

Abstract
Small Ubiquitin‐like Modifier 1 (SUMO1) is an essential protein for many cellular functions, including regulation, signaling, etc., achieved by a process known as SUMOylation, which involves covalent attachment of SUMO1 to target proteins. SUMO1 also regulates the function of several proteins via non‐covalent interactions involving the hydrophobic patch in the target protein identified as SUMO Binding or Interacting Motif (SBM/SIM). Here, we demonstrate a crucial functional potential of SUMO1 mediated by its non‐covalent interactions with α‐Synuclein, a protein responsible for many neurodegenerative diseases called α‐Synucleinopathies. SUMO1 hinders the fibrillation of α‐Synuclein, an intrinsically disordered protein (IDP) that undergoes a transition to β‐structures during the fibrillation process. Using a plethora of biophysical techniques, we show that SUMO1 transiently binds to the N‐terminus region of α‐Synuclein non‐covalently and causes structural compaction, which hinders the self‐association process and thereby delays the fibrillation process. On the one hand, this study demonstrates an essential functional role of SUMO1 protein concerning neurodegeneration; it also illustrates the commonly stated mechanism that IDPs carry out multiple functions by structural adaptation to suit specific target proteins, on the other. Residue‐level details about the SUMO1‐α‐Synuclein interaction obtained here also serve as a reliable approach for investigating the detailed mechanisms of IDP functions.
Keywords: aggregation, intrinsically disordered proteins, non‐covalent interaction, small ubiquitin‐like modifiers, SUMO1, transient interactions, α‐Synuclein, α‐Synucleinopathies
1. INTRODUCTION
Small ubiquitin‐like modifier (SUMO) is a ~100 amino acid protein that belongs to the ubiquitin‐like proteins (UBLs) family and is present in the nucleus of the cell (Hay, 2005). The four sub‐categories of SUMO in humans: SUMO1/2/3/4 have a characteristic ubiquitinfold, ββαβαβ and the C‐terminal Gly‐Gly motif (Bayer et al., 1998). SUMOs regulate various cellular functions such as cell cycle control, inflammation, nuclear transport, oncogenesis, regulation of mitochondrial dynamics, chromatin structure, signal transduction, transcription, DNA repair, and response to virus functions (Hay, 2005; Geiss‐Friedlander and Melchior, 2007; Saitoh and Hinchey, 2000) by post‐translational modification (PTM) of the target proteins via covalent conjugation to its target, called SUMOylation or via non‐covalent interaction. SUMOylation has both positive and negative effects on the protein–protein interaction (Scheschonka et al., 2007). Attachment of SUMO1 could lead to masking of the binding site of the specific interacting protein and thus can lead to disruption of protein–protein interaction (Klenk et al., 2006). The regulation of biochemical pathways by SUMO occurs via non‐covalent interactions between the hydrophobic core of SUMO1 (β2 and α1‐helix) and the hydrophobic region of the target protein known as SUMO interacting/binding motifs (SIM/SBM) (Dorval et al., 2007; Hecker et al., 2006; Song et al., 2005). SIMs generally consist of a stretch of hydrophobic amino acid residues adjacent to the acidic amino acids or serines and threonines with some variations to promote the affinity of the SUMO and SIMs via polar/electrostatic interactions (Song et al., 2004). SIM/SBM sequence determines the specificity and orientation (parallel and anti‐parallel) of the bound paralogues of SUMO (Hecker et al., 2006; Song et al., 2005; Song et al., 2004). SUMO1 also plays an important role in neuron‐specific functions and is essential for CNS development (Loriol et al., 2012) and therefore, tight regulation of the SUMO modification in the central nervous system (CNS) is essential for neuronal differentiation, survival, and hence neuronal cell viability, connectivity, and function (Krumova and Weishaupt, 2013). Therefore, any dysfunction or neurological disorder would be affected by SUMO interaction. One such disorder in which SUMO1 seems to play an important role is the second most prevalent neurodegenerative disorder, Parkinson's disease.
Parkinson's disease (PD) is pathologically characterized by the presence of Lewy bodies and Lewy neurites, predominantly rich in aggregates of protein α‐Synuclein (α‐Syn) (Goedert, 2001; Spillantini et al., 1998). α‐Syn is an intrinsically disordered presynaptic protein that plays a crucial role in synaptic vesicle trafficking, regulation of neurotransmitters release, and plasticity of neurons. However, the exact mechanism for the native function of α‐Syn is still unknown (Auluck et al., 2010; Lashuel et al., 2013; Sulzer and Edwards, 2019). Misfolding of α‐Syn and toxic intermediate species are responsible for neurodegenerative diseases termed as α‐Synucleinopathies which include PD, Dementia with Lewy's Bodies (DLB), and Multiple System Atrophy (MSA). The conformational transition of amyloidogenic α‐Syn occurs from its disordered monomeric state to cross‐β‐sheet‐rich structures (amyloid‐like fibrils) through helical intermediates such as oligomers and protofibrils during the aggregation. On‐pathway intermediates confer toxicity leading to the death of dopaminergic cells (Jo et al., 2000; Eliezer et al., 2001; Kim et al., 2009; Comellas et al., 2012). Various external and intrinsic factors regulate the transition of α‐Syn to different conformations during aggregation or self‐association, and thus the toxicity. Intrinsic factors include diffusion and point mutations (Krüger et al., 1998; Nishioka et al., 2017; Pasanen et al., 2014; Polymeropoulos et al., 1997; Proukakis et al., 2013; Yoshino et al., 2017; Zarranz et al., 2004), whereas external factors are pH, temperature, and PTMs such as phosphorylation (Paleologou et al., 2010; Sato et al., 2011), O‐glycosylation (Marotta et al., 2015), ubiquitination (Hasegawa et al., 2002), nitration (Hodara et al., 2004), and SUMOylation (Krumova et al., 2011). SUMOylation of α‐Syn by SUMOs regulates the extracellular sorting of vesicles by α‐Syn and inter‐neuronal propagation of α‐Syn (pathological characteristic of PD) (Kunadt et al., 2015). SUMO1 is generally localized in the nucleus bound to its target protein, and the amount of free SUMO1 is significantly less. Whereas, SUMO1 in PD exhibit dense co‐localization with α‐Syn in the cytoplasmic inclusions (Pountney et al., 2005). Previously, in vitro SUMOylation at K96 and K102 has been elucidated to prevent α‐Syn fibrillation (Krumova et al., 2011; Dorval and Fraser, 2006; Abeywardana and Pratt, 2015). However, recently, SUMOylation has been shown to promote α‐Syn aggregation by reducing the ubiquitination of α‐Syn (responsible for the clearance of α‐Syn from the cell via proteasomal degradation) further promoting the accumulation of α‐Syn aggregates in the inclusion bodies (Oh et al., 2011; Kim et al., 2011; Rott et al., 2017). Previously, SUMO has been shown to affect the proteins involved in neurodegenerative diseases non‐covalently. SUMO interacts non‐covalently with parkin protein (involved in PD), and this affects its auto‐ubiquitination and nuclear localization (Um and Chung, 2006). Also, SUMO modulates amyloid precursor proteins processing and production of Aβ peptides without SUMOylation (Dorval et al., 2007). Though the covalent modifications of α‐Syn have been studied extensively, non‐covalent interactions between α‐Syn and SUMO1 have not been explored yet, requiring a detailed exploration.
Here, by using NMR spectroscopy along with other biophysical techniques such as analytical ultracentrifugation, small‐angle X‐ray scattering, and molecular dynamics simulations, we demonstrated that the hydrophobic region of α‐Syn is involved in the non‐covalent interaction with SUMO1. This transient interaction between two proteins induced the conformational compaction in α‐Syn, which could be correlated with the alteration in the amyloidogenic/self‐association propensity of α‐Syn in the presence of SUMO1. We also demonstrate that the non‐covalent interaction of SUMO1 has an effect on the backbone dynamics of α‐Syn and its familial mutants, resulting in differential aggregation kinetics in the presence of SUMO1. Our study provides mechanistic insight into the modulation of self‐association of α‐Syn involved in neurodegeneration upon interaction with SUMO1, and this may offer a reliable approach for investigating the role and detailed mechanism of SUMO1 intervention in diseased conditions.
2. RESULTS
Based on the primary structure of α‐Syn, the sequence of α‐Syn is characteristically classified into three prominent regions: 1–60, N‐terminus with amphipathic amino acids; 61–95, non‐amyloid β component (NAC) mostly containing hydrophobic amino acids; and 96–140, negative amino acid rich C‐terminus (Figure S1a). α‐Syn (Figure S1b, PED00024e001) is structurally more compact than the expected random coil conformation due to the existence of long‐range intramolecular interaction between the N‐ and C‐termini, and NAC and C‐terminus regions previously established by paramagnetic resonance enhancement (PRE) and other NMR experiments by various groups (Bertoncini et al., 2005; Dedmon et al., 2005). The electrostatic interactions involving the N‐(mostly positively charged) and C‐terminus (negatively charged) can be explained by the net charge per residue pattern of α‐Syn (Figure S1c). α‐Syn undergoes a series of structural transitions to form oligomeric intermediates and cross β‐sheet fibrillar structure during aggregation (representative; Figure S1d). α‐Syn interacts with many proteins during its biogenesis, translation, and degradation (Hernandez et al., 2020). Any dysregulation in the interaction of α‐Syn with the other proteins would lead to protein misfolding, aggregation, and disease progression. Thus, elucidating the nature of the complexes formed between α‐Syn and its post‐translational modifier protein, SUMO1, would help gain better insight into disease progression.
2.1. Hydrophobic interaction drives the binding of SUMO1 with α‐Syn
We performed NMR experiments in order to delineate the interaction between α‐Syn and SUMO1 and the residues involved therein. A low pH of 6.5 was chosen to mimic the metabolic acidosis condition predominantly present in the brain of patients suffering from neurological disorders, including PD (Ruffin et al., 2014; Chu and Xiong, 2012). 15N‐1H two‐dimensional Heteronuclear Single Quantum Coherence (HSQC) Spectroscopy provides a fingerprint of the protein. Each cross‐peak represents the backbone amide group of one amino acid residue in the polypeptide chain, except for proline. 15N isotopic labeled α‐Syn was titrated with different equivalents of unlabelled SUMO1, and HSQC spectra were recorded for each concentration of SUMO1. The titration did not affect the narrow dispersion of the amide proton (Figure S2a), suggesting that SUMO1 does not induce any stable secondary structure formation and α‐Syn retains its disordered nature. However, the amide cross‐peaks of the residues V37‐A53 exhibited a gradual decrease in the intensity, with no significant perturbation in the chemical shift even at the highest equivalence of SUMO1 (Figure 1a–c). Some C‐terminus residues, Q109‐M116 and E123‐S129, showed a slight decrease in intensity. A relatively higher chemical shift perturbation (CSP) at an equimolar ratio of SUMO1 was observed for these residues (Figure 1c). This could be attributed to the long‐range interaction between the N‐ and C‐terminus of α‐Syn, inducing an indirect effect in the observed CSP (Bertoncini et al., 2005). Similarly, 15N isotope labeled SUMO1 was titrated against unlabelled α‐Syn to identify the residues of SUMO1 interacting with α‐Syn (Figure S2b). Significant perturbations in the amide cross‐peak were observed for residues in the β2 (H35‐K39), α1 (K45‐L47, S50‐Y51, and R54), and loop (M40‐H43) of SUMO1 (Figure 1d,f). These residues and the residues from the β1 (I22‐I27) also exhibited a drastic decrease in intensity (Figure 1e). Significant intensity decay suggests that the residues are in an intermediate exchange regime at the NMR time scale, and the binding is in the micromolar range (Vaynberg and Qin, 2006; Williamson, 2013) between the interacting partners.
