

Abstract
Objectives
The primary mechanism for bone marrow failure in aplastic anemia (AA) is autoimmune hematopoietic stem cell destruction. AA can be cured with antithymocyte globulin (ATG) treatment, and some smaller studies have indicated that the number of regulatory T cells (Tregs) may be predictive of response. Additionally, AA patients appear to have elevated numbers of Th17 cells and bone marrow macrophages, but outcome data are missing.
Methods
We performed immunohistochemistry on bone marrow biopsies from 121 ATG‐treated AA patients and 14 healthy controls, using antibodies against FOXP3 (for Tregs), IL‐17 (for Th17), CD68 (for pan‐macrophages) and CD163 (for M2 type macrophages) to study their possible relation to ATG response and AA prognosis.
Results
AA patients had significantly fewer Tregs and Th17 cells but significantly more macrophages compared with controls. Treg, Th17 and pan‐macrophage cell numbers were not associated with ATG response or differences in survival. Patients with higher levels of M2 macrophages had improved 5‐year overall survival: 79.6% versus 57.4% (p = .017), and this benefit was primarily seen in AA patients with non‐severe disease.
Conclusions
We found that Treg and Th17 cell numbers did not predict ATG response or survival, whereas M2 macrophages may be associated with improved survival.
Keywords: aplastic anemia, immunohistochemistry, macrophages, Tregs
Novelty statements.
What is the new aspect of your work?
We have studied the number of macrophages using immunohistochemistry on bone marrow biopsies from a large aplastic anemia patients' cohort
What is the central finding of your work?
It seems as if the number of macrophages (subtype M2) could have a survival impact
What is (or could be) the specific clinical relevance of your work?
Immunohistochemistry is reproducible in the clinical setting, and the number of macrophages could have the potential to be used as an additional prognostic biomarker
1. INTRODUCTION
The underlying immune mechanisms in acquired aplastic anemia (AA) are still poorly understood. 1 , 2 However, the destruction of hematopoietic stem cells (HSC) and progenitor cells by autoreactive cytotoxic T‐cells, as well as the upregulation of different cytokines, for example, interferon‐γ (IFN‐γ) and tumour necrosis factor‐α (TNF‐α), during this disease are well described 3 , 4 , 5 , 6 , 7 ; IFN‐γ enables the destruction of HSCs via activating T‐cells and the Fas/FasL apoptosis pathway. 8 It was also recently reported that IFN‐γ‐dependent HSC destruction is mediated by macrophages (MΦs), and the depletion of MΦs in a mouse model appeared to rescue HSCs from destruction. 3 , 9 Furthermore, MΦs seem to be responsible for TNF‐α production, thus allowing them to engage cytotoxic T‐cells, triggering IFN‐γ production, which together build an interactive loop. 3 Efforts to find an immunological signature for AA as compared with healthy individuals have identified a lower number of regulatory T‐cells (Tregs) and an increased percentage of T‐helper type 17 (Th17) cells. 10 , 11 , 12 , 13 , 14 First, the number of Tregs at the time of diagnosis has been associated with the response to antithymocyte globulin therapy (ATG), with the non‐responding group having the lowest values. 12 , 13 Second, a deep immunophenotyping analysis of Tregs, using CyTOF mass cytometry techniques, has described two subgroups with distinct phenotypes and functions; patients who had Tregs with a memory/activated phenotype (TregB: high expression of CD95, CCR4 and CD45RO within FOXP3hi, CD127lo Tregs) had a better response to ATG. 15 However, the numbers of ATG‐treated patients in these studies are relatively low (ranging from 11 to 31), and the rather complicated technology, together with presumptive difficulties reproducing the number and proportion of Tregs, make it uncertain if measuring these cells can be a helpful prognostic tool for everyday clinical routine.
We previously reported data on the incidence, treatment and outcome in a nationwide study of all AA patients diagnosed in Sweden in 2000–2011. 16 Over 60% of all patients were primarily treated with immunosuppressive therapy (mainly ATG), and only 10% directly underwent stem cell transplantation (SCT). There were no differences in ATG response rate among the age groups, and ATG‐responsive patients had an excellent overall survival (OS) rate. 17 However, patients with very severe AA responded poorly to both first‐ and second‐line ATG, which translated into worse survival, indicating the need for a different treatment approach, such as earlier SCT intervention. Thus, having reliable prognostic indicators or predictive biomarkers for an ATG response would be very valuable, especially for patients above the age of 40 years, as primary immunosuppressive therapy still is advocated for the majority of patients in this population. 18 , 19
Performing a bone marrow biopsy is required for the diagnosis of AA, as marrow cellularity of <25% and a low peripheral blood count are the cornerstones for diagnosis. Immunohistochemistry is an extensively used standard diagnostic method in pathology, and the enumeration of positively stained cells per field of view is also easily reproducible. There have been only a few small (ranging from 10–27 patients) immunohistochemistry studies on bone marrow biopsies from AA patients, and none of them provided data on disease stage, prognosis, treatment or comparisons with healthy individuals. 20 , 21 , 22 , 23 , 24 Thus, with the aim to study the relation of Tregs, Th17 cells, and MΦs to AA disease severity, ATG response, and prognosis, we performed immunohistochemistry staining on bone marrow biopsy material from a large retrospective cohort (n = 121) of ATG‐treated AA patients.
