

Abstract
Rationale
Emerging data support the existence of a microbial “gut-lung” axis that remains unexplored in bronchiectasis.
Methods
Prospective and concurrent sampling of gut (stool) and lung (sputum) was performed in a cohort of n = 57 individuals with bronchiectasis and subjected to bacteriome (16S rRNA) and mycobiome (18S Internal Transcribed Spacer) sequencing (total, 228 microbiomes). Shotgun metagenomics was performed in a subset (n = 15; 30 microbiomes). Data from gut and lung compartments were integrated by weighted similarity network fusion, clustered, and subjected to co-occurrence analysis to evaluate gut-lung networks. Murine experiments were undertaken to validate specific Pseudomonas-driven gut-lung interactions.
Results
Microbial communities in stable bronchiectasis demonstrate a significant gut-lung interaction. Multibiome integration followed by unsupervised clustering reveals two patient clusters, differing by gut-lung interactions and with contrasting clinical phenotypes. A high gut-lung interaction cluster, characterized by lung Pseudomonas, gut Bacteroides, and gut Saccharomyces, is associated with increased exacerbations and greater radiological and overall bronchiectasis severity, whereas the low gut-lung interaction cluster demonstrates an overrepresentation of lung commensals, including Prevotella, Fusobacterium, and Porphyromonas with gut Candida. The lung Pseudomonas-gut Bacteroides relationship, observed in the high gut-lung interaction bronchiectasis cluster, was validated in a murine model of lung Pseudomonas aeruginosa infection. This interaction was abrogated after antibiotic (imipenem) pretreatment in mice confirming the relevance and therapeutic potential of targeting the gut microbiome to influence the gut-lung axis. Metagenomics in a subset of individuals with bronchiectasis corroborated our findings from targeted analyses.
Conclusions
A dysregulated gut-lung axis, driven by lung Pseudomonas, associates with poorer clinical outcomes in bronchiectasis.
Keywords: bronchiectasis, gut-lung axis, microbiome, mycobiome, metagenomics
At a Glance Commentary
Scientific Knowledge on the Subject
The microbial gut-lung axis, although increasingly recognized in respiratory disease, remains poorly characterized, and its role in bronchiectasis is unknown.
What This Study Adds to the Field
The microbial gut-lung axis in bronchiectasis is described using an integrative multibiome approach that reveals two patient clusters distinguished by their gut-lung interactions. The clusters reveal contrasting clinical phenotypes: a high gut-lung interaction cluster—characterized by lung Pseudomonas, gut Bacteroides, and gut Saccharomyces—is associated with increased exacerbations and greater radiological and overall bronchiectasis severity, whereas a low gut-lung interaction cluster demonstrates an overrepresentation of lung commensals and gut Candida. Experimental murine work reveals an important role for lung Pseudomonas in driving a dysregulated gut-lung axis that associates with poorer outcomes in clinical bronchiectasis.
The human microbiome has a key role in maintaining health, and microbial dysbiosis correlates with the development and progression of disease, including chronic respiratory disease states such as asthma, chronic obstructive pulmonary disease (COPD), cystic fibrosis, and bronchiectasis (1–7). Critical functions of the human microbiome include the processing of nutrients, production of metabolites, protection against invading pathogens, and maintaining immune homeostasis; hence, an understanding of the microbiome-host interaction is key to appreciating its role in health and disease and as a therapeutic target.
Although gut microbiomes are widely studied in a number of clinical settings, emerging work on the lung microbiome in chronic respiratory disease is gaining momentum, including recent descriptions of a holistic multibiome integrative analysis of bacteria, viruses, and fungi in the bronchiectasis airway that associates with clinical correlates, including exacerbations (7–9). Altered gut microbiomes influence distal organ systems, including the brain, kidney, liver, heart, and lung, through direct and indirect interaction of microbes and their associated metabolites (10–14). Several lines of evidence now support the existence of a gut-lung axis with potential roles in chronic respiratory disease that remain incompletely characterized in bronchiectasis (8, 15–19). Such an axis involves microbe-microbe and host-microbe interactions that confer local and distal effects at two concurrent physiologically distinct organ sites. One such example is the observed higher occurrence of bronchiectasis, sometimes with frequent exacerbations, in individuals with inflammatory bowel disease (IBD) (20, 21). Pathogenesis of IBD is shown to link with interactions between intestinal immunity and dysbiosis of the underlying gut microbiome; however, links with the lung microbiome remain to be established (22).
Emerging microbial models assessing interkingdom microbial interaction in bronchiectasis have provided important insight into microbiome-related changes occurring during exacerbations, including the significant alterations in microbial interaction rather than identity that occurs in bronchiectasis (7, 9, 23). An appropriate extension of such integrative approaches, previously applied to lung microbiomes in isolation, is to evaluate this across organ systems such as that provided by the gut-lung axis. Here, using a prospectively recruited cohort of adults with bronchiectasis and using concurrently sampled specimens from the gut and lung, respectively, subjected to integrated multibiome analysis, we investigated whether the gut-lung axis in bronchiectasis provides an enhanced understanding of disease, including better patient stratification. Some of the results of these studies have been previously reported in the form of an abstract (24, 25).
Methods
Study Population
Fifty-seven patients (⩾18 years old, Caucasian Europeans of Italian origin) with non–cystic fibrosis bronchiectasis were prospectively recruited during periods of clinical stability at the Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico (Milan, Italy). All patients had radiological bronchiectasis confirmed by high-resolution computed tomographic scanning of the thorax in accordance with British Thoracic Society guidelines and were identified during outpatient visits at periods of clinical stability and daily sputum production (26). Clinical stability was defined as the absence of new symptoms or change in bronchiectasis therapy with no exacerbations and/or antibiotic use in the preceding 4-week period. Exclusion criteria included other concurrent respiratory diagnoses (asthma or COPD) established by international criteria, including spirometry (27, 28); or any documented gastrointestinal pathology (with endoscopic confirmation), including IBD, gastrointestinal cancer, and/or active gastrointestinal infection. Individuals receiving chemotherapy and those with acute and/or recent infection requiring short-term antibiotic therapy (oral or intravenous) in the 4 weeks preceding outpatient attendance were excluded. Because of the requirement for prolonged antimicrobial therapy, which could affect both lung and gut microbiota, patients with mycobacterial infection were also excluded. (For further details on ethical approval and clinical, radiological, and functional evaluation of all participants, see the online supplement.)
Murine Experiments
Mouse experiments were approved by the institutional review board of Nanyang Technological University, Singapore, in accordance with the guidelines of the Institutional Animal Care and Use Committee (approval numbers: NTU-IACUC: A18089, A21073, and A19032). Male wild-type C57BL/6J mice (aged 8 to 10 wk) were housed in a 12-hour:12-hour light:dark cycle and given ad libitum access to chow diet and water. Mice were exposed to either Pseudomonas aeruginosa (PAO1) intratracheal infection or an equivalent volume of sterile saline (0.9%) under antibiotic-exposed or antibiotic-naive conditions. Imipenem was chosen as a gut microbiome disruptive agent because of its low systemic bioavailability when given orally, allowing the disruption of gut microbiomes with relatively little effect on lung Pseudomonas. Fecal pellets were obtained on day 0 (before imipenem treatment), day 2 (after imipenem treatment), and day 5 (3 days post–PAO1 inoculation; dpi, as described later) and snap frozen in liquid nitrogen before DNA extraction and microbiome analysis.
DNA Extraction and Microbiome Profiling of Lung and Gut
DNA was extracted using the Quant-IT dsDNA Assay Kit (for patient sputum and stool; Thermo Fisher Scientific) and the DNeasy PowerSoil Pro Kit (for mouse stool; QIAGEN). Extracted DNA was quantified by Qubit (Invitrogen) and Nanodrop (Invitrogen). Bacterial and fungal taxonomic profiling was performed by 16S and 18S Internal Transcribed Spacer amplicon sequencing on a MiSeq platform (Illumina), whereas whole genome shotgun (WGS) metagenomic analysis was applied to sputum (lung) and stool (gut) sample sequencing on a HiSeq 2500 platform (Illumina) according to standard library preparation and DNA sequencing protocols (7), in a subset of bronchiectasis patients (n = 15). Details of sequencing and bioinformatic analysis strategies are described in the online supplement. All target amplicon and WGS metagenomic sequencing data have been deposited in the National Center for Biotechnology Information Sequence Read Archive under accession numbers PRJNA740243 (targeted amplicon - human), PRJNA824950 (targeted amplicon - mouse), and PRJNA740243 (WGS).
Statistical Analysis
We assessed distributional differences between groups (clusters, murine-experimental arms, and inhaled corticosteroids and/or macrolide treatment groups) using the paired or unpaired Wilcoxon test (nonparametric) as appropriate for continuous variables (including microbial diversity and continuous clinical variables: number of exacerbations in the previous year, FACED (F: forced expiratory volume in 1 second [FEV1]; A: age; C: chronic colonization by Pseudomonas aeruginosa, E: radiological extension [number of pulmonary lobes affected], and D: dyspnea) score, and Reiff score) and the chi-square test for categorical data (including gender and smoking status). For comparison of three or more groups of continuous variables, we used the Kruskal-Wallis test with Dunn’s post hoc testing and Benjamini-Hochberg correction to account for multiple comparisons (three comparisons - continuous clinical variables; and six comparisons - microbial diversity). As a preprocessing step, microbial datasets from targeted sequencing were filtered to include only microbes present in at least 5% of, or three, patients (whichever is highest) and at least at 1% abundance level. Microbial datasets from metagenomic sequencing were filtered to include only microbes confirmed using BLAST confirmatory analysis, as previously described (7). Microbial diversity from samples were assessed with the Shannon diversity index and visualized as box plots using R (version 3.6.3). Microbial composition was normalized using relative abundance and visualized by stacked bar plots, and principal coordinates analysis was performed using Bray-Curtis dissimilarity. We assessed differences in microbial composition using PERMANOVA (permutational multivariate analysis of variance) on patient dissimilarity matrices derived using Bray-Curtis dissimilarity indexes. Differential abundance analysis of microbes was performed using the LefSe (linear discriminant analysis effect size) webtool with default parameters (29). All statistical analysis was performed using custom scripts written in R and Python, and P values <0.05 were considered statistically significant (https://github.com/Jayanth-kumar5566/Lung-Gut_Study).
