

Summary
Mitophagy is a form of selective autophagy that disposes of superfluous and potentially damage-inducing organelles in a tightly controlled manner. While the machinery involved in mitophagy induction is well known, the regulation of the components is less clear. Here, we demonstrate that TNIP1 knock out in HeLa cells accelerates mitophagy rates, and that ectopic TNIP1 negatively regulates the rate of mitophagy. These functions of TNIP1 depend on an evolutionarily conserved LIR motif as well as an AHD3 domain, which are required for binding to the LC3/GABARAP family of proteins and the autophagy receptor TAX1BP1, respectively. We further show that phosphorylation appears to regulate its association with the ULK1 complex member FIP200, allowing TNIP1 to compete with autophagy receptors, providing a molecular rationale for its inhibitory function during mitophagy. Taken together, our findings describe TNIP1 as a negative regulator of mitophagy that acts at the early steps of autophagosome biogenesis.
eTOC :
Le Guerroué et al show that TNIP1 acts as an inhibitor of mitophagy by binding with TAX1BP1 and FIP200 in a competitive manner. While binding to TAX1BP1 is essential for its inhibitory role, they further demonstrate that phosphorylation regulates binding to FIP200, providing a molecular rationale for TNIP1’s function.
Graphical Abstract
Intro
Macroautophagy, one of the main cellular degradation pathways, sequesters cytosolic components inside a double membrane structure called an autophagosome, before catabolizing its content by fusing to lysosomes. The molecular mechanisms of the formation and maturation processes are well described and revolve around a set of proteins called the ATG conjugation system1. Initially described as a non-selective degradation process triggered when cells face starvation, more recent work shows autophagy can selectively eliminate certain proteins, protein aggregates and organelles2,3. Selective autophagy specifically sequesters cytosolic structures via autophagy receptors. The main feature of these autophagy receptors is their capacity to bind to mATG8 (mammalian ATG8 proteins) as well as FIP200 and to ubiquitin in most cases4. One of the best characterized selective autophagy processes is the degradation of mitochondria via mitophagy5, where autophagy receptors such as NDP52 or OPTN are recruited to mitochondria ubiquitinated by parkin via their ubiquitin-binding domains, allowing the autophagy machinery to assemble and degrade the mitochondria in a wholesale fashion6,7.
TNIP1 (TNFAIP3-interacting protein 1), also called ABIN1 (A20-binding inhibitor of NF-κB activation 1) participates in the NF-κB pathway, where it negatively regulates NF-κB activation, maintaining immune homeostasis8,9. Structurally, TNIP1 possesses a Ub-binding domain (UBAN domain – Ubiquitin binding of ABIN and NEMO) common among ABIN proteins, 3 Abin homology domains (AHD) and a NEMO binding domain (NBD)8,10. Although the AHD3 domain function is currently not described, AHD1 mediates binding with the ubiquitin editing enzyme Tumor necrosis factor alpha-induced protein 3 (TNFAIP3, A20) and AHD4 mediates the interaction with OPTN11. Being involved in immune responses, dysregulation of TNIP1 was shown to be implicated in various human diseases through a number of genome-wide associated studies (GWAS)12. TNIP1 single nucleotide polymorphisms (SNP) have been strongly associated with autoimmune diseases13 and cross ethnic genetic studies identified the GPX3-TNIP1 locus to associate with amyotrophic lateral sclerosis (ALS)14. However, a later study concluded that this locus was less likely to contribute to ALS risk15. In addition, a recent GWAS study identified TNIP1 as the locus of a newly identified risk allele for Alzheimer’s disease (AD)16.
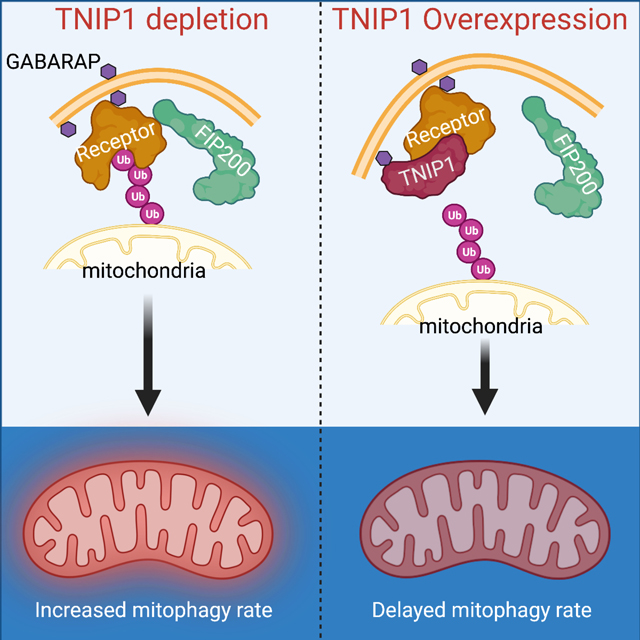
Here we describe a role of TNIP1 as a LC3/GABARAP-interacting protein, that triggers mitophagy when ectopically targeted to mitochondria, and that endogenously negatively inhibits early stages of mitophagy. Furthermore, we show that physical interactions of TNIP1 with LC3/GABARAP proteins and TAX1BP1 drive mitophagy inhibition. This inhibition appears to be regulated by phosphorylation of TNIP1, promoting binding with FIP200 for a counteracting effect on inhibition. Our study suggests that TNIP1 thus acts as a security check to fine tune the rate of mitophagy.
Results
Ectopic localization of TNIP1 to the mitochondria induces mitophagy.
TNIP1 possesses a UBAN domain similar to that in OPTN17,18 and was suggested to be an autophagy substrate19. Most autophagy receptors are defined by a ubiquitin-binding domain and an LC3 interacting region (LIR). We thus investigated if TNIP1 contained an LIR motif. In silico searches of LIR20 motif using the bioinformatic tool iLIR21 website revealed two potential canonical LIR domains in TNIP1 (Figure 1A). A Blast alignment with 4 other higher eukaryote TNIP1 proteins showed that LIR2 is conserved but not LIR1 (Supp Figure 1A). In order to determine whether TNIP1 LIR1 and LIR2 are functional LIR domains, we mutated LIR1 Phe83 and Leu86 to Ala and LIR2 Phe125 and Val128 to Ala (Supp Figure 1B). We also made a LIR1 and LIR2 double mutant construct that carries all 4 mutations. Lysates of HeLa cells transfected with TNIP1 wild type (WT), LIR1 mutant, LIR2 mutant and LIR1+LIR2 mutant were subjected to GST pull down experiments, an assay previously used to assess binding to mATG8 proteins (i.e. MAP1LC3A, MAP1LC3B, MAP1LC3C, GABARAP, GABARAPL1 and GABARAPL2)22,23. As a positive control, SQSTM1/p62 was demonstrated to bind strongly to all mATG8 proteins (Supp Figure 1C). Conversely, as a negative control we employed the TNIP family member TNIP3 where no LIR motif was detected when using iLIR search and accordingly no LC3/GABARAP binding was observed (Supp Figure 1D). TNIP1, on the other hand, was able to bind to all mATG8 proteins, with an apparent preference for LC3C and GABARAP, and a weaker binding to LC3A, LC3B and GABARAPL2 (Figure 1B). LIR1 mutations diminished LC3C binding while LIR2 mutations diminished GABARAP binding. LC3A and LC3B binding was lost with both constructs. However, the double LIR1+LIR2 mutations resulted in a total loss of all 6 mATG8 binding indicating that TNIP1 is a bona fide mATG8 binding protein, where LIR1 seems to preferentially bind LC3C and LIR2 to GABARAP, although both LIR seem to be able to compensate for each other’s loss of function.
Fig. 1. Ectopic localization of TNIP1 to the mitochondria induces mitophagy.

(A) Schematic representation of TNIP1 and its 2 identified putative LIR motifs.
(B) HeLa cells stably expressing HA-TNIP1 wild type, LIR1 mutant, LIR2 mutant or LIR1+LIR2 double mutant co-immunoprecipitated (co-IP) with purified recombinant GST tagged mATG8 proteins and subjected to immunoblot analysis (IB).
(C) Schematization of CID experiment.
(D) HeLa cells stably expressing mito-mKeima, FRB-FIS1 and FKBP-GFP-TNIP1 WT or UBAN mutant (D472N) were treated with Rapalog for 24 h and subjected to FACS analysis. Left, representative FACS plot. Right, bar graph representing data as mean ± SEM obtained from 3 independent replicates.
(E) HeLa cells stably expressing mito-mKeima, FRB-FIS1 and FKBP-GFP-TNIP1 WT, LIR1 mutant, LIR2 mutant or LIR1&LIR2 double mutant were treated with Rapalog for 24 h and subjected to FACS acquisition. Left, representative FACS plot. Right, bar graph representing data as mean ± SEM obtained from 3 independent replicates.
(F) Top, schematic representation of the TNIP family TNIP1, TNIP2 and TNIP3. Bottom, HeLa cells stably expressing mito-Keima, FRB-FIS1 and FKBP-GFP-TNIP1, FKBP-GFP-TNIP2 or FKBP-GFP-TNIP3 were treated with Rapalog for 24 h and subjected to FACS acquisition. Left, representative FACS plot. Right, bar graph representing data as mean ± SEM obtained from 3 independent replicates.
Next, we took advantage of the chemically induced dimerization (CID) assay in cells expressing mitochondrial-targeted mKeima (mt-mKeima)24 (Figure 1C). We fused the protein FRB to the C-terminus tail of the outer mitochondrial protein FIS1 and TNIP1 to the soluble protein FKBP. Adding the small molecule Rapalog induces dimerization of FRB and FKBP, conditionally localizing TNIP1 to the mitochondria. The A/C heterodimerizer Rapalog is an analog of the autophagy-induction drug Rapamycin, however, Rapalog’s interaction is specific for mutated FRB and is thus unable to inhibit mTor and therefore incapable of inducing autophagy25. We then gauge mitophagy using Fluorescent Activated Cell Sorting (FACS) by measuring the ratio of mt-mKeima excited by 488 nm or 561 nm to determine mitochondria in neutral and acidic pH environments, respectively. Because cells are gated to the same intensity levels for the mKeima and GFP channels, we ultimately measure cells with the same level of overexpressed protein26. Interestingly, localizing TNIP1 to mitochondria for 24 h induced robust mitophagy, and the UBAN loss of function mutation (D472N)27 was able to trigger mitophagy to levels similar to WT TNIP1, indicating that the ubiquitin binding domain of TNIP1 is not necessary for inducing mitophagy when TNIP1 is artificially targeted to mitochondria (Figure 1D). We next investigated whether the LIR motifs have any role in TNIP1 to triggered mitophagy. While LIR1 mutations had a relatively minor effect and retained an ability to substantially trigger mitophagy, CID with LIR2 mutant displayed an approximately 55% defect compared to WT TNIP1, indicating the importance of the LIR2 domain in TNIP1 mitophagy activity (Figure 1E). No additional effect was observed when using the LIR1+LIR2 double mutant construct. Immunocytochemistry recapitulated the mitophagy FACS data, with LIR1 showing no defect in recruiting LC3B, while LIR2 mutant construct showed a significant defect but still retained the ability to recruit LC3B to the mitochondria. LIR1+LIR2 double mutant, however, showed no additional defect in LC3B recruitment (Supp Figure 1E). On the other hand, GABARAP recruitment showed a different pattern, with LIR1 and LIR2 constructs showing no defect in recruitment, while the double LIR1+LIR2 construct displayed a small but significant decrease in recruitment (Supp Figure 1F). Based on these results we hypothesize that TNIP1’s function on mitochondria does not depend only on binding with mATG8 proteins, but probably also interacts with other autophagy machinery proteins as the LIR1 and LIR2 double mutant construct is still able to trigger mitophagy. An interesting observation is the clumping of the mitochondria upon Rapalog treatment as is typically observed upon mitochondrial depolarization in Parkin-expressing cells, due to the clustering of p62/SQSTM128. A similar mechanism might occur when ectopically expressing TNIP1 to the mitochondria.
