

SUMMARY
Germline histone H3.3 amino acid substitutions, including H3.3G34R/V, cause severe neurodevelopmental syndromes. To understand how these mutations impact brain development, we generated H3.3G34R/V/W knock-in mice and identified strikingly distinct developmental defects for each mutation. H3.3G34R-mutants exhibited progressive microcephaly and neurodegeneration, with abnormal accumulation of disease-associated microglia and concurrent neuronal depletion. G34R severely decreased H3K36me2 on the mutant H3.3 tail, impairing recruitment of DNA methyltransferase DNMT3A and its redistribution on chromatin. These changes were concurrent with sustained expression of complement and other innate immune genes possibly through loss of non-CG (CH) methylation and silencing of neuronal gene promoters through aberrant CG methylation. Complement expression in G34R brains may lead to neuroinflammation possibly accounting for progressive neurodegeneration. Our study reveals that H3.3G34-substitutions have differential impact on the epigenome, which underlie the diverse phenotypes observed, and uncovers potential roles for H3K36me2 and DNMT3A-dependent CH-methylation in modulating synaptic pruning and neuroinflammation in post-natal brains.
Keywords: H3.3 G34R/V/W, Neurodegeneration, H3K36me2, DNMT3A, CH methylation
Graphical Abstract
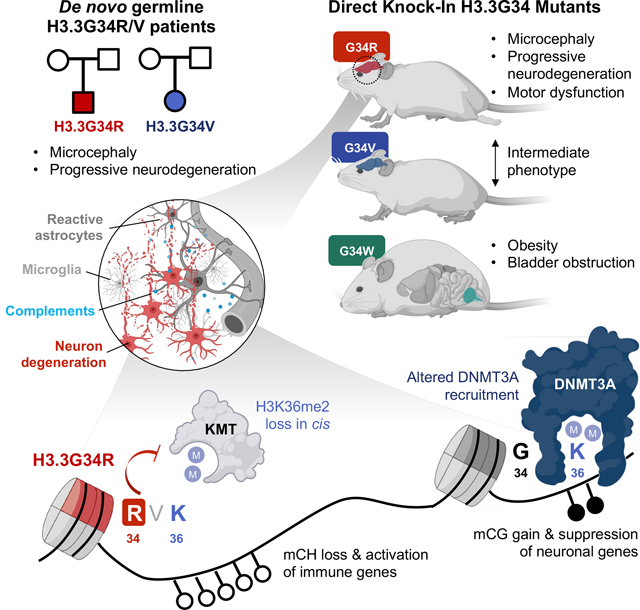
A study exploring the effect of human germline histone H3.3 amino acid mutations in mice provides insights into their differential impact on the epigenome as well as roles for DNA methylation dysregulation in modulating synaptic pruning and neuroinflammation in post-natal brains.
INTRODUCTION
Covalent post-translational modification (PTM) of histones and DNA occur dynamically during development to orchestrate gene expression programs and have essential roles in the mammalian brain from neurulation and synaptogenesis to neurocognition, among other functions 1. The essential role of epigenetic modifications in brain development is further supported by the growing number of neurodevelopmental syndromes attributed to epigenetic dysfunction through mutations of histone-modifying enzymes, DNA methylation modulators, and more recently histones 2,3. The replication-independent histone H3.3 variant, encoded by H3F3A and H3F3B genes, can build up to nearly 90% of the total histone H3 in terminally differentiated postmitotic neurons 4,5. Several de novo germline H3.3 missense mutations in H3F3A or H3F3B were recently reported in patients presenting with varying degrees of neurodegeneration, autism spectrum disorder, and microcephaly 3,6,7, including a patient bearing a H3.3G34 glycine 34 to valine (G34V) substitution. H3.3G34V, in addition to glycine 34 to arginine (G34R) substitutions, were previously identified as somatic drivers in lethal cortical brain tumors of neuronal origin 8–10. Interestingly, somatic G34 substitutions in human diseases are also exclusive to H3.3, including an additional G34 to tryptophan (G34W) substitution identified exclusively in bone tumors of mesenchymal origin 11.
Unlike the global effect mediated by other somatic (K-to-M) substitutions on H3K27/K36 observed in cancer, G34 substitutions on the H3.3 tail act in cis, initially affecting the mutant histone tail. Several studies indicate that H3.3G34-mutants affect PTM of the adjacent K36 residue, potentially impairing its methylation by writers (e.g. SETD2 or NSD2), erasers (e.g. KDM2A/2B), or preventing its recognition by readers (e.g. ZMYND11) 12–15. This initial effect on chromatin precedes more widespread changes 13,16. Importantly, appropriate regulation of PTM in H3K36 is critical in brain development as germline mutations in H3K36 methyltransferases (e.g. SETD2, NSD1/2) as well as the readers (e.g. DNMT3A/3B, ZMYND11) are similarly associated with a wide spectrum of congenital neurodevelopmental syndromes 17,18. This suggests that altered PTM of H3.3K36 or abnormal recognition of this residue due to H3.3G34 mutations may promote the neurodevelopmental phenotype observed in the patient with germline H3.3G34V mutation.
While somatic H3.3G34 mutations have been studied extensively in the context of cancer, their role in the germline and how they affect brain development remain unclear. Also, why H3.3G34W are restricted to mesenchymal tissues while H3.3G34R/V mainly occur in the brain remains unknown. Here, we report an additional de novo germline H3.3G34R patient exhibiting severe progressive developmental delay. To investigate how H3.3G34 mutations affect the epigenome and transcriptome leading to the developmental phenotypes in human disease, we engineered isogenic direct knock-in (DKI) mouse models carrying H3.3G34R, V, W, and control indels at the endogenous H3f3a locus on two distinct genetic mouse backgrounds. We show that H3.3G34R, and to a lesser degree H3.3G34V, affect brain development through progressive neuroinflammation, and further unravel the epigenetic mechanisms promoting these changes and disease phenotype.
RESULTS
Germline H3.3G34R and H3.3G34V mutations in patients lead to severe microcephaly and neurodevelopmental delay
Using whole exome trio-sequencing, we identified a previously unreported patient with de novo germline mutation in H3.3G34R (H3F3B c.103 G>A; p.G34R) showing phenotypic resemblance to the previously described H3.3G34V patient (H3F3B c.104G>T; p.G34V) 3 (Supplemental Table S1). Both patients share major global and neurodevelopmental delay, severe undergrowth with short stature and microcephaly, even when compared to other patients with H3.3 germline mutations (Figure S1A), and behavioral/neurological abnormalities including - feeding difficulties, hand stereotypies, and intractable epilepsy. Neither patient has achieved the ability to walk or speak, while the G34R patient showed regression and decreased social interactivity. This indicates that germline H3.3G34R/V mutations severely impair brain development and neurocognitive functions.
G34R/V/W mice exhibit mutation-specific developmental defects
To understand the developmental consequences of H3.3G34 germline mutations, we generated direct G34R, G34V, and G34W knock-in mice using embryo editing (Figure 1A). Single-guide RNA (sgRNA) targeting the H3f3a locus was co-injected with the Cas9 protein and a G34R/V/W repair template (Key Resource Table) into 2- or 4-cell stage embryos. Edited embryos were then implanted into recipient mothers to obtain mosaic founders (F0), then backcrossed with H3.3 wild-type (WT) mice from two independent strains (C57BL/6J and B6C3F1/Crl) to obtain true heterozygous G1 animals carrying +/− (indel), +/G34R, +/G34V, and +/G34W mutations. Presence of G34 mutations in founders were further confirmed by deep amplicon sequencing (Key Resource Table) and by the global expression of mutant histone proteins throughout the brain and in other somatic tissues (Figure 1B, S1B–C).
Figure 1. Germline knock-in of H3.3G34R/V/W results in distinct developmental defects.
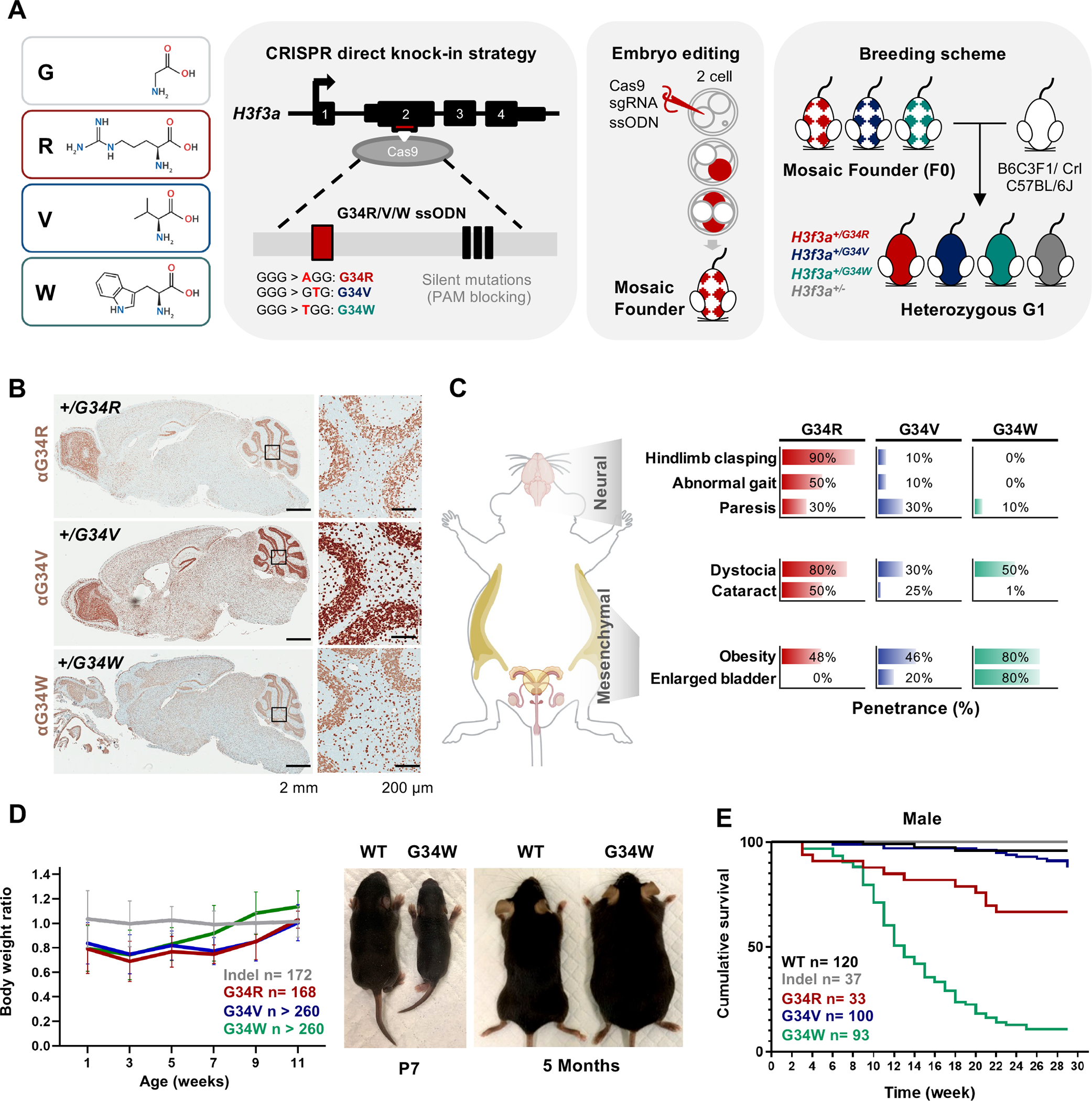
A. Left: amino acid depiction of H3.3 glycine 34 (G, hydrophobic) substitution to arginine (R, basic), valine (V, hydrophobic) or tryptophan (W, aromatic). Right: schematic representation of the derivation of H3.3G34R/V/W direct knock-in mice. Single-guide RNA (sgRNA) targeting exon 2 of H3f3a is represented by the red box, and the ssODN homology-directed repair (HDR) donor sequence is shown inside the gray box. The HDR template contained the G-to-R/V/W nucleotide substitutions at the G34 codon (red box) and the three silent PAM-blocking mutations (black boxes). Cas9 protein, sgRNA, and ssODN were microinjected into 2- to 4-cell embryos to obtain mosaic founder mice (F0) and backcrossed to two genetic strains to obtain true heterozygous generation 1 (G1) founders. B. Immunohistochemistry staining for H3.3G34R/V/W in DKI mutant mice. C. G34R/V/W mice show distinct and overlapping developmental phenotypes. Right: bar chart depicting penetrance of the observed phenotypes for each G34 mutant, > 10 mice were sampled for each genotype and phenotype combination. D. Body weight of G34 mutant and indel mice represented as ratios normalized to WT mice. n: cumulative number of mice. Representative pictures of WT and G34W mice at P7 and 5 months of age. E. Kaplan-Meier curves for cumulative survival rates of male G34 mutants and controls, n: cumulative number of mice.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Histone H3.3 G34R | RevMab | 31-1120, RRID:AB_2716433 |
| Histone H3.3 G34V | RevMab | 31-1193, RRID:AB_2716435 |
| Histone H3.3 G34W | RevMab | 31-1145 RRID:AB_2716434 |
| H3K27me3 | Cell Signaling Technology | 9733, RRID:AB_2616029 |
| H3K27ac | Diagenode | C15410196, RRID:AB_2637079 |
| H3K36me3 | Active Motif | 61021, RRID:AB_2614986 |
| H3K36me2 | Active Motif | 39255 |
| DNMT3A | Abcam | Ab2850 |
| FLAG | Cell Signaling Technology | 8146 |
| Total H3 | Abcam | Ab1791 |
| Calbindin | Millipore Sigma | C9848 |
| Iba1 | Abcam | Ab178846, Ab283346 |
| NeuN | Millipore Sigma | MAB377 |
| GFAP | Cell signalling | 12389S |
| Oct6 | Millipore Sigma | MABN738 |
| C1q | Abcam | Ab182451 |
| C3 | Novus Biologicals | NB200-540 |
| PSD95 | Abcam | Ab18258 |
| NF200 | Millipore Sigma | N4142 |
| Bacterial and Virus Strains | ||
| Biological Samples | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| Recombinant Cas9 | IDT | |
| Critical Commercial Assays | ||
| Deposited Data | ||
| WGBS, ChIP-seq, bulk RNA-seq, single nuceli RNA-seq & ATAC-seq data for murine samples | This paper | GEO: GSE199885 reviewer token: ohybqqkabbmbzcp |
| Experimental Models: Cell Lines | ||
| C3H10T1/2 o/e FLAG-HA-H3.3 WT | Jain et al., 2020 | N/A |
| C3H10T1/2 o/e FLAG-HA-H3.3 G34R | Jain et al., 2020 | N/A |
| C3H10T1/2 o/e FLAG-HA-H3.3 G34W | Jain et al., 2020 | N/A |
| U2-OS FlpIn-T-Rex o/e GFP-FLAG-BIoID2 | This paper | N/A |
| U2-OS FlpIn-T-Rex o/e NLS-GFP-FLAG-BIoID2 | This paper | N/A |
| U2-OS FlpIn-T-Rex o/e H3.3-WT-FLAG-BIoID2 | This paper | N/A |
| U2-OS FlpIn-T-Rex o/e H3.3-G34R-FLAG-BIoID2 | This paper | N/A |
| U2-OS FlpIn-T-Rex o/e H3.3-G34V-FLAG-BIoID2 | This paper | N/A |
| U2-OS FlpIn-T-Rex o/e H3.3-G34W-FLAG-BIoID2 | This paper | N/A |
| DKI H3.3 G34R NPC | This paper | N/A |
| DKI H3.3 G34V NPC | This paper | N/A |
| DKI H3.3 G34W NPC | This paper | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: B6C3F1/Crl | Charles River Laboratories | RRID:IMSR_CRL:031 |
| Mouse: C57BL/6J | The Jackson Laboratory | RRID:IMSR_JAX:000 664 |
| Oligonucleotides | ||
| H3f3a exon 2 targeting sgRNA TAGAAATACCTGTAACGATG | This paper | N/A |
| H3f3a intron 1 targeting gRNA CGCGCTGGCAACGAGCGACT | This paper | N/A |
|
H3f3a G34R ssODN CCGCAAATCCACCGGTGGTAAAGCACCCAGGAAAC AACTGGCTACAAAAGCCGCTCGCAAGAGTGCGCCC TCTACTGGAAGGGTGAAGAAACCTCACCGCTATAG GTATTTCTAAAACGTCGAGCAGTGGGATAGTGTCTA AGCAGTATGTCCGTGTAATTTAACAGGAAGATAGTC ATA |
This paper | N/A |
|
H3f3a G34V ssODN CCGCAAATCCACCGGTGGTAAAGCACCCAGGAAAC AACTGGCTACAAAAGCCGCTCGCAAGAGTGCGCCC TCTACTGGAGTGGTGAAGAAACCTCACCGCTATAG GTATTTCTAAAACGTCGAGCAGTGGGATAGTGTCTA AGCAGTATGTCCGTGTAATTTAACAGGAAGATAGTC ATA |
This paper | N/A |
|
H3f3a G34W ssODN CCGCAAATCCACCGGTGGTAAAGCACCCAGGAAAC AACTGGCTACAAAAGCCGCTCGCAAGAGTGCGCCC TCTACTGGATGGGTGAAGAAACCTCACCGCTATAG GTATTTCTAAAACGTCGAGCAGTGGGATAGTGTCTA AGCAGTATGTCCGTGTAATTTAACAGGAAGATAGTC ATA |
This paper | N/A |
|
H3f3a DKI Sanger genotyping primers: Fwd: CGATAGATGTAATCCGCGCC Rvs: ATCCCACTGCTCGACGTT |
This paper | N/A |
|
H3f3a inducible G34R genotyping primers: 5’ Fwd: CGCGAGCCTCTTAACTGC 5’ Rvs: GTTTACGTCGCCGTCCAG 3’ Fwd: GAGAAGCGCGATCACATGGT 3’ Rvs: TGTGTTTGTGGCTTCGTTCATT |
This paper | N/A |
| Recombinant DNA | ||
| Software and Algorithms | ||
| Trimmomatic v0.32 | Bolger et al., 2014 | http://www.usadellab.org/cms/?page=trimmomatic |
| STAR v2.3.0e | Dobin et al., 2013 | https://github.com/alexdobin/STAR |
| featureCounts v1.4.4 | Liao et al., 2014 | http://bioinf.wehi.edu.au/featureCounts/ |
| DESeq2 v1.14.1 | Love et al., 2014 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| fgsea v1.8.0 | - | https://bioconductor.org/packages/release/bioc/html/fgsea.html |
| BWA-MEM | Li and Durbin et al., 2009 | http://bio-bwa.sourceforge.net/bwa.shtml |
| Picard | Broad Institute | https://broadinstitute.github.io/picard/ |
| IGV | Robinson et al., 2011 | http://software.broadinstitute.org/software/igv/ |
| GenPipes v3.1.2 | Bourgey et al., 2019 | https://bitbucket.org/mugqic/genpipes/src/master/ |
| Homer v4.9.1 | Heinz et al. 2010 | http://homer.ucsd.edu/homer/ |
| VisRSeq v0.9.40 | Younesy et al., 2015 | https://visrsoftware.github.io/ |
| SeqMonk | BabrahamBioinformatics | https://www.bioinformatics.babraham.ac.uk/projects/seqmonk/ |
| MACS | Zhang et al., 2008 | https://github.com/macs3-project/MACS |
| Other | ||
Mice with germline heterozygous H3.3 G34R, V, and W mutations were viable, in contrast to H3.3K27M which produced zygotes that did not live past the four-cell stage 19. Animals carrying indel mutation behaved like WT animals in all in vivo and in vitro tests performed and were included with WT in the control group. As the G34-mutant mice reached adolescence, we observed strikingly distinct phenotypes unique to each G34 substitution (Figure 1C, Supplemental Table S2), that became fully penetrant in adult mice. These phenotypes were fully recapitulated on both genetic mouse backgrounds, indicating they are driven by the introduced G34 mutation. All G34 mutants were born significantly smaller than control mice, and eventually developed obesity in adolescence, with G34W showing even more significant weight gain starting at 8 weeks (Figure 1D). Female G34R mice exhibited difficulties during parturition including dystocia, a phenotype observed to a lesser extent in G34V/W females (Figure 1C). Male G34R mice also displayed a degree of subfertility, due to progressive ataxia and neuromuscular related defects after 3-month of age, which resulted in inability to copulate. Importantly, G34R animals exhibited neurologically associated phenotypes, while G34W showed mesenchymal-related defects, and G34V had an intermediate phenotype between the two mutations (Figure 1C).
