

Abstract
Proteome profiling by activated esters identified >9000 ligandable lysines but they are limited as covalent inhibitors due to poor hydrolytic stability. Here we report our efforts to design and discover a new series of Tunable Amine-Reactive Electrophiles (TAREs) for selective and robust labeling of lysine. The major challenges in developing selective probes for lysine are the high nucleophilicity of cysteines and poor hydrolytic stability. Our work circumvents these challenges by a unique design of the TAREs that form stable adducts with lysine and on reaction with cysteine generate another reactive electrophiles for lysine. We highlight that TAREs exhibit substantially high hydrolytic stability as compared to the activated esters and are non-cytotoxic thus have the potential to act as covalent ligands. We applied these alternative TAREs for the intracellular labeling of proteins in different cell lines, and for the selective identification of lysines in the human proteome on a global scale.
Keywords: bioconjugation, chemoselective, mass sensitivity boosters, protein labelling, traceless
Table of Contents
Tunable and hydrolytically stable electrophiles are reported for the chemo-selective labeling of lysine with mass sensitive probes. These tunable amine-reactive electrophiles (TAREs) were used to label proteins in various subcellular compartments inside cells and for global identification of lysine in the human proteome.
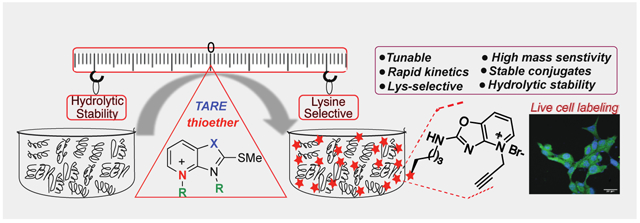
Introduction
Lysine shows rich chemistry through its nucleophilic amine group and is abundant in various active and allosteric sites. Lysines also catalyze multiple reactions and regulate various biological processes.1-3 Moreover, lysines are frequent sites for posttranslational modifications and regulate the structure and functions of proteins.4-7 Lysines have a high frequency in human proteins (~6% of all residues).8 Together, these properties make lysine residues desirable targets with covalent drugs; however, the low nucleophilicity of lysine as compared to cysteine makes selective targeting by covalent probes a highly daunting task. Several small molecule covalent ligands for selective proteome profiling of cysteine has been reported;9-15 however most of the small molecule electrophiles for lysines such as dichlorotriazines,16 imidoesters,17 2-acetyl- or 2-formyl-benzeneboronic acids,18 isothiocyanates,19-20 pyrazolecarboxamidines,21-22 sulfonyl fluorides,23-24 and vinyl sulfonamides25 also react with other amino acids such as serine, tyrosine and cysteine. Sulfonyl acrylate reagent has been reported for the regio- and chemo-selective labeling of lysine on pure protein by using low equivalent of the probe but it has a potential to react with cysteine during profiling.26 Recently, activated esters such as NHS-ester27 and STPyne28 have been explored for ligandability of lysine in the human proteome. Although >9000 ligandable lysine sites have been discovered using NHS-esters26 and STP-esters28, none of these probes act as a covalent ligand/inhibitor for lysine in cells because of poor hydrolytic stability (Figure 1a).27-28 Therefore for the discovery of covalent ligands for lysines, new amine reactive chemotypes are needed that are stable to enzymatic and non-enzymatic hydrolysis, non-cytotoxic and generate stable covalent adducts with lysine. These lysine selective, hydrolytically stable covalent probes would significantly expand the druggable content of the human proteome. With these considerations in mind, we sought to develop new Tunable Amine-Reactive Electrophiles (TAREs) as covalent probes that would retain the advantages of covalent lysine targeting (stable adduct with lysine and high selectivity) without the potential liabilities associated with hydrolysis and cross-reactivity with other nucleophilic amino acids (Figure 1b). In this study, we elucidate specific structural features that underlie selective lysine addition to generate stable adducts, and we apply these principles to the design of lysine-targeted covalent ligands for live cell labeling and proteome profiling. While our paper was under review, a new study exploring varying probes for proteome profiling was published but detailed studies are needed on probes to determine their cytotoxicity, stability inside cells and their suitability to act as covalent ligands for lysine.29 Here, we show that TAREs are non-cytotoxic, exhibit high hydrolytic stability, high activity in live cells and selectively enrich lysine in the human proteome with negligible enrichment of cysteine.
Figure 1.

