

Introduction
Throughout the history of life, evolution has relied on the basic processes of random mutation and natural selection to yield a diverse array of biomolecules with remarkable functions. The field of directed evolution (DE) has long sought to leverage the power of evolution to engineer novel biomolecular functions1,2. The mutation rate of DNA in a typical bacterial, yeast, or human cell is 10-10–10-9 substitutions per base3, or a mutation within a gene of average length (~1 kb) occurring approximately once in every million to 10 million cell divisions. At such low rates of mutation, it is difficult to sample even simple single mutations that improve a gene of interest (GOI) towards a desired function.
DE has traditionally turned to diversity generation in vitro, where high rates of mutation can be imposed on a GOI using error-prone PCR or randomized oligonucleotide pools2. The resulting libraries of GOI variants are then transformed into cells where they are expressed as RNAs and proteins and subjected to selection or screening. Enriched GOI variants serve as templates for the next round of in vitro diversification, transformation and selection or screening, advancing the evolutionary cycle (Figure 1A). While DE has successfully revolutionized biomolecular engineering — particularly fluorescent protein, enzyme and antibody engineering2,4 — its classical reliance on manually staged evolutionary steps limits the accessible depth and scale of evolutionary search. By requiring in vitro GOI diversification, classical DE forfeits the autonomous and decentralized operation of natural evolution and restricts DE campaigns to a few evolutionary cycles at the scale of a few independently evolving populations. To rapidly evolve GOIs while remaining in vivo requires targeting hypermutation to specific genetic material inside the cell while leaving the large host genome alone (Box 1).
Figure 1. Continuous evolution with cellular systems.
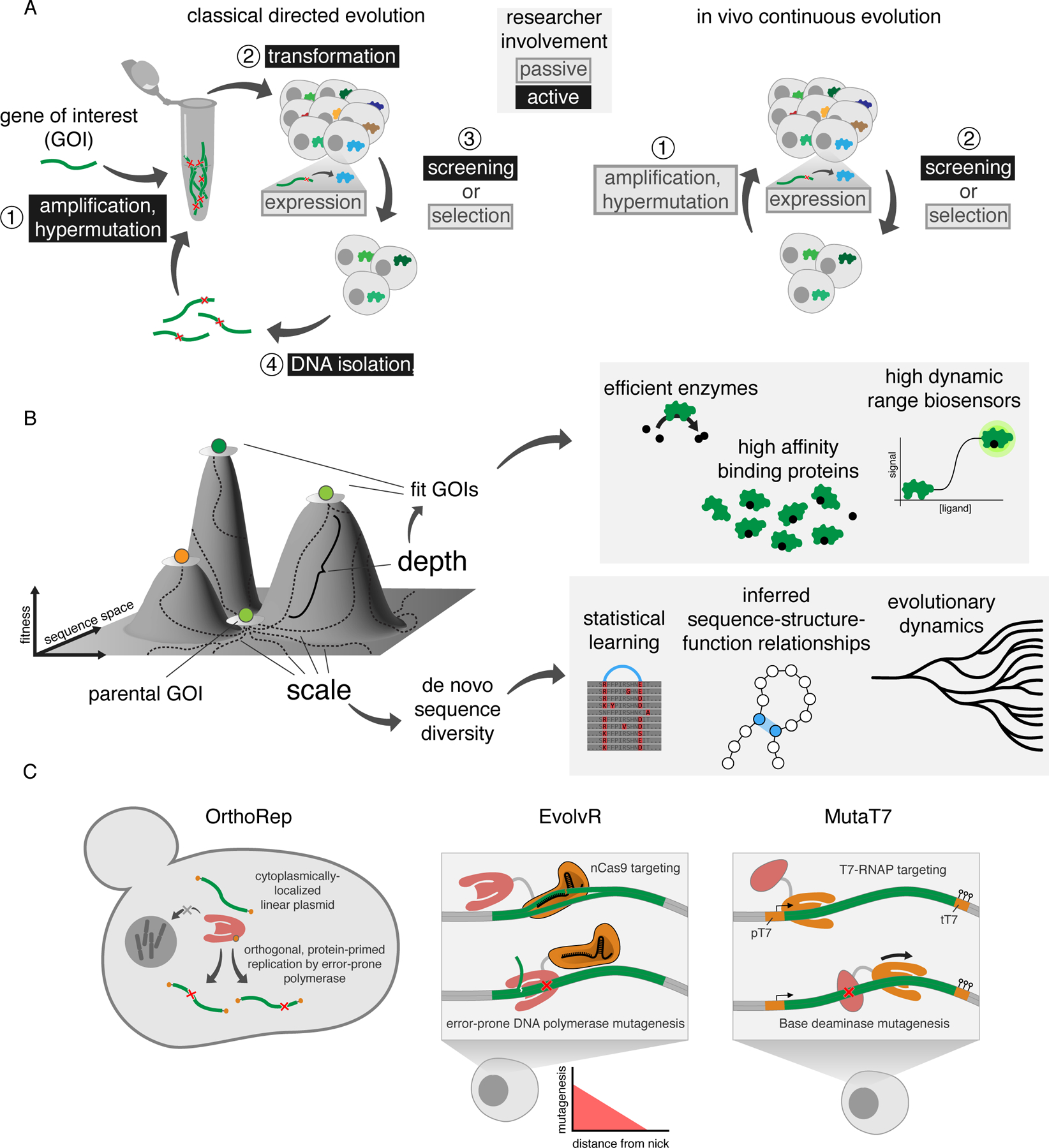
A) Comparison of classical directed evolution and in vivo continuous evolution. Classical directed evolution relies upon discrete steps performed in vitro (for example, hypermutation through error-prone PCR) and in vivo. Transformation, DNA isolation and PCR-based amplification and hypermutation are required to complete classical directed evolution cycles. In contrast, in vivo continuous evolution cycles can be carried out autonomously as cells grow. While amplification and hypermutation require cell culturing and therefore some researcher intervention, this dilution task is trivial enough to be considered passive. B) Depth and scale in continuous evolution enable broad applications. C) Depictions of OrthoRep, EvolvR and MutaT7. Each system achieves targeted hypermutation. OrthoRep and EvolvR both use error-prone DNA replication-based mutagenesis, with the distinction that in EvolvR, error-prone replication is not essential for propagation of the GOI since error-prone replication occurs in addition to replication by host DNA polymerases. MutaT7 utilizes base modification by a nucleotide base deaminase for mutagenesis. All three systems utilize distinct targeting methods. OrthoRep has only been applied in yeast, while both EvolvR and MutaT7 have been used in multiple model organisms. The OrthoRep accessory plasmid p2 is not depicted for simplicity. Note that termination of transcription and hypermutation by MutaT7 systems may be accomplished by terminators (depicted) or dCas9 (not depicted). pT7, T7 RNA polymerase promoter; tT7, T7 RNA polymerase terminator array.
Box 1: Genomic error threshold.
Evolution works best when the mutation rate is high enough to explore sequence space quickly. However, if it is too high, deleterious or lethal mutations are virtually guaranteed in every replication event29,30. This error threshold is determined by the amount of essential DNA-encoded information being replicated. Large genomes encode lots of essential information and have a low error threshold; small genomes have a higher error threshold. From the perspective of a single GOI, high mutation rates are therefore allowed and in fact needed to explore the GOI’s sequence space on laboratory timescales, but these same mutation rates are not tolerable for the cellular genomes which are typically larger by orders of magnitude. To bypass the genomic error threshold, classical DE approaches settle on diversification in vitro, while in vivo continuous evolution strategies specifically target hypermutation to the GOI.
In vivo continuous evolution relies on the construction of targeted hypermutation systems that durably and selectively mutate GOIs inside cells. With such systems, full evolutionary cycles consisting of rapid diversification, selection and amplification can run perpetually and automatically as cells replicate (Figure 1A). New types of biomolecular evolution experiments characterized by extensive search depth and scale are accessible through continuous evolution (Figure 1B). For example, continuous evolution can traverse long mutational pathways along rugged fitness landscapes to reach ambitious biomolecular functions (depth), and researchers can evolve many GOIs in parallel or one GOI in many replicates, making it possible to access larger sets of target functions, probe the rules of evolution and map sequence-function relationships with greater statistical power. These exciting opportunities provide motivation and aspirations for the broad application of the in vivo continuous evolution methods described.
The main approaches for in vivo continuous evolution can be divided into viral systems and cellular systems, differentiated by the unit of selection. In viral systems (Box 2), most prominently phage-assisted continuous evolution (PACE)5, the unit of selection is the virus, so evolvable GOI functions are those that can be linked to viral fitness. In cellular systems, the unit of selection is the cell, so evolvable GOI functions are those that can be linked to cellular fitness. With cells as the unit of selection, such systems are uniquely capable of screens or selections for GOI functions that couple to cell physiology and occur on the timescale of cell metabolism and biology. For this reason, and because viral systems have been extensively reviewed elsewhere6–8, this Primer focuses on cellular systems for in vivo continuous evolution.
Box 2: Virus-based in vivo continuous evolution.
With viral in vivo continuous evolution systems, the desired GOI activity is linked to the fitness of viruses as they propagate through cells. The most successfully applied viral system is PACE, in which the GOI is encoded on a bacteriophage genome engineered to lack essential components for the production of infective virus5,6. The engineered phages are used to infect inducibly-mutagenic Escherichia coli cells containing a genetic selection circuit designed to supply the missing components only if the GOI achieves a desired biomolecular function. In a PACE experiment, phages are serially or continuously diluted in fresh supplies of the mutagenic E. coli, resulting in the continuous evolution of the GOI function. Here, E. coli cells serve as reagents that do not have to replicate and are instead used, discarded and resupplied. Therefore, genome-wide high mutation rates that guarantee lethal mutations in the host genome are allowable as long as those high mutation rates do not destroy the small phage genome containing the GOI. Since the cell is not the replicating unit of selection, acting instead as a reagent whose mutations do not persist through evolutionary cycles, phages are the only entities that accumulate mutations, leading to a form of targeted hypermutation. From its initial publication in 2011, researchers have used PACE to evolve a wide range of targets including RNA polymerases31, biosensors32, proteases33,34, insecticides35, orthogonal tRNA/aminoacyl-tRNA synthetase pairs36 and genome editors37–39, demonstrating PACE’s enormous power. More recently, the PACE architecture has been successfully extended to mammalian cells through the development of adenovirus40 and sindbis virus41 versions that allow for the continuous evolution of mammalian biomolecules, expanding the scope of addressable problems.
Since the early 2000s, researchers have aimed to develop cellular systems for in vivo continuous evolution7–19. In these systems, the GOI is encoded in the cell’s genome, plasmids, or special DNA elements, and molecular machinery is engineered to target the GOI for hypermutation while sparing the rest of the genome. In this Primer, we focus on three recently developed systems: OrthoRep, MutaT7 and EvolvR (Figure 1C). In OrthoRep, a special error-prone DNA polymerase (DNAP) replicates a linear plasmid encoding the GOI. The DNAP/linear plasmid pair is orthogonal to genomic replication such that the error-prone DNAP does not replicate the genome and host DNAPs do not replicate the linear plasmid, thereby achieving targeting13,18. In MutaT7 systems, a nucleobase deaminase is fused to T7 RNA polymerase (T7RNAP). T7RNAP is specifically recruited to a T7 promoter placed next to the GOI and, as T7RNAP transcribes, the deaminase alters the GOI. The term MutaT7, coined in the first publication of such a hypermutation strategy16, serves as an umbrella term for all systems using a similar approach22–26. In EvolvR, an error-prone DNAP is fused to nickase Cas9 (nCas9)27. At a target site dictated by a guide RNA (gRNA), nCas9 makes a single-stranded DNA break from which the error-prone DNAP extends with low fidelity and limited processivity17,28. With these in vivo hypermutation systems in place, if the activity of the GOI is linked to cell survival, simply culturing cells under selection drives the evolution of improved GOI variants.
This Primer is for scientists looking to do their own in vivo continuous evolution experiments where the cell is the unit of selection. We describe experimental considerations, expected results, and successful applications.
Experimentation
There are five basic steps to completing an in vivo continuous evolution experiment (Figure 2): choosing the starting GOI sequences; setting up the hypermutation system; designing the selection; planning and executing the evolution campaign; and collecting and analyzing results (described in the next section). The first three should be approached concurrently to exploit interdependencies.
Figure 2. Generalized steps for carrying out a continuous evolution experiment.

A) a GOI is chosen for its predicted potential to evolve a desired activity. Often, the ability to carry out the desired activity at a low level is a promising starting point, although activities that are similar to the desired activity may be suitable as well. A host cell and a hypermutation system are also chosen at this stage. B) the researcher must design, build and test a selection or screen that is capable of enriching cells encoding GOIs with the desired function. This requires some cell-based mechanism of converting the activity of the GOI into a growth or optical signal, here symbolized as falling dominos. If available, GOI variants with known propensity for the desired activity, or ‘fitness’, may be used to test that the screen or selection is effective. C) the GOI and necessary components of the hypermutation system are encoded in the host cell. D) continuous evolution strategies are applied to evolve the desired activities. Different strategies offer different advantages and disadvantages and should be considered against each other. Labor, resources, feasible population size, durability and other factors can vary depending on the chosen strategy. E) following an evolution experiment that appears successful, GOIs contained in evolved populations should be isolated and characterized via sequencing and functional assays. The chosen approach will depend on the specific demands of the experiment and application.