As mice reached adulthood, G34W male mice died prematurely with only 11% surviving past 7 months of age due to kidney failure secondary to bladder urine retention (Figure S1D). In contrast, G34R and G34V males lived significantly longer, with 67% and 87% surviving past 7 months of age, respectively (Figure 1E), and were often euthanized due to severe ataxia. Female G34 mutants had overall higher survival rates of 77%, 76%, and 93% in G34W, G34R and G34V, respectively (Figure S1E). In summary, we observed tissue specific phenotypes in G34R, G34V, and G34W animals that manifested during adolescence. Obesity was a common phenotype, but most pronounced in G34W mutant mice. Importantly, only G34R mice exhibited severe neurological alterations, while G34W mice had uretero-genital defects and G34V mice had overlapping but milder phenotypes than those observed in G34R and G34W animals.
G34R, but not G34W, mutants show impaired motor functions
Neurologically associated phenotypes (e.g., hindlimb-clasping, abnormal gait, paresis) were fully penetrant in G34R mice, mild to absent in G34V and absent in G34W (Figure 1C). We therefore performed a panel of standard behavioral tests at various developmental time points and compared performance with age-matched control mice. First, we scored hindlimb-clasping, a hallmark of neurological disorders in mice 20,21 and evaluated its progression in G34 mutants. G34V/W and control mice exhibited a normal response of splayed limbs upon suspension (score 0 – 1) across their lifespan. In contrast, G34R mice developed hindlimb-clasping behaviors (score 1–3) by 4 weeks of age and progressed in severity over time with 100% of G34R animals exhibiting severe clasping response by 3 months (Figure 2A). Notably, some G34R mice showed clasping of both hind- and forelimbs (Figure 2A) at 3 months, and this clasping behavior persisted until death. Moreover, involuntary tremor and bat-like posture were observed in middle aged G34R mice. In aged mice, G34R and G34V showed aging-associated paresis (Figure S2A), while this was rarely observed in G34W animals.
Figure 2. G34R/V mice show abnormal motor functions and progressive microcephaly.
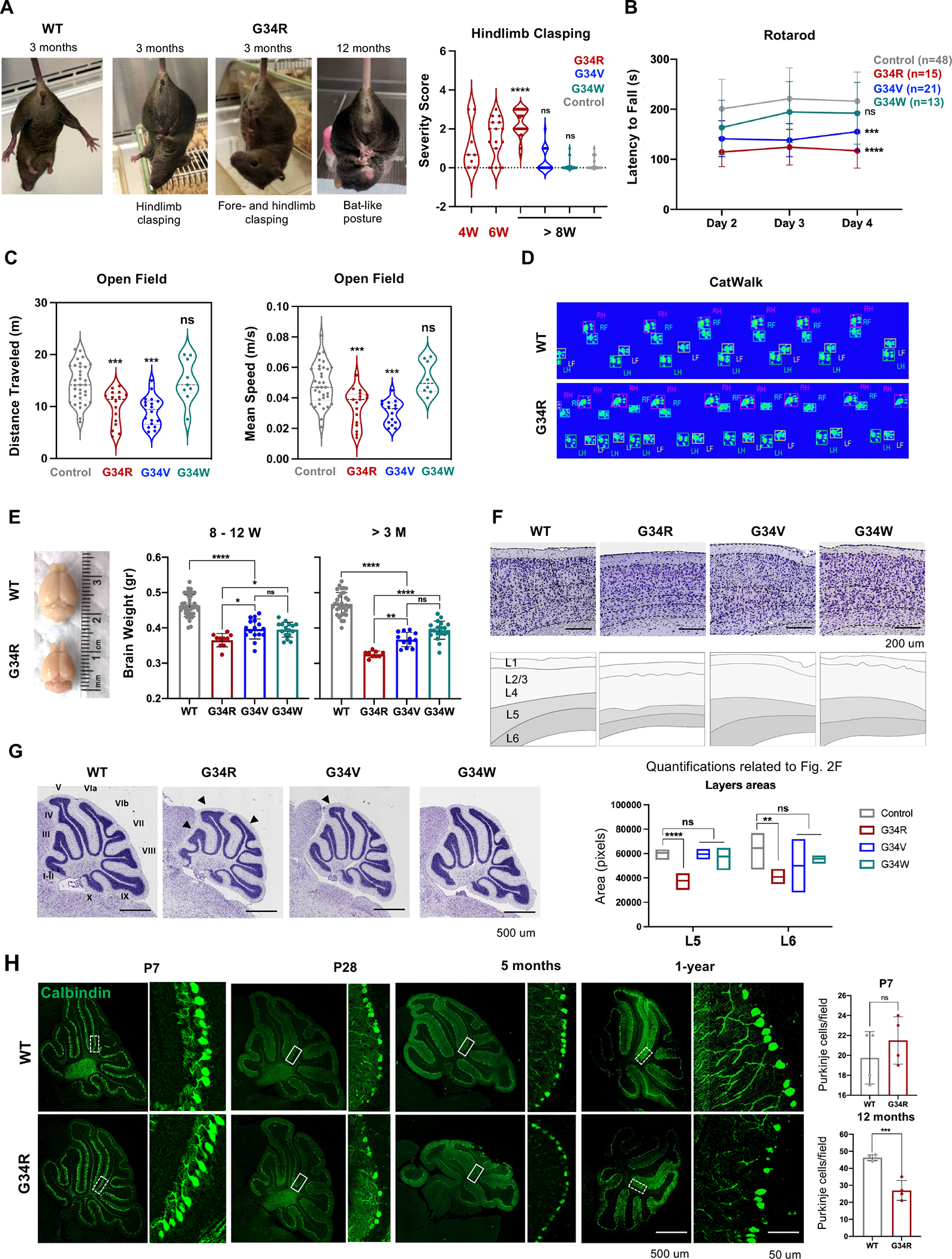
A. Adult WT and G34R mice exhibiting hind- and fore-limb clasping. Right: quantitative measure of hindlimb clasping score for G34R mice at adolescence (< 8 weeks) and adult (> 8 weeks), compared to G34V/W and controls (n > 10). Each mouse is plotted individually. Dashed lines: median. Dotted lines: quartiles. Student’s t-test. **** P < 0.0001 and n.s.: non-significant. B. Adult G34R/V show impaired motor coordination and balance compared to G34W and control mice (n > 10). The data is presented as means ± SD. Two-way ANOVA. ***P < 0.001, **** P < 0.0001 and n.s.: non-significant. C. Adult G34R and G34V mutants show hypoactivity in open field test compared to G34W and control animals (n > 6). Each mouse is plotted individually. Dashed and dotted horizontal lines indicate the median and quartiles, respectively. Student’s t-test. ***P < 0.001 and n.s.: non-significant. D. G34R animals show abnormal gait shown by representative digitized mice footprints, as recorded during the CatWalk test of WT (upper) and G34R (lower) littermates. RF, right front; RH, right hind; LF, left front; LH, left hind. E. Image and brain weight of adult G34 mutant mice showing progressive microcephaly in G34R animals (n > 10). Each mouse is plotted individually and as means ± SD. Student’s t-test. *P < 0.05, **P < 0.01, **** P < 0.0001 and n.s.: non-significant.F. Nissl staining of midsagittal cerebral cortex of control and G34 mutant mice showing thinning of L5/6 in G34R animals. Top panel: Cresyl Violet, bottom panel: scored cortical cell layers. Below, quantification of cerebral cortex layers (L5, layer 5; L6, layer 6) areas of control (n=4) and G34 mutant mice (G34R n=3, G34V n=2, G34W n=3) using the Cresyl Violet (Nissl staining). Line: mean. Student’s t-test. **P < 0.01, **** P < 0.0001 and n.s.: non-significant. G. Representative Nissl staining of midsagittal brain slices of control and G34 mutant cerebellum. Note the simplified cerebellum foliation in G34R brain (black arrowheads). H. Representative confocal images of cerebellar para-sagittal sections from control and G34R mice stained for Calbindin (green), a marker of Purkinje neurons. Left: 10X. Right: 20X. The white hatched areas (inset) of P7 and 12-month cerebellar images were used for quantification (n=4) (upper panel). The result of each mouse is plotted individually and as means ± SD. Student’s t-test. ***P < 0.001 and n.s.: non- significant.
To assess general locomotor activity level and exploration behavior of mutant mice, we performed rotarod and open field tests. G34R mice showed impaired motor coordination and balance in an accelerating rotarod test. At age 4–7 weeks, G34R mice significantly underperformed compared to control mice (Figure S2B). In older mice (8 weeks onward), G34R and G34V, but not G34W mice performed significantly worse compared to controls (Figure 2B), suggesting that G34R/V mutations perturb functions related to coordination and balance. In addition, G34R/V mice showed a significant decrease in traveled distance and walking speed compared to age-matched controls (Figure 2C), while G34W performed similarly to control mice. Finally, we used the Catwalk system to record the digital paw footprints of the unidirectionally-walking mutant and control mice to investigate motor coordination and gait parameters quantitatively. G34R mice displayed multiple placing of the same paw near the same site and a shortening of step length (Figure 2D). We also observed a reduction of hind and front paw stride length 22 (Figure S2C). The frequency of diagonal contacts in G34R mice was also significantly lower than in control animals, leading to an increase in trigonal supports, which also walked with a wider distance between the front paws, an increased forepaws base of support (Figure S2D). In summary, G34R mice exhibit severe neurological deficits after birth, demonstrating clasping of hind limbs, poor locomotion, loss of balance and abnormal gait; finally acquiring a profound tremor and paresis in advanced age, prompting euthanasia. This severe ataxia-like phenotype is observed to a modest extent in G34V but is absent in G34W mice.
G34R causes progressive microcephaly and neurodegeneration
To investigate gross brain anatomy, we measured brain weight and evaluated overall brain structure at various post-natal time points. In early post-natal stage, the brains of all G34-mutant mice were smaller than control littermates, which correlates with reduced body weight in the G34R/V/W mice (Figure 1D, S2E). However, from adolescence (4W – 3M) onwards, the brains of G34R mice, and to a lesser extent G34V, progressively decreased in size relative to those of G34W and control mice (Figure 2E). Similarly, histological examination revealed progressive thinning of deep cortical layers V and VI in G34R compared to G34V/W and control mice brains (Figure 2F, S2F). Cresyl violet staining at different developmental stages revealed abnormal cerebellar structure and simplified foliation patterns in post-natal G34R animals that increases with age (Figure 2G, S2G–H). This morphological difference was less pronounced in G34V, and not observed in G34W animals (Figure 2G, S2H). The hindlimb clasping reflex has been described in several mouse models affected by cerebellar atrophy, and is caused by cell ectopias in the cerebellar cortex combined with Purkinje cells (PCs) loss 23. Immunofluorescence staining with calbindin-D28k in G34R mice revealed a significant loss of PCs compared to WT in 1y adult brain (Figure 2H, S2I). PCs loss was not significant in P7 G34R mice, supporting a model where neuronal loss occurs or increases as mice age. In summary, the G34R mutation, and to a lesser extent G34V but not G34W, induce progressive microcephaly through layer-specific cortical thinning and simplification of cerebellar foliation.
Adult G34R brains show microglia/astrocytes accumulation and neuronal depletion
To identify cell lineage programs perturbed by the G34R mutation and causative of the neurological phenotypes, we performed total RNA-seq on P7 and 10W cortex from G34R mice and matched WT littermates (Figure 3A, Supplemental Table S3). Overall, the extent of transcriptional dysregulation in G34R cortex coincided with the onset of neurological symptoms. G34R P7 neonatal cortex showed modest transcriptional changes, while the 10W adult cortex had a considerably greater number of dysregulated genes and was robustly separated from other genotypes by transcriptomic profiling, reflecting the progressive nature of the neurodegenerative phenotype (Figure 3A).
Figure 3. G34R leads to abnormal accumulation of microglia and depletion of neurons.
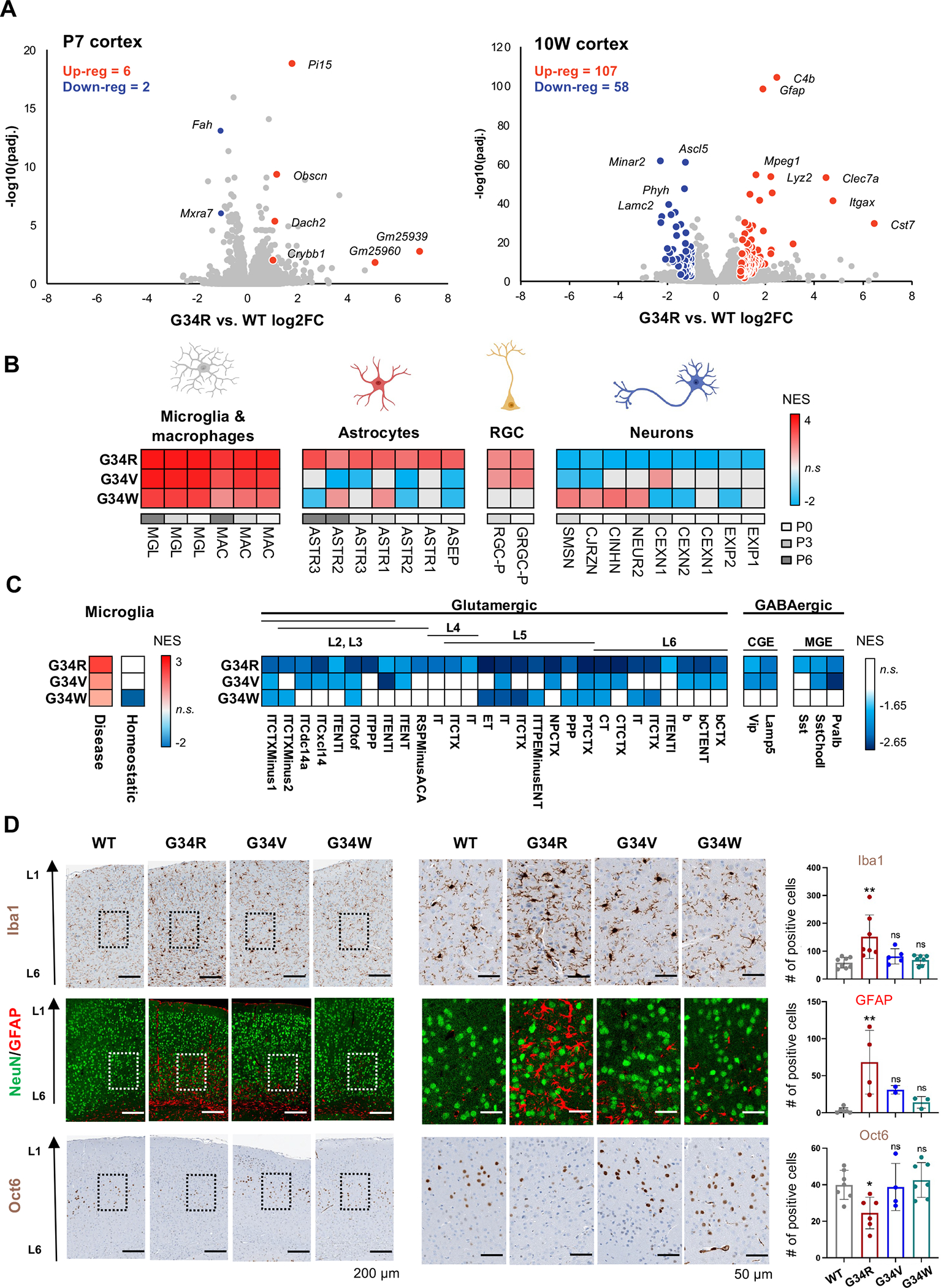
A. Volcano plot depicting differentially expressed genes assayed by bulk transcriptome comparing G34R to WT cortex at P7 (n = 4) and 10W (n = 3). Differentially expressed genes are labelled in red (up-reg.) and blue (down-reg.) using log2FC > 1, adjusted p value < 0.05, normalized mean expression > 100. B. Heatmap of significantly enriched (adjusted p value < 0.05) normalized enrichment scores (NES) of postnatal forebrain cell types of 10W G34 mutants compared with WT (n ≥ 2) by gene set enrichment analysis (GSEA). RGC: radial glial cell. C. G34R shows strong enrichment of disease-associated, but not homeostatic, microglia, and strong depletion of L5/L6 glutaminergic neurons. Heatmap of significantly enriched (adjusted p-value < 0.05) normalized enrichment score (NES) of damage-associated microglia signatures (left) and adult neuronal cell types (right) of G34 mutants compared with WT (n ≥ 2) by gene set enrichment analysis. MGE; medial ganglionic eminence-derived inhibitory GABAergic interneurons, CGE; caudal ganglionic eminence-derived inhibitory GABAergic interneurons. D. G34R cortex shows microglial infiltration and reactive astrocytes, concurrent with depletion of deep cortical neurons. Left: representative immunohistochemical staining of microglia (Iba1) and layer V neuronal marker (Oct6), and double immunofluorescence staining of neurons (NeuN) and astrocytes (GFAP) in cerebral cortex sections from adult mice of control and G34 mutant mice. Right: related quantification of Iba1 (WT n=8, G34R n=7, G34V n=5, and G34W n=7) GFAP (WT n=6, G34R n=4, G34V n=2 and G34W n=3) and Oct6 (WT n=7, G34R n=6, G34V n=4, and G34W n=7). Each mouse is plotted individually and as means ± SD. One-way ANOVA test. *P < 0.05, **P < 0.01, ***P < 0.001, and n.s.: non-significant.
Next, we used cell type specific gene signatures from published single cell RNA sequencing murine atlases 24,25 as input for gene-set enrichment analysis (GSEA) to characterize cell identity programs enriched/depleted in the G34R 10W cortex. All G34 mutant mice showed an enrichment of immune-related gene programs (microglia and macrophages) relative to WT littermates (Figure 3B, S3A), consistent with high expression of canonical microglial genes (e.g. Cd68, Aif1) (Figure S3B). Only G34R mutants showed higher expression of astrocytic gene programs and overexpression of canonical astrocyte markers (e.g. Gfap, Vim) (Figure 3B, S3B). Immunohistochemistry (IHC) staining confirmed accumulation of microglia (Iba1) and reactive astrocytes (GFAP) in the deeper cortical layer of G34R, but showed no significant increase in G34V/W at 10W (Figure 3D, S3C–D). This accumulation was seemingly absent at P7 in the G34R cerebral cortex, consistent with the progressive phenotype observed with age (Figure S3D). To assess whether the infiltrating microglia in G34R cortex correspond to normal homeostatic or disease-associated microglia, we performed gene-set enrichment analysis (GSEA) using a microglia signature 26, which has been consistently associated with neurodegenerative disease. Notably, G34R showed greater enrichment of disease-associated microglia gene expression programs (Figure 3C); suggesting the infiltrating microglial population in G34R is likely due to infiltration of disease-associated microglia rather than an excess of resting homeostatic microglia.
Conversely, G34R were uniquely depleted of neuronal gene programs compared to G34V/W or controls. Using an adult neuronal cell type atlas encompassing both cortical inhibitory and excitatory neurons as input for GSEA 27, G34R mice displayed pronounced depletion across all sampled neuronal signatures, with the strongest depletion observed in layers V and VI glutamatergic excitatory neuron signatures (Figure 3C, S3A). To confirm the depletion of deep layers neurons in cortices of G34R mice, we performed IHC of Oct6 (Pou3f1) which is expressed in cortical layer V 28. As expected, the number of Oct6+ neurons were dramatically reduced in adult G34R cortices relative to control and G34V/W brains (Figure 3D). In contrast, NeuN+ neurons and their distribution in the cortex of G34 mutants at all ages did not differ from WT littermate controls (Figure 3D, S3C). Taken together, transcriptome profiling and IHC show that G34R uniquely causes depletion of neurons and abnormal accumulation of disease-associated immune and glial cells in the mature cortex.