Tunable Amine-Reactive Electrophiles (TAREs) for selective profiling of lysines. (a) Previous work on activated esters for profiling lysine and their limitations. STP- and NHS-esters are commonly used for lysine profiling. (b) TAREs, tunable and hydrolytically stable reagents for selective lysine proteome profiling. Nucleophilic Cys reacts with TAREs to generate another electrophilic thioether probes for selective reaction with lysine to generate stable products. Proposed reaction pathway between lysine and a TARE proceeds by nucleophilic attack of lysine on the heteroaromatic ring of thioether and undergoes SNAr substitution with lysine to generate stable adducts.
Results and Discussion
Design of Covalent TARE: Cysteine vs Lysine
Major challenges in designing the electrophilic covalent probes selective for proteome profiling of lysine are the cross reactivity with nucleophilic cysteine and hydrolytic instability of the probe.27-28 Since the nucleophilicity of Cys is higher than Lys, we cannot avoid the reaction of the electrophilic probe with Cys under physiological conditions. We sought to achieve selective enrichment of lysine by designing electrophilic heteroaromatic thioethers that on reaction with cysteine generate another lysine reactive electrophilic heteroaromatic thioethers that are capable of forming stable adducts with lysine (Figure 1b). Here we display our efforts to tune the selectivity and reactivity of heteroaromatic thioethers thus expanding their utility for the selective profiling of lysine in cells and human proteome. We tune the heteroaromatic thioether probes to make them water soluble, selective for lysine, hydrolytically stable and easy analysis by mass spectrometry. We achieved this by (i) varying different heteroatoms O/S on heteroaromatic ring, (ii) varying the fused aromatic ring (benzene and pyridine) and (iii) by methylation of N-heteroatoms (Figure 2a). We initiated our studies by exploring the reactivity of a panel of TAREs (Figure 2a, Synthesis of probes, Supplementary Fig. 1) with a peptide FKVCF 2a, containing nucleophilic amino acids N-terminus, Lys and Cys, under phosphate buffer (NaP, pH 7.5, 10 mM) at room temperature (Figure 2b). We did not observe the formation of an adduct with heteroaromatic 2-methylthio benzooxazoline probe 1a due to its poor solubility in a buffer and lower reactivity. To increase aqueous solubility, we replaced the benzene ring with pyridine, but the solubility of the probe 2-methylthio pyridoxazoline 1b remained poor. We next carried out the methylation of the pyridine of the 2-methylthio pyridoxazoline 1b to generate the 2-methylthio pyridinium oxazoline ion 1c that substantially increased its reactivity and solubility in buffer (Figure 2a). The reaction of 1c with a peptide FKVCF 2a showed the modification of both Cys and Lys under the reaction conditions as analyzed by LC-MS/MS analysis (90% Figure 2b, Supplementary Fig. 2). To further confirm the chemoselectivity for lysine and cysteine, reactions with probe 1c-yne were performed with peptides PGYAHF and WDQRF containing other nucleophilic amino acid residues such as S, T, Y, W, E and H but no modification of any amino acids was observed (Supplementary Fig. 2). To increase the reactivity and aqueous solubility of 2-methylthio benzooxazoline probe 1a, we further methylated the heteroatom of the azoline ring to generate 2-methylthio benzoN-methyloxazolinium ion 1d. The reaction of 1d with a peptide FKVCF 2a showed the modification of Lys amino acid only with 99% conversion to the modified lysine product. Interestingly, we did not observe any modification of the Cys and the N-terminal residue under the reaction conditions as determined by LC-MS/MS analysis (Figure 2b, Supplementary Fig. 2). In contrast, the reaction of FKVCF 2a with NHS and STP activated esters did not show any selectivity and labeled the N-terminus, cysteine and lysine within 3h under the reaction conditions (Supplementary Fig. 2).
Figure 2.

Tunable and stable TAREs for targeting lysine. (a) Structures of a variety of TAREs with different modifications to interrogate their reactivity and selectivity for lysine. (b) Screening of TAREs to determine high selectivity for lysine by using peptide FKVCF 2a with all the nucleophilic residues. Probe 1d showed high selectivity and reactivity for lysine. (c) Reactivity of cysteine-TARE conjugate towards lysine to generate stable product. (d) Modification of proteins with various TAREs. These proteins do not have any cysteine residue and TAREs showed high selectivity for lysine as analyzed by LC-MS/MS. Reaction Conditions: protein (3 mM in Nap (pH 7.5), probe 1d (10 equiv., 30 mM), 1e (100 equiv., 300mM), room temperature for 1h, detection wavelength 200 nm. For lb and CyC modification, probe 1 equiv. of 1d (3 mM) was used for 1h.
