

Abstract
Background.
Fibrinogen has an established, essential role in both coagulation and inflammatory pathways, and these processes are deeply intertwined in the development of thrombotic and atherosclerotic diseases. Previous studies aimed to better understand the (patho)physiological actions of fibrinogen by characterizing the genomic contribution to circulating fibrinogen levels.
Objective.
Establish an in vitro approach to define functional roles between genes within these loci and fibrinogen synthesis.
Methods.
Candidate genes were selected by proximity to genetic variants associated with fibrinogen levels and expression in hepatocytes and HepG2 cells. HepG2 cells were transfected with siRNAs targeting candidate genes and cultured in the absence or presence of the proinflammatory cytokine interleukin-6. Effects on fibrinogen protein production, gene expression, and cell growth were assessed by immunoblotting, RT-qPCR, and cell counts, respectively.
Results.
HepG2 cells secreted fibrinogen, and stimulation with interleukin-6 increased fibrinogen production 3.4±1.2-fold. In the absence of interleukin-6, siRNA knockdown of FGA, IL6R, or EEPD1 decreased fibrinogen production, and knockdown of LEPR, PDIA5, PLEC, SHANK3, or CPS1 increased production. In the presence of interleukin-6, knockdown of FGA, IL6R, or ATXN2L decreased fibrinogen production. Knockdown of FGA, IL6R, EEPD1, LEPR, PDIA5, PLEC, or CPS1 altered transcription of one or more fibrinogen genes. Knocking down ATXN2L suppressed inducible but not basal fibrinogen production via a post-transcriptional mechanism.
Conclusions.
We established an in vitro platform to define the impact of select gene products on fibrinogen production. Genes identified in our screen may reveal cellular mechanisms that drive fibrinogen production as well as fibrin(ogen)-mediated (patho)biological mechanisms.
Keywords: Fibrinogen, hepatocyte, GWAS, interleukin-6, siRNA
INTRODUCTION
Fibrinogen is a circulating glycoprotein required for hemostasis. Following vascular injury, thrombin proteolytically converts soluble fibrinogen to insoluble fibrin, producing a provisional matrix to help staunch bleeding and support wound healing. Reduced fibrinogen or inability to produce a stable fibrin network in response to injury leads to bleeding that can be life-threatening.1, 2 Conversely, abnormal fibrin formation within vessels (thrombosis) occludes blood flow and causes tissue death. Extravascular deposition of fibrinogen or fibrin, collectively fibrin(ogen), is a powerful proinflammatory signal.3 Abnormalities in fibrin(ogen) quantity and/or quality are associated with many pathologies including hemorrhage, arterial and venous thrombosis/thromboembolism, neurocognitive disorders, obesity, cancer, and infection.2, 4–7 Increased circulating fibrinogen levels are a biomarker for some of these diseases, and studies in mice have shown causal relationships between fibrinogen and the underlying pathologies.2 Elucidating mechanisms that regulate fibrinogen production may improve understanding of how fibrinogen contributes to disease and lead to improved biomarkers and potential therapeutic targets.
Fibrinogen is encoded by 3 genes (FGA, FGB, and FGG) that arose through gene duplication and form a cluster on chromosome 4.8 The corresponding polypeptide chains Aα, Bβ, and γ are synthesized in hepatocytes, where they undergo assembly including inter- and intrachain disulfide bonding and posttranslational modification, and are secreted into circulation as a 340-kDa hexamer (AαBβγ)2. Fibrinogen expression is both constitutive, via a promoter region that binds hepatic nuclear factor-1, and inducible, via an interleukin-6 (IL-6)-responsive element that binds signal transducer and activator of transcription-3 (STAT3) upstream of the promoter.9 IL-6 engagement of the IL-6 receptor increases fibrinogen expression up to 5-fold.10 This increase in circulating fibrinogen may enhance thrombosis risk associated with inflammatory disease.
Heritability of circulating fibrinogen concentrations is estimated to be 34–46%.11, 12 Genome-wide association studies (GWAS) have associated the fibrinogen structural gene cluster and 41 other independent fibrinogen loci with circulating fibrinogen levels.11, 13–23 However, GWAS are limited in their ability to identify the causal gene(s) that underly the association at each locus. Genes within most of these loci have not been experimentally tested to determine if they regulate fibrinogen levels, so causative genes and their functional relationship with fibrinogen are not fully characterized.
Here we established an in vitro system to characterize candidate genes that may alter constitutive and/or inducible production of fibrinogen. Our screen identified several genes for which knockdown significantly changed fibrinogen production.
MATERIALS and METHODS
Identification of candidate genes.
Beginning with 41 loci previously associated with plasma fibrinogen levels11, we identified candidate genes within these loci for functional characterization. To accomplish this we used the Ensembl Variant Effect Predictor (https://useast.ensembl.org/Tools/VEP)24 to identify the gene closest to the index single nucleotide polymorphism (SNP), the most significantly associated genetic variant at the locus. Two loci that did not have a gene within 200 kilobases of the SNP (rs7012814 and rs2420915) were removed from the pipeline. For two loci with genes that were similarly close on either side of the SNP (rs2710804 and rs59104589), we prioritized both genes. The final 41 candidate genes are indicated in Fig 1.
Figure 1. Candidate gene selection.
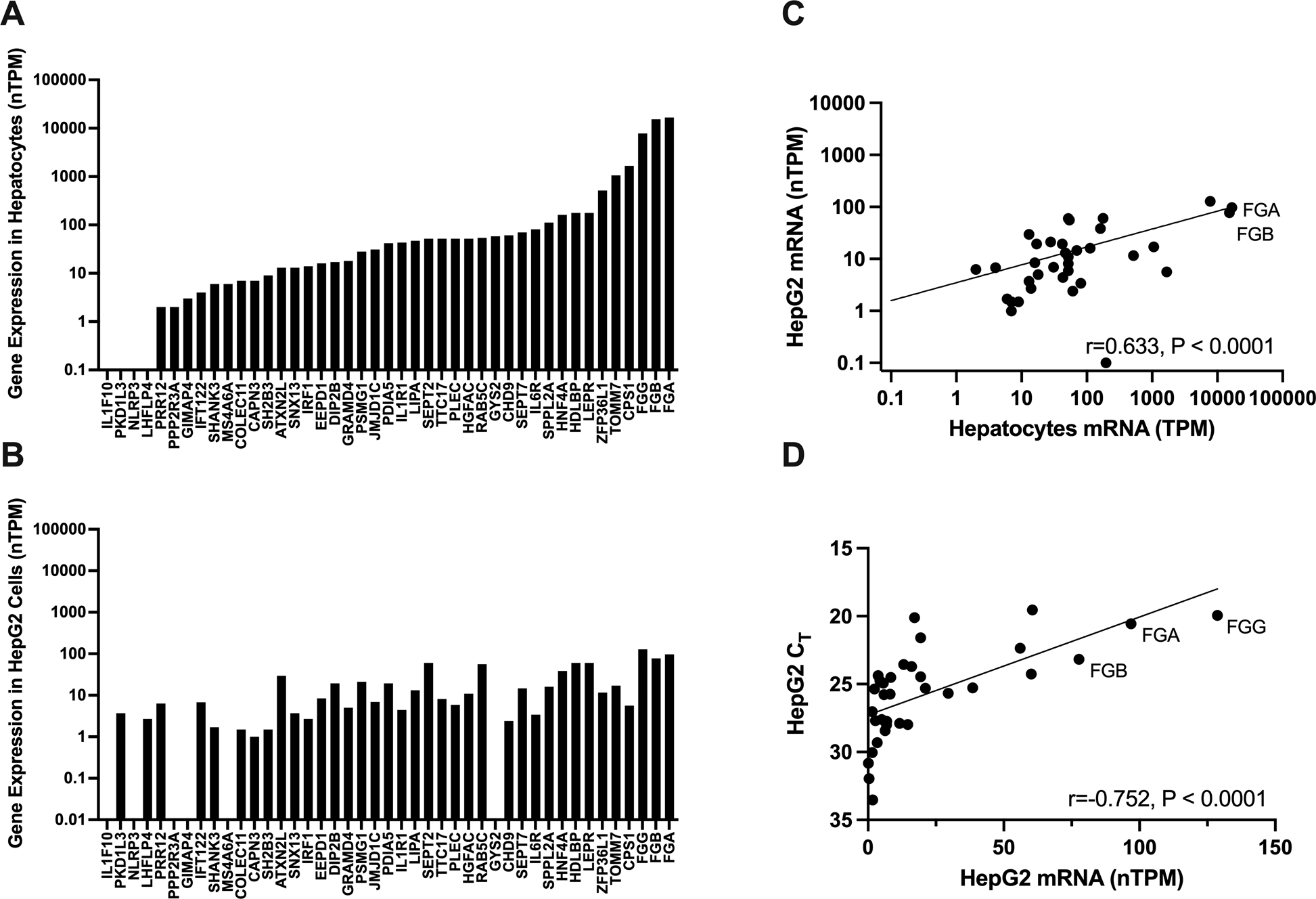
Candidate genes identified by proximity to index single nucleotide polymorphisms were filtered according to their expression in (A) normal hepatocytes and (B) HepG2 cells reported in the Human Protein Atlas. Transcripts per million were normalized within each cell type (nTPM). (C) Spearman correlation between hepatocyte and HepG2 RNA expression. (D) Spearman correlation between HepG2 RNA expression and CT values from in-house HepG2 cells; note that the y-axis is reversed to accommodate the inverse relationship between gene expression and CT value.
