

Abstract
Over the past decade, recognition of the profound impact of the TBX4 (T-box 4) gene, which encodes a member of the evolutionarily conserved family of T-box–containing transcription factors, on respiratory diseases has emerged. The developmental importance of TBX4 is emphasized by the association of TBX4 variants with congenital disorders involving respiratory and skeletal structures; however, the exact role of TBX4 in human development remains incompletely understood. Here, we discuss the developmental, tissue-specific, and pathological TBX4 functions identified through human and animal studies and review the published TBX4 variants resulting in variable disease phenotypes. We also outline future research directions to fill the gaps in our understanding of TBX4 function and of how TBX4 disruption affects development.
Keywords: pulmonary arterial hypertension, lethal lung developmental disorders, TBX4 syndrome
The TBX4 (T-box 4) gene encodes a member of the evolutionarily conserved family of T-box–containing transcription factors. Substantial insights into the developmental and homeostatic role of Tbx4 and its expression dynamics have been established by animal models and human studies, revealing its function as a key regulator of lung branching morphogenesis and hindlimb formation (1, 2). The phenotypic severity of humans TBX4 variants ranges from mild to lethal and includes effects on the respiratory and on skeletal systems (3–7). Heterogeneity in the clinical features and age at presentation observed in affected patients suggests that heterozygous TBX4 abnormalities alone are insufficient to cause the specific phenotypes. Complex biallelic inheritance of coding and noncoding variants within the TBX4 locus have been proposed to explain some differences in disease severity (5, 8), but complete understanding of the variable phenotypic expressivity remains one of the most challenging and important aspects of TBX4 genetics.
Here, we systematically review current knowledge about the TBX4 gene. We summarize data on the cell- and tissue-specific TBX4 expression pattern and discuss how disruption of its function affects development in human and studied model systems. Finally, we characterize the phenotypic spectrum of TBX4 variants and describe current gaps in our understanding of TBX4-related disorders.
TBX4 Gene Location and Structure
The human TBX4 gene is on 17q23.2, head to head with its neighboring paralog TBX2 (9, 10). Phylogenetic analyses have revealed that this gene pair likely arose from duplication in cis and tandem duplication events of a single ancestral gene during vertebrate evolution before the divergence of bony fish and tetrapods (9). The conservation of the physical linkage of this gene cluster throughout vertebrate evolution suggests a key functional role of its physical linkage (9, 11).
The canonical TBX4 transcript (NM_001321120.2) consists of nine exons that encode a 60.3-kD protein with 546 amino acids. The conserved T-box DNA-binding domain is positioned toward the N-terminus and spans amino acid residues 71–251 encoded by a portion of exon 3, exons 4–6, and a part of exon 7 (12). A second transcript isoform (NM_018488.3) contains eight exons, encoding a shorter 545 amino acid protein (60.2 kD) without the alanine residue at position 341 (Figure 1A).
Figure 1.
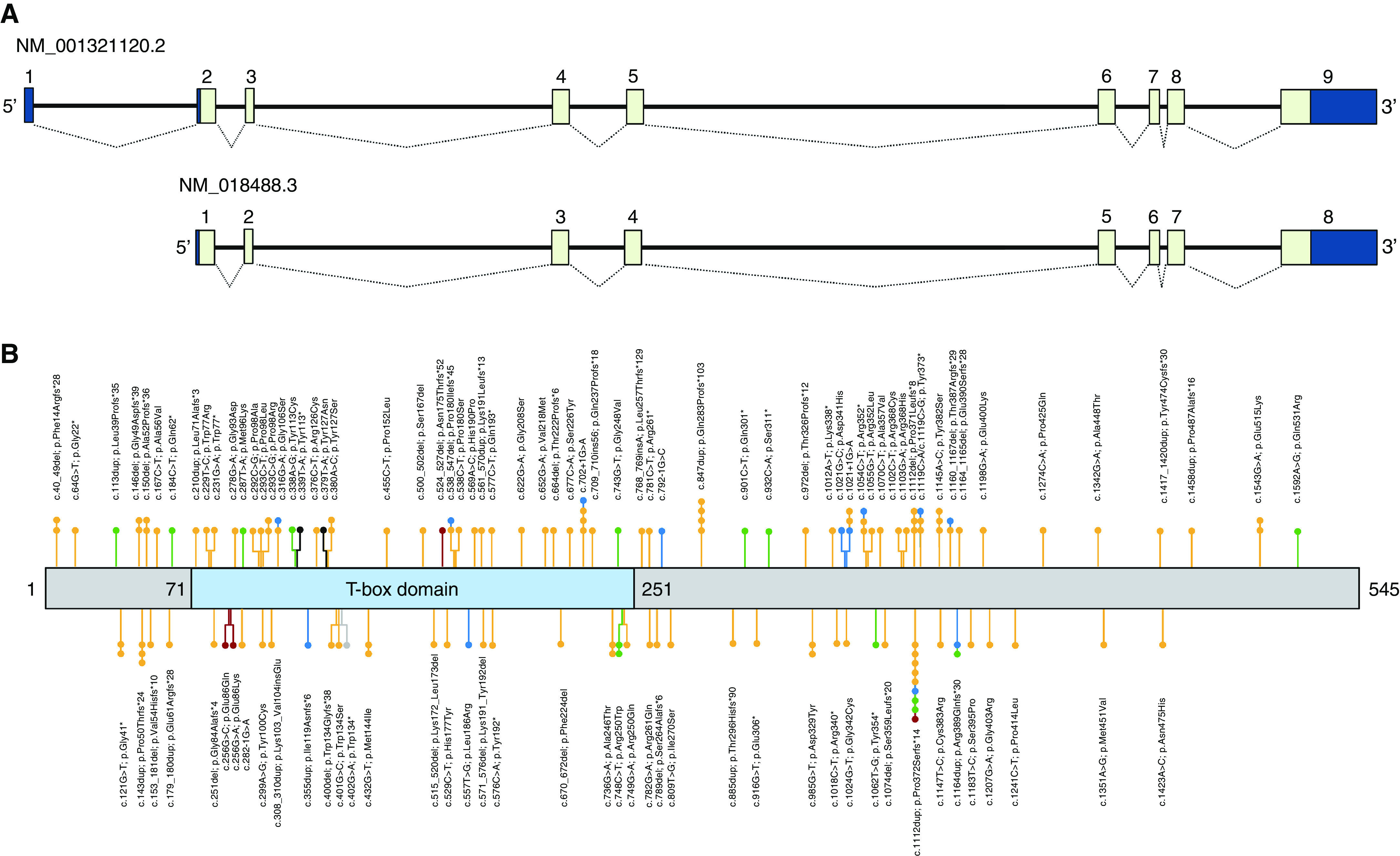
Schematic representation of TBX4 (T-box 4). (A) The upper panel depicts the arrangement of both TBX4 isoforms. Exons and untranslated regions are shown as light and dark blue boxes, respectively. (B) The lower panel depicts the protein with the relative location of the T-box domain (blue). The numbers indicate the positions of amino acid domains. All reported variants with their locations in TBX4 are presented as lollipops. Variants located upstream to amino acids 323-325 could be subjects to degradation by nonsense-mediated decay. Variants identified in patients with pulmonary arterial hypertension (PAH), ischiocoxopodopatellar syndrome (ICPPS), PAH with ICPPS, lethal lung developmental disorders, posterior amelia with pelvic and pulmonary hypoplasia syndrome, and bilateral lung hypoplasia are shown in yellow, green, blue, red, black, and gray, respectively.
Expression and Function of TBX4 in Development
Animal studies, including several mouse, zebrafish, and Xenopus models, have shown that TBX4 expression is dynamic and is restricted to a subset of cell types during development and mature tissues in a highly conserved manner, conferring the framework for our understanding of the role of TBX4 in development and disease in humans.
As documented by mRNA in situ hybridization, immunohistochemistry, and lineage tracing, TBX4 expression is first detected in the lateral plate mesoderm and extraembryonic mesoderm at the primitive streak stage before implantation (13, 14), as well as in mesodermal tissues, including the forelimb, sternum, hindlimb, genital tubercle, urogenital sinus, sinuses, umbilicus, allantois, and placenta (10). In developing chicken embryos, Tbx4 expression has been documented in the lung buds, the trachea, hindlimb, notochord, and body wall surrounding the rostral end of the heart (15). In zebrafish, tbx4 expression has been detected in the developing pelvic fin buds emerging at 3–4 weeks of larval development, as well as in select neuronal populations in the diencephalon and retina (16, 17). The expression pattern of tbx4 in zebrafish is similar to the patterns observed in tetrapods (e.g., birds and mammals), indicating an ancient evolutionary origin of its upstream regulation.