To characterize the Lys coupling product with 1d, a reaction with a model compound hexadecylamine was carried out on a large scale under the optimized conditions. The resulting product was isolated and Lys labeling by 1d was confirmed by NMR (1H and 13C) (Supplementary Fig. 3). In contrast, no product was observed with a peptide Ac-VCF (without Lys), thus reconfirms the high selectivity of 1d for Lys. We hypothesized that this might be due to the high reactivity of the 1d-cysteine conjugate towards lysine to generate a stable product or for the released methanethiol to generate unreacted starting material. To confirm our hypothesis, we synthesized a probe 2-methylthio benzoN-methylthiozolinium ion 1e of lower reactivity by replacing O heteroatom with S in the 2-methylthio benzoN-methyloxazolinium ion 1d. We isolated the small amounts of 1e-Cys-conjugate (VCF-1e) by carrying out the reaction of 1e with a peptide AcVCF and incubated the cysteine-adduct VCF-1e with lysine methylester (Figure 2c, Supplementary Fig. 4). As envisioned, the reaction resulted in the formation of a stable adduct with lysine methylester (Lys-OMe-1e) releasing the free peptide VCF 2b as analyzed by LCMS (Figure 2c, Supplementary Fig. 4). Since 1d-Cys conjugate is highly reactive and difficult to isolate under the aqueous conditions, we synthesized 1d-thio-conjugate under non-aqueous conditions and observed complete modification with lysine methylester within 1 h under physiological conditions (NaP, pH 7.5, 10 mM) at room temperature (Supplementary Fig. 4). These experiments confirmed that adduct obtained by the reaction of Cys with TAREs 1c-1e generated another lysine reactive electrophile thus exhibiting the potential to be selective for lysine profiling in the human proteome. We also incubated 1d-thio-conjugate under buffer (NaP, pH 7.5, 10 mM) at room temperature and observed complete hydrolysis in 1 h (Supplementary Fig. 4) suggesting the selectivity for lysine profiling. The reaction of 1e with a peptide FKVCF 2a modified both the N-terminus and Lys as analyzed by MS/MS (95% Figure 2b, Supplementary Fig. 2). We also synthesized probe 1f of lower reactivity by replacing O heteroatom with S in the azoline ring of 2-methylthio pyridinium oxazoline ion 1c. As anticipated, we detected the modification at Cys of a peptide FKVCF 2a with 1f as analyzed by LC-MS/MS (90% Figure 2b, Supplementary Fig. 2).
Chemoselectivity: Protein Modification
Previous studies with carbon electrophiles such as chloroacetamide, sulfonate esters and aryl halides showed that the reactivity of electrophiles with peptides often vary from their reactivity with proteins due to the unique environment of reactive amino acids in proteins.14,30-31 To evaluate the selectivity and reactivity of probes 1d and 1e with proteins, we carried out the reaction with Myoglobin (Mb) (without Cys Figure 2d).
The multiple modifications of lysines on Mb were observed with both the probes 1d (10 equiv., 1h) and 1e (100 equiv., 12 h) as determined by MS/MS analysis on Mb-1d and Mb-1e protein conjugates (Figure 2d, Supplementary Fig. 5, Supplementary Table 1 and Table 2). The modification of any other reactive amino acids on Mb such as Ser, Thr, Tyr, Trp, Glu, His was not observed even after using excess of probe 1e (100 equiv.) and for longer reaction time (12 h), thus further confirms high reactivity and selectivity of 1d and 1e for lysine. Next, we explored probes 1d for the modification of other proteins such as lactalbumin (lb) and cytochrome C (CyC) and MS/MS analysis confirmed the modification of lysine amino acid only in lb-1d and CyC-1d protein conjugates (Figure 2d and Supplementary Fig. 6). These studies demonstrate that the proteome reactivity of TAREs can be finely adjusted by modulating the electronics of the heteroaromatic azoline ring system, thereby providing ideal tunable electrophiles for chemical proteomic applications. TAREs 1c-1f are charged molecules and increase the ionization of the tagged fragments thus has a potential to enhance their detection sensitivity by MS. We showed that 1d significantly increased the detection sensitivity of the labeled proteolytic fragments of cytochrome C as compared to the unlabeled fragments and STP and NHS-ester modified fragments (Supplementary Fig. 7). The MS results clearly showed that the MS sensitivity of the fragments modified by 1d probes is 100 fold higher than the mass sensitivity of the fragments modified by STP, NHS-ester and unlabeled fragments thus could be of high significance in proteomics studies (Supplementary Fig. 7).