Cell culture and gene silencing.
Human hepatoblastoma (HepG2) cells25 (UNC Lineberger Cancer Center Tissue Culture Facility) were cultured in Minimum Essential Media (MEM, catalog #11095–080, Gibco, Waltham, MA) supplemented with 10% FBS (VWR, Radnor, PA), 0.1 mM non-essential amino acids, and 1 mM sodium pyruvate (Gibco), and passaged using 0.25% Trypsin-EDTA (catalog #25200–056, Gibco). Cells (100,000–150,000 per well) from passages 135–145 were used for transfection experiments. Small interfering RNAs (siRNAs, Silencer Select siRNAs, Thermofisher, Waltham, MA; Supplemental Table 1) were complexed with Lipofectamine RNAi MAX in OptiMEM Reduced Serum Media (RSM, catalog #31985–070, Gibco) for 15 minutes before adding to cells in a 24-well plate (32 nM final) using reverse transfection for 48 hours. The supernatant was replaced with fresh OptiMEM RSM. Fibrinogen-enriched supernatant was collected 24 hours later. For IL-6 treatment, IL-6 (catalog #10395-HNAE, Sino Biological, Houston, TX) was added to fresh OptiMEM RSM (50 ng/mL final) after the 48-hour transfection. For experiments testing STAT3 phosphorylation, cells were reverse transfected for 6 hours, and IL-6 was administered after 4 hours. Viable cells were counted using the Countess II automated cell counter and Trypan Blue (Thermofisher) per the manufacturer’s instructions.
Reverse Transcription Quantitative Polymerase Chain Reaction (RT-qPCR).
Total RNA was extracted from HepG2 cells using an RNeasy Mini Kit (Qiagen, German Town, MD) and RNA concentrations were measured (Nanodrop 2000, Thermofisher) and normalized to ≤ 1 μg. Standard de-salted primers from Integrated DNA Technologies (Newark, NJ) were diluted to 10 μM. Primers from Qiagen (Germantown, MD) were diluted and used per the manufacturer’s instructions. Primer sequences and sources are listed in Supplemental Table 2. PCR master mixes were made by combining primers, cDNA template, RNase-free water, and Qiagen Quantifast SYBR Green Master Mix. Amplification was performed using a QuantStudio 3 RT-qPCR Thermocycler (Thermofisher) with an initial PCR activation step (95°C for 5 minutes) followed by 2-step cycling (denaturation at 95°C for 10 seconds and combined annealing/extension at 60°C for 30 seconds) for 40 cycles. RNA18S and GAPDH were tested as housekeeping genes and IL-6 treatment did not alter the raw cycle threshold value of either gene (data not shown); RNA18S was selected as the housekeeping gene for subsequent experiments in the screen.
Immunoblotting.
Proteins were separated by reducing SDS-PAGE on Biorad Mini-PROTEAN TGX Stain-Free 10% gels (catalog #4568036, Biorad, Hercules, CA) and transferred to a polyvinylidene difluoride membranes. Membranes were blocked for 1 hour using LICOR Intercept Blocking Buffer (catalog #927–60001, Lincoln, NE). Membranes were then probed with rabbit polyclonal primary antibodies: anti-human fibrinogen (1:10,000, catalog #F0111, Dako, Santa Clara, CA), anti-human STAT3 (1:1000, catalog #12640, Cell Signaling Technology, Danvers, MA), anti-human phosphorylated STAT3 Tyr705 (1:1000, catalog #9145, Cell Signaling Technology), or anti-human ataxin-2-like protein (anti-ATXN2L, 1:500, catalog #ab99304, Abcam, Cambridge, MA) as indicated overnight. The membranes were then rinsed 3 times with 20 mM Tris, 150 nM NaCl containing 0.1% Tween (TBS-T) and incubated with goat anti-rabbit secondary antibody (1:10,000, catalog #925–32211, LICOR IRDye 800 CW) for 1 hour. The membranes were rinsed in TBS-T, imaged on a Biorad Chemi-Doc MP Imager, and analyzed to quantify fibrinogen chains. Total protein was measured using Biorad Stain Free Technology as a loading control.26 Unstimulated and stimulated (100 ng/mL IFN-alpha) Hela cell control lysates (catalog #9133, Cell Signaling Technology) were loaded at 10 μL per lane. Phosphorylated STAT3 blots were stripped using NewBlot IR Stripping Buffer (catalog #928–40028, LICOR).
Statistical methods.
Descriptive statistics were summarized using means and standard error of the means. The effects of siRNA treatments on proteins, transcripts, and cells counts were normalized to control siRNA (siNC) treatment within the same experiment. Statistical significance and correlations were tested using Graphpad Prism (version 9.4.0). Treatment groups were compared using a one-way ANOVA with Dunnett’s post-hoc test on normalized log10-transformed (protein) or normalized (mRNA) values to control the family-wise type-I error rate under the significance level of 0.05.
RESULTS
Selection of candidate genes for the functional screen.
Forty-one candidate genes from loci associated with plasma fibrinogen levels11 were prioritized for screening, as described in the Methods. We first characterized expression of these 41 candidate genes in normal hepatocytes using publicly-available RNAseq data in the Human Protein Atlas (Fig 1A). Four genes reported as not expressed in normal hepatocytes (IL1F10, PKD1L3, NLRP3, LHFLP4, <0.1 transcript per million) were excluded from the screen. We then selected HepG2 cells as a model of hepatocyte gene expression because previous studies have shown HepG2 cells are an easily-transfected hepatocyte-derived cell line that exhibits both constitutive and inducible fibrinogen expression.27–30 We used the Human Protein Atlas to characterize expression of the 37 hepatocyte-expressed genes in HepG2 cells and excluded 4 genes reported as not expressed in HepG2 cells (PPP2R3A, GIMAP4, MS4A6A, GYS2, <0.1 transcript per million, Fig 1B). Expression of the remaining 33 gene candidates correlated significantly in hepatocytes and HepG2 cells with few exceptions (Fig 1C). After excluding the gene encoding the fibrinogen Bβ chain FGB, 32 candidate genes were selected for screening. Correlation of HepG2 gene expression data from the Human Protein Atlas and our HepG2 cells confirmed that expression of these candidate genes was consistent with that expected (Fig 1D).
HepG2 cells are a model of constitutive and inducible fibrinogen expression.
To develop the experimental system, we first characterized fibrinogen production in HepG2 cells. As expected, HepG2 cells secreted fibrinogen into their media, modeling constitutive fibrinogen production (Fig 2A–B). Treatment with IL-6 produced a dose-dependent increase in fibrinogen production, modeling inducible expression (Supplemental Fig 1 and Fig 2C); 50 ng/mL IL-6 increased individual fibrinogen chains (Supplemental Fig 1 and Fig 2C) and transcripts (Fig 2D) 3–7-fold, and this concentration was used for subsequent experiments.
Figure 2. HepG2 cells are an experimental model of fibrinogen expression.
