

Abstract
Background
Hypertension is associated with gut dysbiosis, altered intestinal immunity, and gut pathology in animal models and humans. Although these findings have implicated impaired interactions between gut and gut microbiota in hypertension, little is known about the specific functional gut microbes that interact with intestinal mucosa.
Methods and Results
To identify these microbes, we sorted Immunoglobin A (IgA)‐coated (IgA+) and IgA‐noncoated (IgA−) bacteria using a combination of magnetic‐activated cell sorting and fluorescence‐activated cell sorting, and subsequently performed 16 S rRNA gene sequencing (IgA‐SEQ) to determine the microbial composition of IgA+ and IgA− fractions in spontaneously hypertensive rats (SHR) and normotensive Wistar Kyoto rats. We observed a significant decrease in IgA+ bacteria in SHR compared with Wistar Kyoto and a distinct composition of IgA+ and IgA− bacteria between Wistar Kyoto and SHR, showing more IgA‐bound Proteobacteria, Bacteroidetes and Actinobacteria but less of Firmicutes in SHR at the phylum level. We further identified enriched IgA‐coated Romboutsia, Turicibacter, Ileibacterium, and Dubosiella in SHR that were negatively correlated with the various pathways including antigen presentation, immune response, cell junction organization, epithelium development, and defense response to virus.
Conclusions
We demonstrate new IgA‐coated bacteria that participate in host‐microbiota communication in hypertension, suggesting promising therapeutic interventions targeting these bacteria for hypertension management.
Keywords: host‐microbiota communication, hypertension, Immunoglobin A‐coated bacteria
Subject Categories: Hypertension
Nonstandard Abbreviations and Acronyms
- SHR
spontaneously hypertensive rat
Gut microbiota‐host communication modulates host immune responses and thereby blood pressure (BP). 1 The gut functions as a reservoir for gut microbial communities that develop immune cell differentiation and tolerance. Our previous studies established that gut dysbiosis and altered gene expression in gut epithelium are linked to hypertension in spontaneously hypertensive rats (SHR), 2 , 3 , 4 an animal model of hypertension. However, the specific gut commensals that directly interact with the host to contribute to the development of hypertension remain elusive.
Immunoglobin A (IgA) is the first line of defense in dynamic gut‐microbiota communication at the mucosal surface. Homeostatic IgA response depends on a highly polyreactive repertoire to bind a broad subset of microbiota with low affinity. 5 In contrast, T cell‐dependent IgA response is elicited by mucosal microbes with potential pathogenicity. This binding has high affinity and promotes clearance and inhibition to the challenging microbial antigens. The high‐affinity IgA binding to microbial antigens results in immune exclusion, which plays an important role in gut homeostasis by preventing colonization and eliminating harmful bacteria. 6 For example, inflammatory commensals that confer inflammatory bowel disease pathophysiology are identified by high IgA coating. 6 Furthermore, IgA‐coated bacteria in dysbiotic gut are emerging as relevant bacterial communities in the pathogenesis of many diseases (ie, multiple sclerosis, Alzheimer disease, Parkinson disease, diabetes, and obesity, etc). 1 Although limited information is available on IgA‐microbiota interactions in hypertension, a cross‐sectional study with 12 373 participants suggested that higher circulating IgA levels correlated with higher BP. 1 Recently, Saha et al reported that microbiota‐mediated increases in IgA level were associated with a higher BP in SHR. 7 Thus, we tested the hypothesis that the SHR exhibits a distinct IgA‐coated microbial profile that may contribute to altered gut pathology in hypertension.
Methods
The data that support the findings of this study are available from Dr. Tao Yang (tao.yang2@utoledo.edu) upon reasonable request.
