

Abstract
Interleukin-6 (IL-6) is a key immunomodulatory cytokine that affects the pathogenesis of diverse diseases, including autoimmune diseases, chronic inflammatory conditions and cancer. Classical IL-6 signalling involves the binding of IL-6 to the membrane-bound IL-6 receptor α-subunit (hereafter termed ‘mIL-6R’) and glycoprotein 130 (gp130) signal-transducing subunit. By contrast, in IL-6 trans-signalling, complexes of IL-6 and the soluble form of IL-6 receptor (sIL-6R) signal via membrane-bound gp130. A third mode of IL-6 signalling — known as cluster signalling — involves preformed complexes of membrane-bound IL-6–mIL-6R on one cell activating gp130 subunits on target cells. Antibodies and small molecules have been developed that block all three forms of IL-6 signalling, but in the past decade, IL-6 trans-signalling has emerged as the predominant pathway by which IL-6 promotes disease pathogenesis. The first selective inhibitor of IL-6 trans-signalling, sgp130, has shown therapeutic potential in various preclinical models of disease and olamkicept, a sgp130Fc variant, had promising results in phase II clinical studies for inflammatory bowel disease. Technological developments have already led to next-generation sgp130 variants with increased affinity and selectivity towards IL-6 trans-signalling, along with indirect strategies to block IL-6 trans-signalling. Here, we summarize our current understanding of the biological outcomes of IL-6-mediated signalling and the potential for targeting this pathway in the clinic.
Subject terms: Drug development, Molecularly targeted therapy
This Review details the discovery of the interleukin-6 (IL-6) trans-signalling pathway and the subsequent development of biologics that specifically inhibit this pathway. Emerging evidence suggests that specifically targeting IL-6 trans-signalling can reduce pathological disease-promoting activities of IL-6 without blocking the protective actions of IL-6 in infection and tissue repair.
Introduction
In 1986, the group of Tadamitsu Kishimoto cloned a cDNA coding for B cell stimulatory factor 2, which later was renamed interleukin-6 (IL-6)1. The amino acid sequence of IL-6 revealed that other factors such as hepatocyte-stimulating factor2, hybridoma growth factor3, interferon β24 and a 26-kDa protein identified from fibroblasts5 were identical to IL-6, indicating the pleiotropic activity of this cytokine6. Today, IL-6 is seen as one of the major immunomodulatory cytokines controlling health and disease7. Strategies to inhibit global IL-6 signalling based on antibodies and small molecules directed against IL-6, the IL-6 receptor (IL-6R) or downstream signalling effectors such as Janus kinases (JAKs) have entered the clinic and shown success, including as treatments for chronic inflammatory diseases, for COVID-19 and for preventing fatal cytokine storms associated with CAR T cell therapy in patients with cancer8,9. Importantly, our growing understanding of IL-6 biology has led to the description of three distinct modes of IL-6-mediated signalling: classic IL-6 signalling, IL-6 trans-signalling and IL-6 cluster signalling (Fig. 1). In turn, this has guided new strategies to selectively inhibit these specific signalling modes of IL-6. In classic signalling, IL-6 binds to membrane-bound IL-6R (mIL-6R) to induce a signal-transducing homodimer of the glycoprotein 130 (gp130) receptor chain10. In contrast, in the case of IL-6 trans-signalling, soluble forms of IL-6R (sIL-6R), generated either by alternative splicing or limited proteolytic processing, form complexes with IL-6 to activate membrane-bound gp13011,12. Finally, in IL-6 cluster signalling, IL-6–mIL-6R complexes formed on a transmitter cell activate gp130 subunits on a neighbouring receiver cell13,14. Of note, the closest relative of IL-6, IL-11, also signals via gp130 homodimers, but initially binds to the membrane-bound or soluble IL-11 receptor (IL-11R) as an α-receptor. Accordingly, IL-11 classic signalling and trans-signalling have been described in vivo, whereas IL-11 cluster signalling has only principally been shown in cell cultures15–19.
Fig. 1. Principles of IL-6 and IL-11 classic and trans-signalling.

In classic signalling, interleukin-6 (IL-6) and IL-11 bind to membrane IL-6 receptor (mIL-6R) and mIL-11R, respectively, and immediately recruit the high-affinity complex with the signal-transducing glycoprotein 130 (gp130) receptor. Soluble IL-6 receptor (sIL-6R) and sIL-11R can be generated by the proteases ADAM17 ((a disintegrin and metalloproteinase 17) and ADAM10, respectively. In cluster signalling, a transmitter cell presents IL-6–mIL-6R complexes to cells expressing gp130. Cluster signalling has only been described for IL-6, but in principle it is also possible for IL-11. Low-affinity complexes of IL-6–sIL-6R and IL-11–sIL-11R can induce trans-signalling by the formation of high-affinity complexes with gp130. For the sake of simplicity, IL-6 complexes are shown as tetramers (IL-6–IL-6R–2×gp130), although experimental evidence suggest hexamers (2×IL-6–2×IL-6R–2×gp130). The same holds true for IL-11 signalling. Naturally occurring sgp130 might serve as a IL-6 trans-signalling and IL-6 cluster-signalling buffer system (see Box 1).
Although sIL-6R-mediated IL-6 trans-signalling has emerged as the pathological mode of IL-6 signalling underlying various disease states, classic signalling via mIL-6R and the signal-transducing receptor gp130 has homeostatic, protective and acute inflammatory functions. Cluster signalling appears to play a role in the priming of pathogenic T helper 17 (TH17) cells14, but selective inhibitors of IL-6 cluster signalling have not yet been developed. Considering the broad impact of IL-6 trans-signalling on disease, in this Review, we focus on this mode of IL-6 signalling. We first provide a historic overview of the discovery of IL-6 trans-signalling and then we consider the strategies that have been used to selectively interfere with this mode of IL-6 signalling and the potential use of these drugs in the clinic.
The advent of IL-6 trans-signalling
In a seminal article, Bazan hypothesized that cytokines such as growth hormone, prolactin, IL-6, granulocyte stimulating factor and erythropoietin belonged to a four-helical cytokine family20 (Fig. 2). Although the amino acid sequence homology of these proteins is extremely low, they shared structural similarities, which was underlined by a conserved exon–intron organization around the sequences encoding their shared helical structures21.
Fig. 2. Timeline of IL-6 trans-signalling — from discovery to targeted therapies.
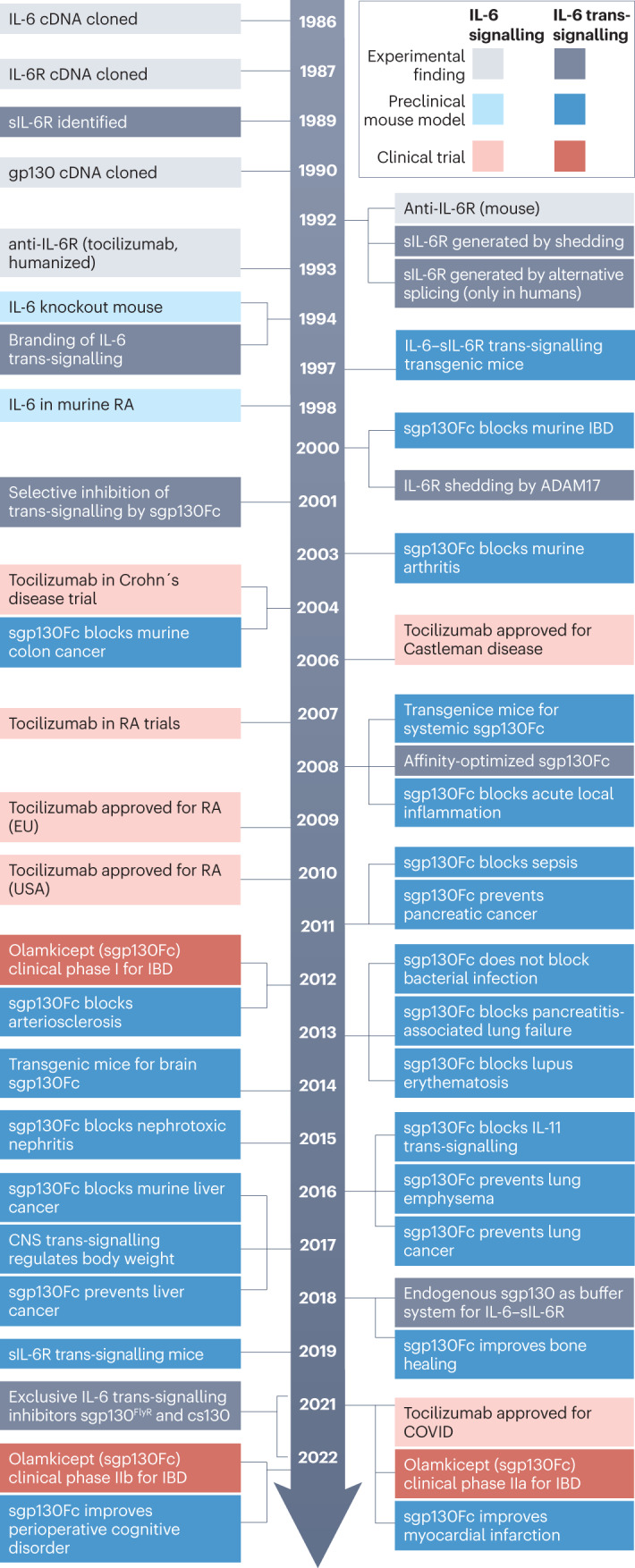
The timeline shows the discovery milestones for interleukin-6 (IL-6) trans-signalling and compares the development of IL-6-targeted therapies for combined classic and trans-signalling inhibition. The key studies defining the IL-6 trans-signalling pathway took place between 1989 and 1994, and preclinical studies began in the early 2000s, with clinical trials for olamkicept starting in 2012. ADAM17, a disintegrin and metalloproteinase 17; CNS, central nervous system; cs130, chimeric soluble gp130; gp130, glycoprotein 130; IBD, inflammatory bowel disease; IL-6, interleukin-6; IL-6R, IL-6 receptor; RA, rheumatoid arthritis; sgp130Fc, soluble gp130–Fc fusion protein; sIL-6R, soluble IL-6R.
When the DNA coding for IL-6R was cloned in 1988, the type I transmembrane protein it encoded appeared to have no signalling potential, and also bound IL-6 with only low affinity22. It emerged in 1989 that classic IL-6 signalling relied on a second protein, gp130. This membrane-bound protein formed a high-affinity receptor complex with mIL-6R and IL-6, whereas no binding of mIL-6R to gp130 could be detected in the absence of IL-610,23. When the structure of the IL-6–mIL-6R–gp130 complex was solved, it became clear that the contribution of IL-6 to the buried area between the IL-6–mIL-6R complex and site II of gp130 formed by the elbow between domain 2 and domain 3 was modestly larger for IL-6 (1272 Å2, site IIa) than for the IL-6R (1078 Å2, site IIb). For the assembly at site III of gp130 formed by domain 1, the contribution of IL-6 (1276 Å2, site IIIa) was substantially larger than that of the IL-6R (473 Å2, site IIIb). The binding energy of IL-6 or IL-6R alone was not sufficient to allow binding to gp13024 (Fig. 3).
Fig. 3. Assembly of the IL-6–IL-6R–gp130 complex via site II and site III.

Hexameric assembly of glycoprotein 130 (gp130)–gp130* with interleukin-6 (IL-6)–soluble IL-6 receptor (sIL-6R) and IL-6*–IL-6R*complexes via site II and site III. The asterisks indicates the second gp130, IL-6 and IL-6R in the assembly of two trimeric complexes. A trimeric complex of gp130 and IL-6–sIL-6R is formed between site IIa of IL-6, site IIb of IL-6R and domain D2–D3 of gp13024. A second trimeric complex of gp130* and IL-6–sIL-6R is formed between site IIIa of IL-6, site IIIb of IL-6R and domain D1 of gp13024.
Further cDNAs of homologous cytokines with partly overlapping but clearly distinct biological activities were cloned21. These included IL-1125, leukaemia inhibitory factor (LIF)26, ciliary neurotrophic factor (CNTF)27, oncostatin M (OSM)28, cardiotrophin 1 (CT1)29, cardiotrophin-like cytokine (CLC)30 and IL-27, the last a dimeric cytokine consisting of a cytokine moiety (p28) and a soluble receptor moiety (EBI3)31. All of these cytokines used gp130 as an obligatory receptor subunit32. Importantly, it was shown that gp130 mRNA is expressed in virtually all cells, with the only exception reported so far being granulocytes33. In contrast, mIL-6R is only expressed by hepatocytes, subsets of epithelial cells and leukocytes8,34, and the expression of specific receptors for IL-11 (IL-11R) and CNTF (CNTFR) is also restricted to only a few cell types35, including cardiomyocytes36, fibroblasts37 and epithelial cells38.
In the early 1990s, our group found that mIL-6R was subject to limited proteolysis, leading to the generation of sIL-6R consisting of the extracellular portion of the IL-6R39,40. The major protease involved in the shedding of sIL-6R during inflammation is the membrane-bound metalloproteinase ADAM17 (a disintegrin and metalloproteinase 17), although ADAM10 is also involved to a minor extent (Fig. 4). The cleavage site in mIL-6R targeted by both of these inflammatory ADAM proteases is close to the membrane, located between Pro355 and Val35641. In humans but not in mice, a soluble form of the IL-6R can also be generated by translation from a differentially spliced mRNA, although this only accounts for about 15% of the sIL-6R found in the blood42,43. Other proteases such as meprin-α and meprin-β44, rhomboid-related protein 245,46, cathepsin S47 and cathepsin G48 are capable of mediating the shedding of the sIL-6R, although the biological importance of these secondary sheddases has not been analysed. The sIL-6R was shown to bind IL-6 with similar affinity as mIL-6R39,40. We demonstrated that cells that express gp130 but not mIL-6R are not responsive to IL-6, but they become responsive to IL-6 in the presence of sIL-6R39,49. This mode of signalling was named IL-6 trans-signalling11. As almost all cells express gp130 on their cell surface34, the switch to IL-6 trans-signalling would make virtually all cells responsive to IL-611.
Fig. 4. Inhibitory principles of IL-6 and IL-11 classic and trans-signalling.

