

Abstract
Background
Proper function of endothelial cells is critical for vascular integrity and organismal survival. Studies over the past 2 decades have identified 2 members of the KLF (Krüppel‐like factor) family of proteins, KLF2 and KLF4, as nodal regulators of endothelial function. Strikingly, inducible postnatal deletion of both KLF2 and KLF4 resulted in widespread vascular leak, coagulopathy, and rapid death. Importantly, while transcriptomic studies revealed profound alterations in gene expression, the molecular mechanisms underlying these changes remain poorly understood. Here, we seek to determine mechanisms of KLF2 and KLF4 transcriptional control in multiple vascular beds to further understand their roles as critical endothelial regulators.
Methods and Results
We integrate chromatin occupancy and transcription studies from multiple transgenic mouse models to demonstrate that KLF2 and KLF4 have overlapping yet distinct binding patterns and transcriptional targets in heart and lung endothelium. Mechanistically, KLFs use open chromatin regions in promoters and enhancers and bind in context‐specific patterns that govern transcription in microvasculature. Importantly, this occurs during homeostasis in vivo without additional exogenous stimuli.
Conclusions
Together, this work provides mechanistic insight behind the well‐described transcriptional and functional heterogeneity seen in vascular populations, while also establishing tools into exploring microvascular endothelial dynamics in vivo.
Keywords: 27 chromatin, endothelial cell, enhancer, Kruppel‐like factor, promoter, transcription
Subject Categories: Endothelium/Vascular Type/Nitric Oxide, Vascular Biology
Nonstandard Abbreviations and Acronyms
- ATAC‐seq
assay for transposase‐accessible chromatin using sequencing
- DEGs
differentially expressed genes
- DKO
double knockout
- EC
endothelial cells
- KLF
Krüppel‐like factor
Clinical Perspective
What Is New?
This work provides novel insights on how microvascular endothelial cells of different organs use the KLF (Krüppel‐like factor) transcription factors to orchestrate programs key to endothelial health.
What Are the Clinical Implications?
Understanding how these factors affect genome‐wide transcription provides insights into diseases of their dysregulation such as atherosclerosis and aneurysmal disease.
The maintenance of an intact vascular network is essential for organismal survival, enabling long distance delivery of oxygen, nutrients, hormones, and immunity to all tissues. In vertebrates, a single layer of endothelial cells (ECs) forms the lining of all vessels and maintains fundamental vascular properties such as selective permeability, blood fluidity, and vasomotor tone. The endothelium is also a dynamic and highly responsive tissue whose function can be altered by biomechanical (eg, blood flow) and biochemical (eg, cytokine) stimuli. Studies over the past decade have led to the appreciation that members of the KLF (Kruppel‐like factor) family of transcription factors play a crucial role in endothelial biology. In particular, 2 members of this family—namely KLF2 and KLF4—are enriched in the endothelium, regulated by flow and cytokines, and have been shown to be critical transcription factors for ECs. 1 , 2 , 3 , 4 , 5 , 6 , 7 , 8 In vivo studies in mice with endothelial‐specific deficiency of either KLF2 or KLF4 demonstrated enhanced susceptibility to vascular diseases such as atherosclerosis, suggesting redundancy. 3 , 5 , 9 However, the strongest evidence in support of this view is derived from studies assessing the effect of compound deletion of both genes. Postnatal deletion of both factors (double knockout [DKO]) in adult endothelium resulted in vascular collapse, leading to 100% mortality within 10 days. 10 Intriguingly, the existence of only 1 allele of either KLF2 or KLF4 rescued survival despite biallelic loss of the other factor. Before death, the DKO animals developed profound vascular leakiness and disseminated intravascular coagulation, indicating a loss of endothelial integrity. Transcriptomic studies revealed massive genome‐wide dysregulation of endothelial gene expression in vivo, yet the underlying molecular mechanisms remain unclear.
KLF2 and KLF4 have been shown in cell‐based studies to directly regulate key endothelial genes, such as endothelial nitric oxide synthase and thrombomodulin via direct binding to promoter regions. However, the breadth of the endothelial transcriptome under control of KLF2/4, as elucidated in the aforementioned DKO study, reached ≈50% to 60% of all transcripts and is unlikely limited only to direct promoter binding. To systematically dissect the molecular mechanisms of KLF2/4 regulation of endothelial function, we used multi‐omics approaches to study how KLF2 and KLF4 regulate endothelial genes in primary ECs from multiple vascular beds. Here, we provide an explanation for the redundancy observed between endothelial KLF2 and KLF4 by demonstrating KLF‐driven enhancer networks that establish core endothelial transcriptional programs that are shared across heart and lung vasculature. In addition to the shared functions of KLF2 and KLF4, we also establish unique binding patterns of these individual factors. Finally, we demonstrate that ECs from different tissues use KLFs uniquely, further establishing tissue‐specific EC heterogeneity.
METHODS
Mice
Endothelial‐specific inducible knockout of KLF2 and KLF4 (DKO) was created as previously described. 10 Briefly, VE‐Cadherin Cre mice with an estrogen receptor responsive element (Cdh5(PAC)‐Cre/ERT2 Mouse, Taconic 13 073, originally from R. Adams, University of Münster, Münster, Germany) were mated to KLF2 floxed or KLF4 floxed mice. To trigger endothelial‐specific deletion of KLF2 and KLF4, 8‐ to 10‐week‐old mice were intraperitoneally injected with tamoxifen (2 mg/25 g) (MP Biomedicals). Cdh5(PAC)‐Cre/ERT2 mice were used as a control group. Five days after tamoxifen injection, cells were harvested from the mice as detailed below.
KLF2‐ and KLF4‐tagged mice were created by Cyagen Inc. CRISPR‐Cas9‐mediated genome engineering was used to knock‐in the 3XFLAG‐AviTag into the endogenous locus of KLF2 or KLF4; 3XFLAG‐AviTag was inserted immediately after the ATG codon (the recombinant protein has an N‐terminal tag). All mice were sequenced for correct insertion of the donor template and off‐target insertion was assayed and not noted.
EC Isolation
Mouse heart and lungs were perfused, excised, minced, digested with type I collagenase, and then were mechanically disrupted and filtered through a 70‐μm cell strainer to get a single cell suspension. Cells were immediately stained for fluorescence‐activated cell sorting analysis. CD31+/CD45‐ cells were isolated as ECs for downstream analyses.
RNA‐Sequencing and Analysis
RNA was extracted from ECs using the Qiagen RNeasy mini prep kit after homogenizing by passing through a shredder column (n=3 mice of each genotype). RNA quantity and quality were checked using a NanoDrop (ThermoFisher) and Fragment Analyzer (Agilent, Santa Clara, CA), respectively. Samples that had an RNA integrity number of 6.8 or higher were used for this study. We used the NEBNext Ultra RNA Library Prep Kit for cells and a Directional RNA Library Prep Kit from Illumnia to generate strand‐specific libraries for ECs. The analyses of the RNA‐seq data sets were performed through the Case Western Reserve University genomics core. Before sequence alignment, trimgalore (version 0.4.3) with cutadapt package (version 1.12) was used for adaptor trimming and to improve data quality. For transcriptome alignment, we used STAR with Gencode reference features. We then mapped sequencing reads to the mouse reference genome (mm10) using STAR aligner. All data sets are deposited under the GEO accession number GSE195928 and are accessible to the scientific community. For single knockout and DKO heart EC samples, data were downloaded from GSE92965 for subsequent analysis. Lung and heart EC data were all reprocessed together using the same pipelines for this article.
DEGs and Functional Annotation
DESeq2 packages were used for differential expression analysis (cutoffs of log2FC=1, P<0.05). Negative binomial distribution and a shrinkage estimator for the distribution's variance were used. False discovery rate was calculated using the Benjamini‐Hochberg method for all comparisons. Variance stabilizing transformation in DESeq2 was used for normalizing count data. The normalized count data were used to create the heatmaps with Complex Heatmap Package in R or with pheatmap. The genes selected for RNA‐seq heatmaps were those with a false discovery rate of ≤0.05.
ATAC‐seq and Analysis
A total of 50000 to 70 000 flow‐sorted live ECs were used to prepare the ATAC‐seq library by preparing nuclei and applying a transposase reaction with 12 cycles of polymerase chain reaction (PCR) amplification (n=2 mice of each genotype). The library was purified using SPRI beads (double‐sided selection, 200‐500bp fragment size). We used Illumina Hi‐Seq system to sequence libraries. After initial quality checking with fastgc (0.11.90, we used TaRGET‐II ATAC‐Seq pipeline (https://github.com/Zhang‐lab) for analysis. TaRGET‐II includes quality trimming using cutadapt and trimmed reads (>36bp minimum alignment length) were mapped against the mm10 genome using Burrows‐Wheeler Aligner). We used de‐duplicated and uniquely mapped reads for peak calling analysis after excluding black‐list regions defined by ENCODE. The candidate peaks were predicted by callpeak implemented in MACS2. We used predicted open chromatin peaks where at least 2 biological replicates were reproducible for the downstream analysis using an irreducible discovery rate cutoff of 0.05. We also applied limma based edgeR method to determine differentially accessible regions; cutoff: log2FC>1.0, counts per million >2, P value <0.01.