Fecal IgA Flow Cytometry and Sorting of IgA + and IgA − Bacteria
Male 14‐week‐old Wistar Kyoto (WKY) rats (mean systolic BP, 132±6 mm Hg) and SHR (mean systolic BP, 209±5 mm Hg) from Charles River were acclimated to housing conditions for 2 weeks. The sample size was n=5 to 7 per group determined based on the power analysis of previous experiments. Two rats per cage were housed in specific pathogen‐free, positive‐ventilated cages with access to irradiated food (#7912; Envigo, Indianapolis, IN), sterile water ad libitum, and autoclaved corn‐cob bedding (Laboratory Supply, Fort Worth, TX). The body weights of WKY and SHR rats were 280±7 g and 270±3 g, respectively. Systolic BP of 13‐week SHR and WKY rats was measured by Tail‐Cuff Plethysmography (Visitech Systems, a newly designed and validated noninvasive computerized tail‐cuff system) with the plate temperature set at 35 °C in an animal surgery operating room. The rats were trained for tail‐cuff BP measurements for 7 days before starting the experiments. The systolic BP of a day per rat was determined by the average of 2 sets of 10 measurements. All studies were approved by the Institutional Animal Care and Use Committee of the University of Florida. Rats were maintained in accordance with the Animal Welfare Act and the Public Health Policy on Humane Care.
As described previously, 6 fecal pellets were mixed at the ratio of 100 mg fecal material per 1 mL PBS in the Fast Prep Lysing Matrix D tubes (with ceramic beads) (MP Biomedicals) and incubated on ice for 1 hour. After incubation, the fecal suspension was homogenized by bead beating for 5 seconds (Minibeadbeater; Biospec) and 100 μL per sample of supernatant was collected after centrifugation (50 g, 15 minutes, 4 °C). Fecal bacteria in the supernatants were washed once with 1 mL PBS containing 1% (w/v) bovine serum albumin (BSA, American Bioanalytical; staining buffer); 1 mL of staining buffer was used to resuspend the washed bacteria; a 20‐μL aliquot (“pre‐sort”) was used for 16 S sequencing analysis; and the rest was collected after centrifugation or 5 minutes (8000g, 4°C) and resuspended in 100 μL blocking buffer (staining buffer containing 20% Normal Mouse Serum, Jackson ImmunoResearch). The resuspended fraction was incubated on ice for 20 minutes and then stained with 100 μL staining buffer containing Goat Anti‐Rat IgA alpha chain (fluorescein isothiocyanate) (1:12.5; Abcam, ab97184) for 30 minutes on ice; 1 mL of staining buffer was used to wash the samples 3 times before flow cytometric analysis or cell separation. Anti‐IgA stained fecal bacteria were incubated in 1 mL staining buffer containing 50 μL Anti‐fluorescein isothiocyanate magnetic activated cell sorting beads (Miltenyi Biotec) at 4 °C for 15 minutes. Magnetic activated cell sorting beads were washed twice with 1 mL staining buffer before being sorted; 50 μL of IgA negative fraction was collected for 16 S sequencing analysis. The IgA‐positive fraction was subjected to fluorescence activated cell sorting (FACSAria; BD Biosciences); 2 million IgA‐positive bacteria were collected from cell sorting for 16 S sequencing analysis.
Bioinformatic Analysis and 16 S rRNA Gene Sequencing
Pre‐sort, IgA‐ and IgA+ samples were used for microbiota analysis. 8 QIAamp PowerFecal DNA kit (QIAGEN, Germantown, MD) was used to extract total bacterial DNA following the manufacturer's protocol. DNA was eluted in Tris‐EDTA (TE) buffer (0.1 mmol/L EDTA, Tris–Hydrogen chloride (HCl) buffer, 10 mmol/L, pH 8.5). NanoDrop (Thermo Fisher, Waltham, MA) was used to determine the DNA concentration. For 16 S library preparation, bacterial DNA was amplified according to the Illumina User Guide for 16 S Metagenomic Sequencing Library Preparation section Preparing 16 S Ribosomal RNA Gene Amplicons for the Illumina MiSeq System (Part # 15044223 Rev. B). The library was followed by clean‐up, normalization, and pooling. Illumina primers used for amplification of the 16 S rRNA gene‐targeted the V3‐V4 region were: TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGCCTACGGGNGGCWGCAG and GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGGGACTACHVGGGTWTCTAAT.
Raw 16 S sequencing data were analyzed by QIIME2 (version 2021.11). 8 The taxonomic assignment was performed using amplicon sequence variants data against SILVA (version 132). The obtained genus feature taxonomical data were further subjected to MicrobiomeAnalyst6. The read depth was 12752.4±15.4 reads per sample. Data were prefiltered to remove any genus feature that was <4 reads or existed in <20% of the samples. For principal coordinate analysis, Bray–Curtis index was used with analysis of similarities. For heatmap, data scaling was done based on total sum scaling. Euclidean and average algorithms were used for distance measurement and clustering, respectively.