2. PATIENTS AND METHODS
2.1. Patients
As previously described, we retrospectively identified all cases of acquired AA diagnosed in Sweden from 2000 to 2011 and obtained detailed clinical and outcome data. 16 Incidence, overall outcome, and results of immunosuppressive therapy with ATG and allogeneic SCT have previously been presented. 17 , 20 The present study was conducted on the subset of AA patients treated with ATG who had available residual bone marrow biopsy samples (n = 121). Healthy bone marrow tissue donors were included as the control group (7 females and 7 males); the median participant age was 52.5 years (range: 22–69 years). The study was approved by the Regional Ethical Review Board in Gothenburg and the Swedish Ethical Review Authority (615‐12 and 2019‐04900).
The diagnosis of AA was confirmed according to the Camitta criteria, 21 and disease severity was classified as follows: severe aplastic anemia (SAA), with two of the following three characteristics: absolute neutrophil count (ANC) <0.5 × 109/L, platelet count <20 × 109/L or reticulocyte count <20 × 109/L; very severe aplastic anemia (VSAA) had the same characteristics as SAA, but with ANC <0.2 × 109/L; and non‐severe aplastic anemia (NSAA) which are patients not fulfilling criteria for SAA or VSAA.
2.2. Immunohistochemistry
Paraffin‐embedded bone marrow tissue blocks were used to produce whole tissue sections with a thickness of 4 μ. After routine deparaffinization, sections were stained with antibodies against CD34, CD68 (both from Agilent, Santa Clara, CA), CD163, FOXP3 (both from BioCare, Pacheco, CA) and IL‐17 (Abcam, Cambridge, UK). The PT Link/PT200 Heat‐Induced Epitope Retrieval (HIER) and Autostainer Link 48 were utilized. These antibodies were selected for the following reasons: CD68 (a pan‐MΦ marker) and CD163 (a marker of MΦ subtype M2) are proteins expressed on monocyte–macrophage lineage cells; forkhead box protein 3 (FOXP3) is a well‐recognized characteristic of Tregs; IL‐17 is a marker for Th17 cells; and CD34 for hematopoietic stem cells. Tonsillar and appendix tissues were used as positive controls. Detailed information on the primary antibodies is provided in Table S1. Sections stained with antibodies against CD34, CD68, CD163, FOXP3 and IL‐17 were scanned at 40× magnification and digitalized with a NanoZoomer S210 (Hamamatsu Photonics, Hamamatsu City, Japan). Enumeration of positively stained cells was performed digitally using NDP.view2 software (Hamamatsu, Japan). Two independent observers, blinded to the AA severity grade, estimated the proportion of positively staining cells and aimed to have a concordance of >80%. Staining was nuclear (FOXP3) or cytoplasmatic (IL‐17) for lymphocytes and was located in the cell membrane (CD34) for stem cells. For CD68 and CD163 staining, cells with a morphological appearance consistent with MΦs (i.e., large cells with abundant cytoplasm with or without filopodia and with detectable medium‐to‐large nuclei) and with a cytoplasmatic staining pattern were considered positive. All cells with positive staining were counted in 10 consecutive high‐power fields (HPF). Microscope Zeiss Axioscope 2 Plus, with the optical field of view (FOV) 23, was used as a standard for digital HPF, with a calculated area of 0.26 mm2 per HPF. In every HPF, the area containing the hematopoietic cells was marked separately with the freehand region tool, and the median bone marrow cellularity was calculated. The ratio of positively stained cells to cellularity was then estimated and used for comparing patients with controls.
2.3. Statistical analyses
We used a Pearson's chi‐squared test and Mann–Whitney U‐test or Kruskal–Wallis test to compare clinical characteristics and immunohistochemical biomarker levels between the groups. We used the Kaplan–Meier method to determine OS; comparisons were based on the log‐rank test. The statistical analyses were performed by using SPSS version 26.