TAREs with Affinity Tags and Hydrolytic Stability
The effective reaction for the enrichment of lysine fragments from a complex mixture should have the ability to attach affinity tags, thus we generated the alkyne and azide functionalized TAREs N3-1e, 1c-yne and 1d-yne (Synthesis of probes, Supplementary Fig. 8). We then evaluated the reactivity of the azide- and alkyne-functionalized TAREs towards proteins such as Mb, lb and CyC. Similar to the model probe studies with 1c-1e, azide- and alkyne-TAREs N3-1e (25 equiv. 12h) and 1c-yne (1equiv., 1h) modified Lys of proteins Mb, lb and CyC under optimized conditions as confirmed by MS/MS (Figure 2d, Supplementary Fig. 9). The modification of lysine residue is independent of the presence of the cysteine residue as observed by the labeling of significant amount of lysine residues (6-7 lysines) on pure proteins myoglobin, cytochrome C and lactalbumin without any cysteine using high equivalents of the probe 1c-yne (50 equiv.) (Supplementary Figure 9).
Next, we compared the reactivity of the two most reactive TAREs 1c-yne and 1d-yne by carrying out the rate studies using a peptide Ac-GKF (GKF 2c). The reactions were monitored after regular intervals of time using HPLC and MS (Figure 3a, Supplementary Fig. 10). The reaction with 1d-yne (k = 307.52 M−1S−1) showed 3-fold higher reactivity than 1c-yne (k =99.27 M−1S−1) (Supplementary Fig. 10). The reaction rate of STPyne (k = 190.92 M−1S−1) with a peptide GKF 2c showed lower reactivity compared to 1d-yne but high reactivity than 1c-yne (Figure 3a, Supplementary Fig. 10). Next, we sought to determine the stability of the reactive TAREs 1c-yne and 1d-yne towards hydrolysis and compare it with hydrolytic stability of NHS-ester and STPyne previously used for lysine profiling.27-28 We incubated probes in aqueous phosphate buffer (pH 7.5) under ambient conditions and monitored after regular intervals of time by HPLC and MS. Although of almost similar reactivity, 1c-yne probe is more stable towards hydrolysis as compared to 1d-yne and only 30% degradation of 1c-yne probe was observed after 6 h and 50% of 1c-yne remained intact even after 12h (Figure 3b, Supplementary Fig. 11). In contrast, NHS-ester showed 90% degradation in 2h and completely degraded within 4h (Figure 3b, Supplementary Fig. 11). STPyne showed 90% degradation in 6h. These studies showed the high stability of TARE 1c-yne as compared to NHS-ester and STPyne thus capable of acting as covalent inhibitors of lysine in cellular environment (Figure 3b, Supplementary Fig. 11). The most reactive TARE probes 1c-yne and 1d-yne are bench stable for multiple weeks as a white solid, demonstrating robust properties for long-term storage.
Figure 3.

Chemoproteomic studies of TAREs by gel-based competitive protein profiling. (a) Rate studies with peptide GKF 2c showed high reactivity with 1d-yne. Each time point represents an average of three independent experiments. (b) Hydrolytic stability studies showed 1c-yne is more hydrolytically stable as compared to other lysine reactive probes. (c) General protocol for proteome profiling by different probes and structure of probes. (d) In-gel fluorescence analysis of STPyne (1mM) and 1d probes at different concentrations of 1d (0.5 mM to 5 mM) followed by detection with NHS-Rh for lysine reactivity and IA-Rh for cysteine reactivity. 1d is more selective for lysine as compared to STPyne. (e) In-gel fluorescence analysis of 1c-yne at different concentrations (0 to 1 mM) showed reactivity with both lysine and cysteine. Click gel assay of 1c-yne and cysteine reactive IA-aky probes with cell lysate using fluorescent-biotin azide to determine the total labeled proteins. High labeling of cell lysate was observed with 1c-yne. (f) Click gel assay of 1d-yne at different concentrations (0.1 mM to 1 mM) and STPyne (0.1 mM) probes with cell lysate using biotin-azide and streptavidin blot to determine the total labeled proteins. Dose-dependent labeling was observed for 1d-yne with banding pattern similar to STPyne.