Mammalian cells possess 3 TNIP homologs, TNIP1, TNIP2 and TNIP3, sharing certain ABIN homology domains (AHDs) that characterize them (Figure 1F). CID and FACS analysis with TNIP2 and TNIP3 revealed that they were not able to induce mitophagy as substantially as TNIP1, indicating the singular capacity of TNIP1 in mitophagy. To further characterize the important domains of TNIP1 other than LIR2, we carried out CID with versions of TNIP1 missing individual AHD domains (Figure 2A and Supp Figure 2A). Looking at their localization, all constructs displayed an even, cytosolic distribution in puncta similar to the full-length construct (Supp Figure 2B). AHD1 deletion had no effect on mitophagy, suggesting that binding to A20 is not necessary for its capacity in mitophagy. TNIP1 with the AHD3 deletion showed a strong reduction in its ability to induce mitophagy, while deletion of AHD4 domain in TNIP1 showed moderate inhibitory effect on mitophagy (Figure 2B). Since we discovered that the LIR2 mutant and AHD3 deletion mutant TNIP1 displayed the strongest deficiency in mitophagy, we combined both and made an AHD3 deletion and LIR2 mutant construct and observed that this construct completely abolished TNIP1’s ability to induce mitophagy (Figure 2C). We conclude that the LIR2 motif and AHD3 domain of TNIP1 are both involved and possibly have distinct roles in mitophagy. Both N-terminal and C-terminal fragments of TNIP1 (Figure 2D) proved incapable of inducing mitophagy, indicating that more than just LIR and AHD3 domains are required for mitophagy, and likely a fully folded protein is necessary.
Fig. 2. IR2 and AHD3 domain of TNIP1 are essential for its role in mitophagy.

(A) Schematic representation of TNIP1 and the AHD mutant constructs.
(B) HeLa cells stably expressing mito-mKeima, FRB-FIS1 and FKBP-GFP-TNIP1 Full Length (FL) or FKBP-GFP-TNIP1 mutants ΔAHD1, ΔAHD3 and ΔAHD4 were treated with Rapalog for 24 h and subjected to FACS analysis. Left, representative FACS plot. Right, bar graph representing data as mean ± SEM obtained from 3 independent replicates.
(C) HeLa cells stably expressing mito-mKeima, FRB-FIS1 and FKBP-GFP-TNIP1 Full Length, LIR2 mutant, ΔAHD3 or LIR2 mutant and ΔAHD3 were treated with Rapalog for 24 h and subjected to FACS acquisition. Left, representative FACS plot. Right, bar graph representing data as mean ± SEM obtained from 3 independent replicates.
(D) Top, schematic representation of full length TNIP1 or the C-terminal and N-terminal part of TNIP1. Bottom, HeLa cells stably expressing mito-Keima, FRB-FIS1 and FKBP-GFP-TNIP1 full length or FKBP-GFP-TNIP1 N-terminal or C-terminal were treated with Rapalog for 24 h and subjected to FACS acquisition. Left, representative FACS plot. Right, bar graph representing data as mean ± SEM obtained from 3 independent replicates.
(E) HeLa cells stably expressing mito-Keima, FRB-FIS1 and FKBP-GFP-TNIP1 in wild type cells, PentaKO (SQSTM1, NBR1, NDP52, TAX1BP1 and TAX1BP1 KO cells), FIP200 KO, WIPI2KO or A20 KO cells were treated with Rapalog for 24 h and subjected to FACS analysis. Bar graph representing data as mean ± SEM obtained from 3 independent replicates.
(F) HeLa cells stably expressing mito-Keima, FRB-FIS1 and FKBP-GFP-ATG16L1, FKBP-GFP-WIPI2 or FKBP-GFP-A20 in wild type cells or TNIP1 KO cells were treated with Rapalog for 24 h and subjected to FACS analysis. Bar graph representing data as mean ± SEM obtained from 3 independent replicates.
To determine the molecular mechanisms of TNIP1 in mitophagy, we performed CID-induced mitophagy in different cellular backgrounds: FIP200 KO, WIPI2 KO, A20 KO and pentaKO24 cells (5KO: NDP52, OPTN, TAX1BP1, SQSTM1 and NBR1). 5KO cells were previously described to be deficient in mitophagy, owing to the dependence on recruitment of autophagy receptors to recruit the autophagy machinery. To our surprise, TNIP1 was able to mediate mitophagy in 5KO cells at a level similar to that in WT cells (Figure 2E and Supp Figure 2C). In FIP200 KO and WIPI2 KO cells, no mitophagy was observed (Figure 2E and Supp Figure 2C), accordingly with their essential roles as general autophagy machineries. Because of the tight role TNIP1 plays with A20 in the NF-κB pathway, we explored mitophagy activation in an A20 KO background (Figure 2E and Supp Figure 2C). Consistent with the results seen with the AHD1 deletion mutant, A20 KO did not alter the mitophagy response, and revealing a new activity of TNIP1 distinct from prior work on its role in inflammation.
To clarify the role of TNIP1 requirement in mitophagy, we conducted different CID experiments in TNIP1 KO cells. Unfortunately, due to the size of FIP200, we could not produce a functioning FKBP-GFP-FIP200 construct. Consequently, we assessed whether other autophagy machinery proteins (i.e., WIPI2 and ATG16L1), as well as A20 were able to trigger mitophagy when placed on mitochondria in WT or TNIP1 KO cells. As previously reported, CID with ATG16L1 showed a strong mitophagy response in control cells7. TNIP1 KO did not impair that response, and even slightly enhanced it, although not significantly (Figure 2F and Supp Figure 2D). Similarly, CID with WIPI2 displayed a robust mitophagy response and this response was significantly stronger in TNIP1 KO cells (Figure 2F and Supp Figure 2D). Finally, CID with A20 displayed a weak mitophagy response that was abolished in TNIP1 KO cells (Figure 2F and Supp Figure 2D). This could be explained by a weak recruitment of TNIP1 mediated by A20 to the mitochondria, and the subsequent recruitment of the autophagy machinery. In light of these results, we conclude that TNIP1-dependent mitophagy induction does not depend on autophagy receptors, but relies on early autophagy machinery. However, TNIP1 is dispensable for these autophagy machinery proteins to induce mitophagy.
TNIP1 is a negative regulator of mitophagy
The CID results showed that TNIP1 has the potential to trigger mitophagy and allowed us to identify the domains of TNIP1 required for this function. Since TNIP1 possesses a ubiquitin-binding domain as well as a LIR motif, we speculated that it could act as an autophagy receptor. To test this hypothesis, we compared TNIP1 to known autophagy receptors in mitophagy flux assays. When subjected to mitophagy induced by mitochondrial depolarization, receptors are degraded in an autophagy-specific manner, together with their cargo. Blocking autophagosomal degradation with the V-ATPase inhibitor Bafilomycin A1 (BafA1) is a common way to estimate substrates specifically degraded in lysosomes. Combining the mitochondrial ATP synthase and complex III inhibitors Oligomycin A and AntimycinA1 (OA), respectively, is an established treatment for triggering mitophagy. Autophagy receptors are selectively degraded during mitophagy, as can be seen with TAX1BP1, NDP52 and OPTN (Supp Figure 3A). Double treatment of cells with BafA1 and OA promotes mitophagy but prevents lysosomal degradation of the encapsulated proteins (Supp Figure 3A). However, similar to the ULK1 complex protein FIP200, TNIP1 levels were not influenced by Bafilomycin or OA treatment. Interestingly, TNIP1 was recently found to be an autophagy substrate in an autophagosome profiling content screening19, and thus would appear to be an autophagy substrate. Consequently, its apparent lack of degradation upon mitophagy or absence of accumulation upon Bafilomycin A1 treatment may be due to a fast turnover rate. Therefore, we performed OA treatments combined with cycloheximide (CHX) to investigate TNIP1 degradation (Supp Figure 3B). Remarkably, TNIP1 seems to be a long-lived protein, as no degradation of TNIP1 was seen after 4 hours of CHX treatment. Interestingly, under steady state conditions, FIP200 is degraded rapidly, but counterintuitively, is slightly stabilized during mitophagy induction. Consequently, TNIP1 is very likely not degraded en masse via mitophagy, as is also the case for FIP200. Immunocytochemistry allowed observation of TNIP1 localization under steady state condition as well as upon mitophagy. To trigger mitophagy, HeLa cells stably expressing HA-Parkin were treated for 4 h with OA (Supp Figure 3C). Under steady state conditions, endogenous TNIP1 appeared as puncta, in the vicinity of the mitochondria, with some colocalization events at the periphery of mitochondria, in a manner reminiscent of autophagy receptors. However, upon OA treatment, TNIP1 was found to coalesce in the perinuclear region and not to substantially relocate to mitochondria, as can be seen with FIP200, contrary to what is seen with the autophagy receptor TAX1BP1, prompting us to reconsider the role of TNIP1 as a mitophagy receptor.
Therefore, we asked if TNIP1 KO cells and such cells rescued with FKBP-GFP-TNIP1 (Figure 3A) show any alteration in PINK1/Parkin mediated mitophagy. HeLa cells expressing mt-mKeima and HA-Parkin were treated for 6 h with OA and analyzed by FACS. This treatment induced an almost complete mitophagy response in WT cells and the same response in TNIP1 KO cells (Figure 3B). In contrast, re-expression of TNIP1 in TNIP1 KO cells showed a substantial defect in mitophagy. These results indicate that TNIP1 may be a negative regulator of mitophagy and prompted us to investigate earlier time points when in mitophagy was submaximal in WT cells. Indeed, at 2 h of OA treatment, we saw a stronger mitophagy induction in TNIP1 KO cells, a response that could be reversed by re-expression of TNIP1 (Figure 3C). To further validate TNIP1’s role in inhibiting mitophagy, we overexpressed TNIP1 in HeLa cells instead of rescuing in TNIP1 KO cells and monitored mitophagy using mt-mKeima and FACS. Similarly, overexpression of TNIP1 in WT HeLa cells induced a robust inhibition of mitophagy (Figure 3D). Over-expression of AHD domain deletion mutants in WT cells was then used to compare to the full length TNIP1 and assess the deletion mutant’s abilities to inhibit mitophagy. While the ΔAHD1 construct showed a mitophagy response similar to WT TNIP1, in accordance with a lack of function in previous CID experiments, ΔAHD3 was less inhibitory, while ΔAHD4 showed an intermediate response compared to ΔAHD3 and WT TNIP1. We also carried out rescue experiments in TNIP1 KO cells with the LIR mutant construct in PINK1-Parkin dependent mitophagy. Similar to the observations with the AHD3 deletion construct, the inhibition of mitophagy was drastically reduced with the LIR mutant construct (Figure 3E). Therefore, we conclude that the LIR motif and the AHD3 domain of TNIP1 are both contributing to its role in inhibiting mitophagy. These results are consistent with the results obtained with CID, where we identified the AHD3 domain and LIR motif as being important for TNIP1 function.
Fig. 3. TNIP1 is a negative regulator of selective autophagy.

(A) IB of HeLa cells, HeLa TNIP1 KO cells and HeLa TNIP1 KO rescued with FKBP-GFP-TNIP1. Green arrow, FKBP-GFP-TNIP1 construct. Black arrow, endogenous TNIP1.
(B) HeLa cells stably expressing mito-mKeima and HA-Parkin in wild type cells, TNIP1 KO cells or TNIP1 KO cells rescued with FKBP-GFP-TNIP1 construct were treated with Oligomycin and Antimycin (O/A) for 6 h and subjected to FACS analysis. Left, representative FACS plot. Right, bar graph representing data as mean ± SEM obtained from 3 independent replicates.
(C) HeLa cells stably expressing mito-mKeima and HA-Parkin in wild type cells, TNIP1 KO cells or TNIP1 KO cells rescued with FKBP-GFP-TNIP1 construct were treated with O/A for 2 h and subjected to FACS analysis. Left, representative FACS plot. Right, bar graph representing data as mean ± SEM obtained from 3 independent replicates.