Lung Morphogenesis
In mice, Tbx4 is expressed throughout the splanchnic mesenchyme during formation of trachea and lung buds in close apposition to epithelial cells in primordial lung buds at embryonic day 9.5 (1). A day later, Tbx4 is induced in tracheal mesenchyme, where it plays an important role in patterning tracheal–bronchial cartilage and smooth muscle (18, 19). The involvement of Tbx4 in respiratory tract formation was also found in chickens; studies showed that Tbx4 expression induced Fgf10 (fibroblast growth factor 10) transcription and lung bud formation (20). Extending this work, both TBX4 and TBX5 have been found to play critical roles in formation of the respiratory tract, where they regulate gene networks in which reciprocal endodermal–mesodermal signaling directs tissue morphogenesis via Wnt, BMP4 (bone morphogenetic protein 4), SHH (Sonic Hedgehog signaling molecule), and FGF10 signaling (1, 5, 21–23) (Figure 2). Mesodermal TBX5 regulates mesodermal Wnt2/2B signaling to endodermal cells to initiate formation of the lung buds. Epithelial cells of the embryonic lung saccules express SHH, activating GLI2 (GLI family zinc finger 2), GLI3, TBX4, and TBX5 in the mesenchyme. TBX4 regulates the precise cell-selective spatial expression of FGF10 by subsets of mesenchymal cells required for normal branching morphogenesis (1, 24). Cell–cell communications are mediated by a complex FGF–SHH feedback loop dependent on ETV4 (ETS variant transcription factor 4) and ETV5, together orchestrating interactions among epithelial, mesenchymal, and endothelial cells (25–27) (Figure 2). In turn, the FGF10 ligand activates the FGFR2-3B (fibroblast growth factor receptor 2-3B) receptors in respiratory epithelial cells to direct migration and proliferation of respiratory tubules during branching morphogenesis. In tracheal mesenchyme, TBX4 is regulated by canonical Wnt signaling and BMP4, which are both required for cartilage and smooth muscle patterning of the conducting airways (19, 28).
Figure 2.

TBX4 (T-box 4) signaling in lung branching. TBX4 is expressed throughout the splanchnic mesenchyme surrounding endodermally derived cells of the primordial lung buds and in fibroblasts, pericytes, and airway smooth muscle cells throughout fetal lung morphogenesis (not shown). Paracrine signals from mesenchymal cells are critical for the proliferation and differentiation of respiratory tubules and pulmonary vasculature. SHH (Sonic Hedgehog signaling molecule) signals from SOX9 (SRY-box transcription factor 9) epithelial cells in the acinar buds activate GLI2/3 in mesenchymal cells inducing FOXF1, which interacts in a complex gene regulatory network with TBX4 to regulate WNT2/2B and spatial expression of FGF10 (fibroblast growth factor 10), and other secreted factors required for initiation and growth of the lung tubules. FGF10 produced by mesenchymal cells activates FGFR2 in SOX9/NKX2-1 epithelial progenitors, which produce VEGFA, required for formation of the pulmonary vasculature. The FGF10–SHH feedback loop that controls the periodicity of lung branching is dependent on ETV4/5. Disruption of the signaling network inhibits branching morphogenesis and vasculogenesis. ETV4/5 = ETS variant transcription factor 4/5; FGFR2 = fibroblast growth factor receptor 2; FOXF1 = forkhead box F1; GLI2/3 = GLI family zinc finger 2/3; NKX2-1 = NK2 homeobox 1; VEGFA = vascular endothelial growth factor A.
TBX4, downstream of SHH and its GLI effectors, is hypothesized to regulate FOXF1 (forkhead box F1), also required for growth and patterning of the pulmonary vasculature and respiratory tubules (23). Studies with transgenic mice based on the Tbx4 lung enhancer showed that angiogenic vessels trigger lung mesenchymal stem cell differentiation and commitment to endothelial progenitor cells, playing a role in the development of the lung vasculature (29). These diverse critical roles of TBX4 in lung and tracheal morphogenesis as well as in pulmonary vasculature development and function are consistent with pulmonary or vascular malformations identified in patients with variants in TBX4 (5, 6, 30). Interestingly, Tbx4-lineage mesenchymal progenitors give rise to fibroblasts, smooth muscle cells, pericytes, and endothelial cells in the adult lung, indicating that TBX4 can also regulate myofibroblast accumulation in lung fibrosis (31).
Although most TBX4 data are derived from studies in mice, analyses of human cells have shown highly specific TBX4 expression in pulmonary fibroblasts, likely regulated by lung-specific superenhancers, providing evidence for the critical role of TBX4 in human lung functioning (32).
Recent single-cell RNA sequencing analyses in humans have also provided new insight into the temporal and cell-specific expression of TBX4 in the developing lung (33). Among 30 major cell types in peripheral lung tissues from infants and adults studied in a reference map of the human lung single-cell RNA expression data, TBX4 expression was detected in pulmonary fibroblasts, airway vascular smooth muscle cells, and pericytes, with highest expression in matrix fibroblasts, a cell type serving as a signaling center required for lung morphogenesis (34).
The role of TBX4 in human lung function is further supported by bulk RNA sequencing studies performed in neonates with prenatal arrest of lung maturation caused by TBX4 mutations. Lung tissues from patients with TBX4 variants are characterized by substantial dysregulation of genes involved in pathways related to the respiratory system, including TMEM100 (transmembrane protein 100), and those with TBX4-binding sites detected in chromatin immunoprecipitation experiments (23, 35).
Limb Development
Hindlimb development, in particular during the first phase of limb formation to establish the hindlimb bud, is the most intensively studied developmental aspect of TBX4. Its activity is critical to initiate the formation of the hindlimb buds, analogous to the involvement of its paralog TBX5 in forelimb formation. In mice, activation of Tbx4 expression in the hindlimb-forming region of the lateral plate mesoderm at the pre–limb bud stages is one of the earliest detectable events that delineates the cells that ultimately form the hindlimb buds (36, 37). As limb development progresses, TBX4 has a more specific role in ensuring the correct formation of the limb connective tissues (muscle and tendons) (38, 39). The hindlimb buds contain all the progenitor cells of the structures of the lower limb and pelvic girdle. The core function of TBX4 and TBX5 is to activate Fgf10 expression in the limb-forming regions, which leads to the establishment of an FGF signaling–based positive feedback loop driving limb bud outgrowth (40). Fgf10-mutant mice lack both forelimbs and hindlimbs; however, rudiments of the pectoral and pelvic girdles do form (41). Nonetheless, the gene regulatory networks that establish Fgf10 expression in the lateral plate mesoderm are different in the forelimb and hindlimb. The forelimb-forming region exclusively requires TBX5 to establish Fgf10 expression; in contrast, dual input from TBX4 and the paired homeodomain transcription factor PITX1 (paired like homeodomain 1) are necessary to initiate the equivalent Fgf10 expression in the hindlimb-forming region (2). PITX1 positively regulates TBX4, which in turn directly regulates FGF10, while PITX1 also has TBX4-independent input into the regulation of Fgf10 that can establish hypomorphic concentrations of FGF10 in the absence of TBX4. Additional input from other transcription factors, such as Isl1 (Islet1), appears to be required for the establishment of normal Fgf10 expression in the hindlimb (42–44). Although the ISL1/LDB (LIM domain binding) proteins are required for Fgf10 expression and are still expressed in the Tbx4/Pitx1 double-mutant mouse, they are not sufficient to rescue Fgf10 expression; consequently, Isl1/Ldb appear to act as obligate coregulators with TBX4 and PITX1 to control Fgf10 expression (2).
Homozygous Tbx4 loss-of-function (LoF) alleles result in a prominent lack of hindlimb structures in mice, Xenopus, and zebrafish (pelvic fins) (45), linking to the ischiocoxopodopatellar syndrome (ICPPS) phenotype observed in patients. Given the reported equivalent roles played by TBX4 and TBX5 in establishing the lower and upper limb buds, it is striking that the spectrum of lower limb defects caused by haploinsufficiency of TBX4 in patients with ICPPS are less severe overall than those seen in Holt-Oram syndrome due to haploinsufficiency of TBX5. A possible explanation for this is some degree of compensation for the absence of TBX4 (in establishing an FGF signaling positive feedback loop) by PITX1, which is also expressed in the hindlimb-forming territory of the lateral plate mesoderm.