ChIPmentation and Analysis
Flow‐sorted endothelial cells were fixed using 1% formaldehyde, and 200 000 cells were lysed and frozen in 100 μL SDS lysis buffer (50 mmol/L Tris/HCl, 0.5% SDS, and 10 mmol/L EDTA) supplemented with 1x cOmplete EDTA‐free protease inhibitor (Roche) (n=3 mice of each genotype, n=6 for heart acetylation of lysine 27 on histone 3 [H3K27ac]). For chromatin immunoprecipitation followed by sequencing (ChIP‐seq), anti‐FLAG (Cell Signaling Technologies, #8146), anti‐Pol II (Sigma, 05–623), anti‐p300 (Cell Signaling Technologies, 54 062), or anti‐H3K27ac (Abcam, ab4729) antibodies were added to Protein A/G‐coupled Dynabeads (Bimake, B23202) in PBS with 0.5% BSA and incubated with rotation for 4 hours at 4 degrees. For 100 000 to 150 000 cells, 25 μL of beads were incubated with 5‐μg antibody per ChIP. Aliquots were diluted with SDS lysis buffer and 100 μL containing the appropriate number of cells were processed. Cells were sonicated for 12 cycles of 30s on/30s off on high power using a Bioruptor Pico (Diagenode). To neutralize the SDS, Triton‐X‐100 was added to a final concentration of 1% along with 50× cOmplete protease inhibitor (final concentration: 1×). Samples were incubated at room temperature for 10 minutes, and 5% aliquots were saved for preparation of input controls. Antibody‐coated Dynabeads were washed with PBS and mixed with cell lysate and incubated overnight at 4 degrees with rotation. Immunoprecipitated chromatin was washed with 150 μL low‐salt buffer (50 mmol/L Tris/HCl, 150 mmol/L NaCl, 0.1% SDS, 0.1% NaDOC, 1% Triton‐X‐100, and 1 mmol/L EDTA), high‐salt buffer (500 mmol/L NaCl), and LiCl buffer (10 mmol/L Tris/HCl, 250 mmol/L LiCl, 0.5% IGEPAL CA‐630, 0.5% NaDOC, and 1 mM EDTA), followed by 2 washes with TE buffer and 2 washes with ice cold Tris/HCl (pH 8, 10 mmol/L). For tagmentation, bead‐bound chromatin was resuspended in 30 μL of tagmentation buffer, 1 μL of transposase (Nextera, Illumina) was added, and samples were incubated at 37 degrees for 10 minutes, followed by 2 washes with low‐salt buffer. For standard reverse crosslinking, chromatin complexes were diluted with 200 μL ChIP elution buffer (10 mmol/L Tris/HCl, 0.5% SDS, 300 mmol/L NaCL, and 5 mmol/L EDTA) and 2 μL of 20 μg/mL proteinase K (Thermo Scientific). Samples were vortexed and incubated overnight at 65 degrees. After reverse crosslinking, 1 μL 20 μg/mL RNase (Sigma) was added and incubated at 37 degrees for 30 minutes. DNA purification was carried out using Qiagen MinElute PCR Purification Kit. 15 μL of PCR master mix and 5 μL of primer mix (Nextera, Illumina) were added to 20 μL of eluted DNA and libraries were amplified. After PCR amplification, library cleanup was done using Agencourt AmPureXP eads (Beckman Coulter) at a ratio of 1:1. DNA concentrations in purified samples were measured using the Qubit dsRNA HS Kit (Invitrogen). Libraries were pooled and single‐end sequenced (50 cycles) using the NextSeq500 platform (Illumina). Analysis was performed as described in the ATAC‐seq pipeline. KLF2 and KLF4 peaks were submitted to search for potential motifs using MEME‐ChIP.
All sequencing was performed with Psomagen Inc (Rockville, MD).
RESULTS
Mapping KLF2 and KLF4 Occupancy Throughout the Endothelial Genome
To uncover precise mechanisms through which KLFs orchestrate endothelial transcription, we sought to define KLF2 and KLF4 chromatin occupancy (Figure 1A). Because of their highly disordered N‐terminus, reliable commercial antibodies targeting KLFs for genomic occupancy studies are lacking. Additionally, there are extensive physiological differences between cell cultured systems and primary ECs with regards to endothelial transcription and function. As such, epitope‐tagged modifications performed in cell lines or ex vivo would not reflect in vivo conditions. As a solution to this issue and to generate a tool for the first in vivo investigation of KLF2 and KLF4 localization, we CRISPR‐engineered 2 mouse lines in which the endogenous locus of KLF2 or KLF4 is N‐terminally tagged with an AviTag/3xFLAG (FLAG‐KLF2 and FLAG‐KLF4, respectively) (Figure S1–S4). To determine the precise location of KLF2/4 binding genome wide, we performed ChIP‐seq of CD31+/CD45‐ cells from hearts and lungs of FLAG‐KLF2 or FLAG‐KLF4 mice. 11 The majority of ECs isolated in this manner are from microvascular beds and, thus, exclude the contribution of large vessel EC heterogeneity. 12 , 13 , 14 Peak calling with the MACS2 pipeline demonstrated that KLF2 or KLF4 occupied around 8600 genomic loci within heart and lung ECs with ≈30% (2544) of these peaks having KLF2/4 overlap present in both tissues (Figure 1B and 1C). Importantly, there were also loci that contained only KLF2 or KLF4 occupancy, demonstrating unique usage of these factors in addition to instances of shared occupancy. Further, we noted tissue‐specific usage of these factors whereby KLF2 or KLF4 would be bound at certain locations in lung ECs but not heart, and vice versa.
Figure 1. KLF2/4 (Krüppel‐like factors 2 and 4) are extensively used transcription factors in multiple endothelial beds whose loss affects chromatin accessibility and transcription.
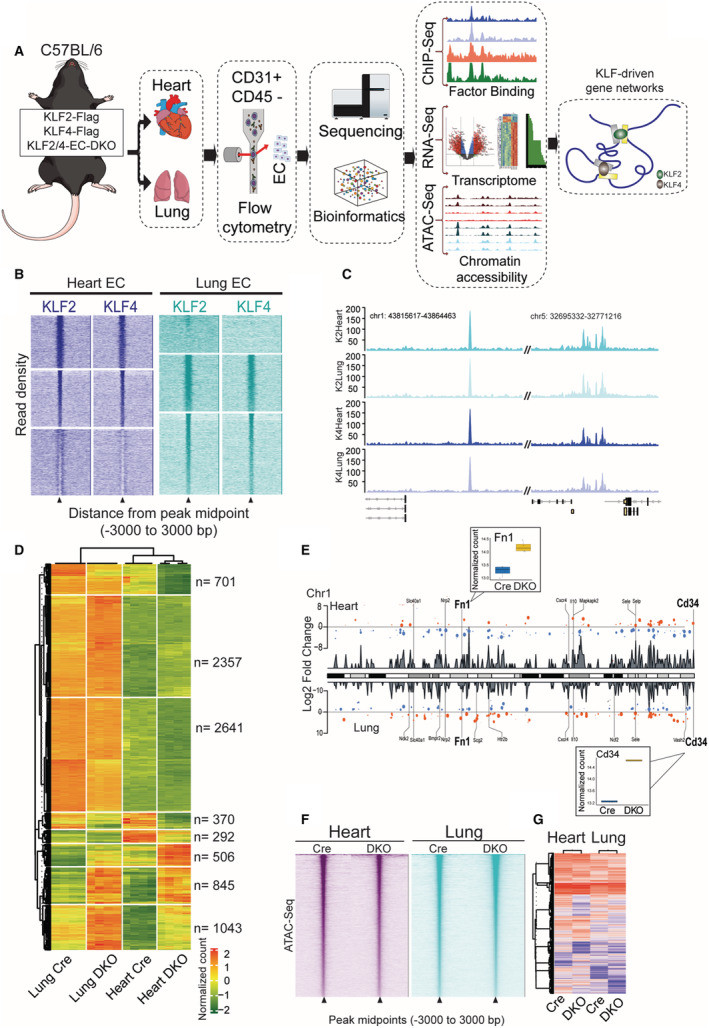
A, Experimental setup: C57BL/6 mice were epitope tagged with 3XFLAG in the endogenous locus of either KLF2 or KLF4. Endothelial cell‐specific double knockout of KLF2 and KLF4 were used in RNA sequencing and assay for transposase‐accessible chromatin using sequencing studies. ECs were flow sorted from heart and lungs and were used for all genome wide analyses. B, Chromatin immunoprecipitation followed by sequencing reads of KLF2 and KLF4 were plotted ±3000 bp from the peak midpoint and clustered using K‐means (K=3). C, Representative tracks of KLF2 and 4 occupancies at 2 loci in heart and lung ECs. D, Differentially expressed genes from RNA‐seq clustered in K=8 in heart and lung ECs of Cre and double knockout mice. E, Representative differentially expressed genes from double knockout heart and lung ECs (chromosome one). F, Assay for transposase‐accessible chromatin using sequencing peak midpoints were sorted highest to lowest and plotted in a window of ±3000 bp. G, Differentially accessible regions from assay for transposase‐accessible chromatin using sequencing are plotted as heatmaps from heart and lung ECs. ATAC‐seq indicates assay for transposase‐accessible chromatin using sequencing; ChIP‐seq, chromatin immunoprecipitation followed by sequencing; Chr1, chromosome 1; DKO, double knockout; EC, endothelial cells; Fn1, fibronectin 1; and KLF, Krüppel‐like factor.
