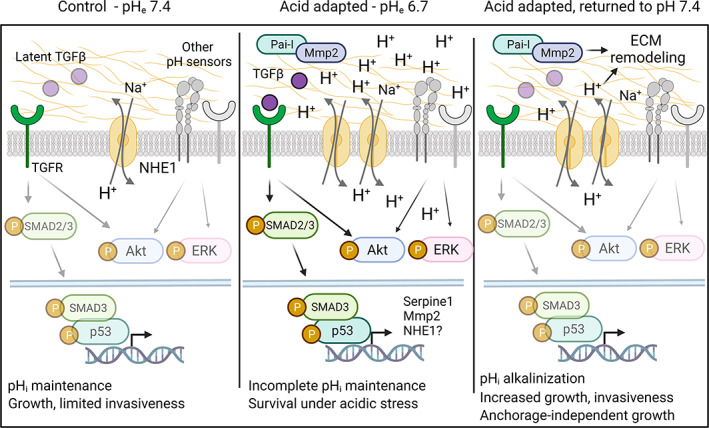
Working hypothesis. Under control conditions (left panel), pHi is maintained by the available acid‐base transport proteins such as NHE1, and noncanonical TGFβ signaling is limited by the sequestration of latent TGFβ. Upon adaptation to acidic pHe conditions (middle panel), acid‐base transporters are upregulated, and noncanonical TGFβ signaling is increased, resulting in increased SMAD2/3‐ and Akt‐ and ERK activity. This leads to upregulation of genes/selection for cells expressing genes allowing survival under these harsh conditions, including NHE1 and p53‐ and SMAD2/3‐dependent genes such as Serpine1 and Mmp2. However, growth and invasiveness are limited due to incomplete pHi maintenance against the strong inward driving force for H+. Other H+ sensing mechanisms are likely also implicated, shown in gray. Upon return of these cells to a more favorable pHe (right panel), the cells are released from this quiescence, pHi alkalinizes due to increased net acid extrusion capacity, and this drives increased proliferation, invasiveness, and anchorage‐independent growth. See text for further details
In conclusion, we demonstrate that PDAC cell 3D‐ and anchorage‐independent growth and invasiveness are potentiated by adaptation to tumor acidosis in an interplay with ECM composition and p53 status. Importantly, the most crucial acid‐induced phenotypes are only observed when cells grown in spheroid‐ or organotypic 3D conditions are returned to normal pH, mimicking conditions of improved vascularization or metastasis. We suggest that such crosstalk between tumor acidosis, ECM and driver mutations is highly relevant to cancer aggressiveness and points to future strategies for acidosis‐targeted therapies in PDAC.
AUTHOR CONTRIBUTIONS
Stine Falsig Pedersen, Albin Sandelin, Dominika Czaplinska and Renata Ialchina developed the hypothesis and designed the experiments, with inputs from Joanna Napp and Frauke Alves. Dominika Czaplinska developed the p53KO cells with help from SPG, and performed IF staining, 2D viability and qPCR experiments. Dominika Czaplinska and Renata Ialchina performed spheroid outgrowth, wound healing, and Boyden chamber assays, Renata Ialchina and Mette Flinck FACS experiments, and Renata Ialchina soft agarose assay, PAS staining, Live/dead analysis, AsPC1 and Panc1 growth data. Henriette Berg Andersen performed RNA isolation, viability assays, and western blotting. Jiayi Yao and Arnaud Stigliani performed statistical analyses. Johs Dannesboe developed AA cell lines and did spheroid viability and growth assays, Xiaoming Chen western blotting, and Jakub Mitrega proliferation assays, Oksana Dmytriyeva and Mette Flinck did IHC experiments. Dominika Czaplinska and Stine Falsig Pedersen wrote the manuscript with inputs from all co‐authors. All authors have seen and approved the final version of the manuscript. The work reported in the paper has been performed by the authors, unless clearly specified in the text.
FUNDING INFORMATION
This work was supported by the Danish Cancer Society (#R204‐A12359 to Albin Sandelin and Stine Falsig Pedersen), the Novo Nordisk Foundation (#NNF19OC0058262 to Albin Sandelin and Stine Falsig Pedersen), the European Union (H2020‐MSCA‐ITN‐2018, grant 813834, to Albin Sandelin, Frauke Alves and Stine Falsig Pedersen) and the Carlsberg Foundation (#CF20‐0491 to Stine Falsig Pedersen).
CONFLICT OF INTEREST
The authors declare that they have no conflicts of interest.
Supporting information
Table S1. Antibodies used in our study.
Figure S1. Adaptation to lower pH does not enhance Panc02 viability in 2D.