TAREs for Gel-based ABPP
Bolstered by the lysine-selectivity and elevated reactivity observed with our TARE-protein and peptide-based labeling studies, we next sought to investigate whether this chemotype would be suitable for activity-based chemoproteomic applications in complex cell lysates. Using a gel-based competitive activity-based protein profiling (ABPP) assay, we first assessed the relative cysteine- and lysine-reactivity of TAREs. HEK293T lysates were first treated with probes 1d, 1c-yne or 1d-yne followed by labeling with a pan-cysteine or the pan-lysine reactive fluorescent probe, iodoacetamide-tetramethylrhodamine (IA-Rh) or NHS-tetramethylrhodamine (NHS-Rh), respectively (Figure 3c, Supplementary Fig. 12). While 1c-yne blocked both cysteine and lysine labeling (Figure 3e), gel-based ABPP analysis of compound 1d suggested improved specificity for lysine (Figure 3d), as indicated by competition of labeling by NHS-Rh but not IA-Rh. By comparison, the canonical lysine-reactive probe STPyne (1 mM) competed labeling of both probes, consistent with off-target cysteine-reactivity, as reported previously.32
As competitive blockage of NHS-Rh labeling was only observed at relatively high concentrations of 1c and 1d (e.g. 5mM for 1d, Figure 3d-3e, Supplementary Fig. 12), our next step was to evaluate the concentration range compatible with probe labeling. For these studies, we turned to an alkyne derivative of 1d, termed 1d-yne. Cell lysates were subjected to labeling by 1d-yne at the indicated concentrations followed by conjugation by CuAAC to biotin-azide, and the labeling was visualized by streptavidin blot (Figure 3f, Supplementary Fig. 12). Gratifyingly, robust concentration dependent labeling was observed with a banding pattern comparable to that of STPyne, consistent with lysine-directed reactivity. Similar labeling intensity was observed for 1 mM of 1d when compared to 100 uM of STPyne, supporting that the 1d TARE is a relatively attenuated warhead.
Intracellular Labeling of Proteins by TARE Probes
The ability of TAREs to profile the lysine proteome was further explored by treating living human cells with 1c-yne. We incubated three cancer cell lines, LNCaP, U87MG and T47D, representing prostate, brain and breast cancers, respectively, with increasing concentrations of 1c-yne, NHS-ester, and STPyne. After 2h, cells were fixed permeabilized, and washed to remove the unreacted probe. Subsequently, reacted probe was conjugated to a fluorophore via CuAAC to directly image the ability of each probe to label proteins within the cell. Confocal fluorescent microscopy confirmed that all three cell lines had taken up 1c-yne, STPyne, and NHS-ester into both the cytosolic and nuclear compartments (Figure 4a, Supplementary Fig. 13) at concentrations ranging from 5 μM to 100 μM (Supplementary Fig. 13). Western blot analysis of LNCaP cells treated with 100 μM of 1c-yne, NHS-ester, and STPyne and subsequent fluorophore labeling with CuAAC demonstrate protein labeling across molecular weights (Figure 4b, Supplementary Fig. 14). The different band intensities may reflect the lysine selectivity of 1c-yne compared to the cross reactivity of NHS-ester and STPyne probes with cysteine. Finally, we examined the rate at which 1c-yne can penetrate and label unique cellular compartments in living cells. Intracellular labeling occurs within the first five minutes at 1c-yne concentrations as low as 100 nM (Figure 4c). These results highlight the efficacy of 1c-yne as a probe for rapid live cell labeling. Next, we carried out cell viability studies with 1c-yne using T47D cells at two different concentrations (5 μM and 20 μM) for 24h. We did not observe any increase in apoptosis/necrosis compared to DMSO control as analyzed by flow cytometer (Supplementary Fig. 15).
Figure 4.

Live cell labeling and amino-acid selectivity in proteome by probe 1c-yne (a) Human LNCAP, U87MG and T47D cells treated with 5 μM 1c-yne, STPyne, or NHSester for 2h followed by fixing the cells, washing of unreacted probes and then conjugation with azide fluorophore tags using CuAAC show labeling in multiple cellular compartments. (b) Western blot fluorescent analysis of LNCAP cells incubated with 100 μM 1c-yne, STPyne, or NHSester for 2h and then conjugated with azide fluorophore tags using CuAAC demonstrates protein labeling across molecular weights. (c) Drug-like concentrations of 1c-yne (100 nM) demonstrate that robust intracellular labeling occurs within 5 min. (d) Cellular lysates are labelled with 1c-yne at different concentrations followed by conjugation with azide-biotin tags (blue) using CuAAC, enrichment of labeled proteins by neutravidin-conjugated beads and digested stepwise with trypsin to yield 1c-yne-labeled peptides for LC-MS analysis. Percentage of unique peptides and proteins labeled on each nucleophilic amino acid by 1c-yne in HEK293T proteome. Probe 1c-yne preferentially enrich lysine residues in human cell proteomes. Data represent means ± standard deviation for two experiments.
