

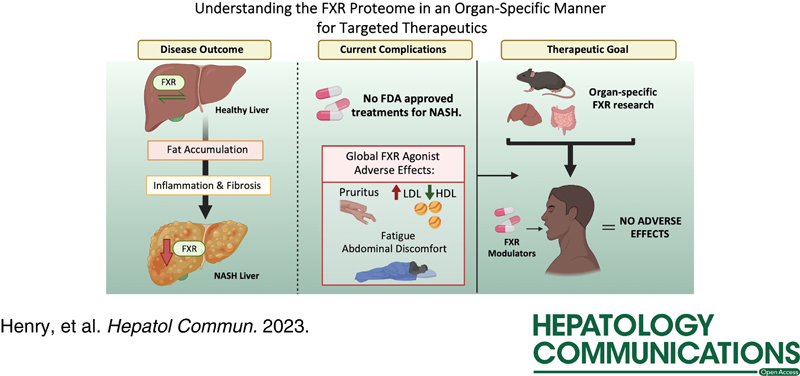
Abstract
NASH is within the spectrum of NAFLD, a liver condition encompassing liver steatosis, inflammation, hepatocyte injury, and fibrosis. The prevalence of NASH-induced cirrhosis is rapidly rising and has become the leading indicator for liver transplantation in the US. There is no Food and Drug Administration (FDA)-approved pharmacological intervention for NASH. The farnesoid X receptor (FXR) is essential in regulating bile acid homeostasis, and dysregulation of bile acids has been implicated in the pathogenesis of NASH. As a result, modulators of FXR that show desirable effects in mitigating key characteristics of NASH have been developed as promising therapeutic approaches. However, global FXR activation causes adverse effects such as cholesterol homeostasis imbalance and pruritus. The development of targeted FXR modulation is necessary for ideal NASH therapeutics, but information regarding tissue-specific and cell-specific FXR functionality is limited. In this review, we highlight FXR activation in the regulation of bile acid homeostasis and NASH development, examine the current literature on tissue-specific regulation of nuclear receptors, and speculate on how FXR regulation will be beneficial in the treatment of NASH.
INTRODUCTION
NAFLD is the most common chronic liver condition in the US, with an estimated 25% of US adults suffering from this disease, particularly simple fatty liver. NAFLD encompasses a spectrum of liver conditions characterized by fat accumulation in the liver, not caused by excessive alcohol consumption, which may develop into NASH. Approximately 20%–25% of the NAFLD population have NASH (5% of US adults). NASH is characterized by hepatocyte ballooning, inflammation, and varying degrees of fibrosis, in addition to steatosis. Excessive fibrosis can lead to cirrhosis, an end-stage liver disease, and increase the risk of HCC.1,2 NASH-induced cirrhosis has become the leading indicator for liver transplantation in the country, and its prevalence is rapidly rising.3–5 The progression from simple fatty liver to NASH is not well elucidated, and although the pathogenesis of NASH has been speculated and theorized, its etiology has yet to be confirmed.6 The current recommended treatment of NASH is lifestyle modification such as diet and exercise with no FDA-approved pharmacologic interventions.
A ligand-activated transcription factor (TF) and type II nuclear receptor (NR), farnesoid X receptor (FXR), has been identified as a clinical target for therapeutic intervention for NASH and other chronic liver diseases. FXR has a wide range of functions that are beneficial in the treatment of NASH, including the reduction of steatosis, inflammation, and fibrosis, through the transcriptional activation and/or suppression of various biological pathways.7 Synthetic steroidal or nonsteroidal agonists of FXR have been developed for the treatment of NASH and are currently undergoing clinical trials, such as obeticholic acid, cilofexor, nidufexor, and tropifexor.8,9 The current FXR agonists activate whole-body FXR and display favorable effects in the treatment of NAFLD/NASH10–12; however, adverse side effects such as pruritus, cholesterol homeostasis imbalance (increases in LDLs and decreases in HDLs), fatigue, and abdominal discomfort have been reported in patients with NASH and other chronic liver diseases after treatment with FXR agonists.9,13 There is an urgent need to determine the tissue-specific role(s) of FXR to prevent adverse effects and to develop targeted and efficacious therapies for NASH patients (Figure 1). This review examines factors that contribute to FXR tissue-specific modulation and their potential effect on the therapeutic development for NASH.
FIGURE 1.
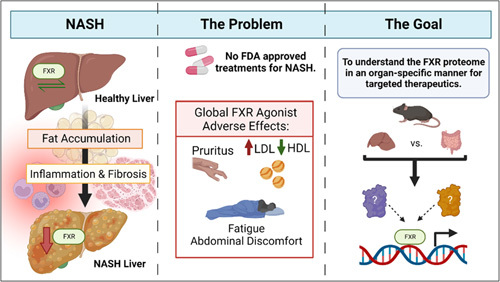
Overall significance. NASH develops following fat accumulation and subsequent hepatic inflammation and scarring. There are currently no approved pharmaceutical therapeutics for NASH patients, and global FXR agonists, currently undergoing clinical trials, demonstrate debilitating adverse effects like pruritus, abnormal cholesterol levels (elevated LDL and decreased HDL), fatigue, and abdominal discomfort. Because of visualized varied FXR functions, it is critical to understand how FXR activation affects NASH development in an organ-specific manner to identify proper therapeutic targets. Abbreviations: FDA, Food and Drug Administration; FXR, farnesoid X receptor.