G34R severely depletes H3.3K36me2/3 in cis compared to G34V/W
We and others have demonstrated perturbation of H3K36 and H3K27 methylation by G34-mutations occurring in cis 10,13,16; however, side-by-side comparisons of H3.3G34 mutations induced in an isogenic background have not been performed. To determine whether similar cis H3K36/K27 methylation imbalances underpinned the phenotypes of G34R/V/W mice, we performed histone mass spectrometry to quantitate PTMs in neural progenitor cells (NPCs) derived from mutant mice fetal brain. We observed a striking depletion of H3K36me2 and H3K36me3 on G34R histone tails compared to their H3.3 WT counterparts (Figure 4A), while G34V/W showed relatively modest changes of H3K36 di-methylation and tri-methylation. Interestingly, H3K27me3 was significantly increased only in G34V and G34W and not in G34R (Figure 4B).
Figure 4. G34R histone tails are depleted of H3K36me2/3 and impaired for DNMT3A recruitment.
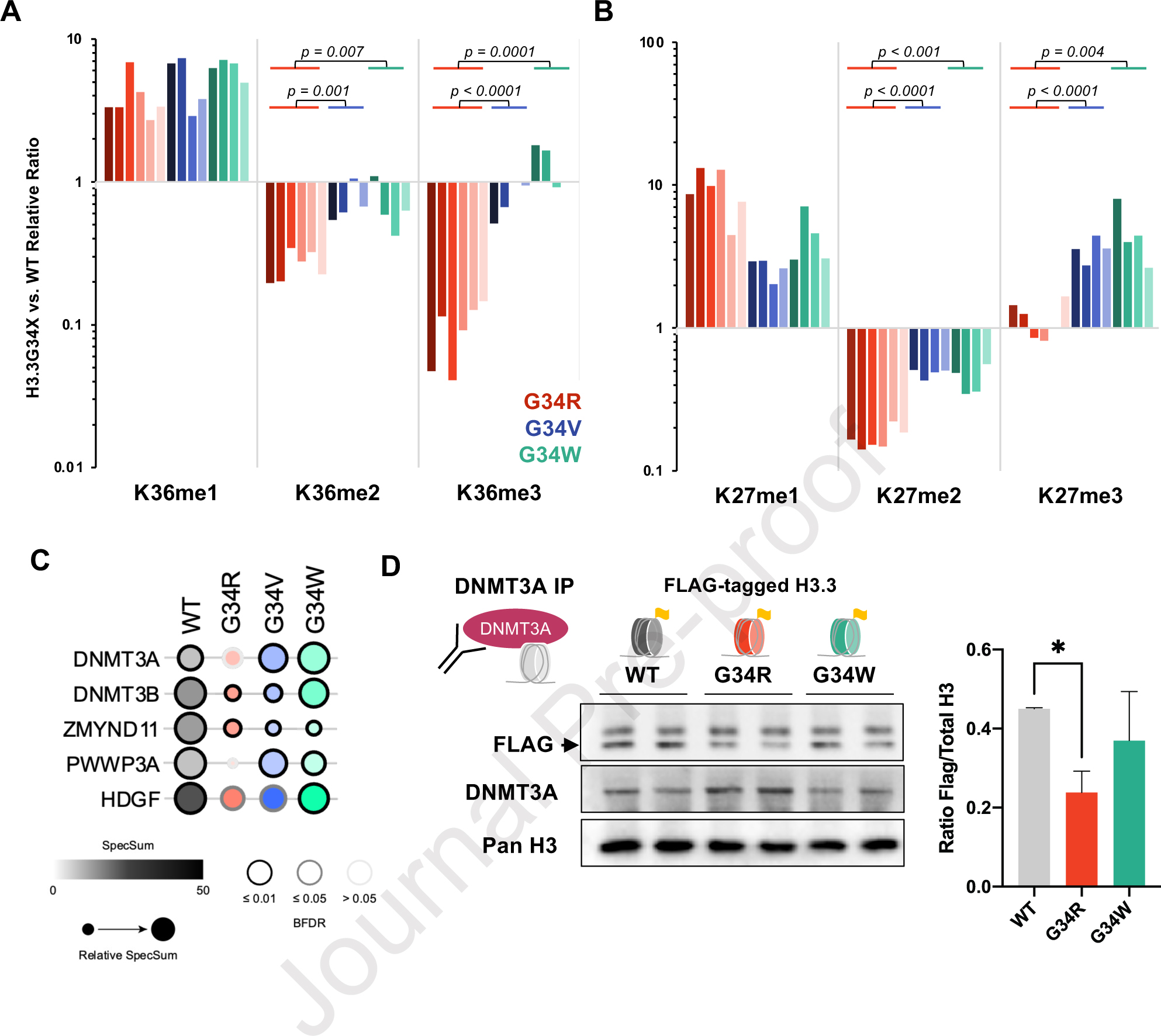
A. Waterfall plot depicting relative cis differences of histone H3K36 methylation levels on G34-mutant H3.3 histone tails compared to non-mutated H3.3 measured by histone PTM mass spectrometry. Histones were extracted from murine DKI G34R/V/W NPCs (3 biological replicate, 2 technical replicates). Note: H3K36 di- and tri-methylation is severely depleted in G34R, compared to G34V/W (p-value depicts significance derived from student pairwise t-test). B. Similar waterfall plot as panel A, depicting H3K27 methylation cis differences on G34-mutant H3.3 histone tails compared to non-mutated H3.3, as measured by histone PTM mass spectrometry. C. Bubble plot depicting BioID interaction frequency of G34-mutant histones (bait) with chromatin modifiers (prey) detected using MS in G34R/V/W and H3.3 WT expressing U2-OS lines (n=2). Note the decrease interaction with DNMT3A and DNMT3B in G34R histones. D. Immunoprecipitation/immunoblot confirmation of decrease DNMT3A (bait) interaction with G34R histones in C3H10T1/2 cell lines exogenously expressing FLAG-tagged H3.3 WT and G34-mutant histones (prey) (n=2). * Denotes p < 0.05, student’s t-test.
H3K36 di- and tri-methylation recruit various chromatin modifiers bearing the PWWP reader domain, including the de novo DNA methyltransferases DNMT3A/B and the transcriptional co-repressor ZMYND11 15,29,30. To determine whether G34-mutations interact differently with chromatin modifiers, we performed BioID on WT and G34 mutants introduced in U2OS cells (Figure 4C, S4, Supplemental Table S4). Known H3K36me modifiers, such as the H3K36me2 di-methyltransferase ASH1L or the H3.3K36me3 reader ZMYND11, showed uniform decreased interaction with G34R, V, and W. On the other hand, DNA methylation complex members showed differential interactions with specific G34 mutation. Specifically, G34R showed decreased interactions with DNMT3A, and G34R/V decreased interactions with DNMT3B, while association with the maintenance methyltransferase DNMT1 was depleted across all G34-mutations. To validate these results, we performed DNMT3A immunoprecipitation in C3H10T1/2 cells manipulated to exogenously express FLAG-tagged H3.3 G34R, G34W, or WT (Figure 4D). Immunoblot of DNMT3A-precipitants with FLAG antibody revealed significantly less G34R nucleosomes associated with DNMT3A compared to WT or G34W. Taken together, these data suggest that G34R mutation specifically hinders H3K36me2/3 deposition in cis and impairs the recruitment of de novo DNA methyltransferase DNMT3A.
G34R restricts DNMT3A deposition and causes mCH hypomethylation
In the developing cortex, expression of Dnmt3a transiently peaks shortly after birth (Figure 5A); whereas Dnmt3b expression is restricted to fetal stages 31,32. Neonatal increase of Dnmt3a expression leads to methylation of non-CG residues (CH) uniquely in neurons 32, which serves a critical neurological function by recruitment of methyl CpG-binding protein 2, MECP2 33. To determine whether decreased recruitment of DNMT3A by G34R disrupts in vivo DNMT3A function in the brain, we performed chromatin immunoprecipitation sequencing (ChIP-seq) for histone modifications and DNMT3A, in addition to whole-genome bisulfite sequencing (WGBS) in G34R and WT post-natal cortex (Figure 5B, Supplemental Table S5). Consistent with its known recruitment by H3K36me2 marked nucleosomes, DNMT3A pervasively localizes to genomic regions enriched for H3K36me2 in the WT cortex (Figure 5B, S5A). In the G34R mutant, H3K36me2 domains were contracted, becoming depleted from intergenic regions and more concentrated at gene bodies. This change in H3K36me2 distribution in G34R was concurrent with focal concentration of DNMT3A to regions with high density of H3K36me2 (Figure 5C) and its decreased deposition in intergenic regions with low H3K36me2 (Figure S5B–C).
Figure 5. G34R re-distribute DNMT3A localization, causing loss of CH methylation at intergenic regions.
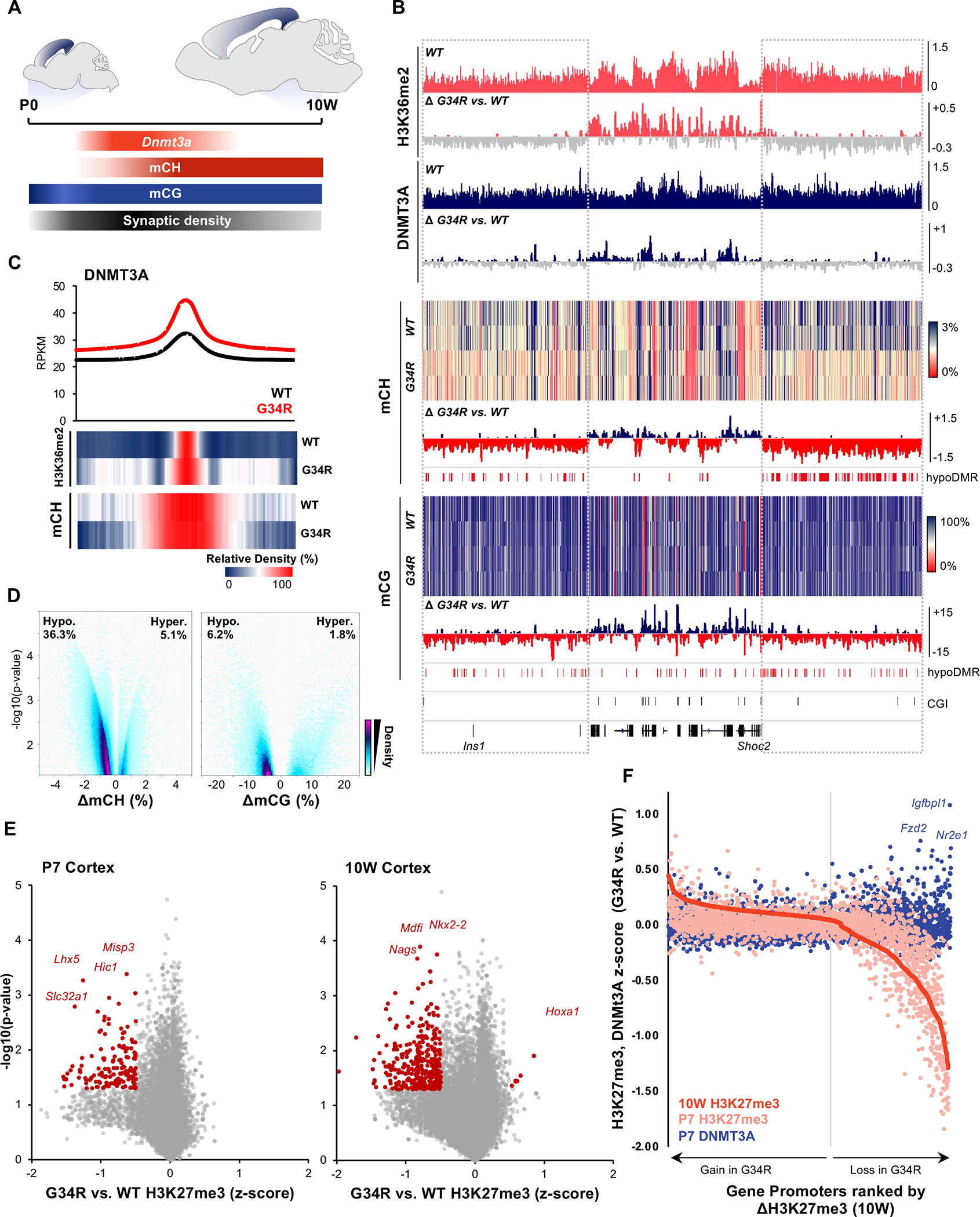
A. Schema depicting post-natal expression pattern of Dnmt3a and accumulation of mCH/mCG levels in the developing murine cortex. B. Genome browser snapshot depicting G34R-mediated epigenomic change in P7 (H3K36me2, DNMT3A) and 10W adult cortex (mCH, mCG). Upper tracks depict the epigenomic feature in WT cortex and change in G34R is depicted below (loss: grey). C. Metaplot of DNMT3A at called peaks in G34R and WT cortex (n=2). Under, heatmap depicting relative enrichment of H3K36me2 and mCH at DNMT3A peaks. D. Volcano plot depicting CH (left) and CG (right) methylation changes from G34R vs. WT (n = 2) in genome-wide 5kb bins. Hypo- and hyper-methylated bins are scored as p < 0.05 using LIMMA. E. Volcano plot depicting loss of H3K27me3 at gene promoters in P7 and 10W cortex of G34R mice (n = 2). F. Gain of DNMT3A occupancy (blue dots) at gene promoters losing the most H3K27me3 (right side of graph) in G34R cortex. Promoters were ranked by descending Z-score for H3K27me3 (10W G34R vs. WT, red), color dots represent corresponding change in H3K27me3 (pink) and DNMT3A (blue) at P7.
We next asked whether G34R alters the DNA methylation landscape. WGBS revealed that G34R mice exhibit striking loss of CH methylation with 36.3% of the genome showing significant hypomethylation, and 5.1% hypermethylation in the adult (10W) cortex (Figure 5D), relative to WT littermates. CG methylation followed similar hypomethylation patterns, albeit to a much lesser extent, with more regions showing hypomethylation (6.2%) than to hypermethylation (1.8%) in G34R. H3K36me2 distribution in G34R was highly correlated with DNMT3A deposition and subsequent CH/CG methylation changes (Figure S5B), with regions losing DNMT3A being depleted of H3K36me2 and losing CH/CG methylation. Consistent with redistribution patterns of H3K36me2 and DNMT3A, hypomethylated regions in G34R cortex are generally intergenic and rich in repetitive elements, with some regions overlapping immune gene clusters (Figure S5D). In contrast, the rare hypermethylated sites in G34R occurred preferentially at CpG islands (Figure S5E), with a subset normally targeted by PRC2-mediated H3K27me3. In mammalian cells, DNA methylation and H3K27me3 are known to exert antagonistic effects, with the two marks rarely coexisting; indeed, we observed marked H3K27me3 loss at genic promoters in G34R mice (Figure 5E). Notably, this H3K27me3 loss is measurable at P7 but becomes more severe in the adult 10W cortex. Intersection of DNMT3A ChIP data further revealed that a subset of promoters exhibiting H3K27me3 loss, such as the Tailless homolog TLX/Nr2e1 and frizzled class receptor 2 Fzd2, concurrently gained DNMT3A in G34R (Figure 5F). Collectively, our data shows that the G34R mutation diminishes H3K36me2 and consequently redistributes DNMT3A from intergenic regions to CpG islands. This change in DNMT3A localization leads to severe loss of CH methylation at gene-poor regions, and aberrant gain of CG methylation at developmental promoters normally regulated by PRC2/H3K27me3.
Dysregulation of mCH and mCG associates with complement activation and decreased neuronal gene expression
To determine the transcriptional effects of DNA methylation alteration in G34R mice, we assessed CH and CG methylation changes across differentially expressed genes in the adult (10W) cortex (Figure 6A). We observed the expected anti-correlation of CH and CG methylation with change in gene expression, consistent with the known transcriptional repressive role of DNA methylation (Figure 6B). Notably, the majority of up-regulated and hypomethylated genes are involved in immune responses, such as complement genes (e.g. C1qa/b/c, C3, C4a, C4b) and inflammatory cytokines (e.g. Ccl3, Ccl4). In the mammalian genome, some immune genes exist as large clusters (e.g. HLA, H-2, Ifna, C1q) that arose evolutionarily from tandem duplication events 34. In the brain, these immune gene clusters are embedded within broad pervasive H3K36me2 domains, showing preferential loss of CH methylation in G34R mice (Figure S5D). Importantly, within these immune clusters, the boundaries of mCH loss extends well beyond up-regulated gene bodies within these clusters (Figure S6B), suggesting this alteration in the methylome is not a consequence of transcriptional activation. Reciprocally, we also observed concurrent gain of promoter CG methylation and down-regulation of neuronal genes (Figure 6A–B), including synaptic protein Nptx2 and neurofilament gene Nefm.
Figure 6. Altered DNA methylation is concurrent with transcriptional dysregulation of immune and neuronal genes.
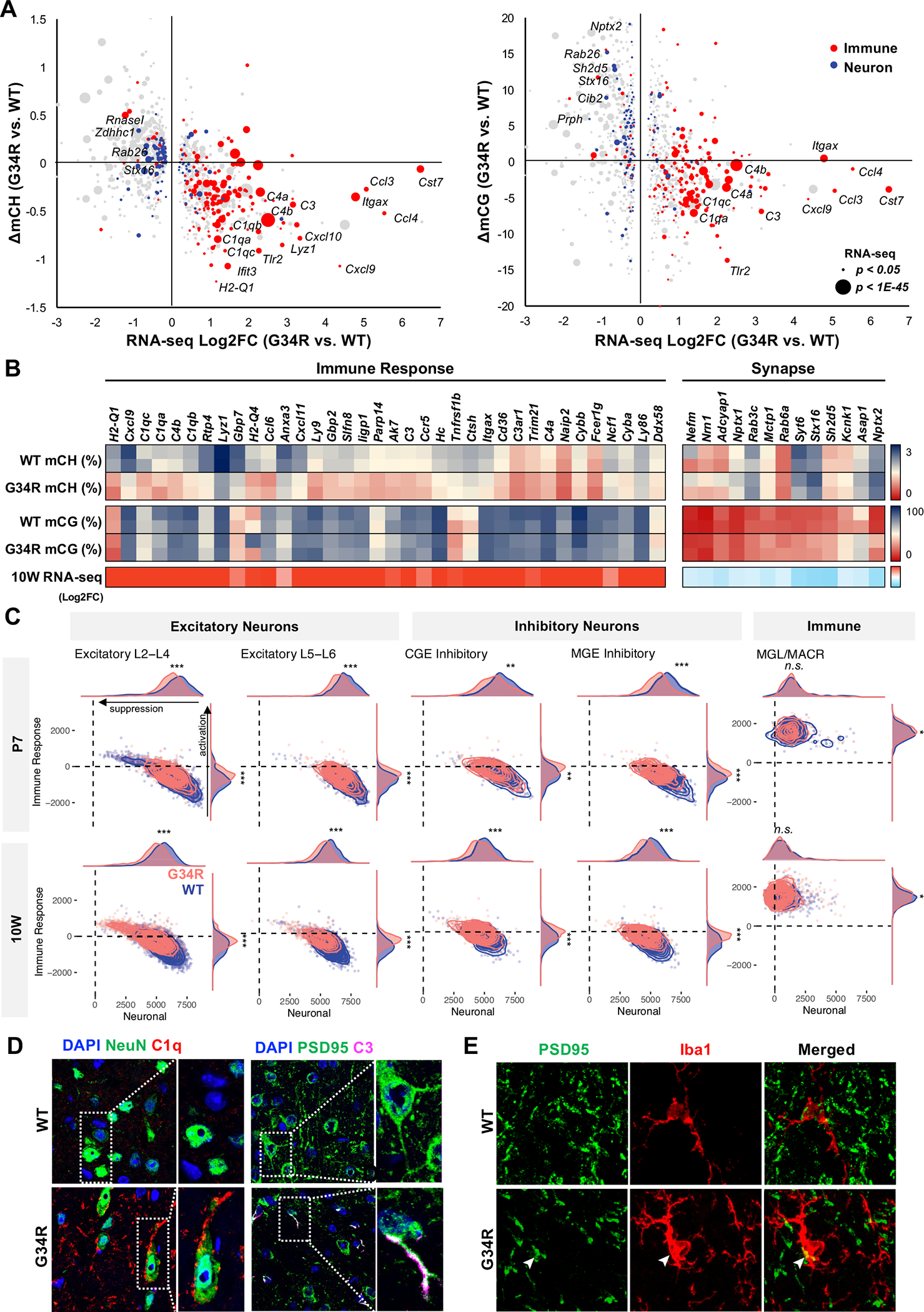
A. Immune genes are CH-hypomethylated and up-regulated, whereas neuronal genes are CG-hypermethylated and downregulated in G34R cortex. 2D bubble plot depicting change of CH (left) and CG methylation (right) correlated with bulk transcriptional dysregulation in 10W G34R cortex (n = 2). Size of bubble depicts significance of expression dysregulation. Gene ontology annotation of DE genes with immune response function in red, whereas genes annotated with synapse are in blue. B. Heatmap depiction of DNA methylation change (% methylation) and transcriptional change (log2FC) in 10W G34R vs. WT cortex. C. G34R neurons show activation of immune genes and suppression of neuronal genes compared to WT neurons. Density plot showing hypomethylated immune gene enrichment score (y-axis) vs. hypermethylated neuronal gene enrichment score (x-axis) in G34R (pink) and WT (blue) neurons and immune cells from P7 (N = 7848 cells; 3809 G34R, 4039 WT) and 10W (n = 17396 cells; 8539 G34R, 8857 WT) single nuclei transcriptome data. *** p < 2.2e-16, ** p < 1e-10, * p < 0.01, Wilcoxon rank sum test with continuity correction. D. Left: immunofluorescent co-staining of neuron NeuN (green) and complement C1Q (red) depicting G34R-specific accumulation of complement in neuron from substantia nigra region of 10-week brain (63x). Right: immunofluorescent co-staining of postsynaptic protein PSD95 (green) and complement C3 (magenta) from cortex region of 10W brain. E. Immunofluorescent co-staining of PSD95 (green) and microglia marker Iba1 (red) from cortex region of 10W brain. White arrowheads mark colocalization of PSD95 with IBA1 (63x).