Classic and trans-signalling of interleukin-6 (IL-6) and IL-11 can be inhibited by antagonistic substances (for example, antibodies and small molecules) directed against the cytokine, the α-receptors, or glycoprotein 130 (gp130)-associated Janus kinases (JAKs). Antibodies to gp130 are currently not in clinical development. Trans-signalling is inhibited by variants of soluble gp130. Sgp130Fc (soluble gp130–Fc fusion protein) inhibits IL-6 and IL-11 trans-signalling, chimeric soluble gp130 (cs130) inhibits IL-6 trans-signalling, and the autoinhibitory N-terminal ADAM17 (a disintegrin and metalloproteinase 17) pro-domain (A17pro) inhibits the release of soluble IL-6 receptor (sIL-6R), prevents IL-6 trans-signalling and might increase classic signalling because mIL-6R level might increase. A selective IL-11 trans-signalling inhibitor has not been described. Selective classic-signalling inhibitors have also not been developed thus far.
To analyse the consequences of IL-6 trans-signalling in vitro and in vivo, the designer cytokine ‘hyper-IL-6’ was developed. This protein consists of IL-6 covalently bound to the sIL-6R via a flexible peptide linker50. Comparing stimulation of cells with IL-6 versus hyper-IL-6 showed that IL-6-mediated stimulation of many cell types, including neural cells51, adult haematopoietic stem cells and embryonic stem cells52,53, depended on the presence of sIL-6R54. Additionally, the presence of sIL-6R augmented the response of mIL-6R-expressing cells to IL-6 in terms of increased signalling intensity, as measured by phosphorylation of STAT3 or cytokine-induced proliferation55,56. As gp130 levels on cells that co-express mIL-6R — including hepatocytes and immune cells — are about 10–20 times higher than mIL-6R levels, stimulation with IL-6 only led to restricted activation of gp130 receptors54. In contrast, stimulation with the complex of IL-6 and sIL-6R activated all gp130 receptors on the cell surface. Moreover, stimulation with IL-6 alone led to the rapid internalization of the IL-6–mIL-6R–gp130 signalling complex, whereas stimulation with the IL-6–sIL-6R complex led to reduced gp130 internalization and therefore longer signalling duration; however, no qualitative differences between intracellular IL-6 classic signalling and trans-signalling pathways (for example, in terms of JAK–STAT and MAPK activation) were detected57,58. We concluded that cells stimulated via IL-6 trans-signalling showed a more sustained IL-6 response with a higher amplitude than cells expressing mIL-6R when stimulated with IL-6 in the absence of sIL-6R54.
To clarify the pathophysiological role of sIL-6R in vivo, we generated mice that expressed the human sIL-6R as a transgene55. As mouse IL-6 does not interact with the human sIL-6R, these mice did not show a discernible phenotype. Importantly, upon injection of human IL-6 into these mice, the presence of sIL-6R prolonged the half-life of IL-6 and led to increased activation of gp130 receptors. Thus, IL-6 target cells — such as hepatocytes — were hypersensitized55. When the human sIL-6R-expressing transgenic mice were bred with human IL-6 transgenic mice to generate IL-6–sIL-6R double-transgenic mice, we observed extramedullary expansion of haematopoietic progenitor cells in the liver that resembled the fetal liver, which is the site of haematopoiesis in the embryo56. The IL-6–sIL-6R double-transgenic mice were markedly smaller than IL-6 or sIL-6R single-transgenic mice and showed enlarged livers and spleens54. These data for the first time demonstrated the difference in outcome between classical IL-6 signalling and trans-signalling, as IL-6 single-transgenic mice showed no liver phenotype even though hepatocytes express the IL-6R and can therefore respond to IL-6 alone56.
Interestingly, extramedullary haematopoiesis and hepatocyte proliferation were not seen in mice that expressed a constitutively active gp130 variant (L-gp130)59 in hepatocytes60, indicating that gp130 stimulation in other liver cell populations contributed to the complex phenotype of the IL-6–sIL-6R double-transgenic mice56. Additionally, we detected hepatocellular hyperplasia in close to 100% of the hepatocytes, resembling liver regeneration61. A dominant role of sIL-6R in liver regeneration was confirmed using recombinant hyper-IL-6 treatment62 or using mice in which mIL-6R was exchanged for sIL-6R63. These transgenic sIL-6R mice showed liver regeneration that was indistinguishable from control mice63.
In summary, the mechanistic separation of IL-6 trans-signalling from classic signalling has provided an opportunity to develop small molecules and antibody-based strategies that either globally block IL-6 activity in vivo64,65, or else selectively block pathological IL-6 trans-signalling11, as will be discussed in the following sections.
Development of IL-6 signalling inhibitors
Studies in IL-6-deficient mice showed that IL-6 contributes to the pathogenesis of many inflammatory diseases, including bacterial and viral infections66, arthritis67,68, experimental colitis69 and multiple sclerosis70. Therefore, IL-6 inhibition emerged as an attractive therapeutic avenue to translate to the clinic. In principle, inhibition of IL-6 signalling can be achieved by the application of antibodies to any component of the receptor signalling complex; blockade of IL-6, IL-6R or gp130 alone is sufficient to completely silence IL-6-mediated signalling (Fig. 4). Small-molecule inhibitors targeting JAKs, which act downstream of gp130, represent another viable approach, and such molecules have been used in the clinic to treat rheumatoid arthritis and other inflammatory diseases71–73. However, JAKs are not exclusively activated by IL-6, so targeting these signalling molecules does not enable selective inhibition of IL-6.
Although expression levels of mIL-6R and gp130 are only marginally upregulated during inflammation, IL-6 levels in the circulation dramatically rise from virtually undetectable (lower picograms-per-millilitre range) in healthy individuals to several nanograms per millilitre in inflammatory diseases and up to micrograms-per-millilitre levels in patients who develop fatal sepsis74. This is an important consideration from a therapeutic point of view, as rather high amounts of antibodies blocking gp130 or the IL-6R (see Box 1) would be needed to saturate all membrane-bound and soluble IL-6 receptors in order to attain a clinical response. In contrast, lower amounts of antibodies that block IL-6 directly might be sufficient, and the application of an IL-6-neutralizing antibody could be rather easily adapted to the amount of IL-6 measured in patient serum samples.
In practice, only antibodies blocking IL-6 or the IL-6R have made their way into the clinic8,75. The fact that gp130 is generally used by IL-6 family cytokines76,77 and that gp130 knockout mice are not viable hindered the clinical development of gp130 antibodies. In 1997, antibodies to gp130, which neutralized IL-6 activity but not the activity of LIF, OSM and CNTF, were described78. Unfortunately, these antibodies were not evaluated in further in vivo studies and were not tested for cross-reactivity to IL-11, IL-27 and CLC.
The first clinical trial for an IL-6-specific inhibitor was performed in a patient with plasma cell leukaemia because IL-6 had previously been identified as a potent growth factor for this cancer79,80. The trial was conducted with a mouse monoclonal antibody to human IL-6 and showed very promising initial results, including suppression of myeloma cell proliferation in the bone marrow and reduced C-reactive protein (CRP) levels81. However, owing to the formation of IL-6–antibody complexes that were not efficiently cleared via the kidney, IL-6 levels, although biologically inactive, accumulated in the circulation and reached high concentrations of up to 14 ng/ml82 in some patients and up to 1.7 µg/ml in a patient who developed sepsis during therapy83. In a second case report, treatment of a patient with Castleman disease with the same antibody led to the resolution of disease symptoms and improved laboratory parameters. However, increased IL-6 levels, probably due to the formation of complexes with the antibody, were detected and the treatment was finally discontinued84. Thus, despite the initial positive clinical response of the two patients, which clearly underlined that IL-6 can the successfully targeted therapeutically in IL-6-driven diseases, this approach was not further pursued.
Instead, the humanized monoclonal antibody tocilizumab, an antibody to IL-6R that prevents binding of IL-6 to the IL-6R, was developed and is currently in clinical use, for example, to treat rheumatoid arthritis, Castleman disease and cytokine release syndrome8. Recently, tocilizumab was shown to improve survival in hospitalized adult patients with COVID-19 with both hypoxia and systemic inflammation9, and was subsequently approved for this indication in several countries. Interestingly, IL-6 levels also slightly increased in patients treated with tocilizumab85,86, which can be explained by lack of IL-6 internalization and degradation by IL-6R-expressing cells87. Interestingly, the reported increases in sIL-6R are much smaller than the increases in IL-6 during anti-IL-6 therapy, because tocilizumab does not form a complex with IL-6, which is therefore still cleared via the kidney. However, sIL-6R levels were increased up to 10-fold in patients treated with tocilizumab (27.7 ± 4.4 ng/ml at baseline versus 251.4 ± 24.7 ng/ml after 6 weeks of tocilizumab treatment) owing to the prolonged half-life of sIL-6R–tocilizumab complexes85. In addition to tocilizumab, the fully humanized anti-IL-6R monoclonal antibody sarilumab was approved for the treatment of rheumatoid arthritis88, whereas the chimeric anti-IL-6 monoclonal antibody siltuximab was approved for patients with idiopathic multicentric Castleman disease8. Several other therapeutics targeting either IL-6 or the IL-6R are currently at different stages of clinical or preclinical development8.
Although inhibition of IL-6 or IL-6R was considered to result in comparable signalling outcomes, a recent publication using a single side-by-side administration protocol for siltuximab and tocilizumab in patients with type 1 diabetes reported differences between the two antibodies89. The authors describe distinct influences on T cell function, because siltuximab but not tocilizumab enhanced the suppression of effector T cells by regulatory T cells89. As this is the first in vivo study addressing this issue, it might simply be related to differences in dosing and pharmacokinetics89. However, it might also, at least in part, be caused by IL-6 family cytokine crosstalk. It has been shown that CNTF90 and the p28 subunit (also known as IL-30) of IL-2791,92 also signal via the IL-6R, and such signals could be blocked by tocilizumab but not by siltuximab. However, both CNTF and p28 have a much lower affinity for the IL-6R compared to IL-6 itself, and a functional role for these other IL-6R ligands in vivo has yet to be established. Crosstalk for IL-6, however, has not been described to date.
Of note, none of these IL-6 signalling inhibitors can discriminate between classic signalling and trans-signalling, and they block both pathways simultaneously. As already mentioned, and described in detail in the next section, IL-6 trans-signalling has turned out to be the pathological arm of IL-6, whereas classic IL-6 signalling often seems to be protective and anti-inflammatory. Therefore, interfering with classic IL-6 signalling might give rise to a negative benefit–risk scenario.
Box 1 The IL-6 buffer in the blood is formed by sIL-6R and sgp130.
In healthy individuals, interleukin-6 (IL-6) levels in the blood are in the range of 1–5 pg/ml. These levels increase during inflammatory states around 1,000 fold21. In sepsis, IL-6 levels of up to several micrograms per millilitre have been reported74. Under homeostatic conditions, levels of soluble IL-6 receptor (sIL-6R) are in the range of 40–75 ng/ml and have been shown to increase between 2-fold and 10-fold during inflammatory diseases21. It is not clear whether sIL-6R levels correlate with disease severity, as this has not been studied systematically. However, recent studies in patients with inflammatory bowel disease203 and COVID-19204 reported conflicting results. Levels of sgp130 in healthy individuals have been observed to be in the range of 250–400 ng/ml. Interestingly, it was first reported that sgp130 was generated by differential splicing205. Unexpectedly, it was recently found that about 50% of the sgp130 in the blood is generated by the protease BACE via limited proteolysis206. Levels of sgp130 remain unaltered during inflammation21 (Fig. 5a,b). IL-6 secreted by cells will bind to the sIL-6R with an affinity of around 1 nM. The complex of IL-6 and sIL-6R binds to sgp130 with considerably higher affinity and will thereby be neutralized94. Thus, sIL-6R and sgp130 in the blood can be seen as a buffer for IL-6, serving as a protection from overstimulation74 (Fig. 5). The capacity of the buffer is determined by the level of the sIL-6R because sgp130 levels exceed sIL-6R levels on the molar level. A single-nucleotide polymorphism has been found in the gene coding for the human IL-6R, which leads to a change of Asp358 to Ala358 close to the ADAM17 cleavage site of the IL-6R protein41. The Ala358 variant of the IL-6R leads to its more efficient cleavage207 and therefore to about 50% higher levels of sIL-6R in the blood182,188. Consequently, carriers of the Ala358 variant of the IL-6R are less susceptible to several inflammatory conditions, such as congestive heart disease, abdominal aortic aneurism and rheumatoid arthritis208. A likely explanation of this phenomenon is the higher buffer capacity of the IL-6 buffer in the blood brought about by the higher levels of sIL-6R209. Disturbance of the buffer has been reported (for example, in patients with type 2 diabetes210) and might contribute to pathophysiology in this and other inflammatory diseases. In our view, the demonstrated beneficial role of sgp130Fc (olamkicept) is on the one hand explained by the selective blockade of IL-6 trans-signalling. On the other hand, the sgp130Fc protein increases the IL-6 buffering capacity, which might be limited in autoimmune diseases.
An IL-6 trans-signalling inhibitor: sgp130Fc
The designer cytokine hyper-IL-6 was the first biological tool to specifically activate cells via IL-6 trans-signalling, but not by classic IL-6 signalling via mIL-6R50. From a molecular three-dimensional model of IL-6 bound to the sIL-6R, we estimated that the distance between the COOH-terminus of IL-6 and the NH2-terminus of the sIL-6R was about 40 Å. We connected the cDNA coding for the sIL-6R to the cDNA coding for IL-6 by a stretch of nucleotides coding for 13 amino acids. These amino acids formed a flexible linker between sIL-6R and IL-6, allowing the natural interaction between the ligand and the receptor50. This designer cytokine enabled the identification of a variety of potential target cells for IL-6 trans-signalling, including neuronal cells51, adult haematopoietic stem cells and embryonic stem cells52,53. A limitation was that these experiments did not prove that IL-6 trans-signalling actually occurred in vivo and drove pathophysiological responses. It came to our notice that most soluble receptors were competitive inhibitors of their cognate membrane-bound versions93 and that the powerful agonistic activity of sIL-6R was a prominent exception. To design an alternative to the antibody IL-6 inhibitor, we generated a fusion protein of the extracellular portion of gp130 fused to the Fc portion of a human IgG1 antibody (Fig. 4). As neither IL-6 nor sIL-6R alone bind to gp130, this fusion protein — sgp130Fc — was a selective inhibitor of IL-6 trans-signalling but did not interfere with classic signalling94. We were rather surprised by this finding, because sgp130Fc interacts not only with IL-6–sIL-6R complexes94 but also with IL-6–mIL-6R complexes13.
This might be explained by the receptor activation mode of IL-6, which might allow binding of naturally occurring sgp130 or sgp130Fc to membrane-bound IL-6–IL-6R complexes in the absence (but not in the presence) of cell surface gp130. Gp130 has been described to assemble as preformed dimers, most likely to enable rapid signal induction after IL-6 binding87,95. It was, however, not analysed whether IL-6R or IL-11R are also included in these preformed receptor complexes. Of note, we recently deciphered the apical and basolateral sorting code of IL-6R, IL-11R and gp130 in polarized cells. Although we have not analysed this directly, our results suggest that IL-6R and IL-11R are either in complex with gp130 or within the same micro-membrane localization, either directly after translocation to the membrane of the endoplasmic reticulum or after transport to the Golgi apparatus96.