FXR
Introduction to NRs
NRs are a family of ligand-activated TFs that regulate various biological processes and functions. There are over 500 members of this superfamily that are further divided into 7 subfamilies or subclasses: NR1 (thyroid hormone-like), NR2 (HNF4-like), NR3 (estrogen-like), NR4 (nerve growth factor IB-like), NR5 (fushi tarazu-F1-like), NR6 (germ cell nuclear factor-like), and NR0 (which do not contain a DNA binding domain).14,15 FXR is an adopted orphan NR1 that is activated by bile acids (BAs). Specifically, the FXR gene (Nr1h4) was first cloned in 1995 from mouse and rat liver,16 and BAs were discovered to be endogenous ligands of FXR in 1999.17 FXR is highly expressed in several organs and cell types, including hepatocytes, kidneys, adrenal glands, enterocytes, and to a lesser extent, HSCs, cholangiocytes, white adipose tissue, and immune cells.18 Six isoforms of FXR have been discovered (FXRα1-4 and FXRβ1-2), with FXRα being greatly expressed in the liver, distal small intestine (ileum), and adrenal glands, and FXRβ in the colon, proximal small intestine (duodenum), and kidney in humans.19–21
FXR and BA regulation
BAs are amphipathic molecules essential in the absorption of dietary fats, cholesterol, and lipid-soluble vitamins (vitamins A, D, E, and K). They are synthesized by hepatocytes through complex and tightly regulated processes involving at least 17 different enzymes22 through 2 major pathways as a result of cholesterol catabolism, the classical and alternative pathways. The classical, also known as the neutral pathway, is initiated with the rate-limiting enzyme cholesterol-7α-hydroxylase (CYP7A1), followed by sterol 12α-hydroxylase (CYP8B1) to yield cholic acid (CA), whereas the alternative, or acidic pathway, consists of sterol-27-hydroxylase (CYP27A1) and 25-hydroxycholesterol 7-alpha-hydroxylase (CYP7B1) to make chenodeoxycholic acid (CDCA).23,24 In mice, CDCA is converted to β-muricholic acid by CYP2C70, which is more hydrophilic and regarded as a strong FXR antagonist.25,26
CA and CDCA are produced in the liver, where they are conjugated with glycine (mainly in humans) or taurine (mainly in mice), which decreases their initial hydrophobicity and increases solubility. Once conjugated, the bile salts are effluxed out of hepatocytes by the bile salt export pump (BSEP) and multidrug resistance-associated protein 2 (MRP2) into the bile canaliculi to be excreted out of the liver through bile ducts. Cholangiocytes, epithelial cells of the bile duct, modify bile salts by diluting and alkalizing bile through bicarbonate or other secreted compounds.27 Bile is stored in the gallbladder in most species until stimulated for release by cholecystokinin, postprandial, into the duodenum through the Sphincter of Oddi for emulsification, digestion, and absorption of lipids in the small intestinal tract. The primary BAs that make up the human BA pool are CDCA and CA, which are converted to secondary BAs, lithocholic acid and deoxycholic acid, respectively, in the gut due to microbial modification.28 BAs also affect the gut microbiota composition, which can in turn alter the BA species pool through a variety of modifications, including deconjugation, dehydroxylation at carbon 7, and oxidation and epimerization, increasing BA diversity.29 Bacteria expressing the bile salt hydrolase gene can cleave glycine and taurine24 from conjugated BAs, and a complex of bacterial enzymes encoded by the Bai operon can further modify BAs into secondary structures that are not toxic to the microbiota population.29,30 Approximately 95% of BAs are reabsorbed in the terminal ileum into enterohepatic circulation through ileal apical sodium-dependent BA transporter (ASBT) and organic solute transporter alpha and beta (OSTα/β).31,32 Circulated BAs enter the liver through hepatic Na+/taurocholate cotransporting polypeptide (NTCP) for conjugated BAs or by organic anion-transporting polypeptide (OATP) for unconjugated BAs. The remaining 5% of BAs can be deconjugated by gut microbes and passively absorbed by the colon (like deoxycholic acid) or excreted from the body through the feces (mainly lithocholic acid).33 Secondary BAs are, in general, more hydrophobic and toxic than primary BAs.34
BAs not only function as critical components of digestion but also as powerful signaling molecules and endogenous ligands of several NRs, including pregnane X receptor and vitamin D receptor, in addition to activating FXR.35 CDCA is the most potent BA activator of FXR, followed by CA, deoxycholic acid, and lithocholic acid.33,36,37 FXR is the main regulator of BA homeostasis and is especially important in activating negative feedback inhibition mechanisms, such as ileal FGF15 in mice, FGF19 in humans, and hepatic small heterodimer partner (SHP) to suppress BA synthesis.
Tissue-specific role of FXR in regulating BA homeostasis
Because of amphipathic chemical properties, BAs behave as detergents and, if not properly regulated, can induce liver injury, inflammation, hepatocyte apoptosis, and cholestasis.38–41 Extensive or chronic liver damage can lead to cholestasis and even malignancy development in patients, making the regulation of BA synthesis a key topic in the field of hepatology.42 FXR is expressed in various organs and cell types such as the pancreas, lungs, kidneys, liver (hepatocytes, cholangiocytes, and stellate cells), and intestine (enterocytes).43–47 FXR function is largely understood in hepatocytes and ileal enterocytes, but its role in other cell types is not fully understood.
Intestinal FXR, specifically in the ileum, is the main regulator of BA synthesis by means of the FXR-FGF15/19 pathway that operates by mechanisms of negative feedback inhibition.48 FXR’s activation in the ileum induces FGF15 secretion in mice49 and FGF19 in humans,50 into the portal vein to the liver where it binds and activates its receptor, FGF receptor 4 along with β-Klotho, in hepatocytes to activate mitogen-activated protein kinase signaling pathways.51,52 FGF receptor 4 activation signaling inhibits the gene expression of Cyp7a1, suppressing the classical pathway of BA synthesis. Intestinal FXR controls BA synthesis, mainly at night, through high Fgf15 expression in the intestine53; however, hepatic FXR regulates BA synthesis through induced expression of Shp, which binds to liver receptor homolog 1 inhibiting Cyp8b1 gene transcription and minorly Cyp7a1.51 Hepatocyte FXR activation also induces the expression of BA efflux transporters, such as Bsep and Ostα/β, in the liver to promote enterohepatic BA circulation and prevent cholestasis.54,55 Because of the lack of FXR specificity for primary and secondary BAs and BA dose-dependent cellular toxicity, the generation of synthetic ligands for FXR activation has been of increased interest.
Pioneer Factors
Regulators of transcription
With genome-based studies becoming especially critical in the study of NASH and other chronic liver diseases, understanding gene expression in an organ-specific manner may hold the key to identifying ideal therapeutic targets. Pioneer factors (PFs), a subset of TFs recognized as the proteins capable of binding condensed chromatin to regulate transcription in a cell-specific manner,56 have recently been recognized as an avenue for chronic liver disease research. Through this process, PFs, and their dynamic expression, implement cell fate and organ development56; however, it has been recently proposed that chromatin opening, and subsequent expression of silent genes, is mediated by PFs and non-PFs alike.57 To target these silent areas in the genome, PFs must recognize their target DNA sequences on the nucleosome.58 Despite these controversies, understanding the organ-specific role of TFs remains a key area of study for drug development.
Forkhead Box A (FOXA) is a family of TFs vital in foregut endoderm for hepatic differentiation and induction of liver-specific genes such as albumin.59 The DNA binding domain of FOXA (“winged helix” structure) resembles the nucleosomal binding domain of the linker histone, causing its displacement and opening of the chromatin.60 The winged helix structure, also known as the forkhead domain, is highly conserved in each isoform. In the liver, FOXA1 and FOXA261 are required for early organ induction,56,62 with FOXA2 deletion being embryonically lethal, whereas another set of PFs known as GATA-binding proteins, specifically GATA-4 and GATA-6,63,64 is redundantly expressed and required for the early organ development from the foregut endoderm. Conditional triple-knockout of Foxa1/2/3 in hepatocytes of adult mice resulted in eventual liver failure 15–20 days after deletion.65 In these mice, hepatocyte nuclear factor 4 alpha (HNF4α) was continually expressed, but there was minimal chromatin accessibility at FOXA-HNF4α cobound sites, confirming that FOXA chromatin manipulation is necessary for adult liver function.65 It has been found that overexpression of GATA-6 in patients with NAFLD resulted in increased HSC activation and subsequent fibrosis.66 Interestingly, hepatocyte-specific deletion of GATA-4, through the albumin promoter, resulted in increased steatosis and insulin resistance in a murine model fed high-fat diet.67 These studies demonstrate the complexity of PFs postdevelopment and highlight an important role for them in the development and progression of steatosis and NASH. There are limited studies investigating PF function in NASH; however, the proteome created by PF and TF binding may provide the key to organ-specific therapeutic targeting.