Chemoproteomic Analysis of Residue Selectivity
To further assess the proteome-wide reactivity profiles of the 1c and 1d TAREs, we turned to mass spectrometry-based chemoproteomics. Cell lysates were subjected to either probe 1c-yne or 1d-yne, labeled proteins were conjugated to biotin-azide via CuAAC and the samples were prepared and analyzed using our SP3 chemoproteomic sample preparation workflow (Supplementary Fig. 16).33 Briefly, TARE-labeled proteomes were subjected to single-pot solid-phase enhanced sample preparation (SP3) decontamination using carboxyl coated magnetic beads. SP3-resin tryptic digest biotinylated peptides were enriched by neutravidin and analyzed by LC-MS/MS. Surprisingly and in contrast with its apparent cysteine-reactivity observed by competitive gel-based studies, 1c-yne showed near complete selectivity for lysine residues (5124 total unique labeled lysine residues and 27 total unique labeled cysteine residues across two biological replicate experiments, Figure 4d, Supplementary Fig. 16 and Supplementary Table 3). Similarly, chemoproteomic analysis of 1d-yne-labeled lysates revealed near-complete lysine selectivity (1595 total unique lysine residues and 54 total unique labeled cysteine residues) across three compound concentrations analyzed.
Lysine specificity was further confirmed through re-search of the 1c-yne datasets using the algorithm MSFragger,34 which includes built in PTMProphet35 for accurate mass modification localization, which identified 1560 unique labeled lysines and 17 unique labeled cysteines (Supplementary Fig. 16, Supplementary Table 3). Consistent with our gel-based analysis, we observed a dose-dependent increase in peptides identified as the concentration of 1d-yne was increased from 100 μM to 1 mM. 1c-yne labeled substantially more peptides (5151) than 1d-yne (1649), in contrast with the aforementioned kinetic analysis that revealed 3-fold higher reactivity for 1d-yne (k = 307.52 M-1S-1) as compared to 1c-yne (k = 99.27 M-1S-1) (Figure 3a). Surprisingly high reactivity of 1c-yne in gel and proteomic studies as compared to 1d-yne might be due to the high hydrolytic stability of 1c-yne (95 % intact in 2h) as compared to 1d-yne (28 % intact in 2h) under the reaction conditions (Figure 3b).
We were intrigued by the lack of detectable cysteine labeling observed for 1c-yne, which was in contrast to our competitive gel-based studies. We speculated that liability of the cysteine adduct, potentially stemming from the reducing CuAAC conditions, might contribute to this observed difference. To confirm this hypothesis, we carried out labeling of a peptide GCF with 1c-yne with and without TCEP under click chemistry conditions. We observed the modification of Cys in a peptide GCF in the absence of TCEP only, no modification of a peptide GCF was observed in the presence of TCEP under click chemistry conditions (Supplementary Fig. 17). To further confirm, we isolated the Cys modified GCF-1c-yne adduct and incubated it in buffer containing TCEP. Within 5 mins, we observed complete decomposition of the GCF-1c-yne adduct to the unchanged peptide GCF (Supplementary Fig. 18).
Reactivity and Selectivity Pattern of Hydrolytically Stable TARE 1c-yne by DFT Calculations
To further assess the observed differences in cysteine- and lysine-reactivity for the TAREs, we next performed DFT analysis on the reaction between 1c-yne and methyl thiolate. Given its hydrolytic stability, observed reactivity with both cysteine and lysine residues, we selected compound 1c-yne as optimal for our DFT studies, aiming to gain insight into the observed differences in the amino acid reactivity profile. The DFT calculations with 1c-yne supported the reversibility of the reaction between 1c-yne and thiol nucleophiles compared to the irreversibility of 1c-yne modification by amine nucleophiles (Figure 5, Supplementary Fig. 19).
Figure 5.

Free energy profiles for SNAr substitution of 1c-yne with methylamine (blue) and methyl thiolate (red) in water, computed at the ωB97X-D/6-311++G(d,p) level of theory in SMD water. DFT method: ωB97X-D/6-311++G(d,p) SMD(H2O)//B3LYP/6-31+G(D) SMD(H2O), with Grimme correction for entropy and Head-Gordon correction for enthalpy in 298.15 K. All energies are in kcal/mol.
