

Abstract
The excitatory neurotransmitter glutamate shapes learning and memory, but the underlying epigenetic mechanism of glutamate regulation in neuron remains poorly understood. Here, we showed that lysine demethylase KDM6B was expressed in excitatory neurons and declined in hippocampus with age. Conditional knockout of KDM6B in excitatory neurons reduced spine density, synaptic vesicle number and synaptic activity, and impaired learning and memory without obvious effect on brain morphology in mice. Mechanistically, KDM6B upregulated vesicular glutamate transporter 1 and 2 (VGLUT1/2) in neurons through demethylating H3K27me3 at their promoters. Tau interacted and recruited KDM6B to the promoters of Slc17a7 and Slc17a6, leading to a decrease in local H3K27me3 levels and induction of VGLUT1/2 expression in neurons, which could be prevented by loss of Tau. Ectopic expression of KDM6B, VGLUT1, or VGLUT2 restored spine density and synaptic activity in KDM6B-deficient cortical neurons. Collectively, these findings unravel a fundamental mechanism underlying epigenetic regulation of synaptic plasticity and cognition.
Keywords: epigenetics, gene regulation, learning and memory, synaptic plasticity, KDM6B
INTRODUCTION
Excitatory neurons control release of the neurotransmitter glutamate from synaptic vesicles to finely modulate synaptic plasticity and cognitive functions in the brain. However, the underlying fundamental mechanisms remain poorly understood. Accumulating evidence has implicated an important role of epigenetic reprogramming in learning and memory. Abnormalities in DNA methylation cause progressive neurologic disorders with intellectual impairment including Rett syndrome1 and alter expression of brain-derived neurotrophic factor required for cognitive functions and long-term memory formation2. It has been previously shown that increased histone acetylation restores learning and memory3 but loss of acetyl lysine 16 of histone H4 in the lateral temporal lobe links to aging and Alzheimer’s disease (AD)4. Trimethyl lysine 27 of histone H3 (H3K27me3) is also elevated in the cortex of AD patients5. However, its significance in cognitive functions remains unknown.
Lysine demethylase 6B (KDM6B) belongs to a Jumonji domain-containing protein family that specifically demethylates H3K27me36–8. Numerous studies have demonstrated a critical role of KDM6B in development, stem cell differentiation, neurogenesis, inflammation, and cancer9–11. KDM6B knockout (KO) mice die at birth due to a respiratory problem12, 13. KDM6B is also involved in neurological diseases. For example, KDM6B promotes blood-spinal cord barrier disruption and neuronal cell death by inducing matrix metalloproteases after spinal cord injury14. KDM6B is induced by ischemic stroke and inhibiting its expression prevents neuronal cell death from stroke15. Recent work showed that genetic variants of the KDM6B gene are associated with neurodevelopmental delays and intellectual disability16, suggesting KDM6B as a risk gene for intellectual disability17. However, little is known about the role of KDM6B in synaptic plasticity and cognitive functions in adult brain under physiological conditions.
In the present study, we identified a physiological role of KDM6B in learning and memory using genetically modified mouse models. KDM6B is mainly expressed in neurons and cooperates with microtubule-associated protein Tau to induce the expression of vesicular glutamate transporter 1 (VGLUT1) and VGLUT2 through its demethylase activity, thereby increasing the dendritic spine density and vesicle number and improving learning and memory in mice. These findings define a fundamental mechanism underlying epigenetic regulation of synaptic activity contributing to learning and memory.
METHODS
Details are provided in Supplementary Materials, including plasmids, animals, co-immunoprecipitation (Co-IP), 5-Bromo-2’-deoxyuridine (BrdU) injection, electrophysiological recording, animal behavioral tests, statistics and reproducibility.
RESULTS
Characterization of the spatial and temporal expression of KDM6B in mouse brain during normal aging process
To analyze the spatial and temporal expression of KDM6B in the murine brain, we first dissected each brain region from 2-month-old mice. We found that Kdm6b mRNA was ubiquitously expressed in the entire mouse brain with relatively high levels in the cortex (Ctx), cerebellum (Cb) and olfactory bulb (OB) (Fig. S1A). Kdm6b mRNA was increased in the cortex during postnatal development with a peak at postnatal day 21 (P21) and then reduced to a relatively stable level during the adulthood until P360 (Fig. S1B). Interestingly, Kdm6b mRNA level in the hippocampus was relatively high at birth (Fig. S1C). Then it was gradually reduced during postnatal development from P0 to P21 and maintained at a relatively stable level during young adulthood but further decreased at P360 (12-month) age (Figs. S1B, C). Similarly, its paralog Kdm6a had a ubiquitous expression pattern in mouse brain with the relatively high levels in the cortex, olfactory bulb, and cerebellum (Fig. S1A). Kdm6a expression peaked at P21 in the cortex and at P14 in the hippocampus but maintained at a relatively stable level during the normal aging process (Figs. S1B, C). Next, we assessed the cell type-specific expression of KDM6B in primary cultures of cortical neurons, astrocytes, and microglial cells. Distinct to Kdm6a that was ubiquitously expressed in neurons, microglia, and astrocytes, Kdm6b was predominantly expressed in neurons (Fig. S1D). The purity of these three cell types was confirmed by their respective markers including NeuN, Iba1 and GFAP (Fig. S1E). These results suggest that KDM6B is mainly expressed in neurons and declined in the hippocampus with age.
Postnatal deletion of KDM6B in excitatory neurons has a minimal impact on mouse brain morphology
To study the function of KDM6B in neurons, we generated KDM6B conditional knockout (cKO) mice by crossing Kdm6bfl/fl mice with transgenic CamKIIα-iCre mice (Figs. S2A, B), where CamKIIα-iCre is highly expressed in excitatory neurons at the cortex and hippocampus postnatally18, 19. Kdm6bfl/fl; CamKIIα-iCre+ mice survived well without obvious abnormality. Kdm6bfl/fl mice were also crossed with Nestin-Cre mice (Fig. S2C), where Nestin-Cre is broadly expressed in brain including both neurons and glial cells20. However, Kdm6bfl/fl;Nestin-Cre+ pups died at birth due to respiratory defects similar to KDM6B KO mice12, 13. In the current study, Kdm6bfl/fl mice crossed with Nestin-Cre mice were mainly used for embryonic neuronal cultures and Kdm6bfl/fl mice crossed with CamKIIα-iCre mice were used as KDM6B cKO models to study in vivo KDM6B functions specifically in excitatory neurons.
We found that Kdm6b mRNA was reduced by about 80–85% in the cortex and hippocampus but not in the cerebellum of KDM6B cKO mice (Fig. S2D), suggesting that KDM6B is mainly expressed in excitatory neurons in the forebrain. Consistently, H3K27me3, a specific substrate of KDM6B6, 7, was increased in the cortex and hippocampus but not in the cerebellum of cKO mice (Figs. S2E, F). As compared with their wild-type (WT) littermates (Kdm6bfl/fl), KDM6B cKO mice appeared to have normal body weight and brain weight at 2 months of age (Figs. S2G–I). Furthermore, no gross abnormality of cortex layers and hippocampus morphology was found in KDM6B cKO mice at 2 months of age (Fig. S2J). The number of NeuN+ neurons at the different layers of the cortex and the different regions of hippocampus was similar between KDM6B cKO and WT mice (Figs. S3A–D). Likewise, Iba1+ microglia and GFAP+ astrocytes were also comparable in the cortex and hippocampus between KDM6B cKO and WT mice (Figs. S3E–J), suggesting that KDM6B deletion in excitatory neurons does not obviously activate microglia or astrocytes in mice. Collectively, these results indicate that KDM6B cKO mice have the intact global structure of the somatosensory cortex and hippocampus with little if any changes of the numbers of neurons, astrocytes, and microglia.
KDM6B cKO impairs learning and memory in mice.
To investigate the physiological consequences of KDM6B ablation in excitatory neurons, we performed a battery of behavioral tests in 2–4-month-old KDM6B cKO mice and their WT littermates. In the open-field test, KDM6B cKO mice showed normal spontaneous activity without obvious anxiety as indicated by similar total moving distance and time spent in the inner zone compared with WT mice (Figs. 1A, C). There was also no difference between KDM6B cKO and WT mice in the total moving distance and time spent in open-arms of the elevated plus maze test (Figs. 1D, E). Moreover, motor coordination, balance, and motor learning skills all appeared to be intact in KDM6B cKO mice as measured by the latency of animal on the accelerating rotarod (Fig. 1F). These behavioral results indicate that KDM6B cKO mice have the normal gross locomotor activity with no signs of anxiety.
Fig. 1. KDM6B cKO mice display learning and memory-associated behavioral deficits.
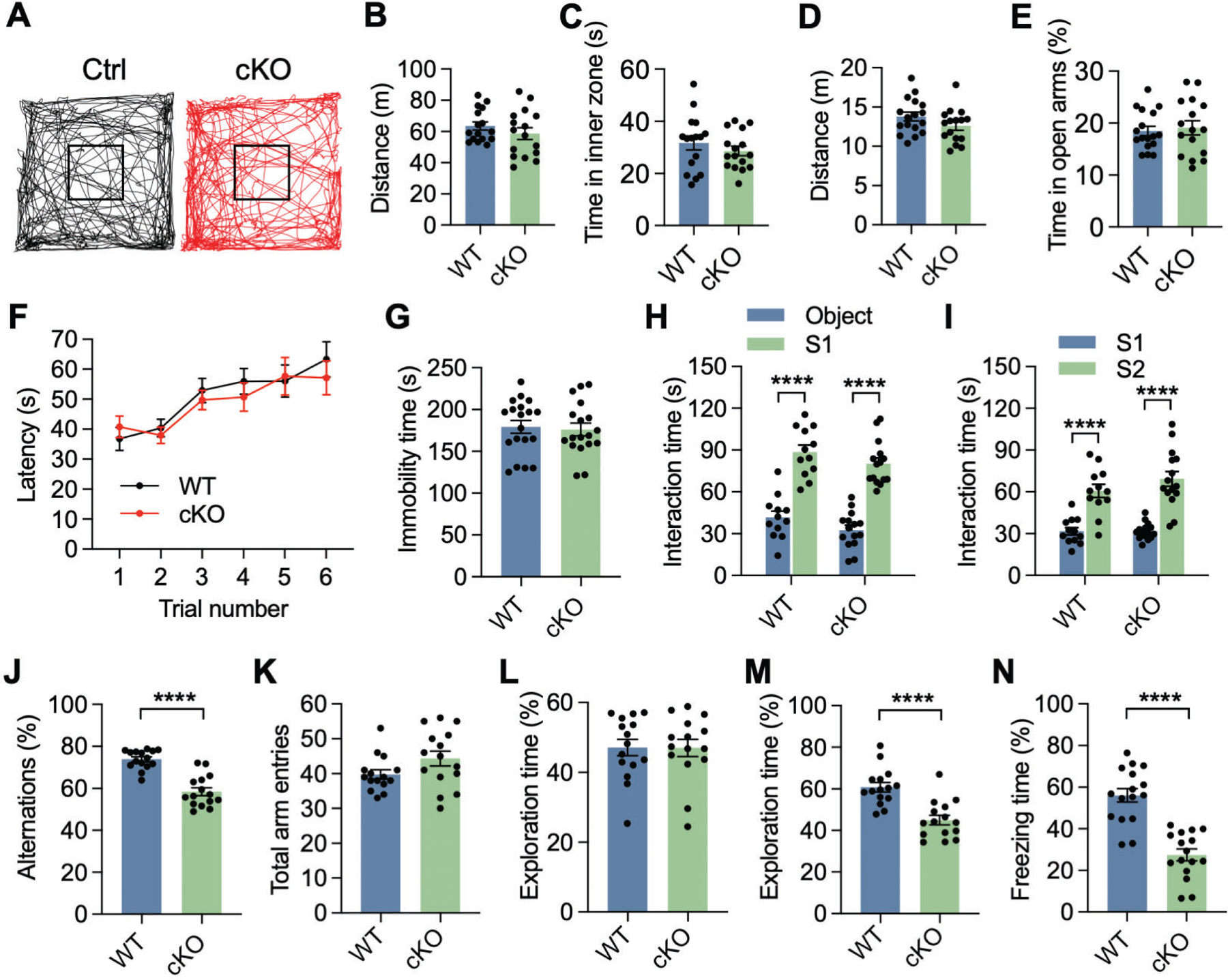
(A) Representative traces of locomotor activity in KDM6B cKO mice and control (Ctrl) littermates during the first 10 min of open field test. (B, C) Open field test showing no difference in the moving distance (B) and duration time in the inner zone (C) between KDM6B cKO and WT mice. n = 16 mice per group. Two-tailed Student’s t test. (D, E) Elevated plus maze test showing no difference in moving distance (D) and duration time in the open arms (E) between KDM6B cKO and WT mice. n = 17 (WT mice) and 16 (KDM6B cKO mice). Two-tailed Student’s t test. (F) Accelerating rotarod test showing no difference in the latency to fall off between KDM6B cKO and WT mice. n = 20 (WT mice) and 19 (KDM6B cKO mice). Two-way ANOVA with Tukey’s multiple comparison test. (G) Tail suspension test showing no difference in the immobility time between KDM6B cKO and WT mice. n = 19 (WT mice) and 18 (KDM6B cKO mice). Two-tailed Student’s t test. (H, I) KDM6B cKO mice display normal social interaction in the three-chamber test. The interaction time in the first (H) and second (I) phases was quantified. S1, mouse 1; S2, stranger mouse. n = 12 (WT mice) and 15 (KDM6B cKO mice). Two-way ANOVA with Sidak’s multiple comparison test. (J, K) KDM6B cKO mice display working memory deficits in the Y maze test. Spontaneous alternations (J) and total arm entries (K) were quantified in KDM6B cKO and WT mice. n = 15 mice per group. Two-tailed Student’s t test. (L, M) KDM6B cKO mice display recognition memory deficits in the novel object recognition test. The percentage of the exploration time on a single object during the familiarization (L) and novel object during the test sessions (M) was quantified. n = 15 mice per group. Two-tailed Student’s t test. (N) KDM6B cKO mice display associative learning deficits in the contextual fear conditioning tests. n = 16 mice for each group. Two-tailed Student’s t test. Data were presented as mean ± s.e.m; ****p < 0.0001.