FOXA2 and BA homeostasis
FOXA2 (previously known as HNF-3β) is essential for murine liver development and remains critical in the adult liver for BA, glucose, and lipid homeostasis.68–71 Foxa2-deficient mice display an accumulation of BAs in the liver (Figure 2).72 Furthermore, FOXA2 has been shown to regulate hyperbilirubinemia in mice and patients with sepsis and acute liver failure by the upregulation of MRP2.73 FOXA2 directly and indirectly regulates the gene expression of hepatic transporters, Oatp2, Mrp2, Mrp3, and Mrp4, and indirectly regulates Cyp3a11 that encodes a key P450 phase I enzyme, contributing to BA accumulation.69,73 Chromatin immunoprecipitation (ChIP) conducted with an anti-FOXA2 antibody on livers with no FOXA2 suggests that Mrp2 and Oatp2 genes are direct targets of FOXA2 in vivo. FOXA2 replaces FXR to maintain the expression of Mrp2 in patients with acute liver failure excluding sepsis.73 In fact, mice with hepatocyte-specific Foxa2 ablation displayed reduced Cyp7a1, Cyp7b1, Cyp8b1, Cyp27a1, and Ntcp gene expression following standard diet feeding, insinuating a key role for Foxa2 in BA regulation.69 It has also been shown that pediatric and adult cholestatic patients have reduced hepatic FOXA2 expression, further exemplifying its importance in liver disease progression.69 FOXA2 and FOXA1 also regulate bile duct and gallbladder development by manipulating chromatin accessibility for glucocorticoid receptor binding.74,75
FIGURE 2.
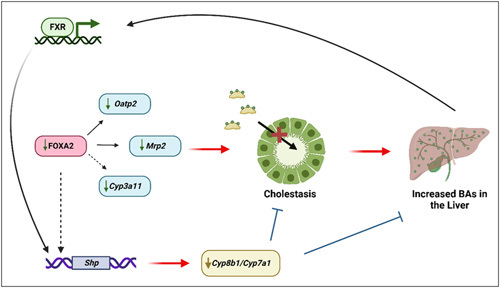
FOXA2 and BA homeostasis. In the liver, FOXA2 directly regulates the expression of genes of transporters Oatp2 and Mrp2 involved in BA transport and, indirectly, Cyp3a11. Deficiency of FOXA2, as seen in cholestatic patients, has decreased the gene expression of Oatp2, Mrp2 (along with Mrp3 and Mrp4, not shown), and Cyp3a11, contributing to cholestasis and increased BA accumulation within the liver. Excess BAs activate FXR, which in turn induces gene expression of Shp to suppress the expression of genes encoding rate-limiting enzymes for BA synthesis, Cyp7a1 and Cyp8b1, to mitigate cholestasis and excess BAs in the liver. FOXA2 is also believed to bind alongside FXR in the upstream regulatory region to elicit these effects. Abbreviations: BA, bile acid; FXR, farnesoid X receptor.
It is possible that BA activation of FXR may acutely activate the transcription of Foxa2.76 In mice, FXR and FOXA2 bind the upstream regulatory region of Shp, with Shp induction decreasing BA production by downregulation of Cyp7a1 transcription (Figure 2).77 The field remains controversial on the actual interactions of FXR and FOXA2. There are several proposed interactions. It is believed that the binding of FOXA2 is dependent on FXR, and FOXA2 may repress FXR transcriptional activity on several genes, including Shp, by a tethering mechanism.78 This proposed mechanism would explain how FOXA2 could regulate FXR tissue-specific functionality. Contrarily, it has been suggested that FXR ligand–directed activation remains FOXA2-independent while its chromatin binding is FOXA2 dependent76,79; however, it has also been shown that FOXA2 is required for ligand-bound FXR DNA binding and activation.78,79 Similarly, FOXA2 occupancy is increased dramatically when FXR is bound by an agonist, leading to the belief that FOXA2 is not bound to DNA before FXR ligand activation. FOXA2 evicts nucleosomes allowing for the opening of chromatin for FXR-binding accessibility and increased transcription. However, FOXA2 is believed to repress the transcriptional activity of FXR appropriate for the maintenance of a particular physiological state. These works suggest an interdependent relationship between FOXA2 and FXR DNA binding during ligand activation (Figure 3). The mechanism of interaction between FXR and FOXA2 is not well understood, and further studies may allow for a deeper understanding of their complex interactions in health and disease.
FIGURE 3.
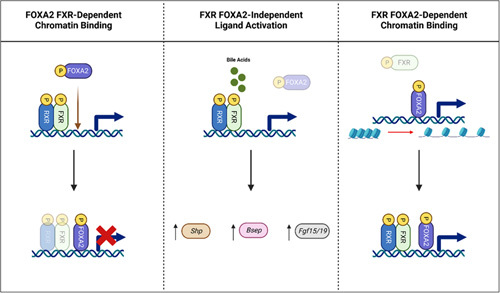
Proposed FXR and FOXA2 interactions. Three proposed FXR and FOXA2 interactions: (1) FOXA2 binding is dependent on FXR. In addition, when bound, FOXA2 may repress FXR (which heterodimerizes with RXR) transcriptional activity. (2) Activation of FXR by ligand-binding (such as BAs) is independent of FOXA2, and activation of FXR leads to transcriptional regulation of various genes such as Shp, Bsep, and Fgf15/19. (3) FXR chromatin binding and activation is dependent on the presence of FOXA2, which displaces histones in highly condensed areas of chromatin for a more open configuration to allow FXR binding. Abbreviations: FOXA1/2, forkhead box A1/2; FXR, farnesoid X receptor.
Importance of FXR tissue specificity in the treatment of NAFLD/NASH
Implications of BAs in NAFLD/NASH progression
Dysregulation of BAs is linked to NASH pathogenesis; therefore, modulating BA homeostasis opens potential therapy of NASH through their signaling effects.80 The ratio of secondary BAs to primary BAs is inversely correlated to the NAFLD Activity Score, indicating a relationship between BA species and disease stage.81,82 Free fatty acid accumulation from diet inhibits Shp expression, leading to decreased repression of Ntcp and Cyp7a1 and continued BA production and accumulation in the liver, which promotes hepatocyte injury and the development of NASH.83,84 Because of the close relationship between the microbiome and BA composition, it has been established that there are differences in microbiome composition between healthy and NAFLD patients. Glycine-metabolizing and taurine-metabolizing bacteria were increased in NAFLD patients, which may help explain the increase in secondary BAs in the BA pool.85 Furthermore, when intestinal microflora composition is altered, conjugated BAs and their metabolites can be increased, which inhibits intestinal FXR signaling leading to reduced BA secretion from the liver and promotion of NAFLD.86
Benefits of tissue-specific FXR activation/inactivation in the treatment of NASH
The current challenge is the design of tissue-specific FXR agonists capable of regulating BA homeostasis, lipid metabolism, and inflammation without off-target effects. Systemic FXR activation is proven to be beneficial in protecting against steatosis, inflammation, and fibrosis because of its activation of FXR. Systemic FXR agonists, such as obeticholic acid, reduce the accumulation of triglycerides in the liver and free fatty acids in mice fed high-fat diet.11 Obeticholic acid also decreases liver inflammation and fibrosis while increasing the risk of pruritus and LDLs.8,87 GW4064, a selective FXR agonist, has been shown to reduce hepatic inflammation in high-fat diet or high-fat, high-cholesterol diet–fed mouse models.10 WAY-362450 decreased fibrosis severity in methionine and choline-deficient mouse models88 and increased VLDL and LDL while decreasing HDL in fructose-fed rats.89 Cilofexor (GS-9674) is beneficial in decreasing steatosis and fibrosis in both mice and humans but increases the risk of pruritus.90,91 Tropifexor (LJN452) is also beneficial in decreasing liver fat and fibrosis while increasing the risk of pruritus and is associated with minor increases in LDLs.9 The benefits and consequences of whole-body FXR agonists demonstrate the importance of understanding mechanisms and/or roles of FXR tissue-specific activation to negate adverse effects in patients with liver diseases.