Loss of Endothelial KLF2 and KLF4 Results in Dramatic Transcriptional Changes With Concurrent Shifts in Chromatin Accessibility
Given the pervasive binding of KLF2 and KLF4 throughout the endothelial genome, we sought to determine the transcriptional effect of loss of these factors. ECs from KLF2‐KLF4 double knockout (DKO) or VE‐Cadherin Cre control (Cre) mice were isolated from the heart and lungs, and we performed RNA‐sequencing (RNA‐seq) (Figure 1A). Because of the loss of viability with endothelial knockout of KLF2 and KLF4, cells were harvested at post‐injection day 5, before loss of viability of these mice. 10 Loss of KLF2/4 from lung and heart ECs resulted in a similar magnitude of effect with approximately 5500 differentially expressed genes (DEGs) in each (Figure 1D). As with KLF2 and KLF4 ChIP‐seq, there were tissue‐specific as well as tissue‐agnostic (shared) DEGs when KLF2/4 were lost (Figure 1D and 1E).
In addition to extensive transcriptional effects, loss of KLF2 and KLF4 is also associated with substantial changes in chromatin accessibility. Assay for transposase‐accessible chromatin using sequencing (ATAC‐seq) was performed on DKO and Cre heart and lung ECs (Figure 1A). Between heart and lung samples, loss of KLF2 and KLF4 was associated with nearly 1000 differentially accessible regions consisting of both gained and lost regions of accessibility (Figure 1F and 1G). Together, these data demonstrate that KLF2 and KLF4 are critical in maintaining a particular accessibility and transcriptional state in ECs and, while there is redundancy between the 2 factors, there are likely factor‐specific and tissue‐specific roles.
Coordinated and Redundant Recruitment of KLFs Mediate Transcription of Central Endothelial Genes
A substantial proportion of KLF2/4 binding sites, as well as DEGs from DKO studies, are shared between heart and lung ECs. Given that KLF2 and KLF4 are, in some aspects, functionally redundant in ECs, we hypothesized that ECs used shared KLF binding to transcriptionally protect core functions. To investigate this, all binding events of KLF2 and KLF4 were annotated to the nearest gene. These gene sets (with duplicates removed) were then analyzed for shared binding of KLF2/4. Of the nearly 5000 non‐duplicated annotated peaks across all ChIP‐seq data sets, 42% (2094) of them are shared by KLF2 and KLF4 across both tissues (Figure 2A). These peaks, which we refer to as “core peaks,” include those annotated closest to genes known to be critical in homeostatic function of ECs such as the genes encoding connexin‐43, cadherin 13, thrombospondin type 1 domain containing 7a, and angiotensin II receptor type 1. Functionally, the KLF2/4‐associated core peaks make up pathways important in EC physiology such as barrier function, cell trafficking of immune cells, and sprouting/migration, confirming phenotypic effects seen via loss of endothelial KLF2/4 (Figure 2B). Among the most enriched pathways is axon guidance, which consists of numerous guidance cues and receptors used in neurological and vascular development and repair such as semaphorins, ephrins, and components of the Slit‐Robo signaling system (Figure 2B).
Figure 2. Coordinated and redundant recruitment of KLFs mediate transcription of central endothelial genes.
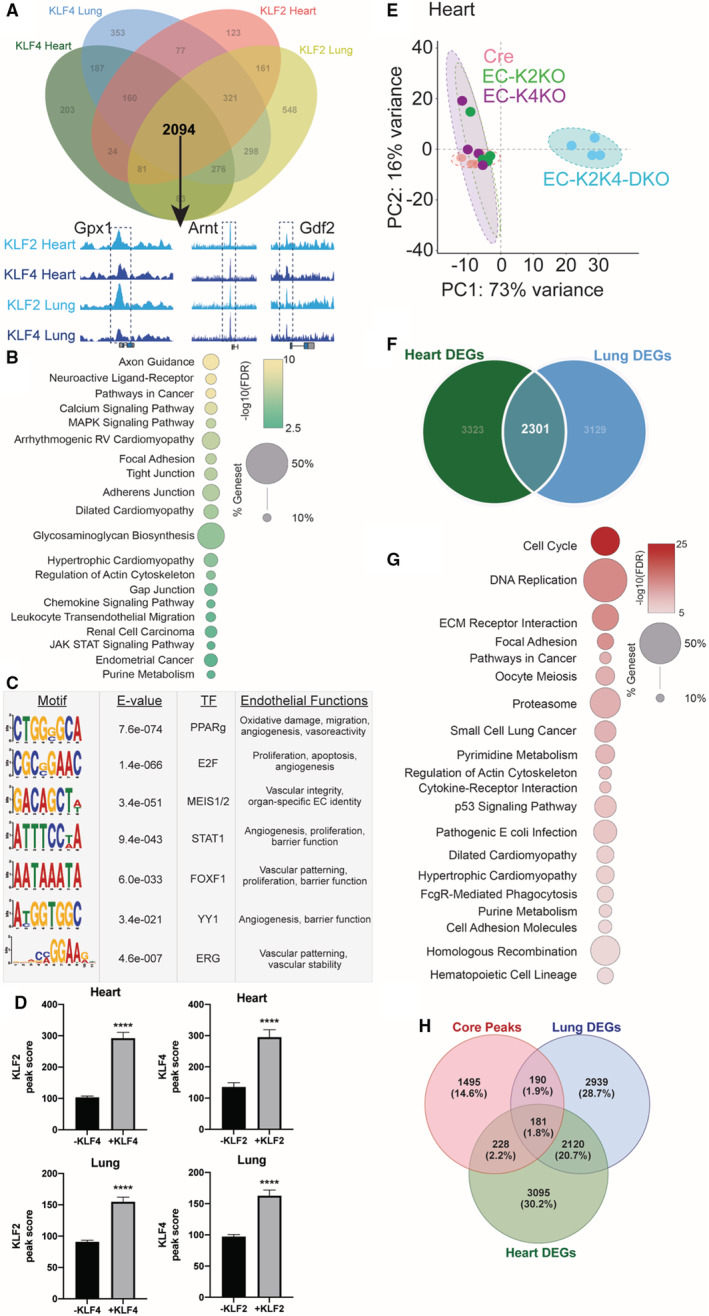
A, Annotated KLF2 and KLF4 chromatin immunoprecipitation followed by sequencing peaks shared in heart and lung endothelial cells make up 2094 “core peaks.” B, Top enriched KEGG pathways from annotated core peak genes demonstrate central endothelial pathways. C, Motif analysis performed surrounding core peaks (±200 bp) demonstrates numerous potential KLF2/4 binding partners important in endothelial function. D, KLF2/4 peak scores in regions with/without the other factor, ****P < 0.001 by unpaired, 2‐tailed Student's t‐test. E, Principal component analysis of RNA‐seq data from heart ECs isolated from either Cre, single knockout, or double knockout mice demonstrating double knockout separation. Each colored circle represents a single sample. F, Overlap of differentially expressed genes from heart and lung double knockout ECs identify 2301 “core genes.” G, Top enriched KEGG pathways from shared core genes demonstrate KLF2/4‐regulated endothelial pathways. H, Overlap of core peaks and differentially expressed genes from heart and lung EC double knockout. Arnt indicates aryl hydrocarbon receptor nuclear translocator; Cre, control; DEGs, differentially expressed genes; E2F, E2 factor; EC, endothelial cell; EC‐K4‐KO, endothelial cell KLF2 knockout; EC‐K2‐KO, endothelial cell KLF2 knockout; EC‐K2K4‐DKO, endothelial cell KLF2 and KLF4 double knockout; ERG, ETS related gene; FDR, false discovery rate; FOXF1, Forkhead box F1; Gdf2, growth differentiation factor 2; Gpx1, glutathione peroxidase 1; KLF, Krüppel‐like factor; MEIS1/2, meis homeobox 1/2; PC1, principal component 1; PC2, principal component 2; PPARg, peroxisome proliferator activated receptor gamma; STAT1, signal transducer and activator of transcription 1; TF, transcription factor; and YY1, Yin Yang 1.
Maintenance of a properly functioning endothelium requires the coordination of numerous transcriptional regulators and cofactors. 15 , 16 While loss‐of‐function studies have demonstrated a crucial role of KLF2 and KLF4 in endothelial health, they are likely to interact with a network of other key endothelial cell transcription factors. To explore which factors may be cooperating with KLFs in the control of endothelial integrity, we performed motif analysis near core peaks. Significant enrichment of several transcription factors with known importance in EC biology was found including ETS‐related gene, meis homeobox 1/2, and signal transducer and activator of transcription 1 (Figure 2C). These factors govern similar functions as those seen in KLF2/4 ChIP‐seq data sets including vascular patterning and barrier function, suggesting a potential KLF‐endothelial cell transcription factors cooperativity that maintains proper endothelial function via the transcriptional regulation of core genes. 17 , 18 , 19 , 20 , 21 , 22 , 23 , 24 , 25 , 26 , 27 , 28 , 29 , 30 , 31 , 32 , 33 , 34
In addition to KLF‐endothelial cell transcription factors interactions, the propensity for KLF2 and KLF4 to bind together at critical endothelial loci suggests a potential cooperativity by these 2 factors. To explore this, we compared the intersection of KLF binding sites to determine if the presence of the other KLF affects binding score. Remarkably, loci with both KLF2 and KLF4 resulted in increased binding score of both factors, regardless of tissue‐of‐origin (Figure 2D). The binding of 1 KLF may enhance the binding of the other, further ensuring proper transcription of key genes in endothelial homeostasis.