Conclusion
In summary, we have systematically designed TAREs that showed high reactivity with both cysteine and lysine but reaction with cysteine generated another lysine reactive electrophile thus these probes are highly selective for the enrichment and formation of stable adducts with lysine only. These TARE chemotypes are stable under hydrolytic conditions as compared to other lysine reactive activated esters such as STPyne and NHS esters thus have the potential to act as covalent ligands for lysine in the cellular environment.
We show that TAREs are highly tunable, where reactivity and selectivity can be varied for desired applications by the addition of different heteroatoms and their methylation. As expected, due to the high electron withdrawing nature of O as compared to S and more electrophilicity and solubility of the charged methylated ions as compared to an uncharged moiety, 1d and its analog 1d-yne were the most reactive, soluble and selective for lysine. We also showed that 1c is more hydrolytic stable as compared to other activated esters. There is no disparity between the reactivity of 1c, 1d and their alkyne derivatives 1c-yne and 1d-yne, and both of them are reactive at low micromolar concentrations. The labeling of both cytoplasmic and nuclear proteins in live cells demonstrate the high permeability, non-toxicity, cellular stability and cellular activity of TARE probes thus indicate its potential as a probe for rapid live cell labeling and covalent inhibitor. Identification of the sites of modification for TAREs showed that both 1c-yne and 1d-yne enriched only lysine peptide fragments from the cell lysate because the cysteine-adducts of both the probes are reactive electrophiles towards lysines and unstable towards TCEP thus unable to be enriched. DFT calculations on 1c-yne support our experimental results showing the reversibility of the reaction between 1c-yne and thiol nucleophiles and the irreversibility of 1c-yne modification by amine nucleophiles. 2-methylthio pyridiniumoxazoline ion 1c and its alkyne analog 1c-yne provides an aromatic, synthetically tractable, non-cytotoxic and hydrolytically stable electrophile to add to the arsenal of lysine-reactive groups available for protein modification. We anticipate that our design of new probes, easy synthesis to varying derivatives and detailed reactivity and selectivity studies with peptides, proteins, proteome and live cells will promote their use in multiple applications in the field of protein modification, bioconjugation, material science, activity based protein profiling and covalent drug discovery for undruggable human proteins.
Supplementary Material
Acknowledgements
This research was supported by NIH (Grant No. 1R35GM133719-01) and Alfred P. Sloan Foundation granted to M.R, Beckman Young Investigator Award to K.M.B and NIGMS System and Integrative Biology 5T32GM008185-33 to L.M.B.
References
- [1].Patricelli MP, Szardenings AK, Liyanage M, Nomanbhoy TK, Wu M, Weissig H, Aban A, Chun D, Tanner S, Kozarich JW, Biochemistry 2007, 46, 350–358. [DOI] [PubMed] [Google Scholar]
- [2].Eliot AC, Kirsch JF, Annu. Rev. Biochem 2004, 73, 383–415. [DOI] [PubMed] [Google Scholar]
- [3].Akcay G, Belmonte MA, Aquila B, Chuaqui C, Hird AW, Lamb ML, Rawlins PB, Su N, Tentarelli S, Grimster NP, Su Q, Nat. Chem. Biol 2016, 12, 931–936. [DOI] [PubMed] [Google Scholar]
- [4].Choudhary C, Weinert BT, Nishida Y, Verdin E, Mann M, Nat. Rev. Mol. Cell. Biol 2014, 15, 536–550. [DOI] [PubMed] [Google Scholar]
- [5].Greer EL, Shi Y, Nat. Rev. Genet 2012, 13, 343–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Zhang K, Dent SY, J. Cell. Biochem 2005, 96, 1137–1148. [DOI] [PubMed] [Google Scholar]
- [7].Mattiroli F, Sixma TK, Nat. Struct. Mol. Biol 2014, 21, 308–316. [DOI] [PubMed] [Google Scholar]
- [8].Tekaia F, Yeramian E, Dujon B, Gene. 2002, 297, 51–60. [DOI] [PubMed] [Google Scholar]
- [9].Shannon DA, Weerapana E, Curr. Opin. Chem. Biol 2015, 24, 18–26. [DOI] [PubMed] [Google Scholar]
