

Previous trials have shown the effects of fluvoxamine alone and inhaled budesonide alone for prevention of disease progression among outpatients with COVID-19. This placebo-controlled, randomized trial, conducted at 12 clinical sites in Brazil, sought to determine whether the combined effect of both drugs would increase treatment effects.
Visual Abstract. Oral Fluvoxamine With Inhaled Budesonide for Treatment of Early-Onset COVID-19.
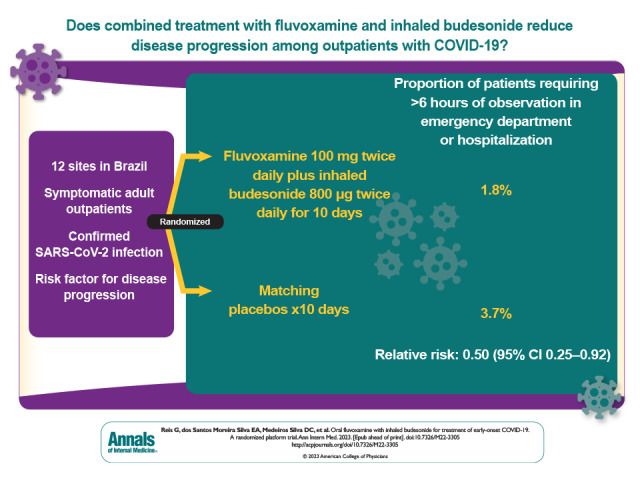
Previous trials have shown the effects of fluvoxamine alone and inhaled budesonide alone for prevention of disease progression among outpatients with COVID-19. This placebo-controlled, randomized trial, conducted at 12 clinical sites in Brazil, sought to determine whether the combined effect of both drugs would increase treatment effects.
Abstract
Background:
Previous trials have demonstrated the effects of fluvoxamine alone and inhaled budesonide alone for prevention of disease progression among outpatients with COVID-19.
Objective:
To determine whether the combination of fluvoxamine and inhaled budesonide would increase treatment effects in a highly vaccinated population.
Design:
Randomized, placebo-controlled, adaptive platform trial. (ClinicalTrials.gov: NCT04727424)
Setting:
12 clinical sites in Brazil.
Participants:
Symptomatic adults with confirmed SARS-CoV-2 infection and a known risk factor for progression to severe disease.
Intervention:
Patients were randomly assigned to either fluvoxamine (100 mg twice daily for 10 days) plus inhaled budesonide (800 mcg twice daily for 10 days) or matching placebos.
Measurements:
The primary outcome was a composite of emergency setting retention for COVID-19 for more than 6 hours, hospitalization, and/or suspected complications due to clinical progression of COVID-19 within 28 days of randomization. Secondary outcomes included health care attendance (defined as hospitalization for any cause or emergency department visit lasting >6 hours), time to hospitalization, mortality, patient-reported outcomes, and adverse drug reactions.
Results:
Randomization occurred from 15 January to 6 July 2022. A total of 738 participants were allocated to oral fluvoxamine plus inhaled budesonide, and 738 received placebo. The proportion of patients observed in an emergency setting for COVID-19 for more than 6 hours or hospitalized due to COVID-19 was lower in the treatment group than the placebo group (1.8% [95% credible interval {CrI}, 1.1% to 3.0%] vs. 3.7% [95% CrI, 2.5% to 5.3%]; relative risk, 0.50 [95% CrI, 0.25 to 0.92]), with a probability of superiority of 98.7%. No relative effects were found between groups for any of the secondary outcomes. More adverse events occurred in the intervention group than the placebo group, but no important differences between the groups were detected.
Limitation:
Low event rate overall, consistent with contemporary trials in vaccinated populations.
Conclusion:
Treatment with oral fluvoxamine plus inhaled budesonide among high-risk outpatients with early COVID-19 reduced the incidence of severe disease requiring advanced care.
Primary Funding Source:
Latona Foundation, FastGrants, and Rainwater Charitable Foundation.