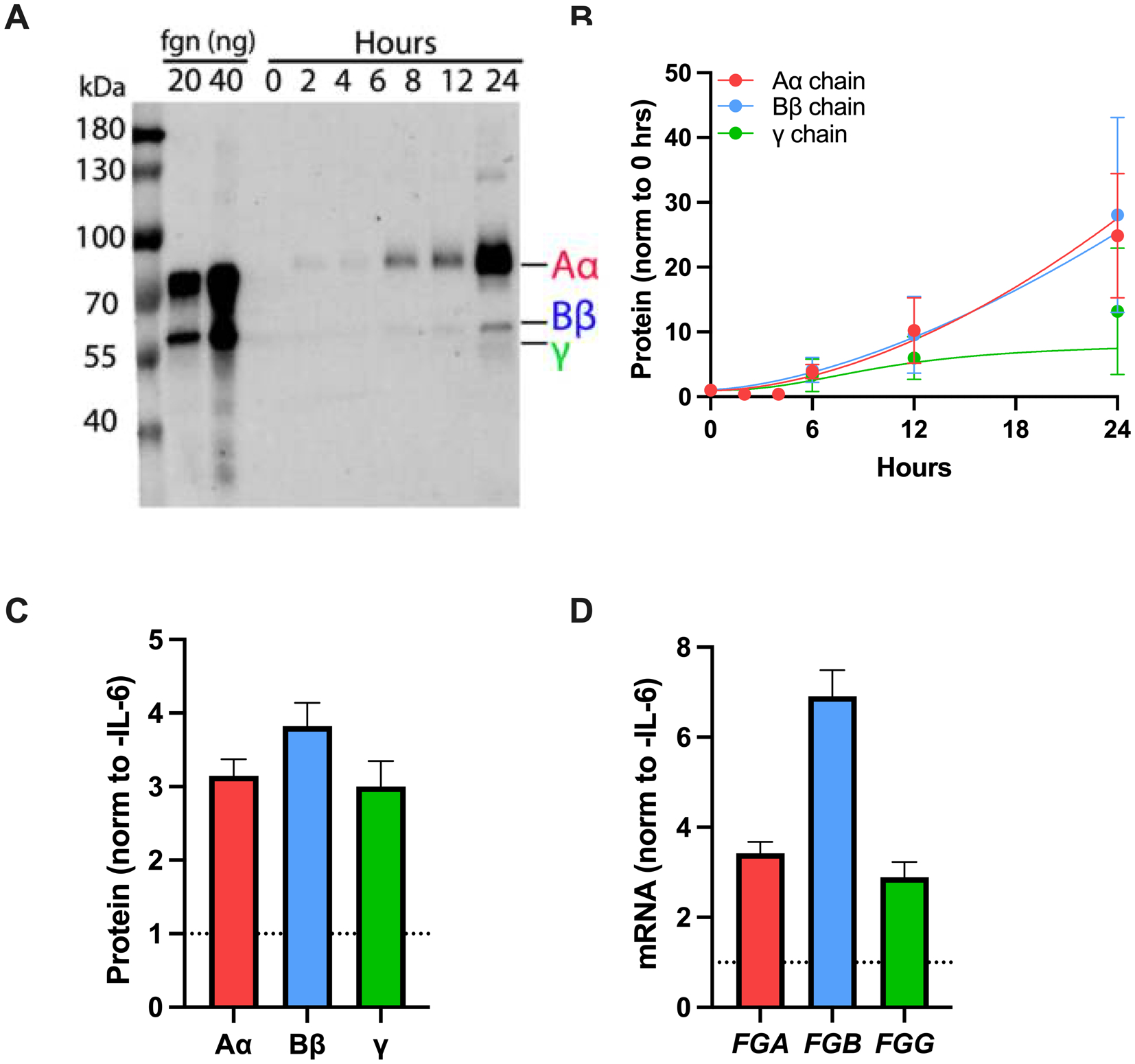
(A) Media was collected from HepG2 cells cultured in the absence or presence of IL-6 for 24 hours. Proteins were separated by reducing SDS-PAGE, transferred to polyvinylidene difluoride membrane, and probed with polyclonal rabbit anti-human fibrinogen and IR-DYE 800CW goat anti-rabbit antibodies. (B) Fibrinogen in the media was quantified by densitometry; symbols indicate mean ± standard error of the mean. (C-D) Effect of IL-6 (50 ng/mL) on fibrinogen (C) protein and (D) mRNA (N=4–6, bars indicate mean + standard error of the mean).
We then developed a workflow in which we treated HepG2 cells with siRNAs targeting the selected candidate genes for 48 hours, replaced media with fresh reduced-serum media in the absence or presence of IL-6, and collected enriched media and cells for analysis by western blot, cell count, and gene expression after 24 hours (Fig 3A). In the absence of IL-6, siRNAs targeting FGA, FGB, or FGG reduced expression of the target gene >90% (Fig 3B) and reduced fibrinogen protein in the enriched media 25–90% (Fig 3C) without altering cell count (Fig 3D). Similarly, in the presence of IL-6, siRNAs targeting FGA, FGB, or FGG reduced expression of the target gene >90% (Fig 3E) and reduced fibrinogen protein in the enriched media 40–90% (Fig 3F) without altering cell count (Fig 3G). In each case, knockdown of an individual gene reduced detection of all three chains in the enriched media, consistent with findings that significant knockdowns of any of the fibrinogen structural genes can limit fibrinogen synthesis.31–33 In all blots, immunodetection of the Aα chain exceeded that of the Bβ or γ chains. However, detection of the Aα chain correlated with both the Bβ and γ chains in both the absence and presence of IL-6 (P<0.0001, Supplemental Figure 2A–B, respectively). Thus, we used FGA knockdown as a control and the Aα chain as a proxy for total fibrinogen protein in subsequent experiments.
Figure 3. Fibrinogen synthesis is decreased by siRNAs targeting the fibrinogen structural genes.
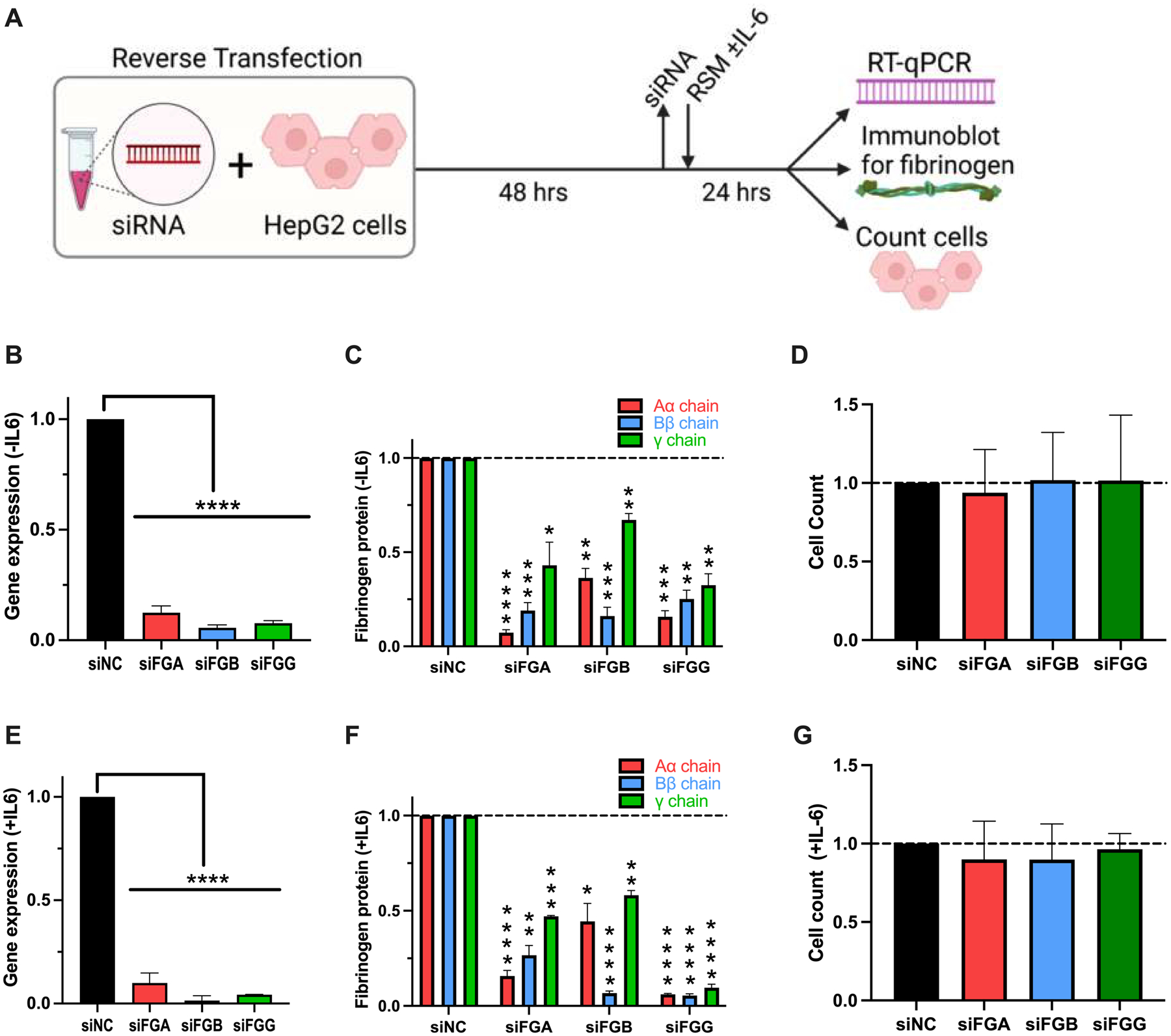
(A) HepG2 cells were treated with siRNAs for 48 hours and then incubated in reduced serum media (RSM) in the (B-D) absence or (E-G) presence of IL-6 for 24 hours. Effects on fibrinogen (B, E) gene expression, (C, F) protein production, and (D, G) cell viability were assessed by immunoblotting, RT-qPCR, and cell counts, respectively. All data were normalized to treatment with control siRNA (siNC), N=3–12 for each siRNA, bars indicate mean + standard error of the mean, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001
Several gene candidates alter constitutive fibrinogen production.