3. RESULTS
3.1. Patients
Detailed patient characteristics are provided in Table 1. The median age was 57 years (range: 2–85 years), and 50.4% of the patients were female; 23 of the patients were < 18 years old, with a median age of 13 (range: 2–18). Regarding AA disease severity, 63% of the patients had SAA or VSAA at the time of diagnosis, and most patients (80%) were treated with rabbit ATG.
TABLE 1.
Patients characteristics
| Patients, N = 121 | Controls, N = 14 | |
|---|---|---|
| Median age (range) | 57 (2–85) | 52.5 (22–69) |
| Sex, female/male | 61/60 | 7/7 |
| Disease severity (VSAA/SAA/NSAA) at diagnosis, n (%) | 32 (26%)/45 (37%)/44 (37%) | |
| Peripheral blood counts | ||
| Neutrophils, ×10*9/L median (range) | 0.5 (0.0–4.0) | |
| Lymphocytes, ×10*9/L median (range) | 1.5 (0.18–5.8) | |
| Reticulocytes, ×10*9/L median (range) | 21 (1–104)* | |
| Treatment | ||
| Rabbit ATG/horse ATG, n (%)** | 97 (80%)/21 (18%) | |
| HSCT, n *** | 28 | |
| VSAA/SAA/NSAA, n (%) | 11 (39%)/11 (39%)/6 (22%) | |
Note: *93 patients, ** For three patients, data on ATG formulation was missing, *** second or third‐line treatment.
Abbreviation: HSCT‐hematopoietic stem cell transplantation.
3.2. Immunohistochemical biomarkers
The median cellularity for the AA patients (n = 121) was 10% (range: 1%–36%), and that for the controls was 48% (range: 37%–63%). There was no difference in median cellularity among AA severity grades: NSAA, 10% (range: 1%–36%); SAA, 10% (3%–30%); and VSAA, 10% (5%–24%) (p = .799). The median number of CD68‐positive cells (patients, n = 119) was 13 (0–66) per HPF, while CD163 staining (patients, n = 108) yielded a median of 24 (range: 1–54) positive cells per HPF. The median FOXP3‐positive cell number (patients, n = 120) per HPF was 0 (range: 0–17). IL‐17 staining was available for 114 patients, with a median of 0 (0–3) positive cells per HPF. A total of 116 patients had available CD34 staining, with a median of 0 (0–3) positive cells per HPF (Figure 1). All median numerical values for patients and controls are presented in Table S2.
FIGURE 1.
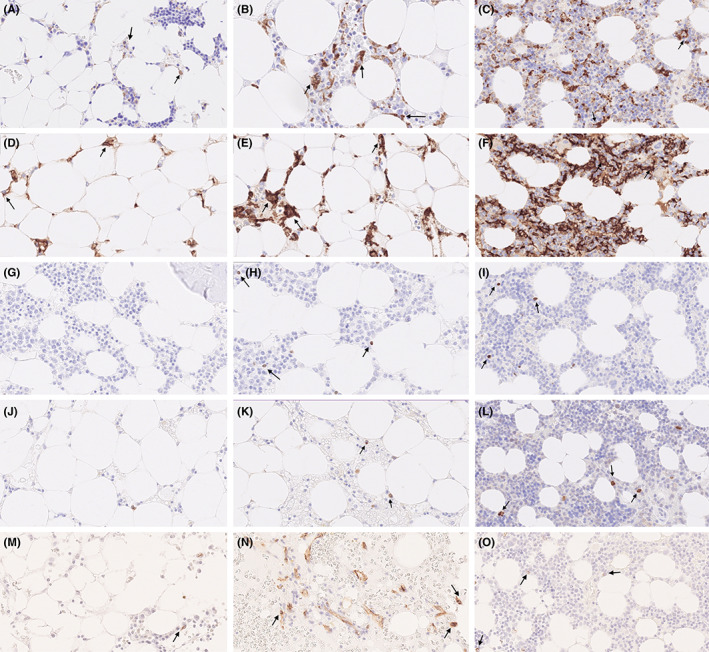
Immunohistochemical staining of CD68, CD163, FOXP3 and IL‐17 protein in BM samples of aplastic anemia patients (magnification: ~40) (A) CD68 < median number; (B) CD68 > median number; (C) CD68 healthy control; (D) CD163 < median number; (E) CD163 > median number; (F) CD163 healthy control; (G) FOXP3 < median number; (H) FOXP3 > median number; (I) FOXP3 healthy control; (J) IL‐17 < median number; (K) IL‐17 > median number; (L) IL‐17 healthy control; (M) CD34 < median number; (N) CD34 > median number; (O) CD34 healthy control.