FIGURE 1.
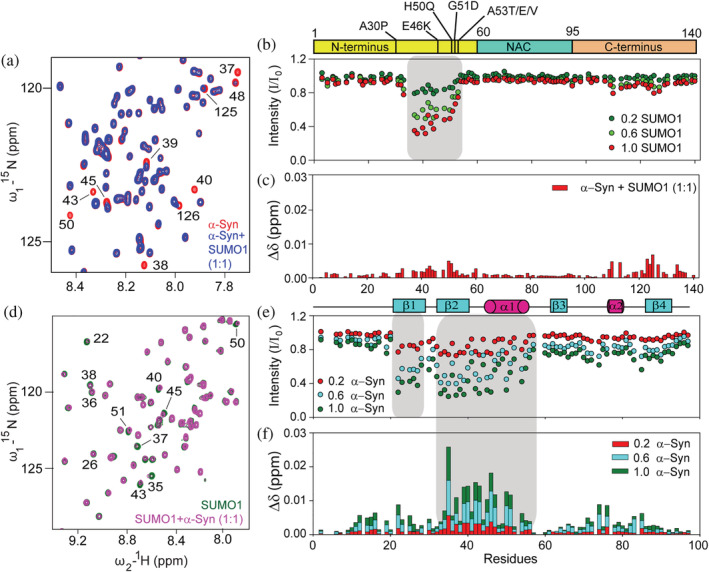
Titration of 15N labeled α‐Syn with SUMO1 and vice versa. (a) Excerpt of overlaid 15N‐1H HSQC spectra of α‐Syn in the absence (red) and presence (blue) of equimolar human SUMO1. The residues exhibiting shifts or reduction in intensities have been labeled. (b) Intensity ratio (I/I 0) of amide cross‐peak of α‐Syn from the 15N‐1H HSQC spectra in the absence (I 0) and presence (I) of different equivalents of SUMO1 (0.2: green; 0.6: light green; and 1.0: red). (c) Chemical shift perturbation of the amide cross‐peaks of α‐Syn in the presence of equimolar SUMO1. (d) Excerpt of overlaid 15N‐1H HSQC spectra of human SUMO1 in the absence (green) and presence (pink) of equimolar α‐Syn. (e) Intensity ratio (I/I 0) of amide cross‐peak of SUMO1 from the 15N‐1H HSQC spectra in the absence (I 0) and presence (I) of different equivalents of α‐Syn (0.2: red; 0.6: cyan; and 1.0: green). (f) Chemical shift perturbation of the amide cross‐peaks of SUMO1 in the presence of different equivalents of α‐Syn (0.2: red; 0.6: cyan; and 1.0: green).
The SUMO1 interacting region in α‐Syn (37‐VLYVGSKTKEGVVHGVA‐53) reveals the presence of two hydrophobic patches, P1: VLYVGS and P2: GVVHGVA, separated by charged residues, KTKE. As reported previously, these regions have a higher propensity for nascent β‐structure conformation in the monomeric state of α‐Syn (Kim et al., 2007). Upon analysis of the residues in the interacting region of α‐Syn, these two hydrophobic patches constitute the two SBMs, SBM1: VLYVGS and SBM2: GVVHGVA. SUMO1 interacts non‐covalently with its substrate through a hydrophobic core (β2 and α1), known as the “code of specificity” on the SUMO1 sequence. The region of SUMO1 interacting with α‐Syn is also the hydrophobic core of the SUMO1. Thus, residues in the patch described above suggest a predominant hydrophobic interaction between SUMO1 and the region of α‐Syn exhibiting the nascent β‐structure. Next, we also determined the effect of the interaction of SUMO1 on α‐Syn at normal physiological pH, and it exhibited the involvement of the same residues in the interaction (Figure S3d–f). This indicates that the decrease in pH does not alter the nature of the interaction between these two proteins.
2.2. SUMO1 and α‐Syn exhibit transient interaction
For quantification of the strength of the interaction, surface plasmon resonance (SPR) was performed. SPR detects the change in mass on the sensor chip by measuring the change in the refractive index. α‐Syn was immobilized on the CH5 chip, and the different concentrations (0.19–400 μM) of SUMO1 were passed over it. Sensogram was recorded at each concentration, and the effect was recorded as a response unit (RU). A distinct pattern of association and dissociation was observed (Figure 2a). The sample exhibited an increase in the response unit with the increase in the concentration of SUMO1. The dissociation constant (K D) was determined to be 9.27 ± 3.23 μM from the response curve obtained by fitting the response at each concentration of SUMO1 by Langmuir isotherm fit (1:1 binding) (Figure 2b).
FIGURE 2.
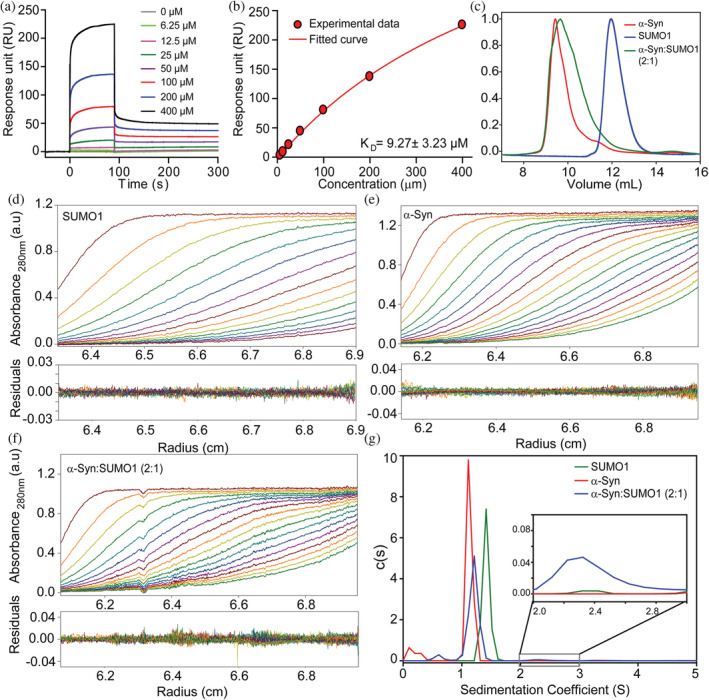
Binding affinity assessment of α‐Syn with SUMO1 and characterization of α‐Syn and SUMO1 complex by size exclusion chromatography and analytical ultracentrifugation. (a) The sensogram was obtained for the α‐Syn at different concentrations of SUMO1 by surface Plasmon resonance. (b) The response unit v/s concentration of SUMO1 fitted by Langmuir isotherm fit (1:1 binding). Overlapped SEC profile of α‐Syn and SUMO1 and its complex formed by α‐Syn: SUMO1 at 2:1 (c). AUC absorbance spectra were obtained at 280 nm for SUMO1 (d), α‐Syn (e), α‐Syn and SUMO1 complex at 2:1 (f) along with the residuals. (g) The sedimentation coefficient obtained from fitting the absorbance spectra for α‐Syn (red, 1.13 S), SUMO1 (green, 1.42 S), α‐Syn and SUMO1 at 2:1 (blue, 1.21 S and 2.43 S, in excerpt).
The complex was isolated to get a better insight into the interaction mechanism between both proteins. α‐Syn and SUMO1 were mixed at different ratios (α‐Syn: SUMO1 = 1:1, and 2:1) and subjected to gel filtration along with the purified proteins (Figure 2c). When we mixed two proteins in an equimolar ratio, three distinct peaks were eluted (Figure S4a, black curve), corresponding to α‐Syn, the complex, and SUMO1 in the order of elution. The SEC data suggests compaction, i.e., the complex formed (represented by the second peak in the SEC curve) is more compact than that of the free α‐Syn and elutes after α‐Syn. For the 2:1 mixture (Figure 2c, green curve), a single broad peak overlaps with that of α‐Syn. However, the shoulder peak represents another population corresponding to the complex formed.
To validate this observation, we employed the analytical ultracentrifugation (AUC) technique which can characterize even weak interactions between proteins (Schuck, 2013). AUC reports on the sedimentation coefficient of the protein or complexes and helps in characterizing the shape and size of the macromolecules (Schuck, 2013). Therefore size, shape, and compactness of α‐Syn, SUMO1, and the complex formed by these proteins at 1:1 and 2:1 were analyzed by AUC (Figures 2d–f and S4b), and the sedimentation coefficients were determined by fitting the absorbance curves obtained at 280 nm (Figures 2g and S4c). α‐Syn and SUMO1 sediments at 1.13 S (M.W., 15.6 kDa) and 1.42 S (M.W., 12.1 kDa) at a frictional ratio of 1.98 and 1.33, respectively (Figure 2g). α‐Syn being an IDP, has a more open structure as compared to that of globular SUMO1 protein and therefore exhibit a higher frictional ratio of 1.98 and sediments slower than that of SUMO1 (Figure 2g). For α‐Syn and SUMO1 complex at equimolar ratio (Figure S4c, black curve), two peaks correspond to two species with sedimentation coefficients 1.27 S (M.W., 14.5 kDa) and 2.18 S (M.W., 32.5 kDa) with 91% and 4.5% population, respectively, at a frictional ratio of 1.67. Also, two peaks corresponding to two species were obtained for the complex of α‐Syn and SUMO1 formed at 2:1 (Figure 2g, green curve). The sedimentation coefficients of both the species are 1.21 S (M.W., 14.3 kDa) and 2.43 S (M.W., 40.5 kDa) with 90% and 2.1% population, respectively, at a frictional ratio of 1.75. The second peak corresponds to the complex formed by two molecules of α‐Syn and one molecule of SUMO1. Reduced frictional ratio and increase in sedimentation coefficient values indicate compaction in α‐Syn. A low complex population indicates transient interaction, which is understandable due to the high K D value in the micromolar range.
To obtain a model for the complex formed and understand the relative orientation of α‐Syn and SUMO1 in the complex, small‐angle X‐ray scattering (SAXS) was carried out. The scattering pattern for the individual proteins SUMO1 (Figure 3a) and α‐Syn (Figure 3b) and for their complexes formed by mixing α‐Syn and SUMO1 at 1:1 and 1:2 were recorded (Figure 3c,d). The scattering curve obtained experimentally was validated by the SAXS curve generated from the PDB structure available for SUMO1 (PDB Id: 1A5R) and ensemble structure for α‐Syn from Protein Ensemble Database (PED; PED00024e001) (Figure S4d,e). Next, the curves for the individual proteins and the complexes were fitted (Figure S4f–i), and the radius of gyration (Rg) was determined by Guinier approximation (Table 1). The D max was determined by the distance distribution (Figure 3e). DAMMIF module of the ATSAS package was used to generate a model for each of the systems (models in the inset of Figure 3a–d). The complexes formed at the equimolar ratio of α‐Syn and SUMO1 have an Rg and D max less than free α‐Syn. However, the complex formed between α‐Syn and SUMO1 at 2:1 ratios is comparable to free α‐Syn in terms of Rg and D max, further indicating the compaction in the complex.
FIGURE 3.