To determine effects of the 32 candidate genes on fibrinogen production, we used siRNA to knock down each gene individually. RT-qPCR confirmed that most siRNAs decreased candidate gene expression >60% (Fig 4A); siRNAs that failed to knock down the candidate gene (CAPN3, HNF4A, TOMM7, HGFAC, Supplemental Figure 3) were removed from the screen, leaving 28 screened genes. Fibrinogen detected in the media significantly decreased with knockdown of IL6R or EEPD1 and increased following knockdown of LEPR, PDIA5, PLEC, SHANK3, or CPS1 (Fig 4B). For genes that significantly altered fibrinogen protein levels, we counted cells to assess effects on cell growth. Cell counts were significantly increased after knockdown of SHANK3 (Fig 4C), suggesting the increase in fibrinogen following siSHANK treatment was due to an increased number of cells rather than a specific effect on fibrinogen synthesis. For the 6 genes that significantly altered fibrinogen protein levels but did not change cell counts (IL6R, EEPD1, LEPR, PDIA5, PLEC, and CPS1), we used RT-qPCR to quantify fibrinogen gene transcripts. Knockdown of each of these genes significantly altered the expression of one or more of the fibrinogen structural genes in directions consistent with the amount of protein detected (Fig 4D–F).
Figure 4. Knockdown of IL6R, EEPD1, LEPR, PDIA5, PLEC, or CPS1 alters constitutive fibrinogen synthesis without changing cell counts.
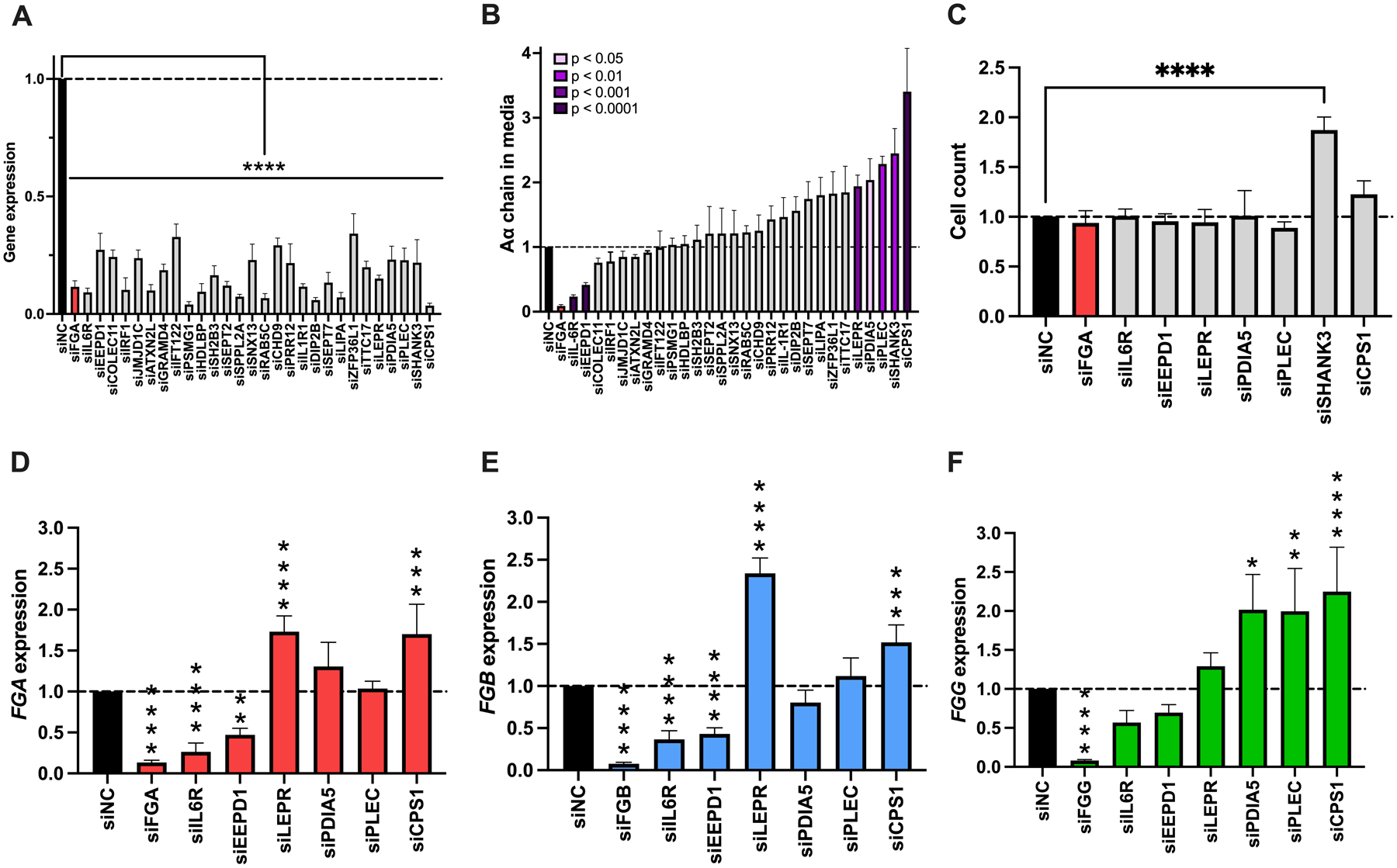
HepG2 cells were transfected with siRNAs against candidate genes in the absence of IL-6, and supernatant and cells were analyzed as in Fig 3. (A) Target gene knockdown. (B) Aα chain in the media. (C) Cell count following treatment with siRNAs that altered fibrinogen synthesis in panel B. (D) FGA, (E) FGB, and (F) FGG expression for treatments that did not alter cell count in panel C. All data were normalized to treatment with control siRNA (siNC), N=4–12 for each siRNA, bars indicate mean + standard error of the mean, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001
Several gene candidates alter inducible fibrinogen production.
To characterize effects of the 28 candidate genes on inducible fibrinogen expression, we used siRNA to knock down each gene in cells stimulated with IL-6 (Fig 5A). Compared to changes seen in the absence of IL-6, fewer siRNAs significantly altered fibrinogen production in the presence of IL-6; however, trends in effects were similar in both the absence and presence of IL-6 (r=0.655, P<0.001, Supplemental Figure 4). Exceptions included knockdowns of ATXNL2, which significantly decreased fibrinogen more in the presence of IL-6 than in the absence of IL-6, and DIP2B and PLEC, which slightly increased fibrinogen more in the presence of IL-6 than in the absence of IL-6 (Supplemental Fig 4). Knockdown of IL6R and ATXN2L significantly decreased fibrinogen production (Fig 5B) without altering cell counts (Fig 5C). Knockdown of IL6R significantly reduced FGA, FGB, and FGG expression. Interestingly, although knockdown of ATXN2L decreased inducible fibrinogen protein production by 80%, this treatment did not decrease FGA, FGB, or FGG gene expression (Fig 5D–F).
Figure 5. Knockdown of IL6R or ATXN2L alters inducible fibrinogen synthesis.

HepG2 cells were transfected with siRNAs against candidate genes in the presence of IL-6, and supernatant and cells were analyzed as in Fig 3. (A) Target gene knockdown. (B) Aα chain in the media. (C) Cell count following treatment with siRNAs that altered fibrinogen synthesis in panel B. (D) FGA, (E) FGB, and (F) FGG expression. All data were normalized to treatment with control siRNA (siNC), N=4–12 for each siRNA, bars indicate mean + standard error of the mean, **P < 0.01, ***P < 0.001, ****P < 0.0001
Knockdown of LEPR does not alter fibrinogen transcription via STAT3 phosphorylation.
To identify potential mechanisms mediating constitutive fibrinogen production, we selected LEPR further study. LEPR encodes the leptin receptor, which regulates adipose tissue mass. Since the leptin receptor signals through STAT334 and since fibrinogen transcription is induced via STAT3 phosphorylation, we hypothesized that leptin receptor knockdown upregulated fibrinogen expression by increasing STAT3 phosphorylation. To test this hypothesis, we measured total and phosphorylated (Tyr705) STAT3 protein after treatment with siLEPR, in the absence and presence of IL-6. As expected, there was no difference in total STAT3 protein with any siRNA treatment in the absence or presence of IL-6 (Fig 6A). Moreover, in the absence of IL-6, cells treated with control siRNA (siNC) or siIL6R did not show STAT3 phosphorylation. In the presence of IL-6, siNC-treated cells showed phosphorylated STAT3 protein, whereas siIL6R-treated cells had substantially reduced STAT3 phosphorylation. Notably, although siLEPR increased fibrinogen transcription in the absence of IL-6 (Fig 4), siLEPR-treated cells did not show STAT3 phosphorylation in the absence of IL-6 (Fig 6A–B). These data suggested leptin receptor-mediated expression of fibrinogen was independent of the canonical STAT3-induced acute phase response.
Figure 6. Knockdown of LEPR does not alter fibrinogen transcription via STAT3 phosphorylation.