(D) HeLa cells stably expressing mito-mKeima, HA-Parkin and overexpressing FKBP-GFP-TNIP1 wild type, ΔAHD1, ΔAHD3 or ΔAHD4 constructs were treated with O/A for 2 h and subjected to FACS analysis. Left, representative FACS plot. Right, bar graph representing data as mean ± SEM obtained from 3 independent replicates.
(E) HeLa TNIP1 KO cells stably expressing mito-mKeima, HA-Parkin and rescued with FKBP-GFP-TNIP1 wild type, LIR2 mutant or ΔAHD3 constructs were treated with O/A for 3 h and subjected to FACS analysis. Left, representative FACS plot. Right, bar graph representing data as mean ± SEM obtained from 3 independent replicates.
(F) IB of HeLa cells stably expressing BFP-Parkin and GFP-TNIP1 WT treated for 15 h, 18 h, 21 h or 24 h with O/A. Right, bar graph representing data as mean ± SEM obtained from 3 independent replicates.
We also performed the same over-expression experiments with TNIP2 and TNIP3 (Supp Figure 3D). While TNIP2 overexpression had no effect, TNIP3 overexpression showed a mild inhibition of mitophagy, but much less than seen with TNIP1 over-expression. This may be explained by the presence of an AHD3 domain in TNIP3 but not in TNIP2.
We next performed a complementary mitophagy assay by monitoring the degradation of the mitochondrial matrix protein MTCO2 (COXII) and outer membrane protein MFN2. In WT cells expressing BFP-Parkin, the outer mitochondrial protein MFN2 is rapidly degraded via the proteasome, while the mitochondrial electron transport chain COXII is degraded at later time points in the lysosome. A robust degradation of COXII was observed after 15 h, progressing further after 24Hrs of mitochondrial depolarization (Figure 3F). When GFP-TNIP1 is overexpressed, the steady state level of COXII is higher than in non-overexpressing cells. 15 h after mitophagy induction, COXII degradation is impaired in GFP-TNIP1 overexpressing cells. 24 h after mitophagy induction, however, COXII levels are similar in non-overexpressing and overexpressing cells, reflecting the observation with the FACS data where TNIP1 reduces the rate of mitophagy but does not abolish it. As we wondered whether TNIP1’s function was specific for mitophagy, we investigated non-selective autophagy. Ectopic expression of TNIP1 did not influence starvation-mediated autophagy as degradation of p62/SQSTM1 was not altered when GFP-TNIP1 was overexpressed (Supp Figure 3E). We also used a modified version of the steady state autophagic flux probe YFP-LC3B_RFP-LC3BΔG reporter29, where autophagic flux can be estimated by measuring the YFP/RFP signal ratio. Because we use the YFP and RFP channels for our imaging experiments, we overexpress TNIP1 with a BFP tag (Supp Figure 4A) and observed that it had no effect on the autophagic flux upon starvation (Supp Figure 4B). We next investigated whether TNIP1 was also involved in inhibition of other selective autophagy pathways by monitoring aggregate clearance using puromycin treatment30. In cells that ectopically express GFP-TNIP1, we observed a defect in aggregate clearance when puromycin was washed out compared to non-overexpressing cells (Supp Figure 4C). Additionally, we explored an alternative mitophagy pathway triggered by iron depletion31. We treated cells with the iron chelator Phenanthroline (Phen) for 16 h and observed the mitophagy response by FACS in WT and TNIP1 KO cells expressing the mt-mKeima reporter. To our surprise and contrary to results seen with mitophagy triggered by membrane depolarization, TNIP1 KO cells displayed a small defect in mitophagy response compared to WT cells (Supp Figure 4D). The lack of inhibition of Phenmediated mitophagy may relate to the lack of ubiquitination in this pathway32, in contrast to Parkinmediated mitophagy and aggrephagy. Together, these data indicate that TNIP1 inhibits some forms of selective autophagy but not non-selective autophagy.
Binding of TAX1BP1 to the AHD3 domain is required for TNIP1’s inhibition of mitophagy
To obtain mechanistic insights into the function of TNIP1 as a negative regulator of selective autophagy, we performed an unbiased mass spectrometry (MS) screen to identify specific interactors of the AHD3 domain. We immuno-precipitated (IP) HA-TNIP1 full length, TNIP1 ΔAHD3 and TNIP1 ΔAHD4 and determined high confidence interaction partners (HCIPs) by MS followed by Comparative Proteomics Analysis Software Suite (compPASS) analysis as previously described33,34 (Figure 4A, Table S1). Gene Ontology (GO) analysis revealed autophagy components as the only significantly enriched class of proteins. In particular, RAB11FIP5, the autophagy receptors TAX1BP1, OPTN and to a smaller extent CALCOCO2 (NDP52) as well as the ULK1 complex component RB1CC1 (FIP200) were the most abundant HCIPs.
Fig. 4. The interaction between the AHD3 domain of TNIP1 and the Zinc-Finger domain of TAX1BP1 is necessary for TNIP1’s mitophagy inhibition.

(A) Left, High Confidence Candidate Interaction Proteins (HCIPs) network of HA-TNIP1 following an HA-pull down and mass spectrometry analysis. The size of the bait represents higher Z score interaction. Right, heat map of the interactors with wild type TNIP1 or ΔAHD3 and ΔAHD4 mutants.
(B) Co-IP and IB of HEK293T cells stably expressing HA-TNIP1 full length, ΔAHD1, ΔAHD3 and ΔAHD4 constructs or HA-TNIP3 using magnetic HA beads.
(C) Top, schematic representation of full length TAX1BP1 and the regions encompassing the coiled-coil domain (CC), SKICH truncation (ΔSKICH) and Zinc-Finger truncation (ΔZinc-Finger) constructs. Bottom, Co-IP and IB of HEK293T cells stably expressing GFP-TNIP1 full length and transiently expressing HA-TAX1BP1 full length, CC, ΔSKITCH and ΔZinc-Finger constructs using magnetic GFP beads.
(D) HeLa WT cells or TAX1BP1 KO stably expressing mito-mKeima, HA-Parkin and overexpressing FKBP-GFP-TNIP1 construct were treated with O/A for 3 h and subjected to FACS analysis. Left, representative FACS plot. Right, bar graph representing data as mean ± SEM obtained from 3 independent replicates.
(E) Co-IP and IB of HeLa TAX1BP1 KO cells stably expressing HA-TNIP1 full length or the ΔAHD1, ΔAHD3, ΔAHD4 constructs using magnetic HA beads. Ø: no overexpressed construct. * Marks unspecific band.
Consistent with previous reports11, we observed that the TNIP1-OPTN interaction occurs via the AHD4 domain by IP/MS and validated this by IP/immunoblotting (Figure 4A and 4B). We also confirmed binding of TNIP1 to TAX1BP135, which we found to require the AHD3 domain. As a control, we used TNIP3, that also possesses an AHD3 domain and observed binding to TAX1BP1 but not to OPTN. To test the possibility that TNIP1’s function in mitophagy might depend on TAX1BP1 binding, we further characterized the interaction between TNIP1 and TAX1BP1. We overexpressed HA-tagged TAX1BP1 full length, coiled-coil (CC), ΔSKICH or Δzinc-finger (ZF) constructs in 293T cells stably expressing GFP-TNIP1 followed by anti-GFP IPs (Figure 4C). These experiments revealed that TNIP1 interacts with full length TAX1BP1 and ΔSKICH TAX1BP1, but not with TAX1BP1 CC and ΔZF constructs indicating that TNIP1 binds via the TAX1BP1 ubiquitin binding domain ZF. We performed FACS experiments to assess the involvement of TAX1BP1 in TNIP1’s role in mitophagy inhibition (Figure 4D). No reduction in mitophagy was observed in TAX1BP1 KO cells compared to WT cells, in accordance to previously published data24. Furthermore, overexpressed TNIP1 in TAX1BP1 KO cells did not inhibit mitophagy to levels similar to those in WT cells. Overall, TNIP1 binds the ZF domain of TAX1BP1 via its AHD3 domain, and this binding is critical for its inhibitory action.
Binding of FIP200 to an evolutionarily conserved LIR motif is required for TNIP1’s inhibition of mitophagy
Other HCIPs in our MS data were FIP200 and other members of the ULK1 complex (ATG13 and ATG101). We confirmed FIP200 binding by IP/Immunoblotting with TNIP1 but not with TNIP3 (Figure 4B). FIP200 binding to TNIP1 potentially explains how CID with TNIP1 induces mitophagy, as it was previously demonstrated that CID with a FIP200-binding peptide was sufficient for mitophagy induction7. Corroborating the MS data that showed the ΔAHD4 mutant seemed to bind more strongly to FIP200 (Figure 4A), the ΔAHD4 mutant displayed a much stronger IP interaction with FIP200 (Figure 4B). We next performed IP/Immunoblotting in TAX1BP1 KO cells and confirmed that TNIP1 was also able to pull down FIP200 (Figure 4E), implying that TNIP1 binding to FIP200 does not depend on TAX1BP1. Surprisingly, the increase of FIP200 binding seen with constructs ΔAHD3 and ΔAHD4 was not observed in TAX1BP1 KO cells, suggesting that TAX1BP1 may mediate this stronger binding. However, while the data in Figure Fig.B was obtained using 293T cells, Figure 4E was obtained using HeLa cells, thus we cannot exclude that this observation could be cell type specific.
SQSTM1/p62 was recently demonstrated to bind to FIP200 through its LIR motif 36. We therefore examined whether TNIP1 would utilize a similar binding mode and asked whether the LIR2 mutant construct bound FIP200 (Figure 5A). Confirming our hypothesis, we could not detect any FIP200 binding with the LIR2 mutant TNIP1. In addition, consistent with our MS analyses (Figure 4A and B), we did not detect co-immunoprecipitation of TNIP1 with WIPI2. We also did not detect binding of N-Terminal and C-terminal deletion constructs of TNIP1 to FIP200 (Supp Figure 5A), which is in line with our CID experiments (Supp Figure 2C). This is somewhat surprising as we expected the N-terminal construct to still bind FIP200 as it retains the LIR motif. Truncating TNIP1 may disturb its folding and thus its ability to bind FIP200. In order to exclude the possibility that TNIP1 could pull down FIP200 via a secondary interaction with mATG8 binding, we performed CID experiments coupled with immunocytochemistry in a mATG8 6KO or TAX1BP1 KO background (Figure 5B and Supp Figure 5B). In WT cells, TNIP1 was able to recruit FIP200 upon Rapalog treatment. The LIR2 mutant TNIP1, however, was less able to recruit FIP200, confirming that this region is necessary for FIP200 binding. Recruitment of FIP200 to mitochondria in mATG8 6KO and TAX1BP1 KO cells was similar as in WT cells and the LIR mutant construct significantly impaired recruitment of FIP200 to the mitochondria. These results confirm that FIP200 binding to TNIP1 is independent of mATG8 proteins as well as TAX1BP1. To further characterize the interaction between TNIP1 and FIP200, we explored the binding region to TNIP1 within FIP200 (Figure 5C). While TNIP1 was able to bind full length and the C-terminal region of FIP200, it was not able to bind the N-terminal part of FIP200 (Figure 5C), prompting us to further map the C-terminal region for the interaction with FIP200. NDP52 was previously shown to interact with the leucine zipper domain of FIP200, while the CLAW domain of FIP200 was recently identified as responsible for binding with p62/SQSTM136. TNIP1 was able to pull-down only the CLAW domain of FIP200, indicating that the CLAW domain is the minimal necessary region for binding to TNIP1 (Figure 5C).
Fig. 5. TNIP1 binds FIP200 via its LIR motif and to the CLAW domain of FIP200.