Full details on specimen collection (sputum and stool), the integrative, cluster, and co-occurrence analysis are detailed in the online supplement. Negative controls (including blanks) are illustrated in Figure E1, and all raw data from this study can be accessed at the National Center for Biotechnology Information Sequence Read Archive with the accession numbers PRJNA740243 (human) and PRJNA824950 (mouse).
Results
To characterize the gut-lung axis in stable bronchiectasis, we assessed concurrently sampled sputum (representing lung) and stool (representing gut) bacterial and fungal microbiomes from individuals with stable bronchiectasis (n = 57) by targeted amplicon sequencing approaches as described (Figure 1). The cohort was predominantly female (78.9%), with a median age of 63 years (interquartile range [IQR]: 54–72) (Table 1). Most patients (69%) had idiopathic bronchiectasis. Following this, the most frequently identified etiologies were immunodeficiency (13%), primary ciliary dyskinesia (9%), and postinfection (7%). Streptococcus and Fusobacterium were the most frequently identified lung bacteria, whereas Bacteriodes was most frequently identified in the gut microbiome (Figure 1A). Candida was the most prevalent fungal taxon identified in both lung and gut mycobiome profiles (Figure 1E). A significantly decreased bacterial (but not fungal) -diversity (P < 0.001) in the lung was identified between paired gut-lung specimens (Figures 1B and 1F), whereas -diversity assessment confirmed significant ecological divergence between anatomical sites (Figures 1C and 1G). After appropriate filtering, direct comparisons revealed greater fungal, as opposed to bacterial, overlap (Figures 1D and 1H).
Figure 1.
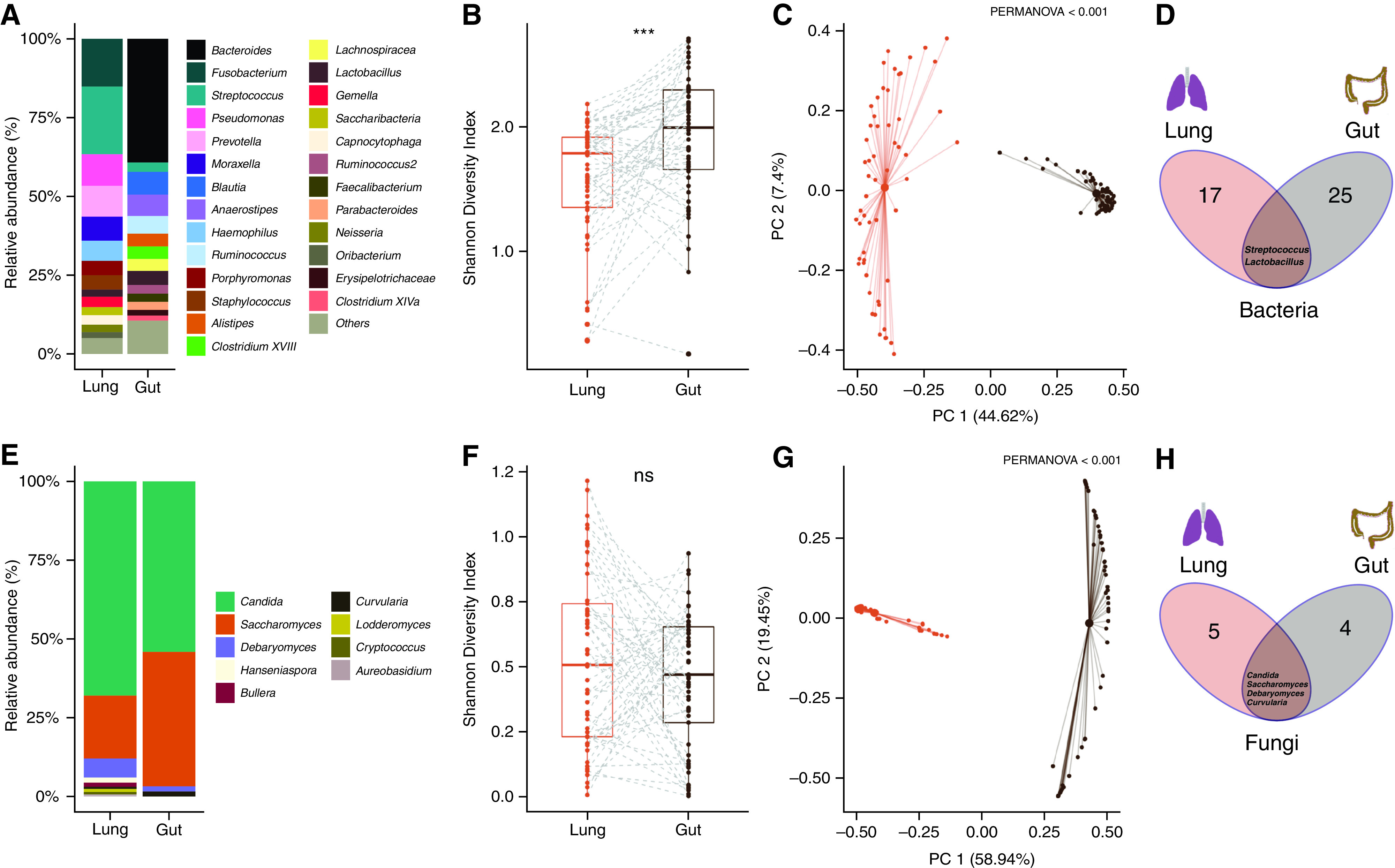
Overview of the lung and gut microbiome in stable bronchiectasis. (A–H) Stacked bar plots represent the (A) bacteriome and (E) mycobiome composition of the lung and gut, respectively. The y-axis represents the relative abundance (%) of microbial taxa (at the genus level) derived using targeted amplicon 16S (bacteria) and Internal Transcribed Spacer (fungal) sequencing approaches applied to sputum (lung) and stool (gut). Paired box plots illustrate (B) bacterial and (F) fungal α-diversity differences between the lung (orange) and gut (black) computed using the Shannon diversity index. Bold lines represent the median diversity, individual dots represent each respective sample, and dotted lines represent the pairing of lung and gut samples (in individual patients). Principal coordinate analysis plots (based on Bray-Curtis dissimilarity) illustrate differences in (C) bacteriome and (G) mycobiome between lung (orange) and gut (black) specimens. Venn diagrams illustrating the overall number of (D) bacterial and (H) fungal genera identified in lung and gut compartments, respectively, with intersections demonstrating overlapping taxa between compartments. ***P < 0.001. ns = nonsignificant; PC 1 = principal coordinates 1; PC 2 = principal coordinates 2.
Table 1.
Demographic Table Summarizing the Stable Bronchiectasis Cohort (Overall and by Cluster)
| Demographic | All (N = 57) |
Cluster 1 (n = 31) |
Cluster 2 (n = 26) |
P value (between Clusters) |
|---|---|---|---|---|
| Sex, n (%) | ns | |||
| Female | 45 (78.9) | 25 (80.6) | 20 (76.9) | — |
| Male | 12 (21.1) | 6 (19.4) | 6 (23.1) | — |
| Age, median (IQR) | 63.00 (54.00–72.00) | 65.00 (55.00–75.00) | 62.50 (54.30–69.80) | ns |
| Disease severity (as FACED score) | 2.00 (2.00–4.00) | 3.00 (2.00–4.00) | 2.00 (1.0–3.00) | 0.0376 |
| Radiological severity (as Reiff score) | 4.00 (3.00–8.00) | 6.00 (4.00–8.00) | 4.00 (2.25–5.00) | 0.0063 |
| BMI | 21.00 (19.00–24.80) | 20.20 (19.00–24.20) | 21.6 (19.30–25.80) | ns |
| Number of exacerbations (in the year preceding study recruitment), median (IQR) | 2.00 (1.00–4.00) | 3.00 (2.00–5.00) | 2.00 (1.00–2.75) | 0.0460 |
| Smoking status, n (%) | ns | |||
| Ex-smoker | 23 (40.4) | 11 (35.5) | 12 (46.2) | — |
| Never smoker | 34 (59.6) | 20 (64.5) | 14 (53.8) | — |
| Lung function (as FEV1% predicted), median (IQR) | 71.00 (62.00–90.00) | 71.00 (59.50–88.50) | 76.50 (67.30–90.80) | ns |
| mMRC dyspnoea score, n (%) | ns | |||
| 0 | 24 (42.1) | 12 (38.7) | 12 (46.2) | — |
| 1 | 23 (40.4) | 11 (35.5) | 12 (46.2) | — |
| 2 | 3 (5.3) | 1 (3.2) | 2 (7.7) | — |
| 3 | 6 (10.5) | 6 (19.4) | 0 (0) | — |
| 4 | 1 (1.8) | 1 (3.2) | 0 (0) | — |
| Chronic macrolide use, n (%) | ns | |||
| No | 49 (86.0) | 28 (90.4) | 21 (80.8) | — |
| Yes | 8 (14.0) | 3 (9.7) | 5 (19.2) | — |
| Inhaled corticosteroid use, n (%) | ns | |||
| No | 35 (61.4) | 22 (71.0) | 13 (50.0) | — |
| Yes | 22 (38.6) | 9 (29.0) | 13 (50.0) | — |
| Total number of courses of oral antibiotics in the 3-yr period preceding study recruitment, median (IQR) | 4.00 (2.00–6.00) | 4.00 (2.25–9.00) | 4.00 (2.00–6.00) | ns |
Definition of abbreviations: BMI = body mass index; FACED = FEV1, age, chronic colonization, extension, and dyspnea; IQR = interquartile range; mMRC = modified Medical Research Council dyspnea scale; ns = nonsignificant.
Demographic table illustrating the study cohort with stable bronchiectasis (n = 57) and P values for significant differences observed between clusters. Demographic data are presented as median value (and IQR) and/or patient n (and %), as appropriate. Significant P values (<0.05) are indicated in bold.