There is an urgent need for accessible and effective treatments for early SARS-CoV-2 infection, especially for patients without access to oral protease inhibitors or effective monoclonal antibodies. Combining interventions with demonstrated effects may improve the effectiveness seen with each individual drug. Fluvoxamine, a selective serotonin reuptake inhibitor, and inhaled budesonide, a corticosteroid, have been evaluated in multiple trials and have shown treatment benefits in phase 2 and 3 trials (1–6), with anti-inflammatory or antiviral effects hypothesized as the potential mechanism behind the treatment effect (7, 8). Fluvoxamine has shown clinical benefits for doses at or above 100 mg twice daily but not at 50 mg twice daily (2, 3, 9). Given the established safety profiles and clinical benefits of fluvoxamine and budesonide in the treatment of COVID-19, we sought to evaluate the potential additive benefits of combining these regimens. Further justification for combining them can be found in the protocol (available at Annals.org).
We conducted a randomized, placebo-controlled, adaptive platform trial at 12 sites in Brazil to evaluate whether fluvoxamine, 100 mg twice daily, plus inhaled budesonide would prevent severe progression of COVID-19 among outpatients with laboratory-documented SARS-CoV-2 infection. A platform trial design allows for agents to be added and tested using a single overarching master protocol (10, 11). Ten different interventions have been evaluated in this platform trial; we report here on the clinical evaluation of oral fluvoxamine plus inhaled budesonide using a concurrent placebo control group.
Methods
Study Design and Oversight
The TOGETHER trial is a randomized adaptive platform trial to investigate the efficacy of repurposed treatments for COVID-19 among high-risk adult outpatients (12). The trial was designed and is conducted in partnership with local public health authorities from 12 participating cities in Brazil to simultaneously test potential treatments for early disease using a master protocol. A master protocol defines prospective decision criteria for discontinuing interventions because of futility, stopping due to superiority over placebo, or adding new interventions. Interventions that have thus far been evaluated include hydroxychloroquine (protocol 1), lopinavir–ritonavir (protocol 1) (13), metformin (protocol 2) (14), ivermectin (protocol 2) (15), fluvoxamine (protocol 2) (2), doxazosin (protocol 2), and pegylated interferon lambda (protocol 2) (16), all versus concurrent placebos. The TOGETHER trial is centrally coordinated by Platform Life Sciences (Vancouver, British Columbia, Canada), with local implementation by Cardresearch (Belo Horizonte, Brazil).
The platform trial began on 2 June 2020, and enrollment into the fluvoxamine–budesonide group began on 6 January 2022. The trial complies with the International Conference on Harmonization Good Clinical Practice guidance as well as local regulatory requirements. Ethical approval was provided by local and national ethics boards in Brazil (CONEP CAAE: 41174620.0.1001.5120, approval letter 5.501.284) and the Hamilton Integrated Research Ethics Board in Canada (approval letter 13390). The full protocol and statistical analysis plan have been published previously (12) and are available at Annals.org. The Adaptive designs CONSORT (Consolidated Standards Of Reporting Trials) Extension statement guided this article (17, 18). The steering committee made all protocol-related decisions, and sponsors had no role in trial conduct, data analysis, or the decision to submit the manuscript for publication.
Setting
The Supplement lists the cities and investigators for the 12 clinical sites in Brazil that participated in the trial. Local investigators, in partnership with local public health authorities, recruited participants at community health facilities (emergency settings, influenza symptom referral centers, and primary care community centers). We used several community outreach strategies, including physical and social media as per local public health authorities, to create awareness of the trial.
Participant Screening
Upon presentation to 1 of the trial outpatient care clinics, potential participants were screened with respect to eligibility criteria. Key inclusion criteria were age 18 years or older; presentation within 7 days of symptom onset to an outpatient care setting with an acute clinical condition consistent with COVID-19; positive rapid test result for SARS-CoV-2 infection; and at least 1 high-risk criterion for deterioration, including age 50 years or older, diabetes mellitus, hypertension requiring medication, cardiovascular disease, lung disease, smoking, obesity (body mass index [BMI] >30 kg/m2), organ transplant, chronic kidney disease (stage IV) or receipt of dialysis, immunosuppressive therapy (≥10 mg of prednisone daily or equivalent), cancer diagnosis within 6 months, or receipt of chemotherapy for cancer. Further inclusion and exclusion criteria are documented in the study protocol (12).
If a patient met the eligibility criteria, study personnel obtained written informed consent in person and performed a rapid antigen test for SARS-CoV-2 (Panbio [Abbott]). Before randomization, study personnel collected data on demographic characteristics, medical history, comorbidities, concomitant medications, exposure to an index case, and score on the World Health Organization (WHO) Clinical Progression Scale (17).