To determine whether CH-hypomethylated immune genes are activated specifically in neurons, we performed single nuclei RNA-seq (snRNA-seq) on G34R and WT cortex at P7 and 10W (Figure 6C, S6C–F, S7A–D). Automated cell annotation using a consensus of four classifiers trained on cortical reference atlases identified well-separated clusters of excitatory- and inhibitory-neurons, as well as non-neuronal clusters of oligodendrocytes, astrocytes, and microglia (Figure S6C–F). Gene expression profiling of each cell population showed large gene expression changes at 10W, while only modest changes were seen at P7 (Figure S6E). Neuronal populations, including L5-L6, and astrocytes were among the most affected cell types; and in line with histology and bulk RNA-seq results, microglia cells showed significant enrichment of disease-associated microglia signature (Figure S7D). Astrocytes, in turn, also showed a reactive phenotype, with increased proportion of GFAP+ cells, significantly higher GFAP expression, and significant enrichment of reactive astrocyte signature genes (Figure S7E) 35. Importantly, G34R neurons showed significant increase in the expression of hypomethylated immune genes, and concurrent decreased expression of hypermethylated neuronal genes relative to WT neurons (Figure 6C, S7B–C). Consistent with mCH being specific to post-mitotic neurons, the upregulation of immune genes was more pronounced in 10W G34R neurons compared to P7, and only weakly observed in non-neuronal cell types such as microglia and astrocytes (Figure S7A).
In mammals, the fetal brain generates a large excess of neurons which is then subsequently pruned after birth 36. Remodeling of synaptic processes and removal of supernumerary neurons is mediated in part by the innate immune system; specifically, components of the complement cascade (e.g. C1qa/b/c, C3, C4) are expressed in newborn neurons to recruit resident microglia and reactive astrocytes 37,38. In mice, this complement-mediated synaptic pruning is initiated shortly after birth and concludes during adolescence 38; notably, Dnmt3a induction follows shortly after this developmental window and its kinetic mirrors the silencing of complement genes. We therefore hypothesized that G34R may impair post-natal H3K36me2/DNMT3A-mediated epigenetic reprogramming, consequently leading to failure in neuronal maturation and inappropriate activation of complement genes. To confirm the expression of complement in G34R neurons, we performed co-staining of complement C1Q and C3, with the neuronal marker NeuN and the postsynaptic density protein PSD95 in adult (10W) WT and G34R brain (Figure 6D). We observed substantial extracellular accumulation of C1Q in the substantia nigra of G34R compared to WT (p < 0.05). More importantly, a subset of G34R neurons showed intracellular staining of C1Q and C3, specifically accumulating as punctate foci in synaptic junctions and dendrites, consistent with the notion that G34R neurons aberrantly express excess complement (Figure 6D, S7F). Conversely, we observed marked depletion of neurofilament protein NF200 in the adult G34R cortex compared to WT controls, in keeping with loss of neuronal processes (Figure S7G). Colocalization of PSD95 and Iba1 further showed loss of PSD95+ neuronal synapses, some of which may be explained by Iba1+ microglia engulfment (Figure 6E, S7H). Collectively, our data suggest that the G34R mutation leads to loss of CH methylation through impaired H3K36me2/DNMT3A deposition at immune genes. This may promote sustained complement expression and further recruitment of reactive microglia and astrocytes in the G34R cortex, which may contribute to the neurological deficits we observe in these mutant mice.
Neuronal expression of H3.3G34R leads to cortical atrophy and neuroinflammation
To study the cell-autonomous effect of G34R mutation on neurons, we generated a conditional H3.3G34R knock-in transgenic mouse model, also designed in the endogenous H3f3a locus (Figure 7A). We targeted intron 1 of H3f3a and knocked-in a cassette containing a splice-acceptor (SA), coding sequence for EGFP, and three poly-A sequences flanked by loxP sites, followed with H3.3G34R cDNA. Upon Cre-mediated recombination, the SA-GFP-polyA sequences are excised resulting in loss of GFP signal and expression of H3.3G34R (Figure 7A).
Figure 7. Neuronal-specific induction of G34R results in neurodegeneration and cortical atrophy.
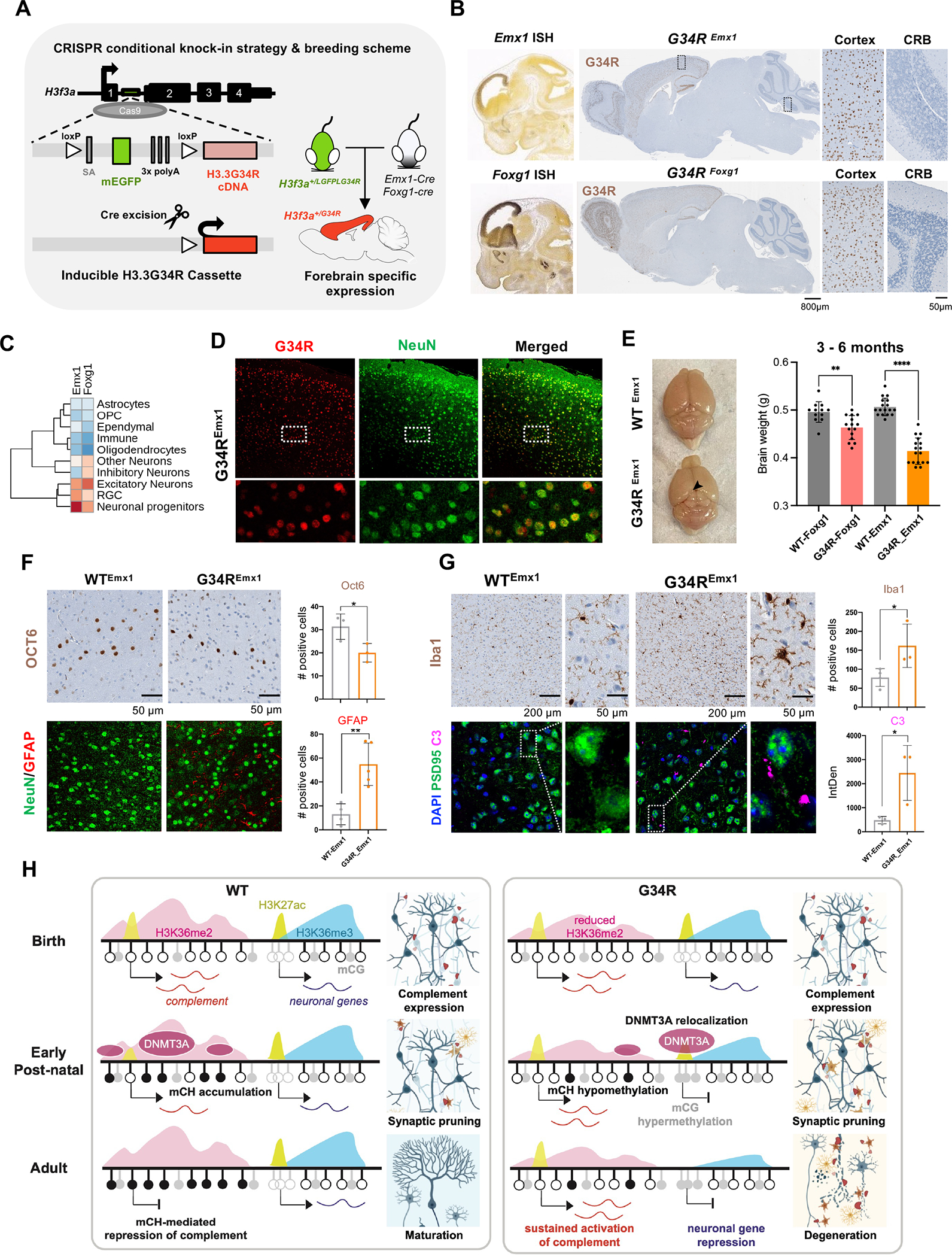
A. Schematic representation of the CRISPR G34R-conditional knock-in strategy. An inducible transgene cassette targeting exon 1 of the H3f3a gene was inserted using Cas9:gRNA approach. Loxp sites flanked an adenovirus splice-acceptor (SA), mEGFP, and 3x polyA sequences, followed by the H3.3G34R cDNA. Carrier H3f3a+/LGFPLG34R mice were crossed with Emx1-Cre or Foxg1-Cre mice to generate heterozygous H3f3a+/G34R mice with cortical-specific expression of G34R. B. In situ hybridization of Emx1 and Foxg1 genes (Allen Brain Atlas). H3.3G34R IHC staining for representative sagittal section of adult mouse brain from G34REmx1 and G34RFoxg1. CRB: cerebellum. C. Expression levels of Emx1 and Foxg1 in murine developing brain atlas. D. Immunofluorescent co-staining of neuronal marker NeuN (green) and G34R (red) in G34REmx1 adult mouse brain (20x). E. G34REmx1 shows cortex atrophy. Representative image and brain weights (means ± SE) of WTEmx1 and the reduced cortical region (arrow) of G34REmx1 mice. Student’s t-test. **P < 0.01, **** P < 0.0001. F. Immunohistochemical staining of layer V neuronal marker (Oct6) (top), neurons (NeuN), and astrocytes (GFAP) (bottom) in WTEmx1 and G34REmx1 (20x). Related quantifications, Oct6 (n = 3) and GFAP (n ≥ 4), means ± SD. Student’s t-test. *P < 0.05, **P < 0.01. G. Immunohistochemical staining of microglial marker (Iba1), neuronal postsynaptic protein PSD95, and complement C3 in adult WTEmx1 and G34REmx1 mice (63x). Related quantifications, Iba1 (n ≥ 3) and C3 (n = 3), means ± SD. Student’s t-test. *P < 0.05. H. Model to illustrate dysregulation of epigenetic reprogramming in the postnatal G34R brain, which may account for neuroinflammation and progressive neurodegeneration.
We then used two well-established Cre drivers, Emx1-Cre 39 and Foxg1-Cre 40, to induce G34R expression in forebrain neurons. Emx1 is mainly expressed in excitatory neuron progenitors and astrocytes 39 in the dorsal neuroepithelium of the fetal brain, whereas Foxg1 is expressed in both excitatory and inhibitory neuron progenitors derived from the dorsal and ventral forebrain, respectively (Figure 7B–C). Immunostaining with the G34R-specific antibody validated cortical expression of G34R in both Cre drivers (Figure 7B). G34R expression pattern was mostly restricted to neurons, as shown by its colocalization with NeuN+ neurons in the cortex of G34REmx1 (Figure 7D). Notably, the induction of H3.3G34R with Foxg1- or Emx1-Cre led to cortical atrophy in adult animals, being more severe in the Emx1-Cre (Figure 7E). Like the DKI model, G34REmx1 mice showed significant depletion of layer V neurons (Oct6+), concurrent with infiltration of reactive astrocytes (GFAP+) and microglia (Iba1+) in the deep cortical layers, when compared to WTEmx1 mice (Figure 7F–G). Finally, to validate the activation of complement in G34R neurons, we co-stained for C3 and PSD95 in adult WTEmx1 and G34REmx1 brains. G34R neurons showed significant accumulation of complement C3 compared to WT (Figure 7G). Taken together, our results indicate that specific expression of H3.3 G34R in neurons leads to complement activation and neuroinflammation, which possibly contribute to the progressive neurodegeneration observed in these animals (Figure 7H).
DISCUSSION
While the pathogenic mechanism of histone mutations has been intensely investigated in the context of cancer, the impact of histone mutations in normal development remains poorly understood. Here, we report patients with germline H3.3G34R and H3.3G34V mutations presenting with severe global developmental delay and worsening neurological symptoms. Our mutant H3.3G34 murine models demonstrate that H3.3G34R, V, W mutations fundamentally impact development of neural and mesenchymal lineages with surprising specificity. G34R and G34W mice demonstrated remarkable phenotypic differences, with G34W mainly affecting tissues of mesodermal origin, and G34R mainly affecting tissues of neuroectodermal origin. This divergence is likely explained by the distinct biochemical properties of these amino acid substitutions on the H3.3 tail, as alluded to by other studies 13,41. Indeed, we show G34 mutations are non-equivalent in their perturbation of cis H3K36 and K27 methylation on the mutant nucleosomes, as G34R uniquely showed severe depletion of both H3K36me2/3 and no gain of H3K27me3, in contrast to G34W. This, in turn, has significant consequences with respect to the differential recruitment of chromatin modifiers in the developing brain.
The neurological phenotypes of G34R mice manifested during adolescence and early adulthood, and progressively worsened with age. The progressive neurodegeneration in G34R mice may be explained by the perturbation of DNMT3A-mediated epigenetic reprogramming during post-natal neuron maturation (Figure 7H). G34R animals show increased expression of DNA hypomethylated immune genes and decreased expression of hypermethylated neuronal genes. At birth, immature neurons induce expression of the classical complement cascade to recruit microglia and reactive astrocytes for synaptic pruning and removal of supernumerary neurons 37,38. We propose a model where these innate immune genes are bookmarked by H3K36me2 for eventual silencing in the normal adult brain. DNMT3A is then transiently expressed in the post-natal brain and is recruited to H3K36me2-enriched regions of the genome to deposit CH methylation, which establishes long-term silencing to conclude synaptic pruning. In the presence of G34R, the pervasive H3K36me2 becomes concentrated focally at genic sites, and is selectively depleted from gene-poor regions and immune gene clusters. Consequently, DNMT3A is redistributed from intergenic and immune gene clusters, and instead localizes to unmethylated CpG islands in G34R mutants. This redistribution leads to aberrant gain of CG methylation at neuronal promoters and loss of CH methylation at intergenic and immune genes. Ultimately, this epigenetic dysregulation leads to transcriptional suppression of canonical neuronal genes, inappropriate activation of complements in G34R adult brain, and concomitant microglial/astrocyte recruitment and neuroinflammation.
Phenotypically, the G34R mice resemble Dnmt3a-depleted mice with regards to hindlimb clasping, abnormal gait, and general hypoactivity 42. Double knockout of Dnmt1 and Dnmt3a in the forebrain post-natal neurons also showed similar upregulation of innate immunity genes, including complement factors C3 and C4 43; whereas protracted Dnmt3a expression in the adult brain leads to repression of neuronal genes, hyperactivity, and seizures 44. Notably, germline DNMT3A mutations in human are associated with divergent Mendelian developmental disorders – Tatton-Brown-Rahman (TBR) and Heyn-Sproul-Jackson (HSJ) Syndromes 45,46. Mutations in the PWWP domain of DNMT3A abolishing H3K36me2/me3 recognition is associated with microcephalic dwarfism in HSJS, in contrast to loss-of-function alleles in TBRS which are associated with overgrowth and macrocephaly. G34R mutation may therefore functionally resemble DNMT3A PWWP mutations, where DNMT3A is redistributed away from H3K36me2-enriched sites, and instead localizes to PRC2/1-regulated developmental promoters 45,47. Whether similar dysregulation of complements or other innate immunity genes underlie these syndromes and other H3.3 neurodevelopmental syndromes warrants further investigations.
In sum, our murine model showcases remarkable developmental differences in H3.3G34R, V, and W mutants, reminiscent of the tissue specificity observed in H3.3G34-mutant diseases. We elucidate G34R-mediated epigenetic dysregulations in the developing brain, potentially underlying the neurological deficits that is unique to this mutation. Finally, our data implicates a novel function of DNMT3A in repression of complement genes following synaptic pruning and has significant therapeutic implications in future investigations of neurodevelopmental diseases.
Limitations of the Study
Our data suggest that the progressive neurodegeneration observed in G34R animals is likely due to decreased H3K36me2 deposition, which in turn disrupts the recruitment of the de novo DNA methyltransferase DNMT3A and the deposition of DNA methylation, including CH methylation, in post-mitotic maturing neurons. However, other mechanisms may also contribute to the epigenetic deregulation promoted by G34R and the phenotype we observe. This may include impaired DNMT3B recruitment following H3K36me3 loss (Figure 4A, 4C), as well as the redistribution of other major epigenetic marks following impaired H3K36 methylation, including H3K27me3 as we see here and as previously suggested by other studies 13,16. Furthermore, whether the disease phenotype is directly caused by persistence of immune gene activation (C3 and C1q) in neurons as we hypothesize, and whether this can be extended to other human neurodegenerative disorders require further studies. These are warranted to demonstrate the causal role of complements and microglia/astrocyte in the neurodegenerative phenotype of G34R mutants. Finally, while we investigated the impact of G34R mostly in brain, our understanding of H3.3G34R/V/W impact in other tissues and especially how G34W mutations result in the mesenchymal phenotypes we observed require additional studies.
STAR*METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to the lead contact, Nada Jabado (nada.jabado@mcgill.ca).
Materials availability
New materials generated in this study, including transgenic mouse models, will be shared by the lead contact upon request.
Data and code availability
All data reported in this paper will be shared by the lead contact upon request.
All sequencing data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
Microscopy data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Patient samples and clinical information
Protocols for this study involving collection of patient samples and information were approved by the Research Ethics and Review Board of McGill University and affiliated Hospitals Research Institutes. Informed consent was obtained from all research participants. Clinical report of germline H3.3G34R and H3.3G34V patients are presented in Table S1.
Animal models
C57BL/6J and B6C3F1/Crl mice were purchased from the Jackson Laboratory and Charles River respectively. Both sexes of mice were used in the experiments. All mice were housed, bred, and subjected to the listed procedures according to the McGill University Health Center Animal Care Committee and in compliance with the Canadian Council on Animal Care guidelines. All procedures and experiments were performed according to the guidelines of the Canadian Council of Animal Care and approved by the Animal Care Committee of the RI-MUHC. The mice were monitored weekly and euthanized at clinical endpoints when recommended by the veterinary and biological services staff members.
Cell lines
Fetal neural progenitor cells (NPC) were isolated from E12.5 forebrain of DKI embryos. Embryos were micro dissected under the loupe microscope in ice-cold HBSS with antibiotics (10% penicillin/streptomycin). Tail snips were taken for genotyping. Meninges was removed from the forebrain/midbrain/hindbrain to isolate the forebrain. Forebrain samples were collected in a 24 well plate with ice-cold HBSS and antibiotics. To derive single cell suspension, forebrain tissues were cut into small pieces and were dissociated mechanically by using pipetting. Next, the single cell suspension was filtered using the 40 μm cell strainer and centrifuged. Cells were then resuspended in mouse NPC medium (STEMCELL Technologies) and plated in 6 well plates (Primaria, Corning). Cells were incubated for 4 days without any disturbance until emergence of neurospheres. After 6–7 days neurospheres measure between 150–200 μm in diameter were carefully removed, and mechanically resuspended into single cells by pipetting. The single cell suspension is then plated onto plates coated with hESC-qualified Matrigel (Corning) in mouse and rat Neurocult™ basal medium (STEMCELL) containing GlutaMax (2mM), SM1 without vitamin A (1x), N2 supplement (1x), mouse EGF (10ng/ml), Human bFGF (10ng/ml), heparin (2 μg/ml), and penicillin/stereptomycin (1x). Media was changed every 2 days and the cells were passaged when they reached 80% of confluency using TrypLE™ express (Thermo Fisher).