Choosing a starting point for evolution
As is true for any DE experiment, choosing the GOI sequences from which to start evolution is a critical step. Important considerations include the activities of the starting sequences and whether strategies to maximize the scale of experimentation should be leveraged initially or after some pilot experimentation.
In classical DE, a typical precondition is that the starting GOI sequence — or at least one member of a library of variants built from the starting sequence — has detectable activity for the function being evolved. While this is also ideal for in vivo continuous evolution, this condition may be relaxed because the population sizes and diversity that can be accumulated through in vivo targeted hypermutation can be much higher than the diversity that can be transformed into cells for selection in classical DE. Nevertheless, characterization of activity in the starting GOI sequence is recommended to begin any evolution experiment.
With in vivo continuous evolution, one can leverage experimental scalability to evolve from multiple starting GOIs in separate experiments or one starting GOI in independent replicates. Collections of different GOI starting points (such as orthologs of an enzyme found in nature or computationally-designed libraries) create distinct opportunities to evolve the desired function42,43. Their separation into independent evolution experiments ensures that GOIs with weak initial activity but high potential to evolve very high fitness are not outcompeted by GOIs with high initial activity but low potential44–46. Likewise, separating a single evolution experiment from one starting GOI into multiple smaller replicates can limit the influence of clonal interference47, allowing the exploration of promising evolutionary paths that depend on early mutations with weak activity improvements29,43. Both strategies for starting point choice can increase the level of evolutionary success and are unique to the capabilities of in vivo continuous evolution.
Setting up the hypermutation system
The cellular hypermutation systems OrthoRep, MutaT7 and EvolvR each have special properties and specific setup requirements. When choosing the best hypermutation system for a particular experiment, it is important to first consider the host cell appropriate for expressing the GOI and the GOI function being evolved. OrthoRep currently only functions in yeast cells whereas MutaT7 and EvolvR systems function in E. coli, yeast and higher eukaryotes. Other aspects requiring consideration include durability and ease of implementation. OrthoRep is unique in that hypermutation of the GOI is enforced and that the host genome does not experience any elevation in mutation rate. These properties make it possible to durably mutate GOIs over prolonged continuous evolution experiments, as discussed in detail in previous literature48. EvolvR and MutaT7 systems are distinguished by their ease of implementation as they rely on standard parts such as nCas9 and expression elements from the T7 RNAP ecosystem.
Special aspects of OrthoRep
OrthoRep is derived from a natural plasmid system found in the yeast Kluveromyces lactis and ported to Saccharomyces cerevisiae13. The natural system is comprised of two linear plasmids in the cytoplasm, p1 (8.9 kb) and p2 (13.4 kb)49. Each plasmid is replicated by its own dedicated DNAP through a unique protein-primed mechanism in which the DNAP recognizes terminal proteins covalently linked to the 5ʹ ends of the plasmids to begin replication. The wildtype p1 plasmid encodes the DNAP that exclusively replicates p1, in addition to a toxin and its antitoxin. The p2 plasmid encodes the DNAP that exclusively replicates p2 ref50, in addition to associated replication components and transcription machinery for cytoplasmic expression from p1 and p2.
In OrthoRep, the p1 DNAP has been engineered to be highly error prone so that GOIs encoded on p1 experience an elevated mutation rate18. Due to the orthogonal replication mechanism, hypermutation is exclusive to p1 and does not affect the genome. The mutation rate of the most error-prone orthogonal DNAP engineered to date is 10-5 substitutions per base, or 100,000-fold above the genomic mutation rate. There are two error-prone orthogonal DNAPs in regular use, available as pAR-Ec633 and pAR-Ec611 on Addgene, and referred to as 633 and 611, respectively. The 633 DNAP contains the mutations L477V, L640Y, I777K, and W814N; and the 611 DNAP contains the mutations I777K and L900S. The 611 DNAP sustains a higher p1 copy number and a lower mutation rate than 633. Since a higher copy number leads to higher expression of the p1-encoded GOI, it is sometimes advisable to use 611.
OrthoRep uses an orthogonal transcription system. The p2 plasmid, which can be considered an accessory plasmid for OrthoRep, encodes an RNAP that recognizes special promoters driving GOIs on p1. Various promoters have been engineered to drive the expression of p1-encoded GOIs at strengths matching moderately-expressed host genes51.
OrthoRep-specific setup
OrthoRep requires the GOI to be integrated into the p1 plasmid. This starts with an S. cerevisiae strain that already harbors p1 and p2, such as strain F102–249,52. Integration cassettes can be designed to replace the DNAP and toxin-antitoxin genes present on wildtype p1 with any GOI alongside an antibiotic or auxotrophy selection marker. By transforming cells with linearized versions of such cassettes and selecting for the integration product, one obtains strains that contain a recombinant p1 with the GOI encoded. Then, an error-prone orthogonal DNAP encoded on a nuclear plasmid (such as pAR-Ec633 or pAR-Ec611) can be transformed into cells and the GOI will undergo autonomously hypermutation. The error-prone orthogonal DNAP can also be transformed concurrently with the p1-integration cassette.
A nuance in this procedure is that p1 is a multicopy plasmid. Therefore, when the GOI is integrated onto p1, resulting cells can carry both wild-type and recombinant p1. Once the error-prone DNAP is added to the cell, the wild-type p1 may no longer be required and can be lost over time. This process can be accelerated by using Cas9/CRISPR to excise wild-type p153 or designing a recombinant p1 that is smaller than wild-type p1 so it has a replicative advantage. It is often the case, however, that wild-type p1 will remain, because it allows for higher p1 copy numbers and higher expression of p1-encoded genes under selection. Although this has not resulted in any issues during continuous evolution experiments, S. cerevisiae strains have been engineered which are devoid of wild-type p1, containing instead a landing pad p1 that encodes only a selection marker and integration flanks for inserting GOIs (unpublished). Such strains use an error-prone orthogonal DNAP (611, the I777K and L900S variant) encoded on a nuclear plasmid or the genome to replicate p1.
OrthoRep is compatible across all S. cerevisiae strains tested53, and there are a variety of strains available upon request that contain a landing pad p1 to receive GOIs. It is also straightforward to transfer recombinant p1 and p2 plasmids from one strain to another by protoplast fusion53. This is recommended if a pre-existing, extensively engineered host strain is needed for selection of the desired GOI function.
Special aspects of MutaT7
MutaT7 systems have been applied in various research organisms, including E. coli (the original MutaT7 system16, eMutaT724 and T7-DIVA23), yeast (TRIDENT25), plants26 and mammalian cells (TRACE22). The main component of these systems is a protein fusion comprising an N-terminal nucleobase deaminase enzyme and T7RNAP. T7RNAP, derived from the T7 bacteriophage54,55, is highly specific for the T7 promoter, a 23 base pair (bp) sequence not native to genomes of standard research organisms, and it can transcribe almost any DNA downstream of its cognate promoter with high processivity56,57. Unlike OrthoRep and EvolvR, which rely on mutagenesis by error-prone DNA polymerases, MutaT7 systems rely on the recruitment of deaminase-T7RNAP fusions to loci adjacent to T7 promoters (Figure 3). Once the T7RNAP domain of a fusion protein recognizes and binds to the T7 promoter, it unwinds a small portion of double-stranded DNA (dsDNA) and initiates transcription. As transcription elongation proceeds, it is the non-template DNA strand that predominantly exists as single-stranded DNA (ssDNA) within the transcription R-loop58 and becomes exposed to the deaminase domain of the fusion protein, resulting in hypermutation (Figure 3C). The template strand usually hydrogen bonds with the nascent RNA and is therefore deaminated somewhat less frequently. The end of the target region of mutagenesis is delineated by a T7 terminator array16 or catalytically dead Cas923 (dCas9) directed with a CRISPR RNA (crRNA) array to block transcriptional elongation.
Figure 3. Mutagenesis mechanisms for in vivo hypermutation systems.

A) Types of nucleotide substitutions. B) and C) Following mutation by DNA replication error (B) or deamination (C), the nucleotide at the corresponding position on the opposite DNA strand is not yet altered. This mismatch will result in a mutation if it is used as a template in a subsequent DNA replication event. Alternatively, the mismatch repair machinery of the cell may correctly repair this mismatch, preventing establishment of the mutation.
Nucleobase deaminases used in MutaT7 systems are either cytidine or adenosine deaminases that accept ssDNA substrates59–64 As their names indicate, nucleobase deaminases catalyze the hydrolysis of exocyclic amines on deoxycytidine (dC) or deoxyadenosine (dA) to generate deoxyuridine (dU) or deoxyinosine (dI), respectively. The resultant dU or dI bases invert the hydrogen bonding properties of the original nucleotides, leading to temporary mismatches at deaminated positions. Unless these deaminated bases are eliminated by DNA repair systems such as uracil DNA N-glycosylase for dU65 or endonuclease V for dI66, these mismatches are resolved as permanent mutations when cellular DNA replication machinery reads dU and dI as deoxythymidine (dT) and deoxyguanosine (dG), respectively67,68. As a result, the deaminase-T7RNAP fusion proteins randomly generate all four possible base pair transition mutations (C•G→T•A, G•C→A•T, T•A→C•G, A•T→G•C) by deaminating the non-template, and somewhat less frequently, template strands. This strand bias can be mitigated by placing T7 promoters on either side of the target region facing inwards and installing terminator arrays just beyond the reciprocal T7 promoters16.
To date, three cytidine deaminases have been used in MutaT7 systems: rat apolipoprotein B mRNA editing catalytic polypeptide 1 (rAPOBEC1)69–71; activation-induced deaminase (AID), required for antibody maturation in the adaptive immune system72,73; and Petromyzon marinus cytidine deaminase 1 (pmCDA1), an AID homolog from sea lamprey74. The adenosine deaminases used so far for in vivo MutaT7-based hypermutation, as first demonstrated in the T7-DIVA platform23, are derived from the E. coli TadA, a tRNA-specific adenosine deaminase that has been evolved to accept ssDNA as a substrate75,76. Although there are now even more active TadA variants (collectively known as the TadA8s)39, these adenosine deaminases have yet to be implemented in the context of MutaT7.
The choice of deaminase will largely depend on the desired mutagenesis profile and the host organism. The original MutaT7 (MutaT7C→T) employed rAPOBEC116, and it was later demonstrated that the pmCDA1-T7RNAP fusion was 7–20 fold more mutagenic in E. coli24. A similar relatively higher mutation rate of pmCDA1-T7RNAP was concurrently observed with the T7-DIVA platform, which showed that the mutagenic activity of different fusions follows the hierarchy of AID < rAPOBEC1 < pmCDA1 in E. coli23. Demonstrating host dependence of base deaminases showed that AID*Δ (a hyperactive mutant of AID)-T7RNAP fusions were more active than rAPOBEC1-T7RNAP fusions in HEK293T cells with TRACE22. For the yeast MutaT7 system, TRIDENT, Cravens and colleagues employed pmCDA1-T7RNAP and also optimized a TadA variant for yeast, yeTadA1.025. In this publication, the group also showed that recruiting DNA repair factors involved in somatic hypermutation to deaminase-T7RNAP fusions can enhance mutagenic diversity by an apparent increase in editing of the template strand.
MutaT7-specific setup
To carry out a MutaT7 experiment, one encodes the GOI on a plasmid or in the genomes of host cells with a T7 promoter as the recognition element to recruit MutaT7 machinery. In E. coli, and in mammalian cells if an internal ribosome entry site is inserted before the GOI, the GOI can be translated directly from the T7 RNA transcript77. The T7 promoter can also be placed adjacent to the GOI in the antisense direction if GOI expression should not be driven from a T7 promoter. To define the endpoint of hypermutation, a T7 terminator array is inserted downstream of the T7 promoter, or a triple crRNA array/dCas9 targeted to the desired endpoint in the GOI can be inserted to limit mutation to a section of the GOI. Once these cloning or genome engineering operations are complete, the mutagenesis machinery (like the deaminase-T7RNAP fusion protein) is introduced. The mutagenesis machinery can be expressed genomically16,25 or from plasmids22–25 and can also be placed under inducible promoters to achieve varying levels of maximum expression and mutagenic activity at controlled times16,22,23.
Importantly, when using a cytidine deaminase, the activity of the DNA repair enzyme uracil-N glycosylase should be neutralized. This enzyme eliminates uracil from DNA to initiate base excision repair, thus suppressing cytidine deaminase-induced mutations. Deletion of the host ung gene (which encodes uracil-N glycosylase) can prevent this activity, as demonstrated in E. coli16,23. Alternatively, the uracil-DNA glycosylase inhibitor (UGI) from bacteriophage PBS2 can be expressed in the host78–82, as was done in eMutaT7 and TRACE.