While the presence of both KLF2 and KLF4 ChIP‐seq peaks in heart and lung ECs suggests a role for these core loci in KLF2/4's role in maintaining critical EC functions, it does not necessarily demonstrate a necessity of KLF2/4 for transcription. Loss‐of‐function experiments would need to be performed to demonstrate a reliance of KLF2/4 binding on transcription of these targets. As previously mentioned, loss of either KLF2 or KLF4 in ECs is not sufficient to generate an acute deleterious vascular phenotype. This is also the case transcriptionally, as RNA‐seq results from either KLF2 or KLF4 single knockout heart ECs are similar to Cre control cells as seen in principal component analysis, while DKO cells exhibit a greater degree of variance from Cre or single knockout cells (Figure 2E). As with core peak occupancy studies, this transcriptional redundancy suggests that certain genes would be disturbed by loss of both KLF2 and KLF4, regardless of tissue of origin. To identify which genes are important in KLF2/4‐mediated transcription in both vascular beds, we compared DEGs from DKO lung ECs to those of DKO heart ECs. Just over one‐quarter of all DEGs identified are shared between lung and heart ECs, indicating considerable overlap in KLF2 and KLF4 transcriptional control across ECs (Figure 2F). Gene set enrichment analysis of these core genes demonstrates a similar functional enrichment as core peaks in critical endothelial functions including adhesion, cardiovascular biology, and housekeeping functions such as nucleotide metabolism and proteostasis (Figure 2G). Confirmatory quantitative PCRs from heart and lung DKO ECs were performed on key genes of endothelial health to verify transcriptional changes (Figure S2). 10 Interestingly, in addition to canonical endothelial programs, loss of KLF2 and KLF4 also impacted genes involved in cell cycling and DNA damage repair, perhaps indicating an intrinsic source of endothelial dysfunction. When comparing KLF2/4 occupancy to DEGs in DKO cells, we note that a subset of core peaks (599, 28.6%) annotate to DEGs from DKO cells, suggesting direct KLF2/4 regulation of local gene transcription (Figure 2H).
KLF2/4 Dynamics Illustrate Tissue‐Specific EC Populations
The heterogeneity of EC populations between tissues has long been appreciated. 35 , 36 , 37 This complexity has been further underscored by the development of single‐cell transcriptomic technology that can identify EC heterogeneity within single tissue types or in response to different stimuli. 24 , 38 , 39 , 40 , 41 , 42 Identification of unique regulators to explain EC transcriptional heterogeneity remains to be fully explored. Although KLF2 and KLF4 exhibit considerable redundancy in shared “core peaks” and “core genes” across endothelial beds from different tissues, we sought to determine if different EC populations use these factors differently. Principal component analysis plots from RNA‐seq data demonstrate substantial variance between the transcription of ECs from Cre lungs compared with Cre hearts, but also a similar difference in DKO cells, indicating that the profound transcriptional effects seen by losing KLF2 and KLF4 are not substantial enough to outweigh tissue‐specific transcriptional differences (Figure 3A). In other words, tissue‐of‐origin affects EC gene expression even more than losing 2 critical transcription factors essential for proper vascular function. While lung and heart ECs from control mice have similar expression patterns of numerous genes, there are distinct populations of genes that are differentially expressed depending on tissue‐of‐origin (Figure 3B). In the context of KLF2/4 deletion, we see similar tissue‐specific discrepancies of the expression of small subsets of genes (Figure 3C, green quadrants). These genes represent those that are uniquely affected by the absence of KLF2 and KLF4 in a tissue‐specific manner. To investigate whether KLF2 and KLF4 have differing roles within ECs from specific tissues, we returned to ChIP‐seq data sets to identify non‐core peaks (ie, binding sites not shared between tissues or factors). Of all annotated genes with a KLF2/4 peak, ≈18% of these exhibit a preference for either KLF2 or KLF4 in heart ECs as demonstrated by relative peak intensities (Figure 3D). A similar distribution was seen in lung ECs (Figure 3D). Interestingly, there is little overlap with how ECs from a particular tissue use a particular KLF (Figure 3D, Venn diagrams). For example, heart ECs use KLF4 distinctly from lung ECs (575 versus 684). Importantly, this further demonstrates that KLF2 and KLF4 are uniquely used by heart and lung ECs, and their loss affects transcription of these EC populations in distinct ways.
Figure 3. Unique KLF2 and 4 use occurs in heart and lung ECs.
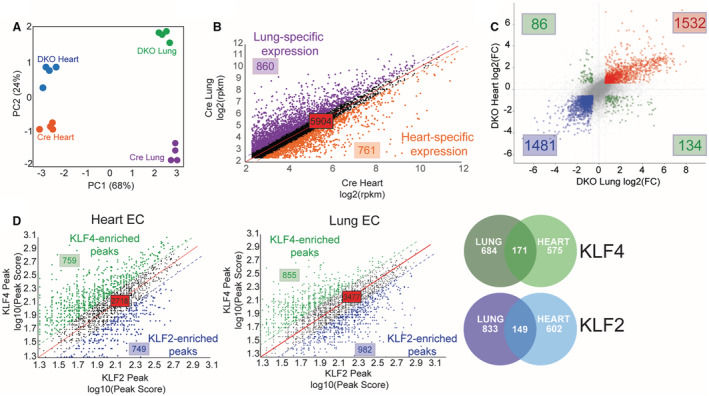
A, Principal component analysis of RNA‐seq data of heart and lung ECs from double knockout mice demonstrating tissue‐specific and genotype‐specific variance. B, Expression patterns of genes from Cre lung and heart ECs demonstrating shared and tissue‐specific expression patterns. C, Four‐way plot from heart and lung double knockout ECs differentially expressed genes. Red dots represent genes that are upregulated in both heart and lung ECs with loss of KLF2/4. Blue dots represent genes that are downregulated in both heart and lung ECs with loss of KLF2/4 and green dots are genes whose expression is opposite between tissue‐of‐origin. D, KLF2 and KLF4 annotated peaks were compared in heart and lung ECs. Peak scores between KLF2 and KLF4 are plotted as a relationship showing KLF4‐enriched (green) and KLF2‐enriched (blue) in each cell type. Venn diagrams demonstrate heart vs lung overlap of KLF4‐ (green) and KLF2‐ (blue) specific genes. DKO indicates double knockout; EC, endothelial cells; FC, fold change; KLF, Krüppel‐like factor; PC1, principal component 1; and PC2, principal component 2.
Promoters Represent a Minority of KLF2/4 Targets But Demonstrate Unique Binding Patterns in a Tissue‐Specific Manner
To explore the mechanism by which KLF2 and KLF4 impact endothelial transcription, we computed the binding distribution of each factor in both lung and heart ECs. Consistently, promoter binding made up ≈7% to 8% of factor binding, irrespective of which KLF and tissue‐type (Figure 4A). The large majority of binding sites occurred in distal intergenic regions or >3 kb downstream of the transcription start site, indicating a potential regulatory role for KLF2 and KLF4 at distal elements. Previous studies have demonstrated direct promoter binding by KLFs to regulate endothelial transcription. To determine the extent of this mechanism, we annotated genes with KLF2 or KLF4 bound no more than 3 kb away from the transcription start site (ie, promoter‐bound). Irrespective of tissue‐of‐origin, ≈25% of all KLF2/4‐bound promoters were associated with differential expression in DKO ECs, with upregulation and downregulation present (Figure 4A). Within lung ECs, >50% of peaks are shared by both factors, again highlighting redundant binding in certain contexts (Figure 4B). In fact, peak scores for lung EC promoter‐bound peaks show a nearly 80% correlation between KLF2 and KLF4 (Figure 4C). In contrast to lung EC promoters, there is little shared binding between KLF2 and KLF4 in heart EC promoters, with only 5% overlap of all KLF2‐ or KLF4‐bound promoters (Figure 4D and 4E). Gene set enrichment analysis of KLF4‐specific heart promoters demonstrate unique functions compared with KLF2‐specific promoters, including core processes (Figure 2) such as adhesion and cell junction (Figure 4D). Finally, to investigate if the presence of KLF2 and KLF4 at promoters was affecting transcriptional activity, we compared ChIP‐seq data to DKO RNA‐seq and ATAC‐seq data. Indeed, KLF2/4 presence at promoters is often associated with open chromatin, while loss of KLF2/4 binding in DKO mice leads to lost accessibility and decreased transcription (Figure 4F). Still, KLF2/4 promoter binding represents a relatively minor contribution to KLF‐mediated transcription as only 2.7% of DEGs had KLF2 and/or KLF4 bound to the promoter (Figure 4G).
Figure 4. Promoters represent a minority of KLF2/4 targets but demonstrate unique binding patterns in a tissue‐specific manner.