Mouse C3H10T1/2 cells expressing FLAG-tagged H3.3 WT, G34R and G34W were maintained in DMEM (Wisent Inc.) supplemented with 10% FBS (Wisent Inc.). Isogenic U2OS FlpIn-T-Rex cells (ThermoFischer Scientific) expressing WT or mutant H3.3-FLAG-BIoID2, GFP-BioID2, or NLS-GFP-BioID2 were maintained in DMEM (Wisent Inc.) supplemented with 10% FBS (Wisent Inc.), 1x penicillin/streptomycin (Wisent Inc.), 75 μg/ml zeocin (Invitrogen), 5 μg/ml blasticidin (Invivogen), and 100 μg/ml hygromycin.
METHOD DETAILS
Generation of direct knock in (DKI) G34 mutant mice
The gRNA as Alt-R crRNA and single-stranded donor templates (ssODNs) (Key Resource Table) along with Alt-R S.p. Cas9 Nuclease (catalog # 1081058) were purchased from IDT. Cas9: gRNA was simultaneously injected with ssODN (50 ng/ul:50 ng/ul :20 ng/ ul) into mouse 2-cell or 4-cell stage embryos from B6C3F1 females that were mated with male B6C3F1 mice. The embryos were transferred into pseudo-pregnant CD-1 females for gestation. The born pups were called as mosaic founder animals. To establish mouse lines with the different H3.3G34 mutations, we crossed all mosaic founder animals with histone H3.3 wild-type (WT) mice (two strains: C57BL/6J and B6C3F1/Crl) and selected 4 different true heterozygous founders (g1), namely: +/− (indel), +/G34R, +/G34V and +/G34W. To genotype the mice, we used PCR to amplify the region flanking the mutation sites in exon 2 of murine H3f3a genomic DNA followed by Sanger sequencing. The sequences of genomic primers for PCR-Sanger-seq listed in supplemental Key Resource Table.
Generation of inducible-G34 mutant mice
Cas9:gRNA was simultaneously injected with the inducible G34R cassette (50 ng/ul:50 ng/ul :20 ng/ ul) into mouse 2-cell stage embryos collected from hybrid B6C3HF1 mice. The cassette consisted of an adenovirus splice-acceptor (adSA), a mEGFP and three poly-A sequences flanked with loxp sites, followed with the H3.3G34R coding sequence. The embryos were transferred into pseudo-pregnant CD-1 females for gestation. The pups born were screened and F0 founder mice were identified. To establish mouse lines with the inducible H3.3G34 mutation, we crossed positive F0 animals with B6C3F1/Crl mice (histone H3.3 wild-type). For genotyping, we used two sets of primers to amplify the insertion site, one set flanking exon 1 and the GFP sequence and the second set flanking GFP sequence and exon 2 of murine H3f3a genomic DNA. The sequences of genomic primers for PCR are listed in supplemental Key Resource Table. To induce the G34R mutation in the telencephalon specifically during development, G34R Flex heterozygous mice were crossed with homozygous Emx1cre/cre (Jax Mouse Strain 005628) and Foxg1-IRES-Cre (Jax Mouse Strain 029690) animals. Foxg1-IRES-Cre allele was inherited maternally to ensure the highest and most consistent pattern levels of Cre activity 40.
Phenotype–tissue characterization
To further investigate affected tissues in an unbiased manner, a cohort of decapitated perfused adult mice, from both sexes and all genotypes, were characterize by the animal examination center IDEXX BioAnalytics (Shari Hamiliton, DVM, Columbia, USA). This dissection/necropsy evaluation focused on examining the requested tissues (heart, duodenum, cecum, rectal/anal junction, colon (distal), colon (proximal), fat (brown), gall bladder, ileum, jejunum, liver, salivary gland parotid, stomach (glandular & no-glandular), spleen, tissue masses/lesions, diaphragm, femur/stifle, muscle, quadriceps, spinal cord, ovary, uterus, vagina/cervix, bulbourethral gland, epididymis, preputial gland, prostate, urethra, penile, testis, lung, kidneys, urinary bladder) and to report any abnormalities found. Supplemental Table S2 shows detailed diagnosis.
Behavioural tests
Hindlimb clasping
For the Hindlimb clasping test, males and females mice were lifted by their tails and the position of their hind limbs was observed and scored for ten seconds in a blinded manner. Unaffected mice consistently splayed their hindlimbs outward away from the abdomen, and this behaviour would be scored 0. A score of 1 was given to mice that pulled one hindlimb hind legs partially toward their abdomen for more than 5s. When this was observed for both hind limbs, the mouse received a score of 2. A score of 3 was assigned to mice that retracted both hind limbs completely for more than 50% of the observation time 20.
Rotarod test
Briefly, males mice were trained to stay on a steady rod during an adaptation period of 30s. Thereafter, the rod started rotating with an acceleration of 4–40 rounds per minute for 300s. The trial ended when the mice fell off the rod or were able to keep standing for 300s. If a mouse clings on the rod and completes a full passive rotation, this is considered a failure in performance similar to falling. The tests were performed for four consecutive days and on each day three test trials were performed.
Open field test
Briefly, adult male mice (3–6 months) were placed in an empty arena, which was novel for the mice, and allowed them to explore this space for five minutes. The movements of the mice were automatically tracked by video tracking to analyze their behavior. All the behaviors were analyzed using the Any-Maze software (Stoelting Co., IL, U.S.A.)
Gait analysis
We used the CatWalk XT gait analysis system (Noldus, The Netherlands) as previously described145. Briefly, each mouse (8-week-old males) walked on an illuminated glass plate of the CatWalk walkway in an unforced manner. The position of each paw was measured and analysed using the Noldus CatWalk XT 8.1 software system and at least three complete runs for each mouse were recorded. Males mice were used.
Brain perfusion fixation/post-fixation
Paraffin-embedded
Males and females mice (from p7 to 12-month-old) were anesthetized and transcardially perfused with cold PBS followed by cold 4% paraformaldehyde (PFA). The brains were post-fixed with 4% PFA for 24–48 hr. Usually the brains were put directly in 30% sucrose/PBS for 12–36 hours. Other tissues were postfixed in 10% formalin for 48–72hr. Following fixation, the samples were dehydrated, embedded in paraffin wax, and sectioned in slices with a thickness of 5μm.
OCT-embedded
Males and females mice (2- to 6-month-old) were anesthetized and brains were collected and fixed in 4% PFA for 24h. The tissue was transferred to 15% sucrose/PBS. The day after, tissue was then incubated in 30% sucrose/PBs for 2–3 hours and embedded in OCT compound, followed by freezing on dry ice. OCT blocks were then sectioned in 10 um-thick slices by using microtome-cryostat machine and kept at −80.
Immunofluorescence staining of paraffin or OCT-embedded tissue sections
Paraffin-embedded
The sections were deparaffinized twice with xylene for 10min, followed by rehydration with decreasing ethanol concentrations. Heat induced antigen retrieval was performed using a decloaking chamber (Biocare Medical) in antigen retrieval buffer. After cooling, the sections were rinsed in PBS-0.03% Triton. The sections were blocked with 5–10% normal goat serum (NGS) in PBS- x Triton (1 hr, room temperature) followed by an overnight incubation at 4°C with diluted primary antibodies (Calbindin 1:200 C9848, GFAP 1:200 12389S, NeuN 1:200 MAB377, G34R 1:200 31-1120-00). The following day, the sections were washed 6 times: 3 times with PBS-0.03% triton (each 5min), 3 times with PBS- x triton (each 5min) on the shaker and the sections were subsequently incubated with AlexaFluor conjugated secondary antibodies for 90 min at room temperature, followed by a final washing 2X in PBS-0.03% Triton 2min on the shaker. The sections were mounted with ProLong™ Gold Antifade (ThermoFisher). The sections were incubated 1 hr at room temperature and stored overnight in 4°C and the results were visualized with a Zeiss LSM780 Laser Scanning Confocal Microscope and IR lasers + OPO (LSM780 IR Laser + OPO).
OCT-embedded
Cryostat sections were placed at room temperature (RT) to thaw for 20 minutes. Sections were then washed three times with TBS for 2 minutes. Sections were permeabilized with PBS containing 0.1% Triton X-100 for 30 minutes, followed by three times washing with 1X TBS for 2 minutes. Sections were blocked in 10% normal goat serum (NGS) in PBS for 1 hr at RT, and then incubated overnight at 4°C with primary antibodies (C3 1:500 NB200-540, C1q 1:500 Ab182451, PSD95 1:500 Ab18258, NeuN 1:200 MAB377). After overnight incubation, sections were washed three times with 1X TBS for 2 minutes and incubated with goat anti-rabbit secondary antibody for 90 minutes at RT. It was followed by three times wash with 1X TBS for 2 minutes and mounted with ProLong™ Gold Antifade Mountant. Sections were incubated for 30 minutes at RT and overnight at 4°C.
Immunohistochemistry (IHC)
Automated IHC on the tissue slides was performed with a Ventana Discovery Ultra. The slides were deparaffinized and rehydrated. Antigen retrieval was done using an EDTA buffer and slides were incubated with primary antibodies (G34R 1:100 RevMab 31–1120, G34V 1:500 RevMab 31–1193, G34W 1:100 RevMab 31–1120, Iba1 1:4000 Ab178846, Oct6 1:1000 MABN738, NF200 1:3500 N4142) After washing, a secondary antibody was added (anti-Mouse or Rabbit/Mse HRP). The DAB kit chromogen was used to detect the signal. IHC was performed at RI-MUHC Histology Platform and the Segal Cancer Centre Research Pathology Facility, Jewish General Hospital.
H&E staining procedure (Autostainer, Leica)
Tissue slides were deparaffinized and rehydrated with graded ethanol to distilled water. The slides were stained with Hematoxylin, defined and incubated in a blue buffer solution, followed by staining with Eosin. The slides again were dehydrated through graded alcohols, cleared in Xylene and mounted.
Cresyl Violet staining (Nissl Staining) procedure
The slides were deparaffinized and rehydrated through graded ethanol to distilled water. Slides were stained with 0.1% cresyl violet solution (3–10 min). Rinsed in distilled water. Slides were dehydrated through graded alcohols, cleared in Xylene and mounted.
Image quantitative analysis
Images were analyzed using ImageJ software. The positively stained cells for each marker were counted using automated method after adjusting the threshold, particle size and circularity. Fluorescence intensity was calculated as the integrated density of the ROI (Region of Interest) minus the background.
Nissl scoring of cortical layers
The neocortical layers were defined based on cellular morphologies and sizes along with cell compaction within each layer. The most superficial layer of the neocortex is layer I which is a cell sparce layer populated by the Cajal-Retzius (CR) neurons. We marked the layer I boundary to encompass these sparsely spaced neurons. In the murine neocortex, layers II and III are fused and comprise of stellate- and pyramidal- neurons respectively. Layer IV comprises of various pyramidal and spiny stellate neurons. All the neurons found in layers IV and II/III were grouped together. To clearly demarcate the end of layer IV and an onset of the cells harbouring layer V, we subsequently binned the large pyramidal neurons of layer V. Finally, layer VI neurons were segregated from those of layer V since layer VI comprises of many small multiform and spindle-like neurons with very few large pyramidal neurons making the distinction between the two layers easy. We used Adobe Illustrator to bin the different layers of the neocortices. The binned images were saved as jpeg and the area per bin was scored using ImageJ. Statistics were generated using PRISM for the area under the respective bin.
Immunoprecipitations and western blot analysis
Mouse MSC C3H10T1/2 cells were transduced with recombinant lentiviruses produced using pCDH-EF1a-MCS-Puro expression vector containing histone transgene, as described previously.13,59 Transduced cells were selected with 1 μg/mL of puromycin for 4 days. The cells (15 × 106) were collected, and cell pellet washed with PBS and lysed with 1 ml NUCLEI EZ PREP NUCLEI (Sigma # NUC-101) and rotated for 30 min at 4°C. Nuclei were collected by centrifugation at 500 × g for five minutes at 4°C. Nuclei were resuspended in 500 ul lysis buffer (50mM Tris-HCL pH 7.6, 100 mM KCL, 2 mM EDTA, 0.1% NP40, 10% Glycerol) and sonicated 3 × 10S on, 30S off, followed by MNase digestion at 37°C in water bath for 7min. Centrifuged at 12000g for 5 min at 4°C. The cell nuclei protein was incubated with 4ul DNMT3A antibody /1 mg protein lysate on rotator overnight at 4°C. Then, washed protein A beads were added to protein-antibody and incubated on rotator for three hours at 4°C. Final IP products were harvested after 3 times wash with 500 ul Lysis buffer supplemented with protease inhibitor cocktail (Roche) and eluted by adding 45 ul 1X laemmli buffer, supplemented with 5% beta-mercaptoethanol, boiling 95°C for 15 min. Western blot analysis was carried out according to standard techniques using anti-FLAG, anti-DNMT3A, and anti-total H3.
QUANTIFICATION AND STATISTICAL ANALYSIS
Histone post-translational modification quantification with nLC/MS
Histone extraction
Histone extracts were prepared for MS analysis as previously described 49. Briefly, acid-extracted histones were treated with propionic anhydride for 15 mins at 37C to derivatize free and mono-methylated lysine residues, after which the samples were dried in a speed-vac and then resuspended in 0.1 M ammonium bicarbonate for a second round of derivatization. Derivatized histones were digested overnight with trypsin at room temperature. At this stage, 2500 fmol of isotope-labeled synthetic peptides modeling the G34V, G34W, and G34R mutant sequences (G34V: KSAPSTGVV*KKPHR; G34W: KSAPSTGWV*KKPHR; G34R: KSAP*STGR + VKKP*HR, * denotes heavy amino acid) were spiked into the respective digested histones (~10 μg) to serve as qualitative standards. Histone peptides were derivatized twice more with propionic anhydride to modify free N-termini and then desalted with C18 stage tips.
Histone LC-MS/MS
Histone peptides were analyzed by LC-MS/MS using data-independent acquisition (DIA), as previously described 49. Parallel reaction monitoring (PRM) was additionally used to analyze WT and mutant peptides containing Lys27 and Lys36 from replication-dependent histone H3 variants (H3.1/H3.2) and the replication-independent histone H3 variant (H3.3). Peptides were resolved on 15–20 cm × 75 μm i.d. columns prepared in-house with C18 resin (2.4 μm) with a chromatography gradient from 2 to 50% solvent B over 45 mins and then from 50 to 95% solvent B over 5 mins using an EASY-nLC 1000 system (Thermo). Water with 0.1% formic acid served as solvent A while 80% acetonitrile with 0.1% formic acid served as solvent B. For each monitored peptide, the highest intensity charge state from 2+ to 4+ was determined and used to construct inclusion lists. WT peptides were included on all inclusion lists, whereas mutant peptides were only included as relevant to the sample genotype. For PRM with a Q-Exactive (Thermo), the MS settings were as follows: positive polarity, 35,000 resolution, 1e6 AGC, 110 ms max injection time, 10 loop count, 2.0 m/z isolation width, 30 NCE, centroid mode. Additionally, a full scan was collected prior to every PRM loop with the settings: positive polarity, 70,000 resolution, 1e6 AGC, 100 ms max injection time, 200–1000 m/z scan range, profile mode. DIA data was analyzed by EpiProfile 50 while PRM data was analyzed by Skyline and R using MS1 peak area.
BioID
Generation of U2-OS G34R/V/W isogenic lines
Mouse H3.3 cDNA was introduced in the Gateway donor plasmid, pDONR223 (ThermoFischer Scientific) for site-directed mutagenesis according to QuickChange protocol (Agilent) to generate G34R, G34V, and G34W constructs. An LR reaction was finally performed to introduce the constructs into pCDNA5-BioID2-FLAG and pCDNA5-FLAG-BioID2 destination plasmids (generously provided by Dr. Anne-Claude Gingras, Lunenfeld-Tanenbaum Research Institute) to express N- and C- terminally tagged mH3.3, respectively. All plasmids were confirmed by Sanger sequencing. U2OS FlpIn-T-Rex cells were then transfected with the constructs using Lipofectamine 3000 (ThermoFisher Scientific) and selected with 100 μg/ml hygromycin to generate stable cell lines.
Protein extraction, streptavidin pull-down, and trypsin digestion
BioID samples were then prepared as previously described 51. Cells were plated in duplicates and induced for 24 hours with 1 μg/ml doxycycline and pulsed with 50 μM biotin (final concentration) during the last 8 hours of induction. Cells were then washed with cold PBS, scrapped, and spun down at 500 × g at 4°C for 5 minutes. Frozen cell pellets were resuspended in ice-cold modified RIPA lysis buffer at a 1:4 pellet weight:volume ratio. Cells were then sonicated and treated with 250 U of TurboNuclease and 10 μg of RNase and rotate (end-over-end). Insoluble fraction was then pelted by centrifuge to yield supernatant containing bait proteins. To pull-down biotin-labelled proteins, the supernatant was incubated with streptavidin agarose beads for 3 hours. Beads were then washed once in RIPA buffer and transferred to a new tube, followed washes (1x SDS-Wash, 2x RIPA-wash, 1x TNNE buffer, 3x ABC buffer containing 50 mM ammonium bicarbonate. Finally, the bead slurry in ABC buffer is incubated with 1 μg trypsin and incubated at 37°C overnight with agitation. Beads were then centrifuged beads (400x g, 2 min) and supernatant containing peptides were eluted with mass spectrometry grade H20. Peptide supernatant were then lyophilize using vacuum centrifugation without heat, and stored at −80°C until ready for mass spectrometry analysis.
LC-MS/MS
For data-dependent acquisition (DDA) LC-MS/MS, affinity purified and digested peptides were analyzed using a nano-HPLC (High-performance liquid chromatography) coupled to MS. For both anti-FLAG AP-MS and BioID, one-quarter of the sample was used. Nano-spray emitters were generated from fused silica capillary tubing, with 100μm internal diameter, 365μm outer diameter and 5–8μm tip opening, using a laser puller (Sutter Instrument Co., model P-2000, with parameters set as heat: 280, FIL = 0, VEL = 18, DEL = 2000). Nano-spray emitters were packed with C18 reversed-phase material (Reprosil-Pur 120 C18-AQ, 3μm) resuspended in methanol using a pressure injection cell. Sample in 5% formic acid was directly loaded at 800nl/min for 20min onto a 100μmx15cm nano-spray emitter. Peptides were eluted from the column with an acetonitrile gradient generated by an Eksigent ekspert™ nanoLC 425, and analyzed on a TripleTOF™ 6600 instrument (AB SCIEX, Concord, Ontario, Canada). The gradient was delivered at 400nl/min from 2% acetonitrile with 0.1% formic acid to 35% acetonitrile with 0.1% formic acid using a linear gradient of 90 min. This was followed by a 15 min wash with 80% acetonitrile with 0.1% formic acid, and equilibration for another 15min to 2% acetonitrile with 0.1% formic acid. The total DDA protocol is 120min. The first DDA scan had an accumulation time of 250ms within a mass range of 400–1800Da. This was followed by 10 MS/MS scans of the top 10 peptides identified in the first DDA scan, with accumulation time of 100 ms for each MS/MS scan. Each candidate ion was required to have a charge state from 2–5 and a minimum threshold of 300 counts per second, isolated using a window of 50mDa. Previously analyzed candidate ions were dynamically excluded for 7 seconds.