Such preformed receptor complexes might prevent sgp130Fc accession and inhibition of classic IL-6 signalling. As already explained, neither IL-6 nor IL-6R alone bind to gp130. Therefore, the first step of activation is the formation of the complex of IL-6 and IL-6R. Thereafter, this complex binds the low-affinity site of gp130, which is formed by the elbow between domain 2 and domain 3 of gp130 (Fig. 3). Only then will contact of the tip of IL-6 to the high-affinity site of gp130 (formed by domain 1 of gp130) be established19. Apparently, once binding of the complex of IL-6 and IL-6R has been established, sgp130Fc has no access to the complex of IL-6 and IL-6R.
This might, however, not be the full explanation. IL-6–IL-6R–gp130 complexes might be additionally stabilized by other, so far unknown factors. For example, it is unclear the extent to which signalling by IL-6–IL-6R–gp130 complexes is initiated directly at the plasma membrane, or if signalling by these complexes is specifically continued or even boosted after endocytosis97. For other cytokines, such as tumour necrosis factor (TNF), signalling from endosomes plays an important role in signal transduction98. At least to some extent, activation of STAT3 involves endosomal trafficking of the receptor complex99.
Having discussed the general features of sgp130 in the inhibition of IL-6 trans-signalling, in the following section we will detail the preclinical and clinical development of the sgp130Fc variant olamkicept.
Preclinical and clinical development of olamkicept
The generation of sgp130Fc (named olamkicept by the World Health Organization in 2016) was a welcome addition to the arsenal of IL-6 inhibitors and has enabled us to define the physiological and pathophysiological roles of IL-6 trans-signalling in numerous mouse models of inflammatory or neoplastic diseases (Table 1 and Fig. 5a–c). The experimental blueprint included comparing global blockade of IL-6 in anti-IL-6-treated mice or IL-6-deficient mice to selective inhibition of IL-6 trans-signalling in sgp130 transgenic mice100 or animals treated with recombinant sgp130Fc. Surprisingly, in many disease models, treatment with sgp130Fc was more beneficial than global blockade by IL-6-neutralizing antibodies. Such disease models included sepsis101, cerulein-induced acute pancreatitis102, bone fracture healing103,104 and myocardial infarction105. In the cerulein-induced acute pancreatitis model, Il6−/− mice showed more inflammation-associated damage in the pancreas than wild-type mice, but no subsequent lung failure. In contrast, sgp130Fc transgenic mice administered with cerulein were completely protected against pancreatic damage and lung failure, and therapeutic treatment of cerulein-administered wild-type mice with sgp130Fc protein attenuated severe acute pancreatitis and led to 100% survival102. Further, in a mouse model for uneventful bone fracture healing, the global blockade of IL-6 with a neutralizing antibody reduced systemic inflammation, the hepatic acute-phase reaction and immune-cell infiltration to the fracture site, but led to delayed fracture healing. In contrast, specific blockade of IL-6 trans-signalling with recombinant sgp130Fc had minor effects on the immune response but did not impede bone fracture healing103,104. In a rat model of reperfused myocardial infarction, treatment with an IL-6-neutralizing antibody did not alter the infarct size, whereas treatment with sgp130Fc reduced the infiltration of neutrophils and mononuclear phagocytes into the myocardium and reduced the infarct size by about 50%, thereby preserving cardiac function 28 days after the myocardial infarction105.
Table 1.
Efficacy of sgp130Fc-mediated blockade of IL-6 trans-signalling in preclinical models of inflammation and cancer
| Year | Disease Model | Study Outcome | Refs. |
|---|---|---|---|
| 2000, 2006, 2009 | Intestinal inflammation | Suppression of inflammatory bowel disease | 69,182,187 |
| 2003, 2006, 2009 | Rheumatoid arthritis | Improvement of established arthritis | 169,188,189 |
| 2004, 2009, 2010, 2018 | Colon cancer | Reduction of tumour load | 182,190,191 |
| 2007, 2008 | Acute local inflammation | Suppression of acute local inflammation | 100,192 |
| 2011 | Pancreatic cancer | Inhibition of tumour progression, reduction of ductal adenocarcinoma | 164 |
| 2011, 2012 | Sepsis | Up to 100% survival in sepsis models | 186,193 |
| 2012 | Mycobacteria infection | No increase of bacterial burden | 194 |
| 2012 | Atherosclerosis | Regression of atherosclerosis plaques | 117 |
| 2013 | Listeria infection | No increase or reduction of bacterial burden | 107 |
| 2013 | Pancreatitis-associated lung failure | 100% survival of severe, acute experimental pancreatitis | 102 |
| 2013 | Lupus erythematosus | Suppression of inflammation and renal pathology | 195 |
| 2015, 2016 | Nephrotoxic nephritis | Amelioration of disease | 196,197 |
| 2016 | Lung emphysema | Improvement of disease by blockade of alveolar apoptosis | 198 |
| 2016 | Lung cancer | Reduction of tumour load | 166 |
| 2017 | Liver cancer | Reduction of tumour load | 199,200 |
| 2018 | Bone healing | Improvement of bone healing | 103,104 |
| 2019 | Abdominal aortic aneurysm | Improved survival | 201 |
| 2021 | Myocardial Infarction | Reduction of infarct size and preserving of cardiac function | 105 |
| 2022 | Perioperative neurocognitive disorder | Prevention of surgery-induced cognitive decline | 202 |
Fig. 5. Endogenous and therapeutic buffer system to limit IL-6 signalling.

The buffer for interleukin-6 (IL-6) in the blood is formed by soluble IL-6 receptor (sIL-6R) and sgp130 (Box 1). a, Under healthy, noninflammatory conditions, the buffer system allows local homeostatic signalling mainly in immune cells and hepatocytes but not in typical tissue cells, smooth muscle cells and endothelial cells. A low level of IL-6 and the buffer system prevents systemic IL-6 signalling. b, Under acute and chronic inflammatory conditions, the buffer system is overloaded and allows systemic IL-6 trans-signalling. c, Therapeutic application of sgp130Fc (soluble gp130–Fc fusion protein) under acute and chronic inflammatory conditions reinforces the buffer system and prevents systemic IL-6 trans-signalling. Coloured shapes have the same meanings as in Fig. 4.
A potential caveat with the use of anti-inflammatory therapies is the common burden of increased frequency of infections, with incidence rates for serious infections (patients with events per 100 patient–years) ranging from 7.59 for the anti-TNF blocker certolizumab pegol, to over 5.45 for tocilizumab, to 2.93 for the JAK inhibitor tofacitinib106. In other words, if a physician treats 100 patients for one year with these inhibitors, approximately 3–8 patients will acquire a serious infection106. This is an important issue because treatment of autoimmune diseases with anti-cytokine drugs need to be continued for years if not for life, leading to an accumulation of the infection risk.
A reduced capacity to cope with infections was also seen in IL-6-deficient mice after Listeria monocytogenes infection. Global blockade of IL-6 activity in mice infected with L. monocytogenes reduced hepatic induction of acute-phase proteins and led to significantly higher titres of bacterial colony-forming units in spleen and liver66,107. In contrast, infected sgp130Fc transgenic mice or infected wild-type mice treated with recombinant sgp130Fc did not show reduced induction of hepatic acute-phase proteins or increased bacterial colony-forming units in spleen and liver107, suggesting that inhibition of IL-6 trans-signalling did not interfere with bacterial defence mechanisms. The beneficial consequences of selectively blocking IL-6 trans-signalling in inflammatory bowel disease (IBD) and cardiovascular disease might point to known effects of trans-signalling on T cells endothelial cells and smooth muscle cells. It has been shown that IL-6-mediated apoptotic resistance in T cells is based on IL-6 trans-signalling69,94. Furthermore, it has been demonstrated that the differentiation of T cells into TH17 cells, which is induced by TGFβ and IL-6, is by far more efficient in the presence of sIL-6R108. Endothelial cells and smooth muscle cells have been shown to express gp130 but not IL-6R109,110. These cells cannot respond to IL-6 alone but can be stimulated by the combination of IL-6 and sIL-6R to express various chemokines and ICAM1. These changes might contribute to the vascular injuries observed in patients with sepsis109,110. Interfering with IL-6 trans-signalling would dampen these effects and thereby act in an anti-inflammatory manner.
These data suggest that the pro-inflammatory activities of IL-6 are predominantly mediated by trans-signalling, whereas classic IL-6 signalling is primarily pro-inflammatory in the acute stage of an inflammatory responses but later acts in a protective, regenerative manner111. Therefore, it might be reasonable to selectively block the IL-6 trans-signalling pathway rather than block IL-6 globally via neutralizing antibodies8,112.
In phase I clinical trials of olamkicept in healthy individuals in 2013 and 2014, no serious adverse events were encountered. A small open-label phase IIa clinical trial was subsequently performed involving 16 patients with IBD (EudraCT No 2016-000205-36). Similar to the results in the phase I study, olamkicept was well tolerated and induced clinical response in 44% and clinical remission in 19% of the patients. Molecular profiling confirmed that clinical remission was associated with distinct changes in mucosal levels of phosphorylated STAT3 in intestinal epithelium and lamina propria of the treated patients113.
In a second double-blind placebo-controlled phase IIb clinical study, 91 patients with moderate-to-severe ulcerative colitis that had shown inadequate response to conventional therapy were enrolled. Olamkicept was well tolerated and showed dose-dependent high efficacy with regards to clinical response, clinical remission and mucosal healing114 (NCT03235752). Most of the patients (94.5%) had previously not been treated with biologics. After 12 weeks with injections every second week, clinical remission was seen in 0% (placebo), 6.7% (olamkicept 300 mg/injection) and 20.7% (olamkicept 600 mg/injection) of the patients. At the same time, mucosal healing occurred in 3.4% (placebo), 10% (olamkicept 300 mg/injection) and 34.5% (olamkicept 600 mg/injection) of the patients115. Based on the positive results of these phase II clinical trials, phase III clinical trials are being planned for the near future.
In addition to IBD, compassionate use of olamkicept has been undertaken in a single patient with very high-risk atherosclerotic cardiovascular disease. After 11 weeks of olamkicept treatment every second week (600 mg/injection), the patient showed reduced arterial wall inflammation116. This was not unexpected because in low-density lipoprotein receptor-deficient (Ldlr−/−) mice on a high-fat, high-cholesterol diet, sgp130Fc treatment not only prevented mouse atherosclerosis but also strongly reduced existing atherosclerotic plaque burden117.
The results with olamkicept in the described clinical trials are encouraging. As mentioned, we have performed numerous preclinical trials in animal models of human autoimmune and neoplastic diseases (Table 1). In all disease models, we noted that specific blockade of IL-6 trans-signalling with the sgp130Fc protein was at least as efficient as treatment with anti-IL-6 or anti-IL-6R monoclonal antibodies, which lead to global blockade of IL-6 activity. However, in animal models of sepsis, pancreatitis, bone fracture healing and myocardial infarction, a clear advantage in the specific blockade of IL-6 trans-signalling was demonstrated over global blockade of IL-6 activity94–103. These diseases are therefore good candidates for further clinical trials with olamkicept.
Taken together, the promising clinical potential of olamkicept as an efficacious anti-inflammatory drug provides the rationale to further refine sgp130Fc for the development of next-generation inhibitors of IL-6 trans-signalling with higher affinity, specificity and bioavailability.
Next-generation inhibitors of IL-6 trans-signalling
Considering that sgp130Fc is derived from the common signal-transducing gp130 receptor used by many members of the IL-6 cytokine family76, it is no surprise that the inhibitory activity of sgp130Fc extends to other family members. This includes IL-11, a cytokine that affects physiological processes including haematopoiesis118, thrombopoiesis119, bone metabolism120, plasma cell proliferation121 and T cell-dependent development of antibody-producing B cells25. Protective effects of IL-11 were described following myocardial infarction122,123, stroke124 and during tissue regeneration125. However, IL-11 also promotes gastric cancer126, and recent work suggests a crucial role of IL-11 in promoting cardiac37, kidney37, liver127–129 and lung fibrosis130. In analogy to the sIL-6R, a soluble form of the IL-11R (sIL-11R) has been detected in human serum19. The sIL-11R has also been detected in tumour tissue from patients with gastric cancer and mouse models of this disease131, which suggests that IL-11 trans-signalling operates in vivo. Furthermore, IL-11 trans-signalling was demonstrated to be induced by the designer cytokine hyper-IL-11 or by naturally occurring IL-11–sIL-11R complexes15–19. However, definitive pathophysiological roles for IL-11 trans-signalling that are distinguishable from IL-6 trans-signalling in vivo remain elusive, the complexities of which might be compounded by differential expression patterns of mIL-11R and mIL-6R. Although only a few cell types express mIL-6R at high levels and all other cell types have either low or no mIL-6R expression at all, the distribution of mIL-11R throughout the body appears to be more balanced. When many cell types already respond to classic signalling, it is difficult to demonstrate an (additional) functional role for trans-signalling.
Importantly, the activity of other gp130-dependent cytokines including OSM, LIF and IL-27 can only be inhibited by more than 100 times higher concentrations of sgp130Fc than those needed to inhibit IL-6 trans-signalling94,132. The promiscuous inhibitory effect of sgp130Fc on several IL-6 family cytokines suggests that its therapeutic use may inadvertently induce side effects. For instance, targeting pathological IL-6 trans-signalling with sgp130Fc could simultaneously inhibit homeostatic and protective functions of other IL-6 family members, such as IL-11. However, it is notable that treatment of patients with IBD with the sgp130 variant olamkicept was found to be safe and efficacious in the above-mentioned phase 2 clinical trials113.
A solution to limiting potential side effects in disease states in which only targeting of pathological IL-6 trans-signalling is desired may be provided by second-generation sgp130Fc variants (Fig. 6). One such variant, sgp130FLYFc, was generated by the substitution of amino acids Q113F or T102Y, Q113F and N114L (FLY) at site III of sgp130Fc. The sgp130FLYFc variant showed enhanced binding affinity for hyper-IL-6 and more potently inhibited IL-6 trans-signalling, but showed strongly diminished inhibition of IL-11 trans-signalling15–17. Interestingly, a gp130 single-nucleotide polymorphism (SNP) (842G>A) — encoding an R281Q substitution at site IIb in the protein — was observed in a patient presenting with craniofacial malformations. This mutation has been associated with selective abrogation of IL-11-mediated responses but intact responses of other gp130-dependent cytokines (including IL-6, IL-27, OSM and LIF)133. Importantly, this discovery guided engineering of the R281Q substitution in sgp130Fc, which selectively augmented specificity for IL-6 inhibition to a level comparable to that observed for the sgp130FLYFc variant. Moreover, the combination of R281Q and FLY mutations in another sgp130Fc variant, sgp130FLYRFc, completely abrogated its inhibitory effects on IL-11 trans-signalling without impacting inhibition of IL-6 trans-signalling16.