Such a model is supported by the results of mouse studies in which the joint homozygous deletions of Tbx4 and Pitx1 produce a hindlimb-less phenotype equivalent to that observed by homozygous deletion of Tbx5 in the forelimb region. After deletion of both Tbx4 and Pitx1, Fgf10 is not expressed, and all hindlimb elements fail to form (2). In the absence of Tbx4, low Fgf10 expression is present, and ultimately distal hindlimb elements are produced, while more proximal elements are missing.
The defects in the most proximal structures of the hindlimb, including the pelvic girdle, appear independent of the role of TBX4 in regulating FGF signaling feedback loops in the forming hindlimb, as these most proximal elements can form in the absence of FGF10. Recent data indicate that the most proximal defects occur as a direct result of a failure in the early differentiation step of chondroprogenitors into chondrocytes (2).
Reported TBX4 Variants in Different Diseases
Most human TBX4-related diseases correlate well with the developmental findings in animal models and include a range of mild to lethal conditions, affecting respiratory and skeletal systems (Figure 1B; see Tables E1 and E2 in the online supplement).
ICPPS
ICPPS is a rare autosomal-dominant skeletal anomaly of the pelvis and feet. The characteristic features associated with ICPPS are patellar aplasia or hypoplasia, disrupted ossification of the ischiopubic junction, increased space between the first and second toes, and short fourth and fifth rays of the feet (3).
The role of TBX4 variants in ICPPS was initially discovered by Bongers and colleagues, who described the putative LoF heterozygous variants in TBX4 in six unrelated families with ICPPS (3). Additional ICPPS-related TBX4 variants have been identified in nine other families (46–48), confirming that haploinsufficiency of TBX4 is causative for ICPPS.
Pulmonary Arterial Hypertension
Pulmonary arterial hypertension (PAH), characterized by proliferative remodeling of the small pulmonary arteries with increased pulmonary pressure and resistance that can lead to right heart failure (49), is another rare and often lethal manifestation associated with TBX4 variants.
In 2010, Ballif and colleagues diagnosed PAH in three of the seven children with a large spectrum of congenital defects caused by a recurrent heterozygous microdeletion on chromosome 17q23.2, including TBX4 (50). Subsequently, other patients with child-onset PAH and the same 17q23.2 microdeletion or heterozygous single-nucleotide variants (SNVs) in TBX4 were reported (4, 24, 51, 52).
Genetic data from large cohorts of pediatric patients with PAH from the Netherlands, France, the United States, the United Kingdom, and other populations showed that rare inherited or de novo heterozygous SNVs, or copy-number variant (CNV) deletions including the TBX4 gene, are enriched in patients with childhood-onset PAH (6, 53–58). The prevalence of the pathogenic TBX4 variants in childhood PAH ranges from 5.6% in the U.S. PAH Biobank (59, 60) to 11.4% in the Dutch National Registry (57).
The link between TBX4 and pulmonary vascular abnormalities has also been confirmed in adulthood-onset PAH, suggesting that after BMPR2 (bone morphogenetic protein receptor type 2), TBX4 variants are among the top genetic causes of PAH in adults (4, 54, 56, 58, 59, 61–67). The frequency of TBX4 variants in adult patients with PAH varies among studied cohorts, ranging from 3% in a French cohort (56) to 0.46% in the U.S. PAH Biobank (59); however, it could be due to different modes of collection of patients with PAH among registries. Notably, it is unknown whether these individuals have adult manifestations of a developmental pulmonary vascular disease, adult presentation of PAH uniquely due to alterations in injury response and repair, or other causes. In these patients, other features of developmental irregularities, including airway branching defects and skeletal abnormalities, have been observed (56).
Together, the data underline that genetic diagnosis of a rare deleterious TBX4 variant or TBX4-containing microdeletion in pediatric PAH is associated with a more complex developmental phenotype: TBX4 syndrome (68). Why, and if, adult-onset PAH represents a distinct phenotype from early childhood–onset TBX4-associated PAH remains unknown.
In addition to PAH, some patients with TBX4 variants also have features of ICPPS or other skeletal anomalies. In the study by Kerstjens-Frederikse and colleagues, all but one of the living individuals with PAH with TBX4 variants had ICPPS features (4). Subsequent analyses also showed a high frequency of skeletal anomalies coexistent with TBX4-related PAH. Galambos and colleagues reported ICPPS with typical foot or other skeletal anomalies in 10 of 19 pediatric patients with PAH (6). A lower frequency (4 of 14) of skeletal malformations associated with PAH was observed in French patients with TBX4 variants (56). One child and one adult individual with TBX4-related PAH and ICPPS have also been reported in the U.S. PAH cohort and a family with German–Bavarian ancestry, respectively (54, 69).
Skeletal and other developmental defects are not routinely assessed as part of a PAH diagnosis, and as such, they may be missed. Thus, chest imaging for severe and diffuse features of pulmonary growth arrest, assessment for congenital heart defects, physical examination of the hands and feet, and radiological assessment of the pelvis and patella are recommended in patients with PAH. In addition, a TBX4 diagnosis predicts potential recurrence of PAH after neonatal persistent pulmonary hypertension of the newborn, and annual screening with echocardiography may be useful (6).
Of note, a recent study provided evidence that in adult patients with PAH without TBX4 pathogenic variants, transcriptional programs involved in lung morphogenesis were epigenetically derepressed in pulmonary artery fibroblasts, leading to overexpression of TBX4 (70). Increased concentration of TBX4 results in myofibroblast accumulation and subsequent pulmonary vascular remodeling, and further studies are needed to clarify the role of TBX4 in PAH pathogenesis in patients without TBX4 variants (70).
Lethal Lung Developmental Disorders
Recently, disruption of the TBX4–FGF10 epithelial–mesenchymal signaling pathway has been reported in patients with acinar dysplasia (AcDys), congenital alveolar dysplasia (CAD), and other unspecified primary pulmonary hypoplasias, which are lethal lung developmental disorders (LLDDs) (5, 30, 71–73). Although clinical and histopathological variation occurs across particular subtypes of LLDD, shortly after birth, most newborns with LLDD present with severe respiratory failure with PAH that is often refractory to therapy (74). LLDDs are associated with 80–90% neonatal mortality; however, whereas newborns with AcDys die within the first hours of life, patients with CAD usually expire within the first weeks or even months after birth (74, 75). TBX4 abnormalities tend to occur more often in newborns with AcDys than those with CAD (74). In 2016, Szafranski and colleagues reported a newborn with AcDys and a heterozygous de novo TBX4 missense variant, suggesting the role of this gene in maldevelopment of human lung (30). Suhrie and colleagues described a patient with CAD with a heterozygous de novo TBX4 frameshift variant (72), and German and colleagues identified a recurrent 17q23.2 CNV deletion, involving TBX4, in a newborn with AcDys (73). De novo recurrent heterozygous missense TBX4 or nonrecurrent CNV deletions on 17q23.1q23.2, involving TBX4, have been subsequently detected in eight additional patients with AcDys, CAD, or primary pulmonary hypoplasias (5, 8). An 8.6-kb intragenic heterozygous frameshifting deletion in TBX4 was identified in siblings with CAD and AcDys spectrum, inherited from their healthy mother (5).
The histology of lungs with TBX4-related AcDys is believed to represent an arrest in the pseudoglandular phase of embryonic and fetal lung development, normally seen from 6–8 to 16 weeks of gestation in humans and dominated by the lack of acinar development with scattered primitive conducting airway profiles surrounded by loose mesenchymal tissue. Features of more advanced lung maturation, including airspace development to form canaliculi and/or saccules with variably thickened interstitium, are seen most commonly in symptomatic infants with TBX4 abnormalities (Figures 3A–3C) (5, 6, 74). These features occur in CAD, which resembles growth arrest in the canalicular to early saccular stage of lung development seen from 16 to 26 weeks of human gestation.
Figure 3.