Mass spectrometry data analysis
For data-dependent acquisition (DDA) LC-MS/MS, affinity purified, and digested peptides were analyzed using a nano-HPLC (High-performance liquid chromatography) coupled to MS. For both anti-FLAG AP-MS and BioID, one-quarter of the sample was used. Nano-spray emitters were generated from fused silica capillary tubing, with 100μm internal diameter, 365μm outer diameter and 5–8μm tip opening, using a laser puller (Sutter Instrument Co., model P-2000, with parameters set as heat: 280, FIL = 0, VEL = 18, DEL = 2000). Nano-spray emitters were packed with C18 reversed-phase material (Reprosil-Pur 120 C18-AQ, 3μm) resuspended in methanol using a pressure injection cell. Sample in 5% formic acid was directly loaded at 800nl/min for 20min onto a 100μmx15cm nano-spray emitter. Peptides were eluted from the column with an acetonitrile gradient generated by an Eksigent ekspert™ nanoLC 425 and analyzed on a TripleTOF™ 6600 instrument (AB SCIEX, Concord, Ontario, Canada). The gradient was delivered at 400nl/min from 2% acetonitrile with 0.1% formic acid to 35% acetonitrile with 0.1% formic acid using a linear gradient of 90 min. This was followed by a 15 min wash with 80% acetonitrile with 0.1% formic acid, and equilibration for another 15min to 2% acetonitrile with 0.1% formic acid. The total DDA protocol is 120min. The first DDA scan had an accumulation time of 250ms within a mass range of 400–1800Da. This was followed by 10 MS/MS scans of the top 10 peptides identified in the first DDA scan, with accumulation time of 100 ms for each MS/MS scan. Each candidate ion was required to have a charge state from 2–5 and a minimum threshold of 300 counts per second, isolated using a window of 50mDa. Previously analyzed candidate ions were dynamically excluded for 7 seconds.
Mass spectrometry data generated were stored, searched, and analyzed using ProHits laboratory information management system (LIMS) platform. Within ProHits, WIFF files were converted to an MGF format using the WIFF2MGF converter and to an mzML format using ProteoWizard (V3.0.10702) and the AB SCIEX MS Data Converter (V1.3 beta). The data was then searched using Mascot (V2.3.02) and Comet (V2016.01 rev.2). The spectra were searched with the human and adenovirus sequences in the RefSeq database (version 57, January 30th, 2013) acquired from NCBI, supplemented with “common contaminants” from the Max Planck Institute (http://maxquant.org/contaminants.zip) and the Global Proteome Machine (GPM; ftp://ftp.thegpm.org/fasta/cRAP/crap.fasta), forward and reverse sequences (labeled “gi|9999” or “DECOY”), sequence tags (BirA, GST26, mCherry and GFP) and streptavidin, for a total of 72,481 entries. Database parameters were set to search for tryptic cleavages, allowing up to 2 missed cleavages sites per peptide with a mass tolerance of 35ppm for precursors with charges of 2+ to 4+ and a tolerance of 0.15amu for fragment ions. Variable modifications were selected for deamidated asparagine and glutamine and oxidized methionine. Results from each search engine were analyzed through TPP (the Trans-Proteomic Pipeline, v.4.7 POLAR VORTEX rev 1) via the iProphet pipeline.
Preparation of mouse cerebral cortex tissue
The murine cortical hemispheres (7-day-old and 10-week-old males) were collected, flash frozen in liquid nitrogen, crushed into powder on dry ice and stored at −80°C until used.
RNA-seq
RNA extraction and library preparation
Total RNA was extracted from pulverized brain tissue (P7 males and females; 10W males) using the Aurum Total RNA Mini Kit (Bio-Rad) according to manufacturer’s instructions. Library preparation was performed with ribosomal RNA (rRNA) depletion according to instructions from the manufacturer (Epicentre) to achieve greater coverage of mRNA and other long non-coding transcripts. Paired-end sequencing was performed on the Illumina HiSeq 4000 platform.
Data processing
Raw reads were trimmed using Trimmomatic v0.32 52. Adaptors and other Illumina-specific sequences were removed using palindrome mode. Next, a four-nucleotide sliding window was used to remove the bases once the average quality within the window fell below 30. Finally, reads shorter than 30 base pairs were dropped. Cleaned reads were aligned to the mouse reference genome build mm10 using STAR 53 v2.3.0e with default settings. Multiple control metrics (Supplemental Table S3) were obtained using FASTQC (v0.11.2).
Gene expression quantification
Gene expression levels were estimated by quantifying reads uniquely mapped to exonic regions defined by ensGene annotation set from Ensembl (GRCm38, n = 39,017 genes) using featureCounts 54 (v1.4.4). Normalization (mean-of-ratios), variance stabilized transformation of the data and differential gene expression analysis were performed using DESeq2 keeping batches separate 55 (v1.14.1). Principal component analysis using the top 10000 most variable genes was performed using base R (v3.5.1). Unless otherwise stated, all reported p-values have been adjusted for multiple testing using the Benjamini-Hochberg procedure.
Assessment of cell lineage gene programs from RNA-seq data
To evaluate gene programs in bulk RNA seq samples, a reference panel of 102 gene signatures was assembled using data from 2 published scRNaseq atlases 24,27 spanning a developmental period from embryonic day 10.5 to postnatal day 6 and adult mouse cortex. For the developmental mouse brain dataset, 100-gene signatures derived from the postnatal cortex were used as reported in the original study. For the adult mouse cortex, 50-gene signatures were downloaded from the UCSC cell browser. To evaluate the disease-associated microglia gene program, the top 100 genes were selected by ranking differentially expressed genes between homeostatic microglia to disease-associated microglia as reported in the original study by fold change and adjusted p-value (Benjamini-Hochberg) 26. Gene set enrichment analysis (GSEA) was performed with these signatures as input, applied to expressed genes in 10W transcriptomes (bulk RNA-seq) ranked using the Negative Binomial Wald test statistic from differential expression analysis, using the fgsea package (v1.8.0). With fgsea, leading edge genes were obtained and normalized enrichment scores (NES) were computed by normalizing enrichment to the average enrichment of 10,000 random gene samples. P values were adjusted using the Benjamini-Hochberg procedure; signatures with adjusted p value < 0.05 were considered significantly enriched or depleted.
Chromatin Immunoprecipitation
Crosslinked ChIP-seq
Crosslinked ChIP-seq was performed as described previously 56. Briefly, 30–50mg pulverized brain tissue (P7 males) fixed with 1% formaldehyde (Sigma), followed by reverse crosslinking with glycine. Fixed tissues were then pelleted and stored at −80 °C until use. To isolate nuclei from tissue, pellets were dounced briefly with syringe and needle (25 5/8 gauge) prior to incubation with nuclei lysis buffer (50 mM Tris-HCl pH 8.0, 10 mM EDTA, 1% (w/v) SDS, 50 mM NaF, 1 mM PMSF, 1 mM Phenylarsine Oxide, 5 mM Sodium Orthovanadate) for 30 mins on ice. Sonication of lysed nuclei was performed on a BioRuptor (Diagenode) for 20 cycles of 30s and 300s off. Samples were checked for sonication efficiency using the criteria of 150–500 bp by gel electrophoresis.
ChIP reaction for DNMT3A was performed as follows: anti-DNMT3A (1:250, ab2850), were incubated with 35 ul protein A Dynabeads at 4 °C for 3 h, then chromatin from ~1–4 million cells was added in RIPA buffer, incubated at 4 °C o/n, washed using buffers from Ideal ChIP-seq Kit (one wash with each buffer, corresponding to RIPA, RIPA + 500 mM NaCl, LiCl, TE), eluted from beads by incubating with Elution buffer for 30 min. at room temperature.
Reverse cross linking took place on a heat block at 65°C for 4 h. ChIP samples were then treated with 2μl RNase Cocktail at 65°C for 30 min followed by 2μl Proteinase K at 65°C for 30 min. Samples were purified with QIAGEN MiniElute PCR purification kit. In parallel, input samples (chromatin from 50,000 cells) were reverse crosslinked, and DNA isolated following the same protocol.
Native ChIP-seq
For histone post-translational modifications, native ChIP were performed as described previously 57 with minor adjustments. Briefly, 30–50mg pulverized brain tissue (P7 and 10W males) was dissociated passing through a syringe (25 5/8 gauge) 20x in douncing buffer (10mM Tris Cl pH7.5, 4mM MgCl2, 1mM CaCl2, and protease inhibitor cocktail (Roche)). Disassociated cells were then digested with 30U of micrococcal nuclease (Worthington) at 37°C for 7 mins, yielding predominantly mono- and di-nucleosomes. Cells were then lysed in ice-cold hypotonic lysis buffer (0.2 mM EDTA, 0.1 mM benzamidine, 0.1 mM PMSF, 1.5 mM DTT, and protease inhibitor cocktail (Roche)) to yield chromatin. Chromatin was then incubated with pre-conjugated antibody:bead complex containing H3K27me3 (CST 9733, 4 μL / IP), H3K27ac (CST 8173, 4 μL / IP), H3K36me3 (Active Motif 61022, 4 μL / IP), or H3K36me2 (Active Motif 39255, 4 μL / IP) complexed to Protein A Dynabeads (Invitrogen, 20 μL / IP) or Anti-Mouse Dynabeads (Invitrogen, 40 μL / IP) rotating at 4°C overnight. Beads were then washed twice in ChIP Wash Buffer (20 mM Tris-HCl pH 8, 0.1% SDS, 1% Triton X-100, 2 mM EDTA, 150 mM NaCl, and protease inhibitor cocktail) and once more in Final Wash Buffer (20 mM Tris-HCl pH 8, 0.1% SDS, 1% Triton X-100, 2 mM EDTA, 500 mM NaCl, and protease inhibitor cocktail). DNA was then eluted in 100mM NaHCO3 and 1% SDS at 65°C for 2h, and purified using phenol:choloroform extraction followed by ethanol precipitation.
ChIP-seq library construction
Library preparation was carried out using KAPA HTP Illumina library preparation reagents (Roche), following manufacturer’s instructions. Briefly, 50 μl of ChIP DNA sample was incubated with 45 μl end repair mix at 20 °C for 30 min followed by Ampure XP bead purification. A tailing: bead bound sample was incubated with 50 μl buffer enzyme mix at 30 °C for 30 min, followed by PEG/NaCl purification. Adaptor ligation: bead-bound sample was incubated with 45 μl buffer enzyme mix and 5 μl of TruSeq DNA adapters (Illumina), for 20 °C 15 min, followed by Ampure XP bead purification. Library enrichment: 12 cycles of PCR amplification. Size selection was performed after PCR using gel excision to collect 200–700 bp fragments. ChIP libraries were sequenced on Illumina NovaSeq 6000 platforms using 50 bp paired-reads.
ChIP-seq data processing
ChIP-seq datasets were processed using the chipseq module of GenPipes v.3.1.2 58. Briefly, raw reads were trimmed using Trimmomatic v0.32 52 to remove adaptor and sequencing-primer associated reads, then aligned to mm10 using bwa-mem v0.7.12 59 with default parameters. PCR duplicate reads as defined by reads with identical mapping coordinates were then collapsed by Picard v2.0.1 to produce uniquely aligned reads. Uniquely aligned reads with mapping quality of 5 or greater were then used to quantify enrichment (q ≥ 5). Visualization tracks were generated using Homer v4.9.1 60 and visualized with IGV Genome Browser 61. Peak calling for DNMT3A was performed using MACS2 62 using broad peak calling strategies. Reads per kilobase mapped (RPKM) was calculated using VisR v0.9.4 63 at genomic tiled 5kb bins or annotated promoters defined as 5kb centred on transcription start site of RefSeq-annotated genes. Differential enrichment between the mutant and WT control were then calculated as Z-scores, with .
Whole Genome Bisulfite Sequencing
WGBS library construction
Whole-genome sequencing libraries were generated from 700 to 1000 ng of genomic DNA derived from brain tissue (10W males) spiked with 0.1% (w/w) unmethylated λ DNA (Promega) previously fragmented to 300–400 bp peak sizes using the Covaris focused-ultrasonicator E210. Fragment size was controlled on a Bioanalyzer DNA 1000 Chip (Agilent) and the KAPA High Throughput Library Preparation Kit (KAPA Biosystems) was applied. End repair of the generated dsDNA with 3′ or 5′ overhangs, adenylation of 3′ ends, adaptor ligation, and clean-up steps were carried out as per KAPA Biosystems’ recommendations. The cleaned-up ligation product was then analyzed on a Bioanalyzer High Sensitivity DNA Chip (Agilent) and quantified by PicoGreen (Life Technologies). Samples were then bisulfite converted using the Epitect Fast DNA Bisulfite Kit (Qiagen) according to the manufacturer’s protocol. Bisulfite-converted DNA was quantified using OliGreen (Life Technologies) and, based on quantity, amplified by 9–12 cycles of PCR using the Kapa Hifi Uracil + DNA polymerase (KAPA Biosystems) according to the manufacturer’s protocol. The amplified libraries were purified using Ampure XP Beads (Beckman Coulter), validated on Bioanalyzer High Sensitivity DNA Chips, and quantified by PicoGreen. Sequencing of the WGBS libraries was performed on the Illumina NovaSeq 6000 system using 150-bp paired-end sequencing.
WGBS data processing
WGBS datasets were processed using the methylseq module of GenPipes v3.1.2 58. Briefly, raw reads were trimmed using Trimmomatic v0.32 52 to remove adaptor and sequencing-primer associated reads, then aligned to bisulfite-converted mm10 genome using Bismark v0.18.11 64 with default parameters. PCR duplicate reads as defined by reads with identical mapping coordinates were then collapsed by Picard v2.0.1 to produce uniquely aligned reads. Methylation status is then called using Bismark v0.18.11 at CpG and CHH genomic contexts to generate visualization tracks. Methylation conversion rate is calculated using spiked λ DNA, which showed > 99.5% conversion on all samples – suggesting methylation call error rate is below 0.5%. Quantitation of mCH and mCG was performed using SeqMonk v1.47.1 (Babraham Bioinformatics) or VisR v0.9.4 63 over tiled genomic 5kb bins, gene promoters (5kb centred on TSS), and gene bodies using RefSeq gene annotations. Differentially methylated CH and CG genomic sites were identified using LIMMA, using p < 0.05 as significance threshold.
Single-nuclei RNA-seq and ATAC-seq (scMultiome) sequencing
Sample handling and library preparation
scMultiome nuclei were prepared using automated nuclei preparation. Pulverized brain samples from P7 and 10W males were subjected to automated nuclei preparation were processed following the Singulator S100 protocol (S2 Genomics). Nuclei were isolated using the Nuclei Isolation kit 1 and Singulator S100 instrument from S2 Genomics. Briefly, 5–30 mg of frozen tissue were put in a pre-cooled nuclei isolation cartridge with RNAse inhibitors. Samples were then processed on the Singulator S100 following the “extended nuclei” protocol. After nuclei preparation, nuclei were centrifuged and washed twice in Diluted Nuclei Buffer (10X Genomics) and counted with the ReadyProbes Cell Viability Blue/Green kit (ThermoFisher Scientific). 16,500 nuclei/sample were loaded on the Chromium Controller (10X Genomics). The 10X libraries (scATAC and sc/snRNA) were then prepared following the manufacturer’s instructions (Next GEM Single Cell Multiome). The 10X libraries were sequenced on the NovaSeq6000 sequencing platform.
Initial processing and quality control of single-nuclei data
Cell Ranger ARC v2.0.0 (10X Genomics) was used (‘count’ option with default parameters) to filter and align raw reads, identify transposase cut sites, detect accessible chromatin peaks, call cells and generate raw count matrices for scMultiome samples. Alignment was performed using the mm10 reference build, coupled with Ensembl transcriptome build GRCh38 v.84. Reads mapping to intronic regions were excluded for the RNA modality.
Quality control and downstream processing was performed using the Signac v1.3.0 65 and Seurat v4.0.0 package 66. QC metrics were computed separately for RNA and ATAC modalities, as described below and jointly used for filtering cells (Supplemental Table S6).
For the RNA modality, cells were filtered on the number of genes, unique molecular identifiers (UMIs), and mitochondrial content (indicative of cell damage). Filtering thresholds were set using a combination of hard cut-offs or based on a distribution of each metric on a per-sample basis (Supplemental Table S6). The minimum number of genes was set to the greater of 400 or two standard deviations below the mean number of genes; the maximum was set to two standard deviations above the mean number of genes. The minimum number of UMIs was set to the greater of 0 or two standard deviations below the mean number of UMIs; the maximum was set to two standard deviations above the mean number of UMIs. The minimum mitochondrial content was set to 0; the maximum was set to the lesser of 5% or two standard deviations below the mean mitochondrial content.
For the ATAC modality, cells were filtered on the number of peaks detected, transcription start site (TSS) enrichment (fold change of reads across a reference set of transcription start sites, relative to regions flanking those sites), and nucleosome signal (expected fragment length periodicity based on a ratio between nucleosome-bound fragments to nucleosome-free fragments). Consistent with the RNA modality, filtering threshold were set using a combination of hard cut-offs or based on a distribution of each metric on a per-sample basis (Supplemental Table S6). The minimum number of peaks detected was set to the greater of 400 or two standard deviations below the mean number of peaks detected; the maximum was set to two standard deviations above the mean number of peaks detected. The minimum TSS enrichment was set to two standard deviations below the mean number of TSS enrichment sites; the maximum was set to two standard deviations above the mean number of TSS enrichment sites. The maximum nucleosome signal was set to 1.5.
Normalization, Dimensionality Reduction and Integration of Samples
For the RNA modality, libraries were scaled to 10,000 UMIs per-cell and log normalized. UMI counts and mitochondrial content was regressed from normalized gene counts and the residuals z-scored gene-wise. Samples were integrated by merging samples without batch correction, followed by scaling to 10,000 UMIs per-cell and log normalization. Variable features were calculated using the variance-stabilizing transform algorithm. Dimensionality reduction was performed using PCA applied to the top 2,000 most variable features. The first 25 principal components were then used as input for projection into two dimensions using uniform manifold approximation and projection (McInnes, 2018), and as input for clustering. Clustering was performed using a shared nearest-neighbor algorithm based on the Louvain algorithm 66 on a k-nearest neighbor graph with k=20 neighbors and resolution 0.2.
Identification of cell types in single nuclei RNA data
Annotation of cell types was performed using an automated, reference-based annotation workflow based on four prediction methods; SciBet 67, SingleCellNet 68, SingleR 69, and Support Vector Machines (SVM). A consensus annotation was assigned when at least two methods agreed. Cell type annotation was performed using two murine brain cell type atlases. Non-neuronal cell-types were annotated using a training reference from embryonic and early postnatal murine brain atlas 24. Next, neuronal cell-types were annotated using the Allen Brain murine cortical and hippocampus atlas 25 as training reference. Only cells with a consensus prediction requiring agreement of at least two methods were used for downstream analysis. For cells annotated as neuronal, only cells which were labeled as neuronal by both reference atlases were considered, using the Allen Brain cell type predictions. Finally, cell labels were aggregated into major cell type classes, with ontology detailed in Supplemental Table S6.
Enrichment of gene lists in single nuclei RNA data
Single-sample gene set enrichment analysis (ssGSEA) was run on a per-cell basis using a gene list for immune response (GO:0006955) filtered on genes which significantly displayed loss of mCH methylation to yield 194 genes, and for synapse (GO:0045202) filtered for genes which significantly displayed gain of mCG methylation to yield 120 genes (Supplemental Table S6).
Differential expression in single nuclei RNA data
Differentially expressed genes (DEGs) between genotypes were computed per cluster of samples integrated by age using the Wilcoxon rank sum test implemented in the FindMarkers function from the Seurat package (logFC.threshold = 0, min.pct = 0.05). Only clusters with at least 20 cells from each genotype were considered. Gene ontology pathway analysis was performed on each cluster’s DEGs with adjusted p-values < 0.05 using the enrichGO function from the clusterProfiler package v3.18.1 70, using all detected genes in the age integration as the universe (OrgDb = org.Mm.eg.db, keyType = “SYMBOL”, ont = “BP”).
Supplementary Material
Supplemental Figure 1. Germline knock-in of H3.3G34R/V/W results in distinct developmental defects, as Related to Figure 1. A. Violin plot showing height, weight, and occipital-frontal circumference (OFC) of human patients with germline H3.3 mutations (n = 47), with severe undergrowth/microcephaly of H3.3G34R (red) and H3.3G34V (blue) highlighted as individual data points. B. Related to Fig. 1B. Negative control immunohistochemistry staining for H3.3G34R/V/W on WT mice. C. Immunoblots of H3.3G34R mutant histone in various somatic tissues from G34R heterozygous mice and WT littermates. D. A representative image of bladder with the urogenital tracts from control and G34W mice. Note the enlarged bladder filled with urine in the G34W. SV: seminal vesicle. E. Kaplan-Meier curves for cumulative survival rates of female G34 mutants and control mice.