Fig. 6. Next-generation IL-6 trans-signalling inhibitors.

a, First-generation sgp130Fc (soluble gp130–Fc fusion protein) is composed of domains D1–D6 of glycoprotein 130 (gp130; blue) fused to a human IgG1 Fc (grey). The first-generation trans-signalling inhibitor efficiently inhibits interleukin-6 (IL-6) and IL-11 trans-signalling and, with lower efficiency, oncostatin M (OSM) and leukaemia inhibitory factor (LIF) signalling. The second-generation sgp130 variants have improved IL-6 trans-signalling activity and/or reduced effects on the signalling of IL-11, LIF and OSM. Point mutations in domains D1 and D3 that improve the activity and specificity of sgp130Fc are indicated by yellow lines. Third-generation inhibitors such as chimeric soluble gp130 (cs130) are composed of the domains D1–D3 of gp130 (blue) fused to the single-domain antibody VHH6 (green), an antibody to the complex of IL-6 and soluble IL-6 receptor (IL-6–sIL-6R) (yellow and red). Bispecific cs130+ variants are based on cs130 fused to VHH6 and a second nanobody (purple) enabling additional functions including SARS-CoV-2-neutralization, inhibition of other inflammatory cytokines or cell-specific targeting. b, Proposed tetrameric and hexameric assembly mode of sgp130 and cs130 with IL-6–sIL-6R complexes. According to Boulanger et al.24, the initial trimeric complex of gp130 and IL-6–sIL-6R is formed between site IIa of IL-6, site IIb of IL-6R and domain D2–D3 of gp130. Subsequently, hexameric complexes are formed by binding of two trimeric IL-6–sIL-6R–sgp130 complexes via site IIIa of IL-6, site IIIb of IL-6R and domain D1 of gp130.
The third generation of trans-signalling inhibitors use alternative overall structural assemblies. Cs130Fc is a chimeric sgp130 molecule containing the cytokine-binding domains D1–D3 of gp130 fused to a noninhibitory single-domain antibody (nanobody, VHH) recognizing an intermolecular, nonlinear epitope of the IL-6–sIL-6R complex, along with the human IgG1 Fc fragment15. Compared to sgp130Fc, cs130Fc is smaller (185 kDa versus 240 kDa), displays enhanced specificity for inhibition of IL-6 trans-signalling (2-fold improved IC50 for IL-6 trans-signalling inhibition, 8-fold reduction in activity against IL-11 trans-signalling), and can be produced without the Fc fragment as an even smaller fully active 75-kDa molecule to enhance bioavailability and tissue penetration (Fig. 6). The improved binding kinetics (mainly koff-rate) and overall activity of cs130Fc relative to the first-generation sgp130Fc is due to its high affinity for the IL-6–sIL-6R complex (mediated via the single-domain antibody VHH6) leading to a more stable association with IL-6 trans-signalling complexes than sgp130Fc15. The potential to further augment the selective inhibitory actions of cs130Fc on IL-6 trans-signalling has been indicated by the incorporation of site III FLY mutations into cs130Fc to generate cs130FLYFc, which enhanced IL-6 trans-signalling blockade by 3-fold versus sgp130Fc, whereas inhibitory effects on IL-11 trans-signalling were completely abrogated15.
Despite the advantages of selectively applying sgp130Fc or its next-generation derivatives in inflammation-associated diseases (including cancer) in which IL-6 trans-signalling contributes to disease pathogenesis, it is likely that other cytokines (for example, TNF, IL-1, IL-17 and IL-23) and/or growth factors (such as TGFβ and amphiregulin) concurrently drive such disease states8. Therefore, the blockade of IL-6 trans-signalling in concert with other known molecular drivers of the disease may provide a more favourable clinical outcome than targeting IL-6 trans-signalling alone. For instance, in mouse models of colitis, rheumatoid arthritis and allergic asthma, simultaneous inhibition of IL-6R and TNF proved more effective than single-target-directed treatment69,134,135. Similarly, dual blockade of IL-6 and IL-17 signalling using a bispecific antibody demonstrated increased efficacy in inflammatory disease models compared to IL-6 or IL-17 blockade alone136.
Dysregulated IL-6 signalling is a hallmark of infections, including SARS-CoV-2, that are characterized by hyperinflammation137–145, with high IL-6 levels in patients with COVID-19 being implicated in the depletion of CD4+ T cells, CD8+ T cells and natural killer cells146. Moreover, recent evidence suggested a key role of IL-6 trans-signalling, but not classic signalling, in the development of hyperinflammatory states in severe COVID-19138,141,142, resulting in respiratory failure and multi-organ damage137. Indeed, increased ADAM17-mediated IL-6R shedding was observed in epithelial cells following SARS-CoV-2 infection or upon overexpression of spike protein, accelerating the inflammatory spiral142. Interestingly, studies on CAR T cell-induced hyperinflammation suggested a rate-limiting role of sIL-6R serum concentrations138. COVID-19-induced dysregulated IL-6 trans-signalling seems to be associated with liver damage141 and abnormalities in the coagulation cascade, including increased tissue factor, thrombin and platelet activity147–149 finally leading to endothelial dysfunction147 and immunothrombotic processes150. Such IL-6 trans-signalling-mediated increases in coagulation factors were efficiently inhibited by sgp130Fc in liver sinusoidal endothelial cells141. These findings, coupled with accumulating evidence that the IL-6R-targeted antibodies tocilizumab and sarilumab demonstrated beneficial effects on survival rates in severe COVID-19 cases151–154, have led to the generation of a bispecific functionalized sgp130Fc variant, c19s130Fc (Fig. 6). This sgp130 variant is composed of cs130Fc functionalized with an additional neutralizing single-domain antibody to the SARS-CoV-2 spike protein, yielding an IL-6 trans-signalling inhibitor with high binding affinity for both IL-6–sIL-6R complexes and the SARS-CoV-2 spike protein155. Importantly, c19s130Fc was shown to inhibit SARS-CoV-2 infection of cells and to improve the therapeutic efficacy of sgp130Fc, indicating that c19s130Fc may provide synergistic therapeutic effects on containing viral infection and the resulting hyperinflammation. Collectively, these observations pave the way for future approaches involving bispecific biologics that target IL-6 trans-signalling and other inflammatory cytokines, such as IL-1 and TNF.
Moreover, the multifunctional type I transmembrane inflammatory protease ADAM17 has emerged as a key regulator of the ectodomain shedding of more than 80 known membrane-tethered substrates, including IL-6R156,157. ADAM17 is expressed in most tissues as a catalytically inactive full-length precursor that is predominantly stored in the endoplasmic reticulum. Although the transcriptional regulation of ADAM17 is static in both physiological and pathological settings, the mechanisms governing ADAM17 surface expression as a mature and biologically active enzyme are complex and remain to be fully elucidated: they involve a series of steps including the cleavage by the furin convertase of the autoinhibitory ADAM17 N-terminal pro-domain in the Golgi apparatus, which triggers its transport to the cell membrane that is dependent on the iRhom intramembrane rhomboid proteases157–160. Considering that upregulated production of sIL-6R and other processed ADAM17 substrates, in particular TNF and EGFR ligands (for example, TGFα and amphiregulin), is a feature of numerous inflammatory conditions (for example, colitis, rheumatoid arthritis, sepsis and pancreatitis) and cancers (such as colorectal, lung and pancreatic cancers), ADAM17 presents as an attractive therapeutic target to simultaneously block inflammatory IL-6 trans-signalling and co-existent disease-associated ADAM17-regulated pathways69,101,102,161–169. This notion is supported by recent studies employing ADAM17-neutralizing monoclonal antibodies that act via several modalities, including allosteric perturbation of the binding ability of its catalytic active site, or steric hindrance leading to competitive or noncompetitive inhibition of protein substrate binding170,171 (Fig. 5b). In mouse models of pancreatic ductal adenocarcinoma or polymicrobial sepsis, the selective antibody-mediated blockade of ADAM17 suppressed the onset and/or progression of disease172,173. Curiously, circulating sIL-6R levels were either unchanged or not investigated in these studies, raising the questions as to whether such antibody-mediating targeting of ADAM17 will be sufficient to indirectly block IL-6 trans-signalling in these disease settings, and whether other IL-6R sheddases, such as ADAM10174 or meprin-α/β174, can substitute for ADAM17 activity.
Importantly, an alternative therapeutic strategy against ADAM17 to target IL-6 trans-signalling has arisen via the autoinhibitory N-terminal pro-domain of ADAM17 (Fig. 4), which serves as a powerful template to generate a recombinant protein that selectively blocks ADAM17 biological activity with high affinity175. The pro-domain of ADAM17 is an independent folding unit comprising ~200 amino acids, which specifically interacts with and inhibits the catalytic domain of ADAM17. Indeed, the ADAM17 pro-domain inhibitor (A17pro), engineered as a stable recombinant protein, exhibits robust therapeutic efficacy in numerous preclinical mouse inflammatory disease and inflammation-associated cancer models, including rheumatoid arthritis, IBD, sepsis, acute pancreatitis, kidney fibrosis and lung adenocarcinoma161,167,175,176. Notably, in acute pancreatitis and lung adenocarcinoma, sIL-6R was the predominant substrate employed by ADAM17 for its pathological activity, and in the preclinical KrasG12D lung adenocarcinoma mouse model, A17pro exhibited greater anticancer activity than sgp130Fc166,167. These observations suggest that inhibitor-based targeting of the ADAM17 protease may provide an effective rationale for targeting inflammatory IL-6 trans-signalling in certain disease settings. From a clinical and safety perspective, it is noteworthy that the above-mentioned ADAM17 pro-domain and antibody-based inhibitors are highly selective for ADAM17, and are therefore advantageous compared to earlier small-molecule inhibitors against ADAM17, which also targeted other related proteases (for example, ADAM10) leading to an unfavourable toxicity profile in the clinic177.
Conclusions and future prospects
Today, IL-6 trans-signalling is recognized as a biologically important independent pathway of cytokine receptor activation. This has led to the clinical development of the first selective inhibitor of IL-6 trans-signalling — olamkicept — as well as to the next-generation inhibitors that we have discussed above. Virtually all cells of the body are responsive to IL-6 trans-signalling. In our opinion, this finding is key to explaining the incredibly far-reaching role of IL-6 in autoimmune diseases, chronic inflammatory conditions and cancer. For more than a decade, antibodies and small molecules that block global IL-6 signalling have served as therapeutic gold standards. However, we will soon know whether selective targeting of IL-6 trans-signalling by olamkicept can show therapeutic benefit while reducing the side effects typically observed with current anti-IL-6 therapies.
In the Western world, at least 0.5% of people have IBD178. Although some biologics have entered the clinic, anti-TNF therapeutics still dominate the treatment regimen for IBD. Therefore, it might seem obvious to select IBD for the first phase II clinical study of olamkicept. In truth, this was a risky decision because IL-6 and IL-6R antibodies failed in phase II clinical trials for IBD due to intolerable side effects, including intestinal perforations179,180, which were also observed for anti-IL-6R therapy for rheumatoid arthritis181. A possible explanation is that classic IL-6 signalling via mIL-6R is involved in the intestinal regeneration response, which is largely blocked in IL-6 knockout mice182 as well as by IL-6 and IL-6R antibodies180,183,184. However, this regenerative response is not affected when only IL-6 trans-signalling is inhibited, as IL-6 activities via mIL-6R are not affected by olamkicept107. Alternatively, it might be that the anti-IL-6 or anti-IL-6R antibodies lead to reduced TH17 responses, which are required for barrier function in the intestine. Blocking IL-17 activity with the antibody secukinumab indicated that IL-17 exhibits protective functions in the intestine185.
We have also delineated second-generation and third-generation therapeutics, based on size-reduced IL-6- selective trans-signalling inhibitors or ADAM17-specific inhibitors with increased bioavailability compared to olamkicept. With the introduction of IL-6 trans-signalling-selective point mutations in sgp130Fc and the size-reduced cs130Fc, inhibition of IL-11 trans-signalling was abrogated. It will be interesting to see whether IL-11 affinity-increasing mutations will lead to IL-11 trans-signalling-selective sgp130 variants, which are needed to identify IL-11 trans-signalling-driven diseases in vivo. The IL-6–sIL-6R binding VHH6 nanobody, which we used in cs130Fc, showed that composite epitopes exist, making the generation of inhibitory trans-signalling nanobodies another future prospect. Size-reduced trans-signalling inhibitors will ease the generation of bispecific drugs, for instance to simultaneously block IL-6 trans-signalling and TNF signalling or enable cell-type- restricted inhibition of IL-6 trans-signalling. This, however, also comprises strategies for the cell-type-restricted abrogation of IL-6R shedding to specifically prevent the local production of inflammatory sIL-6R. Despite the obvious focus on the clinical utility of targeting IL-6 trans-signalling, we also note that in the future, it will be of interest to generate a selective IL-6 classic-signalling inhibitor, not only as a tool but eventually leading to alternative clinical applications.
With respect to pharmacokinetics and pharmacodynamics, we realized from our mouse studies that far less sgp130Fc was needed to completely block IL-6 trans-signalling than to inhibit global IL-6 signalling. Albeit not addressed in the initial phase II clinical studies, this might open up the appealing opportunity to eventually reduce olamkicept dosing for patients who experience an unfavourable ratio of therapeutic benefit to side effects. Moreover, only limited dosing of sgp130 was able to prevent fatal sepsis in mice186, independently suggesting that lower amounts of sgp130 in comparison to standard antibody doses might be beneficial and should be included in future clinical trial design, once olamkicept has been approved for therapy.
We consider the role of endogenous sgp130 as a safety system to limit overshooting systemic IL-6 trans-signalling activities (see Box 1 and Fig. 5). From this, it follows that therapeutic sgp130Fc assists the natural solution to overshooting local IL-6 trans-signalling. Therefore, limiting the pharmaco-dosing practically may define sgp130Fc as a pioneer in the development of natural therapeutics.