In the tail suspension test, KDM6B cKO mice displayed a similar immobility time compared with WT mice (Fig. 1G), suggesting that KDM6B cKO mice do not have a depressive-like behavior. Next, social interaction of KDM6B cKO and WT mice was assessed by the three-chamber sociability and social novelty test. In the sociability phase, both KDM6B cKO and WT mice showed a higher preference for the social partner (S1) than the empty cage (Fig. 1H). In the social novelty phase, a novel partner (S2) was introduced into the chamber to replace the empty cage. KDM6B cKO mice displayed a higher preference for the novel partner S2, as WT mice did (Fig. 1I). These results indicate that KDM6B cKO mice have normal social memory with no signs of depressive-like behavior.
To determine whether KDM6B cKO impairs learning and memory, KDM6B cKO mice were subjected to Y maze, object recognition, and contextual fear conditioning tests that are often used to study spatial working memory, recognition memory, and associative learning, respectively21–23. In the Y maze test, KDM6B cKO mice showed a reduction in spontaneous alternation of the arm entries (Fig. 1J), while the number of total arm entries was comparable (Fig. 1K). In the familiarization phase of object recognition test, both KDM6B cKO and WT mice showed a similar preference for two identical objects (Fig. 1L). However, KDM6B cKO mice did not display a preference for the novel object as indicated by a reduced exploration time than that of WT mice in the test phase when a novel object was introduced into the chamber to replace one of two objects (Fig. 1M). In the fear conditioning test, mice were first trained in a conditioning chamber for 120 s habituation and then subjected to parings of conditioned stimulus (an auditory cue) and an unconditioned stimulus with three sets of electric foot shocks (2 s, 0.5 mA, inter-trial interval of 60–90 s). On the next day, mice were placed back to the same conditioning chamber for 5 minutes but without foot shocks. Freezing responses were significantly reduced in KDM6B cKO mice (Fig. 1N), indicating impaired associative learning. These observations reveal learning and memory deficits in KDM6B cKO mice.
KDM6B cKO reduces vesicle numbers and synaptic activity in mice
Dendritic complexity is critical for building the neuronal circuit, receiving inputs and transforming signals into cognitive learning and spatial memory24. To determine whether KDM6B loss alters neuron dendritic complexity, thereby causing cognitive dysfunction, we cultured KDM6B WT and cKO cortical neurons from Kdm6bfl/fl and Kdm6bfl/fl;Nestin Cre+ embryos at E18 respectively and compared the axon length of KDM6B KO and WT neurons at day in vitro (DIV) 3. The length of Tau-labeled axon was comparable between KDM6B KO and WT neurons (Figs. 2A, B). The dendritic arborization complexity was traced in KDM6B KO and WT neurons at DIV18 by Image J. Sholl analysis revealed that the dendritic complexities of KDM6B KO neurons were not different from those of WT neurons (Figs. 2C, D).
Fig. 2. KDM6B cKO reduces synaptic activity and vesicle numbers in mice.
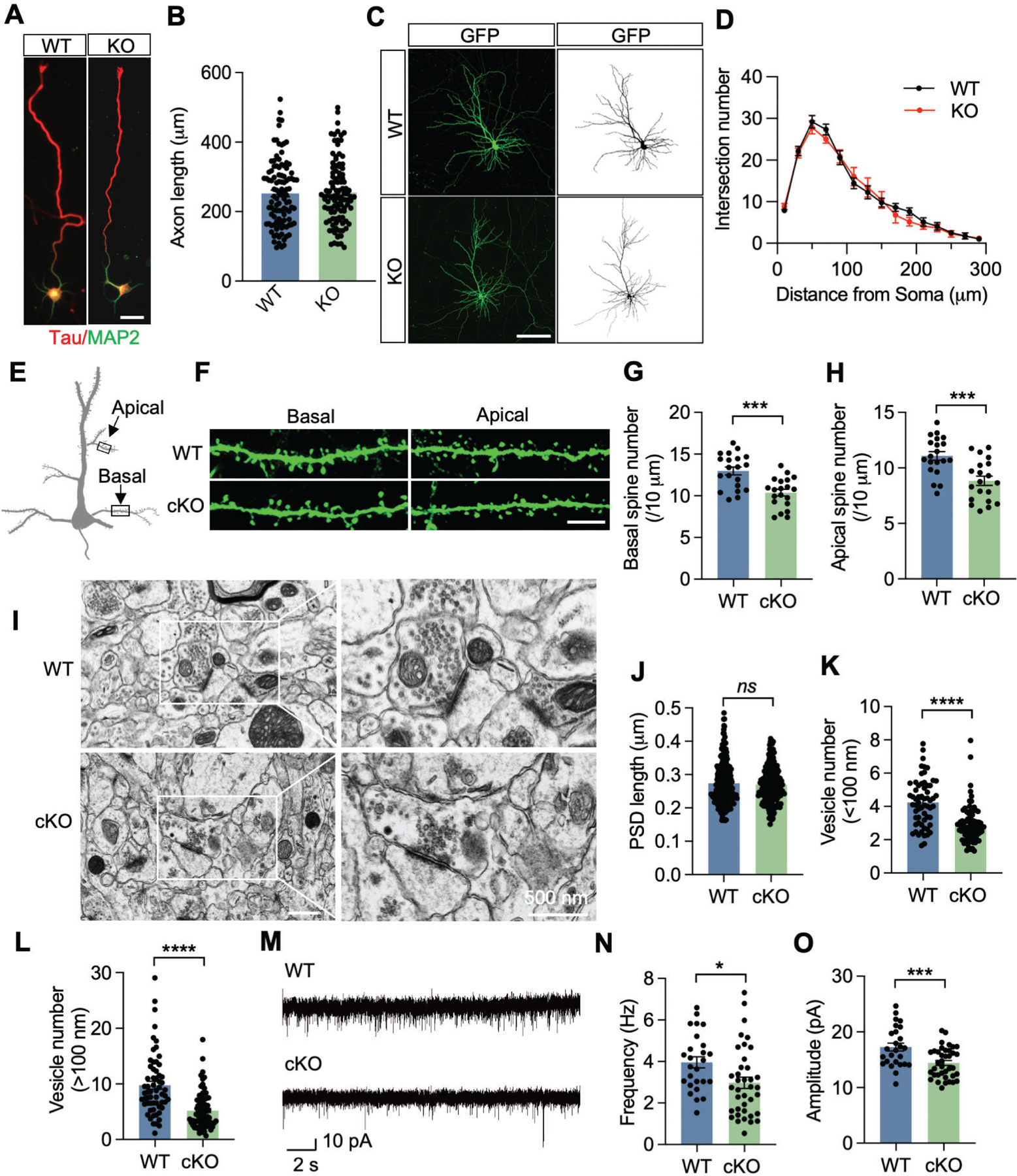
(A, B) Representative images of primary WT and KDM6B KO cortical neurons at day 3 in vitro (DIV 3, A). Axon was labeled with anti-Tau antibody (red), dendrites was labeled with anti-MAP2 antibody (green). Scale bar, 20 μm. The axon length was quantified (B). Axons of neurons were defined as the longest neurite at that time. n = 105 (WT) and 106 (KDM6B KO). Two-tailed Student’s t test. (C, D) Representative images of primary WT and KDM6B KO cortical neurons at DIV 18 (C). Neurons were transfected at DIV 6 with GFP plasmid and immunostained with anti-GFP antibody at DIV 18 (left). The binary picture (right) was used for Sholl analysis at 20 μm concentric circles around the soma (D). Scale bar, 100 μm. n = 16 neurons for each group. Two-tailed Student’s t test. (E-H) Scheme (E) and representative GFP staining images (F) of basal and apical dendrites of layer 5 pyramidal neurons from WT and KDM6B cKO mice crossed with Thy1-GFP mice. Scale bar, 5 μm. The spine numbers in basal and apical dendrites were quantified in G and H, respectively. n = 20 neurons from 3 WT or KDM6B cKO mice. Two-tailed Student’s t test. (I) Representative electron microscope images of the synaptic contacts with presynaptic vesicles and postsynaptic densities in WT and KDM6B cKO mouse brain. Scale bar, 500 nm. (J) Quantitative analysis of the PSD length. n = 220 synapses from 3 WT mice. n = 211 synapses from 3 KDM6B cKO mice. Two-tailed Student’s t test. ns, not significant. (K, L) Quantitative analysis of synaptic vesicle number in the proximal (K) or distant (L) active zone. n = 61 synapses from 3 WT mice. n = 82 synapses from 3 KDM6B cKO mice. Two-tailed Student’s t test. (M) Representative traces of sEPSCs in layer V pyramidal neurons from WT and KDM6B cKO mice. (N, O) Quantification of sEPSC frequency (N) and amplitude (O). n = 26 cells from 3 WT mice, n = 38 from 5 KDM6B cKO mice; Two tailed Student’s t test. Data were presented as mean ± s.e.m. *p < 0.05; ***p < 0.001; ****p < 0.0001.