Genome-wide ChIP-seq technologies have allowed insight into tissue-specific gene expression of FXR in mice.92 There was only an 11% overlap between liver and intestinal FXR-binding sites in mice, suggesting underlying regulation of FXR tissue-specific functionality.92 Activation of hepatic FXR has been shown to be a protective mechanism against hepatic steatosis.93 It was determined that when feeding hepatic FXR knockout and intestinal FXR knockout mice a high-cholesterol diet, the hepatic FXR deficiency exacerbated hepatic steatosis while intestinal FXR deficiency did not.93 Hepatic FXR also inhibits lipogenesis by inducing SHP expression, which suppressed sterol regulatory element-binding protein 1c and its downstream lipogenic target genes.94 In addition, hepatic FXR is important in modulating hepatic inflammation, specifically by inhibiting NF-κB, an important inflammatory modulator.7 In vitro, FXR inhibited NF-κB activation in HepG2 cells and primary hepatocytes.7 In vivo, FXR knockout mice treated with LPS had greater induction of hepatic proinflammatory mediators, such as cyclooxygenase-2 and inducible nitric oxide synthase, compared with the control group, implicating an anti-inflammatory characteristic of hepatic FXR.
The activation and inhibition of intestinal FXR have been beneficial in the treatment of NASH in rodents. The benefits of the inhibition of intestinal FXR have been attributed to the microbiome-intestine-liver ceramide axis.95 Ceramides are intracellular signals for apoptosis96 and also increase sterol regulatory element-binding protein 1c activity in the liver, which promotes lipogenesis.97 Intestinal FXR has been shown to increase the expression of genes involved in ceramide synthesis.86,97 Mice fed a high-fat diet treated with a bile salt hydrolase inhibitor, caffeic acid phenethyl ester, displayed reduced intestinal FXR activity and ceramide synthesis. Treatment with caffeic acid phenethyl ester lowered average body weight and improved insulin sensitivity and glucose tolerance.98 The reduction in ceramide levels also reduced hepatic endoplasmic reticulum stress. It is also known that caffeic acid phenethyl ester activates the cAMP-CREB pathway, which may be the mode of action for bile salt hydrolase gene downregulation. Benefits of activating intestinal FXR, outside of BA synthesis regulation, include improvements in energy metabolism. Mice fed control or high-fat diet treated with fexaramine, an intestinal-specific FXR agonist, demonstrated increased energy expenditure, reduced body weight and body fat mass, decreased systemic inflammation and glucose production, and increased brown adipose tissue mass when compared with vehicle-treated mice on the high-fat diet.99 Because of the complex responses of tissue-restricted FXR activation, identifying tissue-specific or cell-specific modulators of FXR is required to develop safe and effective therapies for NAFL and NASH patients.
Tissue specificity of NRs
TF complexes in NASH
It is widely accepted that TFs work in a complex network for the regulation of gene transcription and repression, which can become altered in diseased states.100,101 HNF4α is a well-studied TF highly enriched in the liver and is important for maintaining liver function and mature hepatocyte function. C57BL/6J mice overexpressing human HNF4α exhibited protective effects against diet-induced NASH, whereas loss of HNF4α displayed opposite effects.102 The explained mechanisms involve transcriptional regulation of BA, lipolytic, and p53 signaling pathways. Restoration of HNF4α through mRNA delivery improves the functionality of fibrotic primary hepatocytes isolated from mice and humans.103 HNF4α also interacts with other TFs to elicit liver protective effects. It has been shown that HNF4α is required for activating transcription factor 3 (ATF3)-associated improvement of steatohepatitis.104 Mice fed a high-fat diet displayed increased hepatic carbohydrate-responsive element-binding protein and the inclusion of fructose to a high-fat diet increases both carbohydrate-responsive element-binding protein and Srbep1 expression.105 In addition, it has been found that zinc fingers and homeoboxes 2 (ZHX2), and its downstream target protein PTEN, are suppressed in murine models of NASH and in steatotic hepatocyte culture.106 Hepatocyte-specific deletion of ZHX2 exacerbated murine NASH phenotype, whereas hepatocyte-specific overexpression ameliorated hepatic steatosis, lipid accumulation, and liver fibrosis and inflammation through increased expression of PTEN.106 A case for cellular programming through TF regulation in NASH has also been speculated during fetal development.107 Pups born to female rats with 50% food restriction during pregnancy and nursed by control dams had reduced hepatic peroxisome proliferator-activated receptor (PPAR)–α and -γ until 9 months of age, which may indicate a complex and developmentally linked role for PFs and TFs in NASH development.108 Together, these studies allow for the speculation that FXR expression and function in NASH may be disrupted through protein complex dysregulation; however, the varied expression of FXR through the body, and lack of FXR proteome knowledge, make it a difficult target for study.
Therapeutics of tissue-specific NRs
Cell-specific modification of the functions of 1 NR, estrogen receptor (ER), has been successful in the development of efficacious and safer medicines in the tissue-specific treatment of diseases. ERs are type I NRs whose tissue specificity has allowed researchers to design cell-specific agonists and antagonists, which have been extensively reviewed.109,110 Three types of predominant ERs have been discovered and characterized: ERα, ERβ, and an estrogen G protein–coupled ER (GPER1) with 2 main signaling mechanisms, genomic and nongenomic.109 Studies of estrogens and ERs in cancer, like breast and ovarian, have provided seminal knowledge on targeted drug development of selective ER modulators (SERMs) and identification of xenoestrogens to control ER function in a tissue-specific manner.109,110 Tamoxifen is a widely used SERM that can serve as an ERα agonist in uterine tissue and antagonist in breast tissue for the treatment of patients with breast cancer.109 Tamoxifen exerts its inhibitory function in breast tissue by interacting with ERα to shift the side chain to block coactivator binding111; however, its weak activator function for uterine ERα has been shown to increase endometrial proliferation and carcinogenesis.109,110
Chronic liver diseases, like NASH, with no FDA-approved therapeutic treatment, have benefitted from liver-specific NR targeting like thyroid hormone receptor–β (TR-β).112 TR-β is a nonsteroidal type I NR with extensive effects on metabolism, including body weight and LDL reduction and increased hepatic fatty acid β-oxidation on activation by thyroid hormones.113 Selective modulation of TR-β in the liver by promising drugs such as resmetirom (MGL-3196) and VK2809 (MB07811) have shown to be beneficial in NASH patients participating in phase 2 studies. Resmetirom significantly reduced hepatic fat after 12 and 36 weeks of treatment in NASH patients, whereas VK2809 significantly reduced liver fat content in treated NAFLD patients compared with a placebo on 12 weeks of administration.114,115 The ability of these compounds to specifically activate the TR-β isoform in the liver is critical to minimize any potential off-target effects of TR-β agonism in the central nervous system and hypothalamic-pituitary-thyroid axis, as TR-β has been demonstrated to affect remyelination.116 In addition, activating liver-specific TR-β minimizes side effects that occur in the heart and bone, which express TR-α. Through the tissue-specific effects of ER and TR-β treatments, the existence of unique tissue-specific FXR function is a promising avenue to investigate pharmacological strategies that can be implemented in the treatment of NASH.