The CD68 and CD163 positively stained cell number to cellularity ratios were significantly higher for AA patients than for healthy controls (p = .036 for CD68; p = .029 for CD163) (Figure 2A–E). For FOXP3 and IL‐17, the positively stained cell number to cellularity ratios were lower in AA patients than in healthy controls (p = .001 and p < .001, respectively). Regarding CD34, no statistical differences were found between AA patients and controls.
FIGURE 2.
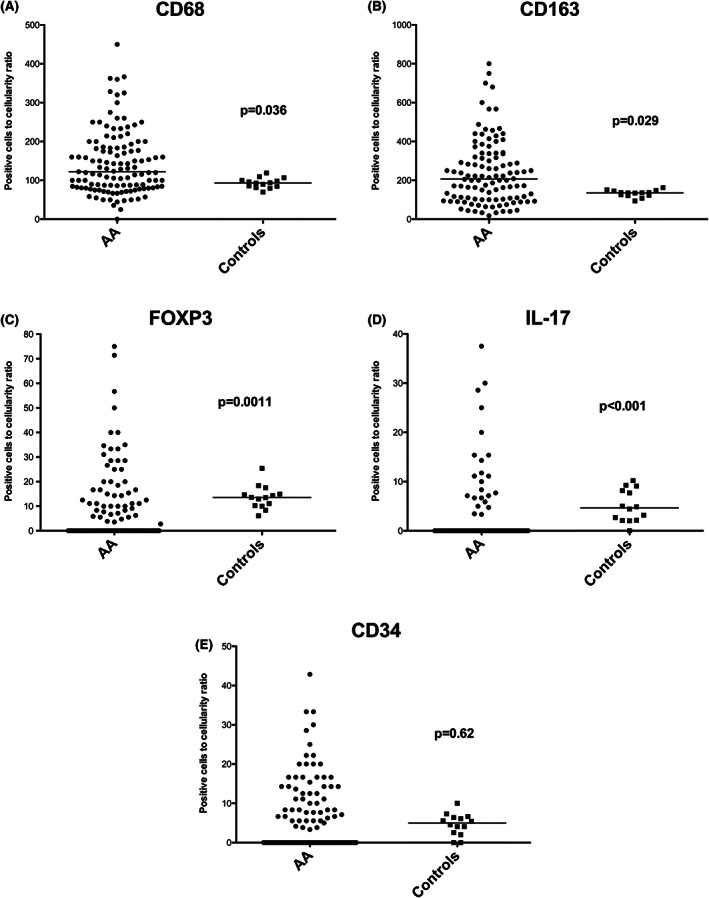
Scatterplots of positive cells to cellularity ratio. (A) CD68 values comparison with controls; (B) CD163 values comparison with controls; (C) FOXP3 values comparison with controls; (D) IL‐17 values comparison with controls; (E) CD34 values comparison with controls
For simplicity, as all AA severity grades had similar median cellularities, only the median number of positively stained cells was used for comparing AA subgroups, and we found no differences in FOXP3, IL‐17, CD34, CD68 or CD163. All numerical values are listed in Table S3. We found a medium‐to‐strong correlation between the number of CD68‐ and CD163‐positive cells, with a Spearman correlation coefficient of 0.442 (p < .001) (Figure S1).
3.3. ATG responsiveness
Fifty‐nine (48.8%) patients responded to first‐line ATG treatment. When dividing patients into two groups according to their median numbers of positively stained cells for FOXP3, IL‐17, CD34, CD68 or CD163, we found no significant differences in ATG response. For FOXP3, 36 (53.7%) patients in the <1 positively stained cells per HPF group responded, compared with 22 (41.5%) in the ≥1 positively stained cells per HPF group (p = .202). For IL‐17, 49.5% (n = 45) responded in the <1 positively stained cells per HPF group, compared with 47.8% (n = 11) in the ≥1 positively stained cells per HPF group (p = .538). For CD34, 29 (48.3%) patients in <1 positively stained cells per HPF group and 26 (46.4%) patients in the ≥1 positively stained cells per HPF group responded (p = .855). For CD68, 27 patients (47.4%) responded in the below‐median CD68 group compared with 31 patients (50%) in the above‐median group (p = .855). Lastly, a similar trend was observed for CD163: 25 patients (46.3%) versus 27 patients (50%) responded in the under and over median groups, respectively (p = .847).
Likewise, we found no differences in ATG response among the AA severity grades when divided according to their FOXP3, IL‐17, CD34, CD68 and CD163 median values. When analysing the ATG response according to the patient absolute reticulocyte count (ARC) and absolute lymphocyte count (ALC), we found that those with an ARC of ≥25 × 109/L had a better response (response/no response, 29/15; 65.9%) than those with an ARC of <25 × 109/L (16/33; 32.7%) (p = .002), but for ALC (<1 or ≥ 1 × 109/L) there was no difference in response (p = .887).