(A) Co-IP and IB of HEK293T cells stably expressing HA-TNIP1 full length or the ΔAHD1, ΔAHD3, ΔAHD4 and LIR2 mutant constructs using magnetic HA beads.
(B) HeLa wild type, TAX1BP1 KO and LC3/GABARAP 6KO cells stably expressing FRB-FIS1 and FKBP-GFP-TNIP1 WT or LIR2m were treated for 24 h with Rapalog and stained for endogenous FIP200 before immunofluorescence acquisition on a confocal microscope. Quantifications as mean ± SEM of Pearson correlation coefficient representing colocalization between FIP200 and GFP. See Fig S5B for representative images.
(C) Top, schematic representation of full length FIP200 and the regions encompassing the N-terminal and C-terminal domains as well as the minimal leucine zipper (LZ) and Claw (CLW) domains. Bottom, Co-IP and IB of HEK293T cells stably expressing GFP-TNIP1 full length and transiently expressing HA-FIP200 full length, N-terminal, C-terminal, LZ and CLW constructs using magnetic GFP beads. The zone in dotted lines was exposed longer to reveal the binding to the CLAW domain (lower part).
Phosphorylation of the FIR motif regulates TNIP1’s binding to FIP200 CLAW domain
A consensus FIP200 Interacting Region (FIR) core motif present in several autophagy receptors was recently reported, showing that this FIR motif overlapped with their LIR motif37 and that phosphorylation of a residue immediately preceding the FIR core motif increases binding to the CLAW domain of FIP200. Careful examination of the LIR motif of TNIP1 showed one threonine and two serine are immediately preceding the LIR motif (Supp Figure 1A). Furthermore, these serine proximal to the LIR domain were recently found to be phosphorylated, resulting in an increased binding to LC3s38. We thus hypothesized that TNIP1 interacts with FIP200 in a phosphor-FIR-dependent manner and that allows it to compete with autophagy receptors for interaction with FIP200. To test this, we generated FITC-labeled peptides unmodified or with either a phosphorylated S122 (pS122) or phosphorylated S123 (pS123) and monitored their ability to bind recombinant CLAW domain using fluorescence polarization (FP) as previously demonstrated for the FIR of ER-phagy receptor protein CCPG1. Phosphorylation at either site on TNIP1 displayed a stronger binding to the CLAW domain, with a KD of 46μM for unmodified TNIP1 versus a KD of 9.7μM for the pS122 peptide and 3.27μM for the pS123 peptide (Figure 6A). Using unlabeled TNIP1 peptides, we performed a competition assay with the FITC-labeled pCCPG1 FIR peptide for the binding to the CLAW domain. TNIP1 pS123 peptide was able to compete with CCPG1 for the binding with the CLAW domain, with an absolute IC50 of 70.16μM (Figure 6B). This suggests that TNIP1 acts as a competitive inhibitor and provides an explanation of how TNIP1 inhibits mitophagy, by competing with autophagy receptors for the binding with the CLAW domain. Of note, the mitophagy receptor OPTN was previously measured with a KD of 306μM for unmodified and 11.5μM when phosphorylated37, indicating a lower affinity for the CLAW domain than TNIP1, suggesting that TNIP1 would outcompete OPTN for binding with the CLAW domain. To our knowledge, TAX1BP1 binding to the CLAW domain of FIP200 has not been estimated. To validate that full length TNIP1 binds the CLAW domain of FIP200, we purified recombinant TNIP1 protein and performed dynamic light scattering (DLS) analysis. When TNIP1 (red) and CLAW (blue) were measured alone, peaks around 2.5nm were observed, with a broader distribution for purified TNIP1 alone. Mixing CLAW and TNIP1 at equal mass-ratios lead to the observation of a second larger peak at 5.5nm (purple), indicating that TNIP1 and CLAW bind together. Adding the pFIR of CCPG1 in excess (100μM) prevented TNIP1 binding to the CLAW domain, reducing the formation of the 5.5nm complex, shifting the apparent peak to 3.1nm (green) (Supp Figure 6A). We noticed the formation of a precipitate upon mixing TNIP1 and the CLAW domain, suggesting larger particle formation than is typically measured by DLS. We thus assessed the turbidity, allowing us to characterize the formation of larger particles when TNIP1 and CLAW were mixed, confirming the formation of an insoluble complex. Adding excess of pFIR CCPG1 peptide resulted in perturbation of this complex by competition binding and partially prevented this precipitation (Supp Figure 6B).
Fig. 6. TNIP1 binds FIP200 via its FIR motif and is regulated by a phosphorylation upstream of FIR.

(A) Fluorescence polarization of unmodified and phosphorylated FIR peptides of TNIP1 and CCPG1 binding to the CLAW domain of FIP200 with increasing concentrations of CLAW. Error bars represent the standard deviation measured across 3–6 independent experiments for each condition.
(B) Competition of the TNIP1 peptides for the displacement of CCPG1 peptides from binding with the CLAW domain of FIP200. Error bars represent the standard deviation measured across four independent experiments for each condition.
(C) HeLa cells stably expressing mito-mKeima, HA-Parkin and overexpressing GFP-TNIP1 WT or AAA constructs were treated with O/A for 4 h and subjected to FACS analysis. Left, representative FACS plot. Right, bar graph representing data as mean ± SEM obtained from 3 independent replicates.
(D) Co-IP and IB of HeLa cells stably expressing GFP-TNIP1 WT or AAA constructs using magnetic GFP beads.
(E) Co-IP and IB of HeLa cells stably expressing HA-TNIP1 AAA mutant with purified recombinant GST tagged mATG8 proteins.
(F) Co-IP and IB of HEK293T cells stably expressing HA-TNIP1 treated for 6 h with the TBK1 inhibitor GSK8612 and/or O/A for 4 h using magnetic HA beads.
(G) Schematic representation of the proposed model of the molecular function of TNIP1. Upon mitophagy stimulation, autophagy receptors are bound to the ubiquitinated mitochondria, and subsequently recruit the ULK1 complex via FIP200 binding, starting the autophagosome formation. Before autophagosomal closure, TNIP1 is targeted to the autophagosome via LC3/GABARAP binding and later activated by TBK1, increasing its binding affinity for FIP200 and competing with the receptors for FIP200 binding, ultimately resulting in release of FIP200 from the autophagosome. Mitophagy can thus proceed and FIP200 can be recycled at another location on the growing phagophore. In a parallel, separate event, excess of TNIP1 inhibits mitophagy by binding TAX1BP1 via AHD3/ZF domains and TAX1BP1 cannot bind ubiquitinated substrates as its UB-binding domain is occupied by TNIP1, as represented in the graphical abstract.
We next performed FACS experiments to assess the contribution of the phosphorylation to the function of TNIP1 by using a phospho-dead mutant, where Thr121, Ser122 and Ser123 residues were replaced by Ala (TNIP1-AAA). Ectopic expression of TNIP1-AAA displayed an even stronger mitophagy inhibition than WT indicating that phosphorylation counteracts TNIP1’s inhibitory effect on mitophagy (Figure 6C). This likely occurs through FIP200, as IP/immunoblotting experiments revealed a weaker binding of TNIP1-AAA to FIP200 compared to WT TNIP1 (Figure 6D). We additionally performed GST pull-down of TNIP1-AAA mutant and observed that, while LC3B binding was reduced, no such loss of binding to GABARAP was observed compared to the LIR2 mutant (Figure 6E vs Figure 1B), indicating that TNIP1-AAA likely impairs binding to FIP200 but not to GABARAP. In an attempt to mimic phosphorylated TNIP1, we mutated residues TSS_121–123_EDD and performed IP/immunoblotting. Unfortunately, these mutations did not function as phospho-mimetic and did not recapitulate the FP data and lacked stronger binding to FIP200 (Supp Figure 6C). We also tried using single mutations, but neither S122E nor S123E displayed phospho-mimicking abilities (Supp Figure 6D). TANK-Binding Kinase 1 (TBK1) regulates several autophagy receptors in the autophagy pathway7. Furthermore, TBK1 was previously shown to be recruited to the TNIP1-TAX1BP1 complex35. We thus investigated whether TBK1 was involved in phosphorylating TNIP1. Similar to results seen with the TNIP1-AAA, cells treated with the TBK1 inhibitor displayed weaker binding to FIP200, implying that TBK1 is likely the kinase responsible for phosphorylating TNIP1 (Figure 6F). Lastly, in order to compare the affinity of TNIP1’s FIR peptide for FIP200-CLAW and mATG8 proteins, we performed FP using purified mATG8 proteins (Supp Figure 6E). As seen with FIP200-CLAW, phosphorylated FIR displayed a stronger binding with all six mATG8 proteins than unmodified TNIP1, with the strongest binding observed with LC3C and GABARAP, as observed using GST pulldowns.
Overall, we here show that TNIP1 is a negative regulator of selective mitophagy, by interacting with autophagy receptors and competing with them for binding with FIP200 via a FIR domain. We therefore speculate that FIR phosphorylation by TBK1 modulates the affinity of TNIP1 by increasing binding to FIP200, displacing the complex FIP200-autophagy receptor to a complex comprised of FIP200 and TNIP1 (Figure 6G).
Discussion
Here, we investigated the involvement of TNIP1 in autophagy and identify it as an inhibitor of mitophagy. Our findings have important implications for our understanding of the regulation of early events of mitophagy induction, ULK1 complex recycling at the forming autophagosome, and the development of neurodegenerative diseases.
One of the key findings of this report is the identification of a LIR motif in TNIP1 suggesting that TNIP1 could be driven to the forming autophagosomes by binding to the LC3/GABARAP proteins, and at the same time, allosterically binding to TAX1BP1 via the zinc finger domain, preventing the latter from binding to ubiquitinated cargos, occasioning a defect in mitophagy (See graphical abstract). One of the remaining unelucidated aspects of selective autophagy is the seemingly small amount of the ULK1 complex proteins degraded compared to autophagy receptors. A mechanism must thus take place to displace the complex from the closing autophagosome. Based on the results reported here, we speculate that upon mitophagy activation, releasing FIP200 from binding with autophagy receptors via competition with phosphorylated TNIP1 removes FIP200 from the autophagosome, ultimately allowing FIP200 to be available for further expansion of the autophagosome (Figure 6G). The MS data with the ΔAHD3 and ΔAHD4 mutants showing a stronger interaction with FIP200 could reflect this binding competition between TNIP1, TAX1BP1 (or OPTN) and FIP200, as it seems that losing the domains responsible for binding to the autophagy receptors enhances FIP200 interaction. Additionally, the FIR-binding deficient mutant (AAA mutant) seems to bind more strongly to TAX1BP1 than the WT construct (Supp Figure 6C), exemplifying the binding competition between FIP200 and TAX1BP1.
Although to our knowledge this is the first time that a negative function of mitophagy is described for TNIP1, it was recently shown to be a signal-induced autophagy receptor in the context of inflammation39. Additionally, phosphorylation of serine residues upstream of the LIR motif by TBK1, resulting in an increased binding to LC3 proteins was recently described38. Another study implicated TNIP1 as a modulator of mitophagy40, where its loss resulted in a lower relative mitophagy. However, the apparent discrepancy between this study and our present results can be explained by the fact that the authors looked at a late stage of mitophagy where not much change can be observed, as we observed in Figure 4A.
To date, very few proteins have been characterized as inhibitors of autophagy. The deubiquitinating enzyme (DUB) USP30 was shown to oppose Parkin by removing ubiquitin moieties from mitochondria41. Most other proteins have been identified to negatively regulate bulk autophagy and not selective autophagy. We also show that the role of TNIP1 in inhibition is not solely observed in mitophagy, but in aggrephagy as well, a type of selective autophagy that was previously shown to mainly rely on TAX1BP130. These results are potentially very interesting considering the importance of aggregate clearance in neurodegenerative diseases and the recent findings implicating mitochondrial dysfunction in neurodegenerative disease (ND), and more particularly in Parkinson’s disease (PD) and AD. One can hypothesize that loss of TNIP1’s function in mitophagy regulation could be a factor leading to NDs. On the other hand, for PD, efforts are being made to activate mitophagy. One strategy seen as amenable to pharmacologic manipulation is to inhibit the proteins that inhibit mitophagy such as USP30. One could consider pharmacologic inhibition of TNIP1 as an alternate approach.