Having determined composition, diversity, and overlap between gut and lung microbiomes, we next assessed for potential gut-lung interaction by co-occurrence analysis to reveal microbial association networks (interactomes) that contain several significantly correlated interaxis interactions (Figures 2A and 2B). To further probe these interactions, we defined microbes (as nodes) on the basis of alternate network metrics, as previously described (7). Briefly, microbes were classified as busy (i.e., node degree: microbes with a higher number of direct interactions with other microbes), critical (i.e., stress centrality: microbes that are key to maintaining the network’s integrity), and influential (i.e., betweenness centrality: microbes that influence other microbes within the network, including indirectly) (7). Using this approach, lung Streptococcus and Prevotella and gut Bacteroides and Lactobacillus were most busy, critical, and influential, demonstrating individual importance within the gut-lung network (Figure 2B). Although these organisms were found to be the busiest, most critical, and influential microbes in the overall gut-lung network (Figure 2B), these measures, on the basis of the overall (direct and indirect) interaction network metrics, go beyond simple direct overlap between the gut and lung compartments (Figures 1D and 1H).
Figure 2.
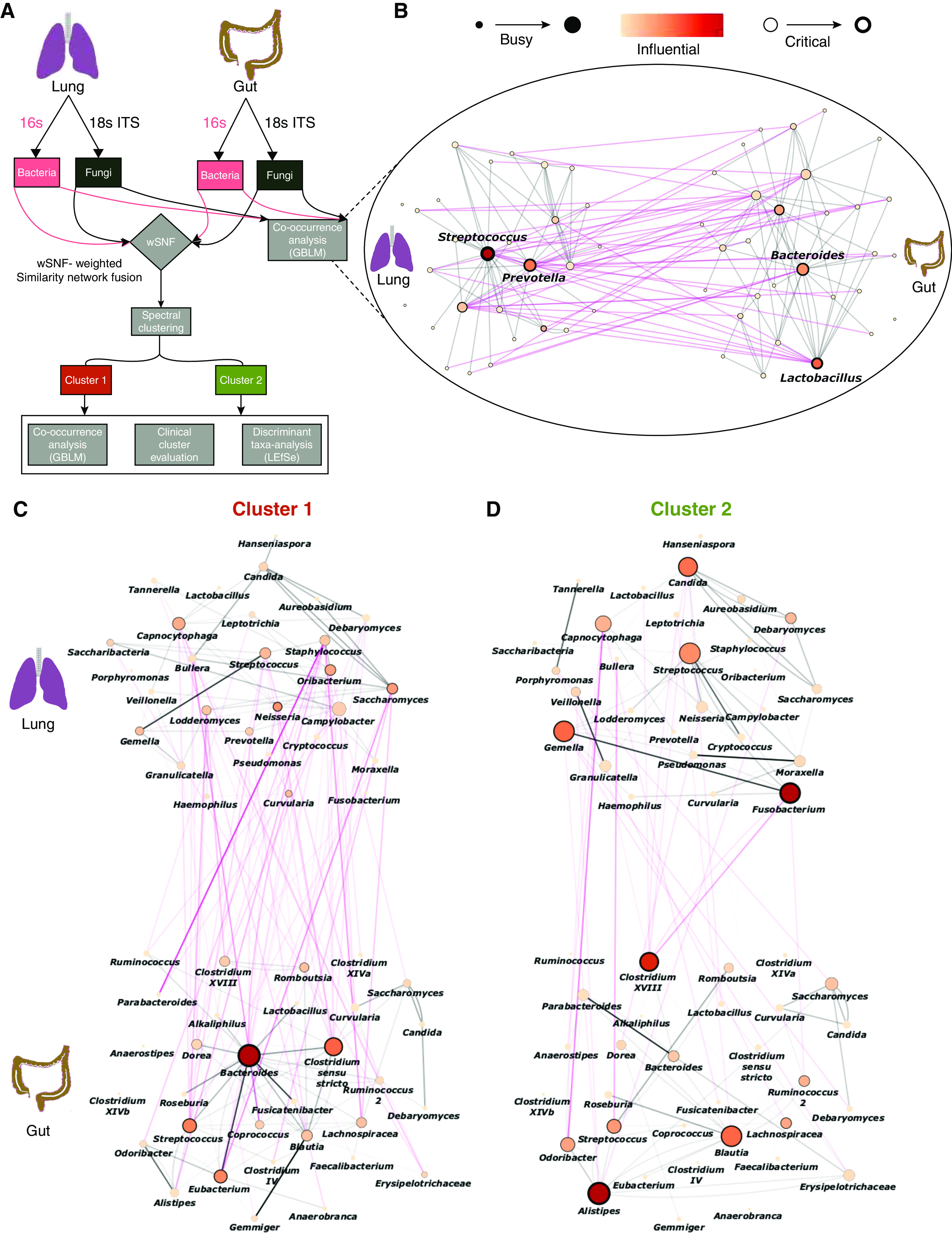
The gut-lung interactome allows patient stratification in stable bronchiectasis. (A–D) In (A): an overview of the analytical approach to evaluating the gut-lung axis in bronchiectasis. The computational workflow implemented in this study includes the application of co-occurrence analysis using the generalized boosted linear model (GBLM) to bacteriome and mycobiome profiles derived from lung and gut compartments, respectively, to obtain the gut-lung interactome network. Four multibiomes were integrated using wSNF followed by spectral clustering to derive unsupervised patient groups. Differentially abundant taxa between clusters are evaluated using LEfSe and clinical evaluation of the respective clusters performed. Cluster-specific interactome networks were generated using the GBLM, and network plots illustrating interactions (as edges) between microbes (indicated as individual nodes) are illustrated for (B) the overall study cohort, (C) cluster 1, and (D) cluster 2, respectively. Only significantly correlated interactions (i.e., P < 0.001) are illustrated. Gut-lung interactions are represented by pink lines (edges). Microbes within gut-lung networks are classified as busy (i.e., node degree: microbes with a higher number of direct interactions with other microbes), critical (i.e., stress centrality: microbes that are key to maintaining the network’s integrity), and/or influential (i.e., betweenness centrality: microbes that influence other microbes within the network, including indirectly), and those with the highest calculated network metrics are highlighted by size, width, and node coloration, respectively, in the presented network plots (7). ITS = internal transcribed spacer; LEfSe = Linear discriminant analysis Effect Size; wSNF = weighted similarity network fusion.
Having identified potential gut-lung interactions in stable bronchiectasis, we next evaluated whether clinically relevant patient groups (differentiated by gut-lung interaction) exist. To achieve this, we first integrated bacteriome and mycobiome profiles from the gut and lung, respectively (i.e., four microbiome datasets per patient; total, 228 microbiomes) using weighted similarity network fusion (7, 9). Weightage of each microbiome dataset for the integration was assigned on the basis of its respective taxonomic richness, reflective of its information content and established by previously methodology (i.e., lung bacteriome 19 genera: 32.2%; lung mycobiome nine genera: 15.2%; gut bacteriome 27 genera: 45.8%; and gut mycobiome four genera: 6.8%) (7, 9). Spectral clustering of the integrated network resolved two patient groups, with an average silhouette score of 0.73 and a cluster robustness of 78.2% (based on 100 bootstrap iterations), indicating strong cluster consistency and high robustness (Figure 2A). The two patient clusters each demonstrate significantly distinct gut-lung axes in terms of their interactomes, exemplified by interaction metrics for lung Fusobacteria, lung Candida, and gut Bacteroides (Figures 2C and 2D). Cluster 1, however, demonstrated an overall increased gut-lung interaction, suggesting that microbial interactions (rather than abundance) distinguish relevant patient strata in bronchiectasis. Cluster 1 exhibited a clinically worse profile, characterized by increased exacerbations (P = 0.046) and greater radiological and overall bronchiectasis severity (indicated as Reiff, P = 0.0063; and FACED scores, P = 0.038, respectively) (Figures 3A–3C). It is important to note that, when only lung microbiomes (bacteriomes and mycobiomes) were integrated and clustered, the derived clinical associations were not apparent, indicating the importance of considering the gut microbiome (Figure E2). It is interesting that multibiome integration using only gut microbiomes (bacteriomes and mycobiomes) reveal three patient clusters, each with distinct (but significantly less apparent) clinical correlates, further confirming the important association between the gut and lung compartments (Figure E3).
Figure 3.