Glossary
- ADAM
(A disintegrin and metalloproteinase). Proteases in a family of single-pass transmembrane proteins structurally related to snake venom disintegrins that contain both catalytic inactive and active members; in particular, ADAM10 and ADAM17 have been characterized over the last decades as the principal proteases for important membrane-anchored proteins such as EGFR ligands, Notch receptors, IL-6R and TNF.
- Castleman disease
A spectrum of heterogeneous lymphoproliferative disorders characterized by common lymph node histopathologic features (including abnormal germinal centre architecture with prominent follicular dendritic cell), with IL-6 overproduction considered as a key mechanism (particularly for multicentric Castleman disease).
- Cerulein
A oligopeptide of ten amino acids long that shows homology to cholecystokinin, stimulates smooth muscle cells and is used to induce pancreatitis in experimental animal models.
- Listeria monocytogenes
A facultative anaerobic, Gram-positive bacterium that reproduces inside the host cells and, when ingested via contaminated food, causes the human disease listeriosis, a severe illness characterized by meningitis, encephalitis and sepsis.
- Single-domain antibody (nanobody)
An antibody fragment consisting of a single monomeric variable domain (VH) naturally occurring in the Camelidae family or synthetically derived from the heavy chain of an antibody, combining high antigen affinity with the absence of complement-dependent or cell-mediated cytotoxicity due to the lack of a constant (Fc) region.
- STAT3
(Signal transducer and activator of transcription 3). A transcription factor that is latent associated with the signal-transducing β-receptor gp130 and translocates to the nucleus after being phosphorylated at tyrosine 705 in response to several cytokines and growth factors, including IL-6 and IL-11.
- TNF
(Tumour necrosis factor). A transmembrane protein that can act as either a pro-inflammatory cytokine or an important adipokine that promotes insulin resistance, which can further either be cleaved into a soluble form and signal pro-inflammatory and apoptotic via TNFR1, or act as a membrane-bound cytokine anti-inflammatory and cell proliferation-promoting fashion mainly through signalling via TNFR2.
Author contributions
The authors contributed equally to all aspects of the article.
Peer review
Peer review information
Nature Reviews Immunology thanks S. Kang, M. Neurath and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Funding
C.G. was supported by grants from the Deutsche Forschungsgemeinschaft (No. 125440785, SFB877, A10 and A14). J.S. was supported by grants from the Deutsche Forschungsgemeinschaft (SCHE907/5-1 and SCHE907/6-1). S.R.-J. was supported by the Deutsche Forschungsgemeinschaft, Bonn, Germany (CRC877 (Proteolysis as a Regulatory Event in Pathophysiology; project A1), CRC841 (Liver inflammation; project C1), German Cancer Aid (Deutsche Krebshilfe), German-Israeli-Foundation (GIF) and the Cluster of Excellence ‘Precision Medicine in Chronic Inflammation’. B.J.J. was supported by a United States Department of Defense Lung Cancer Research Program Idea Development Award (LC170062).
Competing interests
C.G. has received a research grant from Corvidia Therapeutics (Waltham, MA, USA) and has acted as a consultant/speaker for AbbVie and NovoNordisk. J.M.M. and J.S. declare that they have applied for a patent covering cs130Fc and c19s130Fc. J.M.M. has acted as a consultant for Ferring Pharmaceuticals. S.R.-J. has acted as a consultant and speaker for AbbVie, Chugai, Genentech Roche, Regeneron, Pfizer and Sanofi. He also declares that he is an inventor on patents owned by CONARIS Research Institute, which develops the sgp130Fc protein olamkicept together with Ferring Pharmaceuticals and I-Mab Biopharma. S.R.-J. has stock ownership in CONARIS. B.J.J. declares no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Hirano T, et al. Complementary DNA for a novel human interleukin (BSF-2) that induces B lymphocytes to produce immunoglobulin. Nature. 1986;324:73–76. doi: 10.1038/324073a0. [DOI] [PubMed] [Google Scholar]
- 2.Gauldie J, Richards C, Harnish D, Lansdorp P, Baumann H. Interferon β2/B-cell stimulatory factor type 2 shares identity with monocyte-derived hepatocyte-stimulating factor and regulates the major acute phase protein response in liver cells. Proc. Natl Acad. Sci. USA. 1987;84:7251–7255. doi: 10.1073/pnas.84.20.7251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brakenhoff JP, de Groot ER, Evers RF, Pannekoek H, Aarden LA. Molecular cloning and expression of hybridoma growth factor in Escherichia coli. J. Immunol. 1987;139:4116–4121. doi: 10.4049/jimmunol.139.12.4116. [DOI] [PubMed] [Google Scholar]
- 4.Zilberstein A, Ruggieri R, Korn JH, Revel M. Structure and expression of cDNA and genes for human interferon-beta-2, a distinct species inducible by growth-stimulatory cytokines. EMBO J. 1986;5:2529–2537. doi: 10.1002/j.1460-2075.1986.tb04531.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Haegeman G, et al. Structural analysis of the sequence coding for an inducible 26-kDa protein in human fibroblasts. Eur. J. Biochem. 1986;159:625–632. doi: 10.1111/j.1432-1033.1986.tb09931.x. [DOI] [PubMed] [Google Scholar]
- 6.Kang S, Narazaki M, Metwally H, Kishimoto T. Historical overview of the interleukin-6 family cytokine. J. Exp. Med. 2020;217:e20190347. doi: 10.1084/jem.20190347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jones S, Jenkins B. Recent insights into targeting the IL-6 cytokine family in inflammatory diseases and cancer. Nat. Rev. Immunol. 2018;18:773–789. doi: 10.1038/s41577-018-0066-7. [DOI] [PubMed] [Google Scholar]
- 8.Garbers C, Heink S, Korn T, Rose-John S. Interleukin-6: designing specific therapeutics for a complex cytokine. Nat. Rev. Drug Discov. 2018;17:395–412. doi: 10.1038/nrd.2018.45. [DOI] [PubMed] [Google Scholar]
- 9.Group RC. Tocilizumab in patients admitted to hospital with COVID-19 (RECOVERY): a randomised, controlled, open-label, platform trial. Lancet. 2021;397:1637–1645. doi: 10.1016/S0140-6736(21)00676-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Taga T, et al. Interleukin-6 triggers the association of its receptor with a possible signal transducer, gp130. Cell. 1989;58:573–581. doi: 10.1016/0092-8674(89)90438-8. [DOI] [PubMed] [Google Scholar]
- 11.Rose-John S, Heinrich PC. Soluble receptors for cytokines and growth factors: generation and biological function. Biochem. J. 1994;300:281–290. doi: 10.1042/bj3000281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chalaris A, Garbers C, Rabe B, Rose-John S, Scheller J. The soluble interleukin 6 receptor: generation and role in inflammation and cancer. Eur. J. Cell Biol. 2011;90:484–494. doi: 10.1016/j.ejcb.2010.10.007. [DOI] [PubMed] [Google Scholar]
- 13.Lamertz L, et al. Soluble gp130 prevents interleukin-6 and interleukin-11 trans-presentation but not intracellular autocrine responses. Sci. Signal. 2018;11:eaar7388. doi: 10.1126/scisignal.aar7388. [DOI] [PubMed] [Google Scholar]
- 14.Heink S, et al. Trans-presentation of IL-6 by dendritic cells is required for the priming of pathogenic TH17 cells. Nat. Immunol. 2017;18:74–85. doi: 10.1038/ni.3632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Heise D, et al. Selective inhibition of IL-6 trans-signaling by a miniaturized, optimized chimeric soluble gp130 inhibits TH17 cell expansion. Sci. Signal. 2021;14:eabc3480. doi: 10.1126/scisignal.abc3480. [DOI] [PubMed] [Google Scholar]
- 16.Berg AF, et al. Exclusive inhibition of IL-6 trans-signaling by soluble gp130(FlyR)Fc. Cytokine X. 2021;3:100058. doi: 10.1016/j.cytox.2021.100058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lokau J, Garbers Y, Grotzinger J, Garbers C. A single aromatic residue in sgp130Fc/olamkicept allows the discrimination between interleukin-6 and interleukin-11 trans-signaling. iScience. 2021;24:103309. doi: 10.1016/j.isci.2021.103309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dams-Kozlowska H, et al. A designer hyper interleukin 11 (H11) is a biologically active cytokine. BMC Biotechnol. 2012;12:8. doi: 10.1186/1472-6750-12-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lokau J, et al. Proteolytic cleavage governs interleukin-11 trans-signaling. Cell Rep. 2016;14:1761–1773. doi: 10.1016/j.celrep.2016.01.053. [DOI] [PubMed] [Google Scholar]
- 20.Bazan JF. Haemopoietic receptors and helical cytokines. Immunol. Today. 1990;11:350–354. doi: 10.1016/0167-5699(90)90139-Z. [DOI] [PubMed] [Google Scholar]
- 21.Rose-John S. Interleukin-6 family cytokines. Cold Spring Harb. Perspect. Biol. 2018;10:a028415. doi: 10.1101/cshperspect.a028415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yamasaki K, et al. Cloning and expression of the human interleukin-6 (BSF-2/IFNβ 2) receptor. Science. 1988;241:825–828. doi: 10.1126/science.3136546. [DOI] [PubMed] [Google Scholar]
- 23.Hibi M, et al. Molecular cloning and expression of an IL-6 signal transducer, gp130. Cell. 1990;63:1149–1157. doi: 10.1016/0092-8674(90)90411-7. [DOI] [PubMed] [Google Scholar]
- 24.Boulanger M, Chow D, Brevnova E, Garcia K. Hexameric structure and assembly of the interleukin-6/IL-6 α-receptor/gp130 complex. Science. 2003;300:2101–2104. doi: 10.1126/science.1083901. [DOI] [PubMed] [Google Scholar]
- 25.Paul SR, et al. Molecular cloning of a cDNA encoding interleukin 11, a stromal cell-derived lymphopoietic and hematopoietic cytokine. Proc. Natl Acad. Sci. USA. 1990;87:7512–7516. doi: 10.1073/pnas.87.19.7512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gearing DP, et al. Molecular cloning and expression of cDNA encoding a murine myeloid leukaemia inhibitory factor (LIF) EMBO J. 1987;6:3995–4002. doi: 10.1002/j.1460-2075.1987.tb02742.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stockli KA, et al. Molecular cloning, expression and regional distribution of rat ciliary neurotrophic factor. Nature. 1989;342:920–923. doi: 10.1038/342920a0. [DOI] [PubMed] [Google Scholar]
- 28.Malik N, et al. Molecular cloning, sequence analysis, and functional expression of a novel growth regulator, oncostatin M. Mol. Cell Biol. 1989;9:2847–2853. doi: 10.1128/mcb.9.7.2847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pennica D, et al. Expression cloning of cardiotrophin 1, a cytokine that induces cardiac myocyte hypertrophy. Proc. Natl Acad. Sci. USA. 1995;92:1142–1146. doi: 10.1073/pnas.92.4.1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Elson GC, et al. CLF associates with CLC to form a functional heteromeric ligand for the CNTF receptor complex. Nat. Neurosci. 2000;3:867–872. doi: 10.1038/78765. [DOI] [PubMed] [Google Scholar]
- 31.Pflanz S, et al. WSX-1 and glycoprotein 130 constitute a signal-transducing receptor for IL-27. J. Immunol. 2004;172:2225–2231. doi: 10.4049/jimmunol.172.4.2225. [DOI] [PubMed] [Google Scholar]
- 32.Gearing DP, et al. The IL-6 signal transducer, gp130: an oncostatin M receptor and affinity converter for the LIF receptor. Science. 1992;255:1434–1437. doi: 10.1126/science.1542794. [DOI] [PubMed] [Google Scholar]
- 33.Wilkinson A, et al. Granulocytes are unresponsive to IL-6 due to an absence of gp130. J. Immunol. 2018;200:3547–gp3555. doi: 10.4049/jimmunol.1701191. [DOI] [PubMed] [Google Scholar]
- 34.Kishimoto T, Kang S. IL-6 Revisited: from rheumatoid arthritis to CAR T cell therapy and COVID-19. Annu. Rev. Immunol. 2022;40:323–348. doi: 10.1146/annurev-immunol-101220-023458. [DOI] [PubMed] [Google Scholar]
- 35.Uhlén M, et al. Proteomics. Tissue-based map of the human proteome. Science. 2015;347:1260419. doi: 10.1126/science.1260419. [DOI] [PubMed] [Google Scholar]
- 36.Kimura R, et al. Identification of cardiac myocytes as the target of interleukin 11, a cardioprotective cytokine. Cytokine. 2007;38:107–115. doi: 10.1016/j.cyto.2007.05.011. [DOI] [PubMed] [Google Scholar]
- 37.Schafer S, et al. IL-11 is a crucial determinant of cardiovascular fibrosis. Nature. 2017;552:110–115. doi: 10.1038/nature24676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ropeleski M, Tang J, Walsh-Reitz M, Musch M, Chang E. Interleukin-11-induced heat shock protein 25 confers intestinal epithelial-specific cytoprotection from oxidant stress. Gastroenterology. 2003;124:1358–1368. doi: 10.1016/S0016-5085(03)00282-8. [DOI] [PubMed] [Google Scholar]
- 39.Müllberg J, et al. The soluble interleukin-6 receptor is generated by shedding. Eur. J. Immunol. 1993;23:473–480. doi: 10.1002/eji.1830230226. [DOI] [PubMed] [Google Scholar]
- 40.Müllberg J, Schooltink H, Stoyan T, Heinrich PC, Rose-John S. Protein kinase C activity is rate limiting for shedding of the interleukin-6 receptor. Biochem. Biophys. Res. Commun. 1992;189:794–800. doi: 10.1016/0006-291X(92)92272-Y. [DOI] [PubMed] [Google Scholar]
- 41.Riethmueller S, et al. Proteolytic origin of the soluble human IL-6R in vivo and a decisive role of N-glycosylation. PLoS Biol. 2017;15:e2000080. doi: 10.1371/journal.pbio.2000080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lust J, et al. Isolation of an mRNA encoding a soluble form of the human interleukin-6 receptor. Cytokine. 1992;4:96–100. doi: 10.1016/1043-4666(92)90043-Q. [DOI] [PubMed] [Google Scholar]