Heterogeneous histologic appearance with variable degree of lung development arrest seen in patients with TBX4 (T-box 4) gene deficiency. (A) Lung biopsy (hematoxylin and eosin [H&E], 4×) at 4 months of age shows markedly underdeveloped lung parenchyma characterized by extensive interstitial thickening, canalicular airspace development, and bronchiolar structures with abnormally close proximity to the pleura (arrow). These features are most consistent with developmental arrest in the late canalicular stage of fetal lung development. (B) Lung biopsy (trichrome, 4×) at 3 months of age shows marked vascular remodeling, including pulmonary arterial vessel wall muscularization and intimal thickening (arrows). (C) Lung biopsy (H&E, 4×) at 2 months shows maldeveloped lung with variably thickened interstitium and large simplified saclike airspaces (asterisks). These features are most consistent with developmental arrest at the early saccular stage of fetal lung development. (D) Lung transplant sections (H&E, 10×) at 18 years show abnormally formed lung with back-to-back bronchiolar profiles, absent alveolar formation, and patchy interstitial fibrosis. A moderately remodeled pulmonary artery with muscular wall thickening is also shown (arrow).
Interestingly, features of less severe interstitial lung disease or abnormal lung growth have been also observed in other patients with TBX4-associated PAH (6, 71, 76). The lung histology of patients who undergo biopsy and/or transplantation later in childhood or adolescence shows evidence of compromised airway and/or alveolar growth, characterized by variations of alveolar simplification and back-to-back bronchiolar structures with minimal or no intervening alveolar formation (Figure 3D). Vascular maldevelopment and remodeling, including wall thickening of pulmonary arteries, lymphatic and pleural vessels, as well as formation of plexiform lesions, are significant features. The bronchial vascular system is expanded, with congested and dilated bronchial veins and capillaries. Prominent intrapulmonary bronchopulmonary anastomoses have also been noted. In addition, evidence of patchy mesenchymal maldevelopment, including foci of metaplastic bone formation, interstitial capillary proliferation, and pleural and/or interstitial muscularization and fibrosis have been described (6).
Although SNVs and CNVs of TBX4 confer a risk of AcDys, CAD, or other primary pulmonary hypoplasias, the heterogeneity of clinical features associated with TBX4 abnormalities suggests that the heterozygous variants involving TBX4 are incompletely penetrant. A model of biallelic inheritance in which both TBX4 coding and noncoding variants in trans in the lung-specific enhancer of TBX4 contribute to LLDD has been proposed recently, as discussed in the next section.
Posterior Amelia with Pelvic and Pulmonary Hypoplasia Syndrome
In 2019, Kariminejad and colleagues reported two unrelated consanguineous Iranian families with fetuses affected with autosomal recessive posterior amelia with pelvic and pulmonary hypoplasia syndrome (PAPPAS; Mendelian Inheritance in Man [MIM] #601360) due to homozygous TBX4 missense and nonsense variants inherited from heterozygous parents with ICPPS (7). In an Indian family with a fetus affected by PAPPAS, biallelic TBX4 variants were identified, as inherited from the consanguineous parents with mild ICPPS and heterozygous nonsense TBX4 variants (77). These results show that biallelic variants in the TBX4 gene are associated with a severe prenatally lethal syndromic phenotype.
Congenital Clubfoot
Congenital clubfoot is one of the most common congenital malformations that affects bones, muscles, connective tissue, and vascular or neurological structures in limbs (78). The etiology of clubfoot includes environmental and genetic factors.
Recurrent (2.2 Mb, 17q23.1q23.2) and nonrecurrent (∼350 kb, 17q23.2) CNV duplications, involving TBX4, have been identified in patients with familial isolated clubfoot (79, 80). Clubfoot deformity has also been rarely reported in association with the recurrent reciprocal 17q23 deletion (MIM #613355) albeit without a clear association to date (81). Of note, one patient with a TBX4 CNV deletion from a PAH cohort described by Galambos and colleagues also has clubfoot (6).
Multigene Deletions versus Discrete Variations in TBX4
Whereas SNVs and CNV deletions involving TBX4 have been described in patients with various abnormalities, it is unclear how the variant type correlates with the phenotype. Frameshift and missense variants in TBX4 have been found in individuals ICPPS and/or PAH, accounting for two-thirds of the associated variants (see Table E1). Recurrent CNV deletions are less frequently observed in ICPPS and PAH and are more common in patients with LLDD and PAH associated with parenchymal abnormalities (see Table E2). A higher prevalence of developmental delay has also been observed among TBX4 CNV carriers (4, 50).
Beyond TBX4
In contrast to the high penetrance of TBX4 variants in ICPPS, segregating TBX4 variants in families with PAH show variable expressivity and incomplete penetrance, suggesting the involvement of other environmental or genetic factors (4, 54, 55). Of note, digenic heterozygous variants involving TBX4 and BMPR2 have been identified in one patient with PAH from Lebanon (82) and another from the PAH Biobank (59). One patient was reported with rare deleterious variants in TBX4 and ACVRL1 (activin A receptor like type 1) (54). The high incidence of PAH in patients with ICPPS implies that families with ICPPS should be informed about the risk of PAH and that patients with PAH should be screened for the presence of ICPPS features (4, 6, 56).
In addition, recent functional assessment of TBX4 variants describing both gain-of-function (GoF) and LoF effects, have revealed that GoF variants are associated with older age at diagnosis of lung disease compared with LoF variants (83). In addition, it has been suggested that variants located in the T-box or nuclear localization domains are associated with earlier onset and increased incidence of interstitial lung disease (83). Studies in families with rare LLDDs have revealed heterozygous TBX4 variants inherited from unaffected parents or parents with milder phenotypes, indicating that the coding heterozygous TBX4 variants alone are not sufficient to cause abnormal lung phenotype and suggesting the presence of additional genetic modifiers. Of note, the probability of being LoF intolerant score (84) for TBX4 is 0.5, indicating its partial tolerance for LoF variants which would be expected to associate with complete LoF of the resultant gene transcript. In addition, LLDD infants studied by Karolak and colleagues have been found to also harbor at least one noncoding SNV mapped in trans to the coding variants involving TBX4 (5). These noncoding variants are located in an intronic predicted lung-specific enhancer region (85) within BCAS3 (BCAS3 microtubule associated cell migration factor) (MIM #607470), approximately 70 kb upstream of TBX4 (5). The cooccurrence of rare coding variants involving TBX4 with the putative hypomorphic noncoding SNVs in trans suggests a complex biallelic model for these diseases (5).
A model of compound inheritance involving a combination of rare coding variants and common coding and noncoding variants has been also described for TBX6-related developmental anomalies of the spine (86). Moreover, a rare noncoding variant in the lung-specific enhancer in trans to the FOXF1 mutated allele has been described, acting as a hypermorph and mitigating the lethal phenotype of alveolar capillary dysplasia with misalignment of the pulmonary veins (87). These studies demonstrate that noncoding modifying variants can act as hyper- or hypomorphs and suggest newer mutational models for disease such as the compound inheritance gene dosage (CIGD) model, blending the effects of coding and noncoding variants (86).
Further supporting the CIGD model, most recently, a heterozygous frameshift variant, c.1112dup (p.Pro372Serfs*14), in TBX4 was described, previously reported in patients with ICPPS or PAH (46, 47), in a three-generation family with mild interstitial lung disease, bronchiolitis obliterans, recurrent pneumothorax, ICPPS, and LLDD and in unaffected individuals (88). In two deceased neonates with LLDD, a noncoding SNV, rs62069651-C, was present in trans to the mutated TBX4 allele within the predicted binding site of nuclear transcription factor, X-box binding–like 1. This variant, absent in other family members with the frameshift mutation, reduces TBX4 promoter activity by 63% in a reporter assay. These findings provide functional evidence for the reported model of complex compound inheritance in which both TBX4 coding and trans noncoding hypomorphic variants in the lung-specific enhancer of TBX4 contribute to LLDD (88).