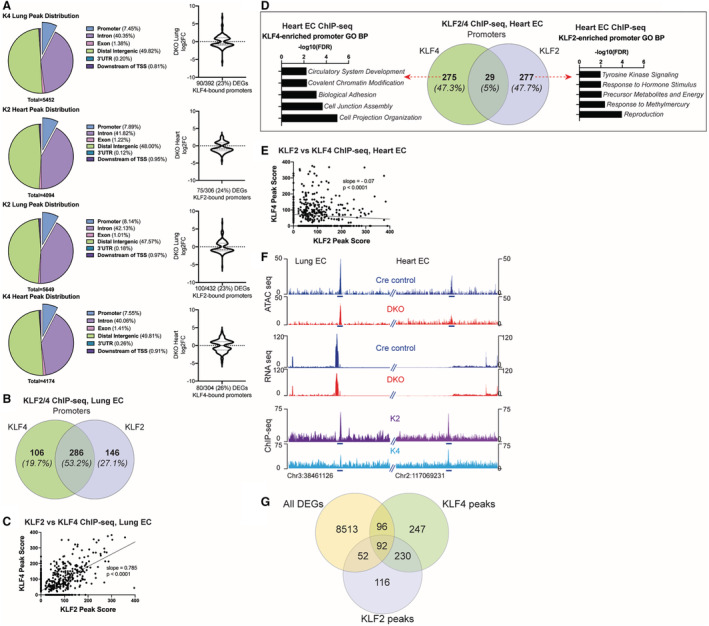
A, Peak distribution of KLF2/4 chromatin immunoprecipitation followed by sequencing peaks in each tissue type. K2=KLF2, K4=KLF4 with quantitation of differential expression of promoter‐bound genes from double knockout mice. B, Venn diagram depicting overlap of target genes whose promoters have KLF2 and/or KLF4 bound in lung endothelial cells. C, Relationship of KLF2 and KLF4 peak scores for peaks annotated to promoters in lung endothelial cells. D, Venn diagram depicting overlap of target genes whose promoters have KLF2 and/or KLF4 bound in heart endothelial cells. Gene ontology biological process analysis of unique KLF4 and KLF2 promoter‐bound genes are shown on their respective sides of the Venn diagram. E, Relationship of KLF2 and KLF4 peak scores for peaks annotated to promoters in heart endothelial cells. F, Browser shot of 2 genomic loci (promoters) from heart and lungs showing the differences in openness of chromatin (assay for transposase‐accessible chromatin using sequencing) between Cre and double knockout mice. Differentially expressed genes from RNA‐seq reads and associated KLF2 and KLF4 chromatin immunoprecipitation followed by sequencing peaks at the same location are also depicted. G, Venn diagram comparing all differentially expressed genes from double knockout heart and lung cells to all KLF4 and KLF2 peaks in both tissues demonstrating a minority of peaks are directly annotated to the promoters of differentially expressed genes. For regression studies, null hypothesis is that the slope of the regression line=1 (exact 1:1 relationship of binding). ATAC‐seq indicates assay for transposase‐accessible chromatin using sequencing; ChIP‐seq, chromatin immunoprecipitation followed by sequencing; DEGs, differentially expressed genes; DKO, double knockout; EC, endothelial cells; K2, KLF2; K4, KLF4; and KLF, Krüppel‐like factor.
Endothelial KLFs Increase Chromatin Accessibility With Factor‐Specific Bias
Opening of chromatin at KLF2/4‐bound promoters suggests that these factors may play a role in maintaining nucleosome positioning. As predicted, loss of KLF2 and KLF4 leads to differential accessibility around genes associated with critical endothelial functions such as EC proliferation, migration, leukocyte adhesion, and junctional proteins (Figure S3A). Making up these processes are core endothelial genes such as hypoxia inducible factor 1a, fms‐related receptor tyrosine kinase 4, semaphorin 5a, and platelet‐derived growth factor beta, which all have increased accessibility in DKO ECs. Conversely, regions that lose accessibility in DKO cells encode genes associated with metabolism and DNA repair, such as cAMP responsive element binding protein 1, MutS homolog 2, and peroxisome proliferator‐activated receptor delta Ppard) (Figure S3A). Changes in accessibility at core endothelial genes, as well as in processes central for post‐mitotic cells, suggests that the absence of KLF2/4 leads to disorganized transcription, which corroborates with changes seen in RNA‐seq analyses.
To determine if the occupancy by KLF2 or KLF4 is concurrently associated with establishing openness throughout genome, we sorted ATAC‐seq peaks by those associated with KLF2 or KLF4 binding. Using data from heart ECs as an exemplar, we noted that KLF2 and KLF4 binding is associated with hundreds of regions of open chromatin (Figure S3B). To investigate the necessity of KLF2/4 to maintain openness, we characterized ATAC peak signatures in heart ECs from DKO mice. Knockout of KLF2 and KLF4 led to the closing of many of these regions (Figure S3B). Of note, this effect was much more striking in KLF2‐bound regions, perhaps suggesting a particularly important role for KLF2 in establishing open chromatin regions.
As with binding and transcriptional studies, KLF2 and KLF4 play unique roles in maintaining chromatin accessibility that is tissue‐specific. While most of the KLF‐bound regions show more closed chromatin in DKO cells, there are examples of KLF binding that leads to less accessible chromatin, an effect that is lost in the DKO cells (Figure S3C). These data demonstrate a complex role for KLF2 and KLF4 in establishing distinct chromatin accessibility landscapes needed for them to exert their transcriptional effects.
KLF2 and KLF4 Occupy Endothelial Enhancers and Maintain Endothelial Transcription
KLF binding throughout the endothelial genome is largely restricted to regions several kilobases away from transcription start site (Figure 4A). This, along with the fact that several thousand genes are changed in DKO cells without proximal binding of KLF2 or KLF4, suggests that these factors may be establishing distal regulatory elements such as enhancers to mediate transcription. To explore this, we compared KLF2 and KLF4 binding to that of H3K27ac, p300, and RNA polymerase II (Pol II. H3K27ac is a mark of active enhancers, while p300 and Pol II are non‐specific factors also found at enhancers. 43 In both heart and lung ECs, KLF2 and KLF4 colocalized with each of these markers in several hundred locations, with KLF2 in heart ECs demonstrating a particularly high binding to enhancer regions (Figure 5A through 5C). As noted in Figure 5B, there is also considerable overlap in KLF2 and KLF4 enhancer binding, irrespective of tissue‐of‐origin, further suggesting a role for redundancy in this system.
Figure 5. KLFs (Krüppel‐like factors) mediate endothelial transcription largely through the orchestration of enhancer‐driven transcriptional hubs.
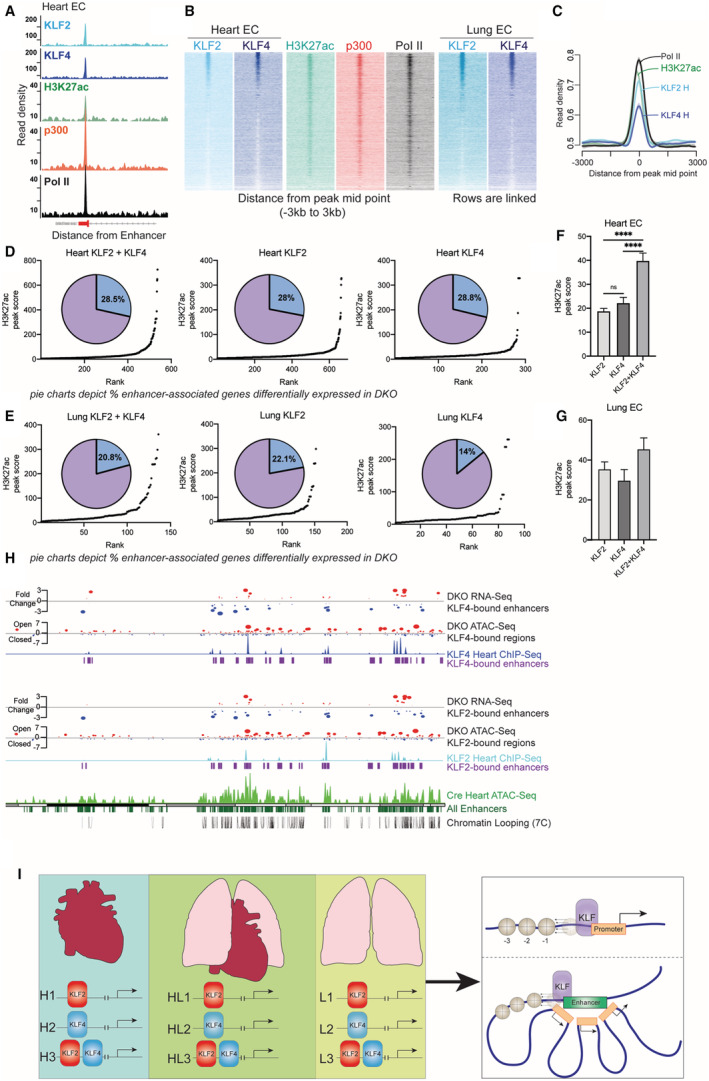
A, Colocalization of KLF2 and KLF4 with enhancer marks (H3K27ac, p300, Pol II) at a canonical enhancer region in heart endothelial cells. B, Genome wide distribution of enhancers (peak midpoints) sorted from highest to lowest occupancy and aligned to enhancer marks, showing highest enrichment of KLF2 and KLF4. C, Composite plots showing quantification of enhancer binding of KLF2 and KLF4 along with enhancer marks. D and E, Hockey stick plot of putative enhancers at sites of KLF2/4 binding based on H3K27ac signal. Pie charts depicting proportion of genes of these enhancers that are differentially expressed in double knockout cells. F and G, Comparing average H3K27ac peak scores for enhancers bound with KLF2, KLF4, or KLF2+KLF4 in heart and lung endothelial cells. ****P<0.0001 by 1‐way ANOVA with Tukey multiple comparisons test. H, Integrative analysis showing the relationship between gene expression, enhancer proximity, chromatin accessibility, and KLF binding. I, Model of KLF‐regulated transcription in different endothelial cell beds (H, heart; L, lung; HL, heart and lung). There is tissue‐specific binding of KLF2, KLF4, and KLF2+KLF4 (H1‐3, L1‐3) as well as shared binding patterns (HL1‐3). KLFs promote chromatin accessibility when bound to promoters and enhancers (right side of model). ATAC‐seq indicates assay for transposase‐accessible chromatin using sequencing; ChIP‐seq, chromatin immunoprecipitation followed by sequencing; DKO, double knockout; EC, endothelial cells; H3K27ac, acetylation of lysine 27 on histone 3; p300, KLF, Krüppel‐like factor; p300 histone acetyltransferase; and Pol II, RNA polymerase II.