Supplemental Figure 2. G34R/V mice show abnormal motor functions and progressive microcephaly, as Related to Figure 2. A. Representative image of G34R mouse exhibiting paresis at 12 months. B. Performance of G34 mutant and control mice (4–7 weeks; n > 9) on rotarod test. The data is presented as means ± SD. The statistical significance assessed using two-way ANOVA. **** P < 0.0001 and n.s.: non-significant compared to control mice. C. CatWalk measurements of stride length of control (n=10) and G34R (n=6) mice at 8 weeks. The data is presented as means ± SE. Student’s t-test. *P < 0.05, **P < 0.01, ***P < 0.001, **** P < 0.0001 and n.s.: non-significant compared to control. D. CatWalk measurements (BOS: base of support). The graphs show the static data from control (n=10) and G34R (n=6) mice at 8 weeks. The data is presented as means ± SD. Student’s t-test. *P < 0.05, **P < 0.01, ***P < 0.001, **** P < 0.0001 and n.s.: non-significant compared to control. E. Brain-to-body weight ratio of G34 mutant and control mice at P21–30 and n > 5 brains per genotype were used. Each mouse is plotted individually and as means ± SD. The statistical significance assessed using Student’s t-test. *P < 0.05, **P < 0.01, ***P < 0.001, **** P < 0.0001 and n.s.: non-significant. F. Representative images of midsagittal brain slices of control and G34R mutant mice stained with Cresyl Violet (Nissl Staining) at 12 months of age. G, H. Representative images of midsagittal brain slices of control and G34 mutant mice stained with Cresyl Violet (Nissl staining) at P7 and P21. Red arrowheads indicate poorly developed lobules or abnormal fissures in the cerebellum of mutant animals. I. Three-dimensional confocal image of Purkinje cells labelled with Calbindin-D28K in adult G34R and WT littermate.
Supplemental Figure 3. G34R leads to abnormal accumulation of microglia and depletion of neurons, as Related to Figure 3. A. Enrichment plot of microglia, astrocyte, and neuronal signatures in 10W cortex of G34-mutant mice. B. Box-whisker plot depicting elevated microglial and astrocytic markers gene expression in 10W G34R cortex compared to G34V/W and WT. C. Quantification of NeuN in WT and G34-mutant 10W cortex (WT n=6, G34R n=4, G34V n=2 and G34W n=3), as related to Fig. 3D. Each mouse is plotted individually and as means ± SD. One-way ANOVA test. *P < 0.05, **P < 0.01, ***P < 0.001, **** P < 0.0001 and n.s.: non-significant compared to control. D. Representative immunohistochemistry and immunofluorescent staining demonstrating lack of change in microglia (Iba1), astrocyte (GFAP), and neurons (NeuN, Oct6) at P7 in G34R and WT cortex.
Supplemental Figure 4. G34R/V/W histones show differential interaction with chromatin modifiers, as Related to Figure 4. Bubble plot depicting BioID interaction frequency of G34-mutant histones with known chromatin modifiers using N-terminal (left) tagging strategy in G34R/V/W and H3.3 WT expressing U2-OS lines (n=2). Also, see Supplemental Table S4 for all significant interactors.
Supplemental Figure 5. G34R re-distribute DNMT3A localization, causing loss of CH methylation at intergenic regions, as Related to Figure 5. A. 2D scatterplot depicting correlation between DNMT3A enrichment (x-axis) and various chromatin modifications (y-axis) in WT P7 cortex. Individual points are genome-wide 5kb bins. B. Parallel coordinate plot depicting concordant epigenetic change in G34R vs. WT cortex, anchoring based on DNMT3A change. 5kb bins were stratified into 6 groups based on DNMT3A change, with red line representing bins that showed DNMT3A-loss in G34R, while blue lines representing bins with DNMT3A gain. G34R shows concordant. loss of H3K36me2, mCH and mCG in DNMT3A-deplete genomic regions. Solid line: median. Shaded area: 25% and 75% quartiles. C. Stacked heatmap centered on DNMT3A-peaks in G34R cortex, related to Fig. 5C. Note DNMT3A-peaks are concurrently marked with H3K36me2 and mCH. D. Genome browser snapshot depicting preferential loss of DNMT3A binding and mCH at interferon immune gene cluster on chr4 in G34R cortex. E. Stacked heatmap depicting loss of H3K27me3 in P7 and 10W G34R cortex. F. Enrichment of genomic sequence features overlapping hypermethylated regions in G34R cortex.
Supplemental Figure 6. Altered DNA methylation is concurrent with transcriptional dysregulation of immune and neuronal genes, as Related to Figure 6 A. Left: heatmap depicting CG-methylation difference in WT vs. G34R cortex at differentially methylated genic promoters. Right: corresponding gene ontology enrichment of hypermethylated genes (top) and hypomethylated genes (bottom). B. Left: Genome browser snapshot of C1qa/b/c and C3 locus showing CH hypomethylation. Right: Genome browser snapshot of Nptx2 locus showing gain of DNA methylation. C, D. UMAP plots of single nuclei RNA-seq of G34R and WT cortex at P7 (C, N = 6926 G34R, N = 7776 WT) and at 10W (D, N = 15641 G34R, N = 17315 WT). Cells are colored by cell type annotation (top), and cells from each cluster with greater than 250 cells and fewer than 20% unlabeled cells are highlighted (bottom). Labels refer to the predominant cell type (at least 25% of the cluster) and number of cells in parentheses. E. For clusters as in C and D, the cluster proportion of predominant cell types, the cluster proportion of each genotype, and the number of differentially expressed genes (DEG) between genotypes with adjusted p-value < 0.05 are shown for P7 (top) and 10W (bottom). F. Distribution of expression for representative cell type marker genes (rows) in major cell types (columns) at 10W. EXT L2-L4, excitatory neurons layers 2/3 and 4; EXT L4-L5, excitatory neurons layers 5 and 6; Other EXT, other excitatory neurons; CGE INH, caudal ganglionic eminence-derived inhibitory GABAergic interneurons; MGE, medial ganglionic eminence-derived inhibitory GABAergic interneurons; Other INH, other inhibitory neurons; OPC, oligodendrocyte precursor cells; OL, oligodendrocytes; Mgl, microglia; Macr, macrophages.
Supplemental Figure 7. Altered DNA methylation is concurrent with transcriptional dysregulation of immune and neuronal genes, as Related to Figure 6 A. Density plot showing hypomethylated immune gene enrichment score (y-axis) vs. hypermethylated neuronal gene enrichment score (x-axis) in G34R (pink) and WT (blue) neuronal and glial cell types from P7 (N = 5008 cells; 2277 G34R, 2731 WT) and 10W (N = 13194 cells; 5900 G34R, 7294 WT) single nuclei transcriptome data. *** p < 2.2e-16, ** p < 1e-10, * p < 0.01, Wilcoxon rank sum test with continuity correction. B. Differential expression (DE) analysis at 10W between G34R (positive log FC) and WT (negative log FC). Hypermethylated neuronal genes (synapse) are colored in red. C. Top GO pathways ranked by adjusted p-value enriched in the 10W DE genes with adjusted p-value < 0.05. D. Distribution of mean DAM signature expression in the 10W microglia and macrophage cluster 9, with cells grouped by genotype (left). DE analysis between G34R (positive log FC) and WT (negative log FC) in cluster 9, with DAM genes colored in red (right). E. Proportion of Gfap+ cells in astrocyte cluster 3 at 10W. Dots represent proportions in each replicate (left). DE analysis between G34R (positive log FC) and WT (negative log FC) in astrocyte cluster 3, with Gfap labeled (middle). Distribution of mean pan-reactive astrocyte signature expression in astrocyte cluster 3, with cells grouped by genotype (right). F. Quantification of C1q and C3 fluorescence (integrated density: IntDen) in WT and G34R mice. Each mouse is plotted individually and as means ± SD. Student’s t-test. *P < 0.05, **P < 0.01, ***P < 0.001, **** P < 0.0001 and n.s.: nonsignificant. Analysis includes n=4 (C1q) and n=5 (C3) mice per genotype and n=2 images per mouse. G. Representative images of immunohistochemical staining of neurofilament protein (Nf200) in sagittal sections from 12 months age of control and G34R mutant mice. Quantification of Nf200 staining of control (n=6) and G34R mutant mice (n=5). The data is presented as means ± SD. Student’s t-test. *P < 0.05, **P < 0.01, ***P < 0.001, **** P < 0.0001 and n.s.: non- significant compared to control mice. H. Fig. 6D and E related quantification of PSD95 (20X) and PSD95 Iba1 colocalization. For PSD95 Iba1 colocalization graph, each mouse (WT n=8 and G34R n=7) is plotted individually and as means ± SD. Analysis includes n=10 Iba+ cells per mouse. For PSD95 IntDen graph, each mouse (WT n=3 and G34R n=4) is plotted individually and as means ± SD. Analysis includes n=2 replicates of confocal microscopy images (20X) per mouse. Student’s t-test. *P < 0.05, **P < 0.01, ***P < 0.001, **** P < 0.0001 and n.s.: non- significant compared to control mice.
Supplemental Table S1. Extended Information on de novo germline H3.3G34R/V patients, related to Figure 1.
Supplemental Table S2. Detailed characterization of G34R/V/W DKI mice performed by IDEXX BioAnalytics, related to Figure 1.
Supplemental Table S3. Analysis of Bulk RNA-Seq Profiles in G34 DKI mice, related to Figures 3.
Supplemental Table S4. Analysis of BioID interaction frequency of G34-mutant histones, Related to Figures 4.
Supplemental Table S5. Analysis of WGBS in G34R DKI mice, Related to Figures 5.
Supplemental Table S6. Analysis of Single-nuclei RNAseq and ATACseq (scMultiome) sequencing in G34R DKI mice, Related to Figures 6.
HIGHLIGHTS.
Germline H3.3G34R/V/W mutations cause distinct developmental defects;
G34R mice show severe neurological deficits, microcephaly, and neurodegeneration;
G34R decreased H3K36me2 and impairing recruitment of DNA methyltransferase DNMT3A;
DNA Methylation dysregulation is associated with immune activation & neuronal loss
ACKNOWLEDGEMENTS
This work was supported by a Large-Scale-Applied-Research-Project from Génome Quebec (GQ), Genome Canada, with the support of the Ontario Research Fund through funding provided by the Government of Ontario to N.J., C.L.K. and L.G. Additional support came from grant #25056 from the Cancer Research Society, GQ and Ministère de l’Économie et de l’Innovation du Québec. Additional funding support was provided by Fondation Charles-Bruneau and National Institutes of Health (NIH) grant P01-CA196539 (to N.J.), R01- R01CA255369 (to L.G.), R01-CA266978 and R35-GM138181 (to C.L.); Canadian Institutes of Health Research (CIHR) grants MOP-286756 and FDN-154307 (to N.J.) and PJT-156086 (to C.L.K.); Canadian Cancer Society grant 705182 and 705799 (to L.G.), a Fonds de Recherche du Québec – Santé (FRQS) salary award (to C.L.K. and L.G.); NSERC RGPIN-2016-04911 (to C.L.K.); CFI Leaders Opportunity Fund 33902 (to C.L.K.); and Genome Canada Science Technology Innovation Centre, McGill Genome Centre, Compute Canada Resource Allocation Projects MJD-574-AC and WST-164-AB. Data analyses were enabled by computing and storage resources provided by Compute Canada and Calcul Québec. Mass spectrometry data was generated at the Network Biology Collaborative Centre (NBCC), a facility supported by Canada Foundation for Innovation funding (Grant CFI 33474), the Ontario Government, Genome Canada and Ontario Genomics (OGI-139). N.J. is a member of the Penny Cole Laboratory and holds a Canada Research Chair (CRC) Tier 1 in Pediatric Oncology from CIHR; A-C.G. is a CRC for Functional Proteomics. C.C.L.C. is supported by a fellowship from RI-MUHC sponsored by Toronto Dominion Bank and Alex’s Lemonade Stand Foundation. B.A.G is funded by NIH grants CA196539 and AI118891, and the St. Jude Chromatin Consortium. E.I.C. is supported by CIHR (PJT-159683), the Natural Sciences and Engineering Research Council of Canada (NSERC, RGPIN-2016-05559), the Cancer Research Society, and the Garron Family Cancer Centre. S.M.M. is supported by a University of Toronto Open Fellowship. J.A.F. and C.C.G. were financed by the National Council for Scientific and Technological Development (CNPq)/Brazil and Coordination for the Improvement of Higher Education Personnel (CAPES)/Brazil Scholarship PDSE 88881.187783/2018-01. Funding is acknowledged from the Crohn’s and Colitis Foundation (RFA 598467 to P.J.L.) and the NIH (T32CA009140 to P.J.L.). Some figures were created using BioRender.com.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Ronan JL, Wu W, and Crabtree GR (2013). From neural development to cognition: unexpected roles for chromatin. Nat Rev Genet 14, 347–359. 10.1038/nrg3413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bjornsson HT (2015). The Mendelian disorders of the epigenetic machinery. Genome Res 25, 1473–1481. 10.1101/gr.190629.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bryant L, Li D, Cox SG, Marchione D, Joiner EF, Wilson K, Janssen K, Lee P, March ME, Nair D, et al. (2020). Histone H3.3 beyond cancer: Germline mutations in Histone 3 Family 3A and 3B cause a previously unidentified neurodegenerative disorder in 46 patients. Sci Adv 6. 10.1126/sciadv.abc9207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lowe BR, Maxham LA, Hamey JJ, Wilkins MR, and Partridge JF (2019). Histone H3 Mutations: An Updated View of Their Role in Chromatin Deregulation and Cancer. Cancers (Basel) 11. 10.3390/cancers11050660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maze I, Wenderski W, Noh KM, Bagot RC, Tzavaras N, Purushothaman I, Elsasser SJ, Guo Y, Ionete C, Hurd YL, et al. (2015). Critical Role of Histone Turnover in Neuronal Transcription and Plasticity. Neuron 87, 77–94. 10.1016/j.neuron.2015.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maver A, Cuturilo G, Ruml SJ, and Peterlin B (2019). Clinical Next Generation Sequencing Reveals an H3F3A Gene as a New Potential Gene Candidate for Microcephaly Associated with Severe Developmental Delay, Intellectual Disability and Growth Retardation. Balkan J Med Genet 22, 65–68. 10.2478/bjmg-2019-0028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Okur V, Chen Z, Vossaert L, Peacock S, Rosenfeld J, Zhao L, Du H, Calamaro E, Gerard A, Zhao S, et al. (2021). De novo variants in H3–3A and H3–3B are associated with neurodevelopmental delay, dysmorphic features, and structural brain abnormalities. NPJ Genom Med 6, 104. 10.1038/s41525-021-00268-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schwartzentruber J, Korshunov A, Liu XY, Jones DT, Pfaff E, Jacob K, Sturm D, Fontebasso AM, Quang DA, Tonjes M, et al. (2012). Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 482, 226–231. 10.1038/nature10833. [DOI] [PubMed] [Google Scholar]
- 9.Wu G, Broniscer A, McEachron TA, Lu C, Paugh BS, Becksfort J, Qu C, Ding L, Huether R, Parker M, et al. (2012). Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat Genet 44, 251–253. 10.1038/ng.1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen CCL, Deshmukh S, Jessa S, Hadjadj D, Lisi V, Andrade AF, Faury D, Jawhar W, Dali R, Suzuki H, et al. (2020). Histone H3.3G34-Mutant Interneuron Progenitors Co-opt PDGFRA for Gliomagenesis. Cell 183, 1617–1633 e1622. 10.1016/j.cell.2020.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Behjati S, Tarpey PS, Presneau N, Scheipl S, Pillay N, Van Loo P, Wedge DC, Cooke SL, Gundem G, Davies H, et al. (2013). Distinct H3F3A and H3F3B driver mutations define chondroblastoma and giant cell tumor of bone. Nat Genet 45, 1479–1482. 10.1038/ng.2814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cheng Z, Cheung P, Kuo AJ, Yukl ET, Wilmot CM, Gozani O, and Patel DJ (2014). A molecular threading mechanism underlies Jumonji lysine demethylase KDM2A regulation of methylated H3K36. Genes Dev 28, 1758–1771. 10.1101/gad.246561.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jain SU, Khazaei S, Marchione DM, Lundgren SM, Wang X, Weinberg DN, Deshmukh S, Juretic N, Lu C, Allis CD, et al. (2020). Histone H3.3 G34 mutations promote aberrant PRC2 activity and drive tumor progression. Proc Natl Acad Sci U S A 117, 27354–27364. 10.1073/pnas.2006076117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Voon HPJ, Udugama M, Lin W, Hii L, Law RHP, Steer DL, Das PP, Mann JR, and Wong LH (2018). Inhibition of a K9/K36 demethylase by an H3.3 point mutation found in paediatric glioblastoma. Nat Commun 9, 3142. 10.1038/s41467-018-05607-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wen H, Li Y, Xi Y, Jiang S, Stratton S, Peng D, Tanaka K, Ren Y, Xia Z, Wu J, et al. (2014). ZMYND11 links histone H3.3K36me3 to transcription elongation and tumour suppression. Nature 508, 263–268. 10.1038/nature13045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Khazaei S, De Jay N, Deshmukh S, Hendrikse LD, Jawhar W, Chen CCL, Mikael LG, Faury D, Marchione DM, Lanoix J, et al. (2020). H3.3 G34W Promotes Growth and Impedes Differentiation of Osteoblast-Like Mesenchymal Progenitors in Giant Cell Tumor of Bone. Cancer Discov 10, 1968–1987. 10.1158/2159-8290.CD-20-0461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zaghi M, Broccoli V, and Sessa A (2019). H3K36 Methylation in Neural Development and Associated Diseases. Front Genet 10, 1291. 10.3389/fgene.2019.01291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Janssen SM, and Lorincz MC (2021). Interplay between chromatin marks in development and disease. Nat Rev Genet. 10.1038/s41576-021-00416-x. [DOI] [PubMed] [Google Scholar]
- 19.Pathania M, De Jay N, Maestro N, Harutyunyan AS, Nitarska J, Pahlavan P, Henderson S, Mikael LG, Richard-Londt A, Zhang Y, et al. (2017). H3.3(K27M) Cooperates with Trp53 Loss and PDGFRA Gain in Mouse Embryonic Neural Progenitor Cells to Induce Invasive High-Grade Gliomas. Cancer Cell 32, 684–700 e689. 10.1016/j.ccell.2017.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guyenet SJ, Furrer SA, Damian VM, Baughan TD, La Spada AR, and Garden GA (2010). A simple composite phenotype scoring system for evaluating mouse models of cerebellar ataxia. J Vis Exp 2010;39:1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lalonde R, and Strazielle C (2011). Brain regions and genes affecting limb-clasping responses. Brain Res Rev 67, 252–259. 10.1016/j.brainresrev.2011.02.005. [DOI] [PubMed] [Google Scholar]
- 22.Liu Y, Ao LJ, Lu G, Leong E, Liu Q, Wang XH, Zhu XL, Sun TF, Fei Z, Jiu T, et al. (2013). Quantitative gait analysis of long-term locomotion deficits in classical unilateral striatal intracerebral hemorrhage rat model. Behav Brain Res 257, 166–177. 10.1016/j.bbr.2013.10.007. [DOI] [PubMed] [Google Scholar]
- 23.Cendelin J (2014). From mice to men: lessons from mutant ataxic mice. Cerebellum Ataxias 1, 4. 10.1186/2053-8871-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jessa S, Blanchet-Cohen A, Krug B, Vladoiu M, Coutelier M, Faury D, Poreau B, De Jay N, Hebert S, Monlong J, et al. (2019). Stalled developmental programs at the root of pediatric brain tumors. Nat Genet 51, 1702–1713. 10.1038/s41588-019-0531-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yao Z, Liu H, Xie F, Fischer S, Adkins RS, Aldridge AI, Ament SA, Bartlett A, Behrens MM, Van den Berge K, et al. (2021). A transcriptomic and epigenomic cell atlas of the mouse primary motor cortex. Nature 598, 103–110. 10.1038/s41586-021-03500-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, David E, Baruch K, Lara-Astaiso D, Toth B, et al. (2017). A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 169, 1276–1290 e1217. 10.1016/j.cell.2017.05.018. [DOI] [PubMed] [Google Scholar]
- 27.Yao Z, van Velthoven CTJ, Nguyen TN, Goldy J, Sedeno-Cortes AE, Baftizadeh F, Bertagnolli D, Casper T, Chiang M, Crichton K, et al. (2021). A taxonomy of transcriptomic cell types across the isocortex and hippocampal formation. Cell 184, 3222–3241 e3226. 10.1016/j.cell.2021.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ubhi K, and Price J (2005). Expression of POU-domain transcription factor, Oct-6, in schizophrenia, bipolar disorder and major depression. BMC Psychiatry 5, 38. 10.1186/1471-244X-5-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Baubec T, Colombo DF, Wirbelauer C, Schmidt J, Burger L, Krebs AR, Akalin A, and Schubeler D (2015). Genomic profiling of DNA methyltransferases reveals a role for DNMT3B in genic methylation. Nature 520, 243–247. 10.1038/nature14176. [DOI] [PubMed] [Google Scholar]