SUMMARY
BAs are instrumental in fat and lipid digestion and in the activation of numerous metabolic pathways; however, their accumulation in tissues can lead to cell damage. FXR, the master regulator of BA homeostasis, is critical in suppressing BA synthesis by negative feedback pathways and promoting BA transport therefore decreasing the risk of developing cholestasis and liver injury. NASH is one of the most common liver diseases, and cholestasis contributes to NASH development and progression into irreversible ailments. The regulation of BAs has been a key therapeutic strategy to maintain a healthy state in NASH patients. Whole-body FXR agonism often results in adverse effects such as pruritus and elevated serum LDL. Because of its ubiquitous expression, understanding and manipulating cell-specific FXR function may be the key for developing NASH therapeutics. PFs such as FOXA2 provide a novel area of study that contributes to underlying mechanisms determining tissue-restricted FXR modulation. After previous studies on NRs like ERα and TR-β, we are hopeful that the discovery of the tissue-specific transcriptional function of FXR will allow us to examine the targeted therapeutic approaches for NASH and other liver diseases.
FUNDING INFORMATION
This work was supported by the National Institutes of Health (Grants: ES007148, ES029258, DK122725, GM135258, AND GM093854), the Department of Veteran Affairs (BX002741), and the Rutgers University Center for Lipid Research.
CONFLICTS OF INTEREST
The authors have no conflicts to report.
Footnotes
Abbreviations: ASBT, apical sodium-dependent bile acid transporter; ATF3, activating transcription factor 3; BA, bile acid; BSEP, bile salt export pump; CA, cholic acid; CDCA, chenodeoxycholic acid; ChIP, chromatin immunoprecipitation; CYP27A1, sterol-27-hydroxylase; CYP7A1, cholesterol-7α-hydroxylase; CYP7B1, 25-hydroxycholesterol 7-alpha-hydroxylase; CYP8B1, sterol 12α-hydroxylase; ER, estrogen receptor; FDA, Food and Drug Administration; FOXA1/2, forkhead box A1/2; FXR, farnesoid X receptor; GPER1, G protein–coupled receptor; HNF4α, hepatocyte nuclear factor 4 alpha; MRP2, multidrug resistance-associated protein 2; NR, nuclear receptor; NTCP, Na+/taurocholate cotransporting polypeptide; OATP, organic anion-transporting polypeptide; OSTα/β: organic solute transporter alpha and beta; PF, pioneer factor; PPAR, peroxisome proliferator-activated receptor; SERM, selective estrogen receptor modulators; SHP, small heterodimer partner; SERMs, selective ER modulators; TF, transcription factor; TR-β, thyroid hormone receptor–β; ZHX2, zinc fingers and homeoboxes 2
Contributor Information
Zakiyah Henry, Email: zh196@scarletmail.rutgers.edu.
Vik Meadows, Email: vik.meadows@rutgers.edu.
Grace L. Guo, Email: guo@eohsi.rutgers.edu.
REFERENCES
- 1.Ludwig J, Viggiano TR, McGill DB, Oh BJ. Nonalcoholic steatohepatitis: Mayo Clinic experiences with a hitherto unnamed disease. Mayo Clin Proc. 1980;55:434–8. [PubMed] [Google Scholar]
- 2.Gottlieb A, Canbay A. Why bile acids are so important in non-alcoholic fatty liver disease (NAFLD) progression. Cells. 2019;8:1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shirazi F, Wang J, Wong RJ. Nonalcoholic steatohepatitis becomes the leading indication for liver transplant registrants among US adults born between 1945 and 1965. J Clin Exp Hepatol. 2020;10:30–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Noureddin M, Vipani A, Bresee C, Todo T, Kim IK, Alkhouri N, et al. NASH leading cause of liver transplant in women: updated analysis of indications for liver transplant and ethnic and gender variances. Am J Gastroenterol. 2018;113:1649–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Younossi Z, Anstee QM, Marietti M, Hardy T, Henry L, Eslam M, et al. Global burden of NAFLD and NASH: trends, predictions, risk factors and prevention. Nat Rev Gastroenterol Hepatol. 2018;15:11–20. [DOI] [PubMed] [Google Scholar]
- 6.Buzzetti E, Pinzani M, Tsochatzis EA. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism. 2016;65:1038–48. [DOI] [PubMed] [Google Scholar]
- 7.Wang YD, Chen WD, Moore DD, Huang W. FXR: a metabolic regulator and cell protector. Cell Res. 2008;18:1087–95. [DOI] [PubMed] [Google Scholar]
- 8.Neuschwander-Tetri BA, Loomba R, Sanyal AJ, Lavine JE, Van Natta ML, Abdelmalek MF, et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): a multicentre, randomised, placebo-controlled trial. Lancet. 2015;385:956–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hernandez ED, Zheng L, Kim Y, Fang B, Liu B, Valdez RA, et al. Tropifexor-mediated abrogation of steatohepatitis and fibrosis is associated with the antioxidative gene expression profile in rodents. Hepatol Commun. 2019;3:1085–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ma Y, Huang Y, Yan L, Gao M, Liu D. Synthetic FXR agonist GW4064 prevents diet-induced hepatic steatosis and insulin resistance. Pharm Res. 2013;30:1447–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gai Z, Visentin M, Gui T, Zhao L, Thasler WE, Hausler S, et al. Effects of farnesoid X receptor activation on arachidonic acid metabolism, NF-kB signaling, and hepatic inflammation. Mol Pharmacol. 2018;94:802–11. [DOI] [PubMed] [Google Scholar]
- 12.Briand F, Brousseau E, Quinsat M, Burcelin R, Sulpice T. Obeticholic acid raises LDL-cholesterol and reduces HDL-cholesterol in the diet-induced NASH (DIN) hamster model. Eur J Pharmacol. 2018;818:449–56. [DOI] [PubMed] [Google Scholar]
- 13.Fiorucci S, Di Giorgio C, Distrutti E. Obeticholic acid: an update of its pharmacological activities in liver disorders. Handb Exp Pharmacol. 2019;256:283–95. [DOI] [PubMed] [Google Scholar]
- 14.Xiao X, Wang P, Chou KC. Recent progresses in identifying nuclear receptors and their families. Curr Top Med Chem. 2013;13:1192–200. [DOI] [PubMed] [Google Scholar]
- 15.Weikum ER, Liu X, Ortlund EA. The nuclear receptor superfamily: a structural perspective. Protein Sci. 2018;27:1876–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Forman BM, Goode E, Chen J, Oro AE, Bradley DJ, Perlmann T, et al. Identification of a nuclear receptor that is activated by farnesol metabolites. Cell. 1995;81:687–93. [DOI] [PubMed] [Google Scholar]
- 17.Makishima M, Okamoto AY, Repa JJ, Tu H, Learned RM, Luk A, et al. Identification of a nuclear receptor for bile acids. Science. 1999;284:1362–5. [DOI] [PubMed] [Google Scholar]
- 18.Lefebvre P, Cariou B, Lien F, Kuipers F, Staels B. Role of bile acids and bile acid receptors in metabolic regulation. Physiol Rev. 2009;89:147–91. [DOI] [PubMed] [Google Scholar]
- 19.Huber RM, Murphy K, Miao B, Link JR, Cunningham MR, Rupar MJ, et al. Generation of multiple farnesoid-X-receptor isoforms through the use of alternative promoters. Gene. 2002;290:35–43. [DOI] [PubMed] [Google Scholar]
- 20.Zhang Y, Kast-Woelbern HR, Edwards PA. Natural structural variants of the nuclear receptor farnesoid X receptor affect transcriptional activation. J Biol Chem. 2003;278:104–110. [DOI] [PubMed] [Google Scholar]