Special aspects of EvolvR
The EvolvR system is comprised of a Cas9 nickase (nCas9)27 fused to a low-fidelity DNAP17. EvolvR diversifies GOIs by recruiting error-prone DNAP activity to single-stranded breaks generated by nCas9 at locations dictated by gRNAs. After nCas9 nicks and dissociates from its target sequence, the fused nick-translating error-prone DNAP initiates DNA extension from the 3ʹ end of the nick, displacing the incumbent strand while unidirectionally generating substitution errors according to the polymerase’s error rate. Unlike OrthoRep and MutaT7, EvolvR can target any locus with an adjacent protospacer adjacent motif (PAM) site without the need for prior engineering of the target sequence. As long as the target site remains sufficiently intact for recognition by the gRNA-Cas9 complex, hypermutation will continuously occur.
As EvolvR relies on nCas9 kinetics and DNA polymerization for generating mutations, its exact substitution rate and window length are modular and are determined by the properties of nCas9 and the error-prone DNAP. In its initial design, EvolvR was composed of a nCas9 (Streptococcus pyogenes Cas9 containing a D10A mutation) fused to the N terminus of a low-fidelity variant of E. coli DNAP I (PolI) harboring the mutations D424A, I709N and A759R (PolI3M)10,17. Different variants of EvolvR have been created by changing the fused DNAP to meet different needs. To increase the targeted hypermutation rate, a more error-prone PolI containing mutations F742Y and P796H in addition to those in PolI3M was developed (PolI5M)17. To increase the length of the editing window, several variants of the more processive bacteriophage Phi29 DNA polymerase (Phi29) were tested. While using Phi29 increased the targeted window length, it also reduced mutation rates. To increase the length of the editing window while maintaining a high mutation rate, the EvolvR variant nCas9-PolI3M-TBD (thioredoxin-binding domain of bacteriophage T7 DNA polymerase) was constructed; the TBD domain was previously shown to increase the processivity of PolI when inserted into the thumb domain of PolI in the presence of thioredoxin from E. coli83. The EvolvR variants nCas9 (D10A)-PolI3M or nCas9 (D10A)-PolI5M have been used in most of the experiments carried out with EvolvR so far.
EvolvR was initially developed in E. coli and more recently extended to S. cerevisiae28, providing both prokaryotic and eukaryotic model microbes as hosts for continuous evolutions with EvolvR.
EvolvR-specific setup
To use EvolvR, the first step is to design gRNAs to recruit EvolvR to target GOIs for hypermutation. A unique advantage of EvolvR is that one can target endogenous loci in addition to GOIs introduced exogenously in plasmids or integrated into the host genome. Mutations introduced by EvolvR occur at the highest frequency between the nCas9 (D10A)-generated nick and 20–40 bps 3ʹ of the nick, so the desired hypermutation region should be placed within ~20 bps of the gRNA spacer region. If the region of interest is longer than ~40 nt, the region of interest can be tiled with additional gRNAs. In this case, we recommend targeting the same strand, as the expression of two gRNAs that nick separate strands at nearby genomic locations generates double strand breaks, which are lethal in E. coli and may lead to abolishment of targeting in other organisms. Nicking the same strand at adjacent locations avoids these double strand break problems.
To express the components of EvolvR, distinct expression cassettes for nCas9-DNAP and gRNAs are included on a plasmid and transformed into the organism of interest. When porting EvolvR into a different strain or organism, we recommend testing different expression strengths of nCas9-DNAP to achieve maximal mutation rates with minimal global mutation rates. For example, in S. cerevisiae, a panel of promoters driving EvolvR expression was tested, including pREV1, pRET2, pRPL18B, pTEF1 and pTDH3 in order of increasing promoter strength28. Among them, the highest mutation rate was already reached at pTEF1 expression levels while the stronger promoter pTDH3 yielded higher off-target mutagenesis rates without higher on-target mutation rates.
Selection design
To take full advantage of in vivo continuous evolution, we recommend setting up selections that link the desired GOI function to cell fitness and/or survival. With survival-based selections, evolution experiments simply involve the serial culturing of cells under selection. Another viable approach is to link GOI function to an optical output for high-throughput screening via fluorescence activated cell sorting (FACS). The use of high-throughput screening breaks the cycle of continuous evolution into discontinuous steps, but even so, in vivo hypermutation allows staged cycles of diversification, selection and amplification to occur in a highly streamlined fashion.
A detailed discussion of selection design is beyond the scope of this Primer, but here we describe some basic principles. Evolution generally works best when selection pressure for the desired GOI activity can be increased over time. Therefore, selection strength should ideally be titratable, for example by altering the concentration of a chemical in the growth medium. The selection should also exhibit a high dynamic range so that higher activity is distinguishable from lower activity across the relevant range. The upper end of the selection, not just the fitness landscape on which the GOI evolves, will limit the possible results of the experiment. Mock selection experiments, in which GOI variants of known fitness are pooled and selection is applied without mutagenesis, can be used to confirm that the selection is capable of enriching fitter GOI variants and serve as a benchmark to evaluate the dynamic range of selection.
Sometimes, it can be helpful to select for an intermediate function that can act as a stepping stone to the final desired function. For example, in the continuous evolution of T7 RNAP to recognize new promoter sequences, hybrid promoters containing only some parts of the target promoter sequence were used as stepping stones5,84.
The durability of selection is an especially important consideration for in vivo continuous evolution. Since cells are the unit of selection and the typical continuous evolution experiment involves passaging cells over many generations under selection, opportunities to cheat compound. There are many paths that cells with cheater mutations can take to evade selection, like improving survival independent of GOI function; these cheater paths need to be mitigated and controlled for. The danger of potential cheater mutations outside the GOI is generally mitigated by the fact that the GOI is hypermutating, giving it most of the opportunity to satisfy the selected function. Still, before embarking on a full evolution experiment, it can be helpful to start with some small pilot experiments to determine an appropriate selection schedule that does not yield frequent cheaters.
Negative selections, in which undesired individuals are actively suppressed in the population, should also be given consideration when an undesired GOI activity can be selected for by the primary, positive selection. Though negative selections have not yet been demonstrated in cellular continuous evolution systems, they have been employed in both traditional directed evolution and in the viral continuous evolution system PACE to engineer specificity in tRNAs85, RNA polymerases31 and proteases34, and will likely be similarly instrumental in future continuous evolution campaigns in cellular systems.
Evolution campaign
There are several considerations for executing an evolution campaign. Within a cell culture, the mutational diversity of the GOI is determined by the size of the culture, the time throughout which mutations have been accumulating and the mutation spectrum and rate of the hypermutation system. The specifics depend on the chosen hypermutation system (Supplementary table 1), but generally, the larger the culture size and the longer mutations are allowed to accumulate, the higher the coverage of sequence space at any given point during evolution86. Another aspect to consider is the number of experimental replicates. Evolving several spatially separated populations at the same time can lead to several useful solutions because each replicate population may follow different evolutionary trajectories47,87. Additionally, this can prevent rare cheaters from dominating the experiment, since cheater mutations are stochastic and may not occur in every replicate. There is a practical tradeoff between the culture size and the number of replicates, however, so these should be balanced based on the culture size needed to achieve reasonable diversity and the expected benefits of many replicates. Finally, continuous evolution campaigns can be run with complex selection histories such as alternating phases of selection and neutral drift or even alternating selection environments, both of which can act to maximize the crossing of fitness valleys in the search for superior optima43,88.
During the evolution campaign, the only hands-on demands of the researcher are to tune the selection parameters and keep the cells propagating by diluting them into fresh media. For a hands-free approach, these steps can be automated in a continuous culture bioreactor, a vessel that maintains a culture with an equal inflow and outflow of media. The possibilities of automated culturing have been expanded with eVOLVER (not to be confused with the hypermutation system EvolvR), an open-source platform to continuously culture tens to hundreds of separate populations under independently controlled growth and selection conditions89,90. With eVOLVER, the researcher can program a closed feedback loop to adjust the selection pressure on each evolving population based on its measured growth rate or other parameters. In this way, each population is challenged or allowed to drift based on the fitness it has achieved. In addition to enabling automation, this can outperform a predetermined selection schedule that in some cases could lead to extinction or a sub-optimal fitness plateau during GOI evolution36,90.
Results
Data collection and analysis of results can be divided into sequencing and functional validation steps. While these are both required in all cases, which of these steps is more the focus depends on the goal of the project. If the primary goal is to obtain an applicable biomolecular function, low throughput sequencing methods, thorough characterization of evolved GOI fitness and functional studies on evolved biomolecules are usually sufficient. If the goal is to understand the space of accessible evolutionary outcomes, high-throughput sequencing and high-throughput functional enrichment assays that rank a large number of evolved variants in order of fitness are used. Here, there is also a unique synergy possible with computational approaches, especially machine learning, where large datasets comprising diverse evolutionary outcomes are used to train probabilistic models91–95. If the goal is to study the principles and rules of evolution itself, it may be necessary to carry out high-throughput sequencing and high-throughput functional enrichment assays across multiple time-points of an evolution experiment. We discuss these basic types of analyses and results for in vivo continuous evolution experiments below.
Sequencing
Sanger sequencing of heterogenous sequences produces mixed signals at nucleotides that are mutated in >10% of the population. Therefore, Sanger sequencing of populations at time-points throughout evolution is an easy way to identify the most common evolutionary pathways of a population. Sanger traces are context dependent, so to obtain accurate estimates of the frequency of particular mutations in a population, one must compare Sanger sequencing traces of an evolved population to traces of the wild-type sequence with a computational tool such as QSVanalyzer96. This provides estimates of population-level mutation frequency similar in accuracy to high-throughput sequencing, but with much lower labor requirements and cost for few samples. Due to the aggregated nature of this data, using it to identify which mutations appear in the same sequence is challenging, but possible; the most common genotypes in a population can be identified with frequent Sanger sequencing time-points97. Identifying rare mutational pathways using this method becomes arduous and expensive with many time-points and/or populations, and therefore lacks the scale necessary to take full advantage of the information on complex epistatic interactions and residue covariation that laboratory evolution is capable of providing36,92.
Unlike Sanger sequencing, high-throughput sequencing can detect low-frequency mutations and linkage among mutations on a large scale. DNA for high-throughput sequencing is generated by PCR amplification of targets from distinct samples using primer-appended barcodes to demarcate different evolutionary cultures or time-points. These can then be combined prior to sequencing preparation and subsequently demultiplexed during analysis steps, saving in preparation and sequencing costs. Additionally, clonal populations used to seed replicate evolution experiments may be uniquely barcoded at the start of evolution to enable multiplexing prior to DNA isolation and PCR.
There are several high-throughput sequencing platforms that are viable options for continuous evolution sequencing projects98–100. The choice of which to use depends on both the length of the target gene and the project goals. Short-read high-throughput sequencing platforms are sufficient if the GOI under evolution is less than ~450 bp in length or if long-range mutation correlations are not of interest, in which case subsections of the GOI can be sequenced independently.
Total sequencing yields and sequencing error rate vary across sequencing platforms and methods of library construction. Illumina®’s short-read sequencing platform MiSeq™ produces sequencing yields up to 15 Gb, read lengths of ~500 bp, and a raw read accuracy of 99.5% or roughly five sequencing errors per 1 kb100,101. Longread sequencing platforms provided by Pacific Biosciences (PacBio®) and Oxford Nanopore Technologies (ONT) offer sequencing yields of 50 Gb and contiguous read lengths of more than 10 kb, but with accuracy ranging from 90–98%99,102,103. The relatively low accuracy of raw sequencing reads, which typically guarantees several errors per sequence, can still be valuable for certain applications. For instance, in engineering-focused applications, high-throughput mutant data coupled with Sanger sequencing of selected clones can reveal the most common and most consequential mutations. Methods to process sequencing data to obtain higher accuracy at the cost of read depth can also be considered.
Higher accuracy than that of raw reads are achievable by combining multiple independent reads of the same original sequence to form a consensus sequence. This is typically accomplished either through circular consensus, or through unique molecular identifier (UMI) consensus sequencing. For circular consensus, a template is circularized prior to amplification, resulting in concatemeric reads containing multiple copies of the original linear template to form a consensus sequence. For UMI consensus, UMIs composed of random DNA barcodes are appended to the template prior to amplification and sequencing, and consensus sequences are derived from reads with the same or similar UMIs.
These error correction methods allow long-read sequencing platforms to compete with short-read sequencing platforms that have higher raw read accuracy. PacBio® is currently the more accessible long-read sequencing platform due to its standardized error correction procedures104, but recent examples of error correction methods for ONT sequencing data demonstrates the potential utility of this platform103,105,106, particularly considering the relatively low cost of sequencing device ownership, which can facilitate rapid data generation.
High-throughput sequencing datasets can be analyzed using freely available tools for the several necessary steps such as demultiplexing (Axe107), alignment (Minimap2108) and variant calling (VarScan2109). When multiplexing samples, care should be taken to prevent or measure the degree of template switching, which can result in erroneous demultiplexing assignments. Data processing pipelines such as Breseq110 unify some of these steps and improve reproducibility.
Validating evolved activities
A successful continuous evolution experiment will produce a population of GOI variants that satisfy the selection and/or screening conditions. However, cheater mutations that circumvent the selection and/or screen are possible, and deleterious on-target mutations generated toward the end of the experiment may not have had sufficient time to be purged from the population. The expected phenotypes for individual GOI variants must be confirmed in host cells that have not been subject to selection. This can be accomplished through PCR amplification of GOI variants, cloning into a plasmid backbone in library format, then transforming this library into host cells. In such uniform fresh strain backgrounds, phenotypic differences will be representative of GOI function. Variants can then be evaluated fairly, ideally using multiple distinct assays.