Correlation Analysis
Correlation analyses were performed on the normalized abundance of fecal IgA‐SEQ data and gene expression values from RNA‐seq data. 4 Briefly, total RNA was extracted from colonic organoids of SHR and WKY rats with RNeasy Plus Mini Kit (Qiagen). Total RNA with 28 S/18 S >1 and RNA integrity numbers 7.1 to 9.3 were used for RNA‐Seq library construction. The cDNA was generated using SMART‐Seq HT kit (Takara Bio). RNA‐Seq libraries were constructed with Nextera DNA Flex Library Prep kit and Nextera DNA Unique Dual Indexes Set A (Illumina) and sequenced on NovaSeq6000 (Illumina) at the University of Florida NextGen DNA Sequencing Core Facility (at 2×150 bp, aiming for 50 million paired end reads per sample). TapeStation was used to analyze the size distribution revealing an average DNA fragment size of 600 bp (range, 150–1500 bp) in the library. Quantitative polymerase chain reaction assays were performed for library quantification. RNA seq analysis was performed using CLC genomics workbench (Qiagen 21.0.4). Data were normalized to reads per kilobase of transcript per million mapped reads. False discovery rate‐corrected P value <0.05 was used as the criterion for significance. The 2 sets of data come from the same rats. Pearson correlation coefficients (r) were generated for all samples regardless of group using MATLAB software with a cutoff value of 0.811 (n=5, P<0.05) and displayed using pixel maps as described previously. 1 The Gene Ontology database was used for annotating the pathways of the differentially expressed genes.
Results
Flow cytometric analysis of rat fecal bacteria stained with anti‐rat IgA‐fluorescein isothiocyanate showed a significant decrease in IgA‐coated (IgA+) bacteria in SHR compared with WKY (Figures 1A and 1B). We subsequently isolated IgA+ and IgA‐noncoated (IgA−) bacteria using a combination of magnetic‐activated cell sorting and fluorescence‐activated cell sorting. The sorted bacteria were subjected to 16 S rRNA gene sequencing (IgA‐SEQ) to determine the microbial composition of IgA+ and IgA− fractions in the WKY and SHR. Principal coordinates analysis (PCoA) demonstrated a clear separation between SHR and WKY rats, regardless of IgA binding (Figure 1C). Distinct clusters were found between IgA+ WKY and IgA+ SHR (Figure 1C). Additionally, the IgA− and IgA+ microbial profiles of the SHR rats were clearly separated (Figure 1C). SHR showed increased Proteobacteria, Bacteroidetes and decreased Firmicutes in pre‐ and IgA− fractions, compared with WKY (Figure 1D). Furthermore, Proteobacteria and Actinobacteria were significantly more bound in the SHR whereas IgA‐bound Firmicutes was reduced (Figure 1E).
Figure 1. Immunoglobulin A (IgA)‐coated bacteria are specifically enriched in spontaneously hypertensive rat (SHR) vs Wistar Kyoto (WKY) stools.