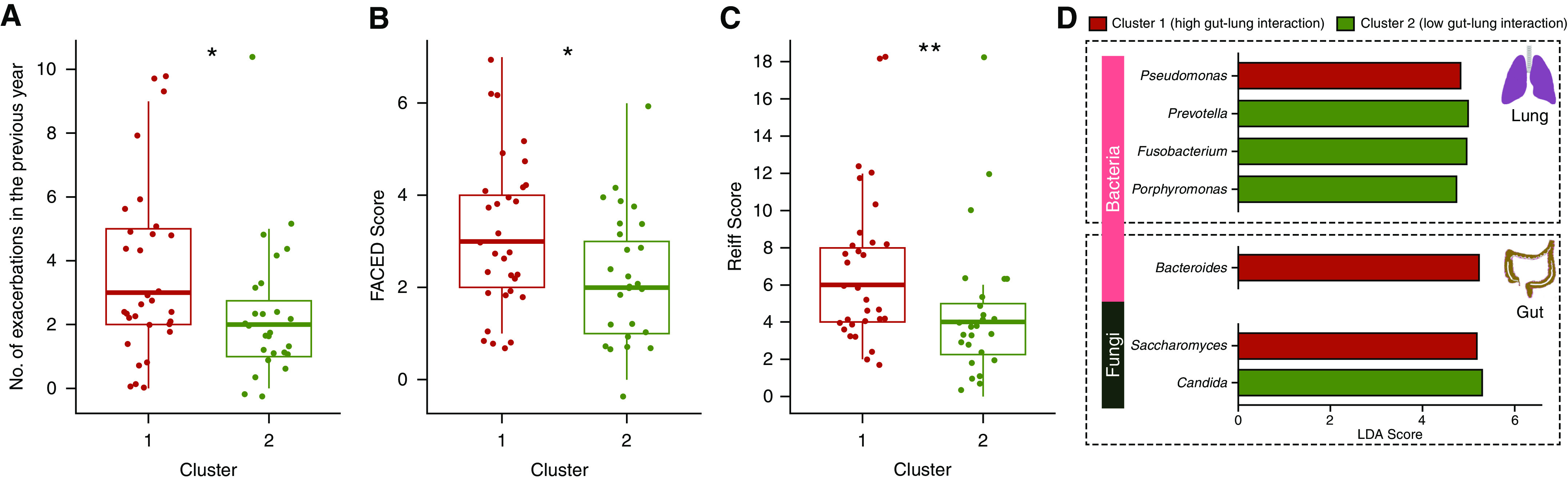
Clinical and microbiome differences between gut-lung interactome–defined patient clusters. (A–D) Box plots illustrating differences in (A) exacerbation frequency, (B) disease severity (as FACED [FEV1, age, chronic colonization, extension, and dyspnea] score) (48) and (C) radiological severity (as Reiff score) (49) between the derived identified patient clusters. Cluster 1 (high gut-lung interaction) is indicated in red, and cluster 2 (low gut-lung interaction) is indicated in green, as derived by spectral clustering of integrated gut-lung multibiomes. (D) Bar plots representing differentially abundant bacterial (light pink) and fungal (dark green) taxa of the lung (top) and gut (bottom) between the high and low gut-lung interaction clusters. Significantly increased taxa in cluster 1 (high gut-lung interaction) and cluster 2 (low gut-lung interaction) are highlighted as red and green bars, respectively. The x-axis represents the linear discriminant analysis (LDA) score, and the y-axis significant taxa with LDA score > 0. *P < 0.05; **P < 0.01. ns = nonsignificant.
No significant influence of inhaled corticosteroid and/or chronic macrolide use or frequent exacerbator status was found in relation to the assessed microbiomes (Figures E4 and E5). Of note, lung bacterial β-diversity is altered in individuals receiving inhaled corticosteroid and/or macrolides (Figure E4), and fungal β-diversity differs in frequent exacerbators (Figure E5).
Microbiome composition and diversity differed between clusters (Figure E6). Key microbes within the gut-lung axis discriminated individuals in cluster 1 (high gut-lung interaction) from cluster 2 (low gut-lung interaction), including a significantly increased lung Pseudomonas, gut Bacteroides, and gut Saccharomyces in the high gut-lung interaction cluster (cluster 1) (Figure 3D). Detecting increased Pseudomonas in the bronchiectasis lung in relation to poor clinical outcome remains consistent with the established literature (20, 30–32); however, our concurrent gut microbiome analyses potentially uncover a role for gut Bacteroides and Saccharomyces in this relationship. In contrast, the low gut-lung interaction cluster (cluster 2) exhibits increased lung commensals and/or pathobionts, including Prevotella, Fusobacterium, and Porphyromonas in concert with gut Candida (Figure 3D). Taken together, integrative analyses assessing the gut-lung axis in bronchiectasis identify a clinically worse patient subgroup, with distinct gut and lung microbiome communities and demonstrable network characteristics associating with clinically relevant bronchiectasis phenotypes, which remains undetected if either lung or gut microbiomes are assessed in isolation, emphasizing the relevance of the gut-lung axis.
Having identified a demonstrable association between the gut-lung axis and clinical features of bronchiectasis, including the role of lung Pseudomonas in determining a deleterious bronchiectasis phenotype (i.e., it represents a key lung determinant in the high gut-lung interaction cluster characterized by poor clinical outcome), we sought to assess this phenomenon in an appropriate experimental system. Given our clinical observations, we hypothesized that the presence of P. aeruginosa in the airway exerts an influence on the gut microbiome. To test this, we isolated the effect of Pseudomonas airway infection on the gut microbiome by using a mouse model of P. aeruginosa (PAO1) infection. This allowed the assessment of changes in the composition and network configuration of the gut microbiome directly linked to the airway challenge of an established bronchiectasis pathogen. As no representative animal model of bronchiectasis exists, we used 8- to 10-week-old male wild-type C57BL/6J mice infected intratracheally (lung) with Pseudomonas aeruginosa (PAO1). To better recapitulate the clinical situation, including antibiotic exposure, we performed the experiment in the presence and absence of antibiotic (imipenem) treatment (Figure 4A). Fecal pellets were collected in each experimental arm (on day 5) and gut bacteriomes and mycobiomes were evaluated with minimal evidence of procedural and/or sequencing contamination (Figures E1G and E1H). Imipenem is a broad-spectrum β-lactam antibiotic with activity against a range of aerobic and anaerobic Gram-positive and Gram-negative bacteria, including Pseudomonas (33). Nonetheless, we selected this agent as it demonstrates low systemic bioavailability when given orally. Hence, our experimental design (i.e., oral administration) allows an effect on the gut microbiome with relatively little effect on lung Pseudomonas. The effects of imipenem on the murine gut microbiomes are illustrated in Figure E7. Bacteriome and mycobiome profiles across the four experimental arms were determined (Figures 4B and 4C), and although no differences in bacterial (P = 0.13) or fungal (P = 0.17) α-diversity were observed, significant effects on bacterial (P = 0.03) and fungal (P = 0.009) β-diversity were detected (Figure E8). P. aeruginosa (PAO1) airway infection exhibited a direct effect on murine gut microbiome architecture, supporting a lung-gut interaction. To evaluate the impact of Pseudomonas (lung) infection on the gut microbiome, we split organisms into those unaffected and (independently) affected by antibiotic (imipenem) treatment (Figures 4D, 4E, and E7). This allows for a clearer appreciation of any additive effect of P. aeruginosa (lung) infection on organisms in the latter group. It is interesting that discriminant taxonomic analysis failed to identify major change to any specific gut microbe associated with P. aeruginosa (lung) infection. In contrast, assessment of the gut interactome revealed distinct alterations in microbial interactions after P. aeruginosa infection, based on network metrics that were impervious to the effects of imipenem (Figure 4D). These include Ruminococcus, Intestinomonas, and Eisenbergiella (Figure 4D). Critically, Blautia, Alistipes, and Bacteroides demonstrate enhanced network metrics after P. aeruginosa (lung) infection that were abrogated in the presence of antibiotic (imipenem) treatment (Figure 4E). Taken together, this suggests that several gut-lung interactions may be mediated by the presence of P. aeruginosa in the lung and are potentially altered after antibiotic intervention targeting the gut microbiome (Figures 4D and 4E). These results thus highlight several gut microbial interactions directly influenced by airway infection (lung-gut) while demonstrating the abrogation of others through antimicrobial alteration of the gut (gut-lung), consistent with the operation of such effects across a bidirectional gut-lung axis.
Figure 4.
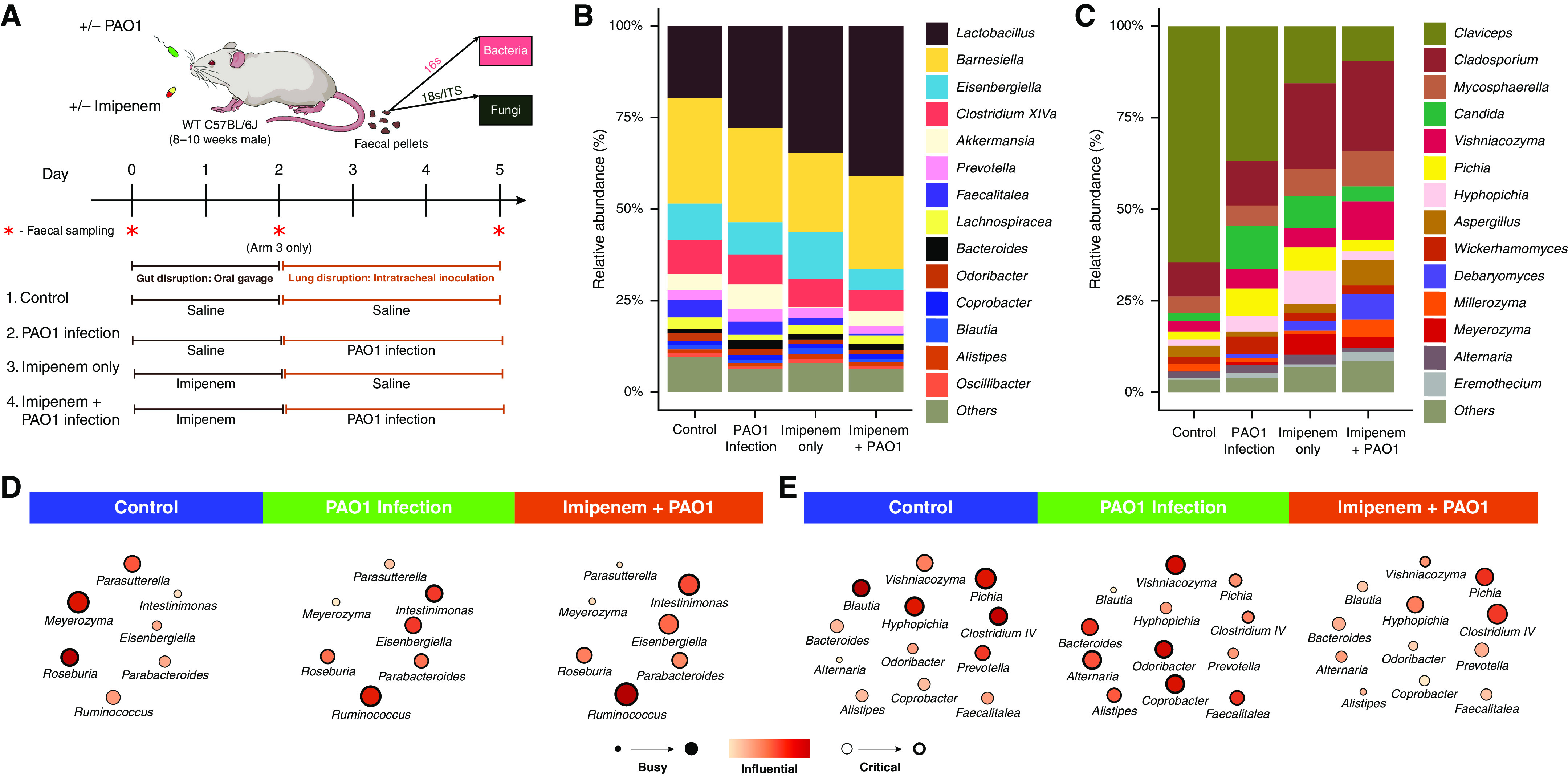
Assessment of gut microbiome dynamics in a murine model of lung Pseudomonas aeruginosa (PAO1) infection. (A) Schematic illustration of the overall experimental design. Twenty-four mice were subjected to four experimental treatment arms (n = 6 per arm). Mice received either a saline control (1 + 2) or antibiotic treatment (imipenem) (3 + 4) by oral gavage for 2 days before either intratracheal delivery of normal saline (1 + 3) or PAO1 inoculation (2 + 4). Bacteriome and mycobiome profiles were characterized by 16S and ITS sequencing approaches derived from fecal pellets obtained at the experimental endpoint (day 5). In addition, assessment of bacteriome and mycobiome profiles pre- and postantibiotic treatment on day 0 and day 2 in treatment arm 3 (indicated by red asterisk) was performed. (B and C) Stacked bar plots illustrate the (B) gut bacteriome and (C) gut mycobiome composition in all four experimental arms (day 5). (D and E) Network plots illustrating key taxa from the mouse gut interactome splitting organisms (D) affected by PAO1 infection independent of antibiotic (imipenem) pretreatment to those (E) affected by PAO1 infection and abrogated by antibiotic (imipenem) pretreatment (see also Figure E7). Microbial genera are represented as nodes indicated as busy (i.e., node degree: microbes with a higher number of direct interactions with other microbes), critical (i.e., stress centrality: microbes key to maintaining the network’s integrity), and/or influential (i.e., betweenness centrality: microbes that influence other microbes within the network, including indirectly), and these network metrics are highlighted by size, width, and node coloration, respectively, in the presented network plots (7). WT = wild-type.