Changes in density and shape of dendritic spines are also associated with learning and memory25. To determine whether KDM6B regulates spine density to alter synaptic functions critical for learning and memory, we crossed KDM6B cKO and WT mice with Thy1-GFP mice26 and quantified GFP-labeled spines in cortex layer V pyramidal neurons. The spine density of both basal and apical dendrites was significantly reduced in KDM6B cKO mice compared with WT mice (Figs. 2E–H).
Synaptic vesicles store and release neurotransmitters critical for synaptic activity and control cognitive functions27. Thus, we examined the synapse structure and vesicle numbers using transmission electron microscope. Analysis of the vesicle number in the presynapse of cortical layer V neurons revealed a significant reduction in KDM6B cKO mice, whereas the length of post synaptic density (PSD) was comparable in KDM6B cKO and WT mice (Figs. 2I–L). We next assessed whether KDM6B cKO alters the synaptic activity in the brain by measuring the spontaneous excitatory postsynaptic currents (sEPSC) in layer V pyramidal neurons. The amplitude and frequency both were significantly decreased in brain slices prepared from KDM6B cKO mice (Figs. 2M–O), indicating impaired synaptic activity in KDM6B cKO mice. In contrast, KDM6B deficiency failed to alter the intrinsic electrophysiological properties of neurons including resting membrane potential, input resistance, and action potential firing in response to injected currents (Figs. S4A–C), indicating that the changes in sEPSC are likely not action potential dependent. To further determine whether the reduced synaptic transmission is due to altered neurotransmitter receptors, we isolated the PSD from the cortex of KDM6B cKO and WT mice. The levels of NMDA receptors (GluN1, GluN2A, and GluN2B) and AMPA receptors (GluA1 and GluA2) were not affected by KDM6B cKO in the PSD fraction (Fig. S4D, E). The purity of the PSD fraction was confirmed by the expression of PSD95 and synaptophysin (SYP) (Figs. S4D, E). These results exclude a role of the composition of both NMDA and AMPA receptor subunits as the cause of KDM6B cKO-induced loss of synaptic activity.
Adult hippocampal neurogenesis has been implicated to modulate learning and memory in mice28. KDM6B has been known to play an important role in postnatal olfactory bulb neurogenesis10. To test whether KDM6B deletion interferes with adult neurogenesis causing cognitive dysfunction, we injected BrdU (100 mg/kg, i.p. injection/6 h) into 2-month-old KDM6B cKO and WT mice. Mouse brains were collected 12 hours after injection. The number of BrdU+ cells was similar in the dentate gyrus of KDM6B cKO mice compared with that of WT mice (Figs. S5A, B). Likewise, a cell proliferation marker Ki67 and an immature neuron marker doublecortin (Dcx) were equally expressed in the hippocampus of WT and KDM6B cKO mice (Figs. S5A, C, D). These results exclude a role of hippocampal neurogenesis as the cause of KDM6B loss-mediated learning and memory deficits in mice.
Collectively, these findings indicate that KDM6B plays an essential role in maintenance of vesicle number, spine density, and synaptic activity in excitatory neurons.
KDM6B controls VGLUT1 and VGLUT2 expression in neurons through its demethylase activity
To address molecular mechanisms by which KDM6B regulates the vesicle number, spine density, synaptic activity, and cognitive functions, we performed an unbiased whole-genome RNA-seq in primary KDM6B KO and WT neuron cultures, which were prepared from Kdm6bfl/fl and Kdm6bfl/fl;Nestin-Cre+ embryos at E18. 150 genes were significantly downregulated while 112 genes were upregulated in KDM6B KO neurons (FDR < 0.05, logCPM > 0, fold change > 1.5. Figs. 3A, B, Source Data 1). Since KDM6B functions as a demethylase of histone H3K27me3, a repressor marker for gene transcription9, 10, we mainly focused on downregulated genes in KDM6B KO neurons. The gene ontology analysis of these downregulated genes identified enrichment of pathways involving regulation of transport and vesicle mediated transport (Fig. 3C, Source Data 2). Importantly, a synaptic vesicle transporter gene, Slc17a6 (encoding VGLUT2), was the top hit among downregulated genes in KDM6B KO neurons (Fig. 3A). Slc17a7 (encoding VGLUT1), another synaptic vesicle transporter gene, was also repressed in KDM6B KO neurons (Fig. 3A). qRT-PCR analysis confirmed that Slc17a7 and Slc17a6 were significantly reduced in KDM6B KO neurons (Fig. 3D), which validated bulk RNA-seq results. In contrast, synaptophysin and PSD95 mRNAs were not regulated by KDM6B KO in cortical neurons (Fig. 3D), indicating the specific regulation of VGLUT1/2 by KDM6B. Consistently, protein expression of VGLUT1/2 was downregulated in the cortex and hippocampus but not cerebellum from KDM6B cKO mice (Figs. 3E–I). Together, these findings indicate that KDM6B controls the expression of VGLUT1/2 in cortical and hippocampal neurons both in vitro and in vivo.
Fig. 3. Identification of VGLUT1 and VGLUT2 as KDM6B main target genes.
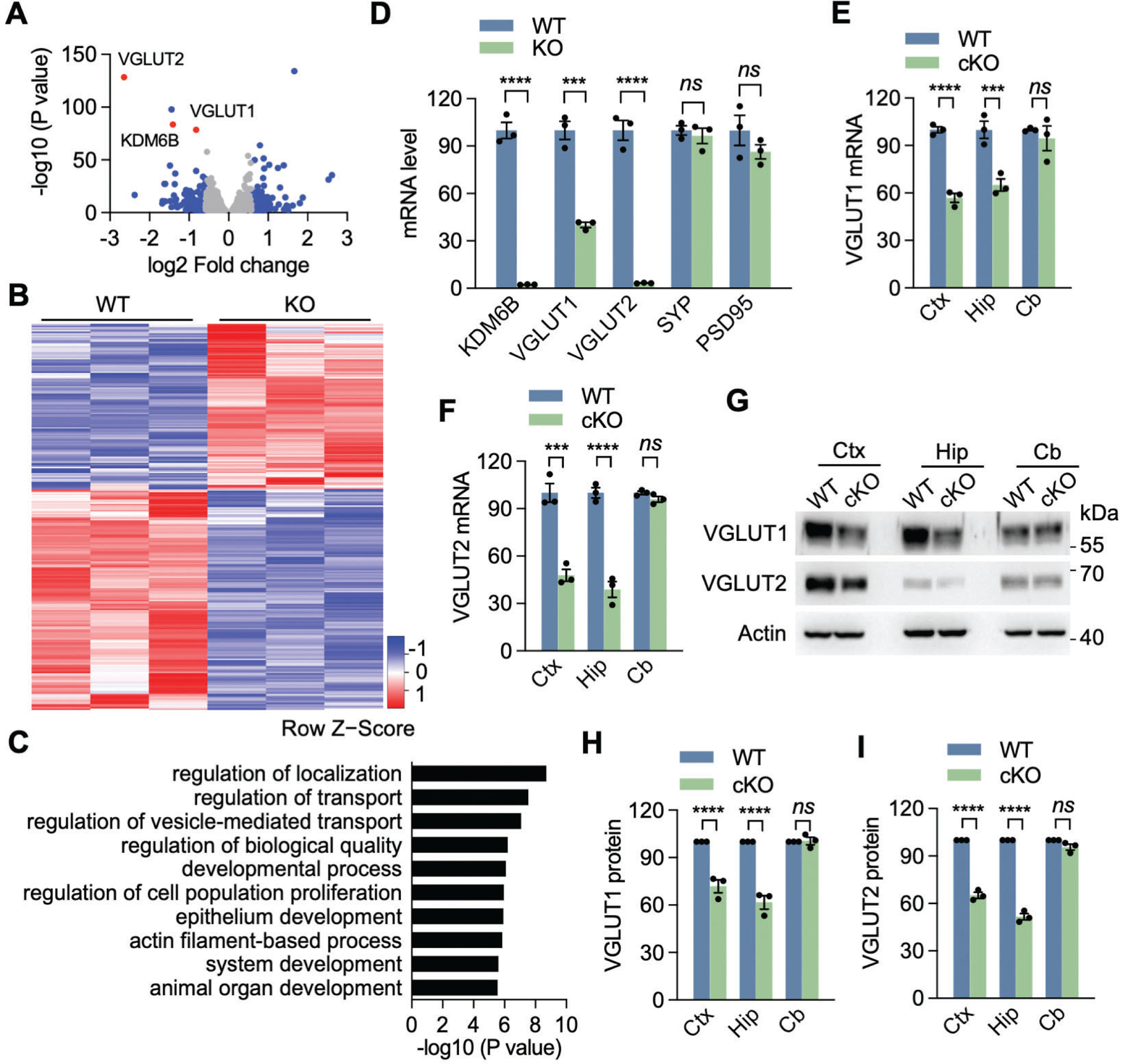
(A) Volcano plot showing differentially expressed genes in KDM6B KO neurons compared with WT neurons. Each dot represents a gene. Blue dots are up- or down-regulated genes with more than 1.5-fold change. Three selected genes are denoted and highlighted in red. (B) Heatmap showing expression levels (in z scores) of up- or down-regulated genes with more than 1.5-fold change in KDM6B KO neurons by RNA-seq. n = 3. (C) Gene ontology analysis of downregulated genes in KDM6B KO neurons using PANTHER classification system. The top 10 enriched biological process terms are shown. The full list of enriched biological process terms can be found in Source Data 2. (D) qRT-PCR analysis of KDM6B, VGLUT1, VGLUT2, SYP and PSD95 expression in KDM6B KO and WT neurons. n = 3 biological replicates in each group. Two-tailed Student’s t test. ns, not significant. (E, F) qRT-PCR analysis of VGLUT1 (E) and VGLUT2 (F) expression in KDM6B cKO and WT mice. Ctx, cortex; Hip, hippocampus; Cb, cerebellum. n = 3 mice each group. Two-way ANOVA with Sidak’s multiple comparison test. (G-I) Immunoblot analysis of VGLUT1 and VGLUT2 protein expression in KDM6B cKO and WT mice. The intensity of VGLUT1 (H) and VGLUT2 (I) bands was quantified and normalized to β-actin. n = 3 mice each group. Two-way ANOVA with Sidak’s multiple comparison test. Data were presented as mean ± s.e.m. **p < 0.01. ***p < 0.001; ****p < 0.0001.
To determine whether the demethylase activity of KDM6B is required for VGLUT1/2 expression, we infected cultured cortical neurons with lentivirus encoding WT JmjC domain of KDM6B (KDM6B-C-WT, aa 1144–1682) or catalytically inactive mutant H1390A29. In line with a previous study showing that this C-terminal KDM6B truncate mimics the function of full-length KDM6B protein8, we found that KDM6B-C-WT phenocopied full-length KDM6B and was sufficient to demethylate H3K27me3 in transfected 293T cells (Figs. S6A, B). In contrast, H1390A mutant (KDM6B-C-mut) failed to do so (Figs. S6A, B). The functional activity of KDM6B-C-WT and KDM6B-C-mut on H3K27me3 demethylation was further confirmed in primary neurons (Figs. S7A, B). Expression of KDM6B-C-WT significantly increased both mRNA and protein levels of VGLUT1 and VGLUT2 in cortical neurons (Figs. 4A–C, S7C–E). H3K27me3 but not histone H3 levels were decreased when KDM6B-C-WT was expressed in neurons (Figs. 4A, D). In contrast, KDM6B-C-mut failed to influence the expression of VGLUT1, VGLUT2, and H3K27me3 in cortical neurons (Figs. 4A–C, S7C–E), indicating that the expression of VGLUT1 and VGLUT2 requires the KDM6B demethylase activity. Importantly, KDM6B-C-WT but not KDM6B-C-mut restored the expression of VGLUT1 and VGLUT2 as well as H3K27me3 in KDM6B KO neurons (Figs. 4A–D, S7C–E). These results reveal that KDM6B controls the expression of VGLUT1 and VGLUT2 in neurons through its H3K27me3 demethylase activity.