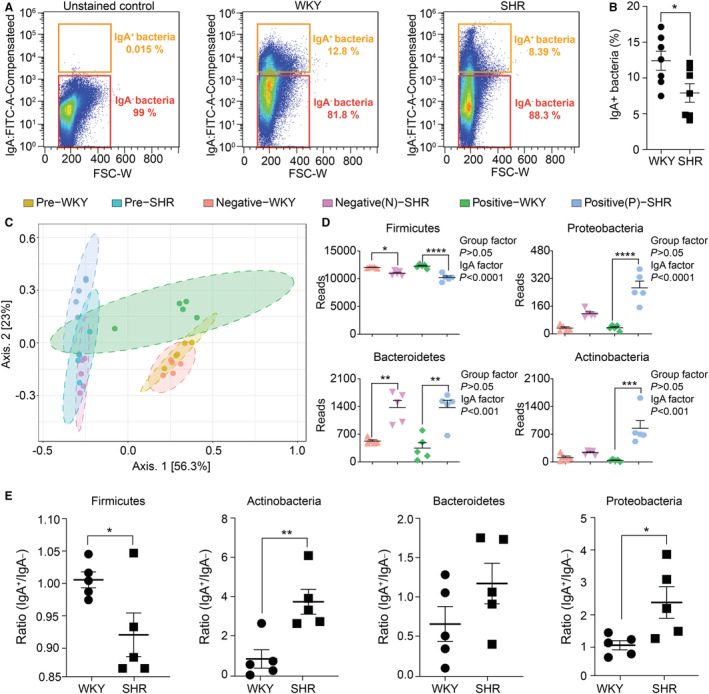
Fecal bacteria isolated from stool samples from WKY (n=7, mean systolic blood pressure, 132±6 mm Hg) and SHR (n=7, mean systolic blood pressure, 209±5 mm Hg) rats were stained with fluorescein isothiocyanate‐conjugated anti‐rat IgA to perform fecal IgA flow cytometry analysis and sort of IgA+ and IgA‐ bacteria. 6 Next, pre‐sort, IgA‐ and IgA+ samples were used for 16 S rRNA gene sequencing and bioinformatic analysis. 8 A, Representative IgA‐staining of fecal bacteria, unstained control (left), WKY (middle), and SHR (right). The unstained control is a 1:1 mix of fecal bacteria of both WKY and SHR. B, Cumulative data, percentage of IgA‐coated, and noncoated bacteria in stool of WKY and SHR rats determined by flow cytometry (mean±SEM), n=7/group. *P<0.05, unpaired t‐test. C, Two‐dimensional principal coordinate analysis plot of total, IgA‐, and IgA+ gut microbiota in WKY and SHR. In analysis of similarities analysis, R=1, P<0.01 for IgA‐ vs IgA+ SHR; R=0.54, P<0.01 for IgA‐ vs IgA+ WKY; R=0.84; P<0.01 for IgA+ WKY vs IgA+ SHR; n=5/group. D, The reads of IgA‐ and IgA+ gut microbiota of major phyla in WKY and SHR; n=5/group. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001 in 2‐way ANOVA followed by Sidak multiple comparison. E, The comparison of IgA+/IgA‐ ratio of 4 major phyla between WKY and SHR. Lower ratio indicates less preference of IgA binding, while higher ratio indicates more preference of IgA binding; n=5/group.*P<0.05, **P<0.01 in unpaired t‐test for Figure 1E. FITC‐A indicates fluorescein isothiocyanate‐area; FSC‐W, forward scatter‐width; IgA, immunoglobulin A; SBP, systolic blood pressure; SHR, spontaneously hypertensive rat; and WKY, Wistar Kyoto.
To further differentiate microbiota specifically bound by IgA in SHR, a heatmap was generated. Two groups of bacteria were identified: (1) bacteria identified in red font were abundant in WKY and SHR gut microbiota but were not bound by endogenous IgA in either WKY or SHR (Figure 2), suggesting that these bacteria either do not generate IgA responses or were not recognized by IgA. (2) Despite low abundances in both WKY and SHR, bacteria identified in green font were coated only by endogenous IgA in the SHR (Figure 2), indicating their crucial roles in inducing host IgA responses in the SHR, but not WKY. Importantly, no bacteria were identified to be preferentially bound by IgA in the WKY, but not SHR (Figure 2).
Figure 2. Heatmap was generated using the reads assigned at the genus level.