3.4. Survival
The 5‐year OS for all the study patients was 67.8%. When the cohort was divided according to their FOXP3, IL‐17, CD34 or CD68 median value, no differences in OS were found. However, when analysing survival according to the CD163 median subgroups (<24 and ≥ 24 positively stained cells per HPF), we found a survival benefit for the ≥24 subgroup; they had an OS of 79.6% (<24 OS: 57.4%; p = .017) (Figure 3A). When dividing patients according to AA severity, we also found a significant difference between the CD163 median subgroups in NSAA patients (OS: 57.9% vs. 89.5%, p = .035) (Figure 3B); in the SAA and VSAA patients, the corresponding numbers were 66.7% versus 80.0% (p = .348) and 42.9% versus 66.7% (p = .245), respectively (Figure 3C–D).
FIGURE 3.
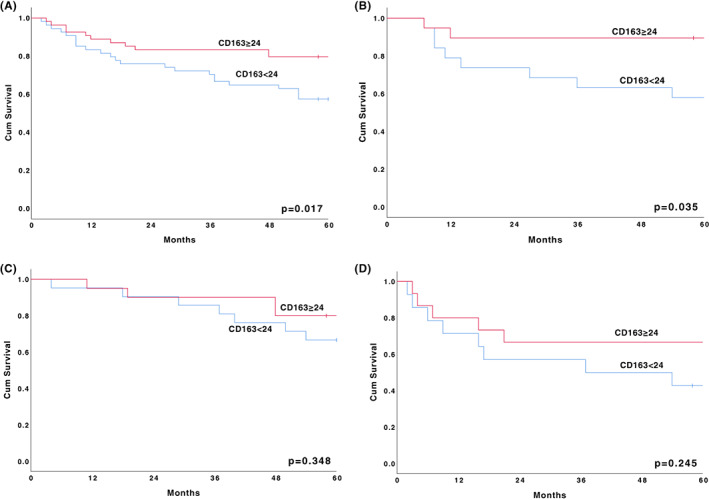
The 5‐year survival according to median value of CD163 (<24 and ≥24 positively stained cells per HPF). (A) All patients; (B) Patients with NSAA at diagnosis; (C) Patients with SAA at diagnosis; (D) Patients with VSAA at diagnosis
For patients with an ARC of ≥25 × 109/L (n = 49), we found no OS difference compared with patients with an ARC of <25 × 109/L (n = 44) (OS: 77.3% vs. 63.3%; p = .141). Similar results were seen for the ALC subgroups. Patients with an ALC of ≥1 × 109/L had a 5‐year OS of 71.6%, whereas patients with an ALC of ≥1 × 109/L had an OS of 53.8% (p = .113).
4. DISCUSSION
In this population‐based cohort study on AA patients treated with first‐line ATG, performing immunohistochemistry on stored bone marrow biopsies revealed that AA patients had fewer Tregs (FOXP3 positive cells) and Th17 cells, but significantly more MΦs compared with controls. We could not associate Treg, Th17 and CD68 positive (pan‐macrophage marker) cell numbers with ATG response or survival. Nevertheless, patients with higher levels of CD163 positive cells (M2 MΦs) had improved 5‐year overall survival, and this benefit was primarily seen in NSAA patients.
So, even though our AA patients had fewer Tregs we found no difference in the number of Tregs between AA severity groups, and no correlation between Tregs and ATG response or OS. Tregs are believed to play a central role in autoimmune disease immunopathogenesis by suppressing autoreactive T cells. 22 Their number has been reported to be depressed in AA; in 2007, a National Institute of Health group analysed Tregs in peripheral blood from 20 AA patients using flow cytometry and found that the absolute numbers of circulating Tregs were lower than those in healthy controls. 11 Other groups have shown similar results, with decreased Treg numbers in the bone marrow as well. 12 , 23 Additionally, Treg function appears to be impaired in AA patients, as illustrated by the reversed ratio of Tregs between peripheral blood and bone marrow, indicating a compromised migratory ability, 23 along with a reduced capacity to suppress effector T cells 12 and inhibit IFN‐γ‐producing T cells. 23 In adult patients, the Treg count at the time of diagnosis has been reported to be lower in the VSAA group than in the NSAA group 12 ; however, in children, no significant difference was observed among the NSAA, SAA and VSAA groups. 13 Furthermore, the likelihood of responding to the treatment was higher for patients with a higher number of Tregs at the time of diagnosis. 12 , 13 Both activated Tregs 12 and the activated/memory type Tregs that predominate in the ATG‐responding AA patients have been found to have bright FOXP3. 15 As we used immunohistochemistry instead of flow cytometry and did not evaluate the Treg functional state, we cannot evaluate the activation state for the positively stained Tregs. Unlike other studies, our patients were treated mostly with rabbit ATG and generally had lower response rates compared with horse ATG‐treated patients. Nevertheless, our data suggest that the number of Tregs in the bone marrow of AA patients seems not to be predictive of response to ATG therapy.