- 43.Schumacher N, et al. Shedding of endogenous interleukin-6 receptor (IL-6R) is governed by a disintegrin and metalloproteinase (ADAM) proteases while a full-length IL-6R isoform localizes to circulating microvesicles. J. Biol. Chem. 2015;290:26059–26071. doi: 10.1074/jbc.M115.649509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Arnold P, et al. Meprin metalloproteases generate biologically active soluble interleukin-6 receptor to induce trans-signaling. Sci. Rep. 2017;7:44053. doi: 10.1038/srep44053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Johnson N, et al. Quantitative proteomics screen identifies a substrate repertoire of rhomboid protease RHBDL2 in human cells and implicates it in epithelial homeostasis. Sci. Rep. 2017;7:7283. doi: 10.1038/s41598-017-07556-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Koch L, et al. Interleukin-11 (IL-11) receptor cleavage by the rhomboid protease RHBDL2 induces IL-11 trans-signaling. FASEB J. 2021;35:e21380. doi: 10.1096/fj.202002087R. [DOI] [PubMed] [Google Scholar]
- 47.Flynn C, et al. Cathepsin S provokes interleukin-6 (IL-6) trans-signaling through cleavage of the IL-6 receptor in vitro. Sci. Rep. 2020;10:21612. doi: 10.1038/s41598-020-77884-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bank U, et al. Selective proteolytic cleavage of IL-2 receptor and IL-6 receptor ligand binding chains by neutrophil-derived serine proteases at foci of inflammation. J. Interferon Cytokine Res. 1999;19:1277–1287. doi: 10.1089/107999099312957. [DOI] [PubMed] [Google Scholar]
- 49.Mackiewicz A, Schooltink H, Heinrich PC, Rose-John S. Complex of soluble human IL-6-receptor/IL-6 up-regulates expression of acute-phase proteins. J. Immunol. 1992;149:2021–2027. doi: 10.4049/jimmunol.149.6.2021. [DOI] [PubMed] [Google Scholar]
- 50.Fischer M, et al. I. A bioactive designer cytokine for human hematopoietic progenitor cell expansion. Nat. Biotechnol. 1997;15:142–145. doi: 10.1038/nbt0297-142. [DOI] [PubMed] [Google Scholar]
- 51.Marz P, Otten U, Rose-John S. Neural activities of IL-6-type cytokines often depend on soluble cytokine receptors. Eur. J. Neurosci. 1999;11:2995–3004. doi: 10.1046/j.1460-9568.1999.00755.x. [DOI] [PubMed] [Google Scholar]
- 52.Audet J, Miller CL, Rose-John S, Piret JM, Eaves CJ. Distinct role of gp130 activation in promoting self-renewal divisions by mitogenically stimulated murine hematopoietic stem cells. Proc. Natl Acad. Sci. USA. 2001;98:1757–1762. doi: 10.1073/pnas.98.4.1757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Viswanathan S, Benatar T, Rose-John S, Lauffenburger DA, Zandstra PW. Ligand/receptor signaling threshold (LIST) model accounts for gp130-mediated embryonic stem cell self-renewal responses to LIF and HIL-6. Stem Cell. 2002;20:119–138. doi: 10.1634/stemcells.20-2-119. [DOI] [PubMed] [Google Scholar]
- 54.Peters M, Muller AM, Rose-John S. Interleukin-6 and soluble interleukin-6 receptor: direct stimulation of gp130 and hematopoiesis. Blood. 1998;92:3495–3504. doi: 10.1182/blood.V92.10.3495. [DOI] [PubMed] [Google Scholar]
- 55.Peters M, et al. The function of the soluble interleukin 6 (IL-6) receptor in vivo: sensitization of human soluble IL-6 receptor transgenic mice towards IL-6 and prolongation of the plasma half-life of IL-6. J. Exp. Med. 1996;183:1399–1406. doi: 10.1084/jem.183.4.1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Peters M, et al. Extramedullary expansion of hematopoietic progenitor cells in interleukin (IL)-6-sIL-6R double transgenic mice. J. Exp. Med. 1997;185:755–766. doi: 10.1084/jem.185.4.755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Peters M, et al. In vivo and in vitro activities of the gp130-stimulating designer cytokine hyper-IL-6. J. Immunol. 1998;161:3575–3581. doi: 10.4049/jimmunol.161.7.3575. [DOI] [PubMed] [Google Scholar]
- 58.Schaper F, Rose-John S. Interleukin-6: biology, signaling and strategies of blockade. Cytokine Growth Factor Rev. 2015;26:475–487. doi: 10.1016/j.cytogfr.2015.07.004. [DOI] [PubMed] [Google Scholar]
- 59.Stuhlmann-Laeisz C, et al. Forced dimerization of gp130 leads to constitutive STAT3 activation, cytokine-independent growth, and blockade of differentiation of embryonic stem cells. Mol. Biol. Cell. 2006;17:2986–2995. doi: 10.1091/mbc.e05-12-1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schumacher N, et al. Cell-autonomous hepatocyte-specific GP130 signaling is sufficient to trigger a robust innate immune response in mice. J. Hepatol. 2021;74:407–418. doi: 10.1016/j.jhep.2020.09.021. [DOI] [PubMed] [Google Scholar]
- 61.Schirmacher P, et al. Hepatocellular hyperplasia, plasmacytoma formation, and extramedullary hematopoiesis in interleukin (IL)-6/soluble IL-6 receptor double-transgenic mice. Am. J. Pathol. 1998;153:639–648. doi: 10.1016/S0002-9440(10)65605-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hecht N, et al. Hyper-IL-6 gene therapy reverses fulminant hepatic failure. Mol. Ther. 2001;3:683–687. doi: 10.1006/mthe.2001.0313. [DOI] [PubMed] [Google Scholar]
- 63.Fazel Modares N, et al. IL-6 trans-signaling controls liver regeneration after partial hepatectomy. Hepatology. 2019;70:2075–2091. doi: 10.1002/hep.30774. [DOI] [PubMed] [Google Scholar]
- 64.Choy E, et al. Translating IL-6 biology into effective treatments. Nat. Rev. Rheumatol. 2020;16:335–345. doi: 10.1038/s41584-020-0419-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kang S, Narazaki M, Metwally H, Kishimoto T. Historical overview of the interleukin-6 family cytokine. J. Exp. Med. 2020;2020:5. doi: 10.1084/jem.20190347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kopf M, et al. Impaired immune and acute-phase responses in interleukin-6-deficient mice. Nature. 1994;368:339–342. doi: 10.1038/368339a0. [DOI] [PubMed] [Google Scholar]
- 67.Ohshima S, et al. Interleukin 6 plays a key role in the development of antigen-induced arthritis. Proc. Natl Acad. Sci. USA. 1998;95:8222–8226. doi: 10.1073/pnas.95.14.8222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Alonzi T, et al. Interleukin 6 is required for the development of collagen-induced arthritis. J. Exp. Med. 1998;187:461–468. doi: 10.1084/jem.187.4.461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Atreya R, et al. Blockade of interleukin 6 trans signaling suppresses T-cell resistance against apoptosis in chronic intestinal inflammation: evidence in crohn disease and experimental colitis in vivo. Nat. Med. 2000;6:583–588. doi: 10.1038/75068. [DOI] [PubMed] [Google Scholar]
- 70.Okuda Y, et al. IL-6-deficient mice are resistant to the induction of experimental autoimmune encephalomyelitis provoked by myelin oligodendrocyte glycoprotein. Int. Immunol. 1998;10:703–708. doi: 10.1093/intimm/10.5.703. [DOI] [PubMed] [Google Scholar]
- 71.Tanaka Y, Luo Y, O’Shea JJ, Nakayamada S. Janus kinase-targeting therapies in rheumatology: a mechanisms-based approach. Nat. Rev. Rheumatol. 2022;18:133–145. doi: 10.1038/s41584-021-00726-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Villarino AV, Kanno Y, O’Shea JJ. Mechanisms and consequences of Jak-STAT signaling in the immune system. Nat. Immunol. 2017;18:374–384. doi: 10.1038/ni.3691. [DOI] [PubMed] [Google Scholar]
- 73.Schwartz DM, et al. JAK inhibition as a therapeutic strategy for immune and inflammatory diseases. Nat. Rev. Drug Discov. 2017;16:843–862. doi: 10.1038/nrd.2017.201. [DOI] [PubMed] [Google Scholar]
- 74.Waage A, Brandtzaeg P, Halstensen A, Kierulf P, Espevik T. The complex pattern of cytokines in serum from patients with meningococcal septic shock. Association between interleukin 6, interleukin 1, and fatal outcome. J. Exp. Med. 1989;169:333–338. doi: 10.1084/jem.169.1.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Giraldez MD, Carneros D, Garbers C, Rose-John S, Bustos M. New insights into IL-6 family cytokines in metabolism, hepatology and gastroenterology. Nat. Rev. Gastroenterol. Hepatol. 2021;18:787–803. doi: 10.1038/s41575-021-00473-x. [DOI] [PubMed] [Google Scholar]
- 76.Garbers C, et al. Plasticity and cross-talk of interleukin 6-type cytokines. Cytokine Growth Factor. Rev. 2012;23:85–97. doi: 10.1016/j.cytogfr.2012.04.001. [DOI] [PubMed] [Google Scholar]
- 77.Grötzinger J, Kurapkat G, Wollmer A, Kalai M, Rose-John S. The family of the IL-6-type cytokines: specificity and promiscuity of the receptor complexes. Proteins. 1997;27:96–109. doi: 10.1002/(SICI)1097-0134(199701)27:1<96::AID-PROT10>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 78.Liautard J, et al. Specific inhibition of IL-6 signalling with monoclonal antibodies against the gp130 receptor. Cytokine. 1997;9:233–241. doi: 10.1006/cyto.1996.0159. [DOI] [PubMed] [Google Scholar]
- 79.Kawano M, et al. Autocrine generation and requirement of BSF-2/IL-6 for human multiple myelomas. Nature. 1988;332:83–85. doi: 10.1038/332083a0. [DOI] [PubMed] [Google Scholar]
- 80.Klein B, et al. Paracrine rather than autocrine regulation of myeloma-cell growth and differentiation by interleukin-6. Blood. 1989;73:517–526. doi: 10.1182/blood.V73.2.517.517. [DOI] [PubMed] [Google Scholar]
- 81.Klein B, et al. Murine anti-interleukin-6 monoclonal antibody therapy for a patient with plasma cell leukemia. Blood. 1991;78:1198–1204. doi: 10.1182/blood.V78.5.1198.1198. [DOI] [PubMed] [Google Scholar]
- 82.Lu ZY, et al. High amounts of circulating interleukin (IL)-6 in the form of monomeric immune complexes during anti-IL-6 therapy. Towards a new methodology for measuring overall cytokine production in human in vivo. Eur. J. Immunol. 1992;22:2819–2824. doi: 10.1002/eji.1830221110. [DOI] [PubMed] [Google Scholar]
- 83.Lu Z, et al. Overall interleukin-6 production exceeds 7 mg/day in multiple myeloma complicated by sepsis. Cytokine. 1993;5:578–582. doi: 10.1016/S1043-4666(05)80007-9. [DOI] [PubMed] [Google Scholar]
- 84.Beck JT, et al. Brief report: alleviation of systemic manifestations of Castleman’s disease by monoclonal anti-interleukin-6 antibody. N. Engl. J. Med. 1994;330:602–605. doi: 10.1056/NEJM199403033300904. [DOI] [PubMed] [Google Scholar]
- 85.Nishimoto N, et al. Mechanisms and pathologic significances in increase in serum interleukin-6 (IL-6) and soluble IL-6 receptor after administration of an anti-IL-6 receptor antibody, tocilizumab, in patients with rheumatoid arthritis and Castleman disease. Blood. 2008;112:3959–3964. doi: 10.1182/blood-2008-05-155846. [DOI] [PubMed] [Google Scholar]
- 86.Shimamoto K, et al. Serum interleukin 6 before and after therapy with tocilizumab is a principal biomarker in patients with rheumatoid arthritis. J. Rheumatol. 2013;40:1074–1081. doi: 10.3899/jrheum.121389. [DOI] [PubMed] [Google Scholar]
- 87.Flynn CM, et al. Interleukin-6 controls recycling and degradation, but not internalization of its receptors. J. Biol. Chem. 2021;296:100434. doi: 10.1016/j.jbc.2021.100434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Boyce E, Rogan E, Vyas D, Prasad N, Mai Y. Sarilumab: review of a second IL-6 receptor antagonist indicated for the treatment of rheumatoid arthritis. Ann. Pharmacother. 2018;52:780–791. doi: 10.1177/1060028018761599. [DOI] [PubMed] [Google Scholar]
- 89.Speake C, et al. IL-6-targeted therapies directed to cytokine or receptor blockade drive distinct alterations in T cell function. JCI Insight. 2022;2:e159436. doi: 10.1172/jci.insight.159436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Schuster B, et al. Signaling of human ciliary neurotrophic factor (CNTF) revisited. The interleukin-6 receptor can serve as an α-receptor for CTNF. J. Biol. Chem. 2003;278:9528–9535. doi: 10.1074/jbc.M210044200. [DOI] [PubMed] [Google Scholar]
- 91.Garbers C, et al. An interleukin-6 receptor-dependent molecular switch mediates signal transduction of the IL-27 cytokine subunit p28 (IL-30) via a gp130 protein receptor homodimer. J. Biol. Chem. 2013;288:4346–4354. doi: 10.1074/jbc.M112.432955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Yang X, et al. Soluble IL-6 receptor and IL-27 subunit p28 protein complex mediate the antiviral response through the type III IFN pathway. J. Immunol. 2016;197:2369–2381. doi: 10.4049/jimmunol.1600627. [DOI] [PubMed] [Google Scholar]
- 93.Heaney M, Golde D. Soluble cytokine receptors. Blood. 1996;87:847–857. doi: 10.1182/blood.V87.3.847.bloodjournal873847. [DOI] [PubMed] [Google Scholar]
- 94.Jostock T, et al. Soluble gp130 is the natural inhibitor of soluble interleukin-6 receptor transsignaling responses. Eur. J. Biochem. 2001;268:160–167. doi: 10.1046/j.1432-1327.2001.01867.x. [DOI] [PubMed] [Google Scholar]
- 95.Tenhumberg S, et al. gp130 dimerization in the absence of ligand: preformed cytokine receptor complexes. Biochem. Biophys. Res. Commun. 2006;346:649–657. doi: 10.1016/j.bbrc.2006.05.173. [DOI] [PubMed] [Google Scholar]
- 96.Monhasery N, et al. Transcytosis of IL-11 and apical redirection of gp130 is mediated by IL-11α receptor. Cell Rep. 2016;16:1067–1081. doi: 10.1016/j.celrep.2016.06.062. [DOI] [PubMed] [Google Scholar]
- 97.Martinez-Fabregas J, et al. Kinetics of cytokine receptor trafficking determine signaling and functional selectivity. Elife. 2019;8:e49314. doi: 10.7554/eLife.49314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Fritsch J, Zingler P, Särchen V, Heck A, Schütze S. Role of ubiquitination and proteolysis in the regulation of pro- and anti-apoptotic TNF-R1 signaling. Biochim. Biophys. Acta Mol. Cell Res. 2017;1864:2138–2146. doi: 10.1016/j.bbamcr.2017.07.017. [DOI] [PubMed] [Google Scholar]