Figure S2. Immunofluorescence analysis of organotypic Panc02 cultures.
Figure S3. Organotypic growth of AsPC1 and Panc1 cells.
Figure S4. Wound healing and Boyden chamber invasion assays.
Figure S5. Western blot densitometry.
Figure S6. Immunohistochemical staining of Panc02 spheroids.
Figure S7. Validation of Akt and TGFβ pathway interventions.
ACKNOWLEDGEMENTS
We are grateful to Signe Meng, Muthulakshmi Ponniah, and Tanja Larsen for excellent technical assistance, and to Dr Ania Trauzold, University of Kiel, Germany, and Dr Marie Kveiborg, University of Copenhagen, Denmark, for kindly providing us with Panc02 cells and HUVECs, respectively.
Czaplinska D, Ialchina R, Andersen HB, et al. Crosstalk between tumor acidosis, p53 and extracellular matrix regulates pancreatic cancer aggressiveness. Int J Cancer. 2023;152(6):1210‐1225. doi: 10.1002/ijc.34367
Dominika Czaplinska and Renata Ialchina share first authorship.
Albin Sandelin and Stine Falsig Pedersen share last authorship
Funding information Carlsbergfondet, Grant/Award Number: CF20‐0491; FP7 People: Marie‐Curie Actions, Grant/Award Number: 813834; Kræftens Bekæmpelse, Grant/Award Number: R204‐A12359; Novo Nordisk Fonden, Grant/Award Number: NNF19OC0058262
Contributor Information
Albin Sandelin, Email: albin@binf.ku.dk.
Stine Falsig Pedersen, Email: sfpedersen@bio.ku.dk.
DATA AVAILABILITY STATEMENT
Data are available from the authors upon reasonable request.
REFERENCES
- 1. Kleeff J, Korc M, Apte M, et al. Pancreatic cancer. Nat Rev Dis Primers. 2016;2:16022. [DOI] [PubMed] [Google Scholar]
- 2. Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014;74:2913‐2921. [DOI] [PubMed] [Google Scholar]
- 3. Kruiswijk F, Labuschagne CF, Vousden KH. p53 in survival, death and metabolic health: a lifeguard with a licence to kill. Nat Rev Mol Cell Biol. 2015;16:393‐405. [DOI] [PubMed] [Google Scholar]
- 4. Hayashi A, Hong J, Iacobuzio‐Donahue CA. The pancreatic cancer genome revisited. Nat Rev Gastroenterol Hepatol. 2021;18:469‐481. [DOI] [PubMed] [Google Scholar]
- 5. Grochola LF, Taubert H, Greither T, Bhanot U, Udelnow A, Würl P. Elevated transcript levels from the MDM2 P1 promoter and low p53 transcript levels are associated with poor prognosis in human pancreatic ductal adenocarcinoma. Pancreas. 2011;40:265‐270. [DOI] [PubMed] [Google Scholar]
- 6. Ormanns S, Siveke JT, Heinemann V, et al. pERK, pAKT and p53 as tissue biomarkers in erlotinib‐treated patients with advanced pancreatic cancer: a translational subgroup analysis from AIO‐PK0104. BMC Cancer. 2014;14:624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ferrara B, Pignatelli C, Cossutta M, Citro A, Courty J, Piemonti L. The extracellular matrix in pancreatic cancer: description of a complex network and promising therapeutic options. Cancers (Basel). 2021;13(17):4442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Weniger M, Honselmann KC, Liss AS. The extracellular matrix and pancreatic cancer: a complex relationship. Cancers (Basel). 2018;10(9):316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Laklai H, Miroshnikova YA, Pickup MW, et al. Genotype tunes pancreatic ductal adenocarcinoma tissue tension to induce matricellular fibrosis and tumor progression. Nat Med. 2016;22:497‐505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Drifka CR, Loeffler AG, Mathewson K, et al. Highly aligned stromal collagen is a negative prognostic factor following pancreatic ductal adenocarcinoma resection. Oncotarget. 2016;7:76197‐76213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Velez DO, Ranamukhaarachchi SK, Kumar A, et al. 3D collagen architecture regulates cell adhesion through degradability, thereby controlling metabolic and oxidative stress. Integr Biol. 2019;11:221‐234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Boedtkjer E, Pedersen SF. The acidic tumor microenvironment as a driver of cancer. Annu Rev Physiol. 2020;82:103‐126. [DOI] [PubMed] [Google Scholar]
- 13. Gatenby RA, Gillies RJ. A microenvironmental model of carcinogenesis. Nat Rev Cancer. 2008;8:56‐61. [DOI] [PubMed] [Google Scholar]
- 14. Vaupel P, Kallinowski F, Okunieff P. Blood flow, oxygen and nutrient supply, and metabolic microenvironment of human tumors: a review. Cancer Res. 1989;49:6449‐6465. [PubMed] [Google Scholar]