While the occupancy of KLF2 and KLF4 in enhancers is evident, we wished to explore if these binding events had transcriptional consequences, especially because of our evidence that there are situations in which KLFs bind to the genome, but their loss does not affect transcription in those locations. To investigate this, we used tissue EC‐specific H3K27ac ChIP‐seq data (Figure 5A). Ranking heart endothelial H3K27ac peaks based on score identified over 900 putative enhancers (Figure S4A). Of these, around 45% have either KLF2 or KLF4 bound, with 16% having both factors present (Figure S4B). While lung H3K27ac peaks were fewer in our analyses (Figure S4C), 47% of them have KLF2 and KLF4 bound, illustrating the importance of these factors in regulating transcriptional networks in ECs (Figure S4D). The transcriptional effects of this are apparent when comparing KLF enhancer binding with differential expression from DKO cells. Using the Genomic regions enrichment of anotations tool algorithm to annotate cis‐regulatory regions to target genes, 44 we compared annotated targets of enhancers to DEGs from DKO cells of the same tissue. Of all annotated putative enhancers, 28.3% have either KLF2 or KLF4 bound in heart ECs, with 22.7% of lung ECs enhancers having at least 1 KLF present (Figure S4E and S4F). Of putative enhancers with both KLF2 and KLF4 present (“core enhancer peaks”), 28.5% exhibit differential gene expression in DKO mice (Figure 5D). A similar trend is maintained in lung ECs, with 20.8% of lung EC enhancers with both KLF2 and KLF4 bound being differentially expressed (Figure 5E). Functionally, these enhancers regulate genes involved in critical endothelial functions such as adhesion and leukocyte chemotaxis (Figure S4G and S4H). These gene ontology terms are shared by core peaks and core genes in KLF2 and 4 ChIP‐seq annotations (Figure 2), underscoring the central role KLF2 and KLF4 play in maintaining endothelial biology.
Interestingly, these data also provide insight into the relative use of KLF2 and KLF4 in enhancer‐driven transcription. In both heart and lung ECs, KLF2 is more extensively found bound to enhancers (Figure 5D and 5E). Conversely, KLF4 occupies substantially fewer enhancers in both heart and lung ECs. Importantly, however, we noted that the presence of both KLFs substantially increased H3K27ac scores compared with 1 factor alone, suggesting a compounding influence on enhancers (Figure 5F and 5G). Together, these data demonstrate a novel role for KLF2 and KLF4 usage at enhancers to control the transcription of endothelial gene networks.
Finally, to gain a better understanding for the interplay between KLF2 and KLF4 in regulating the endothelial transcriptome, we visualized shared and distinct KLF enhancer networks by integrating all of the data sets. Figure 5H depicts chromosome 15 from heart ECs with annotated KLF2 and KLF4 binding and transcriptional signatures. ATAC‐seq signal from Cre heart ECs aligns with known enhancers, the large majority of which are also bound by KLF2 and/or KLF4. Modeling of short‐range chromatin looping using computational chromosome confirmation capture by correlation of ChIP‐seq at CTCF motifs (7C) 45 demonstrates that KLF2/4 anchor enhancer‐promoter loops throughout the endothelial genome. Loss of KLF2 and KLF4 (DKO) demonstrates profound effects on the chromatin accessibility at these enhancers, leading to changes in transcription in shared and factor‐specific regions. The integration of these data demonstrates the powerful role that KLF2 and KLF4 play in maintaining endothelial transcription via establishing chromatin accessibility, promoting enhancer‐promoter looping, and by binding to promoters, themselves. This work also establishes that KLFs are not always used interchangeably in ECs, as distinct patterns of binding and transcription occur with KLF2 and KLF4. Furthermore, these patterns are not ubiquitous to all ECs, as tissue‐specific usage of these factors occurs (Figure 5I).
DISCUSSION
A large body of work implicates KLFs as key transcriptional regulators of EC biology, but a detailed understanding of how these 2 factors control the EC transcriptome has remained poorly understood. The work here provides significant insights into this issue and advances 3 main conclusions: (1) KLF2 and KLF4 are used interchangeably to maintain critical endothelial functions but also exhibit a previously unappreciated singularity at some binding sites; (2) KLFs impart transcriptional effects on ECs principally through the binding of enhancers throughout the endothelial genome; and (3) tissue‐specific usage of transcription factors may explain endothelial heterogeneity.
In the present work, we identified significant KLF2/4 redundancy in control of gene expression while also demonstrating transcription distinct to each factor. Concordant with previous studies, core EC transcriptional programs (eg, tight junctions, adhesion, guidance cues) are governed by both KLF2 and KLF4, underscoring their importance in maintaining a healthy vascular state. In addition, however, we have uncovered KLF2‐specific and KLF4‐specific binding patterns that each have transcriptional consequences. Importantly, these patterns are also tissue‐specific, raising the possibility that organ‐specific vascular beds use similar transcription factors in unique ways. Although both populations explored were microvascular, lung and heart ECs experience immense differences in flow pressure and oxygenation. Thus, we hypothesize that differences in flow dynamics seen in heart and lung vascular beds necessitates distinct KLF usage to finely tune transcription to the needs of local ECs. Additional exploration will be needed to empirically determine how these dynamics may shift KLF bias. Further, the relative contribution of arteriolar versus venular expression KLF usage within the microvasculature is a potentially exciting avenue of investigation. Given their structural similarity, KLF2 and KLF4 specificity may arise from the recruitment of specific cofactors. Tools such as the KLF2/4 tagged mice will allow for in vivo investigation into binding partners under specific stress conditions as well as under different physiological parameters (eg, local pressure, oxygen tension, environmental cues).
Prior work on KLFs in endothelial biology has focused largely on control of gene transcription through promoter binding. However, recent studies in other systems suggest that KLFs can affect local chromatin accessibility and bind to enhancer elements to orchestrate transcription. 46 , 47 , 48 There is also a growing understanding of how enhancer landscapes differ between EC populations. Recent studies have identified distinct enhancers in arterial/venous systems, in specific disease contexts, and across macrovascular beds in humans. 49 , 50 , 51 The current work integrated these ideas and evaluated how KLFs might use enhancer binding and local chromatin accessibility to establish tissue‐specific microvascular transcriptional programs in homeostasis. To this end, we found that both KLF2 and KLF4 open chromatin and bind to enhancers in heart and lung ECs and use these to affect the transcription of thousands of genes. Alterations of chromatin accessibility with loss of KLF2/4 suggests that these factors play an active role in directing chromatin accessibility at their sites of action. Whether this is through interactions with known chromatin remodeling factors or through previously unappreciated functions of KLFs remains an active area of research. Within stem‐cell biology, the role of KLF4 as a pioneer factor suggests that binding occurs before nucleosome remodeling and the subsequent recruitment of remodeling factors of KLF binding facilitates chromatin accessibility changes. Whether these processes occur in a tissue‐agnostic fashion remains to be seen. Further, KLF2 has not yet shown pioneer factor properties despite its similarity to KLF4, suggesting additional mechanisms of chromatin remodeling at play.
The integration of numerous data sets within this work has allowed us to explore shared/distinct KLF usage between tissue types, opening the door for additional exploration into what governs specificity of these binding events. Indeed, uncovering the manner by which KLFs adopt specificity from one another despite similar amino acid sequences and expression pattern will delineate finetuned context‐dependent transcriptional events.