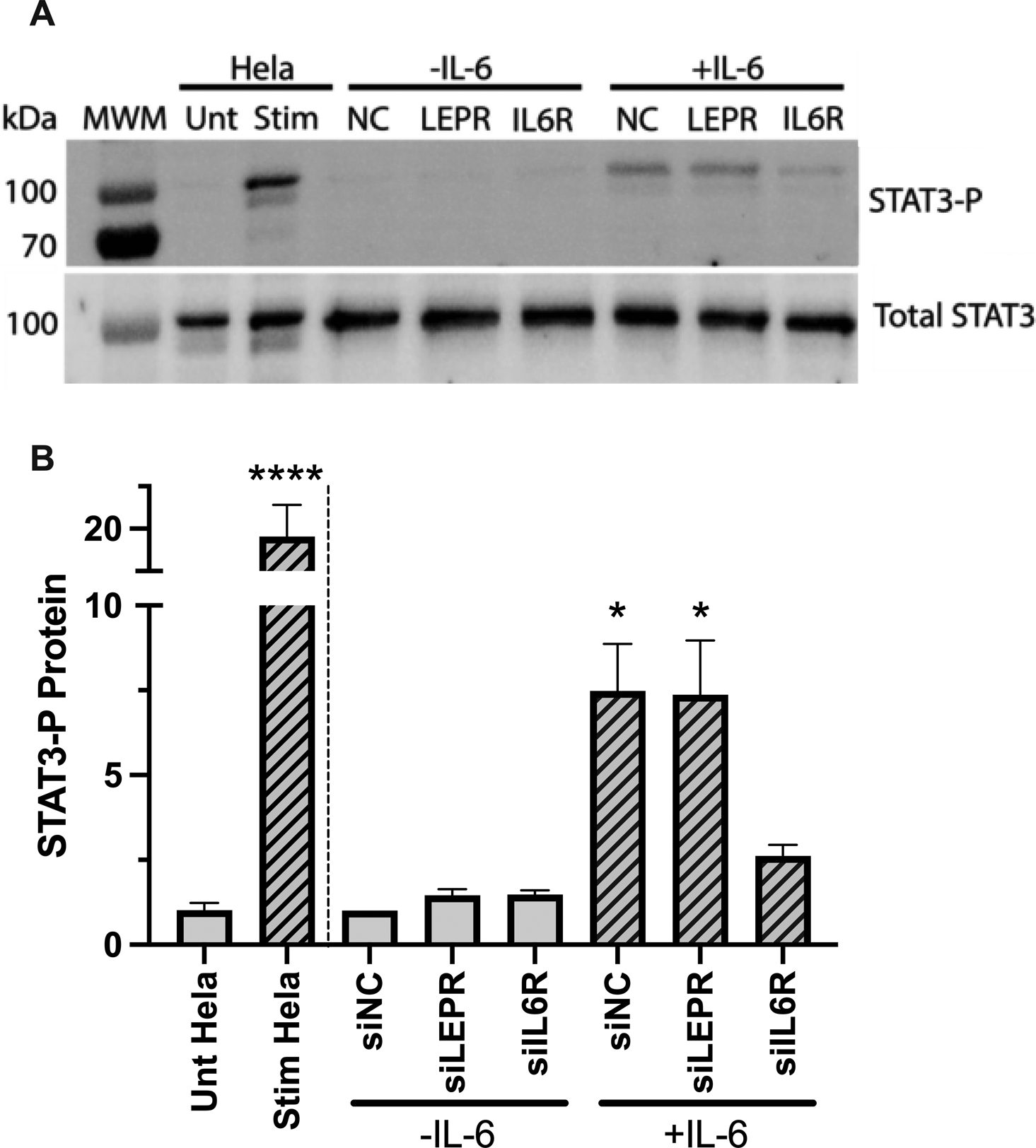
HepG2 cells were transfected with siRNAs against LEPR and IL6R in the absence and presence of IL-6. STAT3 protein (phosphorylated and total) was assessed by immunoblot and quantified by densitometry. N=4, bars indicate mean+standard error of the mean; *P < 0.05, ****P < 0.0001
Loss of ATXN2L increases fibrinogen protein in HepG2 cell lysates.
Finally, we examined ATXN2L, for which the knockdown suppressed inducible but not basal fibrinogen production. ATXN2L encodes ATXN2L, which is implicated in polyribosome assembly and stress granule formation.35–37 We first confirmed that ATXN2L was significantly reduced after siATXN2L treatment (Fig 7A). Since ATXN2L knockdown decreased fibrinogen detected in the supernatant of IL-6-treated cells 90% (Fig 5B) without reducing fibrinogen transcripts (Fig 5D–F), we then lysed siATXN2L-treated cells and probed the lysates for fibrinogen via immunoblot to determine whether fibrinogen was translated but not released from the cells. Control experiments showed that knockdown of FGA reduced Aα chain >90%, and knockdown of FGB or FGG reduced all fibrinogen chains by 40–90% in the cell lysates. Interestingly, knockdown of ATXN2L increased all fibrinogen chains in the cell lysates 1.15–2-fold (Fig 7B), suggesting that fibrinogen chains are translated, and the loss of fibrinogen production seen following ATXN2L knockdown stems from a failure of cells to assemble and/or export hexameric fibrinogen.
Figure 7. Loss of ATXN2L increases fibrinogen protein in HepG2 cell lysates.
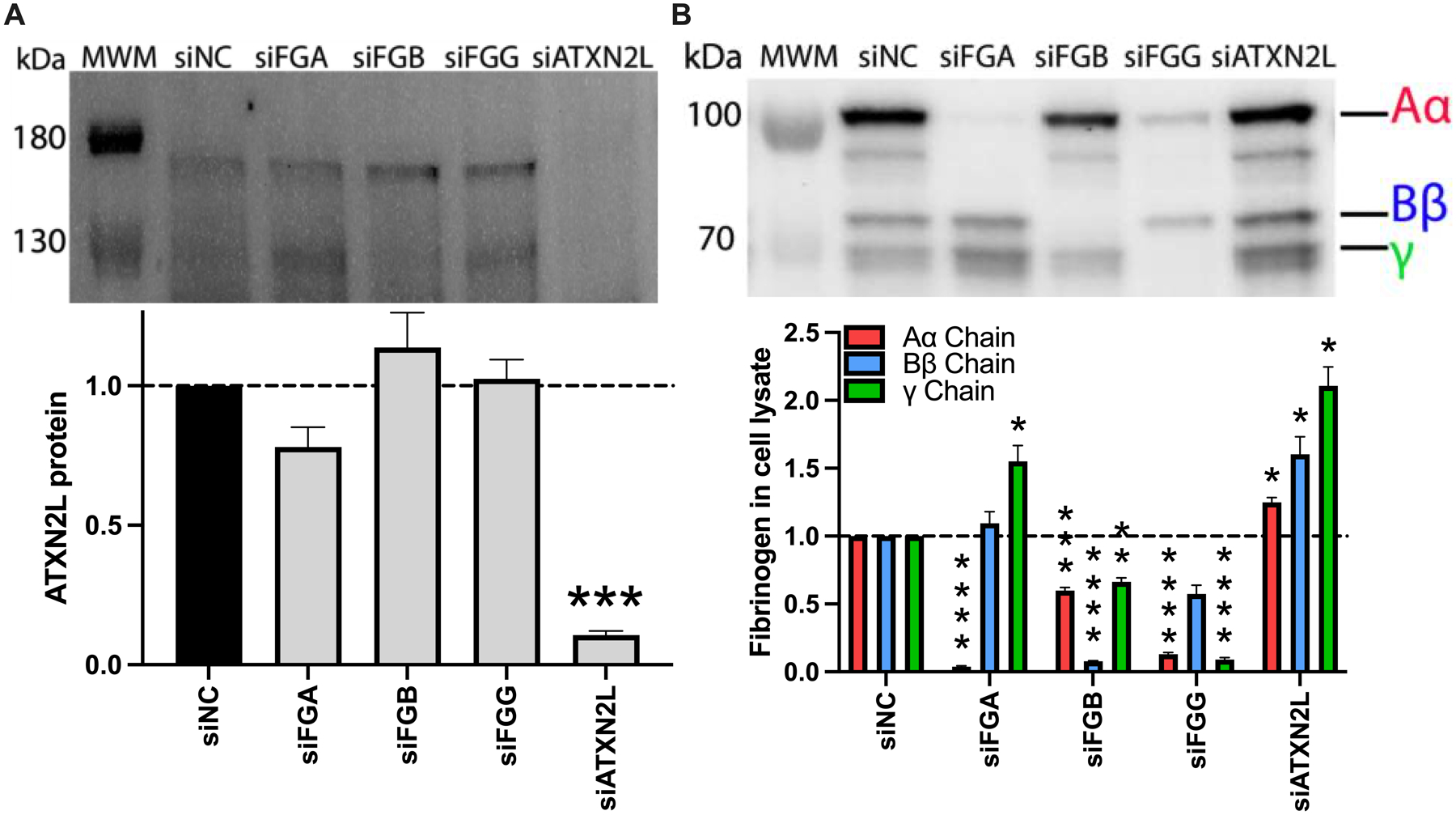
HepG2 cells were transfected with siRNAs against FGA, FGB, FGG, or ATXN2L in the presence of IL-6. (A) ATXN2L protein was visualized by immunoblot and quantified by densitometry (upper band). (B) Fibrinogen was visualized in cell lysates by immunoblot and quantified by densitometry. All data were normalized to treatment with control siRNA (siNC), N=3–4 for each condition, bars indicate mean+standard error of the mean; *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001
DISCUSSION
Defining the mechanisms regulating fibrinogen levels in plasma is critical, because fibrinogen is both a biomarker of, and causative agent in, hemorrhagic, thrombotic, and inflammatory diseases.1–7 Although canonical pathways (e.g., hepatic nuclear factor-1 and STAT3) controlling fibrinogen expression have been characterized, our understanding of the genetic regulation of fibrinogen remains incomplete. Here, we established a straight-forward in vitro platform in which the impact of select gene products on fibrinogen expression can be defined. Using this system, we leveraged discoveries from population-based association studies to identify novel genetic regulators. Genes identified in our screen participate in functional pathways that may reveal not only cellular mechanisms that drive fibrinogen production, but also downstream (patho)biological mechanisms mediated by fibrin(ogen).