Randomization and Trial Interventions
Participants were randomly assigned using a centralized core randomization process handled by an independent, unblinded pharmacist who was not aware of any protocol-related procedures and was contracted specifically for this process. Sites requested randomization via text message to the pharmacist at the coordinating center in order to maintain allocation concealment. Patients were randomly assigned using a block randomization procedure for each participating site, with stratification by age (<50 and ≥50 years). The trial team, site staff, and patients were blinded to treatment allocation. The active drugs and the placebos were packaged in identically shaped bottles and inhalers labeled with letters corresponding to the active group or the placebo group. Only the third-party pharmacist responsible for releasing the randomization was aware of which letter was associated with which drug or placebo.
Data Collection and Participant Follow-up
Our primary outcome was a composite that included emergency setting visits due to clinical worsening of COVID-19 (defined as the participant remaining under observation for >6 hours) or hospitalization due to progression of COVID-19 (worsening of clinical status) and/or suspected COVID-19 complications within 28 days of randomization. The composite end point was chosen because it addresses both hospitalization and a proxy for hospitalization (retention in an emergency setting for COVID-19), as many patients were prevented from being hospitalized by hospital overcapacity during peak waves. This region of Brazil implemented hospital-like services in the emergency settings, with 50 to 80 beds and provision of services that included oxygenation, sedation, multiday stays, and mechanical ventilation. Time was measured from the first medical evaluation to discharge and was derived from medical charts at the clinical sites.
Key secondary outcomes included 1) time to clinical improvement; 2) the number of days with respiratory symptoms; 3) time to hospitalization for any cause or due to COVID-19 progression; 4) mortality from any cause and time to death from any cause; 5) WHO Clinical Progression Scale score; 6) the number of days in the hospital and receiving mechanical ventilation; and 7) adverse events, adverse reactions to the study medications, and the proportion of participants who were nonadherent to the study drugs. All secondary outcomes were assessed up to 28 days after randomization.
Study personnel collected outcome data on days 1, 2, 3, 4, 5, 7, 10, 14, 28, and 60 via in-person contact, telephone contact, or social media applications using videoconferencing. We collected outcome data regardless of whether participants took study medication. In cases of adverse events, unscheduled visits outside clinical care could occur at any time during the treatment period.
Considering the transmission characteristics of SARS-CoV-2 and the isolation recommendations for persons who were positive for infection, we collected limited data on vital signs. Cardiac safety was assessed using 6-lead electrocardiography (KardiaMobile) at the baseline visit. The digital recordings were deidentified and transferred to a central facility (Cardresearch, Belo Horizonte, Brazil) for reading. Oxygen status was assessed using a pulse oximeter for noninvasive arterial oxygen saturation (Spo 2) and pulse (Jumper Medical Equipment), and temperature was measured by research personnel using a standard digital oral thermometer. Mid-turbinate nasal swab kits and sterile recipient storage were provided for collection of a nasopharyngeal swab, sputum, or saliva at days 3 and 7.
All serious and nonserious adverse events were reported to study personnel per local regulatory requirements. Reportable adverse events included serious adverse events, adverse events resulting in discontinuation of study medication, and adverse events deemed to possibly be related to study medication.
Trial Interventions
All participants received usual care for COVID-19 from health care workers at public health facilities. Patients were randomly assigned to fluvoxamine (Luvox [Abbott]; 100 mg twice daily for 10 days) plus inhaled budesonide (800 mcg twice daily for 10 days) or corresponding placebo starting immediately after randomization (day 1). We used both a matched oral placebo and a blinded inhaled placebo. Research personnel provided participants with a welcome video that included information on the trial, the study drugs, adverse events, and follow-up procedures. Clinicians providing usual care in public health facilities typically focus on management of symptoms by providing antipyretics or recommending antibiotics for suspected bacterial pneumonia.
End Point Assessments
Symptom and event assessments were done virtually. Mid-turbinate nasal swabs were self-collected on days 3 and 7 after randomization.
Subgroups
We defined subgroups a priori, as in previous evaluations in our platform trial (2, 13–15). These included age (<50 and ≥50 years), sex, symptom onset (≤3 or >3 days), SARS-CoV-2 vaccination status, and obesity (BMI ≤30 or >30 kg/m2). We applied the ICEMAN (Instrument for assessing the Credibility of Effect Modification Analyses) tool for subgroup credibility (18).