Finally, the work here coupled with our prior work strongly supports the importance of functional redundancy in biological systems. As the vascular system supplies energy to all tissues, the establishment of redundant controls makes intuitive sense and likely offers a greater dynamic range to withstand stress and ensure organismal survival. Prior work from our group and others has shown that noxious stimuli (eg, inflammatory, metabolic, and oxidative), as well as the process of aging, reduce endothelial levels of both KLF2 and KLF4. 52 While genetic studies have shown that endothelial loss of one KLF is compatible with life, animals are rendered susceptible to vascular disease with provocative stress. Restoration of either KLF2 or KLF4 expression in the context of deleterious stimuli, therefore, represents a mechanism to maintain oxygen and nutrient delivery. Failure to maintain proper endothelial function results in vascular compromise, affecting nearly every organ system: endothelial damage resulting in vascular permeability contributes to age‐related dementia and Alzheimer disease, EC‐derived inflammation is widely accepted as a proximal cause of atherothrombotic disease, and recent work in the wake of the COVID‐19 pandemic has identified EC infection and subsequent inflammatory activation as a major contributor to rampant thrombosis seen in these patients. 53 , 54 , 55 , 56 As such, identifying regulatory mechanisms of endothelial function, thus, is of great therapeutic interest that can potentially impact a wide array of diseases.
Sources of Funding
This work was supported by National Institutes of Health grants R01DK111478, R35HL135789, and R01HL086548 (to M.K.J.), R01HL139783 (to R.J.), R01HL142647 (to L.N.), and T32GM007250 and F30HL139014 (to D.R.S.). This work was also supported by American Heart Association‐Allen Frontiers Award (to M.K.J. and R.J.), Leducq Foundation Transatlantic Network of Excellence (to M.K.J. and S.D.V.), AHA Transformative Project Award 18TPA34230033 (to X.L.), the British Heart Foundation FS/1735/32929 (to S.D.V.), and the Burroughs Wellcome Fund (to R.J.).
Disclosures
None.
Supporting information
Figures S1–S4
Supplemental Material is available at https://www.ahajournals.org/doi/suppl/10.1161/JAHA.121.024303
For Sources of Funding and Disclosures, see page 14.
References
- 1. Dekker RJ, Boon RA, Rondaij MG, Kragt A, Volger OL, Elderkamp YW, Meijers JC, Voorberg J, Pannekoek H, Horrevoets AJ. KLF2 provokes a gene expression pattern that establishes functional quiescent differentiation of the endothelium. Blood. 2006;107:4354–4363. doi: 10.1182/blood-2005-08-3465 [DOI] [PubMed] [Google Scholar]
- 2. Sweet DR, Fan L, Hsieh PN, Jain MK. Krüppel‐like factors in vascular inflammation: mechanistic insights and therapeutic potential. Front Cardiovasc Med. 2018;5:1–17. doi: 10.3389/fcvm.2018.00006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zhou G, Hamik A, Nayak L, Tian H, Shi H, Lu Y, Sharma N, Liao X, Hale A, Boerboom L, et al. Endothelial Kruppel‐like factor 4 protects against atherothrombosis in mice. J Clin Investig. 2012;122:4727–4731. doi: 10.1172/JCI66056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. van Thienen JV, Fledderus JO, Dekker RJ, Rohlena J, van IJzendoorn GA, Kootstra NA, Pannekoek H, AJG H. Shear stress sustains atheroprotective endothelial KLF2 expression more potently than statins through mRNA stabilization. Cardiovasc Res. 2006;72:231–240. doi: 10.1016/j.cardiores.2006.07.008 [DOI] [PubMed] [Google Scholar]
- 5. Hamik A, Lin Z, Kumar A, Balcells M, Sinha S, Katz J, Feinberg MW, Gerzsten RE, Edelman ER, Jain MK. Kruppel‐like factor 4 regulates endothelial inflammation. J Biol Chem. 2007;282:13769–13779. doi: 10.1074/jbc.M700078200 [DOI] [PubMed] [Google Scholar]
- 6. Parmar KM, Larman HB, Dai G, Zhang Y, Wang ET, Moorthy SN, Kratz JR, Lin Z, Jain MK, García‐cardeña G. Integration of flow‐dependent endothelial phenotypes by Kruppel‐like factor 2. J Clin Invest. 2006;116:49–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Senbanerjee S, Lin Z, Atkins GB, Greif DM, Rao RM, Kumar A, Feinberg MW, Chen Z, Simon DI, Luscinskas FW, et al. KLF2 is a novel transcriptional regulator of endothelial proinflammatory activation. J Exp Med. 2004;199:1305–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dekker RJ, van Soest S, Fontijn RD, Salamanca S, de Groot PG, VanBavel E, Pannekoek H, Horrevoets AJ. Prolonged fluid shear stress induces a distinct set of endothelial cell genes, most specifically lung Krüppel‐like factor (KLF2). Blood. 2002;100:1689–1698. doi: 10.1182/blood-2002-01-0046 [DOI] [PubMed] [Google Scholar]
- 9. Atkins GB, Wang Y, Mahabeleshwar GH, Shi H, Gao H, Kawanami D, Natesan V, Lin Z, Simon DI, Jain MK. Hemizygous deficiency of kruppel‐like factor 2 augments experimental atherosclerosis. Circ Res. 2008;103:690–693. doi: 10.1161/CIRCRESAHA.108.184663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sangwung P, Zhou G, Nayak L, Chan ER, Kumar S, Kang D‐w, Zhang R, Liao X, Lu Y, Sugi K, et al. KLF2 and KLF4 control endothelial identity and vascular integrity. JCI Insight. 2017;2:e91700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chavkin NW, Walsh K, Hirschi KK. Isolation of highly purified and viable retinal endothelial cells. J Vasc Res. 2021;58:49–57. doi: 10.1159/000510533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Marelli‐Berg FM, Peek E, Lidington EA, Stauss HJ, Lechler RI. Isolation of endothelial cells from murine tissue. J Immunol Methods. 2000;244:205–215. doi: 10.1016/S0022-1759(00)00258-1 [DOI] [PubMed] [Google Scholar]
- 13. Pratumvinit B, Reesukumal K, Janebodin K, Ieronimakis N, Reyes M. Isolation, characterization, and transplantation of cardiac endothelial cells. Biomed Res Int. 2013;2013:359412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Luo S, Truong AH, Makino A. Isolation of mouse coronary endothelial cells. J Vis Exp. 2016;113:53985. doi: 10.3791/53985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Park C, Kim TM, Malik AB. Transcriptional regulation of endothelial cell and vascular development. Circ Res. 2013;112:1380–1400. doi: 10.1161/CIRCRESAHA.113.301078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. De Val S, Black BL. Transcriptional control of endothelial cell development. Dev Cell. 2009;16:180–195. doi: 10.1016/j.devcel.2009.01.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. De Silva TM, Li Y, Kinzenbaw DA, Sigmund CD, Faraci FM. Endothelial PPARγ (peroxisome proliferator‐activated receptor‐γ) is essential for preventing endothelial dysfunction with aging. Hypertension. 2018;72:227–234. doi: 10.1161/HYPERTENSIONAHA.117.10799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kotlinowski J, Jozkowicz A. PPAR gamma and angiogenesis: endothelial cells perspective. J Diabetes Res. 2016;2016:8492353. doi: 10.1155/2016/8492353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhou J, Cheng M, Wu M, Boriboun C, Jujo K, Xu S, Zhao TC, Tang YL, Kishore R, Qin G. Contrasting roles of E2F2 and E2F3 in endothelial cell growth and ischemic angiogenesis. J Mol Cell Cardiol. 2013;60:68–71. doi: 10.1016/j.yjmcc.2013.04.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Spyridopoulos I, Principe N, Krasinski KL, Xu S, Kearney M, Magner M, Isner JM, Losordo DW. Restoration of E2F expression rescues vascular endothelial cells from tumor necrosis factor‐alpha‐induced apoptosis. Circulation. 1998;98:2883–2890. doi: 10.1161/01.CIR.98.25.2883 [DOI] [PubMed] [Google Scholar]
- 21. Pillai S, Kovacs M, Chellappan S. Regulation of vascular endothelial growth factor receptors by Rb and E2F1: role of acetylation. Cancer Res. 2010;70:4931–4940. doi: 10.1158/0008-5472.CAN-10-0501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Azcoitia V, Aracil M, Martínez AC, Torres M. The homeodomain protein Meis1 is essential for definitive hematopoiesis and vascular patterning in the mouse embryo. Dev Biol. 2005;280:307–320. doi: 10.1016/j.ydbio.2005.01.004 [DOI] [PubMed] [Google Scholar]
- 23. Hisa T, Spence SE, Rachel RA, Fujita M, Nakamura T, Ward JM, Devor‐Henneman DE, Saiki Y, Kutsuna H, Tessarollo L, et al. Hematopoietic, angiogenic and eye defects in Meis1 mutant animals. EMBO J. 2004;23:450–459. doi: 10.1038/sj.emboj.7600038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sabbagh MF, Heng JS, Luo C, Castanon RG, Nery JR, Rattner A, Goff LA, Ecker JR, Nathans J. Transcriptional and epigenomic landscapes of CNS and non‐CNS vascular endothelial cells. elife. 2018;7:e36187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Battle TE, Lynch RA, Frank DA. Signal transducer and activator of transcription 1 activation in endothelial cells is a negative regulator of angiogenesis. Cancer Res. 2006;66:3649–3657. doi: 10.1158/0008-5472.CAN-05-3612 [DOI] [PubMed] [Google Scholar]