A major strength of our experimental system is that it can be used to interrogate genes that regulate fibrinogen production in basal and/or inflammatory settings. Our observation that IL-6 receptor (IL6R) knockdown decreased fibrinogen expression in IL-6-treated cells is consistent with clinical observations that the IL-6 receptor-blocking antibody tocilizumab reduces fibrinogen38, and provides important validation of the system and operant mechanisms. Finding that IL6R knockdown also reduced fibrinogen production even in the absence of added IL-6 reveals exquisite sensitivity of this pathway to even trace levels of IL-6 carried within the serum. Thus, since IL-6 can be detected at low levels even in healthy people39, IL-6/IL-6 receptor engagement may contribute to circulating fibrinogen levels even in the absence of overt inflammation.
Our screen also identified genes for which the encoded protein participates in metabolic pathways. These include LEPR and CPS1, for which knockdown increased constitutive fibrinogen production. These findings are particularly interesting given the suggested role of fibrin(ogen) in metabolic syndrome and fatty liver disease.2, 5 The leptin receptor (encoded by LEPR) is a primary driver of genetic obesity.40 LEPR knockout mice (db/db) are obese and have markedly elevated fibrinogen.40, 41 Although hyperfibrinogenemia in these mice has been attributed to obesity-induced inflammation, our data suggest this change could stem from the genetic alteration, itself. Interestingly, since neither we nor others have detected leptin or the gene encoding leptin (LEP) in HepG2 cells (data not shown and42), and increased fibrinogen following LEPR knockdown is not associated with STAT3 phosphorylation, these data suggest an independent mechanism drives leptin receptor-dependent induction of fibrinogen. CPS1 (carbamoyl-phosphate synthase 1, encoded by CPS1) is constitutively secreted by liver into bile.43–45 During acute liver injury, CPS1 is released by hepatocytes directly into the blood, facilitating macrophage recruitment to the liver and protecting against acute liver injury.46 Interestingly, fibrinogen drives repair after acute liver injury from acetaminophen overdose by facilitating leukocyte recruitment.47 Thus, changes in CPS1 may alter fibrinogen synthesis to tune this process. The connection between each of these gene products and fibrinogen expression represents key examples that may link fibrinogen to the pathogenesis of liver disease.
Two genes identified in our screen encode proteins with cytoskeletal functions, including PLEC and SEPT7 for which knockdown increased fibrinogen production significantly or non-significantly, respectively, in the absence and/or presence of IL-6. Plectin (encoded by PLEC) connects filament proteins within the cytoskeleton.48 Septin 7 (encoded by SEPT7) is a cytoskeletal filament GTPase required for normal organization of the actin cytoskeleton. Knockdown of a third gene, SHANK3, which encodes a scaffold protein that connects membrane proteins with the actin cytoskeleton, also increased fibrinogen but did so in concert with increased cell count. Although cytoskeletal changes have not previously been linked to fibrinogen production, this association may be relevant in specific settings. For example, liver injury or surgical liver resection each induce hepatic fibrinogen expression, wherein changes in cytoskeletal proteins may provide a signal to upregulate fibrinogen that is required for liver repair.3, 49 SEPT7 was identified from the same SNP as EEPD1 (rs2710804), so we did not expect both of these knockdowns to alter fibrinogen production, or to do this in opposite directions. Localization of the index SNP that identified this locus in a non-coding region between SEPT7 and EEPD1 may imply a biological function for this untranslated sequence.
The protein disulfide isomerase family A member 5 (PDIA5) encoded by PDIA5 is a member of a family of 21 different thiol isomerases that catalyze protein folding and thiol-disulfide exchanges. Several protein disulfide isomerase (PDI) family members have established roles in blood coagulation and platelet function, with both intracellular and extracellular substrates.50 Although PDIA5 has not been specifically implicated in fibrinogen synthesis, PDI, the head of the PDI superfamily, can bind to and oxidize both fibrinogen and fibrin.51 Thus, PDIA5 may also modify fibrin(ogen). However, since knockdown of PDIA5 increased fibrinogen transcripts, it seems more likely that PDIA5 modifies an intracellular protein within the fibrinogen synthetic pathway that negatively regulates fibrinogen transcription. Indeed, PDI can also regulate the activity of several transcription factors, including nuclear factor-kappa B and hypoxia-inducible factor-1α.52 Thus, the relationship between PDIA5 and fibrinogen transcription may involve compensatory changes in other transcription factors that mediate fibrinogen production.
ATXN2L, encoded by ATXN2L, was unique in our findings as the only gene that significantly altered fibrinogen production without changing fibrinogen transcript levels, and did so only in the presence of IL-6. This change in fibrinogen production was accompanied by a subtle but significant increase in intracellular fibrinogen, suggesting the fibrinogen chains can undergo translation, but are not properly assembled and/or exported. Both ataxin 2 and ATXN2L are cytoplasmic proteins with intracellular functions including polyribosome assembly and stress granule and processing body formation.35–37 Interestingly, loss-of-function mutations in ataxin 2 are associated with susceptibility to type I diabetes, obesity, and hypertension in humans, and obesity, dyslipidemia, and insulin resistance in mice.53–56 Whether ATXN2L also mediates these pathologies is an area of future study.
The design of our siRNA-based screen had limitations. First, selection of the candidate genes was based primarily on proximity to the index variant and did not capture genes more than 200 kilobases upstream or downstream from the variant. For some genetic loci, no genes were tested. Second, although cells were transfected with siRNAs for 48 hours, this time period may not have been sufficient to allow certain intracellular proteins with long half-lives to degrade. Unfortunately, information on the half-lives of most of the proteins encoded by our gene candidates are unknown, so we were not able to account for this in our model. Thus, the lack of an effect of certain gene knockdowns on fibrinogen production should be interpreted cautiously. Third, given the number of genes tested, we did not test multiple siRNA for each gene candidate or complement our experiments with strategies to over-express candidate genes. These experiments should be performed in future studies to characterize the dynamic relationships between these genes and fibrinogen production. Fourth, receptivity of HepG2 cells to siRNA transfection made them ideal for an siRNA-based screen; however, replication in primary hepatocytes and in vivo will be important in future studies to characterize the operant mechanisms.27 Finally, our experiments were not designed to identify genes that modify circulating fibrinogen levels through other mechanisms (e.g., altered clearance or paracrine interactions wherein a distally-expressed intermediate protein alters synthetic mechanisms in hepatocytes). Alternate strategies will be required to interrogate non-synthetic effects of genes within candidate loci on circulating fibrinogen levels.
In addition to the new biology illuminated by our study, our findings have potential implications for disease treatment. The established role of fibrin(ogen) as a driver of many pathologies has given rise to the premise that lowering fibrinogen levels may result in reduced disease risk. Consequently, strategies to therapeutically lower fibrinogen levels have been tested in several settings. siRNAs directed against the fibrinogen structural genes reduce constitutive fibrinogen expression by 60–95% and protect against thrombosis.32, 33 In settings in which fibrinogen levels are abnormal from aberrant expression of proteins that regulate fibrinogen synthesis, targeting the causative gene directly could normalize fibrinogen expression. For example, since ATXN2L knockdown specifically decreases inducible fibrinogen production, ATXN2L may be a potential target to alleviate hyperfibrinogenemia and reduce thrombosis in inflammatory settings.
In summary, we established a cell culture system using hepatocyte-derived cells in which genetic regulators of fibrinogen gene induction and protein synthesis can be identified under basal and inflammatory (IL-6-stimulated) conditions. Using this system, we screened 28 genes and identified 7 novel potential genetic regulators of fibrinogen synthesis. Our results provide new insights into the heritability of fibrinogen levels and suggest potential genetic targets for therapeutic modulation of fibrinogen levels. Future studies investigating mechanisms mediating the interaction of these genes with fibrinogen may reveal new (patho)physiologic pathways that influence circulating fibrinogen concentration. Moreover, the successful implementation of our in vitro siRNA screening system presents a method of functional validation for additional fibrinogen-associated loci.
Supplementary Material
ESSENTIALS.