- 15. Mayer A, Vaupel P. Hypoxia, lactate accumulation, and acidosis: siblings or accomplices driving tumor progression and resistance to therapy? Adv Exp Med Biol. 2013;789:203‐209. [DOI] [PubMed] [Google Scholar]
- 16. Kimbrough CW, Khanal A, Zeiderman M, et al. Targeting acidity in pancreatic adenocarcinoma: multispectral optoacoustic tomography detects pH‐low insertion peptide probes in vivo. Clin Cancer Res. 2015;21:4576‐4585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cruz‐Monserrate Z, Roland CL, Deng D, et al. Targeting pancreatic ductal adenocarcinoma acidic microenvironment. Sci Rep. 2014;4:4410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Corbet C, Feron O. Tumour acidosis: from the passenger to the driver's seat. Nat Rev Cancer. 2017;17:577‐593. [DOI] [PubMed] [Google Scholar]
- 19. Estrella V, Chen T, Lloyd M, et al. Acidity generated by the tumor microenvironment drives local invasion. Cancer Res. 2013;73:1524‐1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Corbet C, Bastien E, Santiago de Jesus JP, et al. TGFβ2‐induced formation of lipid droplets supports acidosis‐driven EMT and the metastatic spreading of cancer cells. Nat Commun. 2020;11:454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rohani N, Hao L, Alexis MS, et al. Acidification of tumor at stromal boundaries drives transcriptome alterations associated with aggressive phenotypes. Cancer Res. 2019;79:1952‐1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Flinck M, Kramer SH, Pedersen SF. Roles of pH in control of cell proliferation. Acta Physiol (Oxf). 2018;223:e13068. [DOI] [PubMed] [Google Scholar]
- 23. Pouyssegur J, Franchi A, L'Allemain G, Paris S. Cytoplasmic pH, a key determinant of growth factor‐induced DNA synthesis in quiescent fibroblasts. FEBS Lett. 1985;190:115‐119. [DOI] [PubMed] [Google Scholar]
- 24. Pedersen SF, Novak I, Alves F, Schwab A, Pardo LA. Alternating pH landscapes shape epithelial cancer initiation and progression: focus on pancreatic cancer. Bioessays. 2017;39(6):39. [DOI] [PubMed] [Google Scholar]
- 25. Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR‐Cas9 system. Nat Protoc. 2013;8:2281‐2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lonowski LA, Narimatsu Y, Riaz A, et al. Genome editing using FACS enrichment of nuclease‐expressing cells and indel detection by amplicon analysis. Nat Protoc. 2017;12:581‐603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. O'Rourke KP, Dow LE, Lowe SW. Immunofluorescent staining of mouse intestinal stem cells. Bio Protoc. 2016;6(4):e1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Racordon D, Valdivia A, Mingo G, et al. Structural and functional identification of vasculogenic mimicry in vitro. Sci Rep. 2017;7:6985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Andersen AP, Samsøe‐Petersen J, Oernbo EK, et al. The net acid extruders NHE1, NBCn1 and MCT4 promote mammary tumor growth through distinct but overlapping mechanisms. Int J Cancer. 2018;142:2529‐2542. [DOI] [PubMed] [Google Scholar]
- 30. Andersen AP, Flinck M, Oernbo EK, Pedersen NB, Viuff BM, Pedersen SF. Roles of acid‐extruding ion transporters in regulation of breast cancer cell growth in a 3‐dimensional microenvironment. Mol Cancer. 2016;15:45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Malinda RR, Zeeberg K, Sharku PC, et al. TGFbeta signaling increases net acid extrusion, proliferation and invasion in Panc‐1 pancreatic cancer cells: SMAD4 dependence and link to Merlin/NF2 signaling. Front Oncol. 2020;10:687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wang Y, Zhang Y, Yang J, et al. Genomic sequencing of key genes in mouse pancreatic cancer cells. Curr Mol Med. 2012;12:331‐341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hirschhaeuser F, Menne H, Dittfeld C, West J, Mueller‐Klieser W, Kunz‐Schughart LA. Multicellular tumor spheroids: an underestimated tool is catching up again. J Biotechnol. 2010;148:3‐15. [DOI] [PubMed] [Google Scholar]
- 34. Czaplinska D, Elingaard‐Larsen LO, Rolver MG, Severin M, Pedersen SF. 3D multicellular models to study the regulation and roles of acid‐base transporters in breast cancer. Biochem Soc Trans. 2019;47:1689‐1700. [DOI] [PubMed] [Google Scholar]