- 21.Ramos Pittol JM, Milona A, Morris I, Willemsen ECL, van der Veen SW, Kalkhoven E, et al. FXR isoforms control different metabolic functions in liver cells via binding to specific DNA motifs. Gastroenterology. 2020;159:1853–65. e1810. [DOI] [PubMed] [Google Scholar]
- 22.Rizzolo D, Buckley K, Kong B, Zhan L, Shen J, Stofan M, et al. Bile acid homeostasis in a cholesterol 7alpha-hydroxylase and sterol 27-hydroxylase double knockout mouse model. Hepatology. 2019;70:389–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Russell DW. The enzymes, regulation, and genetics of bile acid synthesis. Annu Rev Biochem. 2003;72:137–74. [DOI] [PubMed] [Google Scholar]
- 24.Baloni P, Funk CC, Yan J, Yurkovich JT, Kueider-Paisley A, Nho K, et al. Metabolic network analysis reveals altered bile acid synthesis and metabolism in Alzheimer’s disease. Cell Rep Med. 2020;1:100138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Takahashi S, Fukami T, Masuo Y, Brocker CN, Xie C, Krausz KW, et al. Cyp2c70 is responsible for the species difference in bile acid metabolism between mice and humans. J Lipid Res. 2016;57:2130–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guo GL, Chiang JYL. Is CYP2C70 the key to new mouse models to understand bile acids in humans? J Lipid Res. 2020;61:269–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hundt M, Basit H, John S. Physiology, Bile Secretion. StatPearls; 2022. [PubMed] [Google Scholar]
- 28.Li T, Chiang JY. Bile acid signaling in metabolic disease and drug therapy. Pharmacol Rev. 2014;66:948–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guzior DV, Quinn RA. Review: microbial transformations of human bile acids. Microbiome. 2021;9:140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhu Y, Liu H, Zhang M, Guo GL. Fatty liver diseases, bile acids, and FXR. Acta Pharm Sin B. 2016;6:409–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dawson PA, Hubbert M, Haywood J, Craddock AL, Zerangue N, Christian WV, et al. The heteromeric organic solute transporter alpha-beta, Ostalpha-Ostbeta, is an ileal basolateral bile acid transporter. J Biol Chem. 2005;280:6960–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kock K, Brouwer KL. A perspective on efflux transport proteins in the liver. Clin Pharmacol Ther. 2012;92:599–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chavez-Talavera O, Tailleux A, Lefebvre P, Staels B. Bile acid control of metabolism and inflammation in obesity, type 2 diabetes, dyslipidemia, and nonalcoholic fatty liver disease. Gastroenterology. 2017;152:1679–94. e1673. [DOI] [PubMed] [Google Scholar]
- 34.Li T, Apte U. Bile acid metabolism and signaling in cholestasis, inflammation, and cancer. Adv Pharmacol. 2015;74:263–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shin DJ, Wang L. Bile acid-activated receptors: a review on FXR and other nuclear receptors. Handb Exp Pharmacol. 2019;256:51–72. [DOI] [PubMed] [Google Scholar]
- 36.Fiorucci S, Antonelli E, Rizzo G, Renga B, Mencarelli A, Riccardi L, et al. The nuclear receptor SHP mediates inhibition of hepatic stellate cells by FXR and protects against liver fibrosis. Gastroenterology. 2004;127:1497–512. [DOI] [PubMed] [Google Scholar]
- 37.Liu J, Lu H, Lu YF, Lei X, Cui JY, Ellis E, et al. Potency of individual bile acids to regulate bile acid synthesis and transport genes in primary human hepatocyte cultures. Toxicol Sci. 2014;141:538–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Perez MJ, Briz O. Bile-acid-induced cell injury and protection. World J Gastroenterol. 2009;15:1677–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hofmann AF, Hagey LR. Bile acids: chemistry, pathochemistry, biology, pathobiology, and therapeutics. Cell Mol Life Sci. 2008;65:2461–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li M, Cai SY, Boyer JL. Mechanisms of bile acid mediated inflammation in the liver. Mol Aspects Med. 2017;56:45–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ibrahim S, Dayoub R, Krautbauer S, Liebisch G, Wege AK, Melter M, et al. Bile acid-induced apoptosis and bile acid synthesis are reduced by over-expression of augmenter of liver regeneration (ALR) in a STAT3-dependent mechanism. Exp Cell Res. 2019;374:189–97. [DOI] [PubMed] [Google Scholar]
- 42.Sagnelli E, Macera M, Russo A, Coppola N, Sagnelli C. Epidemiological and etiological variations in hepatocellular carcinoma. Infection. 2020;48:7–17. [DOI] [PubMed] [Google Scholar]
- 43.Nijmeijer RM, Schaap FG, Smits AJ, Kremer AE, Akkermans LM, Kroese AB, et al. Impact of global Fxr deficiency on experimental acute pancreatitis and genetic variation in the FXR locus in human acute pancreatitis. PLoS One. 2014;9:e114393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wu JN, Chen JR, Chen JL. Role of farnesoid X receptor in the pathogenesis of respiratory diseases. Can Respir J. 2020;2020:9137251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fiorucci S, Biagioli M, Zampella A, Distrutti E. Bile acids activated receptors regulate innate immunity. Front Immunol. 2018;9:1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yan N, Yan T, Xia Y, Hao H, Wang G, Gonzalez FJ. The pathophysiological function of non-gastrointestinal farnesoid X receptor. Pharmacol Ther. 2021;226:107867. [DOI] [PubMed] [Google Scholar]
- 47.Anderson KM, Gayer CP. The pathophysiology of farnesoid X receptor (FXR) in the GI tract: inflammation, barrier function and innate immunity. Cells. 2021;10:3206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhu Y, Li F, Guo GL. Tissue-specific function of farnesoid X receptor in liver and intestine. Pharmacol Res. 2011;63:259–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Inagaki T, Choi M, Moschetta A, Peng L, Cummins CL, McDonald JG, et al. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab. 2005;2:217–25. [DOI] [PubMed] [Google Scholar]
- 50.Li J, Pircher PC, Schulman IG, Westin SK. Regulation of complement C3 expression by the bile acid receptor FXR. J Biol Chem. 2005;280:7427–34. [DOI] [PubMed] [Google Scholar]
- 51.Kong B, Wang L, Chiang JY, Zhang Y, Klaassen CD, Guo GL. Mechanism of tissue-specific farnesoid X receptor in suppressing the expression of genes in bile-acid synthesis in mice. Hepatology. 2012;56:1034–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu Y, Cao M, Cai Y, Li X, Zhao C, Cui R. Dissecting the role of the FGF19-FGFR4 signaling pathway in cancer development and progression. Front Cell Dev Biol. 2020;8:95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stroeve JH, Brufau G, Stellaard F, Gonzalez FJ, Staels B, Kuipers F. Intestinal FXR-mediated FGF15 production contributes to diurnal control of hepatic bile acid synthesis in mice. Lab Invest. 2010;90:1457–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Landrier JF, Eloranta JJ, Vavricka SR, Kullak-Ublick GA. The nuclear receptor for bile acids, FXR, transactivates human organic solute transporter-alpha and -beta genes. Am J Physiol Gastrointest Liver Physiol. 2006;290:G476–85. [DOI] [PubMed] [Google Scholar]