Summary and Future Directions
Animal models have provided key insights into TBX4 and its function in vertebrate development, yet numerous answers to more nuanced questions remain. Studying Tbx4 loss in the lungs of mice using genetic manipulations remains challenging, as homozygous Tbx4 knockout is fetal lethal, and heterozygous knockouts have no obvious phenotype without additional stressors. However, as floxed Tbx4 alleles are available, creation of inducible and conditional knockouts is possible and will be necessary to better understand the critical role of Tbx4 at various developmental stages. Such conditional LoF will also be instrumental to address the role of Tbx4 during development as well as for maintenance of normal lung physiology. Complementary, although zebrafish do not form a lung, the deep conservation of the Tbx4 gene and of its paralogs including Tbx5 indicate that this model can provide insights into the basic developmental contribution of Tbx4 to select endothelial and mesenchymal lineages. Understanding the times at which TBX4 function is key for therapeutic intervention. Studies of Tbx4 mutations in animal models in a cell type–specific manner as well as intersection with drug screening and signaling modulation will be critical to considering future genetic therapeutic strategies in humans.
Understanding the true natural history of rare genetic conditions and the frequency of associated features is challenging, in part because of the ascertainment bias of the first reported individuals, as they are usually ascertained and genetically tested on the basis of striking clinical features and/or strong family history. Our challenge is to accelerate the expansion of our knowledge by increasing the number of individuals known to have these rare genotypes by using the power of collaborative international consortia to combine data using standardized and complete clinical evaluations. Increased availability of genetic testing for patients with pulmonary disease, PAH, and orthopedic issues should identify additional mutation carriers. Cascade genetic testing of family members across multiple generations for features of TBX4 syndrome can quickly identify many additional mutation carriers of all ages. Engagement of these families in research should help identify resiliency factors, especially in older individuals who have mutations yet remain symptom free.
Large population-scale genomic medicine efforts (i.e., UK Biobank, the Genomics England Ltd. 100,000 Genomes Project, and the NIH All of Us Research Program [89–91]) provide a completely different method of ascertainment and could identify individuals without classical symptoms or features of TBX4 deficiency (and potentially other new symptoms) who have not come to clinical attention on the basis of orthopedic or pulmonary symptoms. All methods of ascertainment are helpful to identify the full spectrum and frequency of disease manifestations and better understand the age- and sex-related penetrance of each clinical feature.
It will be necessary to allow as many individuals as possible around the world to participate and provide their longitudinal clinical data over time and biospecimens. Such a registry should provide value to the participants individually and collectively and ideally should include free family-based genetic testing, education, and/or genetic counseling to understand the information and how to use it for medical care and health surveillance. However, there can be a reluctance to pursue genetic testing because of feelings of guilt for transmission of a mutation to an affected child or if individuals believe that knowing this information does not provide medical benefit through the ability to prevent, cure, or effectively treat disease.
Among the T-box–associated human diseases resulting from haploinsufficiency, there is a range of manifestations, which most frequently involve the heart and skeletal systems (i.e., Holt-Oram syndrome through TBX5). All T-box haploinsufficiency conditions are similarly associated with variable expressivity and incomplete penetrance. There may be lessons learned from other T-box–associated conditions that will be applicable to TBX4.
What is likely to account for the heterogeneity of manifestations in T-box conditions and in TBX4-associated disease in particular? One source of heterogeneity is allelic differences and the amount of TBX4 expression associated with each allele, which could lead to differences in TBX4 activity. It is currently unclear whether there is a minimal threshold of TBX4 expression required for normal development and whether that threshold is the same in each cell type or tissue. Also unclear remains the critical window of development that requires TBX4 activity and whether TBX4 expression is mainly required during development or also whether its persistent expression is required for maintenance of pulmonary vascular integrity and to prevent disease progression over time. Addressing these possibilities is critical to understanding the therapeutic window for treatment strategies.
Even within a single family with the same TBX4 variant, there is often phenotypic heterogeneity. Factors that could alter TBX4 expression include genetic variation in cis or in trans. Identifying involved genomic modifiers remains challenging unless they are small in number and large in effect, given the limited number of TBX4 mutation carriers. Once clinically characterized cohorts are established, it may be possible to begin exploring genetic and environmental modifiers affecting penetrance and expressivity.
Although there are still many unanswered questions in the richness of TBX4 biology, studying this particular T-box factor gene has the potential to illuminate secrets of development and human disease as proxy for T-box factor function in general. Collaborative research priorities to build international cohorts with human biospecimens and relevant animal models bring the promise to advance our understanding of the variabilities observed with TBX4-associated conditions. TBX4Life (https://tbx4.org) is a global initiative bringing together leading clinicians, geneticists, developmental biologists, and patient families through the common interest in the biology and pathophysiology of TBX4, to help identify TBX4 variant carriers and facilitate TBX4 research. Ultimately, discovering genetic and environmental factors and details that modify disease severity will be instrumental in prevention, prognosis, and treatment.
Acknowledgments
Acknowledgment
The authors thank Anton Morkin, a founder of TBX4Life (https://tbx4.org), for helpful discussion and for making this initiative possible. The authors acknowledge Ms. Anusha Sridharan for preparation of Figure 2. Figures 1 and 2 were generated using BioRender (https://biorender.com).
Footnotes
Supported by a National Science Centre in Poland grant 2019/35/D/NZ5/02896 (J.A.K.) and National Institutes of Health, National Heart, Lung, and Blood Institute grant R01 HL 134802 (E.D.A.).
Author Contributions: J.A.K., C.L.W., C.M., K.B., M.P.O.L., J.W., C.G., P.S., W.K.C., and E.D.A. conceptualized and wrote the manuscript. J.D.W., D.M., M.E., M.P.M., S.H.A., M.P., S.G., N.W.M., A.R.H., F.P., and R.H. edited the manuscript. All authors reviewed and discussed the manuscript during preparation.
This article has an online supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1164/rccm.202206-1039TR on November 11, 2022