At a minimum, evolved variants should be compared to unevolved variants using the same selection or screen for the evolution experiment. This comparison can also be performed in high throughput by using functional enrichment assays where barcoded evolved variants are pooled and subject to growth under selection. Enrichment scores for each variant can then be calculated measuring barcode frequencies before and after selection via high-throughput sequencing111. In such experiments, it is often necessary to first use long-read sequencing to match barcodes to specific GOI sequences and then use short-read sequencing — where greater read numbers are available — to track the enrichment of barcodes. The resulting data provides a measurement of relative fitness for many GOI variants, which can be compared to parental GOI variants present in the library.
If engineering is the primary goal of the study, characterization beyond fitness-based assays should be performed to rule out the possibility that unexpected activities are responsible for fitness differences. Individual GOI variants with high fitness in clonal populations can be isolated and the biomolecules encoded by those variants encode purified for in vitro biochemical studies or biological assays.
Applications
Successful applications of cellular systems for in vivo continuous evolution fall under three categories: studying pathways to drug resistance, enzyme engineering and FACS-based evolution. Potential applications are much broader and we discuss them further in the Outlook section.
Studying pathways to drug resistance
Cellular systems for in vivo continuous evolution can be applied to rapidly uncover drug-resistance mutations in clinically-relevant targets (Figure 4A). EvolvR has been used to target the endogenous E. coli rspE gene for hypermutation to identify novel spectinomycin-resistance mutations in the ribosomal unit S517. A single overnight growth step to diversify the gene led to resistant variants fixed by selection. Similarly, MutaT7C→T was used in E. coli to target an episomal copy of rpsl, which encodes the ribosomal unit S12, to evolve streptomycin-resistant S12 variants after 24 hours of growth/mutagenesis16. Variants of the E. coli dihydrofolate reductase (DHFR) resistant to trimethoprim were also evovled using a continuous evolution approach in a bioreactor.
Figure 4. Application of continuous evolution for studying drug resistance.
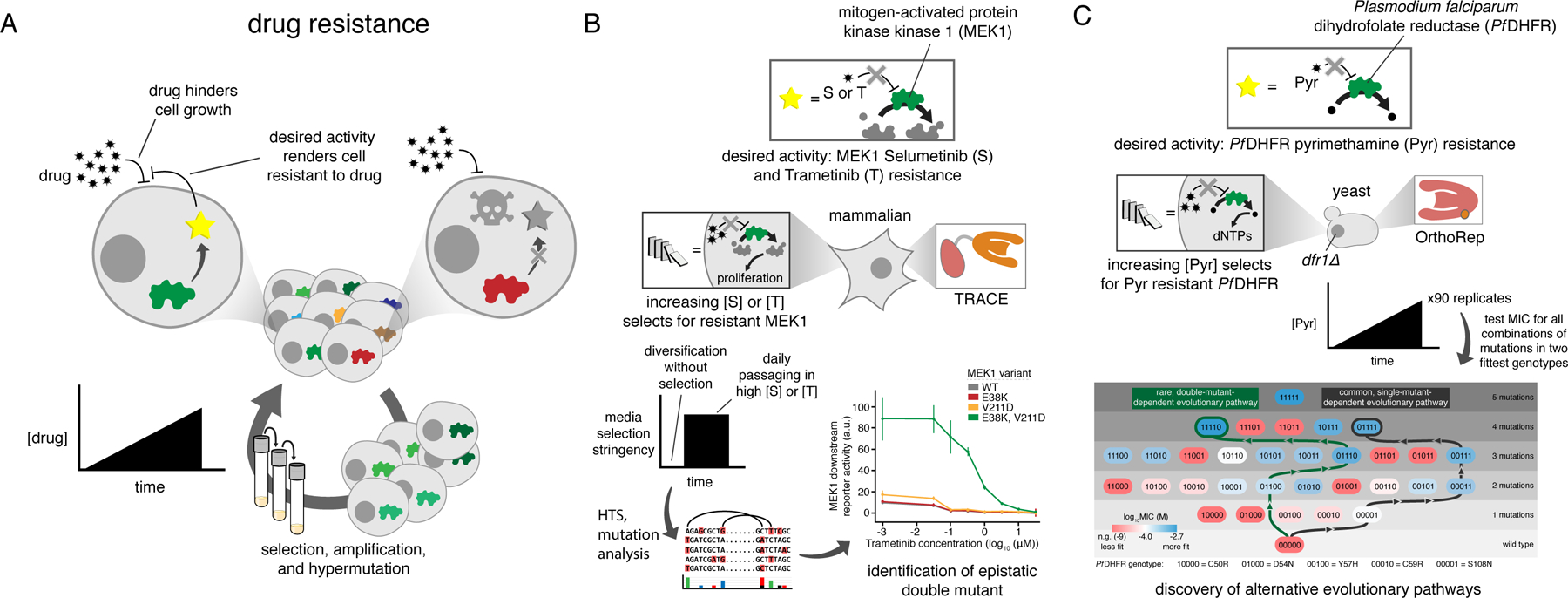
A) Generalized schematic for drug resistance continuous evolution experiments. A cycle with only one step is shown but is meant to represent continuous growth following desired selection schedules, for example serial passaging into media with increasing drug concentration. B) Continuous evolution of MEK1 with the TRACE MutaT7 system. The native activity of MEK1 is essential for proliferation in the mammalian cell line that was used (HEK293T) and is therefore drug selectable. MEK1 was targeted for mutagenesis and grown for a short initial period without selection for drug resistance to generate mutational diversity. Selection was then applied using a static high concentration of either of the two drugs being studied. C) Continuous evolution of DHFR to study drug resistance using OrthoRep. DHFR activity is essential for nucleotide biosynthesis and is therefore selectable in cells deleted for the native enzyme. Ninety replicate cultures were passaged in parallel with gradually increasing drug concentration, and Sanger sequencing of populations revealed two genotypes that commonly become fixed in populations. All combinations of the mutations in these two genotypes were individually cloned and tested in isolation to determine fitness and understand the accessibility of different mutational pathways leading to different outcomes. MIC, minimum inhibitory concentration.
With MutaT7 systems in E. coli, both T7-DIVA and eMutaT7 were used to evolve TEM-1 β-lactamase for resistance to third-generation cephalosporin antibiotics23,24. With T7-DIVA, two iterative cycles of mutagenesis followed by one selection step produced double mutants with a >1000-fold increased minimum inhibitory concentration of ceftazidime. With eMutaT7, serial passaging of batch cultures into increasing antibiotic concentrations was performed, and clones were isolated after 24–32 hours with 9–16 mutations and ~10,000-fold increases in minimum inhibitory concentrations to cefotaxime and ceftazidime. The MutaT7 system TRACE was also used in mammalian cells to identify two functionally correlated mutations in mitogen activated protein kinase kinase 1 (MEK1) that promote resistance to selumetinib and trametinib — two pharmacologically-relevant MEK1 inhibitors22. MEK1 was integrated under a T7 promoter into the genome, diversified and selected for by screening for drug-resistant cells (Figure 4B).
By taking advantage of the accessible depth and scale of in vivo continuous evolution, multiple mutational pathways across complex evolutionary landscapes can be explored. In a demonstration of this, OrthoRep was used to study how dihydrofolate reductase (DHFR) from the malaria-causing parasite P. falciparum (PfDHFR) acquires resistance to pyrimethamine in 90 small volume (0.5 mL) replicates18 (Figure 4C). An engineered yeast strain solely dependent on PfDHFR encoded on the hypermutating p1 plasmid was used. After 13 passages into increasing concentrations of pyrimethamine, 78 replicates adapted to the highest soluble concentration (3 mM) and yielded new highly-resistant variants with 3–6 mutations. Sanger sequencing of each replicate population across time-points showed that multiple mutational pathways in PfDHFR led to resistance. Intricate interplay among adaptive mutational pathways was elucidated and traced to the existence of greedy mutations, sign epistasis, and clonal interference. From these data, population structures and strategies that favor certain pathways over others were predicted and confirmed through additional replicate evolution experiments, providing a template for strategies to control and possibly slow the emergence of resistance.
The MutaT7 system TRIDENT was also used to evolve pyrimethamine-resistant PfDHFR variants in yeast25. In contrast to experiments performed with OrthoRep, the study with TRIDENT observed the dominance of a single mutation (D54N) that conferred resistance to 3 mM pyrimethamine across 180 replicate cultures. In the OrthoRep experiment, 3–6 mutations were necessary to achieve full resistance to pyrimethamine with S108N, C59R, Y57H, and D54N being most dominant18. This is in congruence with what is observed in the field, where multi-mutant variants containing S108N and C59R are common. A possible explanation for why OrthoRep experiments discovered larger mutational sets that more closely match what is found in the field is that evolution experiments with OrthoRep and TRIDENT started from different strengths of PfDHFR expression.
Enzyme engineering
By coupling the activity of an enzyme to cell growth, one can apply in vivo continuous evolution to engineer enzymes towards improved (or potentially different) functions (Fig. 5A). In one example, eMutaT7 was used to evolve DegP, a bacterial heat-shock protease, for increased activity24. A less-active variant of DegP was subjected to continuous evolution by a stepwise increase of temperature during growth cycles. The mutation found at the center of the trimeric DegP was shown to be allosteric, activating DegP without direct interaction with the substrate. In a second example, EvolvR was used to improve the catalytic efficiency of ornithine cyclodeaminase (OCD) for l-proline synthesis from l-ornithine112 (Fig. 5B). A growth-based screen was created in which proline codons in an antibiotic resistance marker were replaced with rare codons, leading to a growth defect that can be rescued by increased l-proline production. After diversifying OCD with EvolvR, variants conferring higher growth were screened for and three mutations in OCD were found that, when combined, improved enzyme activity by 2.4-fold.
Figure 5. Application of continuous evolution for enzyme engineering.
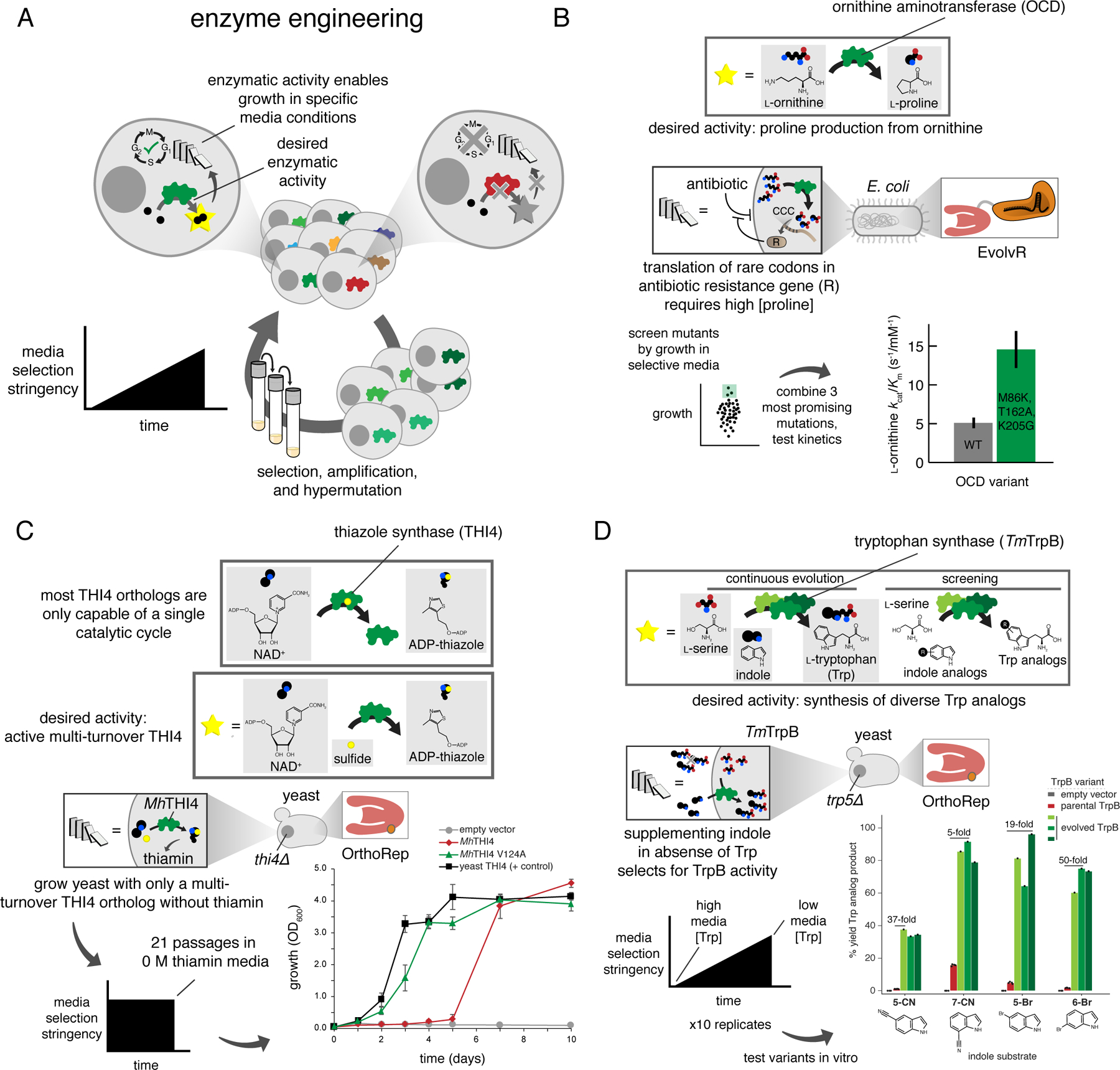
A) Generalized schematic for enzyme engineering through continuous evolution. A cycle with only one step is shown, as experiments are typically carried out using growth-based selections. This means all of the necessary steps for evolution are accomplished by the cell concurrently when provided with a consistent supply of media with increasing selection stringency. Media selection stringency modification is typically accomplished through reducing the concentration of a required nutrient whose production is made to be dependent upon GOI activity. B) Continuous evolution of OCD for improved proline production. Following diversification with EvolvR, clones underwent a single round of screening on the basis of colony size prior to testing for growth. C) Adaptation of a multi-turnover thiazole synthase from the prokaryote Mucinivorans hirudinis for activity in the plant-like cellular environment of yeast. D) Continuous evolution of tryptophan synthase from the thermophile Thermotoga maritima (TmTrpB) to generate enzymes with diverse substrate promiscuities.