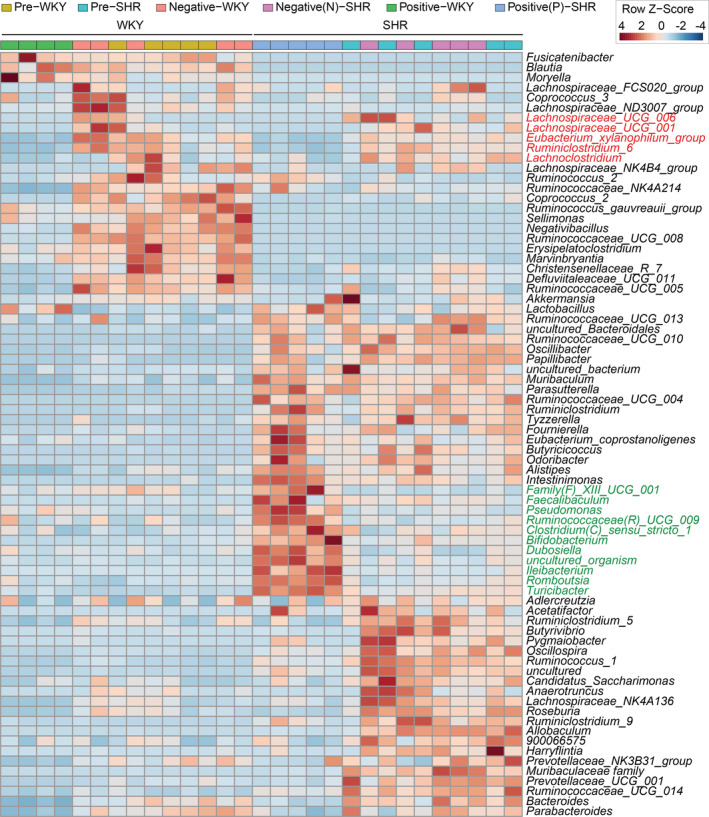
Distance was measured using Euclidean method. Samples were clustered by average linkage (average of the distance of all points in each group) algorithm (n=5/group). SHR indicates spontaneously hypertensive rat; and WKY, Wistar Kyoto.
Pearson correlation analysis of the abundance of IgA+ bacterial genera with RNA‐seq gene expression profiling of gut epithelium data 1 demonstrated that Romboutsia, Turicibacter, Ileibacterium, and Dubosiella were negatively correlated with the various pathways including antigen presentation, immune response, cell junction organization, epithelium development, and defense response to virus (Figure 3).
Figure 3. Correlation analyses were performed on the normalized abundance of immunoglobulin A‐coated bacterial genera data and gene expression values from RNA‐seq data.
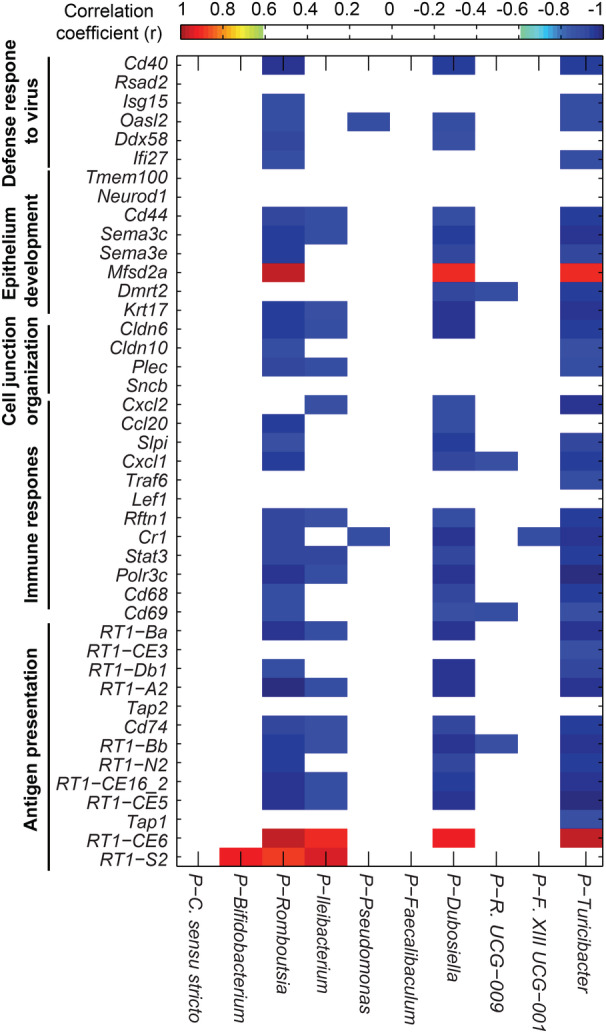
Pearson correlation coefficients (r) were generated for all samples from both groups using MATLAB software with a cutoff value of 0.811 (n=10, P<0.05) and displayed using pixel maps. Red‐highlighted genera are positively correlated, and blue‐highlighted genera are negatively correlated. 4 The heatmap presents the r values of correlation analysis between bacterial abundance and the epithelium gene expression of its corresponding host in both Wistar Kyoto and spontaneously hypertensive rats. C indicates Clostridium; F, family; P, positive; R, Ruminococcaceae; SHR, spontaneously hypertensive rats; and WKY, Wistar Kyoto.