Limitations of the study
Our inability to purify TNIP1 and TAX1BP1 in substantial amounts prevented us from performing competition assays assessing the complex formed by FIP200, TAX1BP1 and TNIP1. Structural studies investigating complex dynamic will be crucial to fully understand TNIP1’s function. Furthermore, the Kd of the binding with all six mATG8 is smaller than measured with FIP200-CLAW domain, implying that the binding of the phosphorylated TNIP1 is stronger with mATG8 proteins than with FIP200, although we only used the CLAW domain of FIP200 and not the full-length protein like we did with mATG8 proteins. This makes interpretation of the phosphorylation of the FIR motif of TNIP1 difficult and therefore we cannot conclude with certainty that phosphorylation of TNIP1 displaces binding with mATG8 toward FIP200, although our pull-down experiments show that AAA mutant loses binding to FIP200 but not to GABARAP, indicating that the phosphorylation more likely influences FIP200 than GABARAP in vivo. Lastly, we unfortunately could not test a phospho-mimicking mutant as this did not seem to bind better to FIP200 as true TNIP1 phosphopeptides do. This is not uncommon for phosphomimicking mutants to not always mimic phosphates, and this was already seen with previous LIR motifs42.
Star Methods
Resource availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Richard Youle (youler@ninds.nih.gov).
Materials Availability
Plasmids and cell lines generated in this study will be available upon request.
High content imaging data is deposited at BioImage Archive and is available with the accession number S-BIAD619. Original Western blot data, microscopy images, raw FACS data, raw MS data, raw FP data and code used in this paper have been deposited at Mendeley Data and are publicly available as of the date of publication at 10.17632/jh7h5cx4yh.1. Mass spectrometry data is deposited at MassIVE and is available at MSV000091090.
No new code has been generated for this study.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Experimental Model and Subject Details
Cell line
HEK293T and HeLa cells were purchased from ATCC. HEK293T and HeLa cells were cultured in Dulbecco’s modified eagle medium (DMEM) with 10% (v/v) Fetal Bovine Serum (FBS) (Sigma), 1 mM Sodium Pyruvate, 2 mM GlutaMAX. All media and supplements were from Thermo Fisher. All cells were tested for mycoplasma contamination every two weeks with PlasmoTest kit (InvivoGen). Reagents used for transfections were, X-tremeGENE 9 (Roche) for sgRNA transfection, or Polyethylenimine (PEI) (PolySciences) for all other transfections. The full list of antibodies and reagents are found in Key Resources Table.
Key Resources Table
| Reagents or Resource | Source | Identifier |
|---|---|---|
| Antibodies | ||
| A20 | Bethyl | A302–633A |
| Actin | CST | 4967S |
| ATG16L1 | CST | 8089 |
| COXII | abcam | ab110258 |
| FIP200 (for immunoblot) | CST | 12436 |
| FIP200 (for immunostaining) | Proteintech | 17250–1-AP |
| GABARAP | Sigma-Aldrich | MABN2303 |
| GAPDH | Sigma-Aldrich | G9545 |
| GFP | nvitrogen | A11122 |
| HA | CST | 3724s |
| HA | biolegend | 901513 |
| HA | Covance | MMS-101P |
| LC3B (for immunoblot) | CST | 2775s |
| LC3B (for immunostaining) | MBL | PM036 |
| MFN2 | House made | N/A |
| NDP52 | CST | 60732S |
| OPTN | proteintech | 10837-I-AP |
| SQSTM1 | abnova | H00008878-M01 |
| TAX1BP1 (for immunoblot) | CST | 5105S |
| TAX1BP1 (for immunostaining) | Sigma-Aldrich | HPA024432 |
| TNIP1 (for immunoblot) | Bethyl | A304–508 |
| TNIP1 (for immunostaining) | proteintech | 15104–1-AP |
| TOM20 | Santa Cruz | SC-11415 |
| TOM20 | Santa Cruz | SC-17764 |
| Ubiquitin FK2 | Biomol International |
PW8810–0500 |
| WIPI2 | abcam | ab105459 |
| Alexa Fluor 488 goat anti-rabbit IgG | Thermofisher | A11008 |
| Alexa Fluor 594 goat anti-rabbit IgG | Thermofisher | A11012 |
| Alexa Fluor 405 goat anti-mouse IgG | Thermofisher | A31553 |
| Alexa Fluor 488 goat anti-mouse IgG | Thermofisher | A21202 |
| Alexa Fluor 594 goat anti-rat IgG | Thermofisher | A11007 |
| Amersham | ECL Rabbit IgG, | HRP-linked |
| Amersham | ECL Mouse IgG, | HRP-linked |
| Chemicals, peptides and recombinant proteins | ||
| A/C heterodimerizer (Rapalog) | Takaro Bio | 635056 |
| antimycin A | Sigma-Aldrich | A8674 |
| Bafilomycin | Cayman Chemical Company |
11038 |
| DAPI | Thermofisher | 62248 |
| Oligomycin | CalBiochem | 495455 |
| Cycloheximide | Cayman Chemical Company |
14126 |
| Puromycin | Invivogen | ant-pr-1 |
| GSK8612 | MedChemExpress | HY-111941 |
| Polybrene | Sigma-Aldrich | H9268 |
| Q-VD | ApexBio | A1901 |
| FBS | R&D Systems | S11550H |
| DMEM | Thermofisher | 31053028 |
| Sodium pyruvate | Thermofisher | 11360070 |
| GlutaMax | Thermofisher | 35050061 |
| Opti-MEM | Thermofisher | 31985062 |
| 0.05% Trypsin-EDTA | Thermofisher | 25300054 |
| BamBanker | Wako | 302–14681 |
| DTT | Sigma-Aldich | D0632 |
| HBSS with calcium and magnesium | Thermofisher | 14025–092 |
| paraformaldehyde 16% solution, | Electron Microscopy Sciences |
15710 |
| polyethylenimine, (PEI 25K) | PolySciences | 23966–1 |
| XtremeGENE9 | Sigma-Aldich | 6365787001 |
| PhosSTOP | Roche | 4906845001 |
| Protease inhibitor cocktail | Roche | 11873580001 |
| Triton X-100 | Sigma-Aldrich | T9284 |
| Tween-20 | Sigma-Aldrich | P7949 |
| 3xHA peptide | Thermofisher | 26184 |
| MS grade trypsin | Promega | V5280 |
| Pierce™ MMTS | Thermofisher | 23011 |
| 1M TEAB | Thermofisher | 90114 |
| RIPA buffer | Thermofisher | 89900 |
| 4X LDS sample buffer | Thermofisher | NP0007 |
| Goat serum | VWR Scientific Products | 76230–088 |
| DAPI Fluoromount-G | SouthernBiotech | 0100–20 |
| Commercial assays | ||
| Amersham ECL Prime WB detection reagent |
GE Healthcare Life Sciences | RPN2232 |
| BCA protein assay | Thermofisher | 23228 |
| SuperSignal West Femto | Thermofisher | 1859022 |
| NEBuilderHiFi DNA assembly master mix | New England BioLabs | M5520A |
| Q5 Hot Start High-Fidelity DNA Polymerase | New England BioLabs | M0493L |
| DNA Ligation kit, Mighty Mix | Takara | 6023 |
| Gateway BP clonase | Thermofisher | 117890–20 |
| Gateway LR clonase | Thermofisher | 11–791-020 |
| GFP-TRAP magnetic beads | Chromotek | gtd-100 |
| Pierce™ Anti-HA Magnetic Beads | Thermofisher | 88837 |
| Pierce™ Glutathione Agarose | Thermofisher | 16102BID |
| S-Trap column | ProtiFi | co2-micro-10 |
| Mycoplasma Detection Kit | Invivogen | rep-mys-100 |
| Pre-treated 1.5mm coverslips | neuVitro | GG-18–1.5-pre |
| 96 well plate glass bottom | DOT Scientific Inc | MGB096–1-2-LG-L |
| Invitrogen Novex NuPAGE 4 12% Bis Tris Protein Gels |
Thermofisher | NP0321BOX |
| Experimental models: Cell lines | ||
| HeLa | ATCC | CCL-2.2 |
| 293T | ATCC | CRL-3216 |
| HeLa FIP200 KO | Vargas et al, 2019 | N/A |
| HeLa WIPI2 KO | Fischer et al, 2020 | N/A |
| HeLa A20 KO | this study | N/A |
| HeLa TNIP1 KO | this study | N/A |
| HeLa autophagy receptor 5KO | Lazarou et al, 2015 | N/A |
| HeLa TAX1BP1 KO | This study | N/A |
| HeLa ATG8 6KO | Nguyen et al, 2016 | N/A |
| Oligonucleotide | ||
| TNIP1 sgRNA GACCCCCTGGCTGAGCTCAC |
This Study | N/A |
| TAX1BP1 sgRNA AAUUGUGUACUAGCAUUCCA |
This Study | N/A |
| A20 sgRNA GGCTGAACAAGTCCTTCCTC |
This Study | N/A |
| Recombinant DNA | ||
| TNIP1 | Harvard medical school | HsCD00377161 |
| pENTR223-TNIP1 | this study | N/A |
| pENTR223-TNIP2 | this study | N/A |
| pENTR223-TNIP3 | this study | N/A |
| pENTR223-TNIP1 LIR1mut | this study | N/A |
| pENTR223-TNIP1 LIR2mut | this study | N/A |
| pENTR223-TNIP1 LIR1+LIR2mut | this study | N/A |
| pDONR223_TNFAIP3_WT | Addgene | 82238 |
| pHAGE-FLAG-HA TNIP1 WT | this study | N/A |
| pHAGE-FLAG-HA TNIP1 LIR1 mutant |
this study | N/A |
| pHAGE-FLAG-HA TNIP1 LIR2 mutant |
this study | N/A |
| pHAGE-FLAG-HA TNIP1 LIR1+LIR2 mutant | this study | N/A |
| pHAGE-FLAG-HA TNIP1 ΔAHD1 |
this study | N/A |
| pHAGE-FLAG-HA TNIP1 ΔAHD3 |
this study | N/A |
| pHAGE-FLAG-HA TNIP1 ΔAHD4 |
this study | N/A |
| pHAGE-FLAG-HA TNIP1 ΔCterminal |
this study | N/A |
| pHAGE-FLAG-HA TNIP1 ΔNterminal |
this study | N/A |
| pHAGE-FLAG-HA TNIP1 AAA | this study | N/A |
| pHAGE-FLAG-HA TNIP1 EDD | this study | N/A |
| pHAGE-FLAG-HA TNIP1 S122A | this study | N/A |
| pHAGE-FLAG-HA TNIP1 S122E | this study | N/A |
| pHAGE-FLAG-HA TNIP1 S123A | this study | N/A |
| pHAGE-FLAG-HA TNIP1 S123E | this study | N/A |
| pHAGE-FLAG-HA TNIP3 WT | this study | N/A |
| pHAGE-FLAG-HA TAX1BP1 WT | Sarraf et al, 2020 | N/A |
| pHAGE-FLAG-HA TAX1BP1 CC | Sarraf et al, 2020 | N/A |
| pHAGE-FLAG-HA TAX1BP1 ΔSKITCH |
Sarraf et al, 2020 | N/A |
| pHAGE-FLAG-HA TAX1BP1 ΔZF |
Sarraf et al, 2020 | N/A |
| pHAGE-FLAG-HA SQSTM1 | this study | N/A |
| pHAGE-FLAG-HA FIP200 ΔCterminal |
Vargas et al, 2019 | N/A |
| pHAGE-FLAG-HA FIP200 ΔNterminal |
Vargas et al, 2019 | N/A |
| pHAGE-FLAG-HA FIP200 LZ | Vargas et al, 2019 | N/A |
| pHAGE-FLAG-HA FIP200 CLAW | Vargas et al, 2019 | N/A |
| pHAGE-GFP TNIP1 WT | this study | N/A |
| pHAGE-GFP TNIP1 AAA | this study | N/A |
| pHAGE-BFP TNIP1 WT | this study | N/A |
| pHAGE-FKBP-GFP-TNIP1 WT | this study | N/A |
| pHAGE-FKBP-GFP-TNIP1 D472N | this study | N/A |