Fig. 4. KDM6B induces VGLUT1 and VGLUT2 expression through its H3K27me3 demethylase activity.
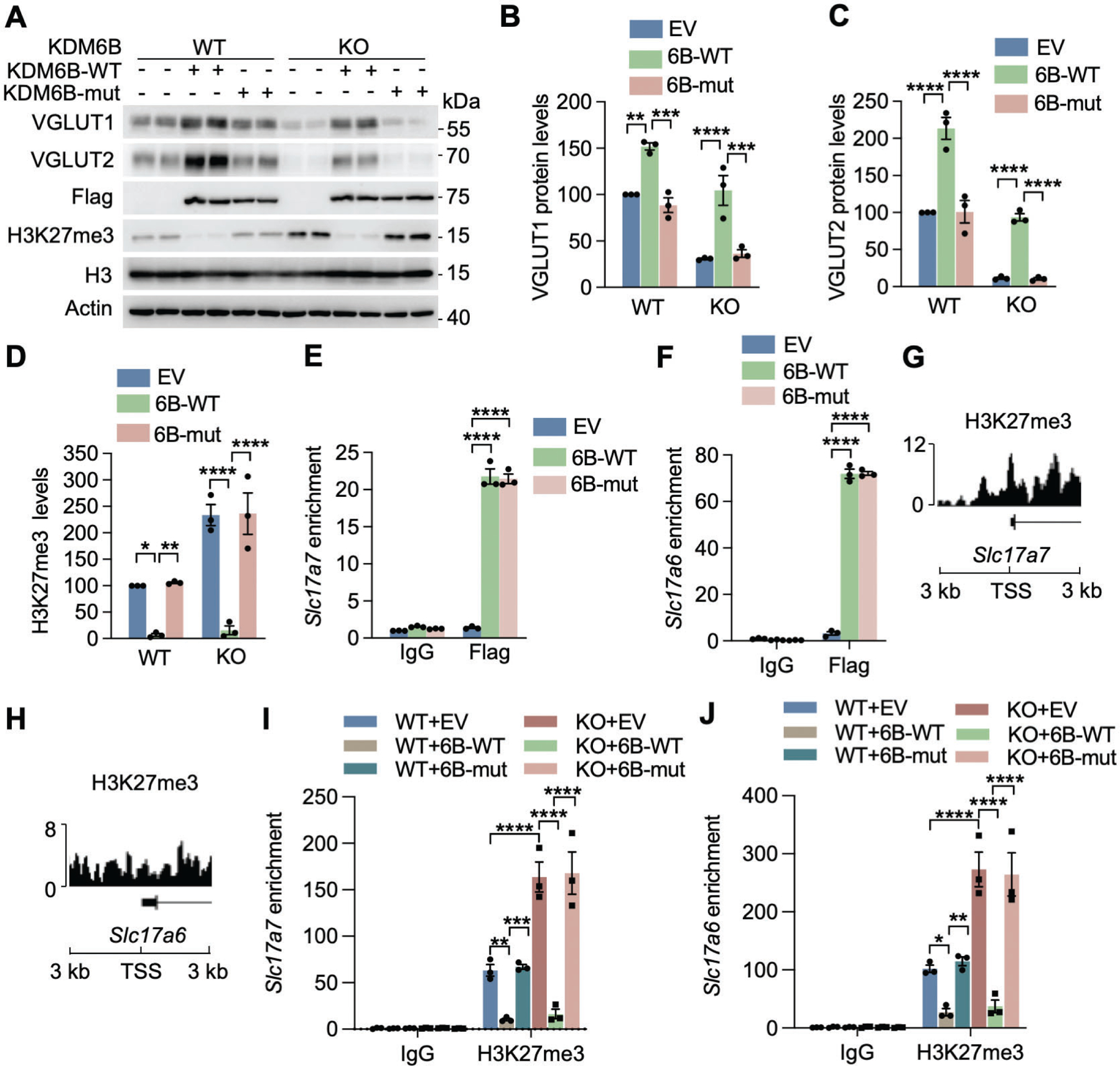
(A-D) Representative immunoblot (A) of VGLUT1 and VGLUT2 expression in WT and KDM6B KO neurons (DIV 14) expressing empty vector (EV) or KDM6B-C-WT or catalytically mutant (mut). The intensity of VGLUT1 (B), VGLUT2 (C), and H3K27me3 (D) bands was quantified and normalized to β-actin. n = 3 biological replicates in each group. One-way ANOVA with Tukey’s multiple comparison test. (E, F) ChIP-qPCR analysis of WT and mut KDM6B enrichment at the promoters of Slc17a7 (E) and Slc17a6 (F) in neurons (DIV 14). n = 3 biological replicates in each group. Two-way ANOVA with Sidak’s multiple comparison test. (G, H) H3K27me3 ChIP-seq peaks at the transcriptional start sites (TSS; ±3 kb) of Slc17a7 (G) and Slc17a6 (H) in neurons. Data were retrieved from GSM2800528. (I, J) ChIP-qPCR analysis of H3K27me3 at the promoters of Slc17a7 (I) and Slc17a6 (J) in WT and KDM6B KO neurons (DIV14) expressing EV or KDM6B-C-WT or mut. n = 3 biological replicates in each group. Two-way ANOVA with Tukey’s multiple comparison test. Data were presented as mean ± s.e.m. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.
To investigate whether KDM6B directly binds to Slc17a6 and Slc17a7 to control their gene transcription, we performed ChIP assay in neurons expressing Flag-KDM6B-C-WT, Flag-KDM6B-C-mut, or empty vector. KDM6B-C-WT and KDM6B-C-mut were equally enriched at promoters of Slc17a6 and Slc17a7 (Figs. 4E, F). ChIP-seq showed enrichment of H3K27me3 at promoters of Slc17a6 and Slc17a7 (Figs. 4G, H), which was confirmed in cortical neurons by ChIP-qPCR (Figs. 4I, J). Importantly, H3K27me3 enrichment at promoters of Slc17a7 and Slc17a6 was diminished in KDM6B-C-WT expressed neurons but significantly increased in KDM6B-KO neurons (Figs. 4I, J). Increased H3K27me3 occupancy in KDM6B KO neurons was reversed by KDM6B-C-WT but not KDM6B-C-mut (Figs. 4I, J). In contrast, KDM6B KO had no effect on H3K27me3 enrichment on Gip, a KDM6B-independent H3K27me3 monovalent gene30, in neurons (Figs. S7F, G), suggesting the specific regulation of VGLUT1/2 by KDM6B-dependent H3K27me3 demethylation. Taken together, these findings indicate that KDM6B binds to promoters of Slc17a6 and Slc17a7 and activates their transcription in neurons through H3K27me3 demethylation.
Forced expression of VGLUT1 and VGLUT2 restores spine density and synaptic activity in KDM6B KO neurons
To determine whether VGLUT1/2 are required for KDM6B-induced reduction of spine density, we performed the rescue experiments in KDM6B KO neurons expressing KDM6B-WT, KDM6B-mut, VGLUT1, or VGLUT2. In line with in vivo results (Figs. 2E–H), KDM6B KO significantly reduced the spine density in neurons, which was rescued by KDM6B-WT but not KDM6B-mut (Figs. 5A, B), suggesting that KDM6B increases the dendritic spine density through its histone demethylase activity. Importantly, expression of VGLUT1 or VGLUT2 similarly restored the dendritic spine density in KDM6B KO neurons (Figs. 5A, B). These results indicate that VGLUT1 and VGLUT2 are the key KDM6B downstream target proteins that are responsible for maintaining proper spine density in neurons.
Fig. 5. KDM6B regulates synapse activity through VGLUT1/2 in neurons.
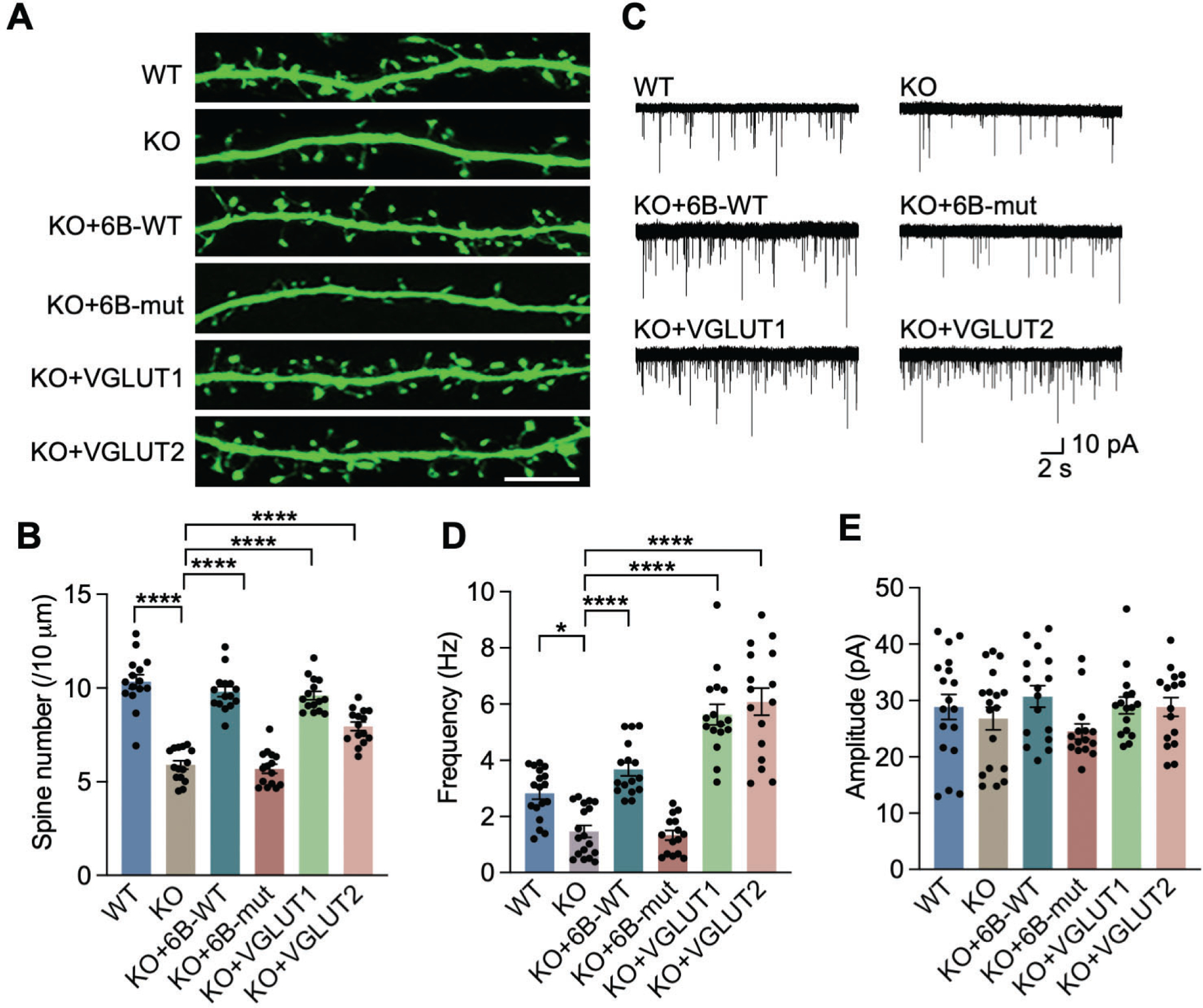
(A, B) Representative images of fragments of dendrites (A) and quantification of spine density (B) in GFP-positive WT and KDM6B KO neurons (DIV 18) expressing KDM6B-C-WT or mut, VGLUT1, or VGLUT2. n = 15 for each group. One-way ANOVA with Tukey’s multiple comparison test. Scale bar, 10 μm. (C) Representative traces of mEPSCs in WT and KDM6B KO neurons expressing KDM6B-C-WT or mut, VGLUT1, or VGLUT2. (D, E) Quantification of mEPSC frequency (D) and amplitude (E). n = 18 cells per group for WT and KO groups; n = 16 cells per group for KO+6B-WT, KO+VGLUT1 and KO+VGLUT2; n = 15 cells for KO+6B-mut group. One-way ANOVA with Tukey’s multiple comparison test. Data were presented as mean ± s.e.m. *p < 0.05; ****p < 0.0001.