Genome-wide association studies associate loci with circulating fibrinogen but causative genes are undefined
Candidate genes were screened using siRNA knockdown in HepG2 cells in the absence/presence of IL-6
Knockdown of IL6R, EEPD1, LEPR, PDIA5, PLEC, and CPS1 altered fibrinogen transcription
Knockdown of ATXN2L decreased fibrinogen production without changing fibrinogen gene transcripts
ACKNOWLEDGEMENTS
The authors thank Dominic Alba for technical support, the UNC Bioinformatics and Analytics Research Collaborative for assistance with Ensembl, and the CHARGE investigators for thoughtful suggestions.
FUNDING
This study was supported by funding from the National Institutes of Health (HL141291 to ASW/ACM, HL126974 to ASW, HL134894 to NLS, and DK122813 to JPL).
Footnotes
SUPPORTING INFORMATION
Supplemental data can be found at the journal website.
CONFLICT OF INTEREST DISCLOSURE
The authors have no competing financial interests to declare.
References
- 1.Casini A, von Mackensen S, Santoro C, Djambas Khayat C, Belhani M, Ross C, Dorgalaleh A, Naz A, Ünal E, Abdelwahab M, Lozeron ED, Trillot N, Susen S, Peyvandi F, de Moerloose P. Clinical phenotype, fibrinogen supplementation, and health-related quality of life in patients with afibrinogenemia. Blood. 2021;137(22):3127–36. [DOI] [PubMed] [Google Scholar]
- 2.Vilar R, J FR, Casini A, Neerman-Arbez M. Fibrin(ogen) in human disease: both friend and foe. Haematologica 2020;105(2):284–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Luyendyk JP, Schoenecker JG, Flick MJ. The multifaceted role of fibrinogen in tissue injury and inflammation. Blood. 2019;133(6):511–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wolberg AS, Campbell RA, Bouck EG, Denorme F, Holle LA, Middelton EA, Blair AM, De Laat B, Schiffman JD, Yost CC, Rondina MT. Arteriosclerosis, Thrombosis, and Vascular Biology COVID-19 and Sepsis Are Associated With Different Abnormalities in Plasma Procoagulant and Fibrinolytic Activity. Arterioscler Thromb Vasc Biol. 2021;41:401–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kopec A, Abrahams S, Thornton S, Palumbo J, Mullins E, Divanovic S, Weiler H, Owens A, Mackman N, Goss A, van Ryn J, Luyendyk J, Flick M. Thrombin promotes diet-induced obesity through fibrin-driven inflammation. The Journal of clinical investigation. 2017;127(8):3152–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Palumbo JS, Kombrinck KW, Drew AF, Grimes TS, Kiser JH, Degen JL, Bugge TH. Fibrinogen is an important determinant of the metastatic potential of circulating tumor cells. Blood. 2000;96(10):3302–9. [PubMed] [Google Scholar]
- 7.Adams GN, Rosenfeldt L, Frederick M, Miller W, Waltz D, Kombrinck K, McElhinney KE, Flick MJ, Monia BP, Revenko AS, Palumbo JS. Colon Cancer Growth and Dissemination Relies upon Thrombin, Stromal PAR-1, and Fibrinogen. Cancer Research. 2015;75(19):4235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pieters M, Wolberg AS. Fibrinogen and fibrin: An illustrated review. Res Pract Thromb Haemost. 2019;3(2):161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fish RJ, Neerman-Arbez M. Fibrinogen gene regulation. Throm Haemost. 2012;108:419–26. [DOI] [PubMed] [Google Scholar]
- 10.Vasse M, Paysant J, Soria J, Ollet JC, Annier JV, Soria C. Regulation of fibrinogen biosynthesis by cytokines, consequences on the vascular risk. Haemostasis. 1996;26(4):33–339. [DOI] [PubMed] [Google Scholar]
- 11.de Vries PS, Chasman DI, Sabater-Lleal M, Chen M-H, Huffman JE, Steri M, Tang W, Teumer A, Marioni RE, Grossmann V, Hottenga JJ, Trompet S, Müller-Nurasyid M, Zhao JH, Brody JA, Kleber ME, Guo X, Wang JJ, Auer PL, Attia JR, et al. A meta-analysis of 120,246 individuals identifies 18 new loci for fibrinogen concentration. Human Molecular Genetics. 2016;25(2):358–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.De Lange M, Snieder H, Ariëns RAS, Spector TD, Grant PJ. The genetics of haemostasis: a twin study. Lancet. 2001;357(9250):101–5. [DOI] [PubMed] [Google Scholar]
- 13.Cronjé HT, Nienaber-Rousseau C, Zandberg L, Chikowore T, de Lange Z, van Zyl T, Pieters M. Candidate gene analysis of the fibrinogen phenotype reveals the importance of polygenic co-regulation. Matrix Biol. 2017;60–61:16–26. [DOI] [PubMed] [Google Scholar]
- 14.Souto JC, Almasy L, Borrell M, Garí M, Martínez E, Mateo J, Stone WH, Blangero J, Fontcuberta J. Genetic determinants of hemostasis phenotypes in Spanish families. Throm Res. 2000;101(13):1546–51. [DOI] [PubMed] [Google Scholar]
- 15.Lovely RS, Yang Q, Massaro JM, Wang J, D’Agostino RB, O’Donnell CJ, Shannon J, Farrell DH. Assessment of genetic determinants of the association of γ’ fibrinogen in relation to cardiovascular disease. Arterioscler Thromb Vasc Biol. 2011;31(10):2345–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dehghan A, Yang Q, Peters A, Basu S, Bis JC, Rudnicka AR, Kavousi M, Chen MH, Baumert J, Lowe GDO, McKnight B, Tang W, De Maat M, Larson MG, Eyhermendy S, McArdle WL, Lumley T, Pankow JS, Hofman A, Massaro JM, et al. Association of novel genetic Loci with circulating fibrinogen levels: a genome-wide association study in 6 population-based cohorts. Circ Cardiovasc Genet. 2009;2(2):125–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jacquemin B, Antoniades C, Nyberg F, Plana E, Müller M, Greven S, Salomaa V, Sunyer J, Bellander T, Chalamandaris AG, Pistelli R, Koenig W, Peters A. Common genetic polymorphisms and haplotypes of fibrinogen alpha, beta, and gamma chains affect fibrinogen levels and the response to proinflammatory stimulation in myocardial infarction survivors: the AIRGENE study. J Am Coll Cardiol. 2008;52(11):941–52. [DOI] [PubMed] [Google Scholar]
- 18.De Maat MPM. Effects of diet, drugs, and genes on plasma fibrinogen levels. Ann N Y Acad Sci. 2001;936:509–21. [DOI] [PubMed] [Google Scholar]
- 19.Hamsten A, De Faire U, Iselius L, Blombäck M. Genetic and cultural inheritance of plasma fibrinogen concentration. Lancet. 1987;2(8566):988–91. [DOI] [PubMed] [Google Scholar]
- 20.Yang Q, Tofler GH, Cupples LA, Larson MG, Feng DL, Lindpaintner K, Levy D, D’Agostino RB, O’Donnell CJ. A genome-wide search for genes affecting circulating fibrinogen levels in the Framingham Heart Study. Thromb Res. 2003;110(1):57–64. [DOI] [PubMed] [Google Scholar]
- 21.Danik JS, Paré G, Chasman DI, Zee RYL, Kwiatkowski DJ, Parker A, Miletich JP, Ridker PM. Novel loci, including those related to Crohn disease, psoriasis, and inflammation, identified in a genome-wide association study of fibrinogen in 17 686 women: the Women’s Genome Health Study. Circ Cardiovasc Genet. 2009;2(2):134–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kathiresan S, Yang Q, Larson MG, Camargo AL, Tofler GH, Hirschhorn JN, Gabriel SB, O’Donnell CJ. Common genetic variation in five thrombosis genes and relations to plasma hemostatic protein level and cardiovascular disease risk. Arterioscler Thromb Vasc Biol. 2006;26(6):1405–12. [DOI] [PubMed] [Google Scholar]
- 23.Sabater-Lleal M, Huang J, Chasman D ea. A Multi-Ethnic Meta-Analysis of Genome-Wide Association Studies in Over 100,000 Subjects Identifies 23 Fibrinogen-Associated Loci but no Strong Evidence of a Causal Association between Circulating Fibrinogen and Cardiovascular Disease. Circulation 2013;128(12):1310–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McLaren W, Pritchard B, Rios D, Chen Y, Flicek P, Cunningham F. Deriving the consequences of genomic variants with the Ensembl API and SNP Effect Predictor. Bioinformatics. 2010;26(16):2069–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.López-Terrada D, Cheung SW, Finegold MJ, Knowles BB. Hep G2 is a hepatoblastoma-derived cell line. Hum Pathol. 2009;40(10):1512–5. [DOI] [PubMed] [Google Scholar]
- 26.Gilda JE, Gomes AV. Stain-Free total protein staining is a superior loading control to b-actin for Western blots. Anal Biochem. 2013;440(2):186–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Redman C, Xia H. Fibrinogen biosynthesis. Assembly, intracellular degradation, and association with lipid synthesis and secretion. Ann N Y Acad Sci. 2001;936(480–495). [PubMed] [Google Scholar]