Discussion
In this study, we used novel IgA‐SEQ analysis to functionally dissect gut microbiota based on their recognition by the host IgA. The immune system is highly involved in the pathogenesis of hypertension. 9 It is known that B cell contributes to the development of hypertension. 10 , 11 Given the close communication between gut microbiota and IgA, we proposed to study the differences in IgA‐coated microbial profiles between WKY and SHR. We demonstrated that, in the SHR, less bacteria were coated by endogenous IgA. At the phylum level, the ratio of IgA+ over IgA− binding of Proteobacteria and Actinobacteria was higher in the SHR than in the WKY. This suggests that Proteobacteria and Actinobacteria were preferentially bound by specific IgA in the SHR, which may result in immune exclusion. In contrast, the differences in Firmicutes and Bacteroidetes abundances between IgA+ WKY and IgA+ SHR are comparable with those between pre‐WKY and pre‐SHR, suggesting no specificity for these 2 phyla by endogenous IgA, which may result in immune inclusion.
To further dissect the function of this gut microbiota‐immune IgA communication, we analyzed the microbial composition at the genus level. Most of these bacteria (ie Faecalibaculum, Clostridium sensu stricto 1, Bifidobacterium, Romboutsia, Turicibacter) identified to be preferentially bound by IgA in the SHR are considered beneficial because of their butyrate‐producing capability and/or negative association with diseases. 12 This prompts the suggestion that their increased binding to the IgA could result in immune exclusion of these beneficial bacteria, promoting intestinal inflammation. Furthermore, the genera Turicibacter, Ileibacterium, and Dubosiella belong to Erysipelotrichaceae, a highly IgA‐coated family associated with inflammation‐related gastrointestinal diseases such as inflammatory bowel disease (IBD) and colorectal cancer. 13 Correlation analysis found 4 IgA‐coated bacteria genera, Romboutsia, Turicibacter, Ileibacterium, and Dubosiella, negatively correlated with genes that belong to multiple homeostasis pathways. The influence of these bacteria on mucosal epithelial development (defense responses to viruses, intestinal cell junctional complexes, etc.) may be fundamental to impaired host‐microbiota communication associated with hypertension‐linked gut pathophysiology. Noteworthy, the correlation did not reach significance within each group of rats, which could be attributable to a smaller sample size or variations of the host. Nonetheless, previous studies have demonstrated the important mutual interactions between gut microbiota and hypertension in the development of hypertension, and one could be an initiator that changes the other. 14 , 15 Our study identified 4 bacterial genera that could be important in driving the overactivation of the immune system in the SHR, which could be targeted for therapeutic purposes.
Sources of Funding
This work was supported by the following grants: National Institutes of Health National Heart, Lung, and Blood Institute grants HL033610, HL110170, and HL132448, and American Heart Association Career Development Award 852969.
Disclosures
None.
For Sources of Funding and Disclosures, see page 6.