Next, we studied whether re-expression of VGLUT1/2 rescues synaptic transmission in KDM6B KO neuron cultures by recording miniature excitatory post-synaptic currents (mEPSC) in the presence of the antagonist of inhibitory gamma aminobutyric acid (GABA)-A receptors picrotoxin (100 μM) as well as tetrodotoxin (TTX, 1 μM) to block action potential as our findings in brain slices revealed that the reduction of frequency and amplitude in EPSCs is likely independent of action potential. Consistent with the results in brain slices (Figs. 2M–O), KDM6B KO significantly decreased the mEPSC frequency in cultured neurons, which was rescued by re-expression of KDM6B-WT but not KDM6B-mut (Figs. 5C, D). Remarkably, re-expression of VGLUT1 or VGLUT2 also fully reversed the reduction of mEPSC frequency conferred by KDM6B KO in neurons (Figs. 5C, D). Although amplitude in mEPSC was not significantly affected by KDM6B or VGLUT1/2 in neurons, there was a trend of amplitude reduction in KDM6B KO and KDM6B-mut neurons, which was reversed by re-expression of KDM6B or VGLUT1/2 (Figs. 5C, E). These findings indicate that VGLUT1/2 are the key proteins that are responsible for KDM6B-mediated synaptic activity in neurons.
Altered H3K27me3 by KDM6A or EZH1/2 fails to regulate VGLUT1 and VGLUT2
KDM6A is a functional homolog of KDM6B and known to demethylate histone H3K27me36. We found that Kdm6a mRNA was increased by about 21% in KDM6B KO neurons (Fig. S8A), but its protein level was not changed (Figs. S8B, C). To determine whether KDM6A has a role in VGLUT1 and VGLUT2 expression similar to KDM6B, we generated KDM6A knockdown (KD) neurons using two independent shRNAs (6A-sh1 and 6A-sh2, Figs. 6A, B). KDM6A KD had no effects on the expression of VGLUT1 and VGLUT2 at both mRNA and protein levels (Figs. 6A–C). KDM6B and H3K27me3 levels were also not altered in KDM6A KD neurons (Figs. 6A–C). We next examined whether overexpression of KDM6A regulates the expression of VGLUT1 and VGLUT2. We constructed a C-terminal KDM6A truncate (6A-C) containing the JmjC domain and its expression was sufficient to reduce H3K27me3 levels in transfected 293T cells (Figs. S8D, E), suggesting that this truncate can mimic the function of full-length KDM6A protein. Overexpression of KDM6A-C induced the expression of VGLUT1 in both WT and KDM6B KO neurons along with loss of H3K27me3, similar to KDM6B-C (Figs. 6D, E). However, KDM6A-C overexpression was not able to upregulate VGLUT2 in WT neurons and also failed to rescue VGLUT2 in KDM6B KO neurons (Figs. 6D, F). Given that VGLUT1 was not completely abolished in KDM6B KO neurons and overexpression of KDM6A also upregulated the expression of VGLUT1, we further examined whether both KDM6A and KDM6B coordinate to control the expression of VGLUT1 in neurons. We generated KDM6A and KDM6B double deficient neurons by transducing KDM6A shRNA lentiviruses into KDM6B KO neurons. We found that KDM6A deletion did not further reduce VGLUT1 and VGLUT2 expression in KDM6B KO neurons (Figs. 6G, H), suggesting that KDM6B is mainly responsible for the expression of VGLUT1/2 in neurons.
Fig. 6. VGLUT1 and VGLUT2 were mainly regulated by KDM6B, but not KDM6A or EZH1/2.
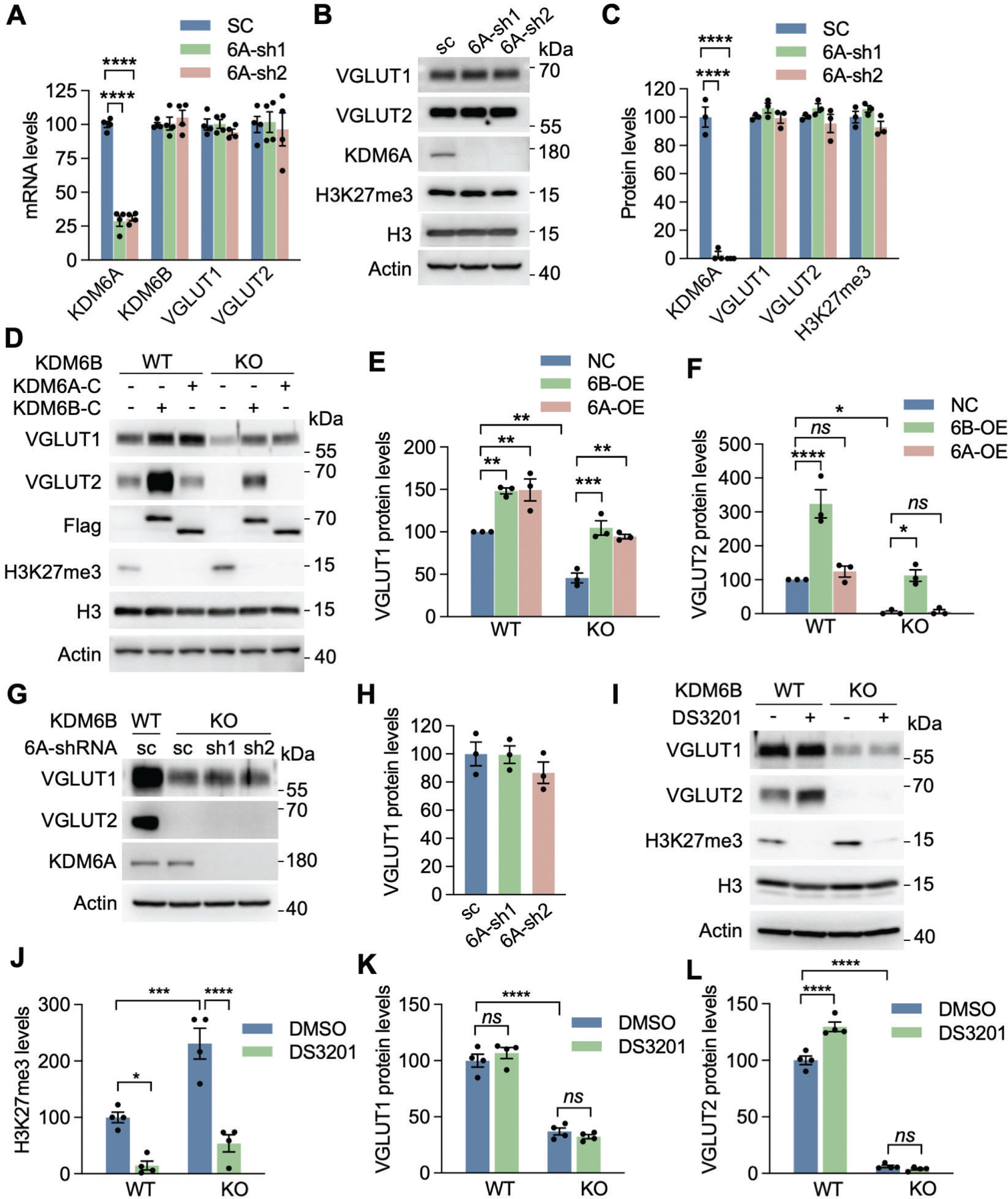
(A) qRT-PCR analysis of KDM6A, KDM6B, VGLUT1, and VGLUT2 in neurons infected with lentivirus encoding scrambled control (SC) shRNA or KDM6A shRNA1 (sh1) or shRNA2 (sh2). n = 4 biological replicates in each group. One-way ANOVA with Dunnett’s multiple comparison test. (B, C) Immunoblot analysis of KDM6A, H3K27me3, VGLUT1, and VGLUT2 in neurons infected with lentivirus encoding SC shRNA or KDM6A sh1 or sh2 (B). The intensity of KDM6A, H3K27me3, VGLUT1 and VGLUT2 bands was quantified and normalized to β-actin (C). n = 3 biological replicates in each group. One-way ANOVA with Dunnett’s multiple comparison test. (D-F) Immunoblot analysis of KDM6A-C, KDM6B-C, H3K27me3, VGLUT1, and VGLUT2 in WT and KDM6B KO neurons expressing KDM6A-C or KDM6B-C (D). The intensity of VGLUT1 (E) and VGLUT2 (F) bands was quantified and normalized to β-actin. n = 3 biological replicates in each group. One-way ANOVA with Tukey’s multiple comparison test. ns, not significant. (G, H) Immunoblot analysis of KDM6A, VGLUT1, and VGLUT2 in WT and KDM6B KO neurons expressing KDM6A sh1 or sh2 (G). The intensity of VGLUT1 bands was quantified and normalized to β-actin (H). n = 3 biological replicates in each group. One-way ANOVA with Tukey’s multiple comparison test. (I-L) Immunoblot analysis of H3K27me3, VGLUT1, and VGLUT2 in WT and KDM6B KO neurons treated for 7 days with or without a EZH1/2 dual inhibitor DS-3201 (I). The intensity of H3K27me3 (J), VGLUT1 (K), and VGLUT2 (L) bands was quantified and normalized to β-actin. n = 4 biological replicates in each group. One-way ANOVA with Tukey’s multiple comparison test. Data were presented as mean ± s.e.m. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.
H3K27me3 levels are also controlled by methyltransferases enhancer of zeste homology (EZH)1 and EZH2 besides demethylases KDM6A and KDM6B. To investigate whether EZH1/2-mediated H3K27me3 regulates the expression of VGLUT1/2, we treated WT and KDM6B KO neurons with a dual EZH1/2 inhibitor DS-3201 (1 μM) or DMSO for 7 days. As expected, H3K27me3 was abolished in DS-3201 treated neurons (Figs. 6I, J). However, DS-3201 treatment had no effect on VGLUT1 expression in both WT an KDM6B KO neurons (Fig. 6I, K). Although a marginal increase (about 30%) in VGLUT2 protein was observed in WT neurons after DS-3201 treatment, DS-3201 treatment did not induce VGLUT2 protein in KDM6B KO neurons (Figs. 6I, L), suggesting that KDM6B rather than EZH1/2 is a primary regulator of VGLUT2. Taken together, these gain- and loss-of function studies indicate that VGLUT1 and VGLUT2 are mainly regulated by KDM6B but not KDM6A or EZH1/2 in neurons.