| pHAGE-FKBP-GFP-TNIP1 LIR1 mutant | this study | N/A |
| pHAGE-FKBP-GFP-TNIP1 LIR2 mutant | this study | N/A |
| pHAGE-FKBP-GFP-TNIP1 LIR1+LIR2 mutant | this study | N/A |
| pHAGE-FKBP-GFP-TNIP2 | this study | N/A |
| pHAGE-FKBP-GFP-TNIP3 | this study | N/A |
| pHAGE-FKBP-GFP-TNIP1 ΔAHD1 |
this study | N/A |
| pHAGE-FKBP-GFP-TNIP1 ΔAHD3 |
N/A | |
| pHAGE-FKBP-GFP-TNIP1 ΔAHD4 |
this study | N/A |
| pHAGE-FKBP-GFP-TNIP1 ΔAHD3+LIR2 mutant |
this study | N/A |
| pHAGE-FKBP-GFP-TNIP1 ΔCterminal |
this study | N/A |
| pHAGE-FKBP-GFP-TNIP1 ΔNterminal |
this study | N/A |
| pHAGE-FKBP-GFP-ATG16L1 | Vargas et al, 2019 | N/A |
| pHAGE-FKBP-GFP-WIPI2 | this study | N/A |
| pHAGE-FKBP-GFP-A20 | this study | N/A |
| pSpCas9-P2A-Puro | Ran et al, 2013 | N/A |
| pHAGE-mtKeima-P2A-HA-Parkin | this study | N/A |
| pHAGE-YFP-LC3B-RFP-LC3BΔG | this study | N/A |
| Software and algorithm | ||
| Fiji | Schindelin et al., 2012 | https://imagej.net/software/fiji/ |
| Prism | https://www.graphpad.com/scientificsoftware/prism/ | |
| FlowJo | Becton, Dickinson and Company; 2023 |
https://www.flowjo.com |
| SnapGene | N/A | www.snapgene.com |
| Image Lab | Biorad | https://www.bio-rad.com/enus/product/image-labsoftware?ID=KRE6P5E8Z |
| BioRender | N/A | https://app.biorender.com/user/signin |
| MATLAB 2022a | N/A | N/A |
Method Details
Knockout line generation using CRISPR/Cas9 gene editing
CRISPR gRNAs were generated to target exon 3 of TNIP1, exon 2 of TNFAIP3 and exon 3 of TAX1BP1. gRNAs were cloned into pSpCas9(BB)-2A-Puro (PX459) V2.0 (Addgene plasmid #62988). To make KO, HeLa cells were transfected with the gRNA plasmid and treated with 1 ug/ml puromycin for 2 days to enrich transfected cells, which were then diluted and placed into 96-well plates for single colonies. Primer set (ggtggaccagcatggagttt and accagggagcttccaactca) were used for PCR screening of TNIP1 KO clones. Primer set (tcagtacccactctctgccttc and ctccaagcctcaatgtgctct) were used for PCR screening of TNFAIP3 KO clones. Primer set (ttatccttgagaaattggatagca and tagtacctaaaaagaaacccactcttc) were used for PCR screening of TAX1BP1 KO clones.
Cloning, mutagenesis and stable cell line generation
For lentiviral constructs, inserts were either amplified by PCR and cloned into pHAGE vector, respectively by Gibson assembly (New England Labs) or Gateway cloning (Thermo Fisher). Deletion mutants were generated using Gibson Cloning. Point mutants were generated by site directed mutagenesis or Gibson cloning. All constructs used or generated in this study were validated by Sanger sequencing and complete plasmid sequences and maps are available upon request. Stable expression of lentiviral constructs in HeLa or HEK293T cells were achieved as follows: lentiviruses were packaged in HEK293T cells by transfecting constructs together with appropriate helper plasmids and PEI. The next day, media was exchanged with fresh media. Viruses were harvested 48 hrs and 72 hrs after transfection and transduced in HeLa or HEK293T cells with 8 μg/mL polybrene (Sigma). Cells were then directly used in experiments or optimized for expression by FACS.
Immunoblot Analyses
Cells seeded into 12-well plates were washed with phosphate buffered saline (PBS) and lysed with RIPA buffer. The protein concentration was measured using a BCA kit. Samples were boiled at 99°C for 5 min. 20–50 μg of protein lysate of each sample was loaded and separated on 4–12% Bis-Tris gels (Thermo Fisher) according to manufacturer’s protocol. Gels were transferred to polyvinyl difluoride membranes and immunostained using specific antibodies. For mitophagy measurements by immunoblotting, cells were treated with 10 μM Oligomycin (Calbiochem), 10 μM Antimycin A (Sigma) and 10 μM QVD (ApexBio) in growth medium at different timepoints indicated in figure legends, prior to western blot analysis. For starvation-induced autophagy, by immunoblotting, cells were washed 3 times with PBS and incubated for the indicated time with HBSS containing calcium and magnesium.
Recombinant protein/peptide production and protein purification
mATG8 proteins (LC3A, LC3B, LC3C, GABARAP, GABARAPL1, GABARAPL2) were first cloned into pENTR vector using GATEWAY cloning. They were cloned into pDEST60 GST vector using LR clonase. GST expression vector were expressed into DL21 (DE3) bacteria. 50ml LB broth was inoculated with 2ml pre-culture of the GST constructs. Bacteria were incubated for ˜1 hr at 37°C with agitation until an optical density of ˜0.6 was reach. 400μM IPTG was used to induce protein production for 4 Hrs. Bacteria were collected and the pellet was lysed with lysis buffer (20mM TRIS-HCl pH7.5, 10mM EDTA, 5mM EGTA, 150mM NaCl, 0.5% NP40, 1% Triton X-100, Benzonase, 1mM DTT, protease inhibitor, 2mg/ml lysozyme). The lysate was shock-freezed with liquid nitrogen before thawing and sonication. The samples were centrifuged and cleared lysate was incubated with 100μl of slurry GST beads, followed by an overnight incubation at 4°C on a rotating shaker. Beads were subsequently washed (20mM TRIS-HCl pH 7.5, 10mM EDTA, 5mM EGTA, 150mM NaCl, 1mM DTT) before exchanging the buffer for the storage buffer (20mM TRIS-HCl pH 7.5, 10mM EDTA, 5mM EGTA, 150mM NaCl, 1mM DTT, 5% glycerol, proteinase inhibitor).
Production of recombinant His6-TEV-FIP200-CLAW domain was purified from E. coli BL21 (DE3) containing the plasmid was grown in TB medium containing 50 μg/L ampicillin, which was shaken at 37 °C to an OD600 of 0.5. The cell culture was cooled down to 20 °C and induced with 1 mM isopropyl-beta-D-thiogalactopyranoside (IPTG) and harvested 16 h later by centrifugation. The cell pellet (≈6 g from 2 L) was suspended in the 30 mL lysis buffer [100 mM Tris (pH 7.4), 500 mM KCl, 5 mM MgCl2, 20 mM imidazole, 5% glycerol, 2 mM β-mercaptoethanol, and protease inhibitor tablet (Roche)]. The cells (held in an ice bath) were lysed by six 30-s pulses of sonication, separated by 2-min intervals. The lysate was centrifuged at 20,000×g for 45 min at 4 °C, and the resulting supernatant was loaded onto a Ni-NTA column [5-mL suspension, preequilibrated with wash buffer (50 mL, 50 mM HEPES [pH 7.4], 150 mM KCl, 5 mM MgCl2, and 20 mM imidazole)] and incubated at 4 °C with rotation for 30 min. The column was then flushed with wash buffer (100 mL), and His6-tagged FIP200-CLAW was eluted by an increased ration of imidazole elution buffer (500 mM imidazole in wash buffer) to wash buffer. Fractions containing the CLAW proteins were combined and concentrated with an Amicon Ultra-15 centrifugal filter unit (nominal molecular weight limit = 10 kDa). The mixture (0.5 mL of 20 mg/mL) was then fractionated with a gel filtration column (Tricorn Superdex 200; GE Healthcare), eluted with GF buffer [20 mM HEPES (pH 7.4), 150 mM KCl, and 1 mM MgCl2] at 0.5 mL/min flow rate, and fractions corresponding to an apparent molecular weight of 15–30 kDa were collected and analyzed by 4–12% SDS/PAGE to evaluate purity (Invitrogen). Fractions that contained FIP200 of ≥95% purity were concentrated to 4 mg/mL, exchanged into storage buffer [20 mM HEPES (pH 7.4), 150 mM KCl, 1 mM MgCl2, 5% glycerol, and 1 mM TCEP], aliquoted, frozen in liquid nitrogen, and stored at −80 °C.
Full-length TNIP1 cDNA was cloned into pFastbac-HTA vector with an N-terminal 6x His tag. Bac-to-Bac® Baculovirus Expression System (Invitrogen) and pFastbac-HTA-6x His-TNIP1 were used to generate recombinant baculoviruses. To express TNIP1, 1L of Sf9 cells (1.5X106 cells/ml) were infected with recombinant TNIP1 baculovirus and incubated in a 28°C-orbital shaker at 100 rpm. After 72 hours of incubation, insect cells were centrifuged at 1000xg for 20 minutes and cell pellets were flash frozen in liquid nitrogen and stored at −80°C. For protein purification, the insect cell pellets containing 6xHis-TNIP1 were resuspended in 25 ml Buffer A (50 mM Tris-HCl pH 8.0, 300 mM NaCl, 10% glycerol, 0.1% Triton X, protease inhibitor cocktail). Cells were lysed using a microfluidizer at 15000 psi. The lysate was centrifuged at 50,000xg at 4°C for 35 minutes to clear cell debris. The supernatant was incubated with 1 mL of Ni-NTA agarose (Qiagen) at 4°C with continuous rotation for 2h. Ni-NTA agarose beads were washed with Buffer A containing 20 mM imidazole and His-TNIP1 was eluted in buffer A with 250 mM imidazole. Subsequently, PD-10 Desalting Columns containing Sephadex G-25 resin was used to remove imidazole from the His-TNIP1 protein solution. His-TNIP1 was further purified by size exclusion chromatography using the Superdex™ 200 Increase 10/300 GL column. Purified His-TNIP1 was aliquoted, flash-frozen in liquid nitrogen, and stored at −80°C. All TNIP1 and CCPG1 peptides used in this study were synthesized in vitro by Genescript.
Immunoprecipitation, GST-pull down, GFP-TRAP and HA beads precipitation
For GFP-TRAP (Chromotek), HA-beads (Pierce) and GST precipitation experiments, HEK293T or HeLa cells seeded in 10cm plates were co-transfected with specific constructs to overexpress proteins of interest for 24 to 48 h, if indicated. IPs were performed following manufacturer’s instructions.