In another enzyme engineering example, OrthoRep was used to evolve the thiamin biosynthesis enzyme THI4 from the anaerobic bacterium Mucinivorans hirudinis (MhTHI4) for plant applications 113,114 (Fig. 5C). Many eukaryotic THI4 orthologs, including those of plants and yeast, use an active-site cysteine residue as a sulfur-donor for the reaction and can thus catalyze only one reaction, making these enzymes energetically costly and a target for replacement with a longer-lived version115. MhTHI4 instead uses a free sulfide as a sulfur-donor and mediates multiple reaction cycles116. However, this ortholog is not fit to function in plants, as it is oxygen-sensitive. To adapt it to function in plant-like conditions, MhTHI4 was encoded onto the OrthoRep system in a yeast strain with the native THI4 deleted. After 21 passages of 9 starting populations, multiple single and double mutants were obtained that improved growth in the absence of thiamin.
OrthoRep combined with the continuous culturing platform eVOLVER was used to adapt an enzyme to a new environment, thereby demonstrating the capabilities of automated continuous culturing89,90. The setup, termed Automated Continuous Evolution (ACE), was used to evolve the thermophilic Thermotoga maritima HisA enzyme (TmHisA) for mesophilic activity in yeast, with implications for industrial biotechnology applications. The evolution of HisA highlights ACE’s potential to realize speed (ACE arrived at HisA solutions hundreds of hours faster than manual batch culture-mediated selection), scale (ACE autonomously managed replicate cultures at >25 mL volumes with frequent, minimal dilutions, minimizing population bottlenecks and independently modulated the histidine concentration of each culture based on feedback from real-time growth rates, maintaining optimal selection across the replicates) and depth (evolution occurred over 600 hours of continuous selection through long mutational pathways ranging from 5 to 18 mutations, suggesting that ACE can traverse relatively complex fitness landscapes that necessitate a large number of small effect mutations to reach desired activity).
In a final example of enzyme engineering, a diverse set of TrpB variants was evolved for substrate promiscuity117 (Figure 5D). TrpB and its allosteric partner TrpA make up tryptophan synthase, which is responsible for the final stages of l-tryptophan (Trp) production118. After receiving indole from TrpA, TrpB synthesizes Trp by coupling the indole to l-serine. TrpB enzymes can also accept indole analogs and readily convert them to Trp analogs, which are useful as biological probes and as scaffolds in the synthesis of pharmaceuticals. Previously, several DE campaigns have been carried out to evolve TrpB to function in the absence of TrpA and expand its substrate scope119–122. Rix et al. reasoned that in vivo continuous evolution could be used to improve and scale this process117. Using OrthoRep, a thermophilic TrpB enzyme was continuously evolved in yeast to complement the biosynthesis of Trp from exogenously supplied indole in several replicates, resulting in highly active TrpB variants containing up to 16 mutations. A panel of more than 60 TrpB variants from 10 independently evolved populations displayed a diverse range of promiscuous activities, with up to 50-fold improvements in activity at mesophile temperatures, despite selecting only for cognate Trp synthesis activity. Not only are these TrpB variants commercially useful, using this new method for de novo generation of enzyme orthologs should be general to the expansion of activities and substrate promiscuity profiles of other biosynthetic enzymes.
FACS-based evolution
In vivo continuous evolution systems can also streamline the engineering of biomolecules with FACS when the desired function is tied to a fluorescent output (Fig. 6A). The simplest application is to evolve a biomolecule that is itself fluorescent, such as a fluorescent protein. In two such examples, the MutaT7 strategy was applied in yeast to evolve a red-shifted variant of mCherry25 (Fig. 6B) and in mammalian cells to shift the emission spectra of blue fluorescent protein to that of green fluorescent protein (GFP) 22.
Figure 6. Continuous evolution applications employing FACS-based screening.
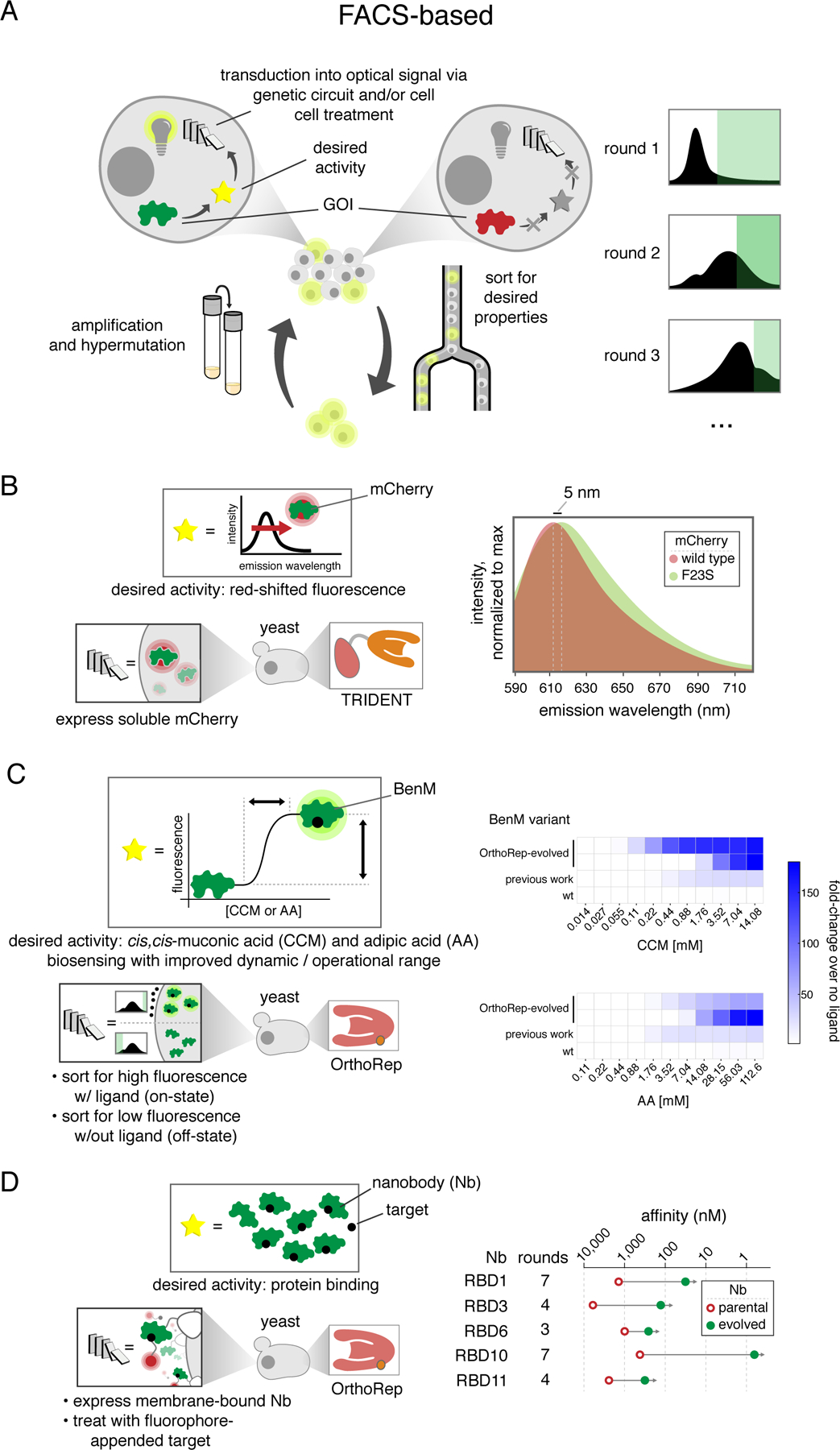
A) Generalized schematic for continuous evolution experiments that employ FACS-based high throughput screening to evolve GOIs with a desired activity. Unlike with selections, screening and amplification must be carried out in two discrete steps. B) Evolution of a red-shifted mCherry using the TRIDENT MutaT7 system. C) Evolution of the cis,cis-muconic acid biosensor BenM to exhibit higher dynamic range and broader operational range. D) Nanobody evolution using OrthoRep for diversification and yeast display for FACS-based screening.
Another way to combine in vivo continuous evolution and FACS is to evolve a GOI whose desired function leads to the expression of a fluorescent reporter gene. For example, a transcription factor biosensor was evolved that transduces the concentration of a desirable small molecule to the expression of GFP. OrthoRep was ised to evolve the allosteric transcription factor BenM for activity in the presence of its cognate ligand, muconic acid, as well as a non-cognate ligand, adipic acid123 (Fig. 6C). With BenM encoded on p1 for targeted hypermutation, 11 evolutionary cycles of yeast culturing were carried out, and positive and negative rounds of FACS were used to enrich cells that expressed GFP only in the presence of the small molecule. The evolved biosensors displayed broad operational ranges of sensitivity to biologically relevant concentrations of muconic and adipic acid, as well as high dynamic ranges up to 180-fold. High-performance biosensors can in turn be used as the readout to evolve synthetic metabolic pathways that more efficiently produce the sensed molecule. In a related example, an enzyme involved in a muconic acid production pathway in yeast was encoded on OrthoRep with BenM as the biosensor to guide selection for higher muconic acid production124.
OrthoRep has also been used to drive the rapid evolution of antibodies in a system termed AHEAD (Autonomous Hypermutation yEast surfAce Display) (Fig. 6D). Here, antibody scaffolds are encoded for yeast surface display from the orthogonal p1 plasmid125. Culturing of the yeast cells results in the self-diversification of the displayed antibodies such that straightforward cycles of yeast growth, induction of surface display and FACS for cells that bind to a labeled antigen generates high-affinity antibody variants over time. In the midst of the COVID-19 pandemic, AHEAD was used to evolve nanobodies with sub-nanomolar affinity and pseudovirus neutralization potency to the receptor binding domain (RBD) of SARS-CoV-2. Starting from a naïve synthetic nanobody library, 8 parental clones were selected with weak binding to RBD, the sequences were transplanted onto p1, and 8 separate evolution experiments were carried out involving cycles of yeast culturing and FACS to affinity mature the parental clones into high-affinity RBD binders. The resulting nanobodies reached subnanomolar binding affinities and neutralization potencies by evolving several-hundred-fold improvements in some cases. The streamlined nature of AHEAD experiments allowed the 8 evolution experiments to be run in parallel, which prevented clonal interference among lineages derived from distinct parents and promoted functional diversity, such as the location of RBD bound, in the set of final binding proteins.
Reproducibility and Data Deposition
Evolution campaign reporting
To ensure reproducibility, researchers undertaking in vivo continuous evolution experiments must report important details of their experimental design as well as how evolved sequences are characterized and annotated. In vivo continuous evolution systems are still under active development, so in addition to reporting the specific system that is used, its precise architecture (exact variants of mutagenic polymerases/enzymes, sequences of gRNAs, genetic modifications to publicly available strains) should also be reported. If feasible, researchers should include the exact sequences of plasmids and modified genomic loci present in strains used for evolution.
The selection used for evolution experiments should be well tested and documented. The exact sequences of GOI starting variants should be reported. How selection is applied during evolution, including the number and volume of cultures, how dilutions are carried out, volume of culture transferred in each passage, the increments used in modifying selection stringency and the criteria used to determine when to increase selection stringency, should all be documented. Additionally, controls used to confirm that evolution is working as expected, such as GOI variants with a single inactivating mutation, should be described.
Evolution outcomes reporting
All GOI variants that are characterized individually should be fully sequenced, even if mutagenesis was targeted only to a particular region of the GOI, and full sequences should be included in publication. Lists of mutations are of course necessary, but for convenience of other researchers and to capture synonymous mutations that may have functional significance, complete sequences should be included as well.
Given the wealth of sequence diversity that continuous evolution can generate to the benefit of future researchers, we encourage high-throughput sequencing data to be collected, properly annotated and publicly deposited. Both raw sequencing data and pre-processed data (for example, that has been demultiplexed or error-corrected) should be deposited on a public database such as the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA), the NCBI BioProject, and/or the European Nucleotide Archive (ENA). Ideally, any analysis performed on high-throughput sequencing data should be easily reproduced, for instance using a version-controlled pipeline that is available for download with clear installation instructions. At a minimum, the analysis steps performed should be carefully described, including all non-default options used for command line tools. Any custom scripts that are critical to the conclusions of a study should be publicly accessible, accompanied by a description of the necessary dependencies.