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1. Arora R, Metzger RJ, Papaioannou VE. Multiple roles and interactions of Tbx4 and Tbx5 in development of the respiratory system. PLoS Genet . 2012;8:e1002866. doi: 10.1371/journal.pgen.1002866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Duboc V, Sulaiman FA, Feneck E, Kucharska A, Bell D, Holder-Espinasse M, et al. Tbx4 function during hindlimb development reveals a mechanism that explains the origins of proximal limb defects. Development . 2021;148:dev199580. doi: 10.1242/dev.199580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bongers EMHF, Duijf PHG, van Beersum SEM, Schoots J, Van Kampen A, Burckhardt A, et al. Mutations in the human TBX4 gene cause small patella syndrome. Am J Hum Genet . 2004;74:1239–1248. doi: 10.1086/421331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kerstjens-Frederikse WS, Bongers EMHF, Roofthooft MTR, Leter EM, Douwes JM, Van Dijk A, et al. TBX4 mutations (small patella syndrome) are associated with childhood-onset pulmonary arterial hypertension. J Med Genet . 2013;50:500–506. doi: 10.1136/jmedgenet-2012-101152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Karolak JA, Vincent M, Deutsch G, Gambin T, Cogné B, Pichon O, et al. Complex compound inheritance of lethal lung developmental disorders due to disruption of the TBX-FGF pathway. Am J Hum Genet . 2019;104:213–228. doi: 10.1016/j.ajhg.2018.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Galambos C, Mullen MP, Shieh JT, Schwerk N, Kielt MJ, Ullmann N, et al. Phenotype characterisation of TBX4 mutation and deletion carriers with neonatal and paediatric pulmonary hypertension. Eur Respir J . 2019;54:1801965. doi: 10.1183/13993003.01965-2018. [DOI] [PubMed] [Google Scholar]
- 7. Kariminejad A, Szenker-Ravi E, Lekszas C, Tajsharghi H, Moslemi A-R, Naert T, et al. Homozygous null TBX4 mutations lead to posterior amelia with pelvic and pulmonary hypoplasia. Am J Hum Genet . 2019;105:1294–1301. doi: 10.1016/j.ajhg.2019.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Karolak JA, Gambin T, Honey EM, Slavik T, Popek E, Stankiewicz P. A de novo 2.2 Mb recurrent 17q23.1q23.2 deletion unmasks novel putative regulatory non-coding SNVs associated with lethal lung hypoplasia and pulmonary hypertension: a case report. BMC Med Genomics . 2020;13:34. doi: 10.1186/s12920-020-0701-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Agulnik SI, Garvey N, Hancock S, Ruvinsky I, Chapman DL, Agulnik I, et al. Evolution of mouse T-box genes by tandem duplication and cluster dispersion. Genetics . 1996;144:249–254. doi: 10.1093/genetics/144.1.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Papaioannou VE. The T-box gene family: emerging roles in development, stem cells and cancer. Development . 2014;141:3819–3833. doi: 10.1242/dev.104471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Horton AC, Mahadevan NR, Minguillon C, Osoegawa K, Rokhsar DS, Ruvinsky I, et al. Conservation of linkage and evolution of developmental function within the Tbx2/3/4/5 subfamily of T-box genes: implications for the origin of vertebrate limbs. Dev Genes Evol . 2008;218:613–628. doi: 10.1007/s00427-008-0249-5. [DOI] [PubMed] [Google Scholar]
- 12. Yi CH, Russ A, Brook JD. Virtual cloning and physical mapping of a human T-box gene, TBX4. Genomics . 2000;67:92–95. doi: 10.1006/geno.2000.6222. [DOI] [PubMed] [Google Scholar]
- 13. Downs KM, Davies T. Staging of gastrulating mouse embryos by morphological landmarks in the dissecting microscope. Development . 1993;118:1255–1266. doi: 10.1242/dev.118.4.1255. [DOI] [PubMed] [Google Scholar]
- 14. Prummel KD, Nieuwenhuize S, Mosimann C. The lateral plate mesoderm. Development . 2020;147:dev175059. doi: 10.1242/dev.175059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gibson-Brown JJ, Agulnik S I, Silver LM, Papaioannou VE. Expression of T-box genes Tbx2-Tbx5 during chick organogenesis. Mech Dev . 1998;74:165–169. doi: 10.1016/s0925-4773(98)00056-2. [DOI] [PubMed] [Google Scholar]
- 16. Tamura K, Yonei-Tamura S, Izpisúa Belmonte JC. Differential expression of Tbx4 and Tbx5 in zebrafish fin buds. Mech Dev . 1999;87:181–184. doi: 10.1016/s0925-4773(99)00126-4. [DOI] [PubMed] [Google Scholar]
- 17. Ruvinsky I, Oates AC, Silver LM, Ho RK. The evolution of paired appendages in vertebrates: T-box genes in the zebrafish. Dev Genes Evol . 2000;210:82–91. doi: 10.1007/s004270050014. [DOI] [PubMed] [Google Scholar]
- 18. Naiche LA, Arora R, Kania A, Lewandoski M, Papaioannou VE. Identity and fate of Tbx4-expressing cells reveal developmental cell fate decisions in the allantois, limb, and external genitalia. Dev Dyn . 2011;240:2290–2300. doi: 10.1002/dvdy.22731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kishimoto K, Furukawa KT, Luz-Madrigal A, Yamaoka A, Matsuoka C, Habu M, et al. Bidirectional Wnt signaling between endoderm and mesoderm confers tracheal identity in mouse and human cells. Nat Commun . 2020;11:4159. doi: 10.1038/s41467-020-17969-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sakiyama J, Yamagishi A, Kuroiwa A. Tbx4-Fgf10 system controls lung bud formation during chicken embryonic development. Development . 2003;130:1225–1234. doi: 10.1242/dev.00345. [DOI] [PubMed] [Google Scholar]
- 21. Cebra-Thomas JA, Bromer J, Gardner R, Lam GK, Sheipe H, Gilbert SF. T-box gene products are required for mesenchymal induction of epithelial branching in the embryonic mouse lung. Dev Dyn . 2003;226:82–90. doi: 10.1002/dvdy.10208. [DOI] [PubMed] [Google Scholar]
- 22. Steimle JD, Rankin SA, Slagle CE, Bekeny J, Rydeen AB, Chan SS-K, et al. Evolutionarily conserved Tbx5-Wnt2/2b pathway orchestrates cardiopulmonary development. Proc Natl Acad Sci USA . 2018;115:E10615–E10624. doi: 10.1073/pnas.1811624115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Karolak JA, Gambin T, Szafranski P, Stankiewicz P. Potential interactions between the TBX4-FGF10 and SHH-FOXF1 signaling during human lung development revealed using ChIP-seq. Respir Res . 2021;22:26. doi: 10.1186/s12931-021-01617-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yoshida Y, Uchida K, Kodo K, Shibata H, Furutani Y, Nakayama T, et al. Genetic and functional analyses of TBX4 reveal novel mechanisms underlying pulmonary arterial hypertension. J Mol Cell Cardiol . 2022;171:105–116. doi: 10.1016/j.yjmcc.2022.07.002. [DOI] [PubMed] [Google Scholar]
- 25. Bellusci S, Furuta Y, Rush MG, Henderson R, Winnier G, Hogan BL. Involvement of Sonic Hedgehog (Shh) in mouse embryonic lung growth and morphogenesis. Development . 1997;124:53–63. doi: 10.1242/dev.124.1.53. [DOI] [PubMed] [Google Scholar]
- 26. Pepicelli CV, Lewis PM, McMahon AP. Sonic Hedgehog regulates branching morphogenesis in the mammalian lung. Curr Biol . 1998;8:1083–1086. doi: 10.1016/s0960-9822(98)70446-4. [DOI] [PubMed] [Google Scholar]
- 27. Herriges JC, Verheyden JM, Zhang Z, Sui P, Zhang Y, Anderson MJ, et al. FGF-regulated ETV transcription factors control FGF-SHH feedback loop in lung branching. Dev Cell . 2015;35:322–332. doi: 10.1016/j.devcel.2015.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Snowball J, Ambalavanan M, Whitsett J, Sinner D. Endodermal Wnt signaling is required for tracheal cartilage formation. Dev Biol . 2015;405:56–70. doi: 10.1016/j.ydbio.2015.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Luo Y, Shi W. Dynamic mapping of lung vascular development [abstract] Am J Respir Crit Care Med . 2016;193:A7293. [Google Scholar]
- 30. Szafranski P, Coban-Akdemir ZH, Rupps R, Grazioli S, Wensley D, Jhangiani SN, et al. Phenotypic expansion of TBX4 mutations to include acinar dysplasia of the lungs. Am J Med Genet A . 2016;170:2440–2444. doi: 10.1002/ajmg.a.37822. [DOI] [PubMed] [Google Scholar]
- 31. Xie T, Liang J, Liu N, Huan C, Zhang Y, Liu W, et al. Transcription factor TBX4 regulates myofibroblast accumulation and lung fibrosis. J Clin Invest . 2016;126:3063–3079. doi: 10.1172/JCI85328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Horie M, Miyashita N, Mikami Y, Noguchi S, Yamauchi Y, Suzukawa M, et al. TBX4 is involved in the super-enhancer-driven transcriptional programs underlying features specific to lung fibroblasts. Am J Physiol Lung Cell Mol Physiol . 2018;314:L177–L191. doi: 10.1152/ajplung.00193.2017. [DOI] [PubMed] [Google Scholar]