- 55.Song X, Chen Y, Valanejad L, Kaimal R, Yan B, Stoner M, et al. Mechanistic insights into isoform-dependent and species-specific regulation of bile salt export pump by farnesoid X receptor. J Lipid Res. 2013;54:3030–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zaret KS, Carroll JS. Pioneer transcription factors: establishing competence for gene expression. Genes Dev. 2011;25:2227–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hansen JL, Loell KJ, Cohen BA. A test of the pioneer factor hypothesis using ectopic liver gene activation. Elife. 2022;11:e73358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cirillo LA, McPherson CE, Bossard P, Stevens K, Cherian S, Shim EY, et al. Binding of the winged-helix transcription factor HNF3 to a linker histone site on the nucleosome. EMBO J. 1998;17:244–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Huppert SS, Iwafuchi-Doi M. Molecular regulation of mammalian hepatic architecture. Curr Top Dev Biol. 2019;132:91–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Iwafuchi-Doi M, Zaret KS. Cell fate control by pioneer transcription factors. Development. 2016;143:1833–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lee CS, Friedman JR, Fulmer JT, Kaestner KH. The initiation of liver development is dependent on Foxa transcription factors. Nature. 2005;435:944–7. [DOI] [PubMed] [Google Scholar]
- 62.Cirillo LA, Lin FR, Cuesta I, Friedman D, Jarnik M, Zaret KS. Opening of compacted chromatin by early developmental transcription factors HNF3 (FoxA) and GATA-4. Mol Cell. 2002;9:279–89. [DOI] [PubMed] [Google Scholar]
- 63.Zhao R, Watt AJ, Li J, Luebke-Wheeler J, Morrisey EE, Duncan SA. GATA6 is essential for embryonic development of the liver but dispensable for early heart formation. Mol Cell Biol. 2005;25:2622–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Holtzinger A, Evans T. Gata4 regulates the formation of multiple organs. Development. 2005;132:4005–14. [DOI] [PubMed] [Google Scholar]
- 65.Reizel Y, Morgan A, Gao L, Lan Y, Manduchi E, Waite EL, et al. Collapse of the hepatic gene regulatory network in the absence of FoxA factors. Genes Dev. 2020;34:1039–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.He L, Yuan H, Liang J, Hong J, Qu C. Expression of hepatic stellate cell activation-related genes in HBV-, HCV-, and nonalcoholic fatty liver disease-associated fibrosis. PLoS One. 2020;15:e0233702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.He L, Wang X, Ding Z, Liu L, Cheng H, Bily D, et al. Deleting Gata4 in hepatocytes promoted the progression of NAFLD via increasing steatosis and apoptosis, and desensitizing insulin signaling. J Nutr Biochem. 2023;111:109157. [DOI] [PubMed] [Google Scholar]
- 68.Mu T, Xu L, Zhong Y, Liu X, Zhao Z, Huang C, et al. Embryonic liver developmental trajectory revealed by single-cell RNA sequencing in the Foxa2(eGFP) mouse. Commun Biol. 2020;3:642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bochkis IM, Rubins NE, White P, Furth EE, Friedman JR, Kaestner KH. Hepatocyte-specific ablation of Foxa2 alters bile acid homeostasis and results in endoplasmic reticulum stress. Nat Med. 2008;14:828–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhou H, Liu R. ER stress and hepatic lipid metabolism. Front Genet. 2014;5:112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wolfrum C, Asilmaz E, Luca E, Friedman JM, Stoffel M. Foxa2 regulates lipid metabolism and ketogenesis in the liver during fasting and in diabetes. Nature. 2004;432:1027–32. [DOI] [PubMed] [Google Scholar]
- 72.Bochkis IM, Shin S, Kaestner KH. Bile acid-induced inflammatory signaling in mice lacking Foxa2 in the liver leads to activation of mTOR and age-onset obesity. Mol Metab. 2013;2:447–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wang S, Feng R, Wang SS, Liu H, Shao C, Li Y, et al. FOXA2 prevents hyperbilirubinaemia in acute liver failure by maintaining apical MRP2 expression. Gut. 2022;38:1619–19. [DOI] [PubMed] [Google Scholar]
- 74.Li Z, White P, Tuteja G, Rubins N, Sackett S, Kaestner KH. Foxa1 and Foxa2 regulate bile duct development in mice. J Clin Invest. 2009;119:1537–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Strazzabosco M. Foxa1 and Foxa2 regulate bile duct development in mice. J Hepatol. 2010;52:765–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bochkis IM, Schug J, Rubins NE, Chopra AR, O’Malley BW, Kaestner KH. Foxa2-dependent hepatic gene regulatory networks depend on physiological state. Physiol Genomics. 2009;38:186–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Li T, Jahan A, Chiang JY. Bile acids and cytokines inhibit the human cholesterol 7 alpha-hydroxylase gene via the JNK/c-jun pathway in human liver cells. Hepatology. 2006;43:1202–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ploton M, Mazuy C, Gheeraert C, Dubois V, Berthier A, Dubois-Chevalier J, et al. The nuclear bile acid receptor FXR is a PKA- and FOXA2-sensitive activator of fasting hepatic gluconeogenesis. J Hepatol. 2018;69:1099–1109. [DOI] [PubMed] [Google Scholar]
- 79.Kain J, Wei X, Reddy NA, Price AJ, Woods C, Bochkis IM. Pioneer factor Foxa2 enables ligand-dependent activation of type II nuclear receptors FXR and LXRalpha. Mol Metab. 2021;53:101291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Fiorucci S, Biagioli M, Sepe V, Zampella A, Distrutti E. Bile acid modulators for the treatment of nonalcoholic steatohepatitis (NASH). Expert Opin Investig Drugs. 2020;29:623–32. [DOI] [PubMed] [Google Scholar]
- 81.Gillard J, Clerbaux LA, Nachit M, Sempoux C, Staels B, Bindels LB, et al. Bile acids contribute to the development of non-alcoholic steatohepatitis in mice. JHEP Rep. 2022;4:100387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Puri P, Daita K, Joyce A, Mirshahi F, Santhekadur PK, Cazanave S, et al. The presence and severity of nonalcoholic steatohepatitis is associated with specific changes in circulating bile acids. Hepatology. 2018;67:534–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Clifford BL, Sedgeman LR, Williams KJ, Morand P, Cheng A, Jarrett KE, et al. FXR activation protects against NAFLD via bile-acid-dependent reductions in lipid absorption. Cell Metab. 2021;33:1671–84. e1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bechmann LP, Kocabayoglu P, Sowa JP, Sydor S, Best J, Schlattjan M, et al. Free fatty acids repress small heterodimer partner (SHP) activation and adiponectin counteracts bile acid-induced liver injury in superobese patients with nonalcoholic steatohepatitis. Hepatology. 2013;57:1394–406. [DOI] [PubMed] [Google Scholar]
- 85.Jiao N, Baker SS, Chapa-Rodriguez A, Liu W, Nugent CA, Tsompana M, et al. Suppressed hepatic bile acid signalling despite elevated production of primary and secondary bile acids in NAFLD. Gut. 2018;67:1881–91. [DOI] [PubMed] [Google Scholar]
- 86.Jiang C, Xie C, Li F, Zhang L, Nichols RG, Krausz KW, et al. Intestinal farnesoid X receptor signaling promotes nonalcoholic fatty liver disease. J Clin Invest. 2015;125:386–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Polyzos SA, Kountouras J, Mantzoros CS. Obeticholic acid for the treatment of nonalcoholic steatohepatitis: expectations and concerns. Metabolism. 2020;104:154144. [DOI] [PubMed] [Google Scholar]