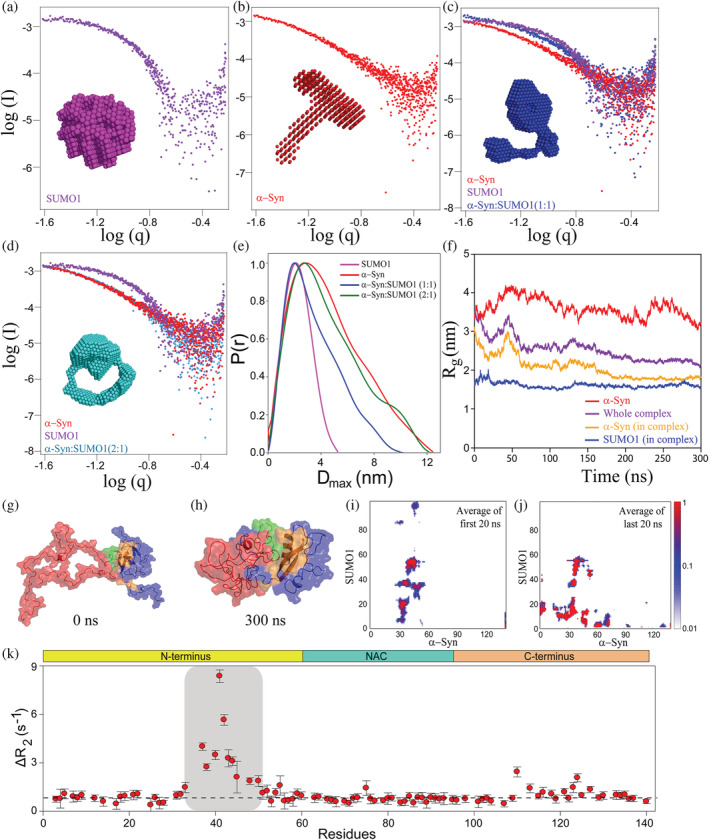
Scattering pattern of α‐Syn and SUMO1 and its complex formed at different ratios by SAXS and molecular dynamic (MD) simulation of docked structure for 300 ns and backbone dynamics of α‐Syn in the presence of SUMO1. Double logarithmic SAXS scattering pattern of SUMO1 (a), α‐Syn (b), and the complex of α‐Syn and SUMO1 obtained by mixing both the proteins at 1:1 (c), 2:1 (d). (e) The distance distribution, P(r) v/s D max curve for the complexes. (f) The radius of gyration of α‐Syn (yellow), and SUMO1 (purple) in the complex, and whole complex (blue), and free α‐Syn (red) over the period of simulation, that is, 300 ns. (g) The structure of the complex was obtained by docking of α‐Syn (red) (structure taken Protein Ensemble Database, PED; PED00024e001) and SUMO1 (blue, PDB; 1A5R) by HADDOCK. It is also the starting structure for the all‐atom Molecular Dynamics simulation. (h) The structure of the complex after 300 ns of MD simulations. The interacting region of SUMO1 and α‐Syn is highlighted in orange and green, respectively. (i) and (j) represent the average contact map of the first and last 20 ns of the 300 ns MD simulations. (k) The differential plot of R 2 of α‐Syn in the presence of SUMO1 from that in the absence of SUMO1. The ΔR 2 was higher for the residues V37‐H50, E110, E123, and A124 as compared to the remaining residues.
TABLE 1.
Radius of gyration and distance distribution of proteins and complexes obtained by SAXS.
| Proteins | Rg (nm) | D max (nm) |
|---|---|---|
| SUMO1 | 1.76 ± 0.01 | 5.3 |
| α‐Syn | 3.23 ± 0.04 | 12.5 |
| α‐Syn: SUMO1 (1:1) | 2.59 ± 0.11 | 10.2 |
| α‐Syn: SUMO1 (2:1) | 3.14 ± 0.04 | 12.5 |
2.3. Dynamics of interaction between α‐Syn and SUMO1 indicate chemical exchange at the interaction site in α‐Syn
We used an all‐atom Molecular Dynamics (MD) simulation to understand the evolution of transient interaction between α‐Syn and SUMO1 proteins. NMR experiments indicated the regions of the two proteins involved in the transient interaction. From the known information on the interaction domain in two proteins from the experiment, we performed three runs of biased docking using the HADDOCK 2.4 server (Van Zundert et al., 2016). The interacting residues of α‐Syn, V37‐A53 (37‐VLYVGSKTKEGVVHGVA‐53) and of SUMO1, 22‐IKLKVI‐27, 33‐EIHFKVKMTTHLKKLKES‐50, and R54 were used as active residues for the input in docking. Each HADDOCK run generated 40 structures categorized into 10 clusters (4 structures in each cluster) with HADDOCK scores (scoring based on the different types of energies). The variations in the HADDOCK scores of the clusters in the three runs are indicated in Table S1. Out of the generated model structures, the α‐Syn and SUMO1 complex structure with the lowest HADDOCK score and displaying maximum residues (obtained by the NMR experiment) at the interface was selected for further MD simulation. The variation in the structures obtained with respect to the selected complex structure was quantified in terms of all‐atom root mean square deviation (RMSD). A considerable number of structures had all‐atom RMSD <7 Å (Figure S5). The variation observed was mostly due to the flexible region of the SUMO1 and the non‐interacting region of α‐Syn. The selected α‐Syn and SUMO1 complex structure was further subjected to PDB‐ePISA software for the analysis of the residues at the interface of interaction. Residues involved in the hydrogen bond and salt bridge formation at the interface and the distance between the atoms are summarized in Tables S2 and S3.
The selected α‐Syn and SUMO1 complex structure was used as the starting structure for the MD simulation by CHARMM36m forcefield (Figure 3g). The simulation was performed for 300 ns, and the transient contact between the proteins remains stable (Figure S6d). To quantify that, we plotted a contact map of the proteins. In Figure 3i, the average contact map from the first 20 ns of the simulation has been plotted. It exhibits a weak interaction between the residues 80–97 from SUMO1 and residues 30–60 from α‐Syn, a region other than involved in stronger interaction (residues 37–53 (green) from α‐Syn and residues 20–55 (orange) from SUMO1 protein), obtained from the NMR experiments. In Figure 3j, the last 20 ns average contact map of the protein has been plotted, which supports that the specific and transient interaction between the two proteins is stable in the timescale of the simulation. However, other interactions are gained in the N‐terminal tail of SUMO1 with the α‐Syn protein as contacts which could be the effect of force‐field induced interactions. To test the hypothesis that the extra contact obtained for the N‐terminal of SUMO1 and α‐Syn is due to the difference in the force‐field used and validate the force‐field dependence gain of additional contacts, we ran an MD simulation with a short period of time (145 ns) using CHARMM36 forcefield. The average contact map of the first and last 20 ns of the MD simulation run for the α‐Syn and SUMO1 complex by force‐field CHARMM36 was plotted (Figure S6e). Even in this forcefield, the MD simulation run shows that the interacting region (obtained by the NMR experiments) was in contact throughout the simulation; with no other extra contacts. The important claim from the simulations is that the interacting region observed from NMR is stable in the timescale of the simulation. The Rg of two proteins, SUMO1 and α‐Syn in the complex, the whole complex, and α‐Syn from free α‐Syn simulation were plotted. Even though the Rg values seem to be fully converged, there might be other quantities not yet fully converged (Figure 3f). From our simulation, we claim that the transient interactions stemming from NMR in the observed regions are stable enough in the trajectory of 300 ns. The Rg of the SUMO1 protein in the complex remained stable at ~1.5 nm and did not change much as it is a globular protein (Figure 3f, blue curve), and the structure has negligible change. It can also be seen by comparing the intramolecular contact maps of SUMO1 in the complex in the initial structure and the final structure at the end of the simulation (Figure S6a,b).
At the same time, α‐Syn in the complex becomes compact and gains contacts in the complex (Figure S6a,c). The radius of gyration of α‐Syn (Figure 3f, yellow curve) in the complex has reduced from ~3 to ~2 nm and stabilized, thereby decreasing the Rg of the whole complex (Figure 3f, purple curve). The Rg of α‐Syn protein in the complex decreases as intra‐protein contact is gained, which is an effect of α‐Syn interaction with the SUMO1 protein (Figure S6c). The initial open structure of the protein complex can be seen in the snapshot at 0 ns in Figure 3g, and the structure gradually attains compact conformation, as observed in the snapshot at 300 ns (Figures 3f,h and Figure S6a). The gain of contacts in the α‐Syn conformation in the complex is also quantified in the contact map (Figure 3i,j), which plots the average contact map of the first 20 ns and last 20 ns of the complex. The argument of α‐Syn gaining structural compaction due to the effect of the transient interaction between the SUMO1 and α‐Syn is supported by the control simulation of free α‐Syn, that is, in the absence of SUMO1. The Rg of only α‐Syn fluctuates between 3 and 4 nm (Figure 3f, red curve). There is negligible contact developed between the residues of α‐Syn protein in the first and last 20 ns of free α‐Syn simulation (Figure S7b). Snapshots at various times in the simulation of free α‐Syn also show the open structure of the α‐Syn protein (Figure S7a).
To elucidate the effect on local mobility and backbone dynamics of α‐Syn upon SUMO1 binding, 15N longitudinal relaxation rates (R1), 15N transverse relaxation rates (R2), and steady‐state 15N‐{1H} heteronuclear NOE (HET‐NOE) were measured in the presence of 0.5 molar equivalent SUMO1. The R 1 (s−1) and 15N‐{1H} HET‐NOE values give the local mobility of the protein in the nanoseconds to the picoseconds timescale (Cavanagh et al., 1996). The average R 1 values (s−1) of α‐Syn in the absence and presence of SUMO1 were 2.02 ± 0.09 and 2.04 ± 0.11, respectively (Figure S8b). The average value of 15N‐{1H} HET‐NOE of the α‐Syn is 0.188. In the presence of SUMO1, an average value of 15N‐{1H} HET‐NOE is not altered much (0.192) (Figure S8c–e). This indicates that the interaction of SUMO1 does not affect the fast motion of the NH bond vector, i.e., ns‐ps timescale motions. The R 2 (s−1) is sensitive to the milliseconds to microseconds time‐scale motions along with the nanoseconds to picoseconds time scale motions. This was used to study the significant effect of SUMO1 on the local mobility of the α‐Syn (Cavanagh et al., 1996). The differential plot (Figure 3k) of R 2 exhibits a uniform increase for all the residues of α‐Syn, indicating an increase in the size of the α‐Syn in the complex. However, some residues (V37‐T54) of α‐Syn showed line‐broadening and reduction in intensity upon interaction with SUMO1; the average R 2 (s−1) increased from 5.44 ± 0.19 s−1 to 8.04 ± 0.38 s−1 for these residues (Figure 3k, and S8a). The differential plot also shows an increase in the R2 for E110, E123‐A124 indicating that the long‐range interaction between C‐terminus and N‐terminus residues also affects the dynamics. The drastic increase in R 2 values for the interacting residues could be due to the increased rigidity of the backbone upon interaction or due to increased chemical exchange between the bound and free form of α‐Syn.
2.4. SUMO1 delays the aggregation kinetics of α‐Syn
Furthermore, we probed into the effect of the non‐covalent interaction of SUMO1 on the aggregation kinetics of α‐Syn. α‐Syn was allowed to aggregate in the absence and presence of an equimolar ratio of SUMO1. The aggregation of α‐Syn follows a nucleation‐dependent polymerization reaction, wherein it follows a sigmoidal aggregation curve. The protein undergoes a structural transition from monomeric unstructured random coil to oligomers and protofibrils consisting of a mixture of α‐helical and β‐sheets conformation and then to mature fibrils mainly consisting of cross β‐sheets. This transition in the conformation of α‐Syn was monitored by ThT fluorescence spectroscopic assay. ThT dye intercalates between the cross β‐sheets of the fibrils and fluoresces between 460 and 500 nm, with a maximum emission occurring between 480 and 485 nm upon excitation at 450 nm (Naiki et al., 1989). During the lag phase, no ThT fluorescence was observed for α‐Syn, both in the absence and presence of SUMO1 (Figure 4a). α‐Syn shows a lag time of 20 h followed by rapid growth in the amyloid fibril formation (steep increase in the ThT fluorescence intensity) between 20 and 40 h and saturation at 60 h. α‐Syn in the presence of equimolar SUMO1 showed a delayed aggregation with a lag time of 35 h and saturation at 90 h. Thus, the presence of SUMO1 slowed down the aggregation kinetics of α‐Syn and, thereby, formation of the cross β‐structure of α‐Syn.