Briefly, for HA-IP, cells were then lysed using ice cold lysis buffer (25 mM Tris-HCl pH 7.4, 150 mM NaCl, 1% NP-40, 1 mM EDTA, 5% glycerol) supplemented with EDTA-free cOmplete protease inhibitor (Roche). Samples were incubated on ice with intermittent agitation by pipetting for 10 min. Beads were equilibrated using bead resuspension buffer (TBS containing 0.05% Tween-20 Detergent (TBS-T)). Protein lysates were precleared by centrifugation at 4°C for 10 min at 15,000 g. 1% INPUT was collected and clarified lysate was incubated with equilibrated beads for 1–2 hrs at 4°C. Beads were then washed with ice cold TBST Wash buffer 3 to 5 times. Bound proteins were eluted with 2x LDS lysis buffer (Thermo Fisher) in boiling conditions. For GFP-Trap, cells were then lysed using ice cold lysis buffer (10 mM Tris/Cl pH 7.5, 150 mM NaCl, 0.5 mM EDTA, 0.5 % NonidetTM P40 Substitute) supplemented with EDTA-free cOmplete protease inhibitor (Roche). Samples were incubated on ice with intermittent agitation by pipetting for 30 min. Beads were equilibrated using bead dilution buffer (10 mM Tris/Cl pH 7.5, 150 mM NaCl, 0.5 mM EDTA). Protein lysates were precleared by centrifugation at 4°C for 10 min at 15,000 g. Clarified lysates were diluted using dilution buffer, 1% INPUT was collected and lysate was incubated with equilibrated beads for 1–2 hrs at 4°C. Beads were then washed with ice cold Wash buffer (10 mM Tris/Cl pH 7.5, 150 mM NaCl, 0.05 % NonidetTM P40 Substitute, 0.5 mM EDTA) 3 to 5 times. Bound proteins were eluted with 2x LDS lysis buffer (Thermo Fisher) in boiling conditions.
For GST pull down, cells were lysed (50mM TRIS-HCl 7.5, 150mM NaCl, 0.5% NP40, protease inhibitor). Samples were incubated at 4°C with constant agitation for 30 min. Protein lysates were precleared by centrifugation at 4°C for 10 min at 15,000 g. 1% INPUT was collected and clarified lysate was incubated with equilibrated beads overnight at 4°C with agitation. Beads were washed 5 times with wash buffer (20mM TRIS-HCl pH 7.5, 10mM EDTA, 5mM EGTA, 150mM NaCl, 1mM DTT). Bound proteins were eluted with 2x LDS lysis buffer (Thermo Fisher) in boiling conditions.
Live cell imaging
Cells seeded into a 96 well plate (MGB096–1-2-LG-L) were imaged in DMEM on a Nikon Ti2-E microscope with a CSU-W1 spinning disk, Hamamatsu ORCA-FLASH 4.0 sCMOS camera, and 20x objective (NA 0.75) at 37°C, 5% CO2, and humidity. Twelve sites were imaged per well every hour for 24 hours in triplicate wells.
Immunofluorescence microscopy
Cells seeded in 12 well plates with a 1.5mm pre-treated coverslip (neuVitro) were treated as indicated in figure legends. After treatment, cells were rinsed in PBS and fixed with 4% paraformaldehyde at RT for 10 min. Cells were washed with PBS. For immunostaining, cells were first permeabilized with 0.5% Triton X-100 for 10 min at RT and and blocked with 3% goat serum in PBS for 1 h at RT. After, cells were incubated with 0.1% Triton X-100, 3% goat serum in PBS supplemented with antibodies (1:200) 1 h at room temperature or overnight at 4°C. Cells were then washed with PBST (PBS + 0.1% Triton X-100) 3 times and incubated with Alexa 505, 488 or 594-conjugated secondary antibodies (Thermo Fisher). For cells expressing fluorescent tagged proteins, cells were seeded as above. After treatments, cells were fixed as above and washed 3 times with PBS prior to image analysis. Images were taken using 63x oil, DIC objective on an LSM 880 Airyscan microscope (Zeiss).
For quantitative measurement, cells were seeded in a 96 well plate, fixed, blocked and immunostained as described above and imaged on the Nikon spinning disk listed above using a 40-water objective (NA 1.15); For the ubiquitin puncta experiments, sixteen sites were imaged per well in triplicate wells. For recruitment experiments, nine sites were imaged per well in triplicate wells.
Mass spectrometry
After samples were bound to magnetic HA-beads as described above, samples were eluted with elution buffer (2mg/ml HA peptide in TBS) according to manufacturer’s instructions, followed by TCA precipitation. Briefly, 100% TCA was added to eluted peptides (f.c. 20% TCA), vortexed and incubated on ice for 30min before centrifugation. Precipitated peptides were washed with 10% ice cold TCA, followed by 4 washes with ice cold acetone. The tubes were left to air dry before trypsin digestion. Briefly, peptides were re-suspended with solubilizing buffer (5% SDS, 8M urea, 50mM TEAB, pH7.6). DTT was added for f.c. 5mM before incubating at 50°C for 30min. MMTS was added for 45min before adding an aqueous phosphoric acid solution. Meanwhile, S-Trap columns (Protifi) were conditioned with binding buffer (90% methanol, 100mM TEAB, pH7.1). 1μg of trypsin was added to the acidified protein mixture and immediately added into the column containing binding buffer. Centrifugation and washing steps were performed before adding digestion buffer (Trypsin in 50mM TEAB) and incubation at 37°C overnight. Columns were rehydrated with 50mM TEAB before elution using 0.2% formic acid and another elution using 50% acetonitrile containing 0.2% formic acid.
Tryptic digests were analyzed using an orbitrap Fusion Lumos tribrid mass spectrometer interfaced to an UltiMate3000 RSLC nano HPLC system (Thermo Scientific) using data dependent acquisition. Initial protein identification was carried out using Proteome Discoverer (V2.4) software (Thermo Scientific). Search results from Proteome Discoverer were incorporated into Scaffold4 for relative quantification using spectral counting. Samples were compared to ~100 reference IPs using a Java script programmed according to the CompPASS software suite as previously described33,34 and also compared to the CRAPome43. For determination of the TNIP1 interaction network, thresholds for high confidence interaction partners (HCIPs) were top 5% of interactors with highest Z-score and lowest abundance in the CRAPome. Cytoscape was used to visualize the TNIP1 interaction network. To compare WT TNIP1 with TNIP1ΔAHD3 and TNIP1ΔAHD4, total spectral counts for each interactor were first normalized to 1000 and then expressed relative to WT/controls in fold-change (log2) and plotted as heatmap using GraphPad.
Mitophagy assay with mito-mKeima via fluorescence activated cytometer
Stable cell lines were generated to express mito-mKeima, HA-Parkin, FRB-Fis1 and FKBP-GFP-tagged target genes with lentivirus system. 200K cells were seeded in 12-well plates and treated with Rapalog for 24h or by OA at various time points before FACS analysis. Quantification of mtKeima ratio (561/488) were performed as previously described24,26. Briefly, Measurements of lysosomal mt-mKeima were made using dual-excitation ratiometric pH measurements at 488 (pH 7) and 561 (pH 4) nm lasers with 620/29 nm and 614/20 nm emission filters, respectively. For each sample, 50,000 events were collected and subsequently gated for YFP/mt-mKeima doublepositive cells that were DAPI-negative. Data were analysed using FlowJo
Dynamic Light Scattering and Nano Differential Scanning Fluorimetry/Turbidity
Purified full-length TNIP1 and FIP200-CLAW domain at 1 mg/ml in GF buffer (150mM KCl, 20 mM HEPES pH 7.4, 1mM MgCl) were mixed at equal volumes with each other or more GF buffer, or together with the addition of 100uM CCPG1 pFIR peptides. Solutions were incubated at room temperature for 20 minutes before being measured for DLS and nanoDSF for turbidity using a Prometheus Panta (NanoTemper Technologies) in high-sensitivity mode. Experiments were performed from 25–95°C, in 3–4 independent replicates. Data was analyzed in Prometheus Panta analysis software v1.4.2 (Nanotemper Technologies).
Fluorescence polarization
For binding affinity measurements, purified FIP200-CLAW protein was diluted in series with FP-assay buffer (150mM KCl, 20 mM HEPES pH 7.4, 1mM MgCl, and 0.05% Tween-20). Next the protein was mixed 1:1 with FITC-tagged peptides at ~625nM. A set of solutions with each peptide alone (No FIP200-CLAW) was used to normalize FP-measurements. For competition assays, FIP200-CLAW at 4uM and 625nM FITC-CCPG1-pFIR in FP-assay buffer were mixed with increasing concentrations of unlabelled TNIP1 peptides. The mixtures were allowed to incubate 20 minutes at room temperature before being measured in triplicate with a Synergy Neo2 plate reader (BioTek) with FITC-Fluorescence Polarization filter set. Delta mP was calculated by subtracting each measurement from the appropriate peptide-alone control. Concentration vs Delta mP curves were fit for each experiment with a one-site binding model, with the mean ± stand deviation for KD calculated from 3–6 independent experiments. Competition experiments were fit with a one-site competition model.
Image Analysis
Image analysis was performed using custom MATLAB scripts for each experiment. All images were background corrected then cells were segmented based on intensity and size. TNIP1 overexpressing cells were further segmented by BFP signal. For autophagy flux, the average YFP and RFP signal was calculated within the cell area mask for each site. The ratio of YFP over RFP signal was calculated for each site then the median value of all sites assessed for each timepoint. Ubiquitin puncta were segmented by combining multiple masks generated from local background subtraction, high pass filtration, and high intensity then filtered by size. For recruitment experiments, to find only GFP-TNIP1 positive cells, single cells were segmented from each other with a water shedding algorithm, then cells with a total GFP intensity above a manual threshold selected for the cell mask, and finally a Pearson’s Correlation Coefficient calculated between GFP-TNIP1 and either LC3B, GABARAP, or FIP200 within the cell mask.
Quantification and Statistical Analysis
All statistical analyses for FACS were calculated in GraphPad Prism 9. Error bars are expressed as mean ± standard deviation. P values were obtained using a two-way ANOVA with multiple comparison. 0.12 (ns), 0.03 (*), 0.002 (**), <0.001 (***).
Supplementary Material
Table S1: CompPASS analysis of TNIP1 interactors, related to Figure 4.
Highlight:
TNIP1 possesses a LIR motif.
TNIP1 endogenously inhibits mitophagy.
TNIP1 interacts with TAX1BP1 and FIP200.
Phosphorylation by TBK1 regulate TNIP1’s function.
Acknowledgement
The authors would like to thank the laboratory of Michael Lazarou (Monash Biomedicine Discovery Institute) for sharing the LC3/GABARAP KO cells. We would also like to thank all other members of the Youle lab for sharing cell lines and reagents as well as discussions and advice. We further thank Dr Yan Wang from the National Institute of Dental and Craniofacial Research Mass Spectrometry Facility (ZIA DE00075) for performing mass spectrometry analysis. FACS data were acquired using the NINDS facility under the supervision of Dr. Dragan Maric. This work was supported by the Intramural Program of the NINDS and the NIDCR, NIH.
Inclusion and Diversity
We support inclusive, diverse, and equitable conduct of research.