Although direct comparisons between results from the mouse infection model and bronchiectasis cohorts are challenging and have inherent limitations, there are important correlates observed providing evidence of potential gut-lung interrelationships in vivo. When murine Pseudomonas lung infection is considered (independent of antibiotic exposure), alterations to Roseburia, Ruminococcus, and Parabacteriodes are observed in mouse gut bacteriomes (Figure 4D) in line with that seen in our bronchiectasis high gut-lung interaction cluster 1 (Figure 2C). Even more strikingly, in the mouse gut, Bacteroides, a key determinant of the high gut-lung interaction cluster 1 group, demonstrates behavioral change within the mouse network after Pseudomonas lung infection (i.e., it becomes a more busy, influential, and critical microbe), which is then attenuated after antibiotic (imipenem) treatment, independent of the effect of antibiotics alone (Figures 2C, 3D, 4E, and E7). Similar observations can be extended to other microbes from our human bronchiectasis cohorts; for instance, Blautia and Alistipes (Figures 2C, 2D, and 4E). Interestingly, both these genera are (conversely) prominent in the gut of the low gut-lung interaction cluster 2 bronchiectasis group, comparable with the potential benefits attained after antibiotic (imipenem) intervention in the mouse (Figures 2D and 4E). Taken together, our murine infection model reveals, in a controlled setting, the key role that lung Pseudomonas has in potentially influencing the gut microbiome and, importantly outlines key microbial interrelationships (e.g., with gut Bacteroides) that occur between the two compartments.
Having determined the importance and clinical relevance of the gut-lung axis in bronchiectasis, we next sought to better understand species-level and functional variability between our patient clusters using a metagenomics approach in a subset of individuals (i.e., n = 7 from cluster 1 and n = 8 from cluster 2, respectively). This confirmed prior patterns observed in our targeted analysis and identified lung P. aeruginosa and Moraxella catarrhalis as predominant organisms in cluster 1 (high gut-lung interaction) and increased Haemophilus influenzae and Streptococcus pneumoniae in cluster 2 (low gut-lung interaction). Discriminant analysis further identified the oral commensal P. melaninogenica as significantly reduced in cluster 1 (Figure 5A). Gut metagenomic profiling by discriminant analysis (linear discriminant analysis effect size) reveals a marked reduction of Collinisella aerofaciens in cluster 1, with an increased abundance of several Bacteroides and Bifidobacterium species (Figure 5A). Further generalized boosted linear model–derived network analyses identified multiple potential gut-lung interactions involving four major bronchiectasis pathogens: P. aeruginosa, S. pneumoniae, H. influenzae, and M. catarrhalis in addition to other interactions across the gut-lung axis (Figure 5B). Assessing gut interaction networks (by cluster group) reveals that the high gut-lung interaction group (cluster 1) is characterized by increased network connectivity of several Bacteroides and Bifidobacterium species relative to cluster 2 (low gut-lung interaction group), supporting prior observations from our targeted human and murine studies (Figures 5C and 5D). Both clusters exhibit altered network configurations of Streptococci, where S. parasanguinus exerts a greater influence compared with S. thermophilus or S. salivarius in cluster 1 but demonstrates a contrasting pattern in cluster 2 (Figures 5C and 5D). This adds to growing literature associating upper airway commensals with favorable clinical outcomes in respiratory disease (34, 35). Metagenomics also provides additional evidence of the lung Pseudomonas-gut Bacteroides relationship, as observed in our targeted human and murine datasets. Microbial pathway analysis demonstrated that cluster 2 (low gut-lung interaction) is distinguished by an increased abundance of pathways related to bacterial cell wall synthesis, where peptidoglycan maturation was the most discriminatory pathway in the lung and gut. This contrasts cluster 1, where none of the top identified microbial pathways were discriminatory and/or common to gut and lung, suggestive of likely more complex interplay in clinically worse individuals (Figures 5E and 5F).
Figure 5.
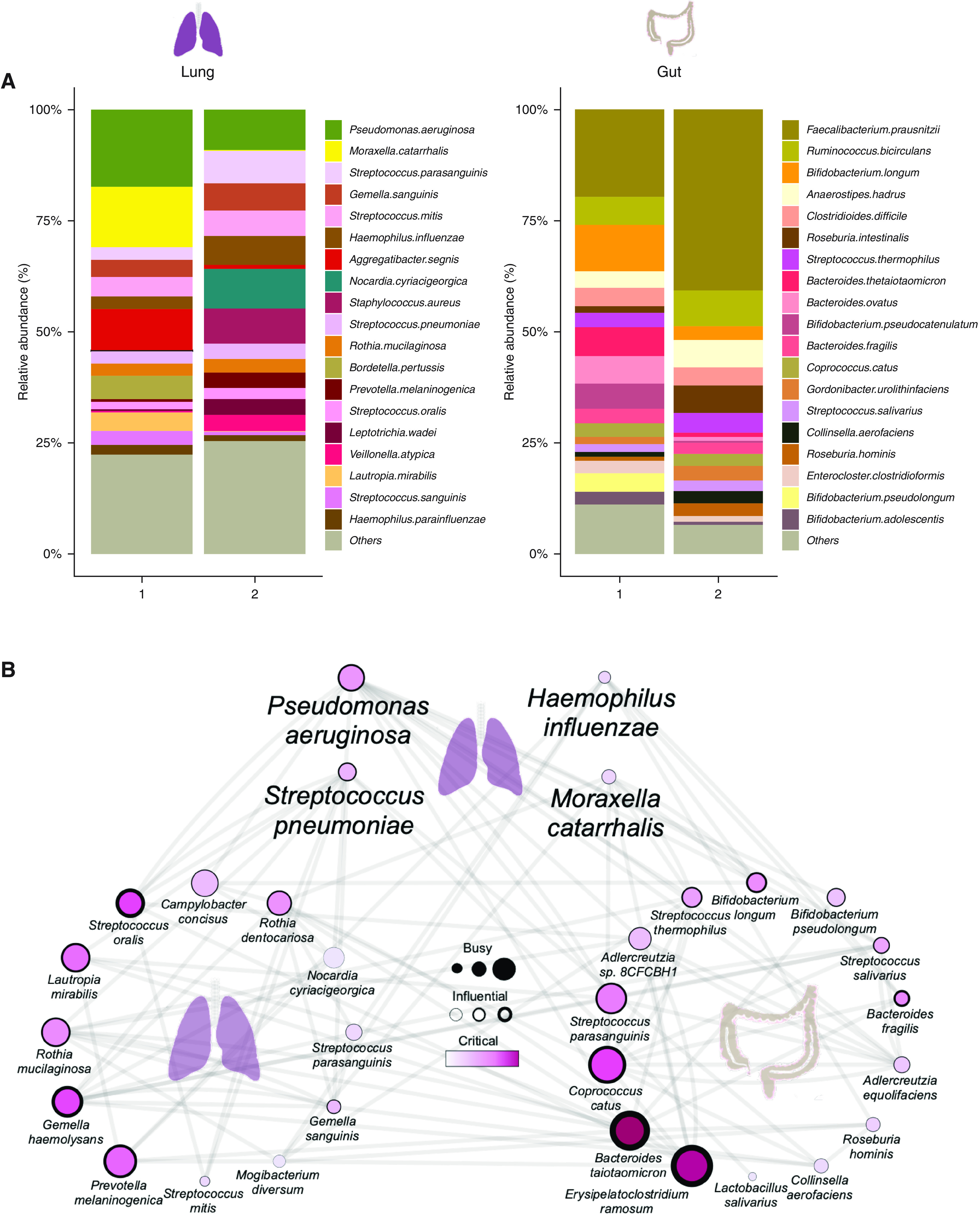
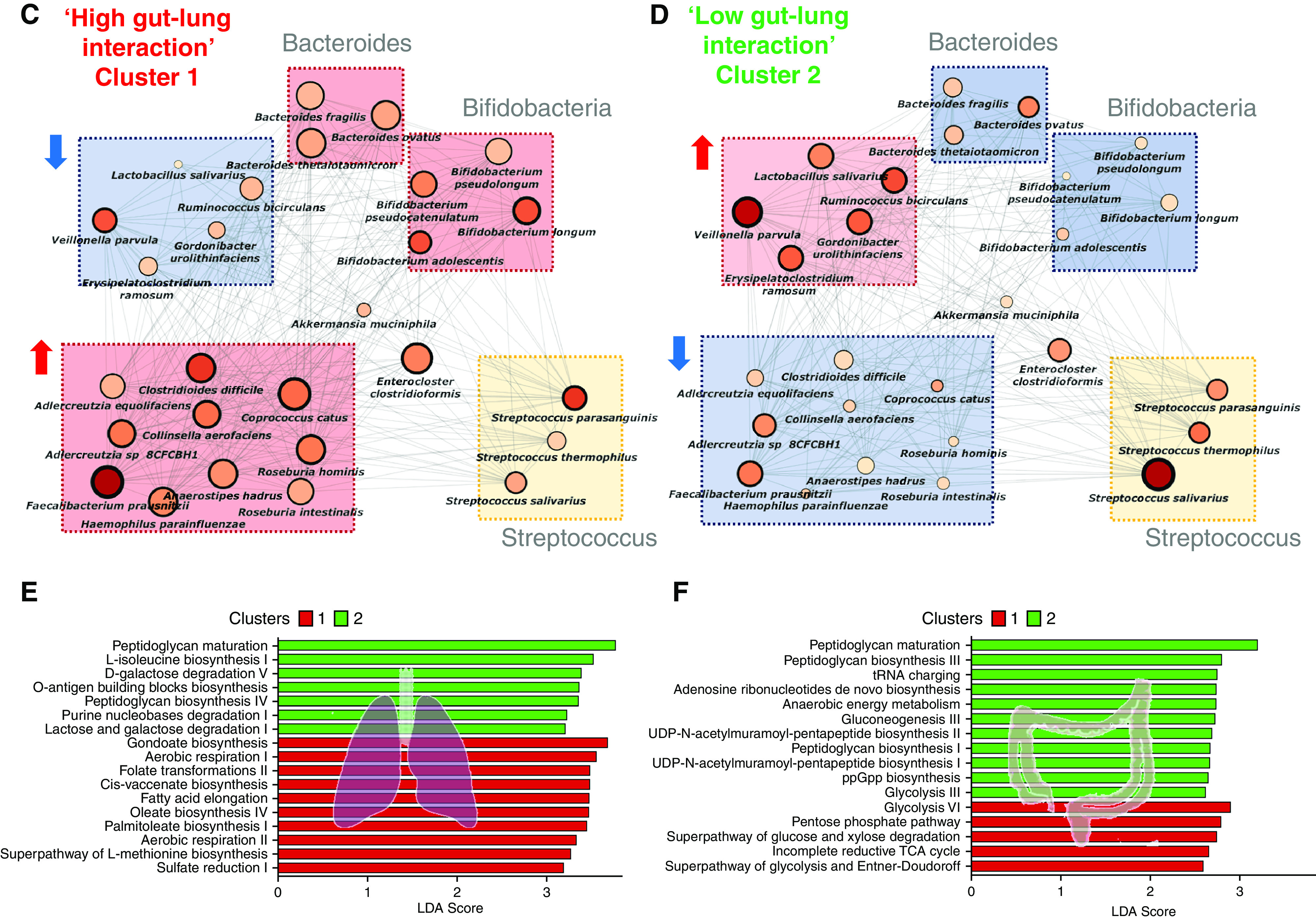
Metagenomic analysis of the gut-lung axis in bronchiectasis. (A) Stacked bar plots illustrating species-level classification of lung (left) and gut (right) microbiome relative-abundance profiles derived by metagenomic sequencing in high (cluster 1, n = 7) and low (cluster 2, n = 8) gut-lung interaction groups. (B) GBLM-derived networks illustrating four key bronchiectasis (bacterial) pathogens and their interactions with other microbes within the bronchiectasis gut-lung interactome. Microbial interactions (edges) are represented as lines (gray), and species as nodes are indicated as busy (i.e., node degree: microbes with a higher number of direct interactions with other microbes), critical (i.e., stress centrality: microbes key to maintaining the network’s integrity), and/or influential (i.e., betweenness centrality: microbes that influence other microbes within the network, including indirectly), and network metrics are highlighted by size, width, and node coloration, respectively, in the presented network plots (7). (C and D) Correlation-based co-occurrence analysis illustrating cluster-based gut microbiome network conformation of the (C) high gut-lung interaction (cluster 1) and (D) low gut-lung interaction (cluster 2), respectively. Species are grouped according to their observed genus-level differential network connectivity within clusters. Genera with three or more representative species-level members (i.e., Bacteroides, Bifidobacteria, and Streptococcus) are highlighted by colored rectangles indicating their respective increased (red), decreased (blue), or neutral (yellow) genus-level network connectivity between clusters. (E and F) Horizontal bar plots illustrating differentially abundant microbial pathways (linear discriminant analysis [LDA] score >2.5) between the high (cluster 1) and low (cluster 2) gut-lung interaction groups in the (E) lung and (F) gut, respectively. The x-axis represents the discriminative score (LDA score), and the y-axis represents specific microbial pathways.