- 88.Zhang S, Wang J, Liu Q, Harnish DC. Farnesoid X receptor agonist WAY-362450 attenuates liver inflammation and fibrosis in murine model of non-alcoholic steatohepatitis. J Hepatol. 2009;51:380–8. [DOI] [PubMed] [Google Scholar]
- 89.Evans MJ, Mahaney PE, Borges-Marcucci L, Lai K, Wang S, Krueger JA, et al. A synthetic farnesoid X receptor (FXR) agonist promotes cholesterol lowering in models of dyslipidemia. Am J Physiol Gastrointest Liver Physiol. 2009;296:G543–52. [DOI] [PubMed] [Google Scholar]
- 90.Liles JT, Karnik S, Hambruch E, Kremoser C, Birkel M, Watkins WJ, et al. Fxr agonism by Gs-9674 decreases steatosis and fibrosis in a murine model of Nash. J Hepatology. 2016;64:S169. [Google Scholar]
- 91.Patel K, Harrison SA, Elkhashab M, Trotter JF, Herring R, Rojter SE, et al. Cilofexor, a nonsteroidal FXR agonist, in patients with noncirrhotic NASH: a phase 2 randomized controlled trial. Hepatology. 2020;72:58–71. [DOI] [PubMed] [Google Scholar]
- 92.Thomas AM, Hart SN, Kong B, Fang J, Zhong XB, Guo GL. Genome-wide tissue-specific farnesoid X receptor binding in mouse liver and intestine. Hepatology. 2010;51:1410–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Schmitt J, Kong B, Stieger B, Tschopp O, Schultze SM, Rau M, et al. Protective effects of farnesoid X receptor (FXR) on hepatic lipid accumulation are mediated by hepatic FXR and independent of intestinal FGF15 signal. Liver Int. 2015;35:1133–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Watanabe M, Houten SM, Wang L, Moschetta A, Mangelsdorf DJ, Heyman RA, et al. Bile acids lower triglyceride levels via a pathway involving FXR, SHP, and SREBP-1c. J Clin Invest. 2004;113:1408–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Gonzalez FJ, Jiang C, Xie C, Patterson AD. Intestinal farnesoid X receptor signaling modulates metabolic disease. Dig Dis. 2017;35:178–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Hannun YA, Obeid LM. Ceramide: an intracellular signal for apoptosis. Trends Biochem Sci. 1995;20:73–77. [DOI] [PubMed] [Google Scholar]
- 97.Jiang C, Xie C, Lv Y, Li J, Krausz KW, Shi J, et al. Intestine-selective farnesoid X receptor inhibition improves obesity-related metabolic dysfunction. Nat Commun. 2015;6:10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Xie C, Jiang C, Shi J, Gao X, Sun D, Sun L, et al. An intestinal farnesoid X receptor-ceramide signaling axis modulates hepatic gluconeogenesis in mice. Diabetes. 2017;66:613–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Fang S, Suh JM, Reilly SM, Yu E, Osborn O, Lackey D, et al. Intestinal FXR agonism promotes adipose tissue browning and reduces obesity and insulin resistance. Nat Med. 2015;21:159–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sun K, Montana V, Chellappa K, Brelivet Y, Moras D, Maeda Y, et al. Phosphorylation of a conserved serine in the deoxyribonucleic acid binding domain of nuclear receptors alters intracellular localization. Mol Endocrinol. 2007;21:1297–311. [DOI] [PubMed] [Google Scholar]
- 101.Cariello M, Piccinin E, Moschetta A. Transcriptional regulation of metabolic pathways via lipid-sensing nuclear receptors PPARs, FXR, and LXR in NASH. Cell Mol Gastroenterol Hepatol. 2021;11:1519–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Xu Y, Zhu Y, Hu S, Xu Y, Stroup D, Pan X, et al. Hepatocyte nuclear factor 4alpha prevents the steatosis-to-NASH progression by regulating p53 and bile acid signaling (in mice). Hepatology. 2021;73:2251–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Yang T, Poenisch M, Khanal R, Hu Q, Dai Z, Li R, et al. Therapeutic HNF4A mRNA attenuates liver fibrosis in a preclinical model. J Hepatol. 2021;75:1420–33. [DOI] [PubMed] [Google Scholar]
- 104.Xu Y, Hu S, Jadhav K, Zhu Y, Pan X, Bawa FC, et al. Hepatocytic activating transcription factor 3 protects against steatohepatitis via hepatocyte nuclear factor 4alpha. Diabetes. 2021;70:2506–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Softic S, Gupta MK, Wang GX, Fujisaka S, O’Neill BT, Rao TN, et al. Divergent effects of glucose and fructose on hepatic lipogenesis and insulin signaling. J Clin Invest. 2017;127:4059–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Zhao Y, Gao L, Jiang C, Chen J, Qin Z, Zhong F, et al. The transcription factor zinc fingers and homeoboxes 2 alleviates NASH by transcriptional activation of phosphatase and tensin homolog. Hepatology. 2022;75:939–54. [DOI] [PubMed] [Google Scholar]
- 107.Wesolowski SR, Kasmi KC, Jonscher KR, Friedman JE. Developmental origins of NAFLD: a womb with a clue. Nat Rev Gastroenterol Hepatol. 2017;14:81–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Magee TR, Han G, Cherian B, Khorram O, Ross MG, Desai M. Down-regulation of transcription factor peroxisome proliferator-activated receptor in programmed hepatic lipid dysregulation and inflammation in intrauterine growth-restricted offspring. Am J Obstet Gynecol. 2008;199:271. e1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Fuentes N, Silveyra P. Estrogen receptor signaling mechanisms. Adv Protein Chem Struct Biol. 2019;116:135–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Chen P, Li B, Ou-Yang L. Role of estrogen receptors in health and disease. Front Endocrinol (Lausanne). 2022;13:839005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Katzenellenbogen JA, Mayne CG, Katzenellenbogen BS, Greene GL, Chandarlapaty S. Structural underpinnings of oestrogen receptor mutations in endocrine therapy resistance. Nat Rev Cancer. 2018;18:377–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kannt A, Wohlfart P, Madsen AN, Veidal SS, Feigh M, Schmoll D. Activation of thyroid hormone receptor-beta improved disease activity and metabolism independent of body weight in a mouse model of non-alcoholic steatohepatitis and fibrosis. Br J Pharmacol. 2021;178:2412–23. [DOI] [PubMed] [Google Scholar]
- 113.Sinha RA, Bruinstroop E, Singh BK, Yen PM. Nonalcoholic fatty liver disease and hypercholesterolemia: roles of thyroid hormones, metabolites, and agonists. Thyroid. 2019;29:1173–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Harrison SA, Bashir MR, Guy CD, Zhou R, Moylan CA, Frias JP, et al. Resmetirom (MGL-3196) for the treatment of non-alcoholic steatohepatitis: a multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet. 2019;394:2012–24. [DOI] [PubMed] [Google Scholar]
- 115.Loomba R, Neutel J, Mohseni R, Bernard D, Severance R, Dao M, et al. LBP-20-VK2809, a novel liver-directed thyroid receptor beta agonist, significantly reduces liver fat with both low and high doses in patients with non-alcoholic fatty liver disease: a phase 2 randomized, placebo-controlled trial. J Hepatol. 2019;70:e150–1. [Google Scholar]
- 116.Ferrara SJ, Bourdette D, Scanlan TS. Hypothalamic-pituitary-thyroid axis perturbations in male mice by CNS-penetrating thyromimetics. Endocrinology. 2018;159:2733–40. [DOI] [PMC free article] [PubMed] [Google Scholar]