Statistical Analysis
The adaptive design protocol and the master statistical analysis plan (available at Annals.org) provide details on sample size calculation and statistical analyses (12). This platform trial is adaptive and allows for sample size reassessment based on the results of interim analyses. To plan for each group, we assumed a minimum clinical utility of 37.5% (relative risk reduction) to achieve 80% power with a 2-sided type I error rate of 0.05 for a pairwise comparison against the placebo, assuming a control event rate of 15%. This resulted in an initial plan to recruit 681 participants per group. Given the pace at which patients were recruited and the fact that recruitment continued during the interim analysis by the data and safety monitoring committee, our total number of patients was 738 per group. The statistical team conducted planned interim analyses. Stopping thresholds for futility were established if the posterior probability of superiority was less than 40% at the interim analysis. A group can be stopped for superiority if the posterior probability of superiority meets the threshold of 97.6%. Our decision rules were calibrated using simulations to meet the type I error rate.
We applied a Bayesian framework for our primary outcome analysis and a frequentist approach for all sensitivity analyses and secondary outcomes. Performing Bayesian analysis will allow us to report the posterior probability of treatment efficacy at the end of the trial independent of the decisions made along the way. Posterior efficacy for the primary outcome is calculated using the β-binomial model for event rates as detailed in the statistical analysis plan (19), assuming informed priors based on the observational data for both placebo and fluvoxamine. Our analyses were limited to the concurrently randomly assigned population. We assessed subgroup effects according to the preplanned statistical analysis plan. Missing data were not imputed.
We assessed time-to-event outcomes using Cox proportional hazards models, with right-censoring recorded whenever patients were lost to follow-up (including hospitalizations not related to COVID-19) or at the end of the 28-day follow-up if no event was recorded before then. Time-to-event analyses that were not adjusted for competing risks and numerical secondary outcomes were performed using the default Bayesian implementation of the Cox proportional hazards model in the brms library in R. Full details of these methods are provided in the Supplement. We assessed binary outcomes using the β-binomial model for binomial proportions. Model assumptions were evaluated by testing for proportionality (20). Relative risk was calculated using Bayesian β-binomial distribution to estimate the probability of an event in the treatment group divided by the probability of an event in the placebo group. All analyses were performed in accordance with a priori analysis plans using R, version 4.0.3 (R Foundation for Statistical Computing).
Data and Safety Monitoring Committee
A data and safety monitoring committee provided independent oversight for this trial. We planned a final interim analysis of the fluvoxamine–budesonide group based on data up to 6 July 2022. We present follow-up of all patients up to 28 days.
Role of the Funding Source
The funders had no role in the study design; collection, analysis, or interpretation of the data; writing of the manuscript; or the decision to submit the manuscript for publication.
Results
We screened 5374 potential participants for inclusion in this phase of the platform trial (Figure 1). The trial enrolled its first participant on 2 June 2020, and enrollment into the fluvoxamine–budesonide group began on 15 January 2022. By 6 July 2022, 1476 recruited participants had been randomly assigned to fluvoxamine–budesonide (n = 738) or placebo (n = 738). Four additional participants who did not meet the eligibility criteria were erroneously randomly assigned and are not included in the analysis. The median age of the participants was 51 years (range, 18 to 102 years), and 898 (60.8%) were women (Table 1). Most participants (n = 1419 [96.1%]) self-identified as mixed race, 36 (2.4%) self-identified as White, and 20 (1.4%) self-identified as Black or of African heritage. Because the trial is ongoing, we provide descriptive summaries of only participants who were randomly assigned to fluvoxamine–budesonide or placebo during the same period. The groups were generally well balanced with respect to age, BMI, and comorbidities (Table 1). The majority of patients had received at least 1 dose of COVID-19 vaccine, although a slightly higher proportion in the placebo group were unvaccinated compared with the treatment group (3.0% vs. 1.6%). The median number of days with symptoms before randomization was 3 (SD, 1.86).
Figure 1. Participant flow chart.

Table 1.