- 99.German CL, Sauer BM, Howe CL. The STAT3 beacon: IL-6 recurrently activates STAT 3 from endosomal structures. Exp. Cell Res. 2011;317:1955–1969. doi: 10.1016/j.yexcr.2011.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Rabe B, et al. Transgenic blockade of interleukin 6 transsignaling abrogates inflammation. Blood. 2008;111:1021–1028. doi: 10.1182/blood-2007-07-102137. [DOI] [PubMed] [Google Scholar]
- 101.Rakonczay Z, Jr., Hegyi P, Takacs T, McCarroll J, Saluja AK. The role of NF-κB activation in the pathogenesis of acute pancreatitis. Gut. 2008;57:259–267. doi: 10.1136/gut.2007.124115. [DOI] [PubMed] [Google Scholar]
- 102.Zhang H, et al. IL-6 trans-signaling promotes pancreatitis-associated lung injury and lethality. J. Clin. Invest. 2013;123:1019–1031. doi: 10.1172/JCI64931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kaiser K, et al. Pharmacological inhibition of IL-6 trans-signaling improves compromised fracture healing after severe trauma. Naunyn Schmiedebergs Arch. Pharmacol. 2018;391:523–536. doi: 10.1007/s00210-018-1483-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Prystaz K, et al. Distinct effects of IL-6 classic and trans-signaling in bone fracture healing. Am. J. Pathol. 2018;188:474–490. doi: 10.1016/j.ajpath.2017.10.011. [DOI] [PubMed] [Google Scholar]
- 105.George MJ, et al. Selective interleukin-6 trans-signaling blockade is more effective than panantagonism in reperfused myocardial infarction. JACC Basic. Transl. Sci. 2021;6:431–443. doi: 10.1016/j.jacbts.2021.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Strand V, et al. Systematic review and meta-analysis of serious infections with tofacitinib and biologic disease-modifying antirheumatic drug treatment in rheumatoid arthritis clinical trials. Arthritis Res. Ther. 2015;17:362. doi: 10.1186/s13075-015-0880-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Hoge J, et al. IL-6 controls the innate immune response against Listeria monocytogenes via classical IL-6 signaling. J. Immunol. 2013;190:703–711. doi: 10.4049/jimmunol.1201044. [DOI] [PubMed] [Google Scholar]
- 108.Dominitzki S, et al. Cutting edge: trans-signaling via the soluble IL-6R abrogates the induction of FoxP3 in naive CD4+ CD25 T cells. J. Immunol. 2007;179:2041–2045. doi: 10.4049/jimmunol.179.4.2041. [DOI] [PubMed] [Google Scholar]
- 109.Klouche M, Bhakdi S, Hemmes M, Rose-John S. Novel path to activation of vascular smooth muscle cells: up-regulation of gp130 creates an autocrine activation loop by IL-6 and its soluble receptor. J. Immunol. 1999;163:4583–4589. doi: 10.4049/jimmunol.163.8.4583. [DOI] [PubMed] [Google Scholar]
- 110.Romano M, et al. Role of IL-6 and its soluble receptor in induction of chemokines and leukocyte recruitment. Immunity. 1997;6:315–325. doi: 10.1016/S1074-7613(00)80334-9. [DOI] [PubMed] [Google Scholar]
- 111.Rose-John S. The soluble interleukin 6 receptor: advanced therapeutic options in inflammation. Clin. Pharmacol. Ther. 2017;102:591–598. doi: 10.1002/cpt.782. [DOI] [PubMed] [Google Scholar]
- 112.Jones SA, Scheller J, Rose-John S. Therapeutic strategies for the clinical blockade of IL-6/gp130 signaling. J. Clin. Invest. 2011;121:3375–3383. doi: 10.1172/JCI57158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Schreiber S, et al. Therapeutic interleukin-6 trans-signaling inhibition by olamkicept (sgp130Fc) in patients with active inflammatory bowel disease. Gastroenterology. 2021;160:2354–2366.e11. doi: 10.1053/j.gastro.2021.02.062. [DOI] [PubMed] [Google Scholar]
- 114.Chen BL, et al. Olamkicept, an IL-6 trans-signaling inhibitor, is effective for induction of response and remission in a randomized, placebo-controlled trial in moderate to severe ulcerative colitis. Gastroenterology. 2021;161:E28–E29. doi: 10.1053/j.gastro.2021.06.038. [DOI] [Google Scholar]
- 115.Chen B, et al. Efficacy and safety of the IL-6 trans-signalling inhibitor olamkicept: a phase 2 randomized, placebo-controlled trial in moderately to severely active ulcerative colitis. J. Crohns Colitis. 2021;15(Suppl. 1):S041–S122. doi: 10.1093/ecco-jcc/jjab073.040. [DOI] [Google Scholar]
- 116.Schulte DM, et al. Case report: arterial wall inflammation in atherosclerotic cardiovascular disease is reduced by olamkicept (sgp130Fc) Front. Pharmacol. 2022;13:758233. doi: 10.3389/fphar.2022.758233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Schuett H, et al. Transsignaling of interleukin-6 crucially contributes to atherosclerosis in mice. Arterioscler. Thromb. Vasc. Biol. 2012;32:281–290. doi: 10.1161/ATVBAHA.111.229435. [DOI] [PubMed] [Google Scholar]
- 118.Musashi M, Clark SC, Sudo T, Urdal DL, Ogawa M. Synergistic interactions between interleukin-11 and interleukin-4 in support of proliferation of primitive hematopoietic progenitors of mice. Blood. 1991;78:1448–1451. doi: 10.1182/blood.V78.6.1448.1448. [DOI] [PubMed] [Google Scholar]
- 119.Teramura M, Kobayashi S, Yoshinaga K, Iwabe K, Mizoguchi H. Effect of interleukin 11 on normal and pathological thrombopoiesis. Cancer Chemother. Pharmacol. 1996;38(Suppl):S99–S102. doi: 10.1007/s002800051048. [DOI] [PubMed] [Google Scholar]
- 120.Kespohl B, Schumertl T, Bertrand J, Lokau J, Garbers C. The cytokine interleukin-11 crucially links bone formation, remodeling and resorption. Cytokine Growth Factor. Rev. 2021;60:18–27. doi: 10.1016/j.cytogfr.2021.04.002. [DOI] [PubMed] [Google Scholar]
- 121.Anderson KC, et al. Interleukin-11 promotes accessory cell-dependent B-cell differentiation in humans. Blood. 1992;80:2797–2804. doi: 10.1182/blood.V80.11.2797.bloodjournal80112797. [DOI] [PubMed] [Google Scholar]
- 122.Obana M, et al. Therapeutic activation of signal transducer and activator of transcription 3 by interleukin-11 ameliorates cardiac fibrosis after myocardial infarction. Circulation. 2010;121:684–691. doi: 10.1161/CIRCULATIONAHA.109.893677. [DOI] [PubMed] [Google Scholar]
- 123.Tamura Y, Kohno H, Mohri T, Fujio Y, Matsumiya G. The cardioprotective effect of interleukin-11 against ischemia-reperfusion injury in a heart donor model. Ann. Cardiothorac. Surg. 2018;7:99–105. doi: 10.21037/acs.2017.09.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Zhang B, et al. Interleukin-11 treatment protected against cerebral ischemia/reperfusion injury. Biomed. Pharmacother. 2019;115:108816. doi: 10.1016/j.biopha.2019.108816. [DOI] [PubMed] [Google Scholar]
- 125.Allanki S, et al. Interleukin-11 signaling promotes cellular reprogramming and limits fibrotic scarring during tissue regeneration. Sci. Adv. 2021;7:eabg6497. doi: 10.1126/sciadv.abg6497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Ernst M, et al. STAT3 and STAT1 mediate IL-11-dependent and inflammation-associated gastric tumorigenesis in gp130 receptor mutant mice. J. Clin. Invest. 2008;118:1727–1738. doi: 10.1172/JCI34944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Dong J, et al. Hepatocyte-specific IL11 cis-signaling drives lipotoxicity and underlies the transition from NAFLD to NASH. Nat. Commun. 2021;12:66. doi: 10.1038/s41467-020-20303-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Widjaja AA, et al. Redefining IL11 as a regeneration-limiting hepatotoxin and therapeutic target in acetaminophen-induced liver injury. Sci. Transl. Med. 2021;13:eaba8146. doi: 10.1126/scitranslmed.aba8146. [DOI] [PubMed] [Google Scholar]
- 129.Widjaja AA, et al. Inhibiting interleukin 11 signaling reduces hepatocyte death and liver fibrosis, inflammation, and steatosis in mouse models of nonalcoholic steatohepatitis. Gastroenterology. 2019;157:777–792 e714. doi: 10.1053/j.gastro.2019.05.002. [DOI] [PubMed] [Google Scholar]
- 130.Ng B, et al. Interleukin-11 is a therapeutic target in idiopathic pulmonary fibrosis. Sci. Transl. Med. 2019;11:eaaw1237. doi: 10.1126/scitranslmed.aaw1237. [DOI] [PubMed] [Google Scholar]
- 131.Balic J, Garbers C, Rose-John S, Yu L, Jenkins B. Interleukin-11-driven gastric tumourigenesis is independent of trans-signalling. Cytokine. 2017;92:118–123. doi: 10.1016/j.cyto.2017.01.015. [DOI] [PubMed] [Google Scholar]
- 132.Scheller J, Schuster B, Holscher C, Yoshimoto T, Rose-John S. No inhibition of IL-27 signaling by soluble gp130. Biochem. Biophys. Res. Commun. 2005;326:724–728. doi: 10.1016/j.bbrc.2004.11.098. [DOI] [PubMed] [Google Scholar]
- 133.Schwerd T, et al. A variant in IL6ST with a selective IL-11 signaling defect in human and mouse. Bone Res. 2020;8:24. doi: 10.1038/s41413-020-0098-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Kim Y, et al. A dual target-directed agent against interleukin-6 receptor and tumor necrosis factor α ameliorates experimental arthritis. Sci. Rep. 2016;6:20150. doi: 10.1038/srep20150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Namakanova OA, et al. Therapeutic potential of combining IL-6 and TNF blockade in a mouse model of allergic asthma. Int. J. Mol. Sci. 2022;23:3521. doi: 10.3390/ijms23073521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Lyman M, et al. A bispecific antibody that targets IL-6 receptor and IL-17A for the potential therapy of patients with autoimmune and inflammatory diseases. J. Biol. Chem. 2018;293:9326–9334. doi: 10.1074/jbc.M117.818559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Bryce C, et al. Pathophysiology of SARS-CoV-2: the Mount Sinai COVID-19 autopsy experience. Mod. Pathol. 2021;8:1456–1467. doi: 10.1038/s41379-021-00793-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Chen LYC, et al. Soluble interleukin-6 receptor in the COVID-19 cytokine storm syndrome. Cell Rep. Med. 2021;2:100269. doi: 10.1016/j.xcrm.2021.100269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.de la Rica R, Borges M, Gonzalez-Freire M. COVID-19: in the eye of the cytokine storm. Front. Immunol. 2020;11:558898. doi: 10.3389/fimmu.2020.558898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Hadjadj J, et al. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science. 2020;369:718–724. doi: 10.1126/science.abc6027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.McConnell MJ, et al. Liver injury in COVID-19 and IL-6 trans-signaling-induced endotheliopathy. J. Hepatol. 2021;75:647–658. doi: 10.1016/j.jhep.2021.04.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Patra T, et al. SARS-CoV-2 spike protein promotes IL-6 trans-signaling by activation of angiotensin II receptor signaling in epithelial cells. PLoS Pathog. 2020;16:e1009128. doi: 10.1371/journal.ppat.1009128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Xu ZS, et al. Temporal profiling of plasma cytokines, chemokines and growth factors from mild, severe and fatal COVID-19 patients. Signal. Transduct. Target. Ther. 2020;5:100. doi: 10.1038/s41392-020-0211-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Aziz M, Fatima R, Assaly R. Elevated interleukin-6 and severe COVID-19: a meta-analysis. J. Med. Virol. 2020;92:2283–2285. doi: 10.1002/jmv.25948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Zhu J, et al. Elevated interleukin-6 is associated with severity of COVID-19: a meta-analysis. J. Med. Virol. 2021;93:35–37. doi: 10.1002/jmv.26085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Giamarellos-Bourboulis EJ, et al. Complex immune dysregulation in COVID-19 patients with severe respiratory failure. Cell Host Microbe. 2020;27:992–1000.e3. doi: 10.1016/j.chom.2020.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Du F, Liu B, Zhang S. COVID-19: the role of excessive cytokine release and potential ACE2 down-regulation in promoting hypercoagulable state associated with severe illness. J. Thromb. Thrombolysis. 2021;51:313–329. doi: 10.1007/s11239-020-02224-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Levi M, van der Poll T. Two-way interactions between inflammation and coagulation. Trends Cardiovasc. Med. 2005;15:254–259. doi: 10.1016/j.tcm.2005.07.004. [DOI] [PubMed] [Google Scholar]
- 149.Levi M, van der Poll T, ten Cate H, van Deventer SJ. The cytokine-mediated imbalance between coagulant and anticoagulant mechanisms in sepsis and endotoxaemia. Eur. J. Clin. Invest. 1997;27:3–9. doi: 10.1046/j.1365-2362.1997.570614.x. [DOI] [PubMed] [Google Scholar]
- 150.McFadyen JD, Stevens H, Peter K. The emerging threat of (micro)thrombosis in COVID-19 and its therapeutic implications. Circ. Res. 2020;127:571–587. doi: 10.1161/CIRCRESAHA.120.317447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Guaraldi G, et al. Tocilizumab in patients with severe COVID-19: a retrospective cohort study. Lancet Rheumatol. 2020;2:e474–e484. doi: 10.1016/S2665-9913(20)30173-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Jordan SC, et al. Compassionate use of tocilizumab for treatment of SARS-CoV-2 pneumonia. Clin. Infect. Dis. 2020;71:3168–3173. doi: 10.1093/cid/ciaa812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Meleveedu KS, et al. Tocilizumab for severe COVID-19 related illness — a community academic medical center experience. Cytokine X. 2020;2:100035. doi: 10.1016/j.cytox.2020.100035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Gordon AC, Angus DC, Derde LPG. Interleukin-6 receptor antagonists in critically ill patients with Covid-19. Reply. N. Engl. J. Med. 2021;385:1147–1149. doi: 10.1056/NEJMc2108482. [DOI] [PubMed] [Google Scholar]