Tau is a KDM6B coregulator that regulates VGLUT1/2 expression in neurons
To further explore the mechanism of KDM6B-induced VGLUT1/2 expression, we screened KDM6B-interacting proteins in the nuclei of cortical neurons expressing KDM6B-WT-C by immunoprecipitation (IP) assay followed by mass spectrometry analysis (Fig. S9A). Top 11 protein candidates were identified to strongly bind to KDM6B-WT-C (Fig. S9B). Among these, Tau is highly abundant in neurons and has been implicated to regulate VGLUT1 expression in neurons31. Thus, Tau was ranked as a top hit for further studies. Co-IP assay using anti-myc or anti-Flag antibody validated that myc-tagged Tau interacted with Flag-tagged KDM6B-WT-C in transfected 293T cells (Figs. 7A, B). A reciprocal interaction between full-length KDM6B and Tau was also observed in transfected 293T cells (Figs. S9C, D). In contrast, Tau failed to bind to KDM6A in transfected 293T cells (Fig. S9E). These results demonstrate that Tau physically and specifically binds to KDM6B.
Fig. 7. Tau is a coregulator for KDM6B to control VGLUT1 and VGLUT2 expression.
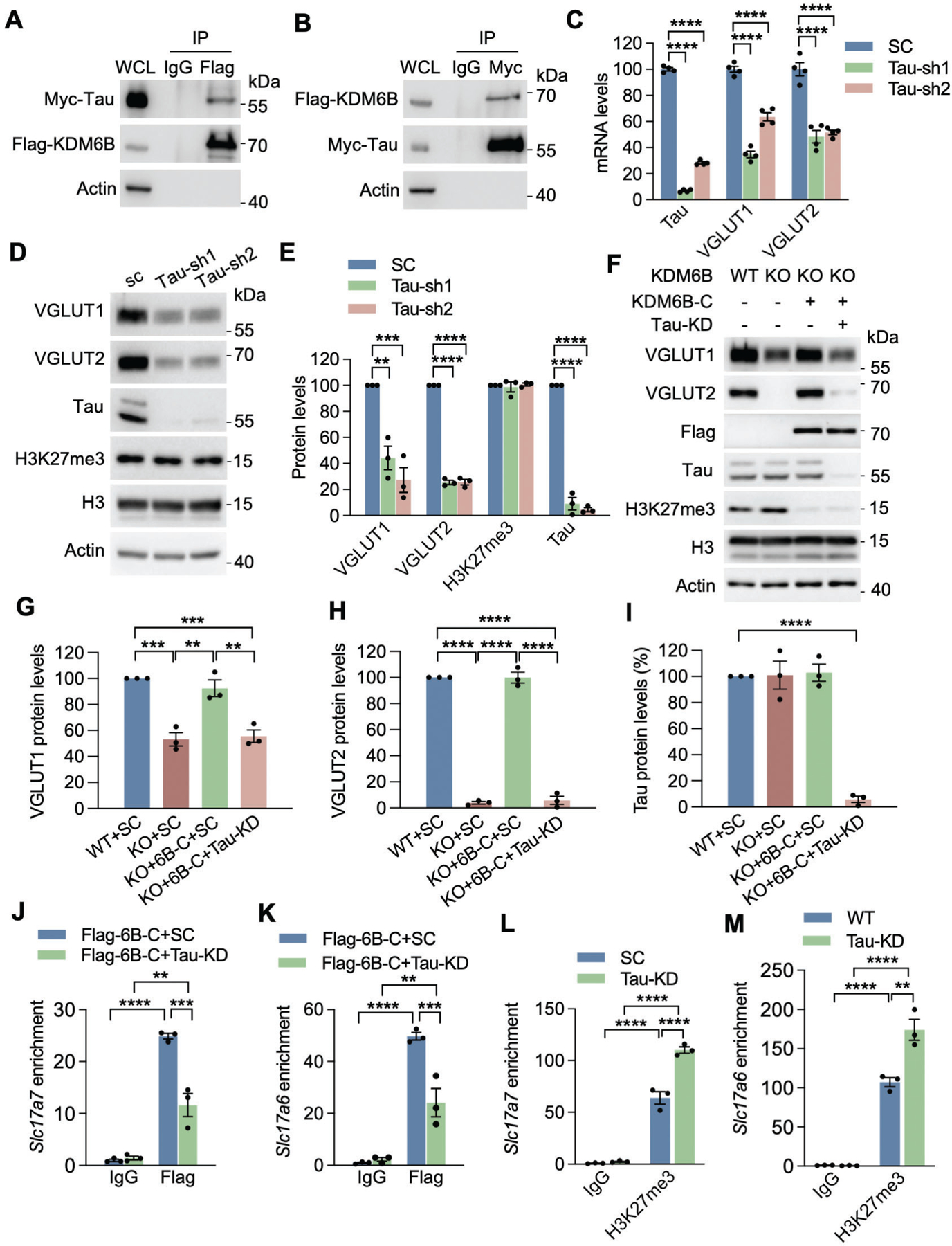
(A) Co-IP with anti-Flag antibody in HEK293T cells transfected with Flag-KDM6B-C and Myc-Tau. WCL, whole cell lysate. (B) Co-IP with anti-Myc antibody in HEK293T cells transfected with Myc-Tau and Flag-KDM6B-C. (C) qRT-PCR analysis of Tau, VGLUT1, and VGLUT2 in neurons infected with lentivirus encoding SC shRNA or Tau shRNA1 (sh1) or shRNA2 (sh2). n = 4 biological replicates. One-way ANOVA with Dunnett’s multiple comparison test. (D, E) Immunoblot analysis of Tau, H3K27me3, VGLUT1 and VGLUT2 proteins in neurons infected with lentivirus encoding SC shRNA or Tau sh1 or sh2 (D). The intensity of Tau, H3K27me3, VGLUT1, and VGLUT2 bands was quantified and normalized to β-actin (E). n = 3 biological replicates. One-way ANOVA with Dunnett’s multiple comparison test. (F-I) Immunoblot analysis of KDM6B-C, Tau, H3K27me3, VGLUT1 and VGLUT2 proteins in WT and KDM6B KO neurons expressing Tau shRNA and/or Flag-KDM6B-C (F). The intensity of VGLUT1 (G), VGLUT2 (H), and Tau (I) bands was quantified and normalized to β-actin. n = 3 biological replicates. One-way ANOVA with Tukey’s multiple comparison test. (J, K) ChIP-qPCR analysis of KDM6B enrichment at the promoters of Slc17a7 (J) and Slc17a6 (K) in WT and Tau KD neurons expressing Flag-KDM6B-C. n = 3 biological replicates. Two-way ANOVA with Tukey’s multiple comparison test. (L, M) ChIP-qPCR analysis of H3K27me3 enrichment at the promoters of Slc17a7 (L) and Slc17a6 (M) in WT and Tau KD neurons. n = 3 biological replicates. Two-way ANOVA with Tukey’s multiple comparison test. Data were presented as mean ± s.e.m. **p < 0.01. ***p < 0.001; ****p < 0.0001.
To assess whether Tau regulates VGLUT1/2 expression, we knocked down endogenous Tau in neurons by transducing lentivirus encoding two independent Tau shRNAs (Figs. 7C–E). Tau KD by either of its shRNAs significantly reduced VGLUT1 and VGLUT2 in neurons at both mRNA and protein levels (Figs. 7C–E). Notably, the global level of H3K27me3 was not changed in Tau KD neurons compared with scrambled control (SC) neurons (Figs. 7D, E). Next, we studied whether Tau is required for KDM6B-induced VGLUT1/2 expression. KDM6B KO neurons were transduced with Tau shRNA lentivirus and/or KDM6B-C lentivirus. As expected, KDM6B KO reduced the expression of VGLUT1/2 in neurons, which was rescued by KDM6B-C (Figs. 7F–H). However, this rescue effect was prevented by Tau KD without H3K27me3 alterations (Figs. 6G–I), indicating that Tau is required for KDM6B-induced VGLUT1/2 expression.
We then examined whether Tau binds to promoters of Slc17a6 and Slc17a7. ChIP-qPCR assays showed that Tau occupied at promoters of Slc17a6 and Slc17a7 in neurons, which was not affected by KDM6B KO (Figs. S9F, G). Remarkably, Tau KD significantly decreased enrichment of KDM6B at promoters of Slc17a6 and Slc17a7 in neurons (Figs. 7J, K). Consistently, H3K27me3 enrichment at promoters of Slc17a6 and Slc17a7 was significantly increased when Tau was knocked down in neurons (Figs. 7L, M), although the global level of H3K27me3 was not affected by Tau KD in neurons (Figs. 7D, E). Taken together, these results indicate that Tau recruits KDM6B to Slc17a6 and Slc17a7 genes to regulate their expression.
DISCUSSION
In this study, we demonstrate that KDM6B is predominantly expressed in neurons and indispensable for maintaining the proper neuronal spine density, synaptic activity, and cognitive functions. KDM6B has been previously known for its role in development and cell differentiation in the brain9, 10. Aberrant development and deficits in synaptic plasticity are primary causes of many neurological disorders. In line with that, KDM6B has been identified as a risk gene for autism17. In this study, KDM6B was selectively knocked out in the excitatory neurons postnatally by crossing with CamKIIα-iCre mice, where iCre expression starts from P0 and results in the best KO effect around P2018, 19, 32. This mouse model allows to study KDM6B functions postnatally other than its roles during development. Indeed, KDM6B cKO mice did not exhibit any obvious developmental abnormality, but displayed reduced spine density and synaptic activity and further developed learning and memory associated behavioral deficits. These findings reveal a novel function of epigenetic modifier KDM6B in the regulation of synaptic plasticity and cognition in mice under physiological conditions.
Our study further uncovered a deep layer mechanism underlying KDM6B-regulated spine density, synaptic activity, and cognitive functions, which requires KDM6B demethylase activity for the expression of its downstream target proteins VGLUT1/2. Glutamate release from excitatory synapses in mammalian brain relies on its vesicular transporters VGLUT1 and VGLUT233, 34. Both VGLUT1 and VGLUT2 play a critical role in the postnatal maturation of pyramidal neuron plasticity, dendritic refinement, and cognition35, 36. Ablation of VGLUTs blocks glutamatergic transmission leading to synaptic failure and cognitive impairment34, 35. Thus, modulation of the expression of VGLUTs remarkably influences homeostasis of the glutamatergic system that is critical for maintenance of normal synaptic functions and the development of neurological disorders. We found that VGLUT1/2 expression is controlled by KDM6B through H3K27me3 demethylation at their promoters. KDM6B cKO mice have impaired learning and memory, similar to VGLUT1 and VGLUT2 deficient mice35, 36. Re-expression of KDM6B or VGLUT1/2 rescues the spine density and synaptic activity in KDM6B KO neurons. These findings reveal that VGLUT1/2 are the key KDM6B downstream targets regulating KDM6B-mediated synaptic plasticity and cognitive functions.