- 33. Wang A, Chiou J, Poirion OB, Buchanan J, Valdez MJ, Verheyden JM, et al. NHLBI LungMap Consortium Single-cell multiomic profiling of human lungs reveals cell-type-specific and age-dynamic control of SARS-CoV2 host genes. eLife . 2020;9:e62522. doi: 10.7554/eLife.62522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Guo M, Du Y, Gokey JJ, Ray S, Bell SM, Adam M, et al. Single cell RNA analysis identifies cellular heterogeneity and adaptive responses of the lung at birth. Nat Commun . 2019;10:37. doi: 10.1038/s41467-018-07770-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Karolak JA, Deutsch G, Gambin T, Szafranski P, Popek E, Stankiewicz P. Transcriptome and immunohistochemical analyses in TBX4- and FGF10-deficient lungs imply TMEM100 as a mediator of human lung development. Am J Respir Cell Mol Biol . 2022;66:694–697. doi: 10.1165/rcmb.2021-0470LE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gibson-Brown JJ, Agulnik SI, Chapman DL, Alexiou M, Garvey N, Silver LM, et al. Evidence of a role for T-box genes in the evolution of limb morphogenesis and the specification of forelimb/hindlimb identity. Mech Dev . 1996;56:93–101. doi: 10.1016/0925-4773(96)00514-x. [DOI] [PubMed] [Google Scholar]
- 37. Logan M, Simon HG, Tabin C. Differential regulation of T-box and homeobox transcription factors suggests roles in controlling chick limb-type identity. Development . 1998;125:2825–2835. doi: 10.1242/dev.125.15.2825. [DOI] [PubMed] [Google Scholar]
- 38. Hasson P, DeLaurier A, Bennett M, Grigorieva E, Naiche LA, Papaioannou VE, et al. Tbx4 and tbx5 acting in connective tissue are required for limb muscle and tendon patterning. Dev Cell . 2010;18:148–156. doi: 10.1016/j.devcel.2009.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Besse L, Sheeba CJ, Holt M, Labuhn M, Wilde S, Feneck E, et al. Individual limb muscle bundles are formed through progressive steps orchestrated by adjacent connective tissue cells during primary myogenesis. Cell Rep . 2020;30:3552–3565.e6. doi: 10.1016/j.celrep.2020.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Nishimoto S, Logan MPO. Subdivision of the lateral plate mesoderm and specification of the forelimb and hindlimb forming domains. Semin Cell Dev Biol . 2016;49:102–108. doi: 10.1016/j.semcdb.2015.11.011. [DOI] [PubMed] [Google Scholar]
- 41. Sekine K, Ohuchi H, Fujiwara M, Yamasaki M, Yoshizawa T, Sato T, et al. Fgf10 is essential for limb and lung formation. Nat Genet . 1999;21:138–141. doi: 10.1038/5096. [DOI] [PubMed] [Google Scholar]
- 42. Kawakami Y, Marti M, Kawakami H, Itou J, Quach T, Johnson A, et al. Islet1-mediated activation of the β-catenin pathway is necessary for hindlimb initiation in mice. Development . 2011;138:4465–4473. doi: 10.1242/dev.065359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Narkis G, Tzchori I, Cohen T, Holtz A, Wier E, Westphal H. Isl1 and Ldb co-regulators of transcription are essential early determinants of mouse limb development. Dev Dyn . 2012;241:787–791. doi: 10.1002/dvdy.23761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Itou J, Kawakami H, Quach T, Osterwalder M, Evans SM, Zeller R, et al. Islet1 regulates establishment of the posterior hindlimb field upstream of the Hand2-Shh morphoregulatory gene network in mouse embryos. Development . 2012;139:1620–1629. doi: 10.1242/dev.073056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Don EK, de Jong-Curtain TA, Doggett K, Hall TE, Heng B, Badrock AP, et al. Genetic basis of hindlimb loss in a naturally occurring vertebrate model. Biol Open . 2016;5:359–366. doi: 10.1242/bio.016295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Oda T, Matsushita M, Ono Y, Kitoh H, Sakai T. A novel heterozygous mutation in the T-box protein 4 gene in an adult case of small patella syndrome. J Orthop Case Rep . 2018;8:85–88. doi: 10.13107/jocr.2250-0685.1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Vanlerberghe C, Jourdain A-S, Dieux A, Toutain A, Callewaert B, Dupuis-Girod S, et al. Small patella syndrome: new clinical and molecular insights into a consistent phenotype. Clin Genet . 2017;92:676–678. doi: 10.1111/cge.13103. [DOI] [PubMed] [Google Scholar]
- 48. Li P, Lan W, Li J, Zhang Y, Xiong Q, Ye J, et al. Identification and functional evaluation of a novel TBX4 mutation underlies small patella syndrome. Int J Mol Sci . 2022;23:2075. doi: 10.3390/ijms23042075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Naeije R, Richter MJ, Rubin LJ. The physiologic basis of pulmonary arterial hypertension. Eur Respir J . 2021;59:2102334. doi: 10.1183/13993003.02334-2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ballif BC, Theisen A, Rosenfeld JA, Traylor RN, Gastier-Foster J, Thrush DL, et al. Identification of a recurrent microdeletion at 17q23.1q23.2 flanked by segmental duplications associated with heart defects and limb abnormalities. Am J Hum Genet . 2010;86:454–461. doi: 10.1016/j.ajhg.2010.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Nimmakayalu M, Major H, Sheffield V, Solomon DH, Smith RJ, Patil SR, et al. Microdeletion of 17q22q23.2 encompassing TBX2 and TBX4 in a patient with congenital microcephaly, thyroid duct cyst, sensorineural hearing loss, and pulmonary hypertension. Am J Med Genet A . 2011;155A:418–423. doi: 10.1002/ajmg.a.33827. [DOI] [PubMed] [Google Scholar]
- 52. Tsoi SM, Jones K, Colglazier E, Parker C, Nawaytou H, Teitel D, et al. Persistence of persistent pulmonary hypertension of the newborn: a case of de novo TBX4 variant. Pulm Circ . 2022;12:e12108. doi: 10.1002/pul2.12108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Levy M, Eyries M, Szezepanski I, Ladouceur M, Nadaud S, Bonnet D, et al. Genetic analyses in a cohort of children with pulmonary hypertension. Eur Respir J . 2016;48:1118–1126. doi: 10.1183/13993003.00211-2016. [DOI] [PubMed] [Google Scholar]
- 54. Zhu N, Gonzaga-Jauregui C, Welch CL, Ma L, Qi H, King AK, et al. Exome sequencing in children with pulmonary arterial hypertension demonstrates differences compared with adults. Circ Genom Precis Med . 2018;11:e001887. doi: 10.1161/CIRCGEN.117.001887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Zhu N, Welch CL, Wang J, Allen PM, Gonzaga-Jauregui C, Ma L, et al. Rare variants in SOX17 are associated with pulmonary arterial hypertension with congenital heart disease. Genome Med . 2018;10:56. doi: 10.1186/s13073-018-0566-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Thoré P, Girerd B, Jaïs X, Savale L, Ghigna M-R, Eyries M, et al. Phenotype and outcome of pulmonary arterial hypertension patients carrying a TBX4 mutation. Eur Respir J . 2020;55:1902340. doi: 10.1183/13993003.02340-2019. [DOI] [PubMed] [Google Scholar]
- 57. Haarman MG, Kerstjens-Frederikse WS, Vissia-Kazemier TR, Breeman KTN, Timens W, Vos YJ, et al. The genetic epidemiology of pediatric pulmonary arterial hypertension. J Pediatr . 2020;225:65–73.e5. doi: 10.1016/j.jpeds.2020.05.051. [DOI] [PubMed] [Google Scholar]
- 58. Zhu N, Swietlik EM, Welch CL, Pauciulo MW, Hagen JJ, Zhou X, et al. Regeneron Genetics Center PAH Biobank Enrolling Centers’ Investigators; NIHR BioResource for Translational Research – Rare Diseases; National Cohort Study of Idiopathic and Heritable PAH. Correction to: rare variant analysis of 4241 pulmonary arterial hypertension cases from an international consortium implicates FBLN2, PDGFD, and rare de novo variants in PAH. Genome Med . 2021;13:106. doi: 10.1186/s13073-021-00915-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Zhu N, Pauciulo MW, Welch CL, Lutz KA, Coleman AW, Gonzaga-Jauregui C, et al. PAH Biobank Enrolling Centers’ Investigators Novel risk genes and mechanisms implicated by exome sequencing of 2572 individuals with pulmonary arterial hypertension. Genome Med . 2019;11:69. doi: 10.1186/s13073-019-0685-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Welch CL, Chung WK. Genetics and genomics of pediatric pulmonary arterial hypertension. Genes (Basel) . 2020;11:E1213. doi: 10.3390/genes11101213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Navas P, Tenorio J, Quezada CA, Barrios E, Gordo G, Arias P, et al. Molecular analysis of BMPR2, TBX4, and KCNK3 and genotype-phenotype correlations in spanish patients and families with idiopathic and hereditary pulmonary arterial hypertension. Rev Esp Cardiol (Engl Ed) . 2016;69:1011–1019. doi: 10.1016/j.rec.2016.03.029. [DOI] [PubMed] [Google Scholar]