Limitations and Optimizations
Host
When deciding which system to use for an in vivo continuous evolution experiment, one clear consideration is the host. OrthoRep has only been demonstrated in yeast; MutaT7 systems have been established in E. coli16,23,24, yeast25, plants26 and mammalian cells22; and EvolvR has been successfully tested in E. coli17 and yeast28. Host choice is typically determined by the biomolecules being evolved — whether they function natively in the host or require host-specific posttranslational modifications, for example — as well as the ease of setting up a reliable genetic or cell-based selection in the various hosts being considered. Other considerations include generation times, population sizes and scale of experimentation possible with different hosts.
OrthoRep, MutaT7 and EvolvR systems are currently being developed and optimized for compatibility with a broader host range. For OrthoRep, it is unknown how difficult it will be to transfer the underlying orthogonal replication machinery into hosts beyond yeast. It may also be possible to boot up OrthoRep in bacteria or mammalian cells by using the DNA replication systems of existing bacterial or mammalian viruses that may be (or engineered to be) orthogonal to host DNA replication126. For MutaT7 and EvolvR systems that already operate in bacteria, yeast and mammalian cells, areas of optimization include addressing host-specific differences in the mismatch repair systems responding to hypermutation, toxicity or burden of the mutagenesis machinery113 (such as deaminase-T7RNAP fusion or nCas9-DNAP fusion), and minimizing cargo size, as in the case of EvolvR, for delivery and stable expression of mutagenesis machinery in mammalian cells.
Hypermutation characteristics
The hypermutation rate of in vivo continuous evolution systems determines how long it takes a cell to sample new GOI sequences at any given time during an evolution experiment. The hypermutation profile determines what types of mutations are sampled. Although it is possible to reach a hypermutation rate that will effectively render any GOI inactive in just one cycle of replication, current in vivo continuous evolution systems are far from this lethal mutagenesis rate. Thus, increasing the mutation rates and expanding the mutational spectrum of OrthoRep, MutaT7 and EvolvR are active areas of research. As it stands, one typically prefers the highest mutation rate and broadest mutational profiles when selecting systems. Since these characteristics have not always been measured in the same way, it is not straightforward to directly compare them across different systems, but we make an attempt in Supplementary table 1.
Another consideration for hypermutation is the level of off-target mutagenesis. Off-target mutagenesis increases the chance of genomic adaptation, mutations in the genetic selection system used to guide evolution, mutations that modulate the hypermutation system itself and mutations that are deleterious to cellular fitness. An advantage of OrthoRep is that there is no measurable mutation rate elevation in the host genome when the GOI is being continuously hypermutated18. This derives from the mechanistic and spatial separation of DNA replication between the orthogonal p1 plasmid and the genome. The error-prone polymerase of EvolvR and the deaminase of MutaT7 cause low but measurable off-target mutagenesis, currently a few-hundred-fold lower than on-target mutagenesis. To achieve low off-target mutagenesis while maximizing on-target mutagenesis in the case of EvolvR and MutaT7, the expression level of the mutation machinery is optimized for each new strain and host.
Finally, an important feature of in vivo continuous evolution is that evolution should be able to occur for extended periods of time, wherein continuous operation of mutation, amplification, and selection cycles explore long mutational paths over many generations. For this to occur, hypermutation must be durable. The durability of hypermutation in OrthoRep is high. Evolution experiments with OrthoRep have been carried out for hundreds of generations with continued evolution18,90. Durability of mutagenesis for MutaT7 and EvolvR systems have not been tested thoroughly but is likely lower than for OrthoRep. This is because the elements recruiting hypermutation machinery, such as the T7 promoter or gRNA target site, can themselves become mutated while still allowing the GOI to be replicated and expressed by host machinery. This may allow the system to reduce its own hypermutation rate over time. The measurable off-target mutation rate of MutaT7 and EvolvR also elevates the chance of mutations in the mutagenesis machinery itself, potentially causing changes in the hypermutation rate over time especially if there is toxicity associated with MutaT7 and EvolvR parts that create selective pressures for their functional degradation. Indeed, reducing burden, toxicity and off-target mutagenesis for MutaT7 and EvolvR are areas of ongoing optimization.
Target size and context
The amount of genetic cargo that can be placed on OrthoRep is up to >20 kb53, although smaller cargo sizes up to ~7 kb are most tractable. MutaT7 systems may tolerate up to >25 kb based on the processivity of T7 RNAP57, although only smaller cargo sizes up to ~2 kb have been tested. The amount of DNA undergoing hypermutation for each targeting gRNA used in EvolvR is under a few hundred bps depending on the EvolvR DNAP used, but one can employ a collection of gRNAs to target multiple loci to expand the effective size of the genetic cargo undergoing mutagenesis. Still, the size of what can be targeted for hypermutation imposes limits for the various in vivo continuous evolution systems.
Another consideration is the context of the target GOI under evolution. EvolvR has the unique benefit that any locus targetable with gRNAs can be the subject of hypermutation. Therefore, genomic loci in their native context can be continuously evolved, preserving native regulation of expression and reducing engineering requirements. MutaT7 can also target host genomic loci, but the loci must first be engineered to contain a T7 promoter. While installing T7 promoters can be relatively trivial if target regions are on plasmids, doing so can be challenging or infeasible if the desired target regions are genomic, although continued innovation in the genome editing field is making genome engineering routine127–129. OrthoRep is restricted to GOIs encoded on the orthogonal p1 plasmid and cannot target genomic loci for hypermutation. Additionally, the cytoplasmic localization of p1 may complicate the evolution of RNAs that function in the nucleus. Some GOIs cannot be evolved with in vivo continuous evolution in general, namely those that are toxic to the host or a GOI whose function cannot be selected or screened for directly in cells.
Finally, targeting only a region of a GOI for evolution (one specific domain in a protein, for example) may be possible with MutaT7 by using dCas9 to terminate MutaT7’s action in the middle of a GOI while still allowing the entire GOI to be expressed, although this technique does not enable exclusion of both termini of a GOI from hypermutation23. EvolvR can also achieve partial mutagenesis of a GOI by using gRNAs corresponding to a small region in a GOI along with nCas9-DNAP fusions where the error-prone DNAP has low processivity. OrthoRep cannot selectively target one part of a GOI for hypermutation because the entire GOI needs to be encoded on the orthogonal p1 plasmid for expression as a single protein product. However, it may be possible to split a GOI into domains that are post-translationally joined, for example by using a split intein130, in which case one domain can be encoded on p1 for hypermutation and the other domain can be encoded on a host plasmid or in the host genome where it is not hypermutated.
Ease of implementation
An advantage of MutaT7 and EvolvR over OrthoRep is their reliance on standard parts. OrthoRep experiments require custom promoters for GOIs and custom genetics for integrating genes onto the cytoplasmic orthogonal p1 plasmid. In contrast, MutaT7 and EvolvR use standard genomic or plasmid engineering techniques to encode GOIs and benefit from a rich ecosystem of T7 RNAP or Cas9/CRISPR parts, making experiments easier to set up. This advantage of MutaT7 and EvolvR over OrthoRep should be balanced against the architectural advantages of OrthoRep in supporting continuous GOI evolution experiments durably over extended periods of time.
Outlook
DE of GOIs has traditionally used iterative cycles of in vitro diversification (such as error-prone PCR) followed by transformation of the resulting GOI mutant libraries into cells for expression and screening or selection. Continuous hypermutation systems bring GOI diversification in vivo, allowing GOIs to evolve autonomously as cells propagate under selection. This dramatically transforms the depth and scale of GOI evolution, accessing new avenues for biomolecular engineering and evolution. The applications section of this Primer summarizes recent work that exploits both the depth and scale of GOI evolution available to in vivo continuous evolution. This includes the traversal of long multi-mutation pathways in the optimization of enzyme function18 and the replicate evolution of enzymes and antibodies to augment the scale at which we gain new GOI functions and sample diverse regions of sequence space117,125. The continued evolution of proteins with new functions at depth and scale will naturally blossom, defining one part of in vivo continuous evolution’s future. We provide three less obvious but equally tantalizing future directions here.
Expansion into multicellular organisms
With the unit of selection having gone from the replicating RNA molecule as in original DE experiments, to a virus (as in the case of PACE5) and now the cell, the scope of functions that we can evolve a GOI to accomplish has dramatically broadened. In essence, the field of continuous evolution has followed an arc where GOI hypermutation has been made possible in the context of increasingly complex units of selection, accessing broader spectrums of function that a GOI can be pressured to evolve. The logical next step in this arc is to bring continuous GOI evolution to multicellular organisms. If in vivo continuous evolution systems can be installed within the cells of a complex animal, we can evolve biomolecules that change the physiology of animals. Short of using an animal as the unit of selection, we can at least carry out cell-based selections in the context of animals where the biomolecular function serves a therapeutic goal occurring in the relevant environment. An example of this would be to continuously evolve receptors encoded in therapeutic T-cells within mouse models of cancer. Naturally, ethical concerns must be carefully assessed before initiating any experiments involving evolution with or in multicellular animals, ranging from less ethically-challenging organisms such as flies and worms to more ethically-fraught mammalian models.
Deep learning and continuous evolution
Concurrent with the development of in vivo continuous evolution has been a revolution in the power of AI, especially deep learning, to navigate the nearly infinite combinatorial space underlying biomolecular engineering93,94,133. Deep learning is only successful when data from which to learn is abundant. By running deep evolution experiments at a scale of thousands of replicates, as is possible with in vivo continuous evolution, we may be able to generate big biomolecular evolution datasets in a systematic manner where the entire evolutionary record is also available. This would allow AI to produce probabilistic sequence-to-function models that can predict and generate new sequences with desired functions and functional improvements. In comparison to techniques like low-throughput classical DE or deep mutational scanning approaches that systematically evaluate the consequences of only one or two mutation variants of a parent sequence, continuous evolution experiments would sample the contours of fitness landscapes through long mutational trajectories at an unprecedented scale. These datasets can train generative deep learning models whose outcomes can even be reloaded into continuous evolution systems for further evolution and divergence, creating a virtuous cycle. Since natural datasets are incomplete and simulation of RNA and protein function is often ineffective, we predict continuous evolution experiments may become the other side of the deep learning coin in the realm of biomolecular engineering.
Going from zero-to-one and one-to-many
The emergence of desired activity where none existed before is a major challenge in the biomolecular engineering field. Strategies that mine diverse gene collections for desired biomolecular functions to bootstrap DE campaigns have acted as an effective solution, but ultimately, the goal is to gain functional sequences from scratch. We call this the zero-to-one goal: going from zero sequences that have any desired activity to one. An approach to the zero-to-one goal that has proven successful in the RNA enzyme and aptamer evolution fields is to start from staggeringly large random sequence libraries (at 1013 variants)134,135. For proteins, such library sizes are traditionally inadmissible because the transformation efficiency of cells can only reach 107–109. With in vivo continuous evolution, diversity is generated directly inside cells, making it possible to bypass transformation efficiency limitations. With sufficiently high mutation rates on a GOI and the large population sizes accessible in a bioreactor, protein libraries that are 1013 in size could conceivably be generated. When such diversity is reached, selection can be imposed to initiate further evolution of low activity sequences in a continuous format.
Another approach to the zero-to-one goal is computational design. The de novo design of desired RNA and protein structures, and to some extent desired functions, has witnessed major advances over the last 20 years93,133,136,137. However, once a de novo design is generated, its activity almost certainly requires improvement, which DE campaigns can address. Perhaps more importantly from a computational design perspective is the value of diverging a de novo design into a much larger set of highly dissimilar variants to be able to map the fitness landscape governing the de novo design. The depth and scale of continuous evolution campaigns may be uniquely capable of achieving this. Indeed, this one-to-many goal — going from one sequence with a desired activity to many — is within the unique purview of in vivo continuous evolution and applies to all cases where we find only one example of a sequence with a desired activity. Besides de novo designed biomolecules and sequences isolated from large random libraries, orphan proteins or ribozymes that could represent a lost epoch of life may be turned into rich families of variants through the power of in vivo continuous evolution. Such efforts may give us a comparative understanding of why well-populated natural RNA and protein families have been so successful and also offer us entry into knowledge-based strategies for engineering de novo138–141, orphan142 and ancient biomolecules143–145. Indeed, in vivo continuous evolution presents many exciting opportunities ahead.
Supplementary Material
Acknowledgements
We thank members of our groups for insightful discussions. This work was funded by NIH NIGMS 1R35GM139513 (C.C.L.); NIH NIGMS 1R35GM136354 (M.D.S); MIT Robert J Silbey Fellowship (A.A.M); MIT School of Science Fund for Future of Science (A.A.M); U.S. Department of Energy, Office of Science, Basic Energy Sciences under Award DE-SC0020153 (A.D.H.); the Innovative Genomics Institute and Laboratory for Genomics Research (J.D., J.H., and D.V.S.); UC Berkeley Miller Basic Research Fellowship (Q.Z.); NIH NIBIB 1R01EB027793 (A.S.K.); DoD Vannevar Bush Faculty Fellowship N00014-20-1-2825 (A.S.K.); NIH NIGMS 1R01GM125887 (F.H.A.); and MICIN-CSIC PTI+ REC-EU SGL2103051 and EU H2020 research and innovation programme FET Open 965018-BIOCELLPHE (L.A.F.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH or other funding agencies.