FIGURE 4.
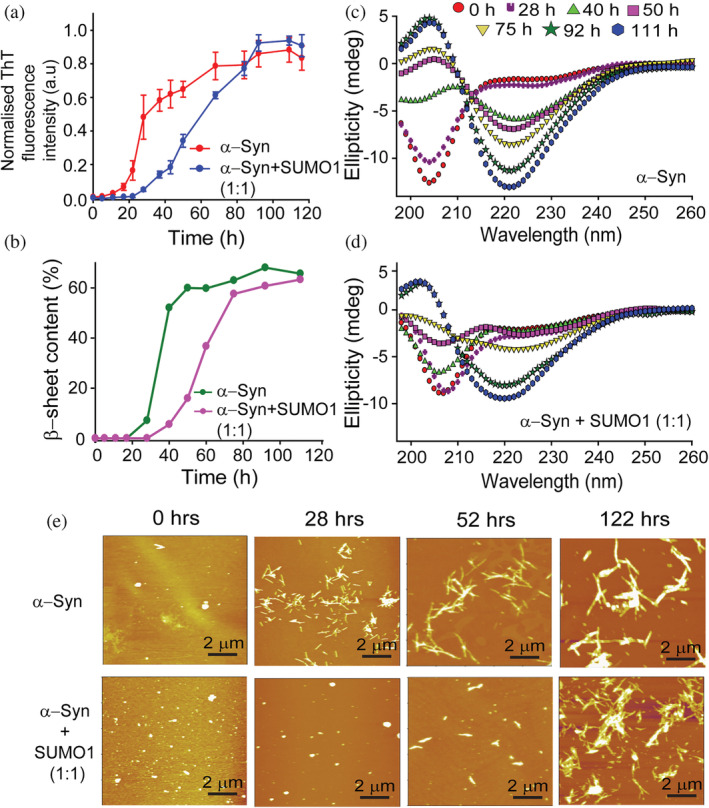
SUMO1 delays the aggregation kinetics of α‐Syn at lower pH. (a) ThT fluorescence of α‐Syn in the absence (red) and presence (blue) of SUMO1 (1:1). (b) The percentage of the β‐sheet in α‐Syn in the absence (green) and presence (pink) of equimolar SUMO1 (extracted from fitting the CD spectra by the Yang model) at different time intervals during aggregation. Far UV‐Circular Dichroism spectroscopy of α‐Syn in the absence (c) and presence (d) of SUMO1 during aggregation to monitor the transition in the secondary structure. (e) Morphological changes in α‐Syn in the absence (upper panel) and presence (lower panel) of SUMO1 during the aggregation were monitored by AFM at different time intervals.
The changes in the secondary structure of α‐Syn were monitored by Circular Dichroism (CD) spectroscopy at different time intervals during the aggregation process. α‐Syn shows minima at 204 nm indicative of random coil conformation at the initiation of aggregation. As the aggregation progressed, α‐Syn showed a prominent minima peak at 220 nm (characteristic of β‐sheet) after 40 h. In contrast, in the presence of SUMO1, a prominent peak at 220 nm was observed only after 92 h (Figure 4b–d). β‐sheet content of α‐Syn, obtained from the deconvolution of CD spectra by Yang's model, indicated the drastic increase in the β‐sheet content during the period, 28–40 h. However, in the presence of SUMO1, the content of the β‐sheets increased during 50–75 h (Figure 4b).
The morphological changes during the aggregation were monitored by atomic force microscopy (AFM). Before the initiation of the aggregation, α‐Syn was present as amorphous aggregates in the absence (Figure 4e, upper panel) and presence (Figure 4e, lower panel) of SUMO1. As the aggregation progressed, at 28 h, short, well‐dispersed fibrils were visible, whereas oligomeric species were detected in the presence of equimolar SUMO1. At 52 h, short fibrils were evident in the presence of SUMO1. α‐Syn after 122 h showed long mature fibrils. The fibrils formed in the presence of SUMO1 were shorter and dense as compared to the fibrils formed in the absence of SUMO1. SUMO1 itself did not aggregate at the above‐mentioned conditions, as shown by ThT fluorescence, CD spectroscopy, and AFM spectroscopy (Figure S9). SUMO1 did not show any significant difference in the ThT fluorescence even after 116 h (Figure S9a). The CD spectra still exhibited minima at 208 and 222 nm even after 111 h indicating the conformation did not change significantly (Figure S9b). Upon morphological characterization at different intervals of time (0, 28, and 122 h) during aggregation, SUMO1 displayed only amorphous morphologies throughout the process (Figure S9c). All the above experiments indicate the delaying effect of SUMO1 on the aggregation process of α‐Syn. A similar delay in the aggregation kinetics of α‐Syn was observed at physiological pH 7.4 (Figure S3a–c).
2.5. SUMO1 interaction with familial mutants of α‐Syn at the SBM binding site does not affect the interaction but does affect the aggregation
Familial mutants of α‐Syn have different aggregation kinetics and fibrillar structure; these mutants exhibited altered onsets and severity of the PD in diverse populations (Zhao et al., 2019; Khalaf et al., 2014; Fares et al., 2014). To investigate whether any familial mutations in the SBM2 of α‐Syn (47‐GVVHGVA‐53) alter the interaction with SUMO1, we performed 15N‐1H HSQC experiments for H50Q and G51D familial mutants of α‐Syn. 15N‐labeled H50Q and G51D α‐Syn were titrated against unlabelled SUMO1. CSP and intensity decay in a residue‐specific manner indicated that the site of interaction is unaltered due to these mutations. We also noticed that the charged residues do not play a major role in the non‐covalent interaction with SUMO1 (Figure S10a,b,d). It could be also validated by the region affected (evident by the perturbation in the chemical shift as compared to wild type α‐Syn) due to H50Q mutation (V48‐G51) and G51D (H50‐K60) (Ranjan and Kumar, 2017). The value of R 2 reporting the backbone dynamics at the interaction site significantly increased, indicating an increase in the rigidity at the interacting region of both the mutants in the SBM2 region (Figure S10c,e).
Furthermore, we checked the effect of another familial mutant, E46K, in which charge reversal occurs from the negatively charged residue, glutamic acid, to the positively charged residue lysine. E46K mutation affects the aggregation kinetics and rearranges the long‐range interactions in α‐Syn (Bhattacharyya et al., 2018). E46K mutation affects the long‐range interaction in the N‐terminus (V40‐H50) and the C‐terminus (D121‐Q134) (Ranjan and Kumar, 2017). Even though E46 is not a part of the two SBM regions in α‐Syn, it is involved in the formation of hydrogen bonds and salt bridges between α‐Syn and SUMO1 in the complex structure obtained by HADDOCK (Figure S11, Tables S2, and S3). In order to determine whether the E46K mutation has any effect on the interaction with SUMO1, HSQC titrations were carried out in a similar manner as that with other mutants at physiological pH, 7.4 (Figure S12b). The interaction of SUMO1 with residues in SBM1 remains unaltered; however, the residues in the second SBM region showed a slight decrease in intensity compared to wild‐type α‐Syn (Figure S12g), suggesting a reduced affinity of SUMO1 and α‐Syn due to E46K mutation. Nonetheless, aggregation kinetics of the E46K mutant was still delayed in the presence of SUMO1 (Figure S12c–f), similar to that in wild‐type α‐Syn. The lag time for aggregation kinetics of the E46K mutant increased from 14 to 32 h in the presence of SUMO1 at pH 7.4 (Figure S12c). It is also evident from the CD spectra that E46K protein at 14 h attains helical conformation, whereas, in the presence of SUMO1, β‐sheet formation initiates at 42 h (Figure S12d,e). Similarly, AFM images disclose the different morphology of the intermediate species of E46K mutant formed in the absence (Figure S12f, left panel) and presence of SUMO1 (Figure S12f, right panel). Nonetheless, the E46K at lower pH (pH 6.5) slightly/non‐significantly accelerates the aggregation kinetics as monitored over the progression of aggregation by ThT fluorescence, CD spectroscopy, and AFM microscopy (Figure 5a–d). The non‐differential effect on the aggregation kinetics of E46K at lower pH could be due to the weaker interaction of both the proteins, as is evident from the shift in the peak position for the residues located at the N‐terminus and C‐terminus (Figure S12a). The reduction in the intensity of the amide cross‐peaks in the SBM2 region was less as compared to that of the SBM1 region at lower pH. This was in accordance with that observed at physiological pH (Figure 5e). Significant CSP was observed for the residues at the N‐terminus (V3‐K6 and H50‐V52) and C‐terminus (E109‐A140), suggesting a weaker interaction for E46K than the wild‐type α‐Syn. Additionally, the effect on the backbone dynamics show a difference in the affinity of SUMO1 at physiological and lower pH. At lower pH, the R 2 of all the amide cross‐peaks of the E46K mutant was slightly less in the presence of 0.5 equivalent of SUMO1 compared to observed ∆R 2 values obtained at physiological pH (Figure S12h). This points toward a decrease in the rigidity of the E46K mutant of α‐Syn (Figure 5g) at lower pH.
FIGURE 5.
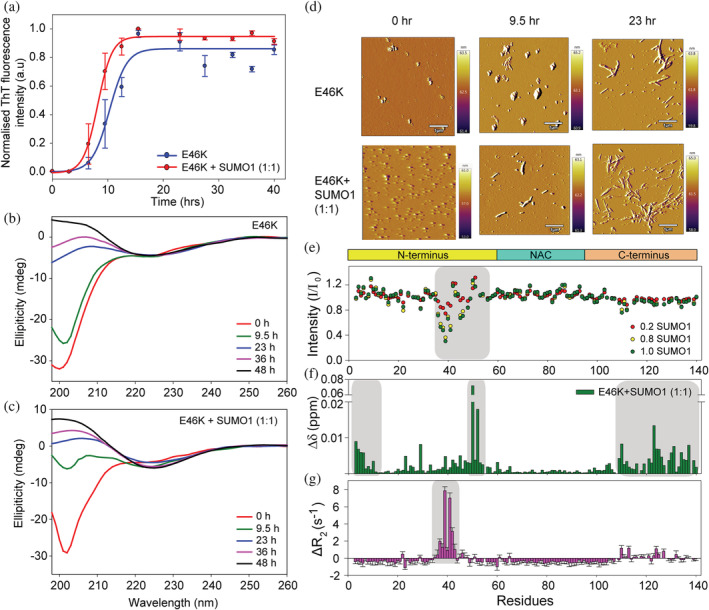
SUMO1 accelerates aggregation kinetics of familial mutant of α‐Syn, E46K at lower pH. (a) ThT fluorescence of E46K in the absence (blue) and presence (red) of SUMO1 (1:1). Far UV‐CD spectroscopy of E46K mutant in the absence (b) and presence (c) of SUMO1 during aggregation to monitor the transition in the secondary structure. (d) Morphological changes in E46K in the absence (upper panel) and presence (lower panel) of SUMO1 monitored by AFM at different time intervals during the aggregation. (e) Intensity profile of amide cross‐peaks of E46K mutant from the 15N‐1H HSQC spectra in presence of equimolar SUMO1. (f) CSP of the amide cross‐peaks of E46K mutant in the presence of equimolar SUMO1. (g) The differential plot of R 2 of E46K mutant in the presence of SUMO1 from that in the absence of SUMO1. The ΔR 2 was significantly positive for the residues in the first SBM (V37‐K43), and some residues in the C‐terminus (E110, L113, A124, and M127) as compared to the remaining residues.