Discussion
The gut microbiome is thought to influence respiratory disease through a gut-lung axis; however, this lacks study in bronchiectasis (8, 16, 19). Our present work begins to address this important knowledge gap by prospectively evaluating patients with bronchiectasis undergoing concurrent gut and lung sampling during disease stability. This approach controls for temporal variation and allows for integrative analysis between organs. Here, we report greater bacterial diversity in the gut compared with lung, in line with prior literature and likely because of the existence of airway disease in these patients (13, 36). Minimal difference in fungal diversity between compartments is observed. The overall low percentage of bacterial versus fungal overlap between the gut and lung does suggest that direct microbial interaction between sites is less likely and that indirect means of interaction, including the secretion of microbial metabolites (e.g., short chain fatty acids) and/or modulation of host immunity—neither assessed in this study—are viable (13, 16, 37). It is important that, in this work, we illustrate the potential clinical utility of concurrently profiling gut and lung microbiomes in bronchiectasis, including the importance of integrated analytics (7, 9, 23). The integration of bacteriomes and mycobiomes (i.e., multibiomes), using methods capable of capturing complex interactions from two distinct anatomical sites, revealed patient stratification into high and low gut-lung interaction groups underpinned by clear differences in their gut-lung interactions. Evaluation of gut and lung multibiomes as separate organ systems notably precludes such strong clinical stratification and demonstrates the importance of the gut-lung axis. Of note, however, some clinical association is evident when only gut multibiomes are considered, underscoring an important and previously unrecognized role for gut microbiota in stratifying bronchiectasis. The high gut-lung interaction bronchiectasis patient cluster was characterized by lung Pseudomonas, gut Bacteroides, and gut Saccharomyces and was associated with increased exacerbations and with greater radiological and overall bronchiectasis severity, whereas the low gut-lung interaction cluster demonstrates an overrepresentation of lung commensals, including Prevotella, Fusobacterium, and Porphyromonas, with gut Candida. This latter group adds further to the increasing body of microbiome literature, indicating that the predominance of upper airway commensals is associated with more favorable clinical outcomes in respiratory disease (34, 35, 38). Microbial commensals are established determinants of the host-immune relationship, demonstrating effector functions reaching far beyond their local environments (13, 16, 39). Although the assessment of conventional microbiome-related metrics such as relative abundance (i.e., microbial identity) and diversity indices did show some variation between patient clusters in our study, our integrated gut-lung interactomes reveal additional insight into microbial interrelationships, a key feature of our novel approach that remains unappreciated if assessing microbial identity alone.
It is interesting that lung Pseudomonas is a key microbial determinant of the high gut-lung interaction cluster associating with adverse clinical outcomes. Although this finding is consistent with the existing bronchiectasis literature (20, 30, 32, 40), our gut-lung assessment reveals a novel relationship with gut Bacteroides. Bacteroides are important gut commensals with potential pathobiont properties and whose increased abundance is linked to IBD (41, 42). In view of the established importance of lung Pseudomonas in clinical bronchiectasis and as a key microbial determinant in our high gut-lung interaction cluster, we next studied lung Pseudomonas in a murine model. Mouse models are successfully used in mechanistic studies elucidating the role of gut microbiomes on distal organ systems; however, notably, no animal model representative of bronchiectasis exists; hence, only an elementary Pseudomonas lung infection model was used (13, 16, 17, 19, 37, 43). Despite inherent differences between human and mouse gut microbiomes, several important associations of lung Pseudomonas infection with gut microbiota were observed, and several matching patterns were seen in the high gut-lung interaction group. These include relationships with gut Roseburia, Ruminococcus, Parabacteriodes, and, critically, Bacteroides—one of the key gut determinants in the high gut-lung interaction cluster. Altered Bacteroides network metrics observed after Pseudomonas lung infection were attenuated after antibiotic treatment, an effect independent of the antibiotic treatment alone and suggestive of the potential for therapeutic manipulation of the gut-lung axis. Alternate organisms, including Blautia and Alistipes, prominent in the low gut-lung interaction group, also demonstrate changes in relation to lung Pseudomonas that are modified by antimicrobial intervention. Our combined human and mouse analysis reveal the importance of appreciating microbial interactions as opposed to microbial identity alone across organ systems and questions whether therapeutic targeting of one organ system gives rise to microbial alterations in another, an important avenue for future work.