- [10].Pace NJ, Weerapana E, ACS Chem. Biol 2013, 8, 283–296. [DOI] [PubMed] [Google Scholar]
- [11].Liu Q, Sabnis Y, Zhao Z, Zhang T, Buhrlage SJ, Jones LH, Gray NS, Chem. Biol 2013, 20, 146–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Ostrem JM, Shokat KM, Nat. Rev. Drug. Discov 2016, 15, 771–785. [DOI] [PubMed] [Google Scholar]
- [13].London N, Miller RM, Krishnan S, Uchida K, Irwin JJ, Eidam O, Gibold L, Cimermancic P, Bonnet R, Shoichet BK, Taunton J, Nat. Chem. Biol 2014, 10, 1066–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Shannon DA, Banerjee R, Webster ER, Bak DW, Wang C, Weerapana E, J. Am. Chem. Soc 2014, 136, 3330–3333. [DOI] [PubMed] [Google Scholar]
- [15].Weerapana E, Wang C, Simon GM, Richter F, Khare S, Dillon MB, Bachovchin DA, Mowen K, Baker D, Cravatt BF, Nature 2010, 468, 790–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Crawford LA, Weerapana E, Mol. Biosyst 2016, 12, 1768–1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Hunter MJ, Ludwig ML, J. Am. Chem. Soc 1962, 84, 3491–3504. [Google Scholar]
- [18].Bandyopadhyay A, Gao J, J. Am. Chem. Soc 2016, 138, 2098–2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Wang X, Di Pasqua AJ, Govind S, McCracken E, Hong C, Mi L, Mao Y, Wu JY-C, Tomita Y, Woodrick JC, Fine RL, Chung F-L, J. Med. Chem 2011, 54, 809–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Zhang Y, Kensler TW, Cho CG, Posner GH, Talalay P, Proc. Natl Acad. Sci. USA 1994, 91, 3147–3150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Musiol H-J, Moroder L, Org. Lett 2001, 3, 3859–3861. [DOI] [PubMed] [Google Scholar]
- [22].Kapp TG, Fottner M, Maltsev OV, Kessler H, Angew. Chem. Int. Ed 2016, 55, 1540–1543; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2016, 128, 1564–1568. [Google Scholar]
- [23].Grimster NP, Connelly S, Baranczak A, Dong J, Krasnova LB, Sharpless KB, Powers ET, Wilson IA, Kelly JW, J. Am. Chem. Soc 2013, 135, 5656–5668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Zhao Q, Ouyang X, Wan X, Gajiwala KS, Kath JC, Jones LH, Burlingame AL, Taunton J, J. Am. Chem. Soc 2017, 139, 680–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Asano S, Patterson JT, Gaj T, Barbas CF 3rd, Angew. Chem. Int. Ed 2014, 53, 11783–11786; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2014, 126, 11977–11980. [Google Scholar]
- [26].Matos MJ, Oliveira BL, Martínez-Sáez N, Guerreiro A, Cal PMSD, Bertoldo J, Maneiro M, Perkins E, Howard J, Deery MJ, Chalker JM, Corzana F, Jiménez-Osés G, Bernardes GJL, J. Am. Chem. Soc 2018, 140, 4004–4017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Ward CC, Kleinman JI, Nomura DK, ACS Chemical Biology 2017, 12, 1478–1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Hacker SM, Backus KM, Lazear MR, Forli S, Correia BE, Cravatt BF, Nat. Chem 2017, 9, 1181–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Abbasov ME, Kavanagh ME, Ichu TA, Lazear MR, Tao Y, Crowley VM, am Ende CW, Hacker SM, Ho J, Dix MM, Suciu R, Hayward MM, Kiessling LL, Cravatt BF, Nat. Chem 2021. 10.1038/s41557-021-00765-4 [DOI] [Google Scholar]
- [30].Adam GC, Sorensen EJ, Cravatt BF, Nat. Biotechnol 2002, 20, 805–809. [DOI] [PubMed] [Google Scholar]
- [31].Weerapana E, Simon GM, Cravatt BF, Nat. Chem. Biol 2008, 4, 405–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Cao J, Boatner LM, Desai HS, Burton NR, Armenta E, Chan NJ, Castellon JO, Backus KM, Anal. Chem 2021, 93, 2610–2618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Yan T, Desai HS, Boatner LM, Yen SL, Cao J, Palafox MF, Jami-Alahmadi Y, Backus KM, Chembiochem 2021, 22, 1841–1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Kong AT, Leprevost FV, Avtonomov DM, Mellacheruvu D, Nesvizhskii AI, Nat. Methods 2017, 14, 513–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Shteynberg DD, Deutsch EW, Campbell DS, Hoopmann MR, Kusebauch U, Lee D, Mendoza L, Midha MK, Sun Z, Whetton AD, Moritz RL, J. Proteome. Res 2019, 18, 4262–4272. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.