Characteristics of Patients in the TOGETHER Trial, by Treatment Allocation

Primary Outcomes
The data and safety monitoring committee met 4 times after the protocol initiation and last met on 6 July 2022, at which time it recommended that the trial stop randomly assigning patients to the fluvoxamine–budesonide group because the comparison had met the prespecified superiority threshold (97.6%) for the primary end point. Overall, 1.8% (13 of 738; 95% credible interval [CrI], 1.1% to 3.0%) of patients randomly assigned to fluvoxamine–budesonide were hospitalized or retained in an acute care emergency setting for 6 hours or longer due to COVID-19 compared with 3.7% (27 of 738; CrI, 2.5% to 5.3%) in the matched placebo group (Table 2). Based on the Bayesian β-binomial model, fluvoxamine–budesonide reduced the composite primary end point of hospitalization or retention in an emergency setting for 6 hours or more due to COVID-19 (relative risk, 0.50 [95% CrI, 0.25 to 0.92]; risk difference, 0.019 [95% CrI, 0.017 to 0.020]) in the intention-to-treat population (Figure 2). The probability that the event rate was lower in the fluvoxamine–budesonide group compared with the placebo group was 98.7%. The number needed to treat was 53.
Table 2.
Outcome Events and Relative Effects of Fluvoxamine–Budesonide Versus Placebo

Figure 2. Subgroup analyses comparing fluvoxamine–budesonide versus matched placebo.

In the prespecified subgroup analyses, we found consistent evidence of treatment benefits with fluvoxamine–budesonide compared with matched placebo. BMI = body mass index; CrI = credible interval; ITT = intention-to-treat.
Table 2 presents findings from secondary outcome analyses. We found that the direction of effect of the combination intervention was consistent across all outcomes. One death occurred in the trial: A patient receiving the combination intervention died 12 days after randomization from a pulmonary cause. We found no substantial difference in health care attendance among active patients (relative risk, 0.64 [95% CrI, 0.36 to 1.11]). In addition, no notable difference in time to recovery was detected between groups (hazard ratio, 1.02 [95% CrI, 0.91 to 1.13]). More adverse events occurred in the intervention group than in the placebo group, but no important differences between groups were detected.
Subgroup Analyses
In the prespecified subgroup analyses, we found no substantial differences in the treatment effect between fluvoxamine–budesonide and placebo among subgroups based on age, sex, number of days since symptom onset, or vaccination status (Figure 2). Estimates across the subgroups were generally consistent with the overall treatment effect.
Discussion
Our study is, to our knowledge, among the first to evaluate a drug combination for treatment of ambulatory patients with COVID-19 in a randomized trial. We found a reduction in the composite end point for COVID-19 disease progression with a combination of oral fluvoxamine, 100 mg twice daily, and inhaled budesonide. Our study builds on several previous trials that evaluated each drug independently. The combined effect seems to offer benefits over individual use of each drug. A difference from prior trials is that our trial was conducted in a population that was approximately 95% vaccinated. Given the safety, tolerability, ease of use, low cost, and widespread availability of these drugs, our findings may be useful for clinicians worldwide who are considering treating outpatients.
Both fluvoxamine and inhaled budesonide have been well described in the medical literature, yet neither has received strong universal recommendations for use by guideline groups. Our results are consistent with those of the earlier trials that evaluated the individual drugs. A meta-analysis that examined fluvoxamine, 100 mg 2 to 3 times per day, estimated a clinically useful reduction in risk for hospitalization of about 25% in unvaccinated persons (4, 21, 22). Low-dose fluvoxamine (50 mg twice daily) did not have a clinical benefit (9), indicating that 100 mg twice daily is probably the minimum effective dose. For inhaled budesonide, 2 randomized trials from early in the pandemic found reductions in time to recovery, hospitalization, and acute care (5, 6). Recently, the ACTIV-6 collaborators found that, compared with placebo, use of inhaled fluticasone furoate was negative for its primary outcome overall in a patient population with a high rate of vaccination similar to that in our study, yet a differential effect was observed by vaccination status (P = 0.020). Among those who were vaccinated, the fluticasone furoate group trended toward faster recovery (hazard ratio, 1.10 [95% CI, 0.95 to 1.28]) than those in the placebo group. This suggests that the protective effect of inhaled corticosteroids in early COVID-19 could be limited to inhaled budesonide as dosed—possibly as a consequence of the drug bioavailability or interaction at the lung epithelium—or may depend on preexisting primed immune status.