Significantly, metagenomics of the gut and lung in a subset of patients validated findings from our targeted analysis, including the identification of P. aeruginosa and several species of Bacteroides in the high gut-lung interaction cluster. In addition, this approach provided additional discriminant commensal species of interest in the low gut-lung interaction bronchiectasis cluster: lung Prevotella melanogenica and gut C. aerofaciens. This is notable, given recent work by Wu and colleagues demonstrating immunoprotective Th17 responses, inducible on commensal P. melanogenica exposure, and which confers protection against an S. pneumoniae challenge (34). As such, their diminished abundance in the clinically adverse high gut-lung interaction cluster appears consistent with a depletion of “beneficial” microbes that may have key roles in immune homeostasis. Conversely, gut C. aerofaciens, implicated in IL-17A signaling, correlates with negative clinical consequence in rheumatoid arthritis, owing to its immune-potentiating effect (44). These reports, alongside our own observations, suggest that microbes in the low gut-lung interaction cluster may engender a proinflammatory response with a net overall immunoprotective effect. It is interesting that increased gut Candida, observed in our low gut-lung interaction cluster, is similarly associated with Th17 airway modulation through gut-lung cross-talk (38). Involvement of immune pathways, induced by lung microbes, with effects at distal sites has recently been shown in multiple sclerosis implicating the lung-brain axis (43). Metagenomics further highlights shifts in microbial metabolic profiles, including enrichment of peptidoglycan maturation in the low gut-lung interaction cluster. Aligned with this, oral administration of peptidoglycan prevents sepsis in response to S. pneumoniae, evidence supporting its positive influence on health (45). Circulating peptidoglycan fragments are traced to gut microbiome constituents and remain important for immune development (45). Whether microbial peptidoglycan fragments, for example, originating from the gut and lung act as tentative intermediary regulators of gut-lung homeostasis in bronchiectasis remains an interesting proposition and one requiring future work incorporating immunological and metabolomic readouts.
Although we report a novel and integrated approach to evaluating the gut-lung axis in bronchiectasis, including clinical relevance, our work has important limitations. First, we used a small cohort from a single site with a cross-sectional design lacking longitudinal follow-up. We, therefore, relied on static data to predict dynamic phenomena. Second, the study relies on small sample sizes, which are partly overcome through data integration and paired analysis. The subgroup comparisons, however, are exploratory at best. Next, although targeted amplicon sequencing is well established, it does have limitations, including primer dependence, low taxonomic coverage, underdevelopment of fungal reference databases, and an underrepresentation of mycobacteria. We overcome this partially by performing metagenomic sequencing in a subset of patients. Furthermore, we did not assess for viruses or viromes in this study, another important microbial kingdom of interest. Although our concurrent gut and lung sampling approach resolves the influence of time-based confounders, other latent confounders may not have been included in our analysis.
Additionally, gut microbiomes are susceptible to other influences, including diet, and no food diaries or information on dietary patterns were collected in this study concerning gut microbiomes. In addition, the persistent long-term effects of antibiotics on gut microbiomes should also be acknowledged, some of which may last beyond the 4-week criterion used in this work. Although the most acute changes to gut microbiomes from antibiotics are observed in the first 8 days after exposure, we cannot out rule the possibility of latent effects, dysbiosis, and/or gut-lung dysfunction that occurred postexposure to antibiotics but preceded the 4-week period before study enrollment.
We also excluded patients with mycobacterial infection who represent an important patient subgroup that should be explored further in future work. Of note, although we excluded COPD in participants on the basis of established spirometry criteria, no systematic assessment of COPD-related changes on chest radiology was performed. Mouse experiments should be interpreted with caution, given the inherent differences between the gut microbiota composition of specific-pathogen–free mice and human subjects. Furthermore, in this work, we cannot exclude potential indirect effects of P. aeruginosa infection on mouse behavior, including food and water consumption (or, possibly, other factors) that may impact gut microbiomes indirectly. Finally, although associative patterns (interactome) between the gut and lung have been identified in this work, they do not delineate the type of interaction (direct or indirect) between microbes. Future mechanistic work, including the assessment of host immunity and incorporating systems biology (i.e., metabolomics and lipidomics), is necessary to assess contributing factors and assign causation.
In conclusion, a dysregulated gut-lung axis driven by lung Pseudomonas occurs in bronchiectasis and associates with poor clinical outcome. Interventional approaches, as seen in ventilator-associated pneumonia, which leverage immunomodulation of this axis warrant future investigation in bronchiectasis (46, 47).
Acknowledgments
Acknowledgment
The authors thank the Academic Respiratory Initiative for Pulmonary Health and the Lee Kong Chian School of Medicine Centre for Microbiome Medicine for collaboration support.
Footnotes
Supported by the Singapore Ministry of Health’s National Medical Research Council Clinician-Scientist Individual Research Grant (MOH-000141) (S.H.C.) and Singapore Ministry of Health’s National Medical Research Council Clinician Scientist Award (MOH-000710) (S.H.C.), the Fondazione IRCCS Cà Granda (RC 2022 – 260-02) (F.B.), and the Engineering and Physical Sciences Research Council through grant EP/T017856/1 (K.T.-A.).
Author Contributions: J.K.N. and S.A.: performance and design of experiments, data analysis and interpretation, statistical analysis, and writing the manuscript. M.M.A.: metagenomic whole genome shotgun and targeted amplicon sequencing analytics, writing the manuscript, statistical analysis, and interpretation. T.K.J. and N.A’.B.M.A.: curation of clinical data, sample preparation, DNA extraction, amplification, and sequencing. F.X.I.: metagenomic whole genome shotgun and targeted amplicon sequencing analytics. H.S.C., Y.S.Y., M.I.G.V., Z.S.L., and J.X.T.L.: performance of animal experiments. F.A., A.G., and F.B.: intellectual contributions, patient recruitment, and procurement of clinical data and specimens. S.H.W., J.J.Y.S., and N.S.T.: oversight of animal experiments and intellectual contributions. K.T.-A.: oversight of mathematical methodology and statistics. S.H.C.: conception and design of overall study and experiments, data analysis and interpretation, statistical analysis, writing the manuscript, and procurement of funding.
This article has an online supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1164/rccm.202205-0893OC on October 26, 2022
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1. Young VB. The role of the microbiome in human health and disease: an introduction for clinicians. BMJ . 2017;356:j831. doi: 10.1136/bmj.j831. [DOI] [PubMed] [Google Scholar]
- 2. Huang YJ, Nariya S, Harris JM, Lynch SV, Choy DF, Arron JR, et al. The airway microbiome in patients with severe asthma: associations with disease features and severity. J Allergy Clin Immunol . 2015;136:874–884. doi: 10.1016/j.jaci.2015.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Layeghifard M, Li H, Wang PW, Donaldson SL, Coburn B, Clark ST, et al. Microbiome networks and change-point analysis reveal key community changes associated with cystic fibrosis pulmonary exacerbations. NPJ Biofilms Microbiomes . 2019;5:4. doi: 10.1038/s41522-018-0077-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sze MA, Dimitriu PA, Hayashi S, Elliott WM, McDonough JE, Gosselink JV, et al. The lung tissue microbiome in chronic obstructive pulmonary disease. Am J Respir Crit Care Med . 2012;185:1073–1080. doi: 10.1164/rccm.201111-2075OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tiew PY, Mac Aogáin M, Chotirmall SH. The current understanding and future directions for sputum microbiome profiling in chronic obstructive pulmonary disease. Curr Opin Pulm Med . 2022;28:121–133. doi: 10.1097/MCP.0000000000000850. [DOI] [PubMed] [Google Scholar]
- 6. Mac Aogáin M, Chotirmall SH. Microbiology and the microbiome in bronchiectasis. Clin Chest Med . 2022;43:23–34. doi: 10.1016/j.ccm.2021.11.002. [DOI] [PubMed] [Google Scholar]
- 7. Mac Aogáin M, Narayana JK, Tiew PY, Ali NABM, Yong VFL, Jaggi TK, et al. Integrative microbiomics in bronchiectasis exacerbations. Nat Med . 2021;27:688–699. doi: 10.1038/s41591-021-01289-7. [DOI] [PubMed] [Google Scholar]
- 8. Budden KF, Shukla SD, Rehman SF, Bowerman KL, Keely S, Hugenholtz P, et al. Functional effects of the microbiota in chronic respiratory disease. Lancet Respir Med . 2019;7:907–920. doi: 10.1016/S2213-2600(18)30510-1. [DOI] [PubMed] [Google Scholar]
- 9. Narayana JK, Mac Aogáin M, Ali NABM, Tsaneva-Atanasova K, Chotirmall SH. Similarity network fusion for the integration of multi-omics and microbiomes in respiratory disease. Eur Respir J . 2021;58:2101016. doi: 10.1183/13993003.01016-2021. [DOI] [PubMed] [Google Scholar]