References
- 1. Richards EM, Li J, Stevens BR, Pepine CJ, Raizada MK. Gut microbiome and neuroinflammation in hypertension. Circ Res. 2022;130:401–417. doi: 10.1161/CIRCRESAHA.121.319816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Yang T, Santisteban MM, Rodriguez V, Li E, Ahmari N, Carvajal JM, Zadeh M, Gong M, Qi Y, Zubcevic J, et al. Gut dysbiosis is linked to hypertension. Hypertension. 2015;65:1331–1340. doi: 10.1161/HYPERTENSIONAHA.115.05315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yang T, Li H, Oliveira AC, Goel R, Richards EM, Pepine CJ, Raizada MK. Transcriptomic signature of gut microbiome‐contacting cells in colon of spontaneously hypertensive rats. Physiol Genomics. 2020;52:121–132. doi: 10.1152/physiolgenomics.00087.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Li J, Richards EM, Handberg EM, Pepine CJ, Raizada MK. Distinct gene expression profiles in colonic organoids from normotensive and the spontaneously hypertensive rats. Cells. 2021;10:1523. doi: 10.3390/cells10061523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bunker JJ, Erickson SA, Flynn TM, Henry C, Koval JC, Meisel M, Jabri B, Antonopoulos DA, Wilson PC, Bendelac A. Natural polyreactive IgA antibodies coat the intestinal microbiota. Science. 2017;358:eaan6619. doi: 10.1126/science.aan6619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Palm NW, de Zoete MR, Cullen TW, Barry NA, Stefanowski J, Hao L, Degnan PH, Hu J, Peter I, Zhang W, et al. Immunoglobulin a coating identifies colitogenic bacteria in inflammatory bowel disease. Cell. 2014;158:1000–1010. doi: 10.1016/j.cell.2014.08.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Saha P, Mell B, Golonka RM, Bovilla VR, Abokor AA, Mei X, Yeoh BS, Doris PA, Gewirtz AT, Joe B, et al. Selective IgA deficiency in spontaneously hypertensive rats with gut dysbiosis. Hypertension. 2022;79:2239–2249. doi: 10.1161/HYPERTENSIONAHA.122.19307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yang T, Mei X, Tackie‐Yarboi E, Akere MT, Kyoung J, Mell B, Yeo J‐Y, Cheng X, Zubcevic J, Richards EM, et al. Identification of a gut commensal that compromises the blood pressure‐lowering effect of ester angiotensin‐converting enzyme inhibitors. Hypertension. 2022;79:1591–1601. doi: 10.1161/HYPERTENSIONAHA.121.18711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Drummond GR, Vinh A, Guzik TJ, Sobey CG. Immune mechanisms of hypertension. Nat Rev Immunol. 2019;19:517–532. doi: 10.1038/s41577-019-0160-5 [DOI] [PubMed] [Google Scholar]
- 10. Chan CT, Sobey CG, Lieu M, Ferens D, Kett MM, Diep H, Kim HA, Krishnan SM, Lewis CV, Salimova E, et al. Obligatory role for B cells in the development of angiotensin II‐dependent hypertension. Hypertension. 2015;66:1023–1033. doi: 10.1161/HYPERTENSIONAHA.115.05779 [DOI] [PubMed] [Google Scholar]
- 11. Dingwell LS, Shikatani EA, Besla R, Levy AS, Dinh DD, Momen A, Zhang H, Afroze T, Chen MB, Chiu F, et al. B‐cell deficiency lowers blood pressure in mice. Hypertension. 2019;73:561–570. doi: 10.1161/HYPERTENSIONAHA.118.11828 [DOI] [PubMed] [Google Scholar]
- 12. Zagato E, Pozzi C, Bertocchi A, Schioppa T, Saccheri F, Guglietta S, Fosso B, Melocchi L, Nizzoli G, Troisi J, et al. Endogenous murine microbiota member Faecalibaculum rodentium and its human homologue protect from intestinal tumour growth. Nat Microbiol. 2020;5:511–524. doi: 10.1038/s41564-019-0649-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Shapiro JM, de Zoete MR, Palm NW, Laenen Y, Bright R, Mallette M, Bu K, Bielecka AA, Xu F, Hurtado‐Lorenzo A. Immunoglobulin a targets a unique subset of the microbiota in inflammatory bowel disease. Cell Host & Microbe. 2021;29:83–93.e3. doi: 10.1016/j.chom.2020.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Santisteban MM, Qi Y, Zubcevic J, Kim S, Yang T, Shenoy V, Cole‐Jeffrey CT, Lobaton GO, Stewart DC, Rubiano A, et al. Hypertension‐linked pathophysiological alterations in the gut. Circ Res. 2017;120:312–323. doi: 10.1161/CIRCRESAHA.116.309006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Vemuri R, Ruggiero A, Whitfield JM, Dugan GO, Cline JM, Block MR, Guo H, Kavanagh K. Hypertension promotes microbial translocation and dysbiotic shifts in the fecal microbiome of nonhuman primates. Am J Physiol Heart Circ Physiol. 2022;322:H474–H485. doi: 10.1152/ajpheart.00530.2021 [DOI] [PMC free article] [PubMed] [Google Scholar]