Footnotes
Competing interests
The authors declare no competing interests.
Related links
Sequence Read Archive (SRA): https://www.ncbi.nlm.nih.gov/sra
NCBI BioProject: https://www.ncbi.nlm.nih.gov/bioproject/
European Nucleotide Archive (ENA): https://www.ebi.ac.uk/ena/browser/home
Supplementary information
Supplementary information is available for this paper at https://doi.org/10.1038/s415XX-XXX-XXXX-X
References
- 1.Arnold FH Design by Directed Evolution. Acc. Chem. Res 31, 125–131 (1998). [Google Scholar]
- 2.Packer MS & Liu DR Methods for the directed evolution of proteins. Nat. Rev. Genet 16, 379–394 (2015). [DOI] [PubMed] [Google Scholar]
- 3.Drake JW, Charlesworth B, Charlesworth D & Crow JF Rates of spontaneous mutation. Genetics 148, 1667–1686 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen K & Arnold FH Engineering new catalytic activities in enzymes. Nat. Catal 3, 203–213 (2020). [Google Scholar]
- 5.Esvelt KM, Carlson JC & Liu DR A system for the continuous directed evolution of biomolecules. Nature. 472, 499–503 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Miller SM, Wang T & Liu DR Phage-assisted continuous and non-continuous evolution. Nat. Protoc 15, 4101–4127 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Morrison MS, Podracky CJ & Liu DR The developing toolkit of continuous directed evolution. Nat. Chem. Biol 16, 610–619 (2020). [DOI] [PubMed] [Google Scholar]
- 8.Hendel SJ & Shoulders MD Directed evolution in mammalian cells. Nat. Methods 18, 346–357 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fabret C et al. Efficient gene targeted random mutagenesis in genetically stable Escherichia coli strains. Nucleic Acids Res 28, 95 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Camps M, Naukkarinen J, Johnson BP & Loeb LA Targeted gene evolution in Escherichia coli using a highly error-prone DNA polymerase I. Proc. Natl. Acad. Sci. U. S. A 100, 9727–9732 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Finney-Manchester SP & Maheshri N Harnessing mutagenic homologous recombination for targeted mutagenesis in vivo by TaGTEAM. Nucleic Acids Res 41, 1–10 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Crook N et al. In vivo continuous evolution of genes and pathways in yeast. Nat. Commun 7, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ravikumar A, Arrieta A & Liu CC An orthogonal DNA replication system in yeast. Nat. Chem. Biol 10, 175–177 (2014). [DOI] [PubMed] [Google Scholar]
- 14.Hess GT et al. Directed evolution using dCas9-targeted somatic hypermutation in mammalian cells. Nat. Methods 13, 1036–1042 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ma Y et al. Targeted AID-mediated mutagenesis (TAM) enables efficient genomic diversification in mammalian cells. Nat. Methods 13, 1029–1035 (2016). [DOI] [PubMed] [Google Scholar]
- 16.Moore CL, Papa LJ & Shoulders MD A Processive Protein Chimera Introduces Mutations across Defined DNA Regions in Vivo. J. Am. Chem. Soc 140, 11560–11564 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Halperin SO et al. CRISPR-guided DNA polymerases enable diversification of all nucleotides in a tunable window. Nature 560, 248–252 (2018). [DOI] [PubMed] [Google Scholar]
- 18.Ravikumar A, Arzumanyan GA, Obadi MKA, Javanpour AA & Liu CC Scalable, Continuous Evolution of Genes at Mutation Rates above Genomic Error Thresholds. Cell 175, 1946–1957.e13 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yi X, Khey J, Kazlauskas RJ & Travisano M Plasmid hypermutation using a targeted artificial DNA replisome. Sci. Adv 7, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yi X, Kazlauskas R & Travisano M Evolutionary innovation using EDGE, a system for localized elevated mutagenesis. PLoS One 15, 1–18 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jensen ED et al. A synthetic RNA-mediated evolution system in yeast. Nucleic Acids Res 49, 1–12 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen H et al. Efficient, continuous mutagenesis in human cells using a pseudo-random DNA editor. Nat. Biotechnol 38, 165–168 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Álvarez B, Mencía M, de Lorenzo V & Fernández LÁ In vivo diversification of target genomic sites using processive base deaminase fusions blocked by dCas9. Nat. Commun 11, 6436 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Park H & Kim S Gene-specific mutagenesis enables rapid continuous evolution of enzymes in vivo. Nucleic Acids Res 49, e32–e32 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cravens A, Jamil OK, Kong D, Sockolosky JT & Smolke CD Polymerase-guided base editing enables in vivo mutagenesis and rapid protein engineering. Nat. Commun 12, 1–12 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Butt H, Luis J, Ramirez M, Mahfouz M & Biology S Synthetic evolution of herbicide resistance using a T7 RNAP-based random DNA base editor. bioRxiv (2021). [DOI] [PMC free article] [PubMed]
- 27.Jinek M et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tou CJ, Schaffer DV & Dueber JE Targeted Diversification in the S. cerevisiae Genome with CRISPR-Guided DNA Polymerase i. ACS Synth. Biol 9, 1911–1916 (2020). [DOI] [PubMed] [Google Scholar]
- 29.Eigen M, McCaskill J & Schuster P Molecular quasi-species. J. Phys. Chem 92, 6881–6891 (1988). [Google Scholar]
- 30.Biebricher CK & Eigen M The error threshold. Virus Res 107, 117–127 (2005). [DOI] [PubMed] [Google Scholar]
- 31.Carlson JC, Badran AH, Guggiana-Nilo DA & Liu DR Negative selection and stringency modulation in phage-assisted continuous evolution. Nat. Chem. Biol 10, 216–222 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pu J, Zinkus-Boltz J & Dickinson BC Evolution of a split RNA polymerase as a versatile biosensor platform. Nat. Chem. Biol 13, 432–438 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Packer MS, Rees HA & Liu DR Phage-assisted continuous evolution of proteases with altered substrate specificity. Nat. Commun 8, 956 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Blum TR et al. Phage-assisted evolution of botulinum neurotoxin proteases with reprogrammed specificity. Science 371, 803–810 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Badran AH et al. Continuous evolution of Bacillus thuringiensis toxins overcomes insect resistance. Nature 533, 58–63 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.DeBenedictis EA et al. Systematic molecular evolution enables robust biomolecule discovery. Nat. Methods 19, 55–64 (2022). [DOI] [PubMed] [Google Scholar]
- 37.Wang T, Badran AH, Huang TP & Liu DR Continuous directed evolution of proteins with improved soluble expression. Nat. Chem. Biol 14, 972–980 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Thuronyi BW et al. Continuous evolution of base editors with expanded target compatibility and improved activity. Nat. Biotechnol 37, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Richter MF et al. Phage-assisted evolution of an adenine base editor with improved Cas domain compatibility and activity. Nat. Biotechnol 38, 883–891 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Berman CM et al. An Adaptable Platform for Directed Evolution in Human Cells. J. Am. Chem. Soc 140, 18093–18103 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.English JG et al. VEGAS as a Platform for Facile Directed Evolution in Mammalian Cells. Cell 178, 748–761.e17 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Khanal A, McLoughlin SY, Kershner JP & Copley SD Differential Effects of a Mutation on the Normal and Promiscuous Activities of Orthologs: Implications for Natural and Directed Evolution. Mol. Biol. Evol 32, 100–108 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zheng J, Payne JL & Wagner A Cryptic genetic variation accelerates evolution by opening access to diverse adaptive peaks. Science 365, 347–353 (2019). [DOI] [PubMed] [Google Scholar]
- 44.Gupta RD & Tawfik DS Directed enzyme evolution via small and effective neutral drift libraries. Nat. Methods 5, 939–942 (2008). [DOI] [PubMed] [Google Scholar]
- 45.Salverda MLM et al. Initial mutations direct alternative pathways of protein evolution. PLoS Genet 7, (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Baier F et al. Cryptic genetic variation shapes the adaptive evolutionary potential of enzymes. eLife 8, 1–20 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lang GI et al. Pervasive genetic hitchhiking and clonal interference in forty evolving yeast populations. Nature 500, 571–574 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rix G & Liu CC Systems for in vivo hypermutation: a quest for scale and depth in directed evolution. Curr. Opin. Chem. Biol 64, 20–26 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gunge N & Sakaguchi K Intergeneric transfer of deoxyribonucleic acid killer plasmids, pGKl1 and pGKl2, from Kluyveromyces lactis into Saccharomyces cerevisiae by cell fusion. J. Bacteriol 147, 155–160 (1981). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Arzumanyan GA, Gabriel KN, Ravikumar A, Javanpour AA & Liu CC Mutually Orthogonal DNA Replication Systems in Vivo. ACS Synth. Biol 7, 1722–1729 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhong Z, Ravikumar A & Liu CC Tunable Expression Systems for Orthogonal DNA Replication. ACS Synth. Biol 7, 2930–2934 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kämper J, Esser K, Gunge N & Meinhardt F Heterologous gene expression on the linear DNA killer plasmid from Kluyveromyces lactis. Curr. Genet 19, 109–118 (1991). [DOI] [PubMed] [Google Scholar]
- 53.Javanpour AA & Liu CC Genetic Compatibility and Extensibility of Orthogonal Replication. ACS Synth. Biol 8, 1249–1256 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chamberlin M, Mcgrath J & Waskell L New RNA polymerase from escherichia coli infected with bacteriophage T7. Nature 228, 227–231 (1970). [DOI] [PubMed] [Google Scholar]
- 55.Tabor S & Richardson CC A bacteriophage T7 RNA polymerase/promoter system for controlled exclusive expression of specific genes. Proc. Natl. Acad. Sci 82, 1074–1078 (1985). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.McAllister WT, Morris C, Rosenberg AH & Studier FW Utilization of bacteriophage T7 late promoters in recombinant plasmids during infection. J. Mol. Biol 153, 527–544 (1981). [DOI] [PubMed] [Google Scholar]
- 57.Thiel V, Herold J, Schelle B & Siddell SG Infectious RNA transcribed in vitro from a cDNA copy of the human coronavirus genome cloned in vaccinia virus. J. Gen. Virol 82, 1273–1281 (2001). [DOI] [PubMed] [Google Scholar]
- 58.Steitz TA The structural changes of T7 RNA polymerase from transcription initiation to elongation. Curr. Opin. Struct. Biol 19, 683–690 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Conticello SG The AID/APOBEC family of nucleic acid mutators. Genome Biol 9, (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gerber AP & Keller W An adenosine deaminase that generates inosine at the wobble position of tRNAs. Science 286, 1146–1149 (1999). [DOI] [PubMed] [Google Scholar]
- 61.Harris RS, Petersen-Mahrt SK & Neuberger MS RNA editing enzyme APOBEC1 and some of its homologs can act as DNA mutators. Mol. Cell 10, 1247–1253 (2002). [DOI] [PubMed] [Google Scholar]
- 62.Navaratnam N & Sarwar R An overview of cytidine deaminases. Int. J. Hematol 83, 195–200 (2006). [DOI] [PubMed] [Google Scholar]
- 63.Cacciamani T et al. Purification of human cytidine deaminase: Molecular and enzymatic characterization and inhibition by synthetic pyrimidine analogs. Arch. Biochem. Biophys 290, 285–292 (1991). [DOI] [PubMed] [Google Scholar]
- 64.Chung SJ, Fromme JC & Verdine GL Structure of human cytidine deaminase bound to a potent inhibitor. J. Med. Chem 48, 658–660 (2005). [DOI] [PubMed] [Google Scholar]
- 65.Lada AG et al. Mutator effects and mutation signatures of editing deaminases produced in bacteria and yeast. Biochem 76, 131–146 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Vik ES et al. Endonuclease v cleaves at inosines in RNA. Nat. Commun 4, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Krokan HE, Drabløs F & Slupphaug G Uracil in DNA - Occurrence, consequences and repair. Oncogene 21, 8935–8948 (2002). [DOI] [PubMed] [Google Scholar]
- 68.Alseth I, Dalhus B & Bjørås M Inosine in DNA and RNA. Curr. Opin. Genet. Dev 26, 116–123 (2014). [DOI] [PubMed] [Google Scholar]
- 69.Hirano KI, Min J, Funahashi T & Davidson NO Cloning and characterization of the rat apobec-1 gene: A comparative analysis of gene structure and promoter usage in rat and mouse. J. Lipid Res 38, 1103–1119 (1997). [PubMed] [Google Scholar]
- 70.MacGinnitie AJ, Anant S & Davidson NO Mutagenesis of apobec-1, the catalytic subunit of the mammalian apolipoprotein B mRNA editing enzyme, reveals distinct domains that mediate cytosine nucleoside deaminase, RNA binding, and RNA editing activity. J. Biol. Chem 270, 14768–14775 (1995). [PubMed] [Google Scholar]
- 71.Scott J, Navaratnam N, Bhattacharya S & Morrison JR The apolipoprotein B messenger RNA editing enzyme. Current Opinion in Lipidology vol. 5 87–93 (1994). [DOI] [PubMed] [Google Scholar]
- 72.Arakawa H, HauschiLd J & Buerstedde JM Requirement of the activation-induced deaminase (AID) gene for immunoglobulin gene conversion. Science 295, 1301–1306 (2002). [DOI] [PubMed] [Google Scholar]
- 73.Muramatsu M et al. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell 102, 553–563 (2000). [DOI] [PubMed] [Google Scholar]