- 10. Gebrayel P, Nicco C, Al Khodor S, Bilinski J, Caselli E, Comelli EM, et al. Microbiota medicine: towards clinical revolution. J Transl Med . 2022; 20:111. doi: 10.1186/s12967-022-03296-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Young RP, Hopkins RJ, Marsland B. The gut-liver-lung axis. Modulation of the innate immune response and its possible role in chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol . 2016;54:161–169. doi: 10.1165/rcmb.2015-0250PS. [DOI] [PubMed] [Google Scholar]
- 12. Yang T, Richards EM, Pepine CJ, Raizada MK. The gut microbiota and the brain-gut-kidney axis in hypertension and chronic kidney disease. Nat Rev Nephrol . 2018;14:442–456. doi: 10.1038/s41581-018-0018-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dang AT, Marsland BJ. Microbes, metabolites, and the gut-lung axis. Mucosal Immunol . 2019;12:843–850. doi: 10.1038/s41385-019-0160-6. [DOI] [PubMed] [Google Scholar]
- 14. Appleton J. The gut-brain axis: influence of microbiota on mood and mental health. Integr Med (Encinitas) . 2018;17:28–32. [PMC free article] [PubMed] [Google Scholar]
- 15. Mac Aogáin M, Baker JM, Dickson RP. On bugs and blowholes: why is aspiration the rule, not the exception? Am J Respir Crit Care Med . 2021;203:1049–1051. doi: 10.1164/rccm.202011-4257ED. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Enaud R, Prevel R, Ciarlo E, Beaufils F, Wieërs G, Guery B, et al. The gut-lung axis in health and respiratory diseases: a place for inter-organ and inter-kingdom crosstalks. Front Cell Infect Microbiol . 2020;10:9. doi: 10.3389/fcimb.2020.00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Noverr MC, Noggle RM, Toews GB, Huffnagle GB. Role of antibiotics and fungal microbiota in driving pulmonary allergic responses. Infect Immun . 2004;72:4996–5003. doi: 10.1128/IAI.72.9.4996-5003.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Russell SL, Gold MJ, Reynolds LA, Willing BP, Dimitriu P, Thorson L, et al. Perinatal antibiotic-induced shifts in gut microbiota have differential effects on inflammatory lung diseases. J Allergy Clin Immunol . 2015;135:100–109. doi: 10.1016/j.jaci.2014.06.027. [DOI] [PubMed] [Google Scholar]
- 19. Budden KF, Gellatly SL, Wood DLA, Cooper MA, Morrison M, Hugenholtz P, et al. Emerging pathogenic links between microbiota and the gut-lung axis. Nat Rev Microbiol . 2017;15:55–63. doi: 10.1038/nrmicro.2016.142. [DOI] [PubMed] [Google Scholar]
- 20. Barker AF. Bronchiectasis. N Engl J Med . 2002;346:1383–1393. doi: 10.1056/NEJMra012519. [DOI] [PubMed] [Google Scholar]
- 21. Stockley RA. Commentary: bronchiectasis and inflammatory bowel disease. Thorax . 1998;53:526–527. doi: 10.1136/thx.53.6.526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ni J, Wu GD, Albenberg L, Tomov VT. Gut microbiota and IBD: causation or correlation? Nat Rev Gastroenterol Hepatol . 2017;14:573–584. doi: 10.1038/nrgastro.2017.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Narayana JK, Mac Aogáin M, Goh WWB, Xia K, Tsaneva-Atanasova K, Chotirmall SH. Mathematical-based microbiome analytics for clinical translation. Comput Struct Biotechnol J . 2021;19:6272–6281. doi: 10.1016/j.csbj.2021.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Narayana J, Fransiskus Xaverius I, Oriano M, Ali N, Jaggi T, Tsaneva-Atanasova K, et al. Microbial dysregulation of the ‘lung-gut’ axis in high-risk bronchiectasis. Am J Respir Crit Care Med . 2021;203:A1223. [Google Scholar]
- 25.Narayana JK, Aliberti S, Mac Aogáin M, Jaggi TK, Ali NABM, Ivan FX, et al. Dysregulation of the microbial ‘gut-lung’ axis in bronchiectasis. Eur Respir J . [DOI] [PMC free article] [PubMed]
- 26. Hill AT, Sullivan AL, Chalmers JD, De Soyza A, Elborn SJ, Floto AR, et al. British Thoracic Society guideline for bronchiectasis in adults. Thorax . 2019;74:1–69. doi: 10.1136/thoraxjnl-2018-212463. [DOI] [PubMed] [Google Scholar]
- 27.Global Initiative for Asthma (GINA) Fontana-on-Geneva Lake, WI: Global Initiative for Asthma; 2021. Global strategy for asthma management and prevention.https://ginasthma.org/reports/ [Google Scholar]
- 28.Global Initiative for Chronic Obstructive Lung Disease (GOLD) Global Initiative for Chronic Obstructive Lung Disease; 2021. https://linkprotect.cudasvc.com/url?a=https%3a%2f%2fgoldcopd.org%2fgold-reports%2fgold-report-2021-v1-0-11nov20_wmv%2f&c=E,1,WgyxkUKT1McyCH8uVx-jHQHQ3BOLs3etlZVtO_3opp2m_ci2PtoELLCK8wiP4xaeUAVlNk4N48ynczd-TKWCVNEzpvxfGYKdl74Tr0X-AsaAX9Q,&typo=1 [Google Scholar]
- 29. Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, et al. Metagenomic biomarker discovery and explanation. Genome Biol . 2011;12:R60. doi: 10.1186/gb-2011-12-6-r60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Finch S, McDonnell MJ, Abo-Leyah H, Aliberti S, Chalmers JD. A comprehensive analysis of the impact of Pseudomonas aeruginosa colonization on prognosis in adult bronchiectasis. Ann Am Thorac Soc . 2015;12:1602–1611. doi: 10.1513/AnnalsATS.201506-333OC. [DOI] [PubMed] [Google Scholar]
- 31. Martinez-García MA, Oscullo G, Posadas T, Zaldivar E, Villa C, Dobarganes Y, et al. Spanish Registry of Bronchiectasis Group of SEPAR (RIBRON) Pseudomonas aeruginosa and lung function decline in patients with bronchiectasis. Clin Microbiol Infect . 2021;27:428–434. doi: 10.1016/j.cmi.2020.04.007. [DOI] [PubMed] [Google Scholar]
- 32. Araújo D, Shteinberg M, Aliberti S, Goeminne PC, Hill AT, Fardon TC, et al. The independent contribution of Pseudomonas aeruginosa infection to long-term clinical outcomes in bronchiectasis. Eur Respir J . 2018;51:1701953. doi: 10.1183/13993003.01953-2017. [DOI] [PubMed] [Google Scholar]
- 33. Papp-Wallace KM, Endimiani A, Taracila MA, Bonomo RA. Carbapenems: past, present, and future. Antimicrob Agents Chemother . 2011;55:4943–4960. doi: 10.1128/AAC.00296-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wu BG, Sulaiman I, Tsay JJ, Perez L, Franca B, Li Y, et al. Episodic aspiration with oral commensals induces a MyD88-dependent, pulmonary T-helper cell type 17 response that mitigates susceptibility to Streptococcus pneumoniae. Am J Respir Crit Care Med . 2021;203:1099–1111. doi: 10.1164/rccm.202005-1596OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rigauts C, Aizawa J, Taylor S, Rogers GB, Govaerts M, Cos P, et al. Rothia mucilaginosa is an anti-inflammatory bacterium in the respiratory tract of patients with chronic lung disease. Eur Respir J . 2022;59:2101293. doi: 10.1183/13993003.01293-2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Grice EA, Segre JA. The human microbiome: our second genome. Annu Rev Genomics Hum Genet . 2012;13:151–170. doi: 10.1146/annurev-genom-090711-163814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Marsland BJ, Trompette A, Gollwitzer ES. The gut-lung axis in respiratory disease. Ann Am Thorac Soc . 2015;12:S150–S156. doi: 10.1513/AnnalsATS.201503-133AW. [DOI] [PubMed] [Google Scholar]
- 38. Bacher P, Hohnstein T, Beerbaum E, Röcker M, Blango MG, Kaufmann S, et al. Human anti-fungal Th17 immunity and pathology rely on cross-reactivity against Candida albicans. Cell . 2019;176:1340–1355.e15. doi: 10.1016/j.cell.2019.01.041. [DOI] [PubMed] [Google Scholar]
- 39. Wypych TP, Wickramasinghe LC, Marsland BJ. The influence of the microbiome on respiratory health. Nat Immunol . 2019;20:1279–1290. doi: 10.1038/s41590-019-0451-9. [DOI] [PubMed] [Google Scholar]
- 40. Aliberti S, Lonni S, Dore S, McDonnell MJ, Goeminne PC, Dimakou K, et al. Clinical phenotypes in adult patients with bronchiectasis. Eur Respir J . 2016;47:1113–1122. doi: 10.1183/13993003.01899-2015. [DOI] [PubMed] [Google Scholar]
- 41. Shanahan F, Ghosh TS, O’Toole PW. The healthy microbiome – what is the definition of a healthy gut microbiome? Gastroenterology . 2021;160:483–494. doi: 10.1053/j.gastro.2020.09.057. [DOI] [PubMed] [Google Scholar]
- 42. Bloom SM, Bijanki VN, Nava GM, Sun L, Malvin NP, Donermeyer DL, et al. Commensal Bacteroides species induce colitis in host-genotype-specific fashion in a mouse model of inflammatory bowel disease. Cell Host Microbe . 2011;9:390–403. doi: 10.1016/j.chom.2011.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hosang L, Canals RC, van der Flier FJ, Hollensteiner J, Daniel R, Flügel A, et al. The lung microbiome regulates brain autoimmunity. Nature . 2022;603:138–144. doi: 10.1038/s41586-022-04427-4. [DOI] [PubMed] [Google Scholar]
- 44. Chen J, Wright K, Davis JM, Jeraldo P, Marietta EV, Murray J, et al. An expansion of rare lineage intestinal microbes characterizes rheumatoid arthritis. Genome Med . 2016;8:43. doi: 10.1186/s13073-016-0299-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wolf AJ, Underhill DM. Peptidoglycan recognition by the innate immune system. Nat Rev Immunol . 2018;18:243–254. doi: 10.1038/nri.2017.136. [DOI] [PubMed] [Google Scholar]
- 46. Morrow LE, Kollef MH, Casale TB. Probiotic prophylaxis of ventilator-associated pneumonia: a blinded, randomized, controlled trial. Am J Respir Crit Care Med . 2010;182:1058–1064. doi: 10.1164/rccm.200912-1853OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Vareille-Delarbre M, Miquel S, Garcin S, Bertran T, Balestrino D, Evrard B, et al. Immunomodulatory effects of Lactobacillus plantarum on inflammatory response induced by Klebsiella pneumoniae. Infect Immun . 2019;87:e00570-19. doi: 10.1128/IAI.00570-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Martínez-García MA, de Gracia J, Vendrell Relat M, Girón RM, Máiz Carro L, de la Rosa Carrillo D, et al. Multidimensional approach to non-cystic fibrosis bronchiectasis: the FACED score. Eur Respir J . 2014;43:1357–1367. doi: 10.1183/09031936.00026313. [DOI] [PubMed] [Google Scholar]
- 49. Reiff DB, Wells AU, Carr DH, Cole PJ, Hansell DM. CT findings in bronchiectasis: limited value in distinguishing between idiopathic and specific types. AJR Am J Roentgenol . 1995;165:261–267. doi: 10.2214/ajr.165.2.7618537. [DOI] [PubMed] [Google Scholar]