Footnotes
Declaration of Interest
The authors declare no financial conflicts and assure that this manuscript is original and has not been published nor is currently under consideration for publication elsewhere.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Mizushima N. (2020). The ATG conjugation systems in autophagy. Curr. Opin. Cell Biol 63, 1–10. 10.1016/J.CEB.2019.12.001. [DOI] [PubMed] [Google Scholar]
- 2.Conway O, Akpinar HA, Rogov V, and Kirkin V. (2019). Selective autophagy receptors in neuronal health and disease. J. Mol. Biol 10.1016/J.JMB.2019.10.013. [DOI] [PubMed] [Google Scholar]
- 3.Gatica D, Lahiri V, and Klionsky DJ (2018). Cargo recognition and degradation by selective autophagy. Nat. Cell Biol 20, 233–242. 10.1038/s41556-018-0037-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Khaminets A, Behl C, and Dikic I. (2016). Ubiquitin-Dependent And Independent Signals In Selective Autophagy. Trends Cell Biol. 26, 6–16. 10.1016/j.tcb.2015.08.010. [DOI] [PubMed] [Google Scholar]
- 5.Vargas JNS, Hamasaki M, Kawabata T, Youle RJ, and Yoshimori T. (2022). The mechanisms and roles of selective autophagy in mammals. Nat. Rev. Mol. Cell Biol, 1–19. 10.1038/s41580-022-00542-2. [DOI] [PubMed] [Google Scholar]
- 6.Padman BS., Nguyen TN., Uoselis L., Skulsuppaisarn M., Nguyen LK., and Lazarou M. (2019). LC3/GABARAPs drive ubiquitin-independent recruitment of Optineurin and NDP52 to amplify mitophagy. Nat. Commun 10, 408. 10.1038/s41467-019-08335-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vargas JNS, Wang C, Bunker E, Hao L, Maric D, Schiavo G, Randow F, and Youle RJ (2019). Spatiotemporal Control of ULK1 Activation by NDP52 and TBK1 during Selective Autophagy. Mol. Cell 10.1016/J.MOLCEL.2019.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mauro C, Pacifico F, Lavorgna A, Mellone S, Iannetti A, Acquaviva R, Formisano S, Vito P, and Leonardi A. (2006). ABIN-1 binds to NEMO/IKKgamma and co-operates with A20 in inhibiting NF-kappaB. J. Biol. Chem 281, 18482–18488. 10.1074/jbc.M601502200. [DOI] [PubMed] [Google Scholar]
- 9.Cohen S, Ciechanover A, Kravtsova-Ivantsiv Y, Lapid D, and Lahav-Baratz S. (2009). ABIN-1 negatively regulates NF-κB by inhibiting processing of the p105 precursor. Biochem. Biophys. Res. Commun 389, 205–210. 10.1016/J.BBRC.2009.08.074. [DOI] [PubMed] [Google Scholar]
- 10.G’Sell RT, Gaffney PM, and Powell DW (2015). Review: A20-Binding Inhibitor of NF-κB Activation 1 Is a Physiologic Inhibitor of NF-κB: A Molecular Switch for Inflammation and Autoimmunity. Arthritis Rheumatol. 67, 2292–2302. 10.1002/art.39245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee Y, Kim J, Kim M-S, Kwon Y, Shin S, Yi H, Kim H, Chang MJ, Chang CB, Kang S-B, et al. (2021). Coordinate regulation of the senescent state by selective autophagy. Dev. Cell 10.1016/J.DEVCEL.2021.04.008. [DOI] [PubMed] [Google Scholar]
- 12.Ramirez VP, Gurevich I, and Aneskievich BJ (2012). Emerging roles for TNIP1 in regulating post-receptor signaling. Cytokine Growth Factor Rev. 23, 109–118. 10.1016/J.CYTOGFR.2012.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shamilov R, and Aneskievich BJ (2018). TNIP1 in Autoimmune Diseases: Regulation of Toll-like Receptor Signaling. J. Immunol. Res 2018, 1–13. 10.1155/2018/3491269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Benyamin B, He J, Zhao Q, Gratten J, Garton F, Leo PJ, Liu Z, Mangelsdorf M, Al-Chalabi A, Anderson L, et al. (2017). Cross-ethnic meta-analysis identifies association of the GPX3-TNIP1 locus with amyotrophic lateral sclerosis. Nat. Commun 8, 611. 10.1038/s41467-017-00471-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.van Rheenen W, van der Spek RAA, Bakker MK, van Vugt JJFA, Hop PJ, Zwamborn RAJ, de Klein N, Westra H-J, Bakker OB, Deelen P, et al. (2021). Common and rare variant association analyses in amyotrophic lateral sclerosis identify 15 risk loci with distinct genetic architectures and neuron-specific biology. Nat. Genet 53, 1636–1648. 10.1038/s41588-021-00973-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wightman DP., Jansen IE., Savage JE., Shadrin AA., Bahrami S., Holland D., Rongve A., Børte S., Winsvold BS., Drange OK., et al. (2021). A genome-wide association study with 1,126,563 individuals identifies new risk loci for Alzheimer’s disease. Nat. Genet 53, 1276–1282. 10.1038/s41588-021-00921-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Herhaus L, van den Bedem H, Tang S, Maslennikov I, Wakatsuki S, Dikic I, and Rahighi S. (2019). Molecular Recognition of M1-Linked Ubiquitin Chains by Native and Phosphorylated UBAN Domains. J. Mol. Biol 10.1016/J.JMB.2019.06.012. [DOI] [PubMed] [Google Scholar]
- 18.Wagner S, Carpentier I, Rogov V, Kreike M, Ikeda F, Löhr F, Wu C-J, Ashwell JD, Dötsch V, Dikic I, et al. (2008). Ubiquitin binding mediates the NF-κB inhibitory potential of ABIN proteins. Oncogene 27, 3739–3745. 10.1038/sj.onc.1211042. [DOI] [PubMed] [Google Scholar]
- 19.Zellner S, Schifferer M, and Behrends C. (2021). Systematically defining selective autophagy receptor-specific cargo using autophagosome content profiling. Mol. Cell 10.1016/J.MOLCEL.2021.01.009. [DOI] [PubMed] [Google Scholar]
- 20.Johansen T, and Lamark T. (2019). Selective Autophagy: ATG8 Family Proteins, LIR Motifs and Cargo Receptors. J. Mol. Biol 10.1016/J.JMB.2019.07.016. [DOI] [PubMed] [Google Scholar]
- 21.Kalvari I, Tsompanis S, Mulakkal NC, Osgood R, Johansen T, Nezis IP, and Promponas VJ (2014). iLIR: A web resource for prediction of Atg8-family interacting proteins. Autophagy 10, 913–925. 10.4161/auto.28260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pankiv S, Clausen TH, Lamark T, Brech A, Bruun J-AA, Outzen H, Øvervatn A, Bjørkøy G, and Johansen T. (2007). p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem 282, 24131–24145. 10.1074/jbc.M702824200. [DOI] [PubMed] [Google Scholar]
- 23.Goold R, McKinnon C, Rabbanian S, Collinge J, Schiavo G, and Tabrizi S. (2013). The LIR motif – crucial for selective autophagy. J Cell Sci 126, 3237–3247. 10.1242/jcs.126128. [DOI] [PubMed] [Google Scholar]
- 24.Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang C, Burman JL, Sideris DP, Fogel AI, and Youle RJ (2015). The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. 524, 309–314. 10.1038/nature14893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bayle JH, Grimley JS, Stankunas K, Gestwicki JE, Wandless TJ, and Crabtree GR (2006). Rapamycin Analogs with Differential Binding Specificity Permit Orthogonal Control of Protein Activity. Chem. Biol 13, 99–107. 10.1016/J.CHEMBIOL.2005.10.017. [DOI] [PubMed] [Google Scholar]
- 26.Wang C. (2020). A Sensitive and Quantitative mKeima Assay for Mitophagy via FACS. Curr. Protoc. Cell Biol 86. 10.1002/cpcb.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nanda SK, Venigalla RKC, Ordureau A, Patterson-Kane JC, Powell DW, Toth RC, Arthur JS, and Cohen P. (2011). Polyubiquitin binding to ABIN1 is required to prevent autoimmunity. J. Exp. Med 208, 1215–1228. 10.1084/jem.20102177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Narendra D, Kane LA, Hauser DN, Fearnley IM, and Youle RJ (2010). p62/SQSTM1 is required for Parkin-induced mitochondrial clustering but not mitophagy; VDAC1 is dispensable for both. Autophagy 6, 1090–1106. 10.4161/auto.6.8.13426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kaizuka T, Morishita H, Hama Y, Tsukamoto S, Matsui T, Toyota Y, Kodama A, Ishihara T, Mizushima T, and Mizushima N. (2016). An Autophagic Flux Probe that Releases an Internal Control. Mol. Cell 64, 835–849. 10.1016/J.MOLCEL.2016.09.037. [DOI] [PubMed] [Google Scholar]
- 30.Sarraf SA, Shah HV, Kanfer G, Pickrell AM, Holtzclaw LA, Ward ME, and Youle RJ (2020). Loss of TAX1BP1-Directed Autophagy Results in Protein Aggregate Accumulation in the Brain. Mol. Cell 10.1016/J.MOLCEL.2020.10.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Allen GFG, Toth R, James J, and Ganley IG (2013). Loss of iron triggers PINK1/Parkin-independent mitophagy. EMBO Rep. 14, 1127–1135. 10.1038/embor.2013.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wilhelm LP., Zapata‐Muñoz J., Villarejo‐Zori B., Pellegrin S., Freire CM., Toye AM., Boya P., and Ganley IG. (2022). BNIP3L / NIX regulates both mitophagy and pexophagy. EMBO J., e111115. 10.15252/embj.2022111115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Werner A, Iwasaki S, McGourty C, Medina-Ruiz S, Teerikorpi N, Fedrigo I, Ingolia N, and Rape M. (2015). Cell-fate determination by ubiquitin-dependent regulation of translation. Nature 525, 523–527. 10.1038/nature14978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sowa ME, Bennett EJ, Gygi SP, and Harper WJ (2009). Defining the Human Deubiquitinating Enzyme Interaction Landscape. 138, 389–403. 10.1016/j.cell.2009.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gao L, Coope H, Grant S, Ma A, Ley SC, and Harhaj EW (2011). ABIN1 Protein Cooperates with TAX1BP1 and A20 Proteins to Inhibit Antiviral Signaling *. Publ. JBC Pap. Press. 10.1074/jbc.M111.283762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Turco E, Witt M, Abert C, Bock-Bierbaum T, Su M-Y, Trapannone R, Sztacho M, Danieli A, Shi X, Zaffagnini G, et al. (2019). FIP200 Claw Domain Binding to p62 Promotes Autophagosome Formation at Ubiquitin Condensates. Mol. Cell 10.1016/J.MOLCEL.2019.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhou Z, Liu J, Fu T, Wu P, Peng C, Gong X, Wang Y, Zhang M, Li Y, Wang Y, et al. (2021). Phosphorylation regulates the binding of autophagy receptors to FIP200 Claw domain for selective autophagy initiation. Nat. Commun 12, 1570. 10.1038/s41467-021-21874-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhou J, Rasmussen NL, Olsvik HL, Akimov V, Hu Z, Evjen G, Kaeser-Pebernard S, Sankar DS, Roubaty C, Verlhac P, et al. (2023). TBK1 phosphorylation activates LIR-dependent degradation of the inflammation repressor TNIP1. J. Cell Biol 222. 10.1083/jcb.202108144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shinkawa Y, Imami K, Fuseya Y, Sasaki K, Ohmura K, Ishihama Y, Morinobu A, and Iwai K. (2022). ABIN1 is a signal‐induced autophagy receptor that attenuates NF‐κB activation by recognizing linear ubiquitin chains. FEBS Lett. 10.1002/1873-3468.14323. [DOI] [PubMed] [Google Scholar]
- 40.Merline R, Rödig H, Zeng-Brouwers J, Poluzzi C, Tascher G, Michaelis J, LopezMosqueda J, Rhiner A, Huber LS, Diehl V, et al. (2022). A20 binding and inhibitor of nuclear factor kappa B (NF-κB)-1 (ABIN-1) - a novel modulator of mitochondrial autophagy. Am. J. Physiol. Physiol 10.1152/ajpcell.00493.2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bingol B, Tea J, Phu L, Reichelt M, Bakalarski C, Song Q, Foreman O, Kirkpatrick D, and Sheng M. (2014). The mitochondrial deubiquitinase USP30 opposes parkin-mediated mitophagy. Nature 510, 370–375. 10.1038/nature13418. [DOI] [PubMed] [Google Scholar]
- 42.Popelka H, and Klionsky DJ (2022). When acidic residues do not mimic phosphorylation: high-affinity binding of the reticulophagy receptor TEX264 to LC3/GABARAP. Autophagy 18, 2515–2518. 10.1080/15548627.2022.2119350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mellacheruvu D., Wright Z., Couzens AL., Lambert J-P., St-Denis NA., Li T., Miteva YV, Hauri S., Sardiu ME., Low TY., et al. (2013). The CRAPome: a contaminant repository for affinity purification–mass spectrometry data. Nat. Methods 10, 730–736. 10.1038/nmeth.2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1: CompPASS analysis of TNIP1 interactors, related to Figure 4.