- 26. Gomez D, Reich NC. Stimulation of primary human endothelial cell proliferation by IFN. J Immunol. 2003;170:5373–5381. doi: 10.4049/jimmunol.170.11.5373 [DOI] [PubMed] [Google Scholar]
- 27. Kwak M, Hong C, Myeong J, Park EYJ, Jeon JH, So I. Gα(i)‐mediated TRPC4 activation by polycystin‐1 contributes to endothelial function via STAT1 activation. Sci Rep. 2018;8:3480. doi: 10.1038/s41598-018-21873-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ren X, Ustiyan V, Pradhan A, Cai Y, Havrilak JA, Bolte CS, Shannon JM, Kalin TV, Kalinichenko VV. FOXF1 transcription factor is required for formation of embryonic vasculature by regulating VEGF signaling in endothelial cells. Circ Res. 2014;115:709–720. doi: 10.1161/CIRCRESAHA.115.304382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bolte C, Flood HM, Ren X, Jagannathan S, Barski A, Kalin TV, Kalinichenko VV. FOXF1 transcription factor promotes lung regeneration after partial pneumonectomy. Sci Rep. 2017;7:10690. doi: 10.1038/s41598-017-11175-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cai Y, Bolte C, Le T, Goda C, Xu Y, Kalin TV, Kalinichenko VV. FOXF1 maintains endothelial barrier function and prevents edema after lung injury. Sci Signal. 2016;9:ra40. [DOI] [PubMed] [Google Scholar]
- 31. Zhang S, Kim JY, Xu S, Liu H, Yin M, Koroleva M, Guo J, Pei X, Jin ZG. Endothelial‐specific YY1 governs sprouting angiogenesis through directly interacting with RBPJ. Proc Natl Acad Sci USA. 2020;117:4792–4801. doi: 10.1073/pnas.1916198117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sade H, Holloway K, Romero IA, Male D. Transcriptional control of occludin expression in vascular endothelia: regulation by Sp3 and YY1. Biochim Biophys Acta. 2009;1789:175–184. doi: 10.1016/j.bbagrm.2009.01.006 [DOI] [PubMed] [Google Scholar]
- 33. De Val S. Key transcriptional regulators of early vascular development. Arterioscler Thromb Vasc Biol. 2011;31:1469–1475. doi: 10.1161/ATVBAHA.110.221168 [DOI] [PubMed] [Google Scholar]
- 34. Shah AV, Birdsey GM, Peghaire C, Pitulescu ME, Dufton NP, Yang Y, Weinberg I, Osuna Almagro L, Payne L, Mason JC, et al. The endothelial transcription factor ERG mediates angiopoietin‐1‐dependent control of Notch signalling and vascular stability. Nat Commun. 2017;8:16002. doi: 10.1038/ncomms16002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ribatti D, Nico B, Vacca A, Roncali L, Dammacco F. Endothelial cell heterogeneity and organ specificity. J Hematother Stem Cell Res. 2002;11:81–90. doi: 10.1089/152581602753448559 [DOI] [PubMed] [Google Scholar]
- 36. Cleuren ACA, van der Ent MA, Jiang H, Hunker KL, Yee A, Siemieniak DR, Molema G, Aird WC, Ganesh SK, Ginsburg D. The in vivo endothelial cell translatome is highly heterogeneous across vascular beds. Proc Natl Acad Sci. 2019;116:23618–23624. doi: 10.1073/pnas.1912409116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Garlanda C, Dejana E. Heterogeneity of endothelial cells. Specific markers. Arterioscler Thromb Vasc Biol. 1997;17:1193–1202. doi: 10.1161/01.ATV.17.7.1193 [DOI] [PubMed] [Google Scholar]
- 38. Jambusaria A, Hong Z, Zhang L, Srivastava S, Jana A, Toth PT, Dai Y, Malik AB, Rehman J. Endothelial heterogeneity across distinct vascular beds during homeostasis and inflammation. elife. 2020;9:e51413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kalucka J, de Rooij L, Goveia J, Rohlenova K, Dumas SJ, Meta E, Conchinha NV, Taverna F, Teuwen LA, Veys K, et al. Single‐cell transcriptome atlas of murine endothelial cells. Cell. 2020;180:764–779.e20. doi: 10.1016/j.cell.2020.01.015 [DOI] [PubMed] [Google Scholar]
- 40. Khan S, Taverna F, Rohlenova K, Treps L, Geldhof V, de Rooij L, Sokol L, Pircher A, Conradi LC, Kalucka J, et al. EndoDB: a database of endothelial cell transcriptomics data. Nucleic Acids Res. 2019;47:D736–D744. doi: 10.1093/nar/gky997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Paik DT, Tian L, Williams IM, Rhee S, Zhang H, Liu C, Mishra R, Wu SM, Red‐Horse K, Wu JC. Single‐cell RNA sequencing unveils unique transcriptomic signatures of organ‐specific endothelial cells. Circulation. 2020;142:1848–1862. doi: 10.1161/CIRCULATIONAHA.119.041433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chen MB, Yang AC, Yousef H, Lee D, Chen W, Schaum N, Lehallier B, Quake SR, Wyss‐Coray T. Brain endothelial cells are exquisite sensors of age‐related circulatory cues. Cell Rep. 2020;30:4418–4432.e4. doi: 10.1016/j.celrep.2020.03.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Andersson R, Sandelin A. Determinants of enhancer and promoter activities of regulatory elements. Nat Rev Genet. 2020;21:71–87. doi: 10.1038/s41576-019-0173-8 [DOI] [PubMed] [Google Scholar]
- 44. McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, Bejerano G. GREAT improves functional interpretation of cis‐regulatory regions. Nat Biotechnol. 2010;28:495–501. doi: 10.1038/nbt.1630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ibn‐Salem J, Andrade‐Navarro MA. 7C: computational chromosome conformation capture by correlation of ChIP‐seq at CTCF motifs. BMC Genomics. 2019;20:777. doi: 10.1186/s12864-019-6088-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Di Giammartino DC, Kloetgen A, Polyzos A, Liu Y, Kim D, Murphy D, Abuhashem A, Cavaliere P, Aronson B, Shah V, et al. KLF4 binding is involved in the organization and regulation of 3D enhancer networks during acquisition and maintenance of pluripotency. Nat Cell Biol. 2019;21:1179–1190. doi: 10.1038/s41556-019-0390-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ilsley MD, Gillinder KR, Magor GW, Huang S, Bailey TL, Crossley M, Perkins AC. Krüppel‐like factors compete for promoters and enhancers to fine‐tune transcription. Nucleic Acids Res. 2017;45:6572–6588. doi: 10.1093/nar/gkx441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Knaupp AS, Buckberry S, Pflueger J, Lim SM, Ford E, Larcombe MR, Rossello FJ, de Mendoza A, Alaei S, Firas J, et al. Transient and permanent reconfiguration of chromatin and transcription factor occupancy drive reprogramming. Cell Stem Cell. 2017;21:834–845.e6. doi: 10.1016/j.stem.2017.11.007 [DOI] [PubMed] [Google Scholar]
- 49. Neal A, Nornes S, Payne S, Wallace MD, Fritzsche M, Louphrasitthiphol P, Wilkinson RN, Chouliaras KM, Liu K, Plant K, et al. Venous identity requires BMP signalling through ALK3. Nat Commun. 2019;10:453. doi: 10.1038/s41467-019-08315-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Payne S, Gunadasa‐Rohling M, Neal A, Redpath AN, Patel J, Chouliaras KM, Ratnayaka I, Smart N, De Val S. Regulatory pathways governing murine coronary vessel formation are dysregulated in the injured adult heart. Nat Commun. 2019;10:3276. doi: 10.1038/s41467-019-10710-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Reyes‐Palomares A, Gu M, Grubert F, Berest I, Sa S, Kasowski M, Arnold C, Shuai M, Srivas R, Miao S, et al. Remodeling of active endothelial enhancers is associated with aberrant gene‐regulatory networks in pulmonary arterial hypertension. Nat Commun. 2020;11:1673. doi: 10.1038/s41467-020-15463-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hsieh PN, Zhou G, Yuan Y, Zhang R, Prosdocimo DA, Sangwung P, Borton AH, Boriushkin E, Hamik A, Fujioka H, et al. A conserved KLF‐autophagy pathway modulates nematode lifespan and mammalian age‐associated vascular dysfunction. Nat Commun. 2017;8:914. doi: 10.1038/s41467-017-00899-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Davignon J, Ganz P. Role of endothelial dysfunction in atherosclerosis. Circulation. 2004;109:Iii27–Iii32. doi: 10.1161/01.CIR.0000131515.03336.f8 [DOI] [PubMed] [Google Scholar]
- 54. Nation DA, Sweeney MD, Montagne A, Sagare AP, D'Orazio LM, Pachicano M, Sepehrband F, Nelson AR, Buennagel DP, Harrington MG, et al. Blood‐brain barrier breakdown is an early biomarker of human cognitive dysfunction. Nat Med. 2019;25:270–276. doi: 10.1038/s41591-018-0297-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Perico L, Benigni A, Casiraghi F, Ng LFP, Renia L, Remuzzi G. Immunity, endothelial injury and complement‐induced coagulopathy in COVID‐19. Nat Rev Nephrol. 2020;17:46–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Varga Z, Flammer AJ, Steiger P, Haberecker M, Andermatt R, Zinkernagel AS, Mehra MR, Schuepbach RA, Ruschitzka F, Moch H. Endothelial cell infection and endotheliitis in COVID‐19. Lancet. 2020;395:1417–1418. doi: 10.1016/S0140-6736(20)30937-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figures S1–S4