- 74.Rogozin IB et al. Evolution and diversification of lamprey antigen receptors: Evidence for involvement of an AID-APOBEC family cytosine deaminase. Nat. Immunol 8, 647–656 (2007). [DOI] [PubMed] [Google Scholar]
- 75.Kim J et al. Structural and Kinetic Characterization of Escherichia coli TadA, the wobble-Specific tRNA deaminase. Biochemistry 45, 6407–6416 (2006). [DOI] [PubMed] [Google Scholar]
- 76.Gaudelli NM et al. Programmable base editing of T to G C in genomic DNA without DNA cleavage. Nature 551, 464–471 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Martínez-Salas E Internal ribosome entry site biology and its use in expression vectors. Curr. Opin. Biotechnol 10, 458–464 (1999). [DOI] [PubMed] [Google Scholar]
- 78.Wang Z & Mosbaugh DW Uracil-DNA glycosylase inhibitor of bacteriophage PBS2: cloning and effects of expression of the inhibitor gene in Escherichia coli. J. Bacteriol 170, 1082–1091 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wang Z & Mosbaugh DW Uracil-DNA glycosylase inhibitor gene of bacteriophage PBS2 encodes a binding protein specific for uracil-DNA glycosylase. J. Biol. Chem 264, 1163–1171 (1989). [PubMed] [Google Scholar]
- 80.Karran P, Cone R & Friedberg EC Specificity of the Bacteriophage PBS2 Induced Inhibitor of Uracil-DNA Glycosylase. Biochemistry 20, 6092–6096 (1981). [DOI] [PubMed] [Google Scholar]
- 81.Bennett SE & Mosbaugh DW Characterization of the Escherichia coli uracil-DNA glycosylase·inhibitor protein complex. J. Biol. Chem 267, 22512–22521 (1992). [PubMed] [Google Scholar]
- 82.Bennett SE, Schimerlik MI & Mosbaugh DW Kinetics of the uracil-DNA glycosylase/inhibitor protein association. Ung interaction with Ugi, nucleic acids, and uracil compounds. J. Biol. Chem 268, 26879–26885 (1993). [PubMed] [Google Scholar]
- 83.Wang Y et al. A novel strategy to engineer DNA polymerases for enhanced processivity and improved performance in vitro. Nucleic Acids Res 32, 1197–1207 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Dickinson BC, Leconte AM, Allen B, Esvelt KM & Liu DR Experimental interrogation of the path dependence and stochasticity of protein evolution using phage-assisted continuous evolution. Proc. Natl. Acad. Sci. U. S. A 110, 9007–9012 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wang L, Brock A, Herberich B & Schultz PG Expanding the Genetic Code of Escherichia coli. Science 292, 498–500 (2001). [DOI] [PubMed] [Google Scholar]
- 86.Szendro IG, Franke J, De Visser JAGM & Krug J Predictability of evolution depends nonmonotonically on population size. Proc. Natl. Acad. Sci. U. S. A 110, 571–576 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Salverda MLM, Koomen J, Koopmanschap B, Zwart MP & de Visser JAGM Adaptive benefits from small mutation supplies in an antibiotic resistance enzyme. Proc. Natl. Acad. Sci. U. S. A 114, 201712999 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Steinberg B & Ostermeier M Environmental changes bridge evolutionary valleys. Sci. Adv 2, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wong BG, Mancuso CP, Kiriakov S, Bashor CJ & Khalil AS Precise, automated control of conditions for high-throughput growth of yeast and bacteria with eVOLVER. Nat. Biotechnol 36, 614–623 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zhong Z et al. Automated Continuous Evolution of Proteins in Vivo. ACS Synth. Biol 9, 1270–1276 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Marks DS et al. Protein 3D structure computed from evolutionary sequence variation. PLoS One 6, (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Stiffler MA et al. Protein Structure from Experimental Evolution. Cell Syst 10, 15–24.e5 (2020). [DOI] [PubMed] [Google Scholar]
- 93.Jumper J et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Yang KK, Wu Z & Arnold FH Machine-learning-guided directed evolution for protein engineering. Nat. Methods 16, 687–694 (2019). [DOI] [PubMed] [Google Scholar]
- 95.Wu Z, Kan SBJ, Lewis RD, Wittmann BJ & Arnold FH Machine learning-assisted directed protein evolution with combinatorial libraries 116, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Carr IM et al. Inferring relative proportions of DNA variants from sequencing electropherograms. Bioinformatics 25, 3244–3250 (2009). [DOI] [PubMed] [Google Scholar]
- 97.Shen MW, Zhao KT & Liu DR Reconstruction of evolving gene variants and fitness from short sequencing reads. Nat. Chem. Biol 17, 1188–1198 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Goodwin S, McPherson JD & McCombie WR Coming of age: Ten years of next-generation sequencing technologies. Nat. Rev. Genet 17, 333–351 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Amarasinghe SL et al. Opportunities and challenges in long-read sequencing data analysis. Genome Biol 21, 1–16 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ravi RK, Walton K & Khosroheidari M MiSeq: A Next Generation Sequencing Platform for Genomic Analysis. in Disease Gene Identification: Methods and Protocols (ed. DiStefano JK) 223–232 (Springer New York, 2018). doi: 10.1007/978-1-4939-7471-9_12. [DOI] [PubMed] [Google Scholar]
- 101.Stoler N & Nekrutenko A Sequencing error profiles of Illumina sequencing instruments. NAR Genomics Bioinforma 3, 1–9 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wang Y, Zhao Y, Bollas A, Wang Y & Au KF Nanopore sequencing technology, bioinformatics and applications. Nat. Biotechnol 39, 1348–1365 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Karst SM et al. High-accuracy long-read amplicon sequences using unique molecular identifiers with Nanopore or PacBio sequencing. Nat. Methods 18, 165–169 (2021). [DOI] [PubMed] [Google Scholar]
- 104.Wenger AM et al. Accurate circular consensus long-read sequencing improves variant detection and assembly of a human genome. Nat. Biotechnol 37, 1155–1162 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zurek PJ, Knyphausen P, Neufeld K, Pushpanath A & Hollfelder F UMI-linked consensus sequencing enables phylogenetic analysis of directed evolution. Nat. Commun 11, 1–10 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Wilson BD, Eisenstein M & Soh HT High-Fidelity Nanopore Sequencing of Ultra-Short DNA Targets. Anal. Chem 91, 6783–6789 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Murray KD & Borevitz JO Axe: Rapid, competitive sequence read demultiplexing using a trie. Bioinformatics 34, 3924–3925 (2018). [DOI] [PubMed] [Google Scholar]
- 108.Li H Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Koboldt DC et al. VarScan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res 22, 568–576 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Deatherage DE & Barrick JE Identification of Mutations in Laboratory-Evolved Microbes from Next-Generation Sequencing Data Using breseq. in Engineering and Analyzing Multicellular Systems: Methods and Protocols (eds. Sun L & Wenying S) vol. 1151 165–188 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Fowler DM & Fields S Deep mutational scanning: A new style of protein science. Nat. Methods 11, 801–807 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Long M et al. Directed Evolution of Ornithine Cyclodeaminase Using an EvolvR-Based Growth-Coupling Strategy for Efficient Biosynthesis of l -Proline. ACS Synth. Biol 9, 1855–1863 (2020). [DOI] [PubMed] [Google Scholar]
- 113.García-García JD et al. Potential for applying continuous directed evolution to plant enzymes: An exploratory study. Life 10, 1–16 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.García-García JD et al. Using continuous directed evolution to improve enzymes for plant applications. Plant Physiol (2021) doi: 10.1093/plphys/kiab500. [DOI] [PMC free article] [PubMed]
- 115.Chatterjee A et al. Saccharomyces cerevisiae THI4p is a suicide thiamine thiazole synthase. Nature 478, 542–546 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Joshi J et al. Structure and function of aerotolerant, multiple-turnover THI4 thiazole synthases. Biochem. J 478, 3265–3279 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Rix G et al. Scalable continuous evolution for the generation of diverse enzyme variants encompassing promiscuous activities. Nat. Commun 11, 1–11 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Dunn MF Allosteric regulation of substrate channeling and catalysis in the tryptophan synthase bienzyme complex. Arch. Biochem. Biophys 519, 154–166 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Buller AR et al. Directed evolution of the tryptophan synthase β-subunit for stand-alone function recapitulates allosteric activation. Proc. Natl. Acad. Sci 112, 14599–14604 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Watkins‐Dulaney E, Straathof S & Arnold F Tryptophan Synthase: Biocatalyst Extraordinaire. ChemBioChem 22, 5–16 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Romney DK, Murciano-Calles J, Wehrmüller JE & Arnold FH Unlocking Reactivity of TrpB: A General Biocatalytic Platform for Synthesis of Tryptophan Analogues. J. Am. Chem. Soc 139, 10769–10776 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Boville CE, Romney DK, Almhjell PJ, Sieben M & Arnold FH Improved Synthesis of 4-Cyanotryptophan and Other Tryptophan Analogues in Aqueous Solvent Using Variants of TrpB from Thermotoga maritima. J. Org. Chem 83, 7447–7452 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Javanpour AA & Liu CC Evolving Small-Molecule Biosensors with Improved Performance and Reprogrammed Ligand Preference Using OrthoRep. ACS Synth. Biol 10, 2705–2714 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Jensen ED et al. Integrating continuous hypermutation with high-throughput screening for optimization of cis,cis-muconic acid production in yeast. Microb. Biotechnol 14, 2617–2626 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Wellner A et al. Rapid generation of potent antibodies by autonomous hypermutation in yeast. Nat. Chem. Biol 17, 1057–1064 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Pezo V et al. Noncanonical DNA polymerization by aminoadenine-based siphoviruses. Science 372, 520–524 (2021). [DOI] [PubMed] [Google Scholar]
- 127.Anzalone AV et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 576, 149–157 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Ling X et al. Improving the efficiency of precise genome editing with site-specific Cas9-oligonucleotide conjugates. Sci. Adv 6, 1–9 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Wang C et al. Microbial single-strand annealing proteins enable CRISPR gene-editing tools with improved knock-in efficiencies and reduced off-target effects. Nucleic Acids Res 49, 1–16 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Stevens AJ et al. Design of a Split Intein with Exceptional Protein Splicing Activity. J. Am. Chem. Soc 138, 2162–2165 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Mills DR, Peterson RL & Spiegelman S An extracellular Darwinian experiment with a self-duplicating nucleic acid molecule. Proc. Natl. Acad. Sci. U. S. A 58, 217–224 (1967). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Wright MC & Joyce GF Continuous in vitro evolution of catalytic function. Science 276, 614–617 (1997). [DOI] [PubMed] [Google Scholar]
- 133.Leman JK et al. Macromolecular modeling and design in Rosetta: recent methods and frameworks. Nat. Methods 17, 665–680 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Beaudry AA & Joyce GF Directed evolution of an RNA enzyme. Science 257, 635–641 (1992). [DOI] [PubMed] [Google Scholar]
- 135.Stoltenburg R, Reinemann C & Strehlitz B SELEX-A (r)evolutionary method to generate high-affinity nucleic acid ligands. Biomol. Eng 24, 381–403 (2007). [DOI] [PubMed] [Google Scholar]
- 136.Torrisi M, Pollastri G & Le Q Deep learning methods in protein structure prediction. Comput. Struct. Biotechnol. J 18, 1301–1310 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Rollins NJ et al. Inferring protein 3D structure from deep mutation scans. Nat. Genet 51, 1170–1176 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Langan RA et al. De novo design of bioactive protein switches. Nature 572, 205–210 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Dou J et al. De novo design of a fluorescence-activating β-barrel. Nature 561, 485–491 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Basanta B et al. An enumerative algorithm for de novo design of proteins with diverse pocket structures. Proc. Natl. Acad. Sci. U. S. A 117, 22135–22145 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Anishchenko I et al. De novo protein design by deep network hallucination. Nature 600, 547–552 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Hadadi N, MohammadiPeyhani H, Miskovic L, Seijo M & Hatzimanikatis V Enzyme annotation for orphan and novel reactions using knowledge of substrate reactive sites. Proc. Natl. Acad. Sci. U. S. A 116, 7298–7307 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Gumulya Y et al. Engineering highly functional thermostable proteins using ancestral sequence reconstruction. Nat. Catal 1, 878–888 (2018). [Google Scholar]
- 144.Xie VC, Pu J, Metzger BPH, Thornton JW & Dickinson BC Contingency and chance erase necessity in the experimental evolution of ancestral proteins. eLife 10, 1–87 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Kaltenbach M et al. Evolution of chalcone isomerase from a noncatalytic ancestor article. Nat. Chem. Biol 14, 548–555 (2018). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.