- 155.Ettich J, et al. A hybrid soluble gp130/spike-nanobody fusion protein simultaneously blocks interleukin-6 trans-signaling and cellular infection with SARS-CoV-2. J. Virol. 2022;96:e0162221. doi: 10.1128/jvi.01622-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Schumacher N, Rose-John S. ADAM17 orchestrates interleukin-6, TNFα and EGF-R signaling in inflammation and cancer. Biochim. Biophys. Acta Mol. Cell Res. 2022;1869:119141. doi: 10.1016/j.bbamcr.2021.119141. [DOI] [PubMed] [Google Scholar]
- 157.Saad MI, Rose-John S, Jenkins BJ. ADAM17: an emerging therapeutic target for lung cancer. Cancers. 2019;11:1218. doi: 10.3390/cancers11091218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Grötzinger J, Lorenzen I, Düsterhöft S. Molecular insights into the multilayered regulation of ADAM17: the role of the extracellular region. Biochem. Biophys. Acta Mol. Cell Res. 2017;1864:2088–2095. doi: 10.1016/j.bbamcr.2017.05.024. [DOI] [PubMed] [Google Scholar]
- 159.Dulloo I, Muliyil S, Freeman M. The molecular, cellular and pathophysiological roles of iRhom pseudoproteases. Open Biol. 2019;9:190003. doi: 10.1098/rsob.190003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Zunke F, Rose-John S. The shedding protease ADAM17: physiology and pathophysiology. Biochem. Biophys. Acta Mol. Cell Res. 2017;1864:2059–2070. doi: 10.1016/j.bbamcr.2017.07.001. [DOI] [PubMed] [Google Scholar]
- 161.Saad MI, et al. Blockade of the protease ADAM17 ameliorates experimental pancreatitis. Proc. Natl Acad. Sci. USA. 2022;119:e2213744119. doi: 10.1073/pnas.2213744119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Schmidt S, et al. ADAM17 is required for EGF-R-induced intestinal tumors via IL-6 trans-signaling. J. Exp. Med. 2018;215:1205–1225. doi: 10.1084/jem.20171696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Zhao X, et al. Inhibiting tumor necrosis factor-alpha diminishes desmoplasia and inflammation to overcome chemoresistance in pancreatic ductal adenocarcinoma. Oncotarget. 2016;7:81110–81122. doi: 10.18632/oncotarget.13212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Lesina M, et al. Stat3/Socs3 activation by IL-6 transsignaling promotes progression of pancreatic intraepithelial neoplasia and development of pancreatic cancer. Cancer Cell. 2011;19:456–469. doi: 10.1016/j.ccr.2011.03.009. [DOI] [PubMed] [Google Scholar]
- 165.Elangovan IM, et al. FOSL1 promotes Kras-induced lung cancer through amphiregulin and cell survival gene regulation. Am. J. Respir. Cell Mol. Biol. 2018;58:625–635. doi: 10.1165/rcmb.2017-0164OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Brooks GD, et al. IL6 Trans-signaling promotes KRAS-driven lung carcinogenesis. Cancer Res. 2016;76:866–876. doi: 10.1158/0008-5472.CAN-15-2388. [DOI] [PubMed] [Google Scholar]
- 167.Saad MI, et al. ADAM17 selectively activates the IL-6 trans-signaling/ERK MAPK axis in KRAS-addicted lung cancer. EMBO Mol. Med. 2019;11:e9976. doi: 10.15252/emmm.201809976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Greenhill CJ GJ, Ernst M, Hertzog PJ, Mansell A, Jenkins BJ. LPS hypersensitivity of gp130 mutant mice is independent of elevated haemopoietic TLR4 signaling. Immunol. Cell Biol. 2012;90:559–563. doi: 10.1038/icb.2011.56. [DOI] [PubMed] [Google Scholar]
- 169.Nowell MA, et al. Therapeutic targeting of IL-6 trans signaling counteracts STAT3 control of experimental inflammatory arthritis. J. Immunol. 2009;182:613–622. doi: 10.4049/jimmunol.182.1.613. [DOI] [PubMed] [Google Scholar]
- 170.Peng L, et al. Molecular basis for the mechanism of action of an anti-TACE antibody. MAbs. 2016;8:1598–1605. doi: 10.1080/19420862.2016.1226716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Kwok H, et al. Development of a ‘mouse and human cross-reactive’ affinity-matured exosite inhibitory human antibody specific to TACE (ADAM17) for cancer immunotherapy. Protein Eng. Des. Sel. 2014;27:179–190. doi: 10.1093/protein/gzu010. [DOI] [PubMed] [Google Scholar]
- 172.Ye J, Yuen SM, Murphy G, Xie R, Kwok HF. Anti-tumor effects of a ‘human & mouse cross-reactive’ anti-ADAM17 antibody in a pancreatic cancer model in vivo. Eur. J. Pharm. Sci. 2017;110:62–69. doi: 10.1016/j.ejps.2017.05.057. [DOI] [PubMed] [Google Scholar]
- 173.Mishra HK, et al. Blocking ADAM17 function with a monoclonal antibody improves sepsis survival in a murine model of polymicrobial sepsis. Int. J. Mol. Sci. 2020;21:6688. doi: 10.3390/ijms21186688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Rahn S, Becker-Pauly C. Meprin and ADAM proteases as triggers of systemic inflammation in sepsis. FEBS Lett. 2022;596:534–556. doi: 10.1002/1873-3468.14225. [DOI] [PubMed] [Google Scholar]
- 175.Wong E, et al. Harnessing the natural inhibitory domain to control TNFα converting enzyme (TACE) activity in vivo. Sci. Rep. 2016;6:35598. doi: 10.1038/srep35598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Kefaloyianni E, et al. ADAM17 substrate release in proximal tubule drives kidney fibrosis. JCI Insight. 2016;1:e87023. doi: 10.1172/jci.insight.87023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Moss M, Minond D. Recent advances in ADAM17 research: a promising target for cancer and inflammation. Mediators Inflamm. 2017;2017:9673537. doi: 10.1155/2017/9673537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Kaplan G. The global burden of IBD: from 2015 to 2025. Nat. Rev. Gastroenterol. Hepatol. 2015;12:720–727. doi: 10.1038/nrgastro.2015.150. [DOI] [PubMed] [Google Scholar]
- 179.Ito H, et al. A pilot randomized trial of a human anti-interleukin-6 receptor monoclonal antibody in active Crohn’s disease. Gastroenterology. 2004;126:989–996. doi: 10.1053/j.gastro.2004.01.012. [DOI] [PubMed] [Google Scholar]
- 180.Danese S, et al. Randomised trial and open-label extension study of an anti-interleukin-6 antibody in Crohn’s disease (ANDANTE I and II) Gut. 2019;68:40–48. doi: 10.1136/gutjnl-2017-314562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Gout T, Ostör A, Nisar M. Lower gastrointestinal perforation in rheumatoid arthritis patients treated with conventional DMARDs or tocilizumab: a systematic literature review. Clin. Rheumatol. 2011;30:1471–1474. doi: 10.1007/s10067-011-1827-x. [DOI] [PubMed] [Google Scholar]
- 182.Grivennikov S, et al. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell. 2009;15:103–113. doi: 10.1016/j.ccr.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Hanioka Y, et al. Exacerbation of ulcerative colitis with tocilizumab: a report of two cases, one with Takayasu arteritis and the other with relapsing polychondritis. Intern. Med. 2021;60:1615–1620. doi: 10.2169/internalmedicine.5215-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Kuhn K, Manieri N, Liu T, Stappenbeck T. IL-6 stimulates intestinal epithelial proliferation and repair after injury. PLoS ONE. 2014;9:e114195. doi: 10.1371/journal.pone.0114195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Hueber W, et al. Secukinumab, a human anti-IL-17A monoclonal antibody, for moderate to severe Crohn’s disease: unexpected results of a randomised, double-blind placebo-controlled trial. Gut. 2012;61:1693–1700. doi: 10.1136/gutjnl-2011-301668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Barkhausen T, et al. Selective blockade of interleukin-6 trans-signaling improves survival in a murine polymicrobial sepsis model. Crit. Care Med. 2011;39:1407–1413. doi: 10.1097/CCM.0b013e318211ff56. [DOI] [PubMed] [Google Scholar]
- 187.Mitsuyama K, et al. STAT3 activation via interleukin 6 trans-signalling contributes to ileitis in SAMP1/Yit mice. Gut. 2006;55:1263–1269. doi: 10.1136/gut.2005.079343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Nowell MA, et al. Soluble IL-6 receptor governs IL-6 activity in experimental arthritis: blockade of arthritis severity by soluble glycoprotein 130. J. Immunol. 2003;171:3202–3209. doi: 10.4049/jimmunol.171.6.3202. [DOI] [PubMed] [Google Scholar]
- 189.Richards PJ, et al. Functional characterization of a soluble gp130 isoform and its therapeutic capacity in an experimental model of inflammatory arthritis. Arthritis Rheum. 2006;54:1662–1672. doi: 10.1002/art.21818. [DOI] [PubMed] [Google Scholar]
- 190.Becker C, et al. TGF-β suppresses tumor progression in colon cancer by inhibition of IL-6 trans-signaling. Immunity. 2004;21:491–501. doi: 10.1016/j.immuni.2004.07.020. [DOI] [PubMed] [Google Scholar]
- 191.Matsumoto S, et al. Essential roles of IL-6 trans-signaling in colonic epithelial cells, induced by the IL-6/soluble-IL-6 receptor derived from lamina propria macrophages, on the development of colitis-associated premalignant cancer in a murine model. J. Immunol. 2010;184:1543–1551. doi: 10.4049/jimmunol.0801217. [DOI] [PubMed] [Google Scholar]
- 192.Chalaris A, et al. Apoptosis is a natural stimulus of IL6R shedding and contributes to the proinflammatory trans-signaling function of neutrophils. Blood. 2007;110:1748–1755. doi: 10.1182/blood-2007-01-067918. [DOI] [PubMed] [Google Scholar]
- 193.Greenhill CJ, et al. IL-6 trans-signaling modulates TLR4-dependent inflammatory responses via STAT3. J. Immunol. 2011;186:1199–1208. doi: 10.4049/jimmunol.1002971. [DOI] [PubMed] [Google Scholar]
- 194.Sodenkamp J, et al. Therapeutic targeting of interleukin-6 trans-signaling does not affect the outcome of experimental tuberculosis. Immunobiology. 2012;217:996–1004. doi: 10.1016/j.imbio.2012.01.015. [DOI] [PubMed] [Google Scholar]
- 195.Tsantikos E, et al. Interleukin-6 trans-signaling exacerbates inflammation and renal pathology in lupus-prone mice. Arthritis Rheum. 2013;65:2691–2702. doi: 10.1002/art.38061. [DOI] [PubMed] [Google Scholar]
- 196.Braun GS, et al. IL-6 Trans-signaling drives murine crescentic GN. J. Am. Soc. Nephrol. 2016;27:132–142. doi: 10.1681/ASN.2014111147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Luig M, et al. Inflammation-induced IL-6 functions as a natural brake on macrophages and limits GN. J. Am. Soc. Nephrol. 2015;26:1597–1607. doi: 10.1681/ASN.2014060620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Ruwanpura SM, et al. Therapeutic targeting of the IL-6 trans-signalling/mTORC1 axis in pulmonary emphysema. Am. J. Respir. Crit. Care Med. 2016;194:201512–2368OC. doi: 10.1164/rccm.201512-2368OC. [DOI] [PubMed] [Google Scholar]
- 199.Bergmann J, et al. IL-6 trans-signaling is essential for the development of hepatocellular carcinoma in mice. Hepatology. 2017;65:89–103. doi: 10.1002/hep.28874. [DOI] [PubMed] [Google Scholar]
- 200.Rosenberg N, et al. Combined hepatocellular-cholangiocarcinoma derives from liver progenitor cells and depends on senescence and IL-6 trans-signaling. J. Hepatol. 2022;77:1631–1641. doi: 10.1016/j.jhep.2022.07.029. [DOI] [PubMed] [Google Scholar]
- 201.Paige E, et al. Interleukin-6 receptor signaling and abdominal aortic aneurysm growth rates. Circ. Genom. Precis. Med. 2019;12:e002413. doi: 10.1161/CIRCGEN.118.002413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Hu J, et al. Interleukin-6 trans-signalling in hippocampal CA1 neurones mediates perioperative neurocognitive disorders in mice. Br. J. Anaesth. 2022;129:923–936. doi: 10.1016/j.bja.2022.08.019. [DOI] [PubMed] [Google Scholar]
- 203.Nikolaus S, et al. Evaluation of interleukin-6 and its soluble receptor components sIL-6R and sgp130 as markers of inflammation in inflammatory bowel diseases. Int. J. Colerectal Dis. 2018;33:927–936. doi: 10.1007/s00384-018-3069-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Rodríguez-Hernández M, et al. Identification of IL-6 signalling components as predictors of severity and outcome in COVID-19. Front. Immunol. 2022;13:891456. doi: 10.3389/fimmu.2022.891456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Diamant M, et al. Cloning and expression of an alternatively spliced mRNA encoding a soluble form of the human interleukin-6 signal transducer gp130. FEBS Lett. 1997;412:379–384. doi: 10.1016/S0014-5793(97)00750-3. [DOI] [PubMed] [Google Scholar]
- 206.Müller S, et al. The Alzheimer’s disease-linked protease BACE1 modulates neuronal IL-6 signaling through shedding of the receptor gp130. Mol. Neurodegener. 2023;18:13. doi: 10.1186/s13024-023-00596-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Garbers C, et al. The interleukin-6 receptor Asp358Ala single nucleotide polymorphism rs2228145 confers increased proteolytic conversion rates by ADAM proteases. Biochim. Biophys. Acta. 2014;1842:1485–1494. doi: 10.1016/j.bbadis.2014.05.018. [DOI] [PubMed] [Google Scholar]
- 208.Ferreira RC, et al. Functional IL6R 358Ala allele impairs classical IL-6 receptor signaling and influences risk of diverse inflammatory diseases. PLoS Genet. 2013;9:e1003444. doi: 10.1371/journal.pgen.1003444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Scheller J, Rose-John S. The interleukin 6 pathway and atherosclerosis. Lancet. 2012;380:338. doi: 10.1016/S0140-6736(12)61246-X. [DOI] [PubMed] [Google Scholar]
- 210.Aparicio-Siegmund S, et al. The IL-6-neutralizing sIL-6R-sgp130 buffer system is disturbed in patients with type 2 diabetes. Am. J. Physiol. Endocrinol. Metab. 2019;317:E411–E420. doi: 10.1152/ajpendo.00166.2019. [DOI] [PubMed] [Google Scholar]