- 62. Gräf S, Haimel M, Bleda M, Hadinnapola C, Southgate L, Li W, et al. Identification of rare sequence variation underlying heritable pulmonary arterial hypertension. Nat Commun . 2018;9:1416. doi: 10.1038/s41467-018-03672-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Wang X-J, Lian T-Y, Jiang X, Liu S-F, Li S-Q, Jiang R, et al. Germline BMP9 mutation causes idiopathic pulmonary arterial hypertension. Eur Respir J . 2019;53:1801609. doi: 10.1183/13993003.01609-2018. [DOI] [PubMed] [Google Scholar]
- 64. Eyries M, Montani D, Nadaud S, Girerd B, Levy M, Bourdin A, et al. Widening the landscape of heritable pulmonary hypertension mutations in paediatric and adult cases. Eur Respir J . 2019;53:1801371. doi: 10.1183/13993003.01371-2018. [DOI] [PubMed] [Google Scholar]
- 65. Hernandez-Gonzalez I, Tenorio J, Palomino-Doza J, Martinez Meñaca A, Morales Ruiz R, Lago-Docampo M, et al. Clinical heterogeneity of pulmonary arterial hypertension associated with variants in TBX4. PLoS ONE . 2020;15:e0232216. doi: 10.1371/journal.pone.0232216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. van den Heuvel LM, Jansen SMA, Alsters SIM, Post MC, van der Smagt JJ, Handoko-De Man FS, et al. Genetic evaluation in a cohort of 126 Dutch pulmonary arterial hypertension patients. Genes (Basel) . 2020;11:E1191. doi: 10.3390/genes11101191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Eichstaedt CA, Saßmannshausen Z, Shaukat M, Cao D, Xanthouli P, Gall H, et al. Gene panel diagnostics reveals new pathogenic variants in pulmonary arterial hypertension. Respir Res . 2022;23:74. doi: 10.1186/s12931-022-01987-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Austin ED, Elliott CG. TBX4 syndrome: a systemic disease highlighted by pulmonary arterial hypertension in its most severe form. Eur Respir J . 2020;55:2000585. doi: 10.1183/13993003.00585-2020. [DOI] [PubMed] [Google Scholar]
- 69. Shrivastava S, Kruisselbrink TM, Mohananey A, Thomas BC, Kushwaha SS, Pereira NL. Rare TBX4 variant causing pulmonary arterial hypertension with small patella syndrome in an adult man. JACC Case Rep . 2021;3:1447–1452. doi: 10.1016/j.jaccas.2021.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Chelladurai P, Kuenne C, Bourgeois A, Günther S, Valasarajan C, Cherian AV, et al. Epigenetic reactivation of transcriptional programs orchestrating fetal lung development in human pulmonary hypertension. Sci Transl Med . 2022;14:eabe5407. doi: 10.1126/scitranslmed.abe5407. [DOI] [PubMed] [Google Scholar]
- 71. Karolak JA, Szafranski P, Kilner D, Patel C, Scurry B, Kinning E, et al. Heterozygous CTNNB1 and TBX4 variants in a patient with abnormal lung growth, pulmonary hypertension, microcephaly, and spasticity. Clin Genet . 2019;96:366–370. doi: 10.1111/cge.13605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Suhrie K, Pajor NM, Ahlfeld SK, Dawson DB, Dufendach KR, Kitzmiller JA, et al. Neonatal lung disease associated with TBX4 mutations. J Pediatr . 2019;206:286–292.e1. doi: 10.1016/j.jpeds.2018.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. German K, Deutsch GH, Freed AS, Dipple KM, Chabra S, Bennett JT. Identification of a deletion containing TBX4 in a neonate with acinar dysplasia by rapid exome sequencing. Am J Med Genet A . 2019;179:842–845. doi: 10.1002/ajmg.a.61096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Vincent M, Karolak JA, Deutsch G, Gambin T, Popek E, Isidor B, et al. Clinical, histopathological, and molecular diagnostics in lethal lung developmental disorders. Am J Respir Crit Care Med . 2019;200:1093–1101. doi: 10.1164/rccm.201903-0495TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Nogee LM. Interstitial lung disease in newborns. Semin Fetal Neonatal Med . 2017;22:227–233. doi: 10.1016/j.siny.2017.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Maurac A, Lardenois É, Eyries M, Ghigna MR, Petit I, Montani D, et al. T-box protein 4 mutation causing pulmonary arterial hypertension and lung disease. Eur Respir J . 2019;54:1900388. doi: 10.1183/13993003.00388-2019. [DOI] [PubMed] [Google Scholar]
- 77. Ranganath P, Perala S, Nair L, Pamu PK, Shankar A, Murugan S, et al. A newly recognized multiple malformation syndrome with caudal regression associated with a biallelic c.402G>A variant in TBX4. Eur J Hum Genet . 2020;28:669–673. doi: 10.1038/s41431-020-0572-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Hordyjewska-Kowalczyk E, Nowosad K, Jamsheer A, Tylzanowski P. Genotype-phenotype correlation in clubfoot (talipes equinovarus) J Med Genet . 2021;59:209–219. doi: 10.1136/jmedgenet-2021-108040. [DOI] [PubMed] [Google Scholar]
- 79. Lu W, Bacino CA, Richards BS, Alvarez C, VanderMeer JE, Vella M, et al. Studies of TBX4 and chromosome 17q23.1q23.2: an uncommon cause of nonsyndromic clubfoot. Am J Med Genet A . 2012;158A:1620–1627. doi: 10.1002/ajmg.a.35418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Peterson JF, Ghaloul-Gonzalez L, Madan-Khetarpal S, Hartman J, Surti U, Rajkovic A, et al. Familial microduplication of 17q23.1–q23.2 involving TBX4 is associated with congenital clubfoot and reduced penetrance in females. Am J Med Genet A . 2014;164A:364–369. doi: 10.1002/ajmg.a.36238. [DOI] [PubMed] [Google Scholar]
- 81. Alvarado DM, Aferol H, McCall K, Huang JB, Techy M, Buchan J, et al. Familial isolated clubfoot is associated with recurrent chromosome 17q23.1q23.2 microduplications containing TBX4. Am J Hum Genet . 2010;87:154–160. doi: 10.1016/j.ajhg.2010.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Abou Hassan OK, Haidar W, Nemer G, Skouri H, Haddad F, BouAkl I. Clinical and genetic characteristics of pulmonary arterial hypertension in Lebanon. BMC Med Genet . 2018;19:89. doi: 10.1186/s12881-018-0608-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Prapa M, Lago-Docampo M, Swietlik EM, Montani D, Eyries M, Humbert M, et al. NIHR BioResource for Translational Research – Rare Diseases, National Cohort Study of Idiopathic and Heritable PAH, PAH Biobank Enrolling Centers’ Investigators First genotype-phenotype study in TBX4 syndrome: gain-of-function mutations causative for lung disease. Am J Respir Crit Care Med . 2022 doi: 10.1164/rccm.202203-0485OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Exome Aggregation Consortium Analysis of protein-coding genetic variation in 60,706 humans. Nature . 2016;536:285–291. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Hnisz D, Abraham BJ, Lee TI, Lau A, Saint-André V, Sigova AA, et al. Super-enhancers in the control of cell identity and disease. Cell . 2013;155:934–947. doi: 10.1016/j.cell.2013.09.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Wu N, Ming X, Xiao J, Wu Z, Chen X, Shinawi M, et al. TBX6 null variants and a common hypomorphic allele in congenital scoliosis. N Engl J Med . 2015;372:341–350. doi: 10.1056/NEJMoa1406829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Szafranski P, Liu Q, Karolak JA, Song X, de Leeuw N, Faas B, et al. Association of rare non-coding SNVs in the lung-specific FOXF1 enhancer with a mitigation of the lethal ACDMPV phenotype. Hum Genet . 2019;138:1301–1311. doi: 10.1007/s00439-019-02073-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Yıldız Bölükbaşı E, Karolak JA, Szafranski P, Gambin T, Murik O, Zeevi DA, et al. Exacerbation of mild lung disorders to lethal pulmonary hypoplasia by a noncoding hypomorphic SNV in a lung-specific enhancer in trans to the frameshifting TBX4 variant. Am J Med Genet A . 2022;188:1420–1425. doi: 10.1002/ajmg.a.62656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Turro E, Astle WJ, Megy K, Gräf S, Greene D, Shamardina O, et al. NIHR BioResource for the 100,000 Genomes Project Whole-genome sequencing of patients with rare diseases in a national health system. Nature . 2020;583:96–102. doi: 10.1038/s41586-020-2434-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. 100,000 Genomes Project Pilot Investigators. Smedley D. Smith KR. Martin A. Thomas EA. McDonagh EM. et al. 100,000 Genomes pilot on rare-disease diagnosis in health care—preliminary report. N Engl J Med . 2021;385:1868–1880. doi: 10.1056/NEJMoa2035790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Sudlow C, Gallacher J, Allen N, Beral V, Burton P, Danesh J, et al. UK Biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med . 2015;12:e1001779. doi: 10.1371/journal.pmed.1001779. [DOI] [PMC free article] [PubMed] [Google Scholar]