H3K27me3 levels have been known to be tightly regulated by EZH2, KDM6A and KDM6B37, 38. Using loss-of-KDM6B, gain-of-KDM6B and rescue studies with re-introduction of both functional KDM6B and demethylase inactive mutant KDM6B H1390A, our results confirmed KDM6B as a dominant epigenetic modifier that regulates the expression of VGLUT1/2 in neurons. While KDM6A is ubiquitously expressed in the central nervous system, knockdown of endogenous KDM6A failed to affect VGLUT1/2 expression, which is consistent with a previous RNA-seq study showing that KDM6A loss in mouse hippocampus by Emx1-Cre has no effect on VGLUT1/2 expression39. Moreover, in KDM6B KO neurons, loss of KDM6A did not further reduce the expression of VGLUT1/2. Interestingly, although overexpression of KDM6A did not have effects on VGLUT2 expression, it indeed increased VGLUT1, suggesting that KDM6A may have a partial redundant function only when it is artificially overexpressed. Likewise, the H3K27 methyltransferases EZH1/2 did not alter VGLUT1/2 expression in KDM6B KO neurons, but inhibition of EZH1/2 modestly increased VGLUT2 expression in KDM6B WT neurons, indicating that EZH1/2 might be weak regulators with a preference to VGLUT2 in the presence of KDM6B as the premise. Nevertheless, these findings indicate that KDM6B, but not KDM6A or EZH2, is a major transcriptional regulator for VGLUT1/2 expression in neurons through local H3K27me3 alteration.
Previous studies reported that EZH2 regulates cognitive functions in mice via controlling adult hippocampal neurogenesis40. In addition, KDM6A has been shown to regulate synaptic plasticity and cognitive functions via controlling expression of neurotransmitter 5-hydroxytrytamine receptor 5B39. However, the role of H3K27me3 in cognition has not been well elucidated. Our study clearly revealed the importance of the demethylase activity of KDM6B in regulation of spine density, synaptic acidity, and cognitive functions, which were confirmed by loss of KDM6B and rescue studies with WT KDM6B and demethylase inactive mutant KDM6B H1390A. Our findings reveal that KDM6B deficiency-induced cognitive deficits are independent of hippocampal neurogenesis and elucidate a novel mechanism of H3K27me3-regulated synaptic plasticity and cognition.
Our current study demonstrated that Tau protein is required for the recruitment of KDM6B to promoters of Slc17a6 and Slc17a7 leading to the expression of VGLUT1 and VGLUT2 in neurons. Tau is a highly abundant microtubule-associated protein in neurons and well-known for its role in the stability of microtubules41. Emerging studies from independent groups have shown that Tau localizes in the nucleus in addition to the cytosol42–45. Tau can directly bind to DNA in vitro determined by electron microscopy and gel electrophoresis mobility shift assay43. Tau-DNA binding is reversible in the presence of histone43. In intact cells, nuclear tau is mainly present at the internal periphery of nucleoli and binds to AT-rich-satellite DNA sequences44. A repressive role of nuclear Tau in ribosomal DNA transcription was also reported45. These combined data indicate that nuclear Tau and its DNA binding property may play a role in normal cellular physiology. Our study here provides direct evidence of Tau protein in gene regulation, which supports a previous study showing that Tau regulates the expression of VGLUT1 in neurons31. We showed that Tau increased KDM6B enrichment but decreased H3K27me3 levels locally on Slc17a6 and Slc17a7 genes. Tau also regulates the clustered distribution of H3K9me346. Thus, our work and others highlight a critical role of Tau in modulation of modified histone levels that regulates gene expression in neurons. We also found that KDM6B KO had no effect on Tau binding to chromatin in neurons, suggesting that Tau binding to DNA is independent of KDM6B. Thus, our working model is that Tau interacts with KDM6B and facilitates the recruitment of KDM6B to promoters of Slc17a6 and Slc17a7 genes, thereby reducing local H3K27me3 levels to enhance VGLUT1/2 expression, leading to increased spine density, synaptic activity, and subsequent cognitive functions (Fig. 8). The precise mechanism of Tau binding to DNA requires further investigations. Nevertheless, our work yields novel molecular insights into the gene regulation functions of Tau in neurons.
Fig. 8. The proposed model for KDM6B function in regulation of synaptic activity and cognition.
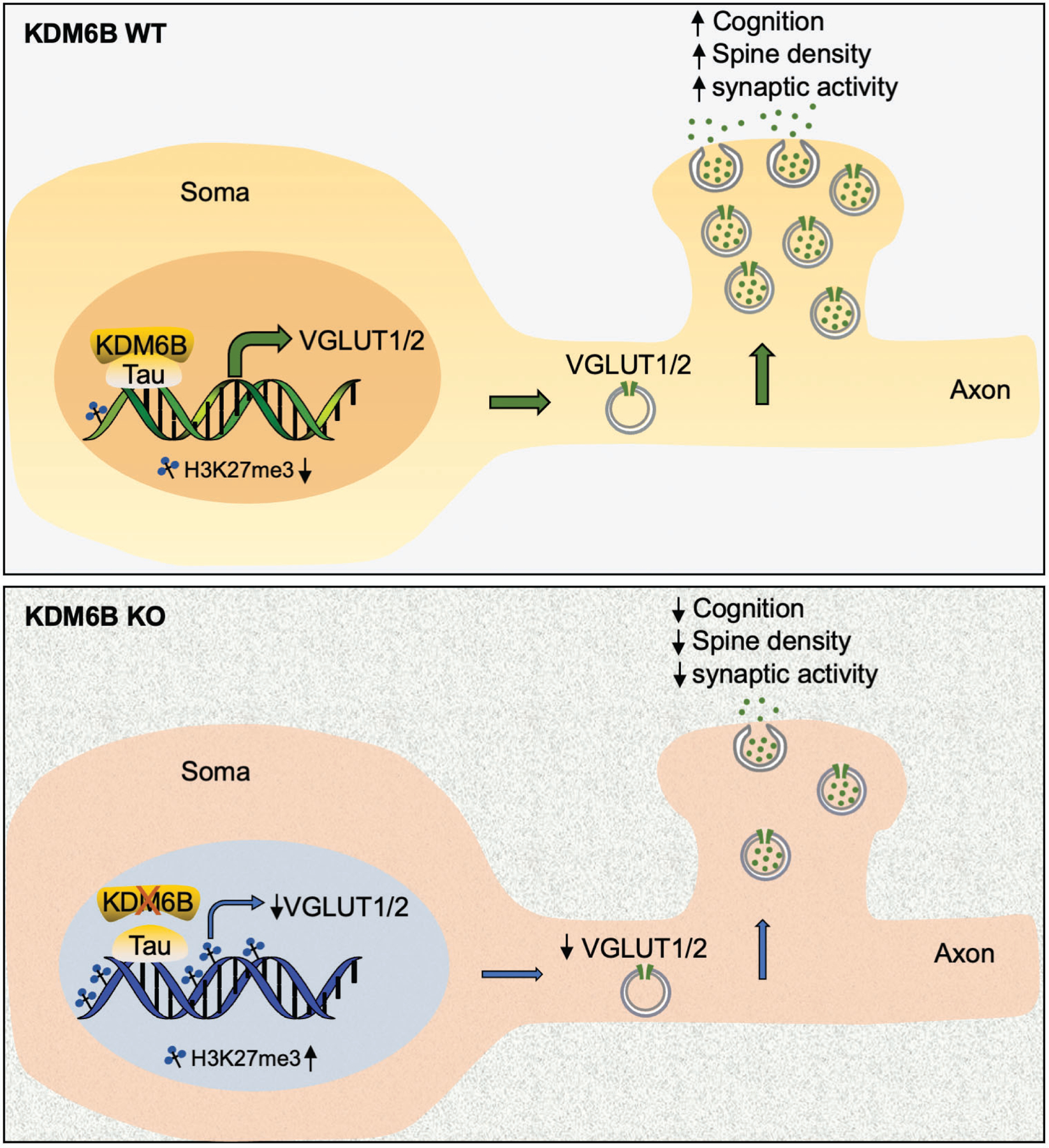
KDM6B interacts with Tau and is recruited by Tau to the promoters of Slc17a6 and Slc17a7, where it demethylates H3K27me3 to induce the expression of VGLUT1/2, leading to increased vesicle number, synaptic activity, dendritic spine density, and animal cognitive functions. KDM6B KO or Tau knockdown reduces VGLUT1/2 expression and vesicle number. Subsequently, it diminishes synaptic activity, spine density, and cognitive functions.
Aggregation of hyperphosphorylated Tau has been recognized as a hallmark of Alzheimer’s disease47. A recent study showed that phosphorylated Tau interacts with nucleoporins of the nuclear pore complex, interferes with the structural and functional integrity of the nuclear pore, and disrupts nucleocytoplasmic transport48. Interestingly, increase of Tau hyperphosphorylation was found to be correlated with reduced O-GlcNAc glycosylation on Tau protein and with diminished Tau nuclear translocation, suggesting that the balance between phosphorylation and O-GlcNAc glycosylation of Tau protein may regulate Tau nuclear localization42. Moreover, aggregated Tau loses the ability to bind to DNA43. These findings suggest that Tau pathology alters Tau nuclear translocation and its physiological role in gene regulation, which may eventually lead to neurotoxicity and neurodegeneration in Alzheimer’s disease.
In conclusion, this study provides evidence for a novel indispensable regulatory machinery of cognition, where the H3K27me3 demethylase KDM6B cooperates with Tau to specifically control VGLUT1 and VGLUT2 expression for proper synaptic transmission and normal neuron functions under physiological conditions. Loss of VGLUT1 and VGLUT2 has been observed in the prefrontal cortex and hippocampus in aging related neurological diseases like Alzheimer’s disease, which has been correlated with cognitive decline49, 50. Our findings on the physiological role of KDM6B in synapse plasticity and cognition may help understand the involvement of KDM6B in neurological diseases and underlying mechanisms.
Supplementary Material
ACKNOWLEDGMENTS
We thank Drs. Woo-ping Ge and Kim Huber for advice on eletrophysology studies, Dr. Stuart H. Orkin at Harvard Medical School for providing Kdm6bfl/fl mice, and UT Southwestern core facilities including electron microscopy core, rodent behavior core, and live cell imaging core for experimental assistance (1P30 CA142543-01). This work was supported by grants from the NIH (R35GM124693, R00NS078049, and R01AG066166) to YW.
Footnotes
Competing interests
The authors declare no competing interests.
Availability of supporting data
The RNA-seq data were deposited at the GEO database with accession number GSE197117.