3. DISCUSSION
The mode of interaction of SUMO with the SBM/SIM motifs of amyloidogenic protein through the hydrophobic core and its implication in amyloid formation is poorly understood (Kerscher, 2007; Doherty et al., 2020). Our study established that SUMO1, through its hydrophobic patch (β2 and α1), non‐covalently interacts with α‐Syn. The interaction induces compaction in α‐Syn and delays its aggregation kinetics. SUMO1 binds at the crucial N‐terminus of α‐Syn in both wild type and its familial mutants at physiological pH. It critically disrupts the β‐hairpin, loosely present in the monomeric α‐Syn. The N‐terminus has been shown to play a critical role in the aggregation and function of α‐Syn. The two specific patches in α‐Syn termed P1 (G36‐S42) and P2 (K45‐E57), have been validated to make multiple intramolecular contacts between the N‐terminus, NAC, and C‐terminus and promote the amyloid formation and hence control aggregation of α‐Syn (Doherty et al., 2020). Hang et al. showed using MD simulations that α‐ Syn and its mutants have an intrinsic propensity to form β‐hairpin in the residue stretch 38–53, where the C‐terminus interacts (Yu et al., 2015). Similarly, studies published by Hoyer's group have highlighted the importance of this β‐hairpin region. Prevention of α‐Syn aggregation by sequestration of the β‐hairpin by β‐wrapin protein AS69 (via intercalation into the β‐hairpin) using in‐vitro NMR experiments (Mirecka et al., 2014) and in neurons (Szegő et al., 2021) further validates the importance of this region in the aggregation. Even the β‐turn segment 44‐TKEG‐47 connecting the two β‐sheets in the β‐hairpin strongly affects the elongation of α‐Syn fibrils. We observed the binding of SUMO1 at the N‐terminus β‐hairpin prone region of α‐Syn consisting of β1 (V37‐K43) and β2 (V48‐T54) connected by a β‐turn (T44‐G47). This interaction delays the aggregation kinetics of α‐Syn due to the sequestration of the β‐hairpin in α‐Syn. The involvement of the hydrophobic core (β2 and α1) of SUMO1 suggests that the interaction between SUMO1 and the region of α‐Syn is dominated by hydrophobic stretches from the nascent β‐structure (Kim et al., 2007). Our observation of the effect of familial single point mutation, E46K, on the interaction of the protein with SUMO1 (note that the E46K mutation hindered the interaction of the SBM2 region with SUMO1), points toward the involvement of other types of interactions, including electrostatic interaction and hydrogen bond formation. Moreover, disruption of these polar/electrostatic interactions and hydrogen bonds affected the ability of SUMO1 to disrupt the self‐association propensity in the E46K mutant of α‐Syn.
On‐pathway oligomers of α‐Syn are more potent and toxic than their other conformations in the aggregation pathways, such as fibrils and monomers (Kim et al., 2009). They result in pore formation in the anionic phospholipids and cause neurological cell death, and various pathways have been proposed over the last two decades for α‐Syn fibrillogenesis (Kim et al., 2009; Comellas et al., 2011; Hellstrand et al., 2013). Various drugs, small molecules, and interacting partners are being discovered to delay or completely inhibit α‐Syn fibrillogenesis or the quick transformation of monomeric species to fibrils. Our study established that SUMO1 has a delaying effect on α‐Syn aggregation kinetics, and this delay could be due to the partial exposure of the hydrophobic patch of SUMO1. Recently, it was shown that peptides of SUMO1 (D15‐Q55) can completely inhibit α‐Syn aggregation (Liang et al., 2021), which could be due to better accessibility of the hydrophobic patch of the SUMO1 with the β‐hairpin prone N‐terminal region of α‐Syn, which is not favored due to steric hindrance in full‐length SUMO1.
In a boarder picture, our biophysical studies, aided by computational experiments, also explore the effect of globular partners on the conformational ensemble of intrinsically disordered proteins (IDPs). The functions of IDPs are based on the potential of the IDPs to interact with multiple partners, i.e., the avidity and promiscuous nature of IDPs (Dunker et al., 2000; Dunker et al., 2002; Brocca et al., 2020; Wright and Dyson, 2015). The disorderliness of the IDPs aids in the interactions with partners either by the “fly‐casting” hypothesis (Scheschonka et al., 2007), or by conformational selection (Klenk et al., 2006); folding upon binding, or induced fit model (Dorval et al., 2007). Upon interactions with the partners, IDPs could either undergo a disorder‐to‐order conformational change at the interaction site or remain disordered and form a fuzzy complex with the interacting partners (reviewed in Uversky, 2018). However, the lack of stable three‐dimensional structures has its downside, as IDPs are more prone to misfolding and lead to various pathological conditions such as cataracts, diabetes, and systemic amyloidosis (reviewed in Chiti and Dobson, 2006). Many IDPs such as Aβ, tau, and α‐Syn are involved in neurodegenerative diseases such as Alzheimer's, tauopathies, and α‐Synucleinopathies. Therefore, studying the complexes formed by these IDPs/IDRs and their interacting partners is crucial for understanding the functions and also disorder‐based interactions leading to the pathophysiological condition.
While the versatility of the IDPs is well recognized, the IDPs and their complexes with other proteins are difficult to categorize due to the disordered nature and variation in the type of interactions. In the last few years, the progress of experimental biophysical techniques such as high‐resolution NMR methods combined with SAXS and other techniques have provided unprecedented insight into mechanistic transitions of IDPs/IDRs (reviewed in Schneider et al., 2019). Here, we investigated the effect of the transient interaction of one such IDP, α‐Syn, with a soluble protein, SUMO1, and showed how a highly soluble globular protein modulates the self‐association propensity of an IDP involved in neurodegenerative disease. In the general context of understanding IDP functions, our results also exemplify experimental approaches to derive residue‐level detail insights into the various interactions and their functional consequences.
4. CONCLUSIONS
α‐Syn is an IDP that aggregates and acquires various conformations leading to the formation of amyloid fibrils. The nucleation/lag phase and the elongation phase during the aggregation are known to be influenced by several factors, including pH, ionic strength of the solution, temperature, point mutation, and post‐translational modification. Covalent modification by SUMO1 in CNS is essential for neuronal cell viability, connectivity, and function. SUMO1 also regulates various biochemical pathways by non‐covalent interactions. We have demonstrated here that SUMO1 interacts non‐covalently with α‐Syn, and induces structural compaction without the introduction of any stable secondary structure in α‐Syn. SUMO1 binds at the two SBM motifs, SBM1 and SBM2, of α‐Syn, which exhibit the propensity to form a β‐hairpin loop in the fibrillar form. Our data indicate transient binding with exchange occurring in the intermediate exchange regime. The SUMO1 interaction delays the aggregation of α‐Syn and affects the backbone dynamics in the millisecond to microsecond time‐scale motion. This study gives a clear insight into the effect of SUMO1 on α‐Syn aggregation. It suggests that other non‐covalent signaling mechanisms of SUMO1 may exist in regulating PD pathogenesis and its onset.
5. METHODOLOGY
5.1. Expression and purification of α‐Syn and its familial mutants
Plasmid pRK172 containing the human α‐Syn gene was transformed and expressed in Escherichia coli BL21 (DE3) cells. Purification of α‐Syn was carried out by a previously well‐established protocol with some modifications (Volles and Lansbury, 2007). For the 15N isotope labeled α‐Syn and its familial mutants E46K, H50Q, and G51D, cells were cultured in M9 minimal media using 15NH4Cl as the sole nitrogen source, and were used for NMR experiments. For unlabeled α‐Syn and its familial mutants, cells were cultured in Luria Bertani (LB) media and were used for the aggregation kinetics studies and other biophysical studies.
5.2. Preparation of α‐Syn and its familial mutants
The lyophilized α‐Syn (in 100 mM ammonium acetate) was buffer exchanged with 20 mM phosphate buffer (PB) containing 50 mM NaCl (pH 6.5/pH 7.4) in a 3 kDa cut‐off filter concentrator (3 kDa MWCO, Millipore) by centrifugation at 1200 g at 4°C. Furthermore, the α‐Syn protein solution was subjected to a pre‐washed centricon YM‐100 filter (100 kDa MWCO, Millipore) and centrifuged at 10,000 g for 20 min at 4°C for separation of low molecular weight (LMW) α‐Syn from the higher aggregates. α‐Syn thus obtained contains majorly monomeric α‐Syn. To confirm the presence of majorly monomers, the purified α‐Syn was subjected to size exclusion chromatography, and a single peak was obtained indicating a monomeric population (data not shown). The pH of α‐Syn in 20 mM PB was verified by a microelectrode pH meter (Cyberscan pH 2100, Eutech instruments). The concentration of the protein was determined by absorbance at 280 nm by spectrophotometer (BIO‐RAD, SmartSpec™ Plus), considering the molar absorptivity to be 5960 M−1 cm−1 for the α‐Syn determined from the ProtParam Software (Gasteiger et al., 2005).
5.3. Expression and purification of SUMO1
Vector pGEX‐4T1 containing the SUMO1 gene was expressed in E. coli BL21 (DE3) cells. The cells were grown to an O.D600nm of 0.8 in the LB media containing 100 μg/mL ampicillin and induced with 0.1 mM IPTG at 28°C for 8 h. The cells were harvested at 7000 g at 4°C for 15 min and resuspended in lysis buffer (20 mM PB, 1 mM EDTA, and 100 mM NaCl, pH 8.0) containing 0.01% Triton X100, 1 mM PMSF, and 1 mg/mL of lysozyme. The cells were sonicated at 5 on/7 off pulse for 20 min and centrifuged at 26,000 g at 4°C for 40 min. The supernatant was incubated with glutathione‐agarose beads for 2 h at 4°C on rotation, and the beads (containing the bound GST‐tagged SUMO1) were washed with lysis buffer. The beads were incubated with thrombin (500 units) at 21°C for 16 h for the proteolytic cleavage of GST from SUMO1 and SUMO1 was eluted. Subsequently, SUMO1 was subjected to size exclusion chromatography using Superdex 75 10/300 GL column (GE Healthcare) and concentrated in an amicon ultra‐15 3 kDa cut‐off centrifugal filter unit (3 kDa MWCO, Millipore) at 4°C. The concentration of SUMO1 was determined by absorbance at 280 nm by Spectrophotometer (BIO‐RAD, SmartSpec™ Plus), considering the molar absorptivity to be 4470 M−1 cm−1 for SUMO1 determined from the ProtParam Software (Gasteiger et al., 2005).