- 28.Ito K, Angata K, Kuno A, Okumura A, Sakamoto K, Inoue R, Morita N, Watashi K, Wakita T, Tanaka Y, Sugiyama M, Mizokami M, Yoneda M, Narimatsu H. Screening siRNAs against host glycosylation pathways to develop novel antiviral agents against hepatitis B virus. Hepatol Res. 2020;50(10):1128–40. [DOI] [PubMed] [Google Scholar]
- 29.Morchang A, Lee RCH, Yenchitsomanus PT, Sreekanth GP, Noisakran S, Chu JJH, Limjindaporn T. RNAi screen reveals a role of SPHK2 in dengue virus-mediated apoptosis in hepatic cell lines. PloS One. 2017;12(11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ren J, Lin J, Dong X, Xu D, Chen Q, Liu Y, Chang Y, Yao J, Han S. Screening of efficient siRNA target sites directed against gatekeeper genes for DNA repair. J Huazhong Univ Sci Technolog Med Sci. 2006;26(6):640–3. [DOI] [PubMed] [Google Scholar]
- 31.Takezawa Y, Matsuda K, Terasawa F, Sugano M, Honda T, Okumura N. siRNA down-regulation of FGA mRNA in HepG2 cells demonstrated that heterozygous abnormality of the Aα-chain gene does not affect the plasma fibrinogen level. Thrombosis research. 2013;131(4):342–8. [DOI] [PubMed] [Google Scholar]
- 32.Hur WS, Paul DS, Bouck EG, Negrón OA, Mwiza JM, Poole LG, Cline-Fedewa HM, Clark EG, Juang LJ, Leung J, Kastrup CJ, Ugarova TP, Wolberg AS, Luyendyk JP, Bergmeier W, Flick MJ. Hypofibrinogenemia with preserved hemostasis and protection from thrombosis in mice with an Fga truncation mutation. Blood. 2022;139(9):1374–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Juang LJ, Hur WS, Silva LM, Strilchuk AW, Francisco B, Leung J, Robertson MK, Groeneveld DJ, La Prairie B, Chun EM, Cap AP, Luyendyk JP, Palumbo JS, Cullis PR, Bugge TH, Flick MJ, Kastrup CJ. Suppression of fibrin(ogen)-driven pathologies in disease models through controlled knockdown by lipid nanoparticle delivery of siRNA. Blood. 2022;139(9):1302–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wauman J, Zabeau L, Tavernier J. The leptin receptor complex: Heavier than expected? Front Endocrinol (Lausanne). 2017;8:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Satterfield TF, Pallanck LJ. Ataxin-2 and its Drosophila homolog, ATX2, physically assemble with polyribosomes. Hum Mol Genet. 2006;15(16):2523–32. [DOI] [PubMed] [Google Scholar]
- 36.Kaehler C, Isensee J, Nonhoff U, Terrey M, Hucho T, Lehrach H, Krobitsch S. Ataxin-2-like is a regulator of stress granules and processing bodies. PloS One. 2012;7(11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ciosk R, DePalma M, Priess JR. ATX-2, the C. elegans ortholog of ataxin 2, functions in translational regulation in the germline. Development (Cambridge, England). 2004;131(19):4831–41. [DOI] [PubMed] [Google Scholar]
- 38.Imamura H, Momohara S, Yano K, Sakuma Y, Nakayama M, Tobimatsu H, Ikari K. Tocilizumab treatment in patients with rheumatoid arthritis is associated with reduced fibrinogen levels and increased blood loss after total knee arthroplasty. Mod Rheumatol. 2018;28(6):976–80. [DOI] [PubMed] [Google Scholar]
- 39.Said EA, Al-Reesi I, Al-Shizawi N, Jaju S, Al-Balushi MS, Koh CY, Al-Jabri AA, Jeyaseelan L. Defining IL-6 levels in healthy individuals: A meta-analysis. J Med Virol. 2021;93(6). [DOI] [PubMed] [Google Scholar]
- 40.Trak-Smayra V, Paradis V, Massart J, Nasser S, Jebara V, Fromenty B. Pathology of the liver in obese and diabetic ob/ob and db/db mice fed a standard or high-calorie diet. Int J Exp Pathol. 2011;2011(6):413–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ernst A, Sharma AN, Elased KM, Guest PC, Rahmoune H, Bahn S. Diabetic db/db mice exhibit central nervous system and peripheral molecular alterations as seen in neurological disorders. Transl Psychiatry. 2013;3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Uhlen M, Zhang C, Lee S, Sjöstedt E, Fagerberg L, Bidkhori G, Benfeitas R, Arif M, Liu Z, Edfors F, Sanli K, von Feilitzen K, Oksvold P, Lundberg E, Hober S, Nilsson P, Mattsson J, Schwenk JM, Brunnström H, Glimelius B, et al. A pathology atlas of the human cancer transcriptome. Science. 2017;357:6352. [DOI] [PubMed] [Google Scholar]
- 43.Nitzahn M, Lipshutz GS. CPS1: Looking at an ancient enzyme in a modern light. Mol Genet Metab. 2020;131(3):289–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Matone A, Scott-Boyer MP, Carayol J, Fazelzadeh P, Lefebvre G, Valsesia A, Charon C, Vervoort J, Astrup A, Saris WHM, Morine M, Hager J. Network Analysis of Metabolite GWAS Hits: Implication of CPS1 and the Urea Cycle in Weight Maintenance. PloS One. 2016;11(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gallego-Durán R, Ampuero J, Pastor-Ramírez H, Álvarez-Amor L, del Campo JA, Maya-Miles D, Montero-Vallejo R, Cárdenas-García A, Pareja MJ, Gato-Zambrano S, Millán R, del Carmen Rico M, Luque-Sierra A, Gil-Gómez A, Rojas Á, Muñoz-Hernández R, García-Lozano M, Aller R, Andrade RJ, García-Monzón C, et al. Liver injury in non-alcoholic fatty liver disease is associated with urea cycle enzyme dysregulation. Scientific Reports 2022 12:1. 2022;12(1):1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Park M, D’Alecy L, Anderson M, Basrur V, Feng Y, Brady G, Kim D, Wu J, Nesvizhskii A, Lahann J, Lukacs N, Fontana R, Omary M. Constitutive release of CPS1 in bile and its role as a protective cytokine during acute liver injury. Proc Natl Acad Sci USA. 2019;116(18):9125–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kopec AK, Joshi N, Cline-Fedewa H, Wojcicki AV, Ray JL, Sullivan BP, Froehlich JE, Johnson BF, Flick MJ, Luyendyk JP. Fibrin(ogen) drives repair after acetaminophen-induced liver injury via leukocyte αMβ2 integrin-dependent upregulation of Mmp12. J Hepatol. 2017;66(4):787–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Svitkina TM, Verkhovsky AB, Borisy GG. Plectin sidearms mediate interaction of intermediate filaments with microtubules and other components of the cytoskeleton. J Cell Biol. 1996;135(4):991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Groeneveld D, Pereyra D, Veldhuis Z, Adelmeijer J, Ottens P, Kopec AK, Starlinger P, Lisman T, Luyendyk JP. Intrahepatic fibrin(ogen) deposition drives liver regeneration after partial hepatectomy in mice and humans. Blood. 2019;133(11):1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Essex DW, Wu Y. Multiple Protein Disulfide Isomerases support Thrombosis. Curr Opin Hematol. 2018;25(5):395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Stopa JD, Baker KM, Grover SP, Flaumenhaft R, Furie B. Kinetic-based trapping by intervening sequence variants of the active sites of protein-disulfide isomerase identifies platelet protein substrates. J Biol Chem. 2017;292(22):9063–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kobayashi Y, Oguro A, Hirata Y, Imaoka S. The regulation of Hypoxia-Inducible Factor-1 (HIF-1alpha) expression by Protein Disulfide Isomerase (PDI). PloS one. 2021;16(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Voisin S, Almén MS, Zheleznyakova GY, Lundberg L, Zarei S, Castillo S, Eriksson FE, Nilsson EK, Blüher M, Böttcher Y, Kovacs P, Klovins J, Rask-Andersen M, Schiöth HB. Many obesity-associated SNPs strongly associate with DNA methylation changes at proximal promoters and enhancers. Genome Med. 2015;7(103). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Auburger G, Sen NE, Meierhofer D, Başak AN, Gitler AD. Efficient Prevention of Neurodegenerative Diseases by Depletion of Starvation Response Factor Ataxin-2. Trends Neurosci. 2017;40(8):507–16. [DOI] [PubMed] [Google Scholar]
- 55.Kiehl TR, Nechiporuk A, Figueroa KP, Keating MT, Huynh DP, Pulst SM. Generation and characterization of Sca2 (ataxin-2) knockout mice. Biochem Biophys Res Commun. 2006;339(1):17–24. [DOI] [PubMed] [Google Scholar]
- 56.Lastres-Becker I, Brodesser S, Lütjohann D, Azizov M, Buchmann J, Hintermann E, Sandhoff K, Schürmann A, Nowock J, Auburger G. Insulin receptor and lipid metabolism pathology in ataxin-2 knockout mice. Hum Mol Genet. 2008;17(10):1465–81. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.