Similar to the previously mentioned study by Sun et al., 3 we found that AA patients had more bone marrow MΦs, of both the CD68 and CD163 subtypes, than did healthy controls. However, we found no difference among AA severity grades. MΦs play a central role in both adaptive and innate immunity and are divided into two main groups: the M1 subtype, which is pro‐inflammatory, and the M2 subtype, which is anti‐inflammatory and controls immune regulation by overexpressing mannose receptor and histocompatibility complex type II and reducing pro‐inflammatory cytokine production. 24 They also have a pathogenic involvement in autoimmune diseases owing to an imbalance in the M1/M2 ratio; a reduced M2 population and increased survival of M1 cells lead to a pro‐inflammatory state. 25 The anti‐inflammatory M2 MΦs have several subsets, one of which (CD163‐expressing M2c) has been suggested to promote Tregs via the production of TGF‐β, but this mechanism is not entirely clear. 24 In an AA mouse model, MΦs produced TNF‐α, and when they engaged T‐effector cells via the TNF‐α receptor, these activated effector cells increased their production of IFN‐γ. 3 Additionally, in another mouse AA model, IFN‐γ‐dependent HSC loss required MΦs, IFN‐γ was necessary for the selective maintenance of bone marrow‐resident MΦs and targeting MΦs both rescued stem cells and improved survival. 9 The bone marrow environment contains tissue‐resident MΦs that sense and respond to IFN‐γ, which stimulates them to produce cytokines, such as TNF‐α. 26 Tissue‐resident MΦs are believed to be embryonically derived and maintained via self‐renewal, which may be another explanation for the persisting MΦs in AA patient bone marrow. 26 , 27 Here, a higher number of CD163‐positive MΦs (M2 subtype) were associated with better survival, and this benefit was most apparent in the NSAA patient group. Regarding AA immunology, the MΦ polarization between M1 and M2 is unclear, but enhanced M2 MΦ activity has been correlated with HSC protection in an SAA mouse model. 9 We cannot explain the potential survival benefit of more CD163/M2 MΦs, mainly seen in the NSAA group, from clinical characteristics, as we did not find any differences in ATG response, age or transplantation rate between the high‐ and low‐CD163 patient groups (Table S4). A higher number of CD163 MΦs could suggest a rudimentary M2 state, indicating an immunosuppressive role. However, we cannot exclude that some other unaccounted variables have influenced the outcome of the NSAA group. Nevertheless, further functional studies are needed to elucidate a possible beneficial role of M2 MΦs in AA.
Regarding Th17 cells, we found that healthy controls had higher numbers of IL‐17‐positive cells compared with AA patients, but without prognostic or predictive impact. Our results contrast with previous data in which a significantly larger population of Th17 (CD3+CD8−IL‐17+) cells was detected with flow cytometry in AA patients than in healthy controls. 10 , 28 There are contradictory data concerning Th17 cells and AA severity grades: Peffault de Latour and co‐workers found a higher number in NSAA than in SAA 10 but a group from King's College in London found higher numbers in SAA. 12 Here, all AA severity grades had very few Th17 cells, without any difference between groups.
As expected, we found very low numbers of CD34 cells, corroborating previous immunohistochemistry AA bone marrow studies. 29 , 30 However, in contrast with Kelaidi and co‐workers who noticed that ATG responders had a somewhat higher percentage of CD34 cells, we found no correlation of CD34 cells with ATG response or survival.
Admittedly, our study has limitations. First, as it was retrospective, we used bone marrow biopsies that were 9–20 years old. However, those biopsies were primarily prepared similarly with formalin fixation and embedding in paraffin wax blocks, and it appears as if most antigens are well preserved over several decades. 31 Second, estimations of positively stained cells were made manually, and even if 80% coherence was achieved, some subjectivity was still possible. Regrettably, apart from bone marrow biopsies no other biobanked material could be identified, so we could not correlate our immunohistochemistry findings with confirmatory analyses using flow cytometry. Third, we have no information on the function of the Tregs or MΦs. Lastly, we used biopsy material instead of bone marrow aspirates. However, this could actually have some advantage because of the low cellularity in AA and the risk of aspirated bone marrow being diluted with peripheral blood.