REFERENCES
- 1.Bienvenu T, Chelly J. Molecular genetics of Rett syndrome: when DNA methylation goes unrecognized. Nat Rev Genet 2006; 7(6): 415–426. [DOI] [PubMed] [Google Scholar]
- 2.Lubin FD, Roth TL, Sweatt JD. Epigenetic regulation of BDNF gene transcription in the consolidation of fear memory. J Neurosci 2008; 28(42): 10576–10586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fischer A, Sananbenesi F, Wang X, Dobbin M, Tsai LH. Recovery of learning and memory is associated with chromatin remodelling. Nature 2007; 447(7141): 178–182. [DOI] [PubMed] [Google Scholar]
- 4.Nativio R, Donahue G, Berson A, Lan Y, Amlie-Wolf A, Tuzer F et al. Dysregulation of the epigenetic landscape of normal aging in Alzheimer’s disease. Nat Neurosci 2018; 21(4): 497–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Persico G, Casciaro F, Amatori S, Rusin M, Cantatore F, Perna A et al. Histone H3 Lysine 4 and 27 Trimethylation Landscape of Human Alzheimer’s Disease. Cells 2022; 11(4): 734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Agger K, Cloos PA, Christensen J, Pasini D, Rose S, Rappsilber J et al. UTX and JMJD3 are histone H3K27 demethylases involved in HOX gene regulation and development. Nature 2007; 449(7163): 731–734. [DOI] [PubMed] [Google Scholar]
- 7.Hong S, Cho YW, Yu LR, Yu H, Veenstra TD, Ge K. Identification of JmjC domain-containing UTX and JMJD3 as histone H3 lysine 27 demethylases. Proc Natl Acad Sci U S A 2007; 104(47): 18439–18444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.De Santa F, Totaro MG, Prosperini E, Notarbartolo S, Testa G, Natoli G. The histone H3 lysine-27 demethylase Jmjd3 links inflammation to inhibition of polycomb-mediated gene silencing. Cell 2007; 130(6): 1083–1094. [DOI] [PubMed] [Google Scholar]
- 9.Iida A, Iwagawa T, Kuribayashi H, Satoh S, Mochizuki Y, Baba Y et al. Histone demethylase Jmjd3 is required for the development of subsets of retinal bipolar cells. Proceedings of the National Academy of Sciences of the United States of America 2014; 111(10): 3751–3756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Park DH, Hong SJ, Salinas RD, Liu SJ, Sun SW, Sgualdino J et al. Activation of neuronal gene expression by the JMJD3 demethylase is required for postnatal and adult brain neurogenesis. Cell reports 2014; 8(5): 1290–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Akizu N, Estaras C, Guerrero L, Marti E, Martinez-Balbas MA. H3K27me3 regulates BMP activity in developing spinal cord. Development 2010; 137(17): 2915–2925. [DOI] [PubMed] [Google Scholar]
- 12.Burgold T, Voituron N, Caganova M, Tripathi PP, Menuet C, Tusi BK et al. The H3K27 demethylase JMJD3 is required for maintenance of the embryonic respiratory neuronal network, neonatal breathing, and survival. Cell reports 2012; 2(5): 1244–1258. [DOI] [PubMed] [Google Scholar]
- 13.Satoh T, Takeuchi O, Vandenbon A, Yasuda K, Tanaka Y, Kumagai Y et al. The Jmjd3-Irf4 axis regulates M2 macrophage polarization and host responses against helminth infection. Nat Immunol 2010; 11(10): 936–944. [DOI] [PubMed] [Google Scholar]
- 14.Lee JY, Na WH, Choi HY, Lee KH, Ju BG, Yune TY. Jmjd3 mediates blood-spinal cord barrier disruption after spinal cord injury by regulating MMP-3 and MMP-9 expressions. Neurobiol Dis 2016; 95: 66–81. [DOI] [PubMed] [Google Scholar]
- 15.Zhang H, Wang J, Huang J, Shi T, Ma X, Luo X et al. Inhibiting Jumoji domain containing protein 3 (JMJD3) prevent neuronal apoptosis from stroke. Exp Neurol 2018; 308: 132–142. [DOI] [PubMed] [Google Scholar]
- 16.Stolerman ES, Francisco E, Stallworth JL, Jones JR, Monaghan KG, Keller-Ramey J et al. Genetic variants in the KDM6B gene are associated with neurodevelopmental delays and dysmorphic features. Am J Med Genet A 2019; 179(7): 1276–1286. [DOI] [PubMed] [Google Scholar]
- 17.De Rubeis S, He X, Goldberg AP, Poultney CS, Samocha K, Cicek AE et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 2014; 515(7526): 209–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Casanova E, Fehsenfeld S, Mantamadiotis T, Lemberger T, Greiner E, Stewart AF et al. A CamKIIalpha iCre BAC allows brain-specific gene inactivation. Genesis 2001; 31(1): 37–42. [DOI] [PubMed] [Google Scholar]
- 19.Dragatsis I, Zeitlin S. CaMKIIalpha-Cre transgene expression and recombination patterns in the mouse brain. Genesis 2000; 26(2): 133–135. [DOI] [PubMed] [Google Scholar]
- 20.Dubois NC, Hofmann D, Kaloulis K, Bishop JM, Trumpp A. Nestin-Cre transgenic mouse line Nes-Cre1 mediates highly efficient Cre/loxP mediated recombination in the nervous system, kidney, and somite-derived tissues. Genesis 2006; 44(8): 355–360. [DOI] [PubMed] [Google Scholar]
- 21.Kraeuter AK, Guest PC, Sarnyai Z. The Y-Maze for Assessment of Spatial Working and Reference Memory in Mice. Methods Mol Biol 2019; 1916: 105–111. [DOI] [PubMed] [Google Scholar]
- 22.Leger M, Quiedeville A, Bouet V, Haelewyn B, Boulouard M, Schumann-Bard P et al. Object recognition test in mice. Nat Protoc 2013; 8(12): 2531–2537. [DOI] [PubMed] [Google Scholar]
- 23.Maren S Neurobiology of Pavlovian fear conditioning. Annu Rev Neurosci 2001; 24: 897–931. [DOI] [PubMed] [Google Scholar]
- 24.Shih TW, Lee LJ, Chang HC, Lin HW, Chang MS. An important role of PHRF1 in dendritic architecture and memory formation by modulating TGF-beta signaling. Sci Rep 2020; 10(1): 10857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pchitskaya E, Bezprozvanny I. Dendritic Spines Shape Analysis-Classification or Clusterization? Perspective. Front Synaptic Neurosci 2020; 12: 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Feng G, Mellor RH, Bernstein M, Keller-Peck C, Nguyen QT, Wallace M et al. Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron 2000; 28(1): 41–51. [DOI] [PubMed] [Google Scholar]
- 27.Alabi AA, Tsien RW. Synaptic vesicle pools and dynamics. Cold Spring Harb Perspect Biol 2012; 4(8): a013680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yau SY, Li A, So KF. Involvement of Adult Hippocampal Neurogenesis in Learning and Forgetting. Neural Plast 2015; 2015: 717958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen S, Ma J, Wu F, Xiong LJ, Ma H, Xu W et al. The histone H3 Lys 27 demethylase JMJD3 regulates gene expression by impacting transcriptional elongation. Genes Dev 2012; 26(12): 1364–1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.von Schimmelmann M, Feinberg PA, Sullivan JM, Ku SM, Badimon A, Duff MK et al. Polycomb repressive complex 2 (PRC2) silences genes responsible for neurodegeneration. Nat Neurosci 2016; 19(10): 1321–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Siano G, Varisco M, Caiazza MC, Quercioli V, Mainardi M, Ippolito C et al. Tau Modulates VGluT1 Expression. J Mol Biol 2019; 431(4): 873–884. [DOI] [PubMed] [Google Scholar]
- 32.Liu S, Zhou M, Ruan Z, Wang Y, Chang C, Sasaki M et al. AIF3 splicing switch triggers neurodegeneration. Mol Neurodegener 2021; 16(1): 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fremeau RT Jr., Kam K, Qureshi T, Johnson J, Copenhagen DR, Storm-Mathisen J et al. Vesicular glutamate transporters 1 and 2 target to functionally distinct synaptic release sites. Science 2004; 304(5678): 1815–1819. [DOI] [PubMed] [Google Scholar]
- 34.Wojcik SM, Rhee JS, Herzog E, Sigler A, Jahn R, Takamori S et al. An essential role for vesicular glutamate transporter 1 (VGLUT1) in postnatal development and control of quantal size. Proceedings of the National Academy of Sciences of the United States of America 2004; 101(18): 7158–7163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.He H, Mahnke AH, Doyle S, Fan N, Wang CC, Hall BJ et al. Neurodevelopmental role for VGLUT2 in pyramidal neuron plasticity, dendritic refinement, and in spatial learning. J Neurosci 2012; 32(45): 15886–15901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Garcia-Garcia AL, Elizalde N, Matrov D, Harro J, Wojcik SM, Venzala E et al. Increased vulnerability to depressive-like behavior of mice with decreased expression of VGLUT1. Biol Psychiatry 2009; 66(3): 275–282. [DOI] [PubMed] [Google Scholar]
- 37.Jiang W, Wang J, Zhang Y. Histone H3K27me3 demethylases KDM6A and KDM6B modulate definitive endoderm differentiation from human ESCs by regulating WNT signaling pathway. Cell research 2013; 23(1): 122–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Das P, Taube JH. Regulating Methylation at H3K27: A Trick or Treat for Cancer Cell Plasticity. Cancers (Basel) 2020; 12(10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tang GB, Zeng YQ, Liu PP, Mi TW, Zhang SF, Dai SK et al. The Histone H3K27 Demethylase UTX Regulates Synaptic Plasticity and Cognitive Behaviors in Mice. Front Mol Neurosci 2017; 10: 267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang J, Ji F, Liu Y, Lei X, Li H, Ji G et al. Ezh2 regulates adult hippocampal neurogenesis and memory. J Neurosci 2014; 34(15): 5184–5199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Barbier P, Zejneli O, Martinho M, Lasorsa A, Belle V, Smet-Nocca C et al. Role of Tau as a Microtubule-Associated Protein: Structural and Functional Aspects. Front Aging Neurosci 2019; 11: 204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lefebvre T, Ferreira S, Dupont-Wallois L, Bussiere T, Dupire MJ, Delacourte A et al. Evidence of a balance between phosphorylation and O-GlcNAc glycosylation of Tau proteins--a role in nuclear localization. Biochim Biophys Acta 2003; 1619(2): 167–176. [DOI] [PubMed] [Google Scholar]
- 43.Hua Q, He RQ. Effect of phosphorylation and aggregation on tau binding to DNA. Protein Pept Lett 2002; 9(4): 349–357. [DOI] [PubMed] [Google Scholar]
- 44.Sjoberg MK, Shestakova E, Mansuroglu Z, Maccioni RB, Bonnefoy E. Tau protein binds to pericentromeric DNA: a putative role for nuclear tau in nucleolar organization. J Cell Sci 2006; 119(Pt 10): 2025–2034. [DOI] [PubMed] [Google Scholar]
- 45.Maina MB, Bailey LJ, Wagih S, Biasetti L, Pollack SJ, Quinn JP et al. The involvement of tau in nucleolar transcription and the stress response. Acta Neuropathol Commun 2018; 6(1): 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mansuroglu Z, Benhelli-Mokrani H, Marcato V, Sultan A, Violet M, Chauderlier A et al. Loss of Tau protein affects the structure, transcription and repair of neuronal pericentromeric heterochromatin. Sci Rep 2016; 6: 33047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Iqbal K, Liu F, Gong CX. Tau and neurodegenerative disease: the story so far. Nat Rev Neurol 2016; 12(1): 15–27. [DOI] [PubMed] [Google Scholar]
- 48.Eftekharzadeh B, Daigle JG, Kapinos LE, Coyne A, Schiantarelli J, Carlomagno Y et al. Tau Protein Disrupts Nucleocytoplasmic Transport in Alzheimer’s Disease. Neuron 2018; 99(5): 925–940 e927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kashani A, Lepicard E, Poirel O, Videau C, David JP, Fallet-Bianco C et al. Loss of VGLUT1 and VGLUT2 in the prefrontal cortex is correlated with cognitive decline in Alzheimer disease. Neurobiol Aging 2008; 29(11): 1619–1630. [DOI] [PubMed] [Google Scholar]
- 50.Jung HY, Yoo DY, Park JH, Kim JW, Chung JY, Kim DW et al. Age-dependent changes in vesicular glutamate transporter 1 and 2 expression in the gerbil hippocampus. Mol Med Rep 2018; 17(5): 6465–6471. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.