- 30.Weinberg DN, Papillon-Cavanagh S, Chen H, Yue Y, Chen X, Rajagopalan KN, Horth C, McGuire JT, Xu X, Nikbakht H, et al. (2019). The histone mark H3K36me2 recruits DNMT3A and shapes the intergenic DNA methylation landscape. Nature 573, 281–286. 10.1038/s41586-019-1534-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Okano M, Bell DW, Haber DA, and Li E (1999). DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 99, 247–257. 10.1016/s0092-8674(00)81656-6. [DOI] [PubMed] [Google Scholar]
- 32.Lister R, Mukamel EA, Nery JR, Urich M, Puddifoot CA, Johnson ND, Lucero J, Huang Y, Dwork AJ, Schultz MD, et al. (2013). Global epigenomic reconfiguration during mammalian brain development. Science 341, 1237905. 10.1126/science.1237905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tillotson R, Cholewa-Waclaw J, Chhatbar K, Connelly JC, Kirschner SA, Webb S, Koerner MV, Selfridge J, Kelly DA, De Sousa D, et al. (2021). Neuronal non-CG methylation is an essential target for MeCP2 function. Mol Cell 81, 1260–1275 e1212. 10.1016/j.molcel.2021.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kelley J, de Bono B, and Trowsdale J (2005). IRIS: a database surveying known human immune system genes. Genomics 85, 503–511. 10.1016/j.ygeno.2005.01.009. [DOI] [PubMed] [Google Scholar]
- 35.Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, Bennett ML, Munch AE, Chung WS, Peterson TC, et al. (2017). Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541, 481–487. 10.1038/nature21029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.De Felipe J, Marco P, Fairen A, and Jones EG (1997). Inhibitory synaptogenesis in mouse somatosensory cortex. Cereb Cortex 7, 619–634. 10.1093/cercor/7.7.619. [DOI] [PubMed] [Google Scholar]
- 37.Stephan AH, Barres BA, and Stevens B (2012). The complement system: an unexpected role in synaptic pruning during development and disease. Annu Rev Neurosci 35, 369–389. 10.1146/annurev-neuro-061010-113810. [DOI] [PubMed] [Google Scholar]
- 38.Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, Micheva KD, Mehalow AK, Huberman AD, Stafford B, et al. (2007). The classical complement cascade mediates CNS synapse elimination. Cell 131, 1164–1178. 10.1016/j.cell.2007.10.036. [DOI] [PubMed] [Google Scholar]
- 39.Gorski JA, Talley T, Qiu M, Puelles L, Rubenstein JL, and Jones KR (2002). Cortical excitatory neurons and glia, but not GABAergic neurons, are produced in the Emx1-expressing lineage. J Neurosci 22, 6309–6314. 20026564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kawaguchi D, Sahara S, Zembrzycki A, and O’Leary DDM (2016). Generation and analysis of an improved Foxg1-IRES-Cre driver mouse line. Dev Biol 412, 139–147. 10.1016/j.ydbio.2016.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brohm A, Schoch T, Grunberger D, Khella MS, Schuhmacher MK, Weirich S, and Jeltsch A (2022). The H3.3 G34W oncohistone mutation increases K36 methylation by the protein lysine methyltransferase NSD1. Biochimie 198, 86–91. 10.1016/j.biochi.2022.03.007. [DOI] [PubMed] [Google Scholar]
- 42.Nguyen S, Meletis K, Fu D, Jhaveri S, and Jaenisch R (2007). Ablation of de novo DNA methyltransferase Dnmt3a in the nervous system leads to neuromuscular defects and shortened lifespan. Dev Dyn 236, 1663–1676. 10.1002/dvdy.21176. [DOI] [PubMed] [Google Scholar]
- 43.Feng J, Zhou Y, Campbell SL, Le T, Li E, Sweatt JD, Silva AJ, and Fan G (2010). Dnmt1 and Dnmt3a maintain DNA methylation and regulate synaptic function in adult forebrain neurons. Nat Neurosci 13, 423–430. 10.1038/nn.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Swahari V, Nakamura A, Hollville E, Stroud H, Simon JM, Ptacek TS, Beck MV, Flowers C, Guo J, Plestant C, et al. (2021). MicroRNA-29 is an essential regulator of brain maturation through regulation of CH methylation. Cell Rep 35, 108946. 10.1016/j.celrep.2021.108946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Heyn P, Logan CV, Fluteau A, Challis RC, Auchynnikava T, Martin CA, Marsh JA, Taglini F, Kilanowski F, Parry DA, et al. (2019). Gain-of-function DNMT3A mutations cause microcephalic dwarfism and hypermethylation of Polycomb-regulated regions. Nat Genet 51, 96–105. 10.1038/s41588-018-0274-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tatton-Brown K, Seal S, Ruark E, Harmer J, Ramsay E, Del Vecchio Duarte S, Zachariou A, Hanks S, O’Brien E, Aksglaede L, et al. (2014). Mutations in the DNA methyltransferase gene DNMT3A cause an overgrowth syndrome with intellectual disability. Nat Genet 46, 385–388. 10.1038/ng.2917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Weinberg DN, Rosenbaum P, Chen X, Barrows D, Horth C, Marunde MR, Popova IK, Gillespie ZB, Keogh MC, Lu C, et al. (2021). Two competing mechanisms of DNMT3A recruitment regulate the dynamics of de novo DNA methylation at PRC1-targeted CpG islands. Nat Genet 53, 794–800. 10.1038/s41588-021-00856-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lewis PW, Muller MM, Koletsky MS, Cordero F, Lin S, Banaszynski LA, Garcia BA, Muir TW, Becher OJ, and Allis CD (2013). Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science 340, 857–861. 10.1126/science.1232245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lund PJ, Kori Y, Zhao X, Sidoli S, Yuan ZF, and Garcia BA (2019). Isotopic Labeling and Quantitative Proteomics of Acetylation on Histones and Beyond. Methods Mol Biol 1977, 43–70. 10.1007/978-1-4939-9232-4_5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yuan ZF, Lin S, Molden RC, Cao XJ, Bhanu NV, Wang X, Sidoli S, Liu S, and Garcia BA (2015). EpiProfile Quantifies Histone Peptides With Modifications by Extracting Retention Time and Intensity in High-resolution Mass Spectra. Mol Cell Proteomics 14, 1696–1707. 10.1074/mcp.M114.046011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lambert JP, Tucholska M, Go C, Knight JD, and Gingras AC (2015). Proximity biotinylation and affinity purification are complementary approaches for the interactome mapping of chromatin-associated protein complexes. J Proteomics 118, 81–94. 10.1016/j.jprot.2014.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bolger AM, Lohse M, and Usadel B (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liao Y, Smyth GK, and Shi W (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- 55.Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Harutyunyan AS, Krug B, Chen H, Papillon-Cavanagh S, Zeinieh M, De Jay N, Deshmukh S, Chen CCL, Belle J, Mikael LG, et al. (2019). H3K27M induces defective chromatin spread of PRC2-mediated repressive H3K27me2/me3 and is essential for glioma tumorigenesis. Nat Commun 10, 1262. 10.1038/s41467-019-09140-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chen CCL, Goyal P, Karimi MM, Abildgaard MH, Kimura H, and Lorincz MC (2018). H3S10ph broadly marks early-replicating domains in interphase ESCs and shows reciprocal antagonism with H3K9me2. Genome Res 28, 37–51. 10.1101/gr.224717.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bourgey M, Dali R, Eveleigh R, Chen KC, Letourneau L, Fillon J, Michaud M, Caron M, Sandoval J, Lefebvre F, et al. (2019). GenPipes: an open-source framework for distributed and scalable genomic analyses. Gigascience 8. 10.1093/gigascience/giz037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Li H, and Durbin R (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38, 576–589. 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, and Mesirov JP (2011). Integrative genomics viewer. Nat Biotechnol 29, 24–26. 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, and Liu XS (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol 9, R137. 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Younesy H, Moller T, Lorincz MC, Karimi MM, and Jones SJ (2015). VisRseq: R-based visual framework for analysis of sequencing data. BMC Bioinformatics 16 Suppl 11, S2. 10.1186/1471-2105-16-S11-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Krueger F, and Andrews SR (2011). Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 27, 1571–1572. 10.1093/bioinformatics/btr167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Stuart T, Srivastava A, Madad S, Lareau CA, and Satija R (2021). Single-cell chromatin state analysis with Signac. Nat Methods 18, 1333–1341. 10.1038/s41592-021-01282-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hao Y, Hao S, Andersen-Nissen E, Mauck WM 3rd, Zheng S, Butler A, Lee MJ, Wilk AJ, Darby C, Zager M, et al. (2021). Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587 e3529. 10.1016/j.cell.2021.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Li C, Liu B, Kang B, Liu Z, Liu Y, Chen C, Ren X, and Zhang Z (2020). SciBet as a portable and fast single cell type identifier. Nat Commun 11, 1818. 10.1038/s41467-020-15523-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tan Y, and Cahan P (2019). SingleCellNet: A Computational Tool to Classify Single Cell RNA-Seq Data Across Platforms and Across Species. Cell Syst 9, 207–213 e202. 10.1016/j.cels.2019.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Aran D, Looney AP, Liu L, Wu E, Fong V, Hsu A, Chak S, Naikawadi RP, Wolters PJ, Abate AR, et al. (2019). Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat Immunol 20, 163–172. 10.1038/s41590-018-0276-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yu G, Wang LG, Han Y, and He QY (2012). clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16, 284–287. 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. Germline knock-in of H3.3G34R/V/W results in distinct developmental defects, as Related to Figure 1. A. Violin plot showing height, weight, and occipital-frontal circumference (OFC) of human patients with germline H3.3 mutations (n = 47), with severe undergrowth/microcephaly of H3.3G34R (red) and H3.3G34V (blue) highlighted as individual data points. B. Related to Fig. 1B. Negative control immunohistochemistry staining for H3.3G34R/V/W on WT mice. C. Immunoblots of H3.3G34R mutant histone in various somatic tissues from G34R heterozygous mice and WT littermates. D. A representative image of bladder with the urogenital tracts from control and G34W mice. Note the enlarged bladder filled with urine in the G34W. SV: seminal vesicle. E. Kaplan-Meier curves for cumulative survival rates of female G34 mutants and control mice.
Supplemental Figure 2. G34R/V mice show abnormal motor functions and progressive microcephaly, as Related to Figure 2. A. Representative image of G34R mouse exhibiting paresis at 12 months. B. Performance of G34 mutant and control mice (4–7 weeks; n > 9) on rotarod test. The data is presented as means ± SD. The statistical significance assessed using two-way ANOVA. **** P < 0.0001 and n.s.: non-significant compared to control mice. C. CatWalk measurements of stride length of control (n=10) and G34R (n=6) mice at 8 weeks. The data is presented as means ± SE. Student’s t-test. *P < 0.05, **P < 0.01, ***P < 0.001, **** P < 0.0001 and n.s.: non-significant compared to control. D. CatWalk measurements (BOS: base of support). The graphs show the static data from control (n=10) and G34R (n=6) mice at 8 weeks. The data is presented as means ± SD. Student’s t-test. *P < 0.05, **P < 0.01, ***P < 0.001, **** P < 0.0001 and n.s.: non-significant compared to control. E. Brain-to-body weight ratio of G34 mutant and control mice at P21–30 and n > 5 brains per genotype were used. Each mouse is plotted individually and as means ± SD. The statistical significance assessed using Student’s t-test. *P < 0.05, **P < 0.01, ***P < 0.001, **** P < 0.0001 and n.s.: non-significant. F. Representative images of midsagittal brain slices of control and G34R mutant mice stained with Cresyl Violet (Nissl Staining) at 12 months of age. G, H. Representative images of midsagittal brain slices of control and G34 mutant mice stained with Cresyl Violet (Nissl staining) at P7 and P21. Red arrowheads indicate poorly developed lobules or abnormal fissures in the cerebellum of mutant animals. I. Three-dimensional confocal image of Purkinje cells labelled with Calbindin-D28K in adult G34R and WT littermate.
Supplemental Figure 3. G34R leads to abnormal accumulation of microglia and depletion of neurons, as Related to Figure 3. A. Enrichment plot of microglia, astrocyte, and neuronal signatures in 10W cortex of G34-mutant mice. B. Box-whisker plot depicting elevated microglial and astrocytic markers gene expression in 10W G34R cortex compared to G34V/W and WT. C. Quantification of NeuN in WT and G34-mutant 10W cortex (WT n=6, G34R n=4, G34V n=2 and G34W n=3), as related to Fig. 3D. Each mouse is plotted individually and as means ± SD. One-way ANOVA test. *P < 0.05, **P < 0.01, ***P < 0.001, **** P < 0.0001 and n.s.: non-significant compared to control. D. Representative immunohistochemistry and immunofluorescent staining demonstrating lack of change in microglia (Iba1), astrocyte (GFAP), and neurons (NeuN, Oct6) at P7 in G34R and WT cortex.
Supplemental Figure 4. G34R/V/W histones show differential interaction with chromatin modifiers, as Related to Figure 4. Bubble plot depicting BioID interaction frequency of G34-mutant histones with known chromatin modifiers using N-terminal (left) tagging strategy in G34R/V/W and H3.3 WT expressing U2-OS lines (n=2). Also, see Supplemental Table S4 for all significant interactors.
Supplemental Figure 5. G34R re-distribute DNMT3A localization, causing loss of CH methylation at intergenic regions, as Related to Figure 5. A. 2D scatterplot depicting correlation between DNMT3A enrichment (x-axis) and various chromatin modifications (y-axis) in WT P7 cortex. Individual points are genome-wide 5kb bins. B. Parallel coordinate plot depicting concordant epigenetic change in G34R vs. WT cortex, anchoring based on DNMT3A change. 5kb bins were stratified into 6 groups based on DNMT3A change, with red line representing bins that showed DNMT3A-loss in G34R, while blue lines representing bins with DNMT3A gain. G34R shows concordant. loss of H3K36me2, mCH and mCG in DNMT3A-deplete genomic regions. Solid line: median. Shaded area: 25% and 75% quartiles. C. Stacked heatmap centered on DNMT3A-peaks in G34R cortex, related to Fig. 5C. Note DNMT3A-peaks are concurrently marked with H3K36me2 and mCH. D. Genome browser snapshot depicting preferential loss of DNMT3A binding and mCH at interferon immune gene cluster on chr4 in G34R cortex. E. Stacked heatmap depicting loss of H3K27me3 in P7 and 10W G34R cortex. F. Enrichment of genomic sequence features overlapping hypermethylated regions in G34R cortex.
Supplemental Figure 6. Altered DNA methylation is concurrent with transcriptional dysregulation of immune and neuronal genes, as Related to Figure 6 A. Left: heatmap depicting CG-methylation difference in WT vs. G34R cortex at differentially methylated genic promoters. Right: corresponding gene ontology enrichment of hypermethylated genes (top) and hypomethylated genes (bottom). B. Left: Genome browser snapshot of C1qa/b/c and C3 locus showing CH hypomethylation. Right: Genome browser snapshot of Nptx2 locus showing gain of DNA methylation. C, D. UMAP plots of single nuclei RNA-seq of G34R and WT cortex at P7 (C, N = 6926 G34R, N = 7776 WT) and at 10W (D, N = 15641 G34R, N = 17315 WT). Cells are colored by cell type annotation (top), and cells from each cluster with greater than 250 cells and fewer than 20% unlabeled cells are highlighted (bottom). Labels refer to the predominant cell type (at least 25% of the cluster) and number of cells in parentheses. E. For clusters as in C and D, the cluster proportion of predominant cell types, the cluster proportion of each genotype, and the number of differentially expressed genes (DEG) between genotypes with adjusted p-value < 0.05 are shown for P7 (top) and 10W (bottom). F. Distribution of expression for representative cell type marker genes (rows) in major cell types (columns) at 10W. EXT L2-L4, excitatory neurons layers 2/3 and 4; EXT L4-L5, excitatory neurons layers 5 and 6; Other EXT, other excitatory neurons; CGE INH, caudal ganglionic eminence-derived inhibitory GABAergic interneurons; MGE, medial ganglionic eminence-derived inhibitory GABAergic interneurons; Other INH, other inhibitory neurons; OPC, oligodendrocyte precursor cells; OL, oligodendrocytes; Mgl, microglia; Macr, macrophages.
Supplemental Figure 7. Altered DNA methylation is concurrent with transcriptional dysregulation of immune and neuronal genes, as Related to Figure 6 A. Density plot showing hypomethylated immune gene enrichment score (y-axis) vs. hypermethylated neuronal gene enrichment score (x-axis) in G34R (pink) and WT (blue) neuronal and glial cell types from P7 (N = 5008 cells; 2277 G34R, 2731 WT) and 10W (N = 13194 cells; 5900 G34R, 7294 WT) single nuclei transcriptome data. *** p < 2.2e-16, ** p < 1e-10, * p < 0.01, Wilcoxon rank sum test with continuity correction. B. Differential expression (DE) analysis at 10W between G34R (positive log FC) and WT (negative log FC). Hypermethylated neuronal genes (synapse) are colored in red. C. Top GO pathways ranked by adjusted p-value enriched in the 10W DE genes with adjusted p-value < 0.05. D. Distribution of mean DAM signature expression in the 10W microglia and macrophage cluster 9, with cells grouped by genotype (left). DE analysis between G34R (positive log FC) and WT (negative log FC) in cluster 9, with DAM genes colored in red (right). E. Proportion of Gfap+ cells in astrocyte cluster 3 at 10W. Dots represent proportions in each replicate (left). DE analysis between G34R (positive log FC) and WT (negative log FC) in astrocyte cluster 3, with Gfap labeled (middle). Distribution of mean pan-reactive astrocyte signature expression in astrocyte cluster 3, with cells grouped by genotype (right). F. Quantification of C1q and C3 fluorescence (integrated density: IntDen) in WT and G34R mice. Each mouse is plotted individually and as means ± SD. Student’s t-test. *P < 0.05, **P < 0.01, ***P < 0.001, **** P < 0.0001 and n.s.: nonsignificant. Analysis includes n=4 (C1q) and n=5 (C3) mice per genotype and n=2 images per mouse. G. Representative images of immunohistochemical staining of neurofilament protein (Nf200) in sagittal sections from 12 months age of control and G34R mutant mice. Quantification of Nf200 staining of control (n=6) and G34R mutant mice (n=5). The data is presented as means ± SD. Student’s t-test. *P < 0.05, **P < 0.01, ***P < 0.001, **** P < 0.0001 and n.s.: non- significant compared to control mice. H. Fig. 6D and E related quantification of PSD95 (20X) and PSD95 Iba1 colocalization. For PSD95 Iba1 colocalization graph, each mouse (WT n=8 and G34R n=7) is plotted individually and as means ± SD. Analysis includes n=10 Iba+ cells per mouse. For PSD95 IntDen graph, each mouse (WT n=3 and G34R n=4) is plotted individually and as means ± SD. Analysis includes n=2 replicates of confocal microscopy images (20X) per mouse. Student’s t-test. *P < 0.05, **P < 0.01, ***P < 0.001, **** P < 0.0001 and n.s.: non- significant compared to control mice.
Supplemental Table S1. Extended Information on de novo germline H3.3G34R/V patients, related to Figure 1.
Supplemental Table S2. Detailed characterization of G34R/V/W DKI mice performed by IDEXX BioAnalytics, related to Figure 1.
Supplemental Table S3. Analysis of Bulk RNA-Seq Profiles in G34 DKI mice, related to Figures 3.
Supplemental Table S4. Analysis of BioID interaction frequency of G34-mutant histones, Related to Figures 4.
Supplemental Table S5. Analysis of WGBS in G34R DKI mice, Related to Figures 5.
Supplemental Table S6. Analysis of Single-nuclei RNAseq and ATACseq (scMultiome) sequencing in G34R DKI mice, Related to Figures 6.
Data Availability Statement
All data reported in this paper will be shared by the lead contact upon request.
All sequencing data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
Microscopy data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.