This is the tenth drug we have evaluated in the TOGETHER trial. Major strengths of the trial include the rapid recruitment and enrollment of patients at high risk for severe COVID-19. The period between first recruitment of a patient receiving this regimen and the final data cut for our trial was 219 days. Our recruitment strategy involves long-term engagement with the local public health system. We successfully administered a matched placebo for both our oral drug and our inhaled drug, and we used a stopping rule based on the primary end point. Our primary outcome is a composite of hospitalization for adjudicated COVID-19 and retention for physician observation for COVID-19 in an emergency setting (4, 21, 22) for longer than 6 hours. Specialized emergency settings were developed to respond to the epidemic in Brazil, and we considered prolonged observation and treatment in these settings to be equivalent in importance to hospitalization, as many patients who typically would have been hospitalized were prevented from being hospitalized because of hospital overcapacity. Our approach helped to identify persons experiencing clinical worsening even if capacity issues prevented them from being hospitalized, given that those kept under observation for 6 hours or longer tended to have more severe symptoms. Unlike many outpatient clinical trials, our study involves direct patient contact via medical students, nurses, and physicians who do at-home visits as well as follow-up via 21st century telecommunications.
Limitations of our trial include the low event rate overall, which is consistent with contemporary trials in a vaccinated population (9). Compared with interventions evaluated in 2021, our composite event rate decreased from an average of 16% in an unvaccinated population to 4.1% in a vaccinated population. This is likely due to the high rate of vaccination in our trial as well as potential previous exposure to SARS-CoV-2 among patients. However, it should be noted that the results of our subgroup analysis suggest that the treatment effect remained significant even after removal of unvaccinated patients from the analysis. As event rates have become lower over the course of the pandemic, it has become more difficult to determine which end points should be prioritized in clinical trials. Although other end points have been suggested, there is no consensus on the optimal outcomes, and regulatory authorities have not provided updated guidance.
Our trial found that the combination of fluvoxamine, 100 mg twice daily, and inhaled budesonide reduced the need for advanced medical care in this high-risk population. The absolute number of serious adverse events associated with this combination therapy was lower than in the placebo group. This study has implications for clinical management of patients globally. Although oral therapeutics for COVID-19 are available in the United States and, to a lesser extent, in other high-income countries, they are predominantly prescribed in elderly populations. These drugs are largely unavailable in low- and middle-income countries. For that reason, the use of repurposed drugs may be an important option for health care providers.
In conclusion, administration of the combination of fluvoxamine, 100 mg twice daily, and inhaled budesonide reduced the rate of COVID-19 progression resulting in prolonged observation in an emergency setting or hospitalization among outpatients with high risk for serious disease.
Supplementary Material
Footnotes
This article was published at Annals.org on 18 April 2023.
A list of the TOGETHER Investigators is provided in the Supplement.
References
- 1. Omi T, Tanimukai H, Kanayama D, et al. Fluvoxamine alleviates ER stress via induction of Sigma-1 receptor. Cell Death Dis. 2014;5:e1332. [PMID: ] doi: 10.1038/cddis.2014.301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Reis G, Dos Santos Moreira-Silva EA, Silva DCM, et al; TOGETHER investigators. Effect of early treatment with fluvoxamine on risk of emergency care and hospitalisation among patients with COVID-19: the TOGETHER randomised, platform clinical trial. Lancet Glob Health. 2022;10:e42-e51. [PMID: ] doi: 10.1016/S2214-109X(21)00448-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lenze EJ, Mattar C, Zorumski CF, et al. Fluvoxamine vs placebo and clinical deterioration in outpatients with symptomatic COVID-19: A randomized clinical trial. JAMA. 2020;324:2292-2300. [PMID: ] doi: 10.1001/jama.2020.22760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Guo CM, Harari O, Chernecki C, et al. Fluvoxamine for the early treatment of COVID-19: A meta-analysis of randomized clinical trials. Am J Trop Med Hyg. 2022;106:1315-20. [PMID: ] doi: 10.4269/ajtmh.21-1310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Guo CM, Harari O, Chernecki C, et al. Fluvoxamine for the early treatment of COVID-19: A meta-analysis of randomized clinical trials. Am J Trop Med Hyg. 2022;106:1315-20. [PMID: ] doi: 10.4269/ajtmh.21-1310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yu LM, Bafadhel M, Dorward J, et al; PRINCIPLE Trial Collaborative Group. Inhaled budesonide for COVID-19 in people at high risk of complications in the community in the UK (PRINCIPLE): a randomised, controlled, open-label, adaptive platform trial. Lancet. 