In conclusion, in this rather large retrospective real‐world study, we immunohistochemically analysed the bone marrow populations of Tregs, Th17 cells, CD34 cells and MΦs in patients treated with first‐line ATG. We found no correlation between cell number and ATG response. To our surprise, the number of M2 MΦs appeared to have a positive impact on survival, especially in NSAA patients. Although further functional studies are needed, our findings suggest that M2 MΦs could be of possible prognostic importance in AA patients.
AUTHOR CONTRIBUTIONS
Krista Vaht, Per Ljungman, Mats Brune and Per‐Ola Andersson conceived and designed the study; Krista Vaht Jonas Brenner and Susanne Bram Ednersson collected and assembled data; Krista Vaht and Per‐Ola Andersson analysed and interpreted data; Krista Vaht and Per‐Ola Andersson wrote the manuscript; and all authors gave final approval of the manuscript.
DISCLOSURES
Per‐Ola Andersson joined the speaker's bureau of AbbVie, Astra Zeneca, Gilead, Janssen and Roche and has been a consultant for Abbvie, Beigene, Incyte, Janssen and Takeda. The remaining authors declare no competing interests.
Supporting information
DATA S1. Supporting Information
ACKNOWLEDGMENTS
This study was supported by grants from the Swedish state under the agreement between the Swedish government and the country councils; the ALF‐agreement (79606), Gothenburg Medical Society, Blodcancerförbundet and Assar Gabrielssons Fund. We thank Katie Oakley, Ph.D., from Edanz (www.edanz.com/ac) for editing a draft of this manuscript.
Vaht K, Brenner J, Ednersson SB, Ljungman P, Brune M, Andersson P‐O. Bone marrow expression of CD68/CD163 macrophages, IL‐17 and FOXP3 cells in aplastic anemia and their relation to prognosis. Eur J Haematol. 2023;110(3):313‐321. doi: 10.1111/ejh.13908
Funding information ALF (79606); Assar Gabrielssons Fund; Blodcancerförbundet; Göteborgs Läkaresällskap
DATA AVAILABILITY STATEMENT
The data supporting the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Young NS. Aplastic anemia. N Engl J Med. 2018;379(17):1643‐1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Young NS, Calado RT, Scheinberg P. Current concepts in the pathophysiology and treatment of aplastic anemia. Blood. 2006;108(8):2509‐2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sun WWZ, Lin Z, Hollinger M, Chen J, Feng X, Young NS. Macrophage TNF‐α licenses donor T cells in murine bone marrow failure and can be implicated in human aplastic anemia. Blood. 2018;132:2730‐2743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zoumbos NCFW, Hsu SM, Goodman S, et al. Analysis of lymphocyte subsets in patients with aplastic anaemia. Br J Hematol. 1984;58:95‐105. [DOI] [PubMed] [Google Scholar]
- 5. Lu J, Basu A, Melenhorst JJ, Young NS, Brown KE. Analysis of T‐cell repertoire in hepatitis‐associated aplastic anemia. Blood. 2004;103(12):4588‐4593. [DOI] [PubMed] [Google Scholar]
- 6. Chen J, Lipovsky K, Ellison FM, Calado RT, Young NS. Bystander destruction of hematopoietic progenitor and stem cells in a mouse model of infusion‐induced bone marrow failure. Blood. 2004;104(6):1671‐1678. [DOI] [PubMed] [Google Scholar]
- 7. Zoumbos N, Gascon P, Djeu J, Young N. Interferon is a mediator of hematopoietic suppression in aplastic anemia in vitro and possibly in vivo. Proc Natl Acad Sci U S A. 1985;82(1):188‐192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Liu CY, Fu R, Wang HQ, et al. Fas/FasL in the immune pathogenesis of severe aplastic anemia. Genet Mol Res. 2014;13(2):4083‐4088. [DOI] [PubMed] [Google Scholar]
- 9. McCabe A, Smith JNP, Costello A, Maloney J, Katikaneni D, MacNamara KC. Hematopoietic stem cell loss and hematopoietic failure in severe aplastic anemia is driven by macrophages and aberrant podoplanin expression. Hematologica. 2018;103(9):1451‐1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. de Latour RP, Visconte V, Takaku T, et al. Th17 immune responses contribute to the pathophysiology of aplastic anemia. Blood. 2010;116(20):4175‐4184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Solomou EE, Rezvani K, Mielke S, et al. Deficient CD4+ CD25+ FOXP3+ T regulatory cells in acquired aplastic anemia. Blood. 2007;110(5):1603‐1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kordasti S, Marsh J, Al‐Khan S, et al. Functional characterization of CD4+ T cells in aplastic anemia. Blood. 2012;119(9):2033‐2043. [DOI] [PubMed] [Google Scholar]