5.4. Nuclear magnetic resonance studies
The NMR data were acquired on a Bruker Avance III 750 MHz spectrometer with a 5 mm TXI probe. All the experiments were carried out in 20 mM phosphate buffer containing 50 mM sodium chloride (pH 6.5 or pH 7.4) at 15°C and H2O/D2O (90:10). Two‐dimensional 15N‐1H correlation heteronuclear single quantum coherent (HSQC) experiments were recorded to study the residue‐specific interaction of SUMO1 with α‐Syn and its familial mutants. Two hundred micromolars of 15N‐labeled α‐Syn and mutants were titrated against different concentrations of SUMO1 (20, 40, 80, 120, 160, and 200 μM), and 15N‐1H HSQC spectra were recorded in the absence and presence of each concentration of SUMO1. Similarly, 200 μM of 15N‐labeled SUMO1 was titrated against different concentrations of wild type α‐Syn (20, 40, 80, 120, 160, and 200 μM), and 15N‐1H HSQC spectra were recorded. The spectra thus obtained were processed by using Topspin 3.5pl6 software (BRUKER) and further analyzed by CCPNmr (Collaborative Computational Project for NMR) software (Vranken et al., 2005). Peaks were assigned using the previously published HNN and (Geiss‐Friedlander and Melchior, 2007; Saitoh and Hinchey, 2000) D‐hNCOCAnH (reduced dimensionality experiment) assignments of α‐Syn (Reddy and Hosur, 2014). The perturbation in the chemical shifts of the amide cross‐peaks due to the interaction was calculated by the following CSP formula (Cavanagh et al., 1996),
where δH and δN are the difference in the chemical shifts of amide proton and nitrogen, respectively. The ratio of the amide cross‐peak intensities, I/I 0 was also calculated where I 0 and I are the intensity of the peaks of the protein in the HSQC in the absence and presence of a particular concentration of the interacting partner, respectively.
5.5. Residue‐specific backbone dynamic studies
The effect of SUMO1 on the residue‐specific backbone dynamics of α‐Syn and its mutants were studied by 15N longitudinal relaxation rates (R 1), 15N transverse relaxation rates (R 2), and steady‐state 15N‐heteronuclear NOE (HET‐NOE) measurements. Four hundred micromolars of 15N‐labeled α‐Syn and its mutants were used for the studies of the dynamics. R 1 was measured with CPMG delays: 10, 50, 120, 250, 400, 580, 750, and 900 ms (120 and 580 ms were repeated twice). The R 2 was measured with different CPMG delays: 16.96, 33.92, 50.88, 67.84, 84.81, 118.72, 136, 153, and 187 ms (50.88 and 136 ms repeated in duplicates). The spectra thus obtained were processed by Topspin 3.5pl6 software (BRUKER). The intensities of the amide cross‐peaks were determined by CCPNmr software and data were exponentially fitted for the individual residues against the different delays. The time constant decay, T 1 and T 2 values along with the errors were determined by using CCPNmr software. The R 1 and R 1 error (ΔR 1) were calculated as follows:
Similarly, the values of R 2 and R 2 error (ΔR 2) were calculated,
where the ΔT 1 and ΔT 2 are the standard deviations obtained by the curve fitting of the T 1 and T 2 data. Steady‐state 15N‐{1H} HET‐NOE was measured with proton saturation of 3 s and a relaxation delay of 2 s. For the spectra without proton saturation, the relaxation delay used was 5 s. Steady‐state 15N‐{1H} HET‐NOE and its error (σ NOE) was calculated by the following formulae,
where I nonsat and I sat are the peak intensities in the absence and presence of proton saturation and σ nonsat and σ sat are the root mean square values of the noises in the spectra for peaks in the absence and presence of the proton saturation.
5.6. Surface plasmon resonance
The interaction strength between the wild type α‐Syn and SUMO1 was determined by the SPR spectroscopy (BIAcore T200, GE Healthcare). CM5 sensor chip which has a carboxymethylated dextran matrix coated on a gold film was used. During immobilization of the ligand, α‐Syn, the derivatized carboxyl groups on the sensor chip undergoes amine‐coupling, that is; it is linked with the amine groups in α‐Syn. An immobilization level of 1600 response units (RU) for α‐Syn (0.5 mg/mL) on the CM5 sensor chip in 10 mM sodium acetate buffer (pH 4.0) was achieved. Increasing concentrations of the analyte, SUMO1 (6.25–400 μM), was passed over immobilized α‐Syn at a flow rate of 30 μL/min with the association and the dissociation time of 90 and 210 s, respectively. The RU of the blank run and the reference surface was subtracted from the response unit of the samples. The RU was plotted against the concentration of SUMO1 and was fitted by a 1:1 Langmuir isotherm fit to determine the dissociation constant.
5.7. Size exclusion chromatography
The complex was prepared by mixing the two proteins, α‐Syn and SUMO1 at different ratios (α‐Syn: SUMO1 = 1:1, and 2:1). To separate the complex formed from the individual proteins, the protein mixture was passed through Superdex™ 75 10/300 GL gel filtration column (Cytiva). The purified α‐Syn and SUMO1 were also passed through the column to determine the elution volume of α‐Syn and SUMO1. The profiles were overlapped to determine the elution volume of the complex formed.
5.8. Analytical ultracentrifugation
We used analytical ultracentrifugation to estimate the fraction of the complex formed between α‐Syn and SUMO1. The concentration of α‐Syn, SUMO1, and its complex formed at different molar ratios (1:1, and 2:1) used for AUC was determined based on its absorbance at 280 nm (A 280nm = 1.0). The sedimentation velocity of α‐Syn, SUMO1, and its complex formed at different molar ratios were measured at 20°C using a Beckman Coulter Optima analytical ultracentrifuge equipped with a UV–Visible detection system. An‐60 Ti four‐hole rotor with a double‐sector cell which had a 12 mm Epon centerpiece and quartz windows, was taken for absorbance measurement. We filled 400–420 μL of protein (right sector) and buffer (left sector) in the sector cells. The samples were first centrifuged at 5000 rpm (2000 g) for 15 min to stabilize the absorbance and check for the leakage of the sample in the assembled cell. It was followed by equilibration for 2 h to attain a stable temperature, and the speed was increased to 45,000 rpm (16,300 g). Absorbance data at 280 nm for the sample displacement profile was collected at an interval of 150 s with a radial increment of 0.001 cm for 375 scans in the continuous scanning mode. The data was analyzed using SEDFIT software (Brown and Schuck, 2006) to obtain the molecular weight, Stokes's radius, frictional ratio, and sedimentation coefficient (s 20w) of the individual proteins and the complexes formed. For all the measurements, the parameters of the solvent condition were corrected to s 20w which is the standard solvent condition in water at 20°C.
5.9. Small‐angle X‐ray scattering
Small‐angle X‐ray scattering (SAXS) data of α‐Syn and SUMO1 and the complexes formed at different molar ratios were collected at Xenocs SAS instrument (model: Xeuss 2.0) using an Eiger R1M with vacuum fed through a set high‐resolution hybrid pixel photon‐counting detector. The scattering pattern was measured with an exposure time of 2.5 h at 298 K. The distance between the sample‐to‐detector was 0.54 m, and the range of scattering vector(s) covered was 0.017–0.59 A−1. The concentration of the proteins and the complexes used for SAXS were 7–8 mg/mL. The scattering pattern of the buffer was also recorded at the same condition. The data of the samples were subtracted from that of the buffer scattering pattern after normalization against the concentration in the PRIMUS software (available in ATSAS 3.0 package) (Manalastas‐Cantos et al., 2021). The radius of gyration was evaluated by the Guinier approximation. Additionally, the maximum dimension (diameter), D max, and interatomic distance distribution function, P(r) were determined by the distance distribution module in PRIMUS. The SAXS scattering pattern was generated for the SUMO1 structure (PDB; 1A5R) and α‐Syn structure (PED; PED00024e001) by CRYSOL software (available in ATSAS 3.0 package) (Manalastas‐Cantos et al., 2021). The simulated scattering data were overlapped and compared with that of the experimental scattering profile.
5.10. HADDOCK docking and molecular dynamics simulations
α‐Syn and SUMO1 complex structure was modeled by biased docking of SUMO1 (PDB; 1A5R) and α‐Syn (PED; PED00024e001) in HADDOCK 2.4 (Van Zundert et al., 2016). For the docking, the residues involved in the interaction determined by NMR experiments were used as active residues. For α‐Syn, residues V37‐A53 (37‐VLYVGSKTKEGVVHGVA‐53) were selected as active residues, and for SUMO1, residues 22‐IKLKVI‐27, 33‐EIHFKVKMTTHLKKLKES‐50, and R54 were used. Three runs of HADDOCK docking were carried out. Each HADDOCK run generated 10 clusters of structures with 4 structures in each cluster. Out of the generated model structures, the α‐Syn/SUMO1 complex structure with the lowest HADDOCK score and displaying maximum residues at the interface was selected for further MD simulation. The obtained complex structures contained one molecule of α‐Syn and one molecule of SUMO1 protein.
All‐atom Molecular Dynamics (MD) simulation was carried out to understand the evolution of interaction between the α‐Syn and SUMO1. The simulation was performed using the MD simulation package NAMD 2.15 (Phillips et al., 2020), and the force field used in the simulation was CHARMM36m (Huang et al., 2017). The structure was solvated within a box with a layer of water 10 Å in each direction for the complex and ionized to neutralize the system of water and protein. The ionized system was energy minimized and then heated gradually to 310 K with an increase of 0.01 K each step. The simulation was performed in 310 K at 1 atm pressure with electrostatic interaction, and the van der Waals interactions cut‐off was given as 12 Å. Runtime of the simulation was 300 ns with a time‐step of 2 fs. We also simulated only α‐Syn protein in a box as a control. The PDB structure of the α‐Syn was taken from the protein ensemble database (PED; PED00024e001) (Lazar et al., 2021). The simulation setup used was similar to the simulation setup of the complex, except the simulation box was layered with water molecules to a 15 Å thickness in each direction from the protein. The simulation was performed for a total of 300 ns, same as the complex. The snapshots were taken from the Visual Molecular dynamics (VMD) software package (Humphrey et al., 1996) and PyMOL.
5.11. Aggregation kinetics of α‐Syn
The aggregation kinetics, secondary‐structural transition, and the temporal changes in the morphology of the α‐Syn and its familial mutant E46K in the absence and presence of equivalent SUMO1 were monitored by ThT fluorescence assay, CD spectroscopy, and AFM, respectively during aggregation. Two hundred micromolars of α‐Syn in 20 mM PB buffer containing 50 mM NaCl and 0.01% sodium azide (pH 6.5 and pH 7.4) were incubated in a vial at 37°C on a rotary shaker for end‐to‐end rotation at 60 rpm. To understand the role of SUMO1 on the aggregation kinetics of α‐Syn and its familial mutant, 200 μM of wild‐type α‐Syn and E46K were incubated with equimolar SUMO1. Two hundred micromolars of SUMO1 were also incubated under identical conditions as a control. The aggregation was initiated at 37°C on a rotary shaker and was continued for 120 h. The aggregation studies were conducted in triplicates.
5.12. Thioflavin T fluorescence assay
During aggregation, 5 μL of the protein solution was taken from the aggregation mixture at different intervals of time and was diluted to 200 μL in the 20 mM phosphate buffer (pH 6.5 and pH 7.4) containing 50 mM NaCl to obtain a concentration of 5 μM. Ten microliters of 1 mM thioflavin T (ThT) solution were added and ThT fluorescence was monitored on a Horiba‐Jobin Yvon fluorimeter. The emission spectrum from 460 to 500 nm was recorded upon excitation at 450 nm. ThT fluorescence at 485 nm (emission maxima) was plotted against the incubation time. The lag time (t lag) for the aggregation of α‐Syn in the absence and presence of SUMO1 was calculated by the equations (Willander et al., 2012):
where y is the ThT fluorescence intensity at a particular point of aggregation curve, y 0 is the ThT fluorescence at t 0 (initiation of aggregation), y max is the maximum ThT fluorescence, k is the rate of aggregation, and t 1/2 is the time at the 50% of the y max.