2021;398:843-855. [PMID: ] doi: 10.1016/S0140-6736(21)01744-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sukhatme VP, Reiersen AM, Vayttaden SJ, et al. Fluvoxamine: A review of its mechanism of action and its role in COVID-19. Front Pharmacol. 2021;12:652688. [PMID: ] doi: 10.3389/fphar.2021.652688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Stanaland BE. Once-daily budesonide aqueous nasal spray for allergic rhinitis: a review. Clin Ther. 2004;26:473-92. [PMID: ] doi: 10.1016/s0149-2918(04)90050-1 [DOI] [PubMed] [Google Scholar]
- 9. Bramante CT, Huling JD, Tignanelli CJ, et al; COVID-OUT Trial Team. Randomized trial of metformin, ivermectin, and fluvoxamine for covid-19. N Engl J Med. 2022;387:599-610. [PMID: ] doi: 10.1056/NEJMoa2201662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Park JJH, Siden E, Zoratti MJ, et al. Systematic review of basket trials, umbrella trials, and platform trials: a landscape analysis of master protocols. Trials. 2019;20:572. [PMID: ] doi: 10.1186/s13063-019-3664-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Woodcock J, LaVange LM. Master protocols to study multiple therapies, multiple diseases, or both. N Engl J Med. 2017;377:62-70. [PMID: ] doi: 10.1056/NEJMra1510062 [DOI] [PubMed] [Google Scholar]
- 12. Reis G, Moreira Silva EAS, Silva DCM, et al. A multi-center, adaptive, randomized, platform trial to evaluate the effect of repurposed medicines in outpatients with early coronavirus disease 2019 (COVID-19) and high-risk for complications: the TOGETHER master trial protocol. Gates Open Research. 2021;5:117. [Google Scholar]
- 13. Reis G, Moreira Silva EADS, Medeiros Silva DC, et al; TOGETHER Investigators. Effect of early treatment with hydroxychloroquine or lopinavir and ritonavir on risk of hospitalization among patients with COVID-19: the TOGETHER randomized clinical trial. JAMA Netw Open. 2021;4:e216468. [PMID: ] doi: 10.1001/jamanetworkopen.2021.6468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Reis G, Dos Santos Moreira Silva EA, Medeiros Silva DC, et al. Effect of early treatment with metformin on risk of emergency care and hospitalization among patients with COVID-19: The TOGETHER randomized platform clinical trial. Lancet Reg Health Am. 2022;6:100142. [PMID: ] doi: 10.1016/j.lana.2021.100142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Reis G, Silva EASM, Silva DCM, et al; TOGETHER Investigators. Effect of early treatment with ivermectin among patients with covid-19. N Engl J Med. 2022;386:1721-1731. [PMID: ] doi: 10.1056/NEJMoa2115869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Reis G, Moreira Silva EAS, Medeiros Silva DC, et al; TOGETHER Investigators. Early treatment with pegylated interferon lambda for covid-19. N Engl J Med. 2023;388:518-528. [PMID: ] doi: 10.1056/NEJMoa2209760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. WHO Working Group on the Clinical Characterisation and Management of COVID-19 infection. A minimal common outcome measure set for COVID-19 clinical research. Lancet Infect Dis. 2020;20:e192-e197. [PMID: ] doi: 10.1016/S1473-3099(20)30483-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Schandelmaier S, Briel M, Varadhan R, et al. Development of the Instrument to assess the Credibility of Effect Modification Analyses (ICEMAN) in randomized controlled trials and meta-analyses. CMAJ. 2020;192:E901-E906. [PMID: ] doi: 10.1503/cmaj.200077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Reis G, Silva E, Silva D, et al. A multi-center, adaptive, randomized, platform trial to evaluate the effect of repurposed medicines in outpatients with early coronavirus disease 2019 (COVID-19) and high-risk for complications: the TOGETHER master trial protocol. Gates Open Research. 2021;5. [Google Scholar]
- 20. Grambsch PM, Therneau TM. Proportional hazards tests and diagnostics based on weighted residuals. Biometrika. 1994;81:515-526. [Google Scholar]
- 21. Lee TC, Vigod S, Bortolussi-Courval É, et al. Fluvoxamine for outpatient management of COVID-19 to prevent hospitalization: A systematic review and meta-analysis. JAMA Netw Open. 2022;5:e226269. [PMID: ] doi: 10.1001/jamanetworkopen.2022.6269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mills EJ, Thorlund K, Ioannidis JP. Calculating additive treatment effects from multiple randomized trials provides useful estimates of combination therapies. J Clin Epidemiol. 2012;65:1282-8. [PMID: ] doi: 10.1016/j.jclinepi.2012.07.012 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.