- 13. Lin S, Hou L, Liu S, et al. Roles of regulatory T cells in the pathogenesis of pediatric aplastic anemia. Pediatr Hematol Oncol. 2019;36(4):198‐210. [DOI] [PubMed] [Google Scholar]
- 14. Yan L, Fu R, Liu H, et al. Abnormal quantity and function of regulatory T cells in peripheral blood of patients with severe aplastic anemia. Cell Immunol. 2015;296(2):95‐105. [DOI] [PubMed] [Google Scholar]
- 15. Kordasti S, Costantini B, Seidl T, et al. Deep phenotyping of Tregs identifies an immune signature for idiopathic aplastic anemia and predicts response to treatment. Blood. 2016;128(9):1193‐1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Vaht K, Goransson M, Carlson K, et al. Incidence and outcome of acquired aplastic anemia: real‐world data from patients diagnosed in Sweden from 2000‐2011. Hematologica. 2017;102(10):1683‐1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Vaht K, Goransson M, Carlson K, et al. Low response rate to ATG‐based immunosuppressive therapy in very severe aplastic anaemia: a Swedish nationwide cohort study. Eur J Hematol. 2018;100(6):613‐620. [DOI] [PubMed] [Google Scholar]
- 18. Killick SB, Bown N, Cavenagh J, et al. Guidelines for the diagnosis and management of adult aplastic anaemia. Br J Hematol. 2016;172(2):187‐207. [DOI] [PubMed] [Google Scholar]
- 19. Marsh JC, Ball SE, Cavenagh J, et al. Guidelines for the diagnosis and management of aplastic anaemia. Br J Hematol. 2009;147(1):43‐70. [DOI] [PubMed] [Google Scholar]
- 20. Vaht K, Goransson M, Carlson K, et al. High graft‐versus‐host disease‐free, relapse/rejection‐free survival and similar outcome of related and unrelated allogeneic stem cell transplantation for aplastic anemia: a Nationwide Swedish Cohort Study. Biol Blood Marrow Transpl. 2019;25(10):1970‐1974. [DOI] [PubMed] [Google Scholar]
- 21. Camitta BM, Rappeport JM, Parkman R, Nathan DG. Selection of patients for bone marrow transplantation in severe aplastic anemia. Blood. 1975;45(3):355‐363. [PubMed] [Google Scholar]
- 22. Wei S, Kryczek I, Zou W. Regulatory T‐cell compartmentalization and trafficking. Blood. 2006;108(2):426‐431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Shi J, Ge M, Lu S, et al. Intrinsic impairment of CD4(+)CD25(+) regulatory T cells in acquired aplastic anemia. Blood. 2012;120(8):1624‐1632. [DOI] [PubMed] [Google Scholar]
- 24. di Benedetto P, Ruscitti P, Vadasz Z, Toubi E, Giacomelli R. Macrophages with regulatory functions, a possible new therapeutic perspective in autoimmune diseases. Autoimmun Rev. 2019;18(10):102369. [DOI] [PubMed] [Google Scholar]
- 25. Ma WT, Gao F, Gu K, Chen DK. The role of monocytes and macrophages in autoimmune diseases: a comprehensive review. Front Immunol. 2019;10:1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Smith JN, Kanwar VS, MacNamara KC. Hematopoietic stem cell regulation by type I and II interferons in the pathogenesis of acquired aplastic anemia. Front Immunol. 2016;7:330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ginhoux F, Guilliams M. Tissue‐resident macrophage ontogeny and homeostasis. Immunity. 2016;44(3):439‐449. [DOI] [PubMed] [Google Scholar]
- 28. Du HZ, Wang Q, Ji J, et al. Expression of IL‐27, Th1 and Th17 in patients with aplastic anemia. J Clin Immunol. 2013;33(2):436‐445. [DOI] [PubMed] [Google Scholar]
- 29. Park M, Park CJ, Jang S, et al. Reduced expression of osteonectin and increased natural killer cells may contribute to the pathophysiology of aplastic anemia. Appl Immunohistochem Mol Morpol. 2015;23(2):139‐145. [DOI] [PubMed] [Google Scholar]
- 30. Kelaidi C, Makis A, Tzotzola V, et al. Severe aplastic anaemia in children: impact of histopathology profile and treatment on very long‐term outcomes. Acta Paediatr. 2021;110(4):1308‐1314. [DOI] [PubMed] [Google Scholar]
- 31. Grillo F, Bruzzone M, Pigozzi S, et al. Immunohistochemistry on old archival paraffin blocks: is there an expiry date? J Clin Pathol. 2017;70(11):988‐993. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
DATA S1. Supporting Information
Data Availability Statement
The data supporting the findings of this study are available from the corresponding author upon reasonable request.