5.13. Circular dichroism spectroscopy
Fifteen microliters of 200 μM of protein solution was aliquoted at different intervals of time and diluted to 200 μL in 20 mM phosphate buffer, pH 6.5 and pH 7.4 containing 50 mM NaCl and 0.01% sodium azide to obtain a concentration of 15 μM. The CD spectra of the diluted samples were acquired over the wavelength 198–260 nm (far UV region) in a 0.1 cm path length quartz cuvette (Starna, Hainault, London) in JASCO‐810 CD spectrometer at room temperature. Three independent readings were recorded, and the signals were averaged. The raw data was subtracted from that of the buffer for the α‐Syn, and the spectra were processed by smoothening using the Spectra Manager software. For the samples containing α‐Syn and SUMO1, the spectra were subtracted by the signal contributed by SUMO1 in the same buffer.
5.14. Atomic force microscopy
The temporal changes in the morphology of the α‐Syn were monitored using AFM (Asylum Research, Santa Barbara, CA). Eight microliters of the protein solution was taken out at different intervals during aggregation and diluted to obtain a final concentration of 40 μM. The protein solution was spotted on a freshly cleaved and smoothened uniform surface of the mica sheet. The samples were incubated for 15 min at room temperature, washed with autoclaved and filtered double‐distilled water, and dried under vacuum for 1–2 h. The imaging was performed at 2–3 randomly selected areas with silicon nitride cantilever in tapping mode at a frequency of 300 kHz.
AUTHOR CONTRIBUTIONS
Rajlaxmi Panigrahi: Conceptualization (equal); formal analysis (lead); investigation (lead); methodology (lead); software (lead); validation (lead); visualization (lead); writing – original draft (lead). Rakesh Krishnan: Formal analysis (supporting); investigation (supporting); software (supporting); validation (supporting); writing – original draft (supporting). Jai Shankar Singh: Methodology (supporting); writing – review and editing (supporting). Ranjith Padinhateeri: Supervision (supporting); writing – review and editing (supporting). Ashutosh Kumar: Conceptualization (equal); funding acquisition (lead); methodology (supporting); supervision (lead); writing – review and editing (lead).
FUNDING INFORMATION
This work was supported by Ramalingaswamy re‐entry fellowship (BT/RLF/Re‐entry/22/2010) from the Department of Biotechnology, and Science and Engineering Research Board (EMR/2016/002798), Government of India to Ashutosh Kumar. Rajlaxmi Panigrahi, Jai Shankar Singh, and Rakesh Krishnan are grateful to MHRD (Government of India) for their fellowship.
CONFLICT OF INTEREST STATEMENT
The authors declare no conflicts of interest.
Supporting information
Appendix S1: Supporting Information
ACKNOWLEDGMENTS
The authors acknowledge HFNMR 750 MHz, Bio‐AFM and Surface Plasmon Resonance, SAXS, and AUC facility, funded by RIFC, IRCC, and IIT Bombay. We thank Dr. Ambuja Navalkar and Dr. Shivangi Shukla for their useful inputs in the analysis of SPR data. We thank Parveen Sehrawat for his aid in setting up the analytical ultracentrifugation experiments. We thank Prof. R.V. Hosur and Dr. Priyatosh Ranjan for the useful suggestions during the preparation of the manuscript. We thank Dr. Mandar Bopardikar for the critical reading of the manuscript and Nitin Kachariya for the critical reading and the suggestions in the preparation of the manuscript and the figures.
Panigrahi R, Krishnan R, Singh JS, Padinhateeri R, Kumar A. SUMO1 hinders α‐Synuclein fibrillation by inducing structural compaction. Protein Science. 2023;32(5):e4632. 10.1002/pro.4632
Review Editor: Carol Beth Post
DATA AVAILABILITY STATEMENT
Data available on request from the authors
REFERENCES
- Abeywardana T, Pratt MR. Extent of inhibition of α‐synuclein aggregation in vitro by SUMOylation is conjugation site‐and SUMO isoform‐selective. Biochemistry. 2015;54:959–61. [DOI] [PubMed] [Google Scholar]
- Auluck PK, Caraveo G, Lindquist S. α‐Synuclein: membrane interactions and toxicity in Parkinson's disease. Annu Rev Cell Dev Biol. 2010;26:211–33. [DOI] [PubMed] [Google Scholar]
- Bayer P, Arndt A, Metzger S, Mahajan R, Melchior F, Jaenicke R, et al. Structure determination of the small ubiquitin‐related modifier SUMO‐1. J Mol Biol. 1998;280:275–86. [DOI] [PubMed] [Google Scholar]
- Bertoncini CW, Jung Y‐S, Fernandez CO, Hoyer W, Griesinger C, Jovin TM, et al. Release of long‐range tertiary interactions potentiates aggregation of natively unstructured α‐synuclein. Proc Natl Acad Sci U S A. 2005;102:1430–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharyya D, Kumar R, Mehra S, Ghosh A, Maji SK, Bhunia A. Multitude NMR studies of α‐synuclein familial mutants: probing their differential aggregation propensities. Chem Commun. 2018;54:3605–8. [DOI] [PubMed] [Google Scholar]
- Brocca S, Grandori R, Longhi S, Uversky V. Liquid–liquid phase separation by intrinsically disordered protein regions of viruses: roles in viral life cycle and control of virus–host interactions. Int J Mol Sci. 2020;21:9045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown PH, Schuck P. Macromolecular size‐and‐shape distributions by sedimentation velocity analytical ultracentrifugation. Biophys J. 2006;90:4651–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavanagh J, Fairbrother WJ, Palmer AG III, Skelton NJ. Protein NMR spectroscopy: principles and practice. California, United States of America: Academic Press; 1996. [Google Scholar]
- Chiti F, Dobson CM. Protein misfolding, functional amyloid, and human disease. Annu Rev Biochem. 2006;75:333–66. [DOI] [PubMed] [Google Scholar]
- Chu X‐P, Xiong Z‐G. Physiological and pathological functions of acid‐sensing ion channels in the central nervous system. Curr Drug Targets. 2012;13:263–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comellas G, Lemkau LR, Nieuwkoop AJ, Kloepper KD, Ladror DT, Ebisu R, et al. Structured regions of α‐synuclein fibrils include the early‐onset Parkinson's disease mutation sites. J Mol Biol. 2011;411:881–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comellas G, Lemkau LR, Zhou DH, George JM, Rienstra CM. Structural intermediates during α‐synuclein fibrillogenesis on phospholipid vesicles. J Am Chem Soc. 2012;134:5090–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dedmon MM, Lindorff‐Larsen K, Christodoulou J, Vendruscolo M, Dobson CM. Mapping long‐range interactions in α‐synuclein using spin‐label NMR and ensemble molecular dynamics simulations. J Am Chem Soc. 2005;127:476–7. [DOI] [PubMed] [Google Scholar]
- Doherty CP, Ulamec SM, Maya‐Martinez R, Good SC, Makepeace J, Khan GN, et al. A short motif in the N‐terminal region of α‐synuclein is critical for both aggregation and function. Nat Struct Mol Biol. 2020;27:249–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorval V, Fraser PE. Small ubiquitin‐like modifier (SUMO) modification of natively unfolded proteins tau and α‐synuclein. J Biol Chem. 2006;281:9919–24. [DOI] [PubMed] [Google Scholar]
- Dorval V, Mazzella MJ, Mathews PM, Hay RT, Fraser PE. Modulation of Aβ generation by small ubiquitin‐like modifiers does not require conjugation to target proteins. Biochem J. 2007;404:309–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunker AK, Romero P, Obradovic Z, Garner EC, Brown CJ. Intrinsic protein disorder in complete genomes. Genome Inform. 2000;11:161–71. [PubMed] [Google Scholar]
- Dunker AK, Brown CJ, Lawson JD, Iakoucheva LM, Obradović Z. Intrinsic disorder and protein function. Biochemistry. 2002;41:6573–82. [DOI] [PubMed] [Google Scholar]
- Eliezer D, Kutluay E, Bussell R, Browne G. Conformational properties of α‐synuclein in its free and lipid‐associated states. J Mol Biol. 2001;307:1061–73. [DOI] [PubMed] [Google Scholar]
- Fares M‐B, Ait‐Bouziad N, Dikiy I, Mbefo MK, Jovičić A, Kiely A, et al. The novel Parkinson's disease linked mutation G51D attenuates in vitro aggregation and membrane binding of α‐synuclein, and enhances its secretion and nuclear localization in cells. Hum Mol Genet. 2014;23:4491–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasteiger E, Hoogland C, Gattiker A, Duvaud SE, Wilkins MR, Appel RD, et al. Protein identification and analysis tools on the ExPASy server. Berlin, Germany: Springer; 2005. [Google Scholar]
- Geiss‐Friedlander R, Melchior F. Concepts in sumoylation: a decade on. Nat Rev Mol Cell Biol. 2007;8:947–56. [DOI] [PubMed] [Google Scholar]
- Goedert M. Alpha‐synuclein and neurodegenerative diseases. Nat Rev Neurosci. 2001;2:492–501. [DOI] [PubMed] [Google Scholar]
- Hasegawa M, Fujiwara H, Nonaka T, Wakabayashi K, Takahashi H, Lee VM‐Y, et al. Phosphorylated α‐synuclein is ubiquitinated in α‐synucleinopathy lesions. J Biol Chem. 2002;277:49071–6. [DOI] [PubMed] [Google Scholar]
- Hay RT. SUMO: a history of modification. Mol Cell. 2005;18:1–12. [DOI] [PubMed] [Google Scholar]
- Hecker C‐M, Rabiller M, Haglund K, Bayer P, Dikic I. Specification of SUMO1‐and SUMO2‐interacting motifs. J Biol Chem. 2006;281:16117–27. [DOI] [PubMed] [Google Scholar]
- Hellstrand E, Nowacka A, Topgaard D, Linse S, Sparr E. Membrane lipid co‐aggregation with α‐synuclein fibrils. PLoS One. 2013;8:e77235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez SM, Tikhonova EB, Karamyshev AL. Protein‐protein interactions in alpha‐synuclein biogenesis: new potential targets in Parkinson's disease. Front Aging Neurosci. 2020;12:72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodara R, Norris EH, Giasson BI, Mishizen‐Eberz AJ, Lynch DR, Lee VM‐Y, et al. Functional consequences of α‐Synuclein tyrosine nitration diminished binding to lipid vesicles and increased fibril formation. J Biol Chem. 2004;279:47746–53. [DOI] [PubMed] [Google Scholar]
- Huang J, Rauscher S, Nawrocki G, Ran T, Feig M, De Groot BL, et al. CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat Methods. 2017;14:71–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph. 1996;14:33–8. [DOI] [PubMed] [Google Scholar]
- Jo E, McLaurin J, Yip CM, George‐Hyslop PS, Fraser PE. α‐Synuclein membrane interactions and lipid specificity. J Biol Chem. 2000;275:34328–34. [DOI] [PubMed] [Google Scholar]
- Kerscher O. SUMO junction—what's your function? New insights through SUMO‐interacting motifs. EMBO Rep. 2007;8:550–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalaf O, Fauvet B, Oueslati A, Dikiy I, Mahul‐Mellier A‐L, Ruggeri FS, et al. The H50Q mutation enhances α‐synuclein aggregation, secretion, and toxicity. J Biol Chem. 2014;289:21856–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HY, Heise H, Fernandez CO, Baldus M, Zweckstetter M. Correlation of amyloid fibril β‐structure with the unfolded state of α‐synuclein. Chembiochem. 2007;8:1671–4. [DOI] [PubMed] [Google Scholar]