- 35. Kirschmann DA, Seftor EA, Hardy KM, Seftor RE, Hendrix MJ. Molecular pathways: vasculogenic mimicry in tumor cells: diagnostic and therapeutic implications. Clin Cancer Res. 2012;18:2726‐2732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Deer EL, Gonzalez‐Hernandez J, Coursen JD, et al. Phenotype and genotype of pancreatic cancer cell lines. Pancreas. 2010;39:425‐435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. White KA, Ruiz DG, Szpiech ZA, et al. Cancer‐associated arginine‐to‐histidine mutations confer a gain in pH sensing to mutant proteins. Sci Signal. 2017;10(495):eaam9931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Padua RA, Warren N, Grimshaw D, et al. The cystic fibrosis delta F508 gene mutation and cancer. Hum Mutat. 1997;10:45‐48. [DOI] [PubMed] [Google Scholar]
- 39. Wilde BR, Ye Z, Lim TY, Ayer DE. Cellular acidosis triggers human MondoA transcriptional activity by driving mitochondrial ATP production. Elife. 2019;8:e40199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Elingaard‐Larsen LO, Rolver MG, Sorensen EE, Pedersen SF. How reciprocal interactions between the tumor microenvironment and ion transport proteins drive cancer progression. Rev Physiol Biochem Pharmacol. 2022;182:1‐38. [DOI] [PubMed] [Google Scholar]
- 41. Kawarada Y, Inoue Y, Kawasaki F, et al. TGF‐β induces p53/Smads complex formation in the PAI‐1 promoter to activate transcription. Sci Rep. 2016;6:35483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yao J, Czaplinska D, Ialchina R, et al. Cancer cell acid adaptation gene expression response is correlated to tumor‐specific tissue expression profiles and patient survival. Cancers (Basel). 2020;12(8):2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Corbet C, Draoui N, Polet F, et al. The SIRT1/HIF2alpha axis drives reductive glutamine metabolism under chronic acidosis and alters tumor response to therapy. Cancer Res. 2014;74:5507‐5519. [DOI] [PubMed] [Google Scholar]
- 44. Moellering RE, Black KC, Krishnamurty C, et al. Acid treatment of melanoma cells selects for invasive phenotypes. Clin Exp Metastasis. 2008;25:411‐425. [DOI] [PubMed] [Google Scholar]
- 45. Cifone MA, Fidler IJ. Correlation of patterns of anchorage‐independent growth with in vivo behavior of cells from a murine fibrosarcoma. Proc Natl Acad Sci U S A. 1980;77:1039‐1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Valente LJ, Tarangelo A, Li AM, et al. p53 deficiency triggers dysregulation of diverse cellular processes in physiological oxygen. J Cell Biol. 2020;219(11):e201908212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Annes JP, Munger JS, Rifkin DB. Making sense of latent TGFbeta activation. J Cell Sci. 2003;116:217‐224. [DOI] [PubMed] [Google Scholar]
- 48. Vander Ark A, Cao J, Li X. TGF‐β receptors: In and beyond TGF‐β signaling. Cell Signal. 2018;52:112‐120. [DOI] [PubMed] [Google Scholar]
- 49. Amith SR, Wilkinson JM, Baksh S, Fliegel L. The Na+/H+ exchanger (NHE1) as a novel co‐adjuvant target in paclitaxel therapy of triple‐negative breast cancer cells. Oncotarget. 2015;6:1262‐1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Pedraz‐Cuesta E, Fredsted J, Jensen HH, et al. Prolactin signaling stimulates invasion via Na(+)/H(+) exchanger NHE1 in T47D human breast cancer cells. Mol Endocrinol. 2016;30:693‐708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Cardone RA, Greco MR, Zeeberg K, et al. A novel NHE1‐centered signaling cassette drives epidermal growth factor receptor‐dependent pancreatic tumor metastasis and is a target for combination therapy. Neoplasia. 2015;17:155‐166. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Antibodies used in our study.
Figure S1. Adaptation to lower pH does not enhance Panc02 viability in 2D.
Figure S2. Immunofluorescence analysis of organotypic Panc02 cultures.
Figure S3. Organotypic growth of AsPC1 and Panc1 cells.
Figure S4. Wound healing and Boyden chamber invasion assays.
Figure S5. Western blot densitometry.
Figure S6. Immunohistochemical staining of Panc02 spheroids.
Figure S7. Validation of Akt and TGFβ pathway interventions.
Data Availability Statement
Data are available from the authors upon reasonable request.