- Kim H‐Y, Cho M‐K, Kumar A, Maier E, Siebenhaar C, Becker S, et al. Structural properties of pore‐forming oligomers of α‐synuclein. J Am Chem Soc. 2009;131:17482–9. [DOI] [PubMed] [Google Scholar]
- Kim YM, Jang WH, Quezado MM, Oh Y, Chung KC, Junn E, et al. Proteasome inhibition induces α‐synuclein SUMOylation and aggregate formation. J Neurol Sci. 2011;307:157–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klenk C, Humrich J, Quitterer U, Lohse MJ. SUMO‐1 controls the protein stability and the biological function of phosducin. J Biol Chem. 2006;281:8357–64. [DOI] [PubMed] [Google Scholar]
- Krüger R, Kuhn W, Müller T, Woitalla D, Graeber M, Kösel S, et al. AlaSOPro mutation in the gene encoding α‐synuclein in Parkinson's disease. Nat Genet. 1998;18:106–8. [DOI] [PubMed] [Google Scholar]
- Krumova P, Weishaupt JH. Sumoylation in neurodegenerative diseases. Cell Mol Life Sci. 2013;70:2123–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krumova P, Meulmeester E, Garrido M, Tirard M, Hsiao H‐H, Bossis G, et al. Sumoylation inhibits α‐synuclein aggregation and toxicity. J Cell Biol. 2011;194:49–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunadt M, Eckermann K, Stuendl A, Gong J, Russo B, Strauss K, et al. Extracellular vesicle sorting of α‐Synuclein is regulated by sumoylation. Acta Neuropathol. 2015;129:695–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lashuel HA, Overk CR, Oueslati A, Masliah E. The many faces of α‐synuclein: from structure and toxicity to therapeutic target. Nat Rev Neurosci. 2013;14:38–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazar T, Martínez‐Pérez E, Quaglia F, Hatos A, Chemes LB, Iserte JA, et al. PED in 2021: a major update of the protein ensemble database for intrinsically disordered proteins. Nucleic Acids Res. 2021;49:D404–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Z, Chan HYE, Lee MM, Chan MK. A SUMO1‐derived peptide targeting SUMO‐interacting motif inhibits α‐Synuclein aggregation. Cell Chem Biol. 2021;28(180–190):180–190.e6. [DOI] [PubMed] [Google Scholar]
- Loriol C, Parisot J, Poupon G, Gwizdek C, Martin S. Developmental regulation and spatiotemporal redistribution of the sumoylation machinery in the rat central nervous system. PLoS One. 2012;7:e33757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manalastas‐Cantos K, Konarev PV, Hajizadeh NR, Kikhney AG, Petoukhov MV, Molodenskiy DS, et al. ATSAS 3.0: expanded functionality and new tools for small‐angle scattering data analysis. J Appl Cryst. 2021;54:343–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marotta NP, Lin YH, Lewis YE, Ambroso MR, Zaro BW, Roth MT, et al. O‐GlcNAc modification blocks the aggregation and toxicity of the protein α‐synuclein associated with Parkinson's disease. Nat Chem. 2015;7:913–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirecka EA, Shaykhalishahi H, Gauhar A, Akgül Ş, Lecher J, Willbold D, et al. Sequestration of a β‐hairpin for control of α‐Synuclein aggregation. Angew Chem Int Ed. 2014;53:4227–30. [DOI] [PubMed] [Google Scholar]
- Naiki H, Higuchi K, Hosokawa M, Takeda T. Fluorometric determination of amyloid fibrils in vitro using the fluorescent dye, thioflavine T. Anal Biochem. 1989;177:244–9. [DOI] [PubMed] [Google Scholar]
- Nishioka K, Hirano M, Stoessl A, Yoshino H, Imamichi Y, Ikeda A, et al. Homozygous alpha‐synuclein a53v in familial parkinson's disease. J Neurol Sci. 2017;381:161. [DOI] [PubMed] [Google Scholar]
- Oh Y, Kim YM, Mouradian MM, Chung KC. Human Polycomb protein 2 promotes α‐synuclein aggregate formation through covalent SUMOylation. Brain Res. 2011;1381:78–89. [DOI] [PubMed] [Google Scholar]
- Paleologou KE, Oueslati A, Shakked G, Rospigliosi CC, Kim H‐Y, Lamberto GR, et al. Phosphorylation at S87 is enhanced in synucleinopathies, inhibits α‐synuclein oligomerization, and influences synuclein‐membrane interactions. J Neurosci. 2010;30:3184–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasanen P, Myllykangas L, Siitonen M, Raunio A, Kaakkola S, Lyytinen J, et al. A novel α‐synuclein mutation A53E associated with atypical multiple system atrophy and Parkinson's disease‐type pathology. Neurobiol Aging. 2014;35:2180.e1–5. [DOI] [PubMed] [Google Scholar]
- Phillips JC, Hardy DJ, Maia JD, Stone JE, Ribeiro JV, Bernardi RC, et al. Scalable molecular dynamics on CPU and GPU architectures with NAMD. J Chem Phys. 2020;153:044130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, et al. Mutation in the α‐synuclein gene identified in families with Parkinson's disease. Science. 1997;276:2045–7. [DOI] [PubMed] [Google Scholar]
- Pountney D, Chegini F, Shen X, Blumbergs P, Gai W. SUMO‐1 marks subdomains within glial cytoplasmic inclusions of multiple system atrophy. Neurosci Lett. 2005;381:74–9. [DOI] [PubMed] [Google Scholar]
- Proukakis C, Dudzik CG, Brier T, MacKay DS, Cooper JM, Millhauser GL, et al. A novel α‐synuclein missense mutation in Parkinson disease. Neurology. 2013;80:1062–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranjan P, Kumar A. Perturbation in long‐range contacts modulates the kinetics of amyloid formation in α‐synuclein familial mutants. ACS Chem Nerosci. 2017;8:2235–46. [DOI] [PubMed] [Google Scholar]
- Reddy JG, Hosur RV. A reduced dimensionality NMR pulse sequence and an efficient protocol for unambiguous assignment in intrinsically disordered proteins. J Biomol NMR. 2014;59:199–210. [DOI] [PubMed] [Google Scholar]
- Rott R, Szargel R, Shani V, Hamza H, Savyon M, Elghani FA, et al. SUMOylation and ubiquitination reciprocally regulate α‐synuclein degradation and pathological aggregation. Proc Natl Acad Sci U S A. 2017;114:201704351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruffin VA, Salameh AI, Boron WF, Parker MD. Intracellular pH regulation by acid‐base transporters in mammalian neurons. Front Physiol. 2014;5:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitoh H, Hinchey J. Functional heterogeneity of small ubiquitin‐related protein modifiers SUMO‐1 versus SUMO‐2/3. J Biol Chem. 2000;275:6252–8. [DOI] [PubMed] [Google Scholar]
- Sato H, Arawaka S, Hara S, Fukushima S, Koga K, Koyama S, et al. Authentically phosphorylated α‐synuclein at Ser129 accelerates neurodegeneration in a rat model of familial Parkinson's disease. J Neurosci. 2011;31:16884–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheschonka A, Tang Z, Betz H. Sumoylation in neurons: nuclear and synaptic roles? Trends Neurosci. 2007;30:85–91. [DOI] [PubMed] [Google Scholar]
- Schneider R, Jensen MR, Blackledge M. Experimental studies of binding of intrinsically disordered proteins to their partners. Intrinsically Disordered Proteins. San Diego, CA, USA: Elsevier; 2019. p. 139–87. [Google Scholar]
- Schuck P. Analytical ultracentrifugation as a tool for studying protein interactions. Biophys Rev. 2013;5:159–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song J, Durrin LK, Wilkinson TA, Krontiris TG, Chen Y. Identification of a SUMO‐binding motif that recognizes SUMO‐modified proteins. Proc Natl Acad Sci U S A. 2004;101:14373–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song J, Zhang Z, Hu W, Chen Y. Small ubiquitin‐like modifier (SUMO) recognition of a SUMO binding motif a reversal of the bound orientation. J Biol Chem. 2005;280:40122–9. [DOI] [PubMed] [Google Scholar]
- Spillantini MG, Crowther RA, Jakes R, Hasegawa M, Goedert M. α‐Synuclein in filamentous inclusions of Lewy bodies from Parkinson's disease and dementia with Lewy bodies. Proc Natl Acad Sci U S A. 1998;95:6469–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulzer D, Edwards RH. The physiological role of α‐synuclein and its relationship to Parkinson's disease. J Neurochem. 2019;150:475–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szegő ÉM, Boß F, Komnig D, Gärtner C, Höfs L, Shaykhalishahi H, et al. A β‐Wrapin targeting the N‐terminus of α‐Synuclein monomers reduces fibril‐induced aggregation in neurons. Front Neurosci. 2021;15:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Um JW, Chung KC. Functional modulation of parkin through physical interaction with SUMO‐1. J Neurosci Res. 2006;84:1543–54. [DOI] [PubMed] [Google Scholar]
- Uversky VN. Intrinsic disorder, protein–protein interactions, and disease. Adv Protein Chem Struct Biol. 2018;110:85–121. [DOI] [PubMed] [Google Scholar]
- Van Zundert G, Rodrigues J, Trellet M, Schmitz C, Kastritis P, Karaca E, et al. The HADDOCK2. 2 web server: user‐friendly integrative modeling of biomolecular complexes. J Mol Biol. 2016;428:720–5. [DOI] [PubMed] [Google Scholar]
- Vaynberg J, Qin J. Weak protein–protein interactions as probed by NMR spectroscopy. Trends Biotechnol. 2006;24:22–7. [DOI] [PubMed] [Google Scholar]
- Volles MJ, Lansbury PT. Relationships between the sequence of α‐synuclein and its membrane affinity, fibrillization propensity, and yeast toxicity. J Mol Biol. 2007;366:1510–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vranken WF, Boucher W, Stevens TJ, Fogh RH, Pajon A, Llinas M, et al. The CCPN data model for NMR spectroscopy: development of a software pipeline. Proteins Struct Funct Bioinform. 2005;59:687–96. [DOI] [PubMed] [Google Scholar]
- Willander H, Presto J, Askarieh G, Biverstål H, Frohm B, Knight SD, et al. BRICHOS domains efficiently delay fibrillation of amyloid β‐peptide. J Biol Chem. 2012;287:31608–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson MP. Using chemical shift perturbation to characterise ligand binding. Prog Nucl Magn Reson Spectrosc. 2013;73:1–16. [DOI] [PubMed] [Google Scholar]
- Wright PE, Dyson HJ. Intrinsically disordered proteins in cellular signalling and regulation. Nat Rev Mol Cell Biol. 2015;16:18–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshino H, Hirano M, Stoessl AJ, Imamichi Y, Ikeda A, Li Y, et al. Homozygous alpha‐synuclein p. A53V in familial Parkinson's disease. Neurobiol Aging. 2017;57:248.e7–248.e12. [DOI] [PubMed] [Google Scholar]
- Yu H, Han W, Ma W, Schulten K. Transient β‐hairpin formation in α‐synuclein monomer revealed by coarse‐grained molecular dynamics simulation. J Chem Phys. 2015;143:12B623_621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarranz JJ, Alegre J, Gómez‐Esteban JC, Lezcano E, Ros R, Ampuero I, et al. The new mutation, E46K, of α‐synuclein causes parkinson and Lewy body dementia. Ann Neurol. 2004;55:164–73. [DOI] [PubMed] [Google Scholar]
- Zhao K, Li Y, Liu Z, Long H, Zhao C, Luo F, et al. Parkinson's disease associated mutation E46K of α‐synuclein triggers the formation of a novel fibril structure. bioRxiv. 2019;11:870758. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1: Supporting Information
Data